First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about. Inspired by the MELiSSA project (Micro-Ecological Life Support System Alternative) from ESA, this project proposes an ecosystem composed of microorganisms and higher plants using their metabolic waste products as a substrate for the next compartment. This project is designed to study the behavior of artificial ecosystems and to develop the technologies required for future regenerative life-support systems in long-duration human space missions, such as lunar bases or missions to Mars. The system comprises five different compartments, each one colonized respectively by anoxygenic thermophilic bacteria, photoheterotrophic bacteria, nitrifying bacteria, photosynthetic bacteria, higher plants, and the human crew. I would like to conceptually integrate these microorganisms and higher plants with a plasmids-based control system, through the use of reporter genes and inducible regulatory elements. This would increase the security (allowing real-time monitoring of metabolics states, for example) and predictability of the system.
Week 02 - Lecture Questions Professor Jacobson The fidelity of DNA replication is governed by DNA polymerase and its associated repair systems. The intrinsic error rate of DNA polymerase, in the absence of proofreading, is approximately 10-4 to 10-5 per nucleotide. In eukaryotes, replicative polymerases utilize 3’ —} 5’ exonuclease activity for proofreading, which enhances fidelity to an error rate of approximately 10-7. When integrated with post-replicative mismatch repair (MMR) mechanisms, the effective error rate is further optimized to roughly 10-9 to 10-10 per nucleotide.Given that the human genome comprises approximately 3.2 x 109 base pairs, replication without these multi-layered fidelity mechanisms would result in a mutational load incompatible with cellular viability. Biological systems mitigate this risk through a hierarchy of safeguards—polymerase proofreading, mismatch repair, and various DNA damage response pathways—ensuring that the mutation rate per genome remains within a range that sustains evolutionary stability and life. A typical human protein consists of approximately 300 to 400 amino acids. Due to the degeneracy of the genetic code—where 64 codons encode 20 amino acids—the theoretical number of DNA sequences capable of encoding a single protein is exceptionally high. However, functional constraints significantly restrict this theoretical diversity. Key limiting factors include:
Week 03 - Python Script for Opentrons Artwork I was not able to write the code entirely by myself. The closest I got was generating concentric circles, wich reminded me of the Argentine “Escarapela” (with the help AI). My original idea, however, was to made an Argentine Mate which I did in https://opentrons-art.rcdonovan.com/ I also did a Cherry!
Week 04 - Part A: Conceptual Questions How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) 500 g of meat has more or less 22% of protein, so 500 g x 0.22 =110 g of protein
Average amino acid ≈ 100 Daltons and 1 Dalton ≈ 1 g/mol, so 100 Da≈100 g/mol, in order to convert grams of protein to moles of amino acids
Week 5 Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation. Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card: Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison. Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Week 6 — Genetic Circuits Part I: Assembly Technologies DNA Assembly Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The Phusion High-Fidelity PCR Master Mix contains several components: Phusion DNA polymerase → a high-fidelity enzyme that synthesizes DNA with very low error rates (With a failure rate 50 times lower than Taq and 6 times lower than Pfu, these polymerases are an excellent choice for cloning and other applications requiring high fidelity), which is critical when amplifying fragments of the amilCP gene. dNTPs (deoxynucleotide triphosphates) → building blocks for new DNA strands MgCl₂ → cofactor necessary for polymerase activity Buffer system → maintains optimal pH and ionic conditions These components work together to ensure accurate and efficient DNA amplification, also Phusion DNA polymerases offer robust performance with short protocol times, even in the presence of PCR inhibitors. They generate higher yields with less enzyme than other DNA polymerases. In this protocol, the master mix is used to amplify amilCP fragments that will later be assembled using Gibson Assembly. What are some factors that determine primer annealing temperature during PCR? Primer annealing temperature depends on: Primer length → longer primers have higher melting temperatures, GC content → higher GC increases stability and raises Tm. Higher melting temperatures are caused due to stronger hydrogen bonding. In this protocol, primers include additional overhangs (20–22 bp) for Gibson Assembly, but only the binding region determines the annealing temperature. The annealing temperature is typically set a few degrees below the melting temperature (Tm) to ensure specific binding. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other. In this protocol, PCR amplify specific regions of the amilCP gene, including mutated regions in the chromophore, allowing precise control over sequence design In contrast, restriction digestion (using PvuII) is used to linearize the pUC19 plasmid backbone. PCR is more flexible and allows introduction of mutations and overlaps, while restriction digestion relies on specific enzyme recognition sites. PCR is preferable for designing new constructs, whereas digestion is useful for preparing existing plasmid backbones.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Traditional genetic circuits primarily rely on Boolean logic (AND, OR, NOT gates), which results in “all-or-nothing” digital responses. Intracellular Artificial Neural Networks (IANNs) offer several distinct advantages:
Non-linear Signal Integration: Unlike Boolean gates that require strict thresholds, IANNs use activation functions (like Hill functions) to process analog chemical gradients, allowing for more nuanced environmental sensing. Weighted Inputs: IANNs allow for “tunable” inputs. By varying promoter strength or ribosome binding site (RBS) efficiency, the cell can assign different weights (w) to various biological signals, prioritizing one metabolite over another. Noise Filtering: Biological environments are inherently “noisy.” The summation and thresholding architecture of a perceptron acts as a natural buffer, preventing the circuit from misfiring due to minor stochastic fluctuations in gene expression. Computational Density: A single-layer IANN can perform complex classifications that would require a much larger and more metabolically taxing combination of traditional logic gates. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal. Application: An engineered E. coli strain that acts as a therapeutic diagnostic tool within the human gut.
Homework Part A: General and Lecturer-Specific Questions General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. The primary advantage lies in the decoupling of the reaction from cellular metabolism. Flexibility: It allows the use of linear DNA, eliminates the need for transformation and host-specific codon optimization, and facilitates the expression of proteins that are toxic to the host. Control of variables: It is an “open” system. You can manipulate buffer composition (pH, ionic strength), add chaperones, modify the Mg2+/K+ ratio, or add specific redox agents for disulfide bond formation in real-time, without the limitations of cellular homeostasis. Use cases: Toxic proteins: Production of proteins that compromise host viability (e.g., antimicrobial peptides or nucleases). Non-canonical amino acid (ncAA) incorporation: Facilitates genetic code expansion via stop codon suppression without competition from endogenous tRNAs.
Week 10 — Advanced Imaging & Measurement Technology Homework: Final Project For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements. What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail. Measurement and Validation Techniques for the Bio-Sticker Controlled Gas Exposure Assays The Bio-Sticker will first be tested in sealed exposure chambers containing precisely known concentrations of target toxic gases, such as ammonia or formaldehyde. These chambers allow accurate simulation of hazardous industrial environments while maintaining strict control over temperature, humidity, and gas concentration. By exposing the engineered fungal Bio-Sticker to increasing concentrations of the target analyte, we can determine its activation threshold, sensitivity, and dynamic range. This approach also enables the generation of dose-response curves, which are essential for calibrating the system and defining the concentration at which the color change becomes visible. Colorimetric Analysis The primary readout of the Bio-Sticker is the visible blue color produced by expression of the chromoprotein AmilCP. Colorimetric analysis will be used to quantify this response objectively. Images of the Bio-Sticker will be captured under standardized lighting conditions, and software such as ImageJ will be used to analyze changes in color intensity. Measurements will focus on RGB (red, green, blue) values and, when applicable, absorbance at the wavelength corresponding to AmilCP. This technique allows precise quantification of signal strength, comparison between samples, and monitoring of signal development over time. Digital Image Analysis In addition to simple colorimetric measurements, digital image processing will be employed to evaluate spatial uniformity, signal progression, and long-term stability of the color response. Time-course imaging can be used to track the kinetics of AmilCP expression after exposure to toxic gases. This enables measurement of response time, persistence of the signal, and any degradation or fading over extended periods. Such analyses are particularly important for assessing practical usability in field conditions. Polymerase Chain Reaction (PCR) PCR will be used to confirm successful integration of the engineered genetic circuit into the Aspergillus nidulans genome. Specific primers will be designed to amplify regions spanning the inserted construct and adjacent genomic sequences. Successful amplification of fragments of the expected size will verify the presence of the biosensing cassette. This serves as an initial molecular confirmation that the strain has been correctly engineered. DNA Sequencing Following PCR confirmation, DNA sequencing will be performed to verify the exact nucleotide sequence of the inserted construct. This step ensures that the promoter, sensing elements, reporter gene (AmilCP), and regulatory sequences have been integrated without mutations, deletions, or rearrangements. Sequence verification is critical to ensure that the genetic circuit will function as intended. Reverse Transcription Quantitative PCR (RT-qPCR) RT-qPCR will be used to measure transcriptional activation of the reporter gene after gas exposure. RNA will be extracted from the fungal cells before and after exposure to target gases, converted into complementary DNA (cDNA), and amplified using gene-specific primers. By comparing transcript levels under different conditions, this technique will quantify the extent to which the sensing circuit is activated. RT-qPCR provides highly sensitive, quantitative insight into gene expression dynamics. Spectrophotometry (Optional) Spectrophotometric analysis may be used to complement image-based measurements. Pigments extracted from fungal samples can be analyzed by measuring absorbance at wavelengths specific to AmilCP. This provides an additional quantitative assessment of chromoprotein production and can be particularly useful for validating colorimetric data. Specificity Testing To ensure selectivity, the Bio-Sticker will be exposed not only to target toxic gases but also to non-target compounds commonly present in industrial environments. By comparing responses across these conditions, we can determine whether the system selectively responds to the intended analyte or produces false positives. This is essential for establishing reliability in real-world applications. Stability and Shelf-Life Testing Long-term performance will be evaluated by monitoring the Bio-Sticker under different storage and environmental conditions. Parameters such as baseline color, response capability, and signal durability will be assessed over time. These studies will determine shelf life, operational stability, and robustness under field deployment conditions. Together, these techniques will provide a comprehensive characterization of the Bio-Sticker, from genetic validation to functional performance, ensuring that it operates as a reliable, low-cost, and easily interpretable biosensor for toxic gas detection in hazardous industrial environments. Homework: Waters Part I — Molecular Weight We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Week 11 — Bioproduction & Cloud Labs Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Unfortunately, I couldn’t contribute but I think it’s a great project that improves creativity and working in teams. The best part of it is there’s a contribution from all over the world. I think for next year we could have a more detailed explanation of the draw-to-made in order to create something specific but with different points of view. For example to create a plate to draw a bacteria and see what happens. I think this would be interesting.
Subsections of Homework
Week 1 HW: Principles and Practices
1) First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
Inspired by the MELiSSA project (Micro-Ecological Life Support System Alternative) from ESA, this project proposes an ecosystem composed of microorganisms and higher plants using their metabolic waste products as a substrate for the next compartment. This project is designed to study the behavior of artificial ecosystems and to develop the technologies required for future regenerative life-support systems in long-duration human space missions, such as lunar bases or missions to Mars.
The system comprises five different compartments, each one colonized respectively by anoxygenic thermophilic bacteria, photoheterotrophic bacteria, nitrifying bacteria, photosynthetic bacteria, higher plants, and the human crew.
I would like to conceptually integrate these microorganisms and higher plants with a plasmids-based control system, through the use of reporter genes and inducible regulatory elements. This would increase the security (allowing real-time monitoring of metabolics states, for example) and predictability of the system.
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
A) Safety
The goal is to guarantee that biotechnological systems used in closed life-support environments do not cause biological, ecological, or health-related harm.
Sub-goals:
-Biological control:
Establish that all microorganisms used in the system are strictly contained within closed bioreactors, with multiple physical and genetic safeguards to prevent unintended survival outside the system.
-Genetic stability and monitoring:
Ensure continuous monitoring protocols to detect mutations, horizontal gene transfer, or loss of function in engineered plasmids and microbial strains over long mission durations.
-Human health protection:
Assess and regulate potential risks to astronaut health, including allergenicity, toxin production, or unintended interactions with the human microbiome in confined environments.
B) Promote responsible and transparent use of synthetic biology
Goal: Ensure that the development of biotechnological life-support systems are governed transparently and responsibly.
Sub-goals:
-Ethical oversight and review:
Require interdisciplinary ethical review (including biologists, engineers, ethicists, and policymakers) before implementing genetically modified organisms in space missions.
-Clear responsibility and accountability:
Define who is responsible for the design, maintenance, and emergency response related to biotechnological failures during long-term missions.
-Open scientific communication:
Promote the publication and sharing of safety data, failures, and best practices to avoid repetition of risks and to foster responsible innovation in space biotechnology.
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Action: Ethical and biosafety protocols
Actors: Academic institutions & research ethics committees
Purpose:
This action proposes to develop a standardized requirement for ethical and biosafety review (chosen by researchers, universities and space agencies) before deploying or publishing biotechnological applications.
Design:
Universities and research institutions must require approval from ethics and biosafety committees. Funding agencies could condition grants on compliance. Researchers must submit risk assessments and mitigation plans.
Assumptions:
Assumes ethics committees have sufficient expertise and resources. Assumes researchers will comply honestly. Training and standardization significantly reduce human error.
Risks of Failure & “Success”:
Failure: Bureaucratic delays could slow innovation.
Success risk: Over-standardization may discourage exploratory or low-risk research.
Action: Incentives for safety-by-design practices
Actors: Biotech companies & funding bodies
Purpose:
Currently, safety features are often added after development. This action encourages integrating safety mechanisms from the design stage.
Design:
Grant programs, tax benefits, or certifications for companies that implement safety-by-design standards. Requires collaboration between engineers, biologists, and policymakers.
Assumptions:
Assumes financial incentives are strong enough to change behavior. Assumes safety-by-design standards can be clearly defined across technologies.
Risks of Failure & “Success”:
Failure: Incentives may be insufficient.
Success risk: Companies may focus on “checking boxes” rather than meaningful safety improvements.
Action: Controlled access and monitoring of biotechnological tools
Actors: Federal regulators & law enforcement
Purpose:
At present, access to certain tools or data may be insufficiently monitored. This action proposes tiered access controls to prevent misuse while allowing legitimate research.
Design:
Regulators define categories of risk. Developers implement user verification, logging, and auditing systems. Law enforcement intervenes only in cases of credible misuse.
Assumptions:
Assumes misuse can be detected through monitoring. Assumes access controls do not excessively burden legitimate users.
Risks of Failure & “Success”:
Failure: Overly strict controls may push users toward unregulated alternatives.
Success risk: Normalization of surveillance could raise privacy and academic freedom concerns.
Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
2
1
1
• By helping respond
2
1
1
Foster Lab Safety
• By preventing incident
1
1
1
• By helping respond
1
1
1
Protect the environment
• By preventing incidents
2
2
2
• By helping respond
3
3
1
Other considerations
• Minimizing costs and burdens to stakeholders
1
2
2
• Feasibility?
1
2
2
• Not impede research
1
1
1
• Promote constructive applications
1
1
1
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
The most important governance option for me would be a combination of the three, emphasizing “Safety-by-design” and “Ethical and biosafety protocols” supported by “Controlled access and monitoring” as a complementary safeguard.
For complex projects such as MELiSSA, it is not enough to have only one governance idea, we need some of them in order to control each step properly during the project.
Safety-by-design is important because it encourages the integration of biosafety from the beginning, for example through the use of plasmids-based mechanisms as a way to control the metabolic pathways in each step.
Ethical and biosafety protocols are more than just formalities; they are tools that ensure shared responsibility and protect scientific integrity through risk prevention and accountability mechanisms.
Prioritizing these governance actions required balancing competing interests. While ‘safety-by-design’ might delay early research and increase budgets, these trade-offs are necessary given the high stakes of life-support failures in space. This strategy relies on the assumption that institutional incentives work and that standards remain consistent across platforms. Despite lingering uncertainties about how space environments affect genetic stability, merging technical guardrails with institutional oversight creates a more resilient framework than relying on a single approach.
Target Audience: This proposal targets international bodies like NASA and ESA, which have the strategic power to align regulations and funding for space biotech.
Ethical Reflection: A core concern is accountability within semi-autonomous systems. In setups like MELiSSA, failures might stem from unpredictable biological behaviors rather than human oversight, blurring the lines of responsibility. Furthermore, we must prevent the ‘silent’ transfer of extreme bio-engineering to Earth without public oversight.
Proposed Actions: We need explicit accountability frameworks, scenario-based ethical reviews for off-Earth missions, and transparent protocols for knowledge sharing. These steps ensure that space biotech evolves safely and ethically."
Note: This assignment was developed with the assistance of an AI language model (ChatGPT, Gemini), used to help structure ideas and refine wording. The concepts and final decisions were critically reviewed and adapted by the author.
Week 2 HW: DNA read, write and edit.
Week 02 - Lecture Questions
Professor Jacobson
The fidelity of DNA replication is governed by DNA polymerase and its associated repair systems. The intrinsic error rate of DNA polymerase, in the absence of proofreading, is approximately 10-4 to 10-5 per nucleotide. In eukaryotes, replicative polymerases utilize 3’ —} 5’ exonuclease activity for proofreading, which enhances fidelity to an error rate of approximately 10-7. When integrated with post-replicative mismatch repair (MMR) mechanisms, the effective error rate is further optimized to roughly 10-9 to 10-10 per nucleotide.Given that the human genome comprises approximately 3.2 x 109 base pairs, replication without these multi-layered fidelity mechanisms would result in a mutational load incompatible with cellular viability. Biological systems mitigate this risk through a hierarchy of safeguards—polymerase proofreading, mismatch repair, and various DNA damage response pathways—ensuring that the mutation rate per genome remains within a range that sustains evolutionary stability and life.
A typical human protein consists of approximately 300 to 400 amino acids. Due to the degeneracy of the genetic code—where 64 codons encode 20 amino acids—the theoretical number of DNA sequences capable of encoding a single protein is exceptionally high.
However, functional constraints significantly restrict this theoretical diversity. Key limiting factors include:
-Codon Usage Bias: Variations in tRNA availability that influence translation efficiency.
-mRNA Secondary Structure: Folding patterns that may impede ribosome binding or elongation.
-GC Content: Extreme ratios that affect both sequence stability and the feasibility of synthesis.
-Regulatory Interference: The unintended presence of cryptic splice sites or premature termination signals.
-Metabolic Burden: High expression levels that may lead to cellular stress or protein misfolding.
Consequently, while the sequence space is vast, the biological context dictates a much narrower range of viable genetic sequences.
Dr. LeProust
Modern oligonucleotide synthesis primarily relies on solid-phase phosphoramidite chemistry. In this process, DNA is synthesized in the 3’ to 5’ direction through iterative cycles of deprotection, coupling, capping, and oxidation.Direct chemical synthesis is currently limited to approximately 150–200 nucleotides. This constraint arises because coupling efficiency is never 100%; as the sequence length increases, the yield of full-length, error-free molecules decreases exponentially. Furthermore, the accumulation of truncated products and point mutations makes the purification of long, high-fidelity oligonucleotides technically prohibitive.To produce longer sequences, such as a 2,000 bp gene, researchers must assemble multiple overlapping short oligonucleotides using enzymatic techniques like PCR assembly or Gibson assembly, followed by sequence verification and cloning.
Animals cannot synthesize certain amino acids de novo and must acquire them through their diet. The ten commonly recognized essential amino acids are: Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, Valine, and Arginine. Notably, lysine is universally essential across all animal species, representing a fundamental and highly conserved metabolic dependency.
George Church
Question 1: The “Lysine Contingency,” a biocontainment framework proposed by George Church, leverages the metabolic dependency on lysine to prevent the unintended proliferation of engineered organisms. By disabling endogenous lysine biosynthesis, the survival of the organism becomes contingent upon an external supply of this amino acid.
The universal necessity of lysine in animals reinforces the robustness of this strategy, as the evolutionary pressure to bypass such a deeply rooted biochemical constraint is significant. However, because many microorganisms possess the innate ability to synthesize lysine, effective biocontainment requires the knockout of redundant pathways and the implementation of multi-layered genetic safeguards. Thus, the lysine contingency is most effective when integrated into a broader, polygenic containment architecture rather than acting as a singular point of failure.
Week 2 - DNA Read, Write and Edit HM
Part 1: Benchling & In-silico Gel Art
By reordering restriction digest lanes of Lambda DNA, I created a symmetrical gel pattern resembling a butterfly!
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
Unfortunately No Lab Access
Part 3: DNA Design Challenge
3.1 Chosen Protein: GFP
I chose Green Fluorescent Protein (GFP) because it is widely used as a reporter protein in molecular biology. Since MELiSSA involves plasmid-based control systems and monitoring metabolic states, GFP represents a practical and symbolic example of how biological systems can be visually tracked in real time.
GFP was originally isolated from Aequorea victoria and is commonly used as a fluorescent marker in genetic engineering experiments.
Using UniProt, I obtained the amino acid sequence for GFP (UniProt ID: P42212).
Amino Acid Sequence: >sp|P42212|GFP_AEQVI Green fluorescent protein OS=Aequorea victoria
MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTT
LSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELK
GTDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIG
DGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK
3.2 Using an online reverse translation tool, I converted the GFP amino acid sequence into a possible coding DNA sequence. Because of codon degeneracy, multiple DNA sequences can encode the same protein. The sequence below represents one possible nucleotide sequence using standard codon usage.
One possible nucleotide sequence: ATGAGCAAAGGTGAAGAACTGTTTACCGGTGTTGTCCCAATTCTGGTTGAATTGGGTGATGGT
AATGGTCATAAATTTTCTGTCTCTGGCGGAGAAGGTGATGCTACCTATAAGCTGACACTGAAA
TTTATTTGCACCACTGGAAAATTGCCAGTTCCATGGCCAACACTGGTTACTACTCTGTCTTAT
GGTGTTCAGTGCTTCTCTCGCTACCCAGATCATATGAAACATGATTTTTTTAAATCTGCCATG
CCAGAGGGTTATGTTCAGGAGCGTACTATTTTTAAAGATGATGGTAATTATAAAACACGTGCT
GAAGTCAAATTTGAAGGTGATACACTGGTAAATCGCATTGAGCTGAAAGGTACCGACTTTAAG
GAAGATGGTAATATTCTGGGTCATAAACTGGAATACAATTATAACTCTCATAATGTCTATATT
ATGGCTGATAAACAGAAGAATGGTATTAAAGTTAATTTTAAAATTCGTCATAATATTGAAGAT
GGTTCTGTTCAGCTGGCTGATCACTACCAGCAGAATACTCCAATTGGAGATGGTCCTGTTCTG
CTGCCAGATAATCACTATCTGAGTACTCAGTCTGCTCTGTCTAAGGATCCAAATGAAAAGCGA
GATCATATGGTTCTGCTGGAATTTGTTACTGCTGCAGGTATTACCCATGGTATGGATGAGCTG
TATAAATAA
3.3 Codon optimization is important to improve protein development in the chosen host organism.
As we know, multiple DNA sequences can encode the same protein due to the degeneracy of the genetic code, but not all codons are used equally in all organisms. This is due to the abundance of tRNA pools.
If a gene contains codons that are rare for the organism, translation may decrease leading to slower protein production or ribosome stalling.
I optimized the codon sequence for Escherichia coli (E. coli) because it grows rapidly, it is inexpensive and has a fully sequenced and well-characterized genome.
Optimizing the gene for E. coli ensures that the codons match the organism’s tRNA abundance, thereby maximizing expression efficiency.
3.4 Cell-Free Protein Expression (In Vitro)
In this method:
The DNA template is added to a reaction mixture containing: RNA polymerase, ribosomes, tARNs, aminoacids, energy sources.
Transcription and translation occur in a test tube without living cells.
The protein is synthesized directly in vitro.
Advantages:
Faster expression
No need to maintain living cells
Useful for toxic proteins
More controllable environment
Limitations:
Higher cost
Typically lower yield than in vivo systems
How DNA Becomes A Protein?
In both systems (cell- dependent or cell-free), the process follows the Central Dogma:
DNA → mRNA → Protein
1)The DNA sequence is transcribed into messenger RNA (mRNA).
2)The ribosome reads the mRNA in codons (sets of three nucleotides).
3)Transfer RNAs (tRNAs) match each codon with the corresponding amino acid.
4)The amino acids are linked together to form a polypeptide chain in a specific site in the ribosome.
5)The polypeptide folds into a functional protein.
Part 4: Prepare a Twist DNA Synthesis Order
For this design, I prepared a linear expression cassette in Benchling containing: Constitutive promoter, ribosome Binding Site (RBS), start codon, codon-optimized GFP coding sequence, 6xHis tag, stop codon, T7 terminator
This cassette would be ordered as a clonal gene through Twist Bioscience.
I would select a high-copy plasmid backbone such as pTwist Amp High Copy, which provides: Ampicillin resistance for selection, high-copy origin of replication and efficient propagation in E. coli
Ordering as a clonal gene would allow direct transformation into E. coli without additional cloning steps, accelerating experimental validation.
Part 5: DNA Read/Write/Edit
5.1 DNA READ
(i) What DNA would you want to sequence and why?
I would like to sequence environmental microbial DNA from closed ecological life-support systems, such as bioreactors used in regenerative environments (similar to MELiSSA-type systems). Specifically, I would sequence microbial community DNA to monitor biodiversity, metabolic stability, and potential pathogenic shifts.
(ii) What sequencing technology would you use and why?
I would use a combination of:
Illumina provides high accuracy short reads, ideal for detecting small mutations and precise taxonomic profiling.
Oxford Nanopore provides long reads, which are useful for assembling genomes, detecting structural variants, and monitoring plasmids or gene clusters.
Using both increases robustness and ecological insight.
Preparation (Essential Steps)
DNA extraction from environmental sample
Fragmentation (if needed for Illumina)
Adapter ligation
PCR amplification (Illumina)
Library preparation
Loading onto flow cell
In closed systems, small microbial imbalances can lead to system instability or health risks. Sequencing allows early detection of contamination, horizontal gene transfer, or harmful mutations. Therefore, DNA sequencing becomes a tool for real-time biosurveillance and ecological control.
Essential Steps of Sequencing Technology
-Illumina (Second-generation)
• DNA fragments attach to flow cell
• Bridge amplification creates clusters
• Sequencing-by-synthesis with fluorescent reversible terminators
• Camera detects fluorescence
• Base calling via signal interpretation
Output:
Short reads (FASTQ files with quality scores)
-Oxford Nanopore (Third-generation)
• DNA passes through nanopore
• Changes in ionic current measured
• Signal processed into nucleotide sequence
Output:
Long reads (FASTQ, real-time data)
5.2 DNA WRITE
(i) What DNA would you want to synthesize and why?
I would synthesize a plasmid-based genetic circuit encoding:
• A fluorescent reporter (e.g., GFP)
• A stress-responsive promoter
• A regulatory element sensitive to metabolic imbalance
The purpose would be to create a biosensor that detects environmental stress inside a microbial ecosystem and produces a measurable fluorescence output.
This construct could function as an early warning system in closed bioreactors.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use commercial gene synthesis through Twist Bioscience
Why?
• High accuracy
• Scalable synthesis
• Codon optimization
• Assembly-ready fragments
Essential Steps of DNA Synthesis
Digital DNA design
Oligonucleotide synthesis
Assembly (e.g., Gibson assembly)
Sequence verification
Plasmid construction
Limitations
• GC-rich or repetitive sequences are difficult
• Length constraints
• Cost increases with size
• Biosecurity screening restrictions
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would edit the GFP gene expressed in E. coli to modify its fluorescence intensity. By introducing targeted mutations into the GFP coding sequence, it is possible to alter protein folding efficiency or chromophore structure, potentially enhancing fluorescence output. This modification would allow better signal detection and improved reporter performance in synthetic biology applications.
(ii) I would use CRISPR-Cas9 genome editing
CRISPR-Cas9 uses a guide RNA (gRNA) designed to match a specific DNA sequence within the GFP gene. The Cas9 enzyme introduces a double-strand break at that location. To introduce a precise modification, a donor DNA template containing the desired mutation would be supplied. The bacterial cell then repairs the break, incorporating the modified sequence.
Essential inputs include: Guide RNA targeting GFP, Cas9 nuclease (plasmid or protein form), Donor DNA template containing the intended mutation and
Competent E. coli cells
Limitations of this method include potential off-target effects, variable editing efficiency, and the need for downstream screening to confirm successful edits.
Note: This assignment was developed with the assistance of an AI language model (ChatGPT, Gemini), used to help structure ideas and refine wording. The concepts and final decisions were critically reviewed and adapted by the author.
Week 03 HW: Lab Automation
Week 03 - Python Script for Opentrons Artwork
I was not able to write the code entirely by myself. The closest I got was generating concentric circles, wich reminded me of the Argentine “Escarapela” (with the help AI).
My original idea, however, was to made an Argentine Mate which I did in https://opentrons-art.rcdonovan.com/
I also did a Cherry!
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Case Study: Automation in Drug Discovery
Paper Title: Improving an Open-Sourced Automated Microplate Assay for our Drug Discovery Process
Authors: M. Yunos Alizai, Brianna N. Davis, and Paul H. Davis (University of Nebraska at Omaha).
In order to discover new medicines (mainly against infections), scientifics must try hundreds of chemical compounds against different pathogens and cells. These assays are performed using manual microplate techniques, which are labor-intensive and highly susceptible to user-associated variations and human error, limiting the speed and the reliability of the drug discovery process.
The solution? In this paper the authors developed an automated wide-spectrum screening assay utilizing the Opentrons liquid handling platform. The robot was programmed to automate the preparation of microplate assays, handling precise liquid transfers for:
a-Compound Screening: Rapidly evaluating the effectiveness of various substances against specific pathogens.
b-Cytotoxicity Testing: Measuring the impact of these compounds on host cell metabolism to determine potential toxicity.
The significance of this study lies in the optimization of an open-source tool to achieve high-throughput screening (HTS) capabilities that were previously reserved for labs with much more expensive, proprietary equipment. Key achievements described in the paper include:
-Scalability: The ability to process a significantly larger number of samples in a reduced timeframe.
-Precision: A marked reduction in human-induced variability, leading to more reproducible data.
-Feasibility: Proving that open-source automation is a robust and viable tool for complex clinical applications in combating infections
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details
Final Project Proposal: Plasmid-Based Autonomous Control Loops for the MELiSSA Ecosystem
The goal of this project is to implement an autonomous biological regulation system within the MELiSSA (Micro-Ecological Life Support System Alternative) framework. By engineering specific plasmids to act as “genetic controllers,” we can regulate metabolic flux and resource production in response to environmental fluctuations (such as CO2 levels or nutrient concentration). This ensures the stability of the artificial ecosystem during long-term space missions.
A central component of this project is the use of GFP (Green Fluorescent Protein) as a reporter. The plasmids will be designed with sensor-promoter systems that trigger GFP expression when specific conditions are met (e.g., a stress-induced promoter).
a) Real-time Monitoring: The fluorescence intensity will serve as a direct proxy for the “health” of a specific compartment (like the cyanobacteria loop).
b) Feedback Loop: Automation tools will be used to measure this fluorescence. If the signal deviates from the setpoint, the system can automatically trigger a corrective action, such as adjusting the flow of nutrients or light intensity.
Automation Tools
The complexity of characterizing these genetic circuits requires high-throughput automation:
a) Opentrons Platform: The OT-2 will be utilized to automate the DNA Assembly (Golden Gate or Gibson Assembly) of the plasmid variants. It will also handle the transformation protocols, ensuring high reproducibility when inserting these controllers into the target microbial strains.
b) Custom 3D-Printed Hardware: To bridge the gap between automation and biology, I will design and 3D-print custom modular tube holders and adapters. These will allow the Opentrons to interface directly with specialized bioreactor sampling tubes, maintaining the required thermal conditions for sensitive enzymes and reagents.
c) Ginkgo Nebula Integration: For large-scale characterization, Ginkgo Nebula will be used to test the plasmids under a vast array of simulated space environments. This high-throughput data will allow for the fine-tuning of the genetic “gain” and “sensitivity” of the controllers before they are deployed in a physical MELiSSA prototype.
By replacing electronic sensors with biological ones (plasmids + GFP), we reduce the reliance on external hardware that can fail in deep space. This “living” control system makes the MELiSSA loop more resilient, self-healing, and inherently integrated into the biological processes it aims to sustain.
Week 4 HW: Protein Design - Part I
Week 04 - Part A: Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
500 g of meat has more or less 22% of protein, so 500 g x 0.22 =110 g of protein
Average amino acid ≈ 100 Daltons and 1 Dalton ≈ 1 g/mol, so 100 Da≈100 g/mol, in order to convert grams of protein to moles of amino acids
110 g % 100 g/mol = 1.1 moles of amino acids in 500 g of meat
To convert moles to number of molecules
Use Avogadro’s number: 6.022×1023 molecules/mol
1.1 moles of amino acids × (6.022×1023) amino acids molecules≈ 6.6×10^23 (600 sextillion amino acids)
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When we eat beef or fish, we do not incorporate their proteins intact. Our digestive enzymes break them into amino acids that enter our metabolic pool.
Our ribosomes synthesize human proteins coded in our DNA sequences
So we recycle the amino acids, not the structure or identity of the organism.
Biological identity is encoded in genetic information, not in dietary proteins.
Why are there only 20 natural amino acids?
The canonical genetic code uses 20 amino acids because evolution optimized for:
Chemical diversity (hydrophobic, polar, charged, aromatic, special cases like Gly and Pro)
Translational efficiency
Error minimization
Adding more amino acids increases: Complexity of aminoacyl-tRNA synthetases and risk of translational errors
There are actually two additional genetically encoded amino acids:
Selenocysteine (21st)
Pyrrolysine (22nd)
But they require specialized insertion machinery.
Evolution settled on 20 as a balance between chemical versatility and system simplicity.
Where did amino acids come from before enzymes and before life?
Several hypotheses:
Atmospheric synthesis
The classic 1953 experiment by Stanley Miller and Harold Urey simulated early Earth conditions and produced amino acids from simple gases and electrical sparks.
Extraterrestrial delivery
The Murchison meteorite contained over 70 amino acids.
Hydrothermal vent chemistry
Mineral-catalyzed reactions at deep-sea vents could generate organic molecules.
Before enzymes, amino acids formed via abiotic chemistry driven by energy sources like UV radiation, lightning, or geothermal heat.
If you make an α-helix using D-amino acids, what handedness would you expect?
Natural proteins use L-amino acids and form right-handed α-helices.
If you build a protein entirely from D-amino acids:
→ The chirality inverts
→ You obtain a left-handed α-helix
Helix handedness is dictated by the stereochemistry of the α-carbon.
Can you discover additional helices in proteins?
Yes. Besides the α-helix, known helices include:
3₁₀ helix
π-helix
Polyproline helix
With non-natural amino acids, we could theoretically design: tighter helices, helices with internal charge networks or metal-stabilized helices
The constraints are geometric (bond angles, sterics) and thermodynamic (free energy minimization).
Why are most molecular helices right-handed?
Because biological proteins are built from L-amino acids.
The stereochemistry of L-amino acids restricts backbone dihedral angles (φ and ψ) such that the energetically favored α-helix is right-handed.
If life had evolved using D-amino acids, helices would predominantly be left-handed.
Molecular chirality propagates upward into macroscopic structure.
Why do β-sheets tend to aggregate?
β-strands expose backbone hydrogen bond donors and acceptors.
When proteins partially unfold:
These groups seek new hydrogen bonding partners.
Intermolecular β-sheet formation occurs.
Extended networks form between molecules.
Additionally:
Alternating hydrophobic side chains promote stacking.
β-sheets are inherently “sticky” when exposed.
a) What is the driving force for β-sheet aggregation?
The main driving forces are:
Intermolecular hydrogen bonding
Hydrophobic interactions
Entropic gain from water release
Formation of extended β-sheet networks lowers free energy.
Aggregation is often thermodynamically favorable once nucleation begins.
Why do many amyloid diseases form β-sheets?
In diseases like Alzheimer’s disease:
Proteins misfold.
Normally buried β-prone sequences become exposed.
They assemble into extended β-sheets.
These stack into amyloid fibrils.
β-sheet architecture allows: Extremely stable cross-β structures, template-based propagation and resistance to degradation
β-sheets represent a deep energy minimum in protein conformational space.
a) Can amyloid β-sheets be used as materials?
Yes! This is a growing area in biomaterials science.
Amyloid fibrils have:
High tensile strength
Self-assembly properties
Chemical stability
Applications include:
Tissue engineering scaffolds
Nanofibers
Biocompatible materials
Conductive biomaterials
The same structural features that cause disease can be harnessed for design.
Part B: Protein Analysis and Visualization
Protein Choice: Human Adenylate Cyclase Type 5 (ADCY5)
Organism: Homo sapiens
UniProt ID: O95622
For easier structural analysis in PyMol, I chose to use the catalytic domain structure, a classic solved structure is: 1CJK
This is the catalytic core of mammalian adenylyl cyclase in complex with Gsα.
Briefly describe the protein you selected and why you selected it.
Adenylate cyclase (AC) is the enzyme that converts:
ATP → cAMP + PPi
cAMP is a second messenger that regulates:
PKA
Ion channels
Gene transcription (CREB pathway)
I selected adenylate cyclase because: It is central to signal transduction, links extracellular signals to intracellular responses, is regulated by G proteins (GPCR signaling) and its catalytic mechanism is structurally well characterized.
Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids. How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs. Does your protein belong to any protein family?
Length: 1261 amino acids (If analyzing only catalytic domain → ~400 residues)
It is a large membrane protein with:
2 transmembrane domains
2 cytosolic catalytic domains (C1 and C2)
Most frequent amino acid: Most frequent: L (129 times)
Homologs? 250 results found in UniProtKB
Adenylate cyclases exist in: Mammals, Insects, Fungi and Bacteria (structurally different class)
You can safely state:
UniProt BLAST reveals thousands of homologous sequences across eukaryotic organisms, reflecting the conserved role of cAMP signaling in evolution.
Family
It belongs to:
Adenylate cyclase family
Nucleotide cyclase superfamily
Class III adenylate cyclases (in mammals)
Class III ACs are evolutionarily conserved catalytic enzymes.
RCSB structure page:
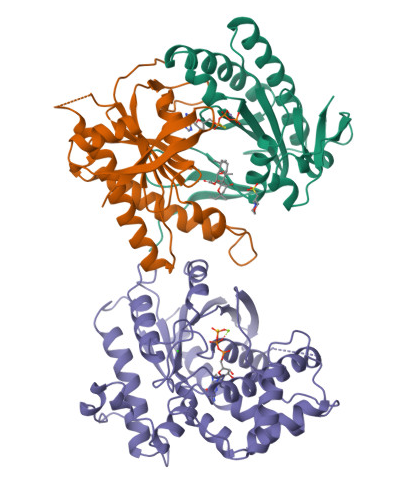
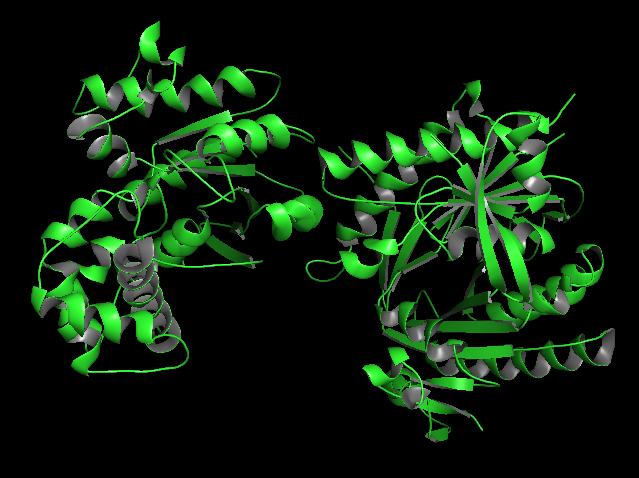
In the structure, three main components can be identified:
At the top, the purple chain corresponds to the regulatory G protein subunit (Gsα).
Below, the green and orange chains represent the two catalytic domains (C1 and C2) of adenylyl cyclase.
In the center of the complex, small molecules can be observed, corresponding to ATP (or an ATP analog) and associated magnesium ions (Mg²⁺), which are required for catalytic activity.
For structural and functional analysis, the most relevant region is the complex formed by the green and orange domains. These two domains together constitute the catalytic core of adenylyl cyclase. The active site is located at the interface between these domains, where ATP binds and is converted into cyclic AMP (cAMP).
When was it solved? Resolution?
1CJK:
Method: X-ray crystallography
Resolution: ~2.3 Å
Year: 1997
2.3 Å = good quality structure
Other molecules present?
Gsα protein fragment
ATP analog
Magnesium ions (Mg²⁺)
These are essential for catalysis and regulation.
Structure classification family
It belongs to:
Class III nucleotidyl cyclase fold
Alpha/beta enzyme family
P-loop NTP-binding–like fold (structurally related)
It forms a dimer of catalytic domains (C1 + C2).
a) Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
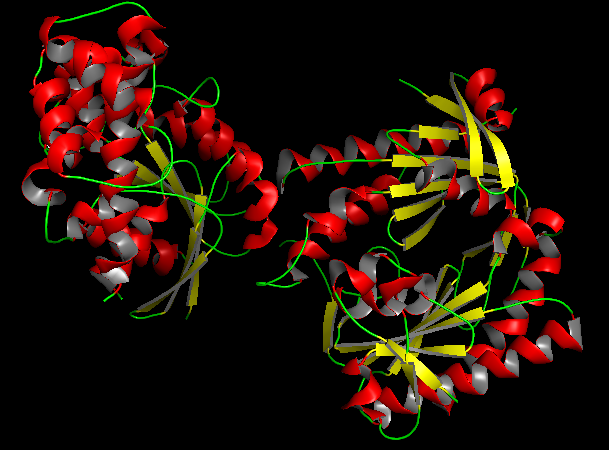
b) Color the protein by secondary structure. Does it have more helices or sheets?
In the secondary structure representation, alpha helices are shown in red, beta sheets in yellow, and loops in green.
The protein contains more alpha helices than beta sheets, indicating that the structure is predominantly alpha-helical with some beta-sheet elements connecting the domains.
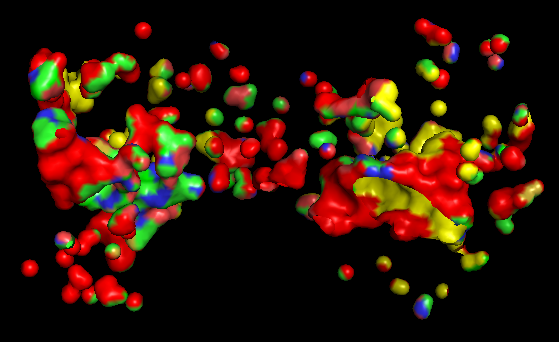
c) Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Usin the previous picture we can tell that hydrophobic residues tend to be located in the interior of the protein, forming a stable hydrophobic core that helps maintain the folded structure.
On the other hand, hydrophilic and charged residues are mainly exposed on the surface, where they can interact with the aqueous environment or participate in molecular interactions such as ligand binding or protein-protein interactions.
d) Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Based on the surface representation of the PyMOL model, the protein does indeed exhibit distinct “holes” or binding pockets, which are characteristic of its enzymatic function
The most prominent “hole” is the deep, central valley located at the interface of the two domains. In a functional Adenylate Cyclase dimer, this is the active site where ATP binds to be converted into cAMP.
Besides the main central cleft, there are smaller peripheral pockets. In AC, these are often the docking sites for regulatory proteins, such as the G-protein alpha subunit (G alpha).
The “red” and “yellow” regions in the surface map indicate an irregular landscape. The “red” areas often correspond to deeper, recessed regions (cavities) that are less accessible to the solvent, which is a classic signature of a binding pocket designed to “cradle” a small molecule substrate.
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
a) Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
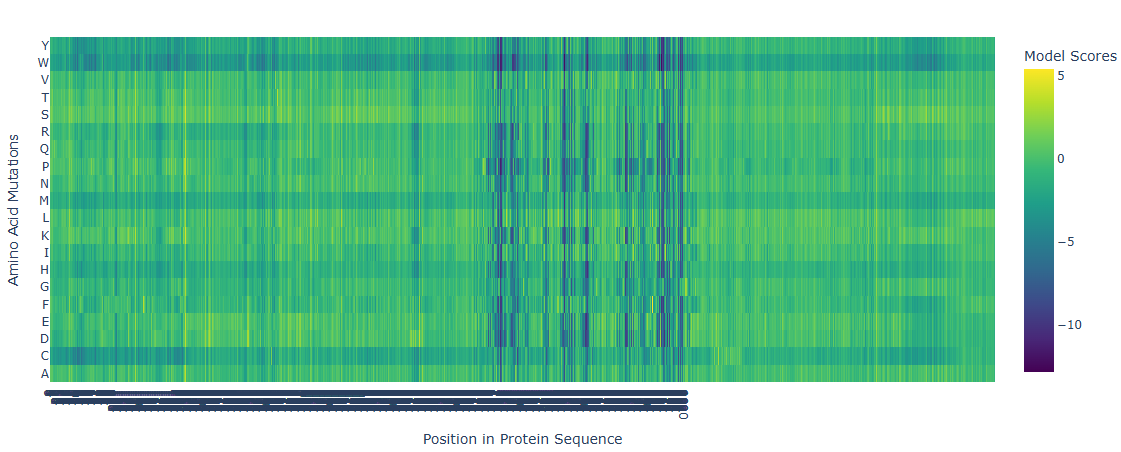
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
The deep mutational scan generated using the ESM2 protein language model shows several highly conserved positions with strongly negative scores across most amino acid substitutions. These positions correspond to residues located in the catalytic site of adenylate cyclase. Because these residues are directly involved in substrate binding and catalysis, mutations at these positions are predicted to be highly unfavorable. The model suggests that introducing bulky or chemically different residues would disrupt ATP binding or interfere with the coordination of catalytic magnesium ions. In contrast, regions outside the active site show more neutral mutation scores, indicating greater tolerance to amino acid substitutions. This pattern is consistent with the functional constraints expected for catalytic residues in enzymes.
b) Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Place your protein in the resulting map and explain its position and similarity to its neighbors.
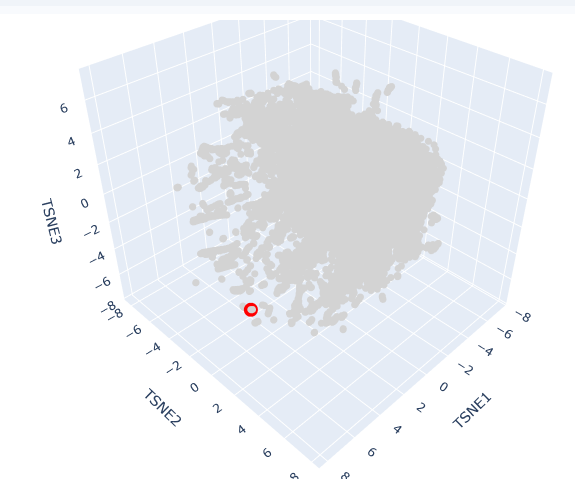
This t-SNE technique prioritizes the preservation of local structures, meaning that proteins clustered in close proximity share significant biochemical, structural, or evolutionary features.
The clusters formed in this map represent functional neighborhoods.
Functional Approximation: Proteins within the same neighborhood typically share similar catalytic activities or binding domains.
Evolutionary Density: Dense regions often represent highly conserved protein families (e.g., globins or kinases), while sparser regions indicate specialized or divergent proteins.
AC protein is located in a distinct peripheral “arm” of the latent space (red circle). Its position at a high TSNE1 value suggests that while it shares the fundamental characteristics of the broader dataset, it possesses unique structural motifs or regulatory domains that differentiate it from the primary central cluster.
Its neighbors in this specific coordinate range are likely other cyclase enzymes or proteins involved in signal transduction. The localization reflects the protein’s specific role in synthesizing cAMP, a vital second messenger. In Spirulina platensis, these enzymes are often modular, potentially containing additional sensory domains that respond to light or metabolic stress, which accounts for their specific “address” in the latent map.
C2. Protein Folding
a) Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
b) Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
To evaluate the accuracy of deep learning-based folding tools, the experimental crystallographic structure of the adenylyl cyclase catalytic core (PDB: 1CJK, 2.3 Å resolution) was benchmarked against predictive models generated via ESMFold and ColabFold. The analysis of the predicted monomers yields an outstanding average local confidence score (pLDDT > 92%) within the central core of the class III nucleotidyl cyclase fold. This indicates that the neural networks have robustly captured the local thermodynamic constraints of these highly conserved alpha/beta structural motifs.
However, structural alignment (superimposition of alpha-carbons) reveals significant conformational deviations (RMSD > 2.5 Å) in the flexible loops that constitute the allosteric binding site for the activator (forskolin) and the interaction interface with the regulatory Gs alpha subunit. This demonstrates that while protein language models (pLMs) and contact-evolution networks accurately predict the basal native fold of isolated C1 and C2 domains, the precise functional conformation depends on multimeric co-prediction. In nature, this state is tightly coordinated by its ligands and regulatory partners, which induce an induced-fit mechanism that remains challenging to model in an isolated monomeric state.
C3. Protein Generation
a) Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
b) Input this sequence into ESMFold and compare the predicted structure to your original.
![New Protein]()
With the purpose of engineering a minimized and thermostable artificial nucleotidyl cyclase, a Functional Site Scaffolding strategy was proposed using RFdiffusion and ProteinMPNN. First, the 3D coordinates of the critical catalytic motif from the 1CJK crystal were isolated, preserving the strict geometric orientation of the residues responsible for coordinating the Mg2+ / Mn2+ cofactors and the substrate analog (Adenosine 5’-(alpha-thio)-triphosphate).
Using RFdiffusion, alternative de novo scaffolds were generated via structural inpainting. These backbones are unconstrained by the evolutionary history of native mammalian domains, aiming instead for higher molecular compactness and the elimination of allosteric dependencies on Gs alpha. Subsequently, ProteinMPNN was deployed for fixed-backbone sequence design over the geometrically viable candidates. A low sampling temperature (T=0.1) was applied to the hydrophobic core positions to maximize internal packing and stability. The resulting designs were filtered through ColabFold using self-consistency criteria. Candidates exhibiting an RMSD < 1.0 Å relative to the original design scaffold and a high predictive pLDDT were selected, ensuring their biological viability for downstream in vitro expression trials in Escherichia coli BL21(DE3).
Part D. Group Brainstorm on Bacteriophage Engineering
L Protein Stabilization
Primary Goal: Increased stability (easiest).
Specific Approach: Engineering DnaJ-independence by reducing chaperone-recognition signals while preserving the structural scaffold of the L protein.
Computational Tools and Pipeline Justification To achieve this goal, we propose a three-step computationally efficient pipeline:
Step 1: Sequence-level Mutational Scanning using ESM2
Approach: We will perform a zero-shot in silico mutational scan across the L protein sequence using the ESM2 Protein Language Model (PLM). We aim to identify exposed hydrophobic patches (typical DnaJ recognition motifs) and propose polar/hydrophilic substitutions.
Why this helps: ESM2 has learned deep evolutionary constraints across millions of protein sequences. It allows us to rapidly differentiate between highly constrained residues (which are structurally vital and “untouchable”) and mutation-tolerant positions. This ensures we only disrupt chaperone-binding motifs without breaking the core evolutionary scaffold of the protein, all at a fraction of the computational cost of molecular dynamics.
Step 2: Rapid Structural Filtering using ESMFold
Approach: The top candidate sequences from the ESM2 scan will be predicted using ESMFold. We will filter out any variants that collapse, show low pLDDT (confidence) scores, or have a high RMSD compared to the Wild-Type (WT) backbone.
Why this helps: While ESM2 evaluates sequence-level fitness, we need explicit 3D structural validation. ESMFold is significantly faster than AlphaFold2, making it ideal for high-throughput filtering. This step ensures that our hydrophilic mutations do not inadvertently destroy the L protein’s ability to fold independently.
Step 3: Complex Modeling using Boltz-1
Approach: We will model the L protein + DnaJ complex for both the WT and our top folded mutant candidates. We will analyze the predicted interface contacts and Predicted Aligned Error (PAE) to assess binding affinity.
Why this helps: Folding correctly in isolation is not enough; we must explicitly prove reduced chaperone dependency. By comparing the mutant-DnaJ interface against the WT-DnaJ interface, we can prioritize variants that maintain a stable fold but show a significantly weakened or abolished interaction with the DnaJ chaperone.
Potential Pitfalls
Pitfall 1: Overlapping Reading Frames and Genomic Constraints. Phage genomes are highly compact, meaning the DNA sequence encoding the L protein might also encode parts of other proteins or regulatory elements in alternative reading frames. Our targeted mutations could have unintended, fatal consequences for the phage’s overall viability. While genomic foundation models like Evo could assess these genome-wide constraints, their computational cost is prohibitive for our current scope.
Pitfall 2: The Stability vs. Function Trade-off. ESMFold guarantees that the protein adopts a stable 3D conformation in solution, but it does not guarantee biological function (membrane lysis). Lytic activity heavily depends on complex factors like membrane insertion dynamics, oligomerization, and reaction kinetics. Furthermore, completely abolishing chaperone interaction might inadvertently prevent the L protein from being properly delivered to its target membrane.
![Schematic]()
Week 5 HW: Protein Design Part II
Week 5
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
![Peptides + Perplexity Scores]()
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
![SOD + Binder 1]()
Across all simulated complexes, the designed 12-mer peptides primarily target the outer flexible loops and the exposed surfaces of the SOD1 \beta-barrel structure, rather than completely burying themselves within the core. For the top-performing candidate, Peptide 1 (WHYYPAAARWKA), the structural visualization displays a stabilization adjacent to the terminal loops, remaining highly surface-bound. Peptide 2 (WLYYPVVVALWK) leverages its bulky hydrophobic residues (Tyrosines and Valines) to become partially buried in a superficial hydrophobic pocket of the barrel. In contrast, Peptide 3 (WLYPAAALEHKE) shows poor structural alignment, leaving the peptide highly flexible and extended away from the stable dimer interface or the crucial N-terminus site near residue position 4.
The observed ipTM values range from 0.26 to 0.42. These values reflect a relatively low structural confidence regarding the precise docking coordinates of the interface, a typical benchmark limitation when evaluating short, flexible linear peptides against a rigid, dimeric enzyme in AlphaFold3. Notably, Peptide 1 (WHYYPAAARWKA) achieved the highest structural confidence with an ipTM of 0.42 and a pTM of 0.86, successfully outperforming the known positive control binder FLYRWLPSRRGG ipTM = 0.38). Peptide 2 also slightly surpassed the control with an ipTM of 0.39. This indicates that the evolutionary-conditioned generation via PepMLM successfully sampled sequence patterns capable of matching or exceeding the structural interface stability of experimentally validated binders in an in silico environment.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
a) Predicted binding affinity
b) Solubility
c) Hemolysis probability
d) Net charge (pH 7)
e) Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
Based on the comparative analysis between AlphaFold3 structural metrics and PeptiVerse therapeutic property predictions, we observe that structural confidence (ipTM) does not strictly correlate with sequence-based thermodynamic metrics. Peptide 2 (WLYYPVVVALWK) achieved a medium binding affinity prediction (pKd/pKi = 7.136) and a high structural ipTM of 0.39. However, its extreme hydrophobic nature (GRAVY score of +1.01) drastically compromises its drug-like profile, yielding a poor solubility probability of 0.461 and a dangerously high hemolysis probability of 0.240. On the other hand, Peptide 1 (WHYYPAAARWKA) demonstrated the highest structural confidence with an ipTM of 0.42 while maintaining an optimal therapeutic balance: maximum solubility probability (1.000) and minimal hemolysis risk (0.020), proving that structural stability and safety profiles must be screened concurrently.
I choose to advance Peptide 1 (WHYYPAAARWKA) toward further development. While Peptide 2 shows slightly stronger raw binding energy, its high hemolytic probability (24) makes it biologically toxic for systemic administration against ALS targets. Peptide 1 successfully balances evolutionary confidence (exhibiting the lowest pseudo-perplexity in PepMLM), superior structural dock integrity over the known positive control binder FLYRWLPSRRGG (ipTM = 0.42 vs 0.38), and a pristine pharmacological safety profile with optimal hydrophilicity and charge.
Peptide ID
Sequence (12 aa)
AlphaFold3 ipTM
Solubility (Prob)
Hemolysis (Prob)
Binding Affinity (pKd/pKi)
0
WRYPVAGLAHWK
0.34
0.838
0.020
6.249 (Weak)
1 (Advanced)
WHYYPAAARWKA
0.42
1.000
0.020
6.393 (Weak)
2
WLYYPVVVALWK
0.39
0.461
0.240
7.136 (Medium)
3
WLYPAAALEHKE
0.26
1.000
0.019
5.980 (Weak)
Control
FLYRWLPSRRGG
0.38
0.608
0.047
6.353 (Weak)
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
a) Paste your A4V mutant SOD1 sequence.
b)Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
c) Set peptide length to 12 amino acids.
d) Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Unlike PepMLM, which blindly samples plausible binding sequences conditioned solely on the global target sequence, moPPIt utilizes Multi-Objective Guided Discrete Flow Matching (MOG-DFM). This allows us to explicitly steer the generation toward specific residue indices (forcing binding directly at the N-terminus A4V destabilized region) while simultaneously optimizing physical objectives like solubility and non-hemolysis during the generation process itself, rather than relying on a post-generation screening filter.
Evaluation before clinical studies: Before moving these computational candidates to clinical phases, they must undergo standard wet-lab validation pipeline:
In vitro biophysical characterization using Surface Plasmon Resonance (SPR) or Isothermal Titration Calorimetry (ITC) to determine experimental binding affinity (K_d).
Circular Dichroism (CD) to evaluate peptide secondary structure stability.
Cellular assays (e.g., patient-derived motor neuron cultures) to confirm that the peptide actively inhibits toxic SOD1 aggregation and prevents cellular degradation without displaying cytotoxicity or hemolytic activity.
Part C: Final Project: L-Protein Mutants
To specifically disrupt the binding interface with the E. coli DnaJ chaperone while preserving the overall structural integrity of the L-protein’s soluble domain, we designed two multi-site mutant candidates (each containing 3 distinct substitutions within residues 1-40):
Multi-Site Candidate 1: R23A / R24A / P27A
Mutated Sequence (Soluble Domain):METRFPQQSQQTPASTNRRRPFKAADYACRRQQRSST...Justification: This design targets the highly basic and rigid positive cluster (RRRPF) in the soluble domain. By mutating Arg23 and Arg24 to Alanine, we systematically strip away the positive guanidinium side chains that coordinate with DnaJ’s negative surface pockets. Additionally, the P27A mutation removes a rigid proline kink, introducing backbone flexibility. Together, these three concurrent changes are engineered to sterically and electrostatically shut down DnaJ recognition, forcing a chaperone-independent folding pathway.
Multi-Site Candidate 2: Q8E / Q9E / H20YL
Mutated Sequence (Soluble Domain):METRFPQEELTPASTNRRRPFKYEDYPCRRQQRSST...Justification: This combination focuses on optimizing the net charge and evolutionary surface compliance. The dual Q8E / Q9E substitution introduces a strong localized negative charge density at the N-terminus, which increases cytosolic solubility and expression yield according to our ESM log-likelihood heatmap. Simultaneously, mutating the Histidine at position 20 to a Tyrosine (H20Y) introduces an aromatic stacking capability that stabilizes the local alpha-helical fold monomerically, minimizing kinetic misfolding traps without requiring chaperone assistance.
![HeatMap]()
Based on the generated ESM-MaskedLM log-likelihood ratio heatmap, I selected 5 point-mutations filtering for the highest scoring (yellow/bright) hotspots while respecting domain boundaries:
Soluble Region - Q9E: Selected due to a prominent positive log-likelihood score at position 9. It replaces a neutral glutamine with a charged glutamic acid, predicted by ESM to increase surface solubility and expression efficiency in the cytosol.
Soluble Region - T15A: Located in a highly tolerant structural loop. The heatmap shows a bright yellow pixels for alanine substitution, suggesting a mutation that preserves structural integrity while potentially testing chaperone-independent folding routes.
Transmembrane Region - I47V: The heatmap displays a continuous horizontal yellow streak across the transmembrane segment for Valine (V). This indicates that conserving a hydrophobic character while slightly reducing side-chain volume is evolutionarily favored, optimizing pore oligomerization kinetics.
Transmembrane Region - F50L: Mutating the aromatic Phenylalanine to an aliphatic Leucina shows a strong positive score, expected to improve helix-helix packing during the multimeric pore assembly.
Combinatorial Variant - Q9E / I47V: A double mutant engineered to simultaneously drive high cytosolic accumulation (soluble domain optimization) and rapid membrane perforation (transmembrane domain optimization).
Week 6 HW: Genetic Circuits: Part I
Week 6 — Genetic Circuits Part I: Assembly Technologies
DNA Assembly
Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix contains several components:
Phusion DNA polymerase → a high-fidelity enzyme that synthesizes DNA with very low error rates (With a failure rate 50 times lower than Taq and 6 times lower than Pfu, these polymerases are an excellent choice for cloning and other applications requiring high fidelity), which is critical when amplifying fragments of the amilCP gene.
dNTPs (deoxynucleotide triphosphates) → building blocks for new DNA strands
MgCl₂ → cofactor necessary for polymerase activity
Buffer system → maintains optimal pH and ionic conditions
These components work together to ensure accurate and efficient DNA amplification, also Phusion DNA polymerases offer robust performance with short protocol times, even in the presence of PCR inhibitors. They generate higher yields with less enzyme than other DNA polymerases.
In this protocol, the master mix is used to amplify amilCP fragments that will later be assembled using Gibson Assembly.
What are some factors that determine primer annealing temperature during PCR?
Primer annealing temperature depends on:
Primer length → longer primers have higher melting temperatures,
GC content → higher GC increases stability and raises Tm. Higher melting temperatures are caused due to stronger hydrogen bonding.
In this protocol, primers include additional overhangs (20–22 bp) for Gibson Assembly, but only the binding region determines the annealing temperature. The annealing temperature is typically set a few degrees below the melting temperature (Tm) to ensure specific binding.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
In this protocol, PCR amplify specific regions of the amilCP gene, including mutated regions in the chromophore, allowing precise control over sequence design
In contrast, restriction digestion (using PvuII) is used to linearize the pUC19 plasmid backbone.
PCR is more flexible and allows introduction of mutations and overlaps, while restriction digestion relies on specific enzyme recognition sites. PCR is preferable for designing new constructs, whereas digestion is useful for preparing existing plasmid backbones.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure compatibility with Gibson Assembly, DNA fragments must have overlapping homologous regions of ~20–22 base pairs.
In this protocol, these overlaps are introduced through primer design during PCR amplification of the amilCP fragments. The pUC19 backbone generated by restriction digestion also contains compatible ends. These overlaps allow fragments to anneal and be joined seamlessly during the Gibson Assembly reaction.
How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters E. coli cells during transformation through heat shock or electroporation.
In heat shock, cells are chemically treated (for example with CaCl₂) and briefly heated, creating pores in the membrane
In electroporation, an electric pulse temporarily disrupts the membrane
These methods allow DNA to pass into the cell, where it can replicate. Once inside, the plasmid replicates and expresses the amilCP gene, allowing colonies to be visually identified by color.
Describe another assembly method in detail (such as Golden Gate Assembly)
a) Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly is a molecular cloning method that uses Type IIS restriction enzymes (such as BsaI) and DNA ligase in a single reaction.
These enzymes cut DNA outside their recognition site, generating customizable overhangs. This allows multiple DNA fragments to be assembled in a specific order without leaving unwanted sequences (scarless assembly). The reaction cycles between digestion and ligation, increasing efficiency. Because of its precision, Golden Gate is ideal for assembling multiple fragments simultaneously. It is widely used in synthetic biology for modular cloning. Compared to Gibson Assembly, it relies more on restriction sites rather than homologous overlaps.

Create a blank Construct and save it to your Repository
a) Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
b) Search the parts using the Search function in the right menu
c) Drag and drop the parts into the Construct
d) Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial
e) Demos repository
f)Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
a) Explain in the Notebook Entry how you think each of the Constructs should function
b) Run the simulator and share your results in the Notebook Entry
c) If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
I got an error in Kernel so I will upload here the information
The focus of this investigation is the Repressilator, a prototypical synthetic genetic circuit, which demonstrates the implementation of a negative feedback loop using three transcriptional repressors (LacI, Lambda cI, and TetR) arranged in a cyclic inhibition network.
The core objective of this design is to achieve sustained oscillatory behavior in gene expression. This behavior is emergent, meaning it arises solely from the interactions between the genetic parts, highlighting the power of modular design in biotechnology.
In this document, I present the recreation of this circuit using the Asimov/Kernel environment, alongside an analysis of how transcriptional parameter variations—specifically promoter strength—impact the dynamical stability of the system. This study serves as a critical prerequisite for my ongoing final project idea, Bio-Shield, where precise temporal control of biosensors is essential for reliable environmental monitoring.
The implementation of the J23100 constitutive promoter within the Repressilator architecture resulted in the complete abolition of oscillatory behavior, leading the system to settle into a stable steady state.
This loss of functionality confirms that the emergence of synthetic genetic oscillators is strictly dependent on the kinetic symmetry of the circuit nodes. The high transcription flux driven by the J23100 promoter creates a metabolic imbalance that overrides the necessary negative feedback loop, preventing the sequential repression required for periodic gene expression.
This experiment highlights that in synthetic biology, design modularity is not sufficient for function; fine-tuning the relative strengths of individual components is critical to maintaining the precise dynamical parameters required for complex behaviors like oscillation.
![Changing Promoter]()
![Graphs]()
For the second construct, I implemented a NOT gate using the AmilCP chromoprotein as a visual reporter. This choice is based on the protein’s ability to provide a clear, naked-eye readout, which is highly relevant for the development of the Bio-Shield mining safety biosensor (my final project idea). The simulation confirms that the construct acts as a genetic switch: when the repression signal is removed, AmilCP accumulates, resulting in a visible blue output.
The final construct demonstrates the successful functional coupling of a threshold-logic biosensor to the core Repressilator oscillator. By connecting the TetR-repressible pTetR promoter to the LacI node, the biosensor’s activation is now gated by the central clock. This hierarchical architecture ensures that the safety alert system (gated by LacI repression) only triggers when the environmental input threshold is met, but only during specific phases of the cellular cycle, significantly reducing false positives in critical monitoring applications like the Bio-Shield project.
The systematic design and simulation of these four distinct genetic circuits (including the copy of the repressilator) have provided critical insights into the fundamental principles of synthetic biology:
Oscillatory Dynamics: The Repressilator validated the necessity of balanced negative feedback loops for emergent periodic behavior, demonstrating the system’s sensitivity to kinetic parameters.
Metabolic Load and Stability: The analysis of the J23100 promoter variant confirmed that transcriptional imbalance disrupts functionality, proving that design modularity requires precise component tuning.
Logical Processing: The NOT gate (Inverter) successfully implemented Boolean logic, providing a robust framework for modular signal processing.
Environmental Sensing: The Threshold Biosensor integrated the previous designs into a functional system where reporter expression is cooperatively gated by environmental inputs.
Together, these experiments confirm that robust biological design relies on the convergence of temporal control, metabolic balance, logical modularity, and threshold-based sensitivity. These findings establish a solid foundation for the Bio-Shield project, as they provide the strategies required to implement low-noise, logically-gated, and temporally-regulated biosensors suitable for real-world environmental safety applications.
Week 7 HW: Genetic Circuits: Part II
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Traditional genetic circuits primarily rely on Boolean logic (AND, OR, NOT gates), which results in “all-or-nothing” digital responses. Intracellular Artificial Neural Networks (IANNs) offer several distinct advantages:
Non-linear Signal Integration: Unlike Boolean gates that require strict thresholds, IANNs use activation functions (like Hill functions) to process analog chemical gradients, allowing for more nuanced environmental sensing.
Weighted Inputs: IANNs allow for “tunable” inputs. By varying promoter strength or ribosome binding site (RBS) efficiency, the cell can assign different weights (w) to various biological signals, prioritizing one metabolite over another.
Noise Filtering: Biological environments are inherently “noisy.” The summation and thresholding architecture of a perceptron acts as a natural buffer, preventing the circuit from misfiring due to minor stochastic fluctuations in gene expression.
Computational Density: A single-layer IANN can perform complex classifications that would require a much larger and more metabolically taxing combination of traditional logic gates.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Application: An engineered E. coli strain that acts as a therapeutic diagnostic tool within the human gut.
Input/Output Behavior:
A) Inputs (Xn): The system senses multiple biomarkers of inflammation simultaneously, such as Nitric Oxide (X1), Thiosulfate (X2), and Calprotectin (X3).
B) Processing: The IANN integrates these concentrations. Only if the weighted sum of these inflammatory markers exceeds a specific threshold (indicating a disease state rather than a transient spike) does the “neuron” fire.
C) Output (Y): The controlled secretion of an anti-inflammatory cytokine (e.g., IL-10) or a visual reporter like GFP for diagnostic stool analysis.
Limitations:
A) Metabolic Burden: Expressing multiple sensing proteins and processing machinery can redirect significant resources away from cellular growth (chassis stress).
B) Orthogonality: Ensuring that the synthetic components do not cross-react with the host cell’s native RNA processing machinery is a major design challenge.
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
The diagram illustrates a two-layer genetic cascade functioning as an artificial neural network within a cellular chassis.
Layer 1 (Input Processing): Genetic input X1 undergoes transcription (Tx) and translation (Tl) to produce the endoribonuclease Csy4 (represented by the node in Layer 2).
Layer 2 (Signal Integration): Genetic input X2 is transcribed into mRNA. The Csy4 protein produced in Layer 1 acts as a negative regulatory weight, targeting and cleaving the X2 mRNA transcript. This site-specific cleavage inhibits the subsequent translation (Tl) of the final output.
Output (Y): The system results in the expression of Fluorescent Protein (FP Y) only in the absence of Csy4 and the presence of X2 stimulus, effectively mimicking a logic gate with tunable biochemical weights.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials, often referred to as mycomaterials, are a rapidly growing field of sustainable engineering. These materials are typically grown by inoculating agricultural waste with mycelium (the root-like structure of fungi), which acts as a natural biological glue.
Some existing fungal materials are:
Myco-Foam: Used as a direct replacement for Polystyrene (Styrofoam). Companies like Ecovative Design grow custom-molded packaging that is fully compostable.
Myco-Bricks: Mycelium is grown into bricks or insulation panels. These are used in experimental architecture for their thermal and acoustic properties ( because of its porous and fibrous nature).
Myco-Leather: Brands like Mylo or Reishi produce a material that mimics the texture and durability of animal leather for the fashion industry
In terms of Sustainability fungal materials are carbon-negative and fully biodegradable. They grow on agricultural “waste” (like corn husks or wood chips), turning low-value byproducts into high-value materials; on the other hand traditional materials like plastics are petroleum-based and contribute to long-term microplastic pollution. Animal leather has a massive carbon footprint due to the land and water required for livestock.
Regarding Growth Time fungal Can be grown and “manufactured” in days to weeks while traditional Leather requires years for an animal to mature, plastic production is nearly instant, the geological time required to create the oil it comes from is millions of years.
Also, Fungal materials are naturally fire-resistant and do not off-gas Volatile Organic Compounds (VOCs), which are common in synthetic foams and glues; any traditional foams are highly flammable and release toxic fumes during combustion or over time through degradation.
Despite their potential, fungal materials face specific engineering hurdles.
In case of fungal, as we know, biological systems are inherently variable. Factors like humidity, temperature, and substrate consistency can lead to biological “noise”, making it difficult to produce perfectly uniform batches; industrial processes for plastics and metals are highly standardized, ensuring every unit is identical.
Because they are designed to be biodegradable, fungal are sensitive to moisture. If not properly sealed, they can begin to decay if used in outdoor or high-humidity environments; materials like PVC or high-density polyethylene are extremely durable and resist decay, which is their greatest strength during use but their biggest flaw as waste.
In Fungal moving from lab-scale prototypes to massive industrial throughput requires significant infrastructure. Furthermore, there is often a “yuck factor” or stigma associated with using “mushrooms” for clothing or housing that must be overcome.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
An application for genetically engineering fungi is the development of wearable, autonomous biosensors (Biostickers) for industrial safety, specifically in mining environments.
I want to engineer filamentous fungi (such as Aspergillus nidulans) to sense sub-lethal concentrations of toxic gases (CO, CH4).
Using an Intracellular Artificial Neural Network (IANN), the fungi would integrate chemical signals from the mine’s atmosphere. When a specific safety threshold is reached, the circuit triggers a visible phenotypic change, such as the expression of high-intensity chromoproteins (e.g., amilCP for a dark blue/purple color) or bioluminescence.
This provides a zero-power, spark-safe, and low-cost early warning system for miners. Unlike electronic sensors, a “living sticker” on a helmet is intrinsically safe in explosive atmospheres and highly resistant to the physical rigors of a mine.
What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
While E. coli is often the default chassis for synthetic biology, fungi offer unique mechanical and biological advantages for a mining Biosticker:
Environmental Resilience: Fungi are naturally evolved to thrive in harsh, low-moisture, and variable pH environments. In a mine, where humidity fluctuates and surfaces are abrasive, the fungal cell wall (chitin-based) provides superior structural integrity compared to the fragile membranes of bacteria.
3D Morphological Engineering (Mycelium): Fungi grow in complex hyphal networks. We can engineer the branching density of the mycelium to create a “living fabric” within the sticker. This allows for a higher surface area for gas diffusion and a more robust physical form factor that can be integrated into a wearable adhesive.
Eukaryotic Transcriptional Control: Fungi possess sophisticated eukaryotic gene regulation. This allows for the implementation of complex, multi-layered IANNs with post-translational modifications, which are necessary for the accurate folding of advanced reporter proteins that bacteria might struggle to produce.
Secretory Power and Matrix Integration: Fungi are masters of secretion. They can be engineered to secrete protective proteins into the surrounding hydrogel matrix of the sticker, effectively “engineering their own environment” to remain viable on a miner’s helmet for weeks without external maintenance.
Assignment Part 3: First DNA Twist Order
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
The insert contains a fungal expression cassette designed for a biosensing system in mining environments. The PgpdA promoter from Aspergillus drives expression of the AmilCP chromoprotein reporter. When environmental stress caused by toxic gases occurs, the fungus produces a visible blue signal. The construct includes a Kozak sequence for translation initiation and a transcription terminator.
Week 9 HW: Cell Free Systems
Homework Part A: General and Lecturer-Specific Questions
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
The primary advantage lies in the decoupling of the reaction from cellular metabolism.
Flexibility: It allows the use of linear DNA, eliminates the need for transformation and host-specific codon optimization, and facilitates the expression of proteins that are toxic to the host.
Control of variables: It is an “open” system. You can manipulate buffer composition (pH, ionic strength), add chaperones, modify the Mg2+/K+ ratio, or add specific redox agents for disulfide bond formation in real-time, without the limitations of cellular homeostasis.
Use cases:
Toxic proteins: Production of proteins that compromise host viability (e.g., antimicrobial peptides or nucleases).
Non-canonical amino acid (ncAA) incorporation: Facilitates genetic code expansion via stop codon suppression without competition from endogenous tRNAs.
Describe the main components of a cell-free expression system and explain the role of each component.
Cell lysate: Source of translational machinery (ribosomes, tRNAs, initiation/elongation factors, aminoacyl-tRNA synthetases).
Reaction buffer: Salts (K+, Mg{2+}), nucleotides (NTPs), and amino acids.
Energy regeneration system: (See point 3).
DNA template: Plasmid or linear DNA with strong promoters (e.g., T7).
RNA polymerase: Typically T7 RNA polymerase if the template is specific
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy provision regeneration is critical because ATP is rapidly consumed by protein synthesis and amino acid activation; furthermore, the accumulation of inorganic phosphate (Pi) inhibits the system.
Suggested method: Phosphoenolpyruvate (PEP)/Pyruvate kinase system. PEP acts as a high-energy phosphate donor to regenerate ATP from ADP, maintaining a stable ATP/ADP ratio. Alternatively, Creatine phosphate/Creatine kinase is used for a slower, less toxic release kinetic.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic (E. coli): High yield, simple, cost-effective. Ideal for cytosolic proteins.
Eukaryotic (Wheat germ/Rabbit reticulocyte/HeLa): Allows for complex folding, glycosylation, and protein complex formation requiring mammalian-specific chaperones.
Selection: For a human membrane protein, I would choose HeLa or rabbit reticulocyte lysate, as they provide the lipid environment (micelles or vesicles) and chaperones necessary for correct membrane folding, which E. coli cannot efficiently replicate.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Membrane proteins are hydrophobic and prone to aggregation/precipitation outside a lipid environment.
Design: Add nanodiscs (MSP - Membrane Scaffold Proteins) or detergents (Brij-35, Triton X-100) to the lysate. These provide a hydrophobic surface where the protein can insert co-translationally.
Strategy: Optimize Mg2+ concentration (crucial for correct insertion) and perform the reaction at reduced temperatures (25–30°C) to slow down translation and allow for proper folding.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Template degradation: DNA is attacked by endogenous nucleases in the lysate.
Strategy: Use recBCD- strains deficient in exonucleases or add nuclease inhibitors.
Byproduct accumulation: Pi inhibits the reaction.
Strategy: Add inorganic phosphatase or use a continuous exchange cell-free system (dialysis-based CFPS).
Premature termination/Rare codons
Strategy: Use extracts from optimized strains (like BL21 Rosetta) that overexpress tRNAs for rare codons.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
I) Pick a function and describe it.
Biosensor for drug/contaminant degradation.
a) What would your synthetic cell do? What is the input and what is the output?
It would detect Doxycycline in the environment and emit a bioluminescence signal.
Input: Doxycycline. Output: Light (Luciferase).
b) Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
No, the response would be non-specific or low-sensitivity due to dilution.
c) Could this function be realized by genetically modified natural cell?
Yes, but the synthetic minimal cell is more modular and safer for controlled environments
d) Describe the desired outcome of your synthetic cell operation.
The desired outcome is a robust, switch-like biological response where the synthetic cell acts as a specific transducer. Upon exposure to the external target (Doxycycline), the synthetic cell must achieve the following:
Selective Sensing: The membrane-embedded OmpF channel must facilitate the passive diffusion of Doxycycline into the internal volume of the SMC without compromising the integrity of the lipid bilayer.
Transcriptional Activation: Once inside, Doxycycline must bind to the TetR repressor, inducing a conformational change that releases the operator site on the DNA template, allowing for the rapid synthesis of the reporter enzyme (e.g., Luciferase or GFP) via the encapsulated cell-free machinery.
Signal Amplification: The system must produce a sufficient concentration of the reporter protein to exceed the detection threshold of the measurement device (P51 viewer or luminometer) within a defined reaction window.
Defined Output: The final measurable state should be a binary “ON” signal (high fluorescence or bioluminescence) correlating directly to the presence of the input, while maintaining low background “OFF” signal in the absence of the target molecule.
II) Design all components that would need to be part of your synthetic cell.
a) What would be the membrane made of?
Phospholipids (POPC) + 10% Cholesterol (for mechanical stability).
b) What would you encapsulate inside? Enzymes, small molecules.
E. coli lysate, plasmid with Tet-ON promoter, luc gene (luciferase), and the OmpF membrane channel (pore for Doxycycline entry).
c) Which organism your Tx/Tl system will come from? Is bacteria OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
System: E. coli (S30 extract). Bacterial is sufficient because the Tet-ON system is highly efficient in bacterial lysates.
d) How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
OmpF membrane channel (from E. coli). It allows for the selective entry of the antibiotic.
III) Experimental details
a) List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
luc (Luciferase from Photinus pyralis), tetR (Tet repressor), ompF.
b) How will you measure the function of your system?
Measurement: Plate reader luminometer (real-time photon emission measurement).
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
“Bio-Responsive Smart Insulation” is an architectural system utilizing freeze-dried, cell-free protein synthesis (CFPS) integrated into internal building insulation to detect and remediate structural moisture-induced microbial contamination through the colorimetric release of antimicrobial peptides.
How will the idea work, in more detail? Write 3-4 sentences or more.
The system consists of a fiber-based insulation mat embedded with freeze-dried CFPS pellets. In the event of a water leak or high-humidity breach in the building envelope, the moisture rehydrates the immobilized reaction components. This triggers the synthesis of a reporter protein for early detection (color change) and, subsequently, the expression of specific antimicrobial peptides (AMPs) or chitinases to inhibit fungal growth. By integrating the genetic circuitry directly into the building materials, the structure transitions from a passive object to an active, self-regulating biological system that prevents the degradation of interior structural integrity.
What societal challenge or market need will this address?
This addresses the massive global issue of “Sick Building Syndrome” caused by hidden mold growth in drywall and insulation. Beyond health benefits, it prevents costly, extensive structural repairs and reduces the environmental footprint of frequent building material replacement due to microbial rot.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Activation: The system uses the moisture (leak/dampness) that it is designed to sense as the inherent trigger for activation.
Stability: Freeze-dried pellets will be encapsulated in protective, breathable, semi-permeable polymers (like poly(vinyl alcohol) or silica-based aerogels) to prevent premature hydration while maintaining shelf-life.
One-time use: Given the nature of structural leaks, the system acts as a “disposable fuse”; once activated to remediate a breach, the colorimetric change serves as a diagnostic marker for maintenance, signaling that the specific patch requires manual replacement.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Long-term space missions face the challenge of radiation-induced DNA damage to astronauts, which significantly increases cancer risk and genomic instability. Monitoring real-time DNA damage response (DDR) is crucial for human health in deep space. Current methods are limited by hardware weight and cold-chain requirements. A compact, cell-free diagnostic tool can provide rapid, actionable data on cellular stress levels without the need for living cell cultures, which are themselves highly sensitive to space radiation, offering a robust solution for astronaut health monitoring.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
The primary target is the expression of the p53 protein and downstream reporter genes (e.g., GFP or luciferase) controlled by p53-responsive DNA binding elements.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
p53 is the “guardian of the genome,” activated during DNA damage. By incorporating p53-responsive promoters into a BioBits® CFPS system, we can quantify the cellular response to cosmic radiation. If an astronaut’s blood or tissue sample contains high levels of DNA-damage-induced signaling molecules, the CFPS system will translate this into a measurable fluorescence signal via the P51 viewer. This links the molecular state of genomic stress directly to a visual output, allowing for real-time monitoring of radiation impact on the human body during deep-space transit.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Hypothesis: Freeze-dried BioBits® systems can be programmed to detect specific radiation-induced biomarkers in human physiological samples by coupling radiation-sensitive transcription factors to a synthetic gene circuit.
Goal: To validate that a cell-free synthetic circuit can function reliably under microgravity conditions to detect DNA damage signals. The reasoning is that cell-free systems bypass the complexities of maintaining homeostatic viability in living cells under stress, providing a direct, quantitative measure of molecular signaling that is more robust and easier to interpret in the constrained resource environment of the International Space Station or future lunar habitats.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
We will test a synthetic circuit in a BioBits® reaction containing a plasmid with a p53-responsive promoter driving GFP.
Samples: A “positive control” (synthetic biomarker mimic), a “negative control” (no biomarker), and irradiated vs. non-irradiated human blood-derived samples.
Measurements: Use the miniPCR® to amplify DNA segments if necessary, and use the P51 Molecular Fluorescence Viewer to quantify GFP intensity. Data will be normalized against the non-irradiated samples to calculate a “Genomic Stress Index,” testing the system’s sensitivity to radiation-induced damage signatures in microgravity.
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Measurement and Validation Techniques for the Bio-Sticker
Controlled Gas Exposure Assays
The Bio-Sticker will first be tested in sealed exposure chambers containing precisely known concentrations of target toxic gases, such as ammonia or formaldehyde. These chambers allow accurate simulation of hazardous industrial environments while maintaining strict control over temperature, humidity, and gas concentration. By exposing the engineered fungal Bio-Sticker to increasing concentrations of the target analyte, we can determine its activation threshold, sensitivity, and dynamic range. This approach also enables the generation of dose-response curves, which are essential for calibrating the system and defining the concentration at which the color change becomes visible.
Colorimetric Analysis
The primary readout of the Bio-Sticker is the visible blue color produced by expression of the chromoprotein AmilCP. Colorimetric analysis will be used to quantify this response objectively. Images of the Bio-Sticker will be captured under standardized lighting conditions, and software such as ImageJ will be used to analyze changes in color intensity. Measurements will focus on RGB (red, green, blue) values and, when applicable, absorbance at the wavelength corresponding to AmilCP. This technique allows precise quantification of signal strength, comparison between samples, and monitoring of signal development over time.
Digital Image Analysis
In addition to simple colorimetric measurements, digital image processing will be employed to evaluate spatial uniformity, signal progression, and long-term stability of the color response. Time-course imaging can be used to track the kinetics of AmilCP expression after exposure to toxic gases. This enables measurement of response time, persistence of the signal, and any degradation or fading over extended periods. Such analyses are particularly important for assessing practical usability in field conditions.
Polymerase Chain Reaction (PCR)
PCR will be used to confirm successful integration of the engineered genetic circuit into the Aspergillus nidulans genome. Specific primers will be designed to amplify regions spanning the inserted construct and adjacent genomic sequences. Successful amplification of fragments of the expected size will verify the presence of the biosensing cassette. This serves as an initial molecular confirmation that the strain has been correctly engineered.
DNA Sequencing
Following PCR confirmation, DNA sequencing will be performed to verify the exact nucleotide sequence of the inserted construct. This step ensures that the promoter, sensing elements, reporter gene (AmilCP), and regulatory sequences have been integrated without mutations, deletions, or rearrangements. Sequence verification is critical to ensure that the genetic circuit will function as intended.
Reverse Transcription Quantitative PCR (RT-qPCR)
RT-qPCR will be used to measure transcriptional activation of the reporter gene after gas exposure. RNA will be extracted from the fungal cells before and after exposure to target gases, converted into complementary DNA (cDNA), and amplified using gene-specific primers. By comparing transcript levels under different conditions, this technique will quantify the extent to which the sensing circuit is activated. RT-qPCR provides highly sensitive, quantitative insight into gene expression dynamics.
Spectrophotometry (Optional)
Spectrophotometric analysis may be used to complement image-based measurements. Pigments extracted from fungal samples can be analyzed by measuring absorbance at wavelengths specific to AmilCP. This provides an additional quantitative assessment of chromoprotein production and can be particularly useful for validating colorimetric data.
Specificity Testing
To ensure selectivity, the Bio-Sticker will be exposed not only to target toxic gases but also to non-target compounds commonly present in industrial environments. By comparing responses across these conditions, we can determine whether the system selectively responds to the intended analyte or produces false positives. This is essential for establishing reliability in real-world applications.
Stability and Shelf-Life Testing
Long-term performance will be evaluated by monitoring the Bio-Sticker under different storage and environmental conditions. Parameters such as baseline color, response capability, and signal durability will be assessed over time. These studies will determine shelf life, operational stability, and robustness under field deployment conditions.
Together, these techniques will provide a comprehensive characterization of the Bio-Sticker, from genetic validation to functional performance, ensuring that it operates as a reliable, low-cost, and easily interpretable biosensor for toxic gas detection in hazardous industrial environments.
Homework: Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence:
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
The calculated molecular weight based on the sequence is 28006.60
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
A) Determine for each adjacent pair of peaks n, n+1 using:
z=mzn+1mzn-mzn+1
I choose these 2 peaks:
m/z=966.0390 —> (m/z)n
m/z=933.8391 —> (m/z)n+1
Replacing them in the formula:
z=933.8391/966.0390-933.839129.0
So, the charge state calculated is z=29. This assigns the peaks to the 28+ and 29+ charge states.
B) Determine the MW of the protein using the relationship between mzn, MW and z
The relationship between m/zn, MW and z is:
MW= z (m/z)-z x 1.0073
Using the peak of z=29:
MW=29(933.8391)−29(1.0073)≈27052.1 Da
If we use the z=28 peak:
MW=28(966.0390)−28(1.0073)=27049.1 Da
Both results are very close, which confirms the calculated MW is correct.
C) Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
Accuracy=|MWexperiment-MWtheory|/MWtheory
Theoretical mass of eGFP is: MW theory=27,053
So:
Accuracy=|27050-27053|/27053=≈1.1×10−4 (≈0.011%)
The resulting molecular weight of eGFP is approximately 27,050 Da, which is in excellent agreement with the theoretical mass of ~27,053 Da, corresponding to an error of only 0.011%.
3) Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
The zoomed peak at m/z≈1473.7 likely corresponds to the 18+ charge state. However, its assignment is less certain because lower charge states at higher m/zm/zm/z often exhibit broader, less well-resolved isotopic distributions and lower signal intensity.
z=27050/1473.7~18.4
Homework: Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
![K and R]()
How many peptides will be generated from tryptic digestion of eGFP?
I) Navigate to https://web.expasy.org/peptide_mass/
II) Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
III) Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
IV) Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
There are 19 peptides generated when we use Trypsin to digest eGFP.
These residues are important because they readily accept protons, generating the multiple charge states observed in your intact protein mass spectrum. More Lys and Arg residues generally allow a protein to carry more positive charges during ESI-MS.
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
Looking at the TIC chromatogram, the labeled peaks between 0.5 and 6.0 min are: 0.61, 0.79, 1.43, 1.80, 1.85, 1.93, 2.17, 2.26, 2.54, 2.78, 3.27, 3.53, 3.59, 3.70, 4.30, 4.48, 4.64, 4.87, 5.06 and 5.43
That gives a total of 20 chromatographic peaks above ~10% relative abundance in that time window
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
The number of observed chromatographic peaks is very close to the number of peptides predicted from the tryptic digest. Using the ExPASy PeptideMass tool, a complete trypsin digestion of eGFP is predicted to generate 19 peptides.
Since approximately 20 chromatographic peaks are observed between 0.5 and 6.0 minutes, the experimental result matches the theoretical prediction quite well. The slight difference is expected and may arise from peptide co-elution, minor impurities, incomplete digestion, or the presence of peptide isoforms such as missed-cleavage products or modified peptides.
Overall, the peptide map is highly consistent with the expected tryptic digestion pattern of eGFP.
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H]+) based on its m/z and z
The m/z of the most abundant peak (the monoisotopic peak) is clearly labeled in the centre of the spectrum and it’s 525.76712
To find z we look at the separation between the isotope peaks in the zoomed-in inset. In MS, isotopes typically differ by approximately 1 Dalton. The distance between these peaks on the x-axis (delta m/z) is defined by the formula:
delta m/z=1/z
First peak (M): 525.76712
Second peak (M+1): 526.25916
Difference (delta m/z): 526.25916 - 525.76712 = 0.49204~ 0.5
Now, solving for z:
z=1/0.5=2
The charge state of this peptide is +2.
The singly charged mass represents the molecule with just one proton added. Since we know the m/z for the +2 state, we can find the neutral mass (M) and then add one proton, or use the following derivation:
The observed m/z for a charge z is:
(m/z)obs=M+z(H+)/z
To find the singly charged form [M+H]+, we can use:
[M+H]+=((m/z)obs x z)-(z-1)1.00727
(Using 1.00727 Da for the mass of a proton)
(m/z)obs x z= 525.76712 x 2= 1051.53424
Now we subtract the extra proton (since z=2, there is one more proton than the [M+H]+ form): 1051.53424 - 1.00727 = 1050.52697
The mass of the singly charged form [M+H]+ is ~ 1050.5270.
(To confirm we can see a smaller peak at mz1050.52438 on the far right of the original spectrum, which confirms this calculation!)
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm. (Recall that Accuracy=MWexperiment-MWtheoryMWtheory)
We need to identify the peptide by comparing the experimental mass derived from the spectrum with the theoretical masses of a tryptic digest of Green Fluorescent Protein (GFP).
Peptide Identification
Using a tool like PeptideMass (ExPASy) for a tryptic digestion of the GFP sequence (allowing for zero missed cleavages and focusing on the monoisotopic mass), we find a match for our experimental [M+H]+ value of ~ 1050.5270 Da.
Sequence: FEGDTLVNR (Residues 157–165 of Aequorea victoria GFP)
Composition: C46H72N13O16
Theoretical Monoisotopic Mass ([M+H]+): 1050.52142 Da
Mass Accuracy Calculation (Error in ppm)
MWexperiment= 1050.5270 Da
MWtheory= 1050.5214 Da (reference value).
Error (ppm)=|1050.5270-1050.5214|/ 1050.5214 x 10^6=5.33 ppm
An error of ~5 ppm is highly characteristic of a high-resolution TOF (Time-of-Flight) analyzer. Interestingly, if we use the secondary peak labeled at m/z ~1050.52438 visible on the right of the original spectrum, the error drops to ~2.8 ppm, which further validates the identification.
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
Based on Figure 6, the percentage of the sequence confirmed by peptide mapping is 88%.
This value represents the sequence coverage, which is the proportion of the protein’s primary amino acid sequence successfully identified through detected peptides. In the provided figure, this information is explicitly stated in two places:
The progress bar in the top-left legend: Identified: 88%
The summary text at the bottom: Chain 1 (88% coverage).
The visualization in Figure 6 uses blue highlighting to show which specific amino acids were “mapped” or detected during the LC-MS analysis. The white gaps in the sequence (such as those seen between residues 61-90 or 121-150) represent segments of the protein that were either not ionized well, were too small/large to be detected, or were lost during the sample preparation process.
A coverage of 88% is considered very high for a bottom-up proteomics experiment, indicating a successful digestion and high sensitivity in the mass spectrometry run.
Bonus Peptide Map Questions
Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c? (HINT: Use your results from Question 2 above to match the peptide molecular weight that is closest to that shown in Figure 5b. Copy and paste its sequence into this tool online to predict the fragmentation pattern based on its amino acid sequence: http://db.systemsbiology.net/proteomicsToolkit/FragIonServlet.html. What is the sequence of the eGFP peptide that best matches the fragmentation spectrum in Figure 5c?
The peptide sequence that best matches the fragmentation spectrum in Figure 5c is:
FEGDTLVNR
(Phenylalanine-Glutamate-Glycine-Aspartate-Threonine-Leucine-Valine-Asparagine-Arginine)
Justification using Fragmentation Ions:
In MS/MS fragmentation, peptides typically break along the backbone, producing y-ions (counting from the C-terminus) and b-ions (counting from the N-terminus). The prominent peaks observed in Figure 5c correspond to the theoretical y-ion series for this sequence:
Ion
Theoretical m/z
Observed in Fig 5c (approx)
y2
289.16
~289
y3
388.23
~388
y4
501.31
~501
y5
602.36
~602
y6
717.39
~717
y7
774.41
~774
y8
903.45
~903
The presence of this nearly complete y-ion series provides high-confidence confirmation that the peptide sequence is indeed FEGDTLVNR.
Does the peptide map data make sense, i.e. do the results indicate the protein is the eGFP standard? Why or why not? Consult with Figure 6, which depicts the % amino acid coverage of peptides positively identified using their calculated mass and fragmentation pattern.
Yes, the results strongly indicate that the protein is the eGFP (enhanced Green Fluorescent Protein) standard.
Sequence Coverage: As shown in Figure 6, the analysis achieved 88% sequence coverage. In proteomics, coverage above 70-80% for a single protein digest is exceptionally high and serves as a “fingerprint” that definitively identifies the protein. It means almost every part of the eGFP sequence was accounted for by detected peptides.
Mass Accuracy: The experimental mass for the FEGDTLVNR peptide showed a very low error (approx. 5.3 ppm). This level of precision is characteristic of high-quality standards measured on high-resolution instruments (like the TOF used here).
MS/MS Validation: The fragmentation pattern (the “fingerprint” of the peptide) in Figure 5c matches the theoretical predictions for a known tryptic fragment of eGFP.
Retention Time & m/z Consistency: The peptide eluted at a specific retention time (2.78 min) and yielded a consistent m/z that matches the eGFP sequence database perfectly.
In summary, the combination of high mass accuracy, high sequence coverage, and matching fragmentation patterns leaves no doubt that the sample is the eGFP standard.
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
7FU Decamer
8FU Didecamer
8FU 3-Decamer
8FU 4-Decamer
The sequence is FEGDTLVNR.
How to confirm: This sequence corresponds to the tryptic fragment of eGFP (residues 157–165).
Theoretical Mass ([M+H]+): 1050.5214 Da.
Fragmentation Pattern: The prominent peaks in Figure 5c match the y-ion series for this sequence (y2 to y8):
y2: 289.2
y3: 388.2
y4: 501.3
y5: 602.4
y6: 717.4
y7: 774.4
y8: 903.5
Q: Does the peptide map data make sense? Does it indicate the protein is the eGFP standard?
Yes, it makes perfect sense for the following reasons:
Sequence Coverage: Figure 6 shows a coverage of 88%. In proteomics, anything over 70% for a single protein digest is a definitive “fingerprint” identity.
Mass Accuracy: The error for the identified peptides (like the one above) is consistently low (approx. 5 ppm), which is the industry standard for high-resolution TOF (Time-of-Flight) instruments like the Waters Xevo G3.
MS/MS Validation: The fragmentation (MS/MS) peaks perfectly match the theoretical cleavage points of the expected eGFP tryptic peptides.
Polypeptide Subunit Name
Subunit Mass
7FU
340 kDa
8FU
400 kDa
Homework: Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Feature
Theoretical
Observed/Measured(LC-MS)
Molecular Weight
28.0066 kDa
27.9820 kDa (approx.)
SequenceCoverage
100%
88% (from Figure 6)
Mass Accuracy
0 ppm
≈5.3
The table you uploaded above the GFP section refers to Keyhole Limpet Hemocyanin (KLH). To identify the oligomeric states in Figure 7 (not shown but usually part of this section), use these calculated masses:
E7FU Decamer: 10 x 340 kDa = 3.4 MDa
8FU Didecamer: 20 x T4000 kDa =8.0 MDa
These extremely high masses are measured using CDMS (Charge Detection Mass Spectrometry), which is why they are included as a contrast to the smaller eGFP protein (28 kDa).
Accuracy=MWexperiment-MWtheory/MWtheory= 0.0056/1050.5214 x 10^6 = 5.33 ppm
Theoretical
Observed/measured on the Intact LC-MS
PPM Mass Error
Molecular weight (kDa)
28 kDa
5.33 ppm
Based on the LC-MS/MS analysis conducted, it can be concluded that the experimental results definitively identify the sample as the eGFP standard.
The high-resolution characterization yielded a sequence coverage of 88%, providing an extensive “molecular fingerprint” that matches the primary structure of the protein. This identification is further supported by the high mass accuracy observed; for instance, the tryptic peptide FEGDTLVNR was detected with a mass error of only 5.33 ppm, which is well within the acceptable range for high-performance TOF (Time-of-Flight) instrumentation.
Furthermore, the MS/MS fragmentation data confirmed the sequence through a clearly defined y-ion series, matching the theoretical predictions for eGFP. The integration of high sequence coverage, precise mass measurements, and consistent fragmentation patterns confirms both the identity and the integrity of the protein standard.
Week 11 HW: Bioproduction & Cloud Labs
Week 11 — Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Unfortunately, I couldn’t contribute but I think it’s a great project that improves creativity and working in teams. The best part of it is there’s a contribution from all over the world.
I think for next year we could have a more detailed explanation of the draw-to-made in order to create something specific but with different points of view. For example to create a plate to draw a bacteria and see what happens. I think this would be interesting.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
It contains the cellular machinery required for gene expression, including ribosomes, tRNAs, translation factors, metabolic enzymes, and aminoacyl-tRNA synthetases. It also provides T7 RNA polymerase, which transcribes DNA templates carrying a T7 promoter into mRNA.
Salts/Buffer
Potassium Glutamate: maintains intracellular-like ionic strength and supports ribosome stability and enzyme activity.
HEPES-KOH pH 7.5: buffers the reaction, keeping the pH stable for optimal transcription and translation.
Magnesium Glutamate: provides Mg²⁺, an essential cofactor for ribosomes, RNA polymerase, ATP-utilizing enzymes, and nucleotide interactions.
Potassium phosphate monobasic and dibasic: form a phosphate buffer pair that stabilizes pH and supplies phosphate for nucleotide phosphorylation and energy metabolism.
Energy / Nucleotide System
Ribose: serves as the sugar backbone precursor for nucleotide biosynthesis via salvage pathways.
Glucose: provides a slow, sustained energy source through glycolysis, enabling long-duration protein synthesis.
AMP, CMP, GMP, and UMP: they are nucleoside monophosphates that are enzymatically converted into their triphosphate forms (ATP, CTP, GTP, and UTP), which are required for RNA synthesis and energy transfer.
Guanine: acts as a precursor for GMP production through the purine salvage pathway, replenishing guanine nucleotide pools.
Translation Mix (Amino Acids)
17 Amino Acid Mix supplies the majority of amino acids required for protein synthesis.
Tyrosine is added separately because it has limited solubility in concentrated amino acid mixtures.
Cysteine is also added separately because it is chemically unstable and readily oxidizes during storage, it also forms disulfure bounds.
Additives
Nicotinamide: supports regeneration of NAD⁺, which is essential for redox balance and sustained metabolic activity in the lysate.
Backfill
Nuclease Free Water: adjusts the final reaction volume while protecting DNA and RNA from nuclease contamination.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour system uses PEP as a high-energy phosphate donor and provides all four nucleotides directly as NTPs, allowing rapid transcription and high initial protein production. In contrast, the 20-hour system relies on glucose for gradual ATP regeneration and supplies nucleotides as monophosphates (NMPs) plus ribose, which the lysate converts into NTPs over time, making the reaction more sustainable and cost-effective for long incubations.
A second key difference is that the long-duration mix simplifies the additive package: it removes components like spermidine, cAMP, folinic acid, and NAD, replacing them with nicotinamide to support prolonged metabolic activity while reducing complexity and cost.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Although GMP is not supplied directly, the lysate contains enzymes of the purine salvage pathway that convert guanine into GMP via guanine phosphoribosyltransferase. GMP is then phosphorylated to GDP and finally GTP, which is the actual substrate required by T7 RNA polymerase for RNA synthesis.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP is engineered for exceptionally robust folding, allowing it to fold efficiently even under suboptimal conditions or when fused to other proteins. Its rapid maturation and high folding efficiency make it one of the brightest and most reliable reporters in cell-free systems.
mRFP1 matures more slowly than most green fluorescent proteins, so fluorescence appears later during incubation. It also has lower intrinsic brightness, which can reduce signal intensity in short or resource-limited cell-free reactions.
mKO2 has relatively fast maturation for an orange fluorescent protein, making it well suited for time-sensitive expression assays. However, its fluorescence is somewhat sensitive to acidic conditions, so pH drift during long incubations can reduce signal.
mTurquoise2 has an exceptionally high quantum yield, making it one of the brightest cyan fluorescent proteins available. However, like many CFPs, its chromophore formation and fluorescence are highly dependent on proper folding and sufficient oxygen availability.
mScarlet-I combines very high brightness with improved maturation kinetics compared with older red fluorescent proteins. Its efficient folding and rapid chromophore formation make it particularly effective for long-term cell-free fluorescence production.
Electra2 is designed for enhanced brightness and/or unique spectral properties, but like many engineered fluorescent proteins, its performance can be sensitive to folding conditions and redox balance. Chromophore maturation is also oxygen-dependent, which can become limiting in dense or long-duration cell-free reactions.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
For mScarlet-I, increasing the concentration of nicotinamide and slightly optimizing magnesium glutamate could improve long-term fluorescence output. Nicotinamide would help sustain NAD⁺ regeneration and metabolic activity over extended incubation, while optimized magnesium would enhance ribosome function and protein synthesis without causing aggregation.
Hypothesis: Increasing nicotinamide and fine-tuning magnesium glutamate will improve mScarlet-I folding, maturation, and sustained expression, resulting in higher total red fluorescence after 36 hours.
Alternatively, for oxygen-dependent proteins such as mTurquoise2 or Electra2, reducing reaction volume or increasing the air-liquid interface could enhance oxygen diffusion, accelerating chromophore maturation and increasing final fluorescence intensity.