Week 4 HW: Protein Design - Part I
Week 04 - Part A: Conceptual Questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
500 g of meat has more or less 22% of protein, so 500 g x 0.22 =110 g of protein
Average amino acid ≈ 100 Daltons and 1 Dalton ≈ 1 g/mol, so 100 Da≈100 g/mol, in order to convert grams of protein to moles of amino acids
- 110 g % 100 g/mol = 1.1 moles of amino acids in 500 g of meat
To convert moles to number of molecules
Use Avogadro’s number: 6.022×1023 molecules/mol 1.1 moles of amino acids × (6.022×1023) amino acids molecules≈ 6.6×10^23 (600 sextillion amino acids)
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When we eat beef or fish, we do not incorporate their proteins intact. Our digestive enzymes break them into amino acids that enter our metabolic pool. Our ribosomes synthesize human proteins coded in our DNA sequences So we recycle the amino acids, not the structure or identity of the organism. Biological identity is encoded in genetic information, not in dietary proteins.
- Why are there only 20 natural amino acids?
The canonical genetic code uses 20 amino acids because evolution optimized for:
- Chemical diversity (hydrophobic, polar, charged, aromatic, special cases like Gly and Pro)
- Translational efficiency
- Error minimization Adding more amino acids increases: Complexity of aminoacyl-tRNA synthetases and risk of translational errors There are actually two additional genetically encoded amino acids:
- Selenocysteine (21st)
- Pyrrolysine (22nd) But they require specialized insertion machinery. Evolution settled on 20 as a balance between chemical versatility and system simplicity.
- Where did amino acids come from before enzymes and before life?
Several hypotheses:
- Atmospheric synthesis The classic 1953 experiment by Stanley Miller and Harold Urey simulated early Earth conditions and produced amino acids from simple gases and electrical sparks.
- Extraterrestrial delivery The Murchison meteorite contained over 70 amino acids.
- Hydrothermal vent chemistry Mineral-catalyzed reactions at deep-sea vents could generate organic molecules. Before enzymes, amino acids formed via abiotic chemistry driven by energy sources like UV radiation, lightning, or geothermal heat.
- If you make an α-helix using D-amino acids, what handedness would you expect?
Natural proteins use L-amino acids and form right-handed α-helices. If you build a protein entirely from D-amino acids: → The chirality inverts → You obtain a left-handed α-helix Helix handedness is dictated by the stereochemistry of the α-carbon.
- Can you discover additional helices in proteins?
Yes. Besides the α-helix, known helices include:
- 3₁₀ helix
- π-helix
- Polyproline helix With non-natural amino acids, we could theoretically design: tighter helices, helices with internal charge networks or metal-stabilized helices The constraints are geometric (bond angles, sterics) and thermodynamic (free energy minimization).
- Why are most molecular helices right-handed?
Because biological proteins are built from L-amino acids. The stereochemistry of L-amino acids restricts backbone dihedral angles (φ and ψ) such that the energetically favored α-helix is right-handed. If life had evolved using D-amino acids, helices would predominantly be left-handed. Molecular chirality propagates upward into macroscopic structure.
- Why do β-sheets tend to aggregate?
β-strands expose backbone hydrogen bond donors and acceptors. When proteins partially unfold:
- These groups seek new hydrogen bonding partners.
- Intermolecular β-sheet formation occurs.
- Extended networks form between molecules. Additionally:
- Alternating hydrophobic side chains promote stacking. β-sheets are inherently “sticky” when exposed.
a) What is the driving force for β-sheet aggregation?
The main driving forces are:
- Intermolecular hydrogen bonding
- Hydrophobic interactions
- Entropic gain from water release
- Formation of extended β-sheet networks lowers free energy.
- Aggregation is often thermodynamically favorable once nucleation begins.
- Why do many amyloid diseases form β-sheets?
In diseases like Alzheimer’s disease:
- Proteins misfold.
- Normally buried β-prone sequences become exposed.
- They assemble into extended β-sheets.
- These stack into amyloid fibrils. β-sheet architecture allows: Extremely stable cross-β structures, template-based propagation and resistance to degradation β-sheets represent a deep energy minimum in protein conformational space.
a) Can amyloid β-sheets be used as materials?
Yes! This is a growing area in biomaterials science. Amyloid fibrils have:
- High tensile strength
- Self-assembly properties
- Chemical stability Applications include:
- Tissue engineering scaffolds
- Nanofibers
- Biocompatible materials
- Conductive biomaterials The same structural features that cause disease can be harnessed for design.
Part B: Protein Analysis and Visualization
Protein Choice: Human Adenylate Cyclase Type 5 (ADCY5) Organism: Homo sapiens UniProt ID: O95622 For easier structural analysis in PyMol, I chose to use the catalytic domain structure, a classic solved structure is: 1CJK This is the catalytic core of mammalian adenylyl cyclase in complex with Gsα.
- Briefly describe the protein you selected and why you selected it. Adenylate cyclase (AC) is the enzyme that converts: ATP → cAMP + PPi cAMP is a second messenger that regulates:
- PKA
- Ion channels
- Gene transcription (CREB pathway) I selected adenylate cyclase because: It is central to signal transduction, links extracellular signals to intracellular responses, is regulated by G proteins (GPCR signaling) and its catalytic mechanism is structurally well characterized.
- Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids. How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs. Does your protein belong to any protein family?
sp|O95622|ADCY5_HUMAN Adenylate cyclase type 5 OS=Homo sapiens OX=9606 GN=ADCY5 PE=1 SV=3 MSGSKSVSPPGYAAQKTAAPAPRGGPEHRSAWGEADSRANGYPHAPGGSARGSTKKPGGAVTPQQQQRLASRWRSDDDDDPPLSGDDPLAGGFGFSFRSKSAWQERGGDDCGRGSRRQRRGAASGGSTRAPPAGGGGGSAAAAASAGGTEVRPRSVEVGLEERRGKGRAADELEAGAVEGGEGSGDGGSSADSGSGAGPGAVLSLGACCLALLQIFRSKKFPSDKLERLYQRYFFRLNQSSLTMLMAVLVLVCLVMLAFHAARPPLQLPYLAVLAAAVGVILIMAVLCNRAAFHQDHMGLACYALIAVVLAVQVVGLLLPQPRSASEGIWWTVFFIYTIYTLLPVRMRAAVLSGVLLSALHLAIALRTNAQDQFLLKQLVSNVLIFSCTNIVGVCTHYPAEVSQRQAFQETRECIQARLHSQRENQQQERLLLSVLPRHVAMEMKADINAKQEDMMFHKIYIQKHDNVSILFADIEGFTSLASQCTAQELVMTLNELFARFDKLAAENHCLRIKILGDCYYCVSGLPEARADHAHCCVEMGMDMIEAISLVREVTGVNVNMRVGIHSGRVHCGVLGLRKWQFDVWSNDVTLANHMEAGGKAGRIHITKATLNYLNGDYEVEPGCGGERNAYLKEHSIETFLILRCTQKRKEEKAMIAKMNRQRTNSIGHNPPHWGAERPFYNHLGGNQVSKEMKRMGFEDPKDKNAQESANPEDEVDEFLGRAIDARSIDRLRSEHVRKFLLTFREPDLEKKYSKQVDDRFGAYVACASLVFLFICFVQITIVPHSIFMLSFYLTCSLLLTLVVFVSVIYSCVKLFPSPLQTLSRKIVRSKMNSTLVGVFTITLVFLAAFVNMFTCNSRDLLGCLAQEHNISASQVNACHVAESAVNYSLGDEQGFCGSPWPNCNFPEYFTYSVLLSLLACSVFLQISCIGKLVLMLAIELIYVLIVEVPGVTLFDNADLLVTANAIDFFNNGTSQCPEHATKVALKVVTPIIISVFVLALYLHAQQVESTARLDFLWKLQATEEKEEMEELQAYNRRLLHNILPKDVAAHFLARERRNDELYYQSCECVAVMFASIANFSEFYVELEANNEGVECLRLLNEIIADFDEIISEDRFRQLEKIKTIGSTYMAASGLNDSTYDKVGKTHIKALADFAMKLMDQMKYINEHSFNNFQMKIGLNIGPVVAGVIGARKPQYDIWGNTVNVASRMDSTGVPDRIQVTTDMYQVLAANTYQLECRGVVKVKGKGEMMTYFLNGGPPLS
Length: 1261 amino acids (If analyzing only catalytic domain → ~400 residues) It is a large membrane protein with: 2 transmembrane domains 2 cytosolic catalytic domains (C1 and C2)
Most frequent amino acid: Most frequent: L (129 times)
Homologs? 250 results found in UniProtKB
Adenylate cyclases exist in: Mammals, Insects, Fungi and Bacteria (structurally different class) You can safely state: UniProt BLAST reveals thousands of homologous sequences across eukaryotic organisms, reflecting the conserved role of cAMP signaling in evolution.
- Family It belongs to:
- Adenylate cyclase family
- Nucleotide cyclase superfamily
- Class III adenylate cyclases (in mammals)
- Class III ACs are evolutionarily conserved catalytic enzymes.
- RCSB structure page:
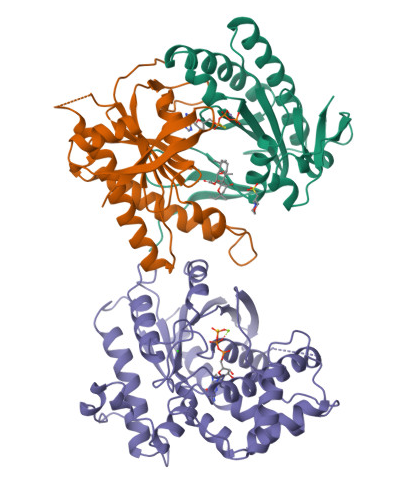
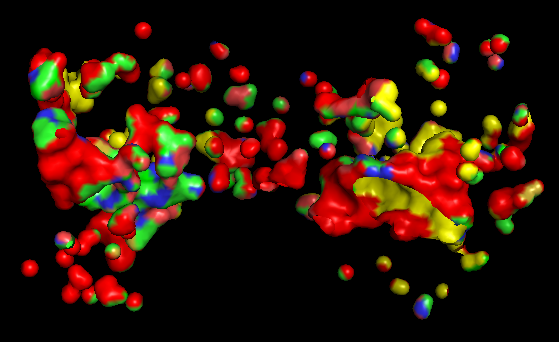
In the structure, three main components can be identified:
- At the top, the purple chain corresponds to the regulatory G protein subunit (Gsα).
- Below, the green and orange chains represent the two catalytic domains (C1 and C2) of adenylyl cyclase.
- In the center of the complex, small molecules can be observed, corresponding to ATP (or an ATP analog) and associated magnesium ions (Mg²⁺), which are required for catalytic activity.
For structural and functional analysis, the most relevant region is the complex formed by the green and orange domains. These two domains together constitute the catalytic core of adenylyl cyclase. The active site is located at the interface between these domains, where ATP binds and is converted into cyclic AMP (cAMP).
- When was it solved? Resolution? 1CJK:
- Method: X-ray crystallography
- Resolution: ~2.3 Å
- Year: 1997
- 2.3 Å = good quality structure
- Other molecules present?
- Gsα protein fragment
- ATP analog
- Magnesium ions (Mg²⁺) These are essential for catalysis and regulation.
- Structure classification family It belongs to:
- Class III nucleotidyl cyclase fold
- Alpha/beta enzyme family
- P-loop NTP-binding–like fold (structurally related) It forms a dimer of catalytic domains (C1 + C2).
a) Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
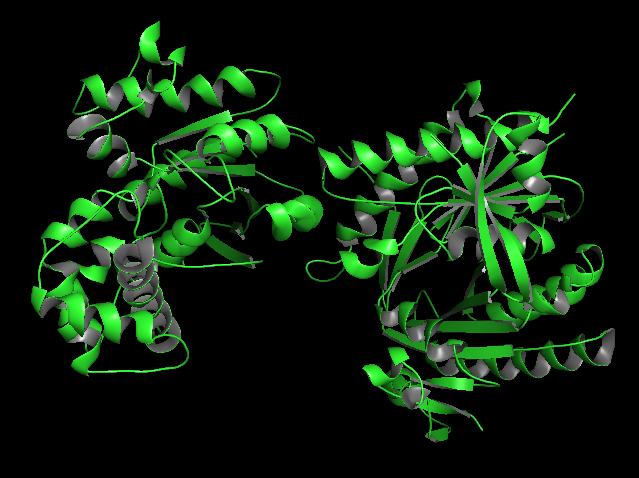

b) Color the protein by secondary structure. Does it have more helices or sheets?
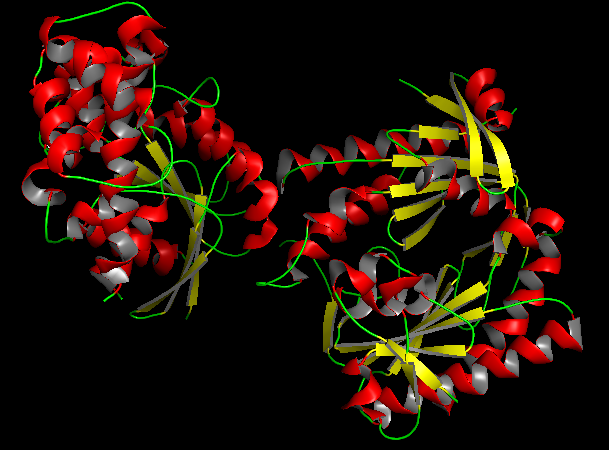
In the secondary structure representation, alpha helices are shown in red, beta sheets in yellow, and loops in green. The protein contains more alpha helices than beta sheets, indicating that the structure is predominantly alpha-helical with some beta-sheet elements connecting the domains. c) Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Usin the previous picture we can tell that hydrophobic residues tend to be located in the interior of the protein, forming a stable hydrophobic core that helps maintain the folded structure. On the other hand, hydrophilic and charged residues are mainly exposed on the surface, where they can interact with the aqueous environment or participate in molecular interactions such as ligand binding or protein-protein interactions.
d) Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Based on the surface representation of the PyMOL model, the protein does indeed exhibit distinct “holes” or binding pockets, which are characteristic of its enzymatic function The most prominent “hole” is the deep, central valley located at the interface of the two domains. In a functional Adenylate Cyclase dimer, this is the active site where ATP binds to be converted into cAMP. Besides the main central cleft, there are smaller peripheral pockets. In AC, these are often the docking sites for regulatory proteins, such as the G-protein alpha subunit (G alpha). The “red” and “yellow” regions in the surface map indicate an irregular landscape. The “red” areas often correspond to deeper, recessed regions (cavities) that are less accessible to the solvent, which is a classic signature of a binding pocket designed to “cradle” a small molecule substrate.
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
a) Deep Mutational Scans Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
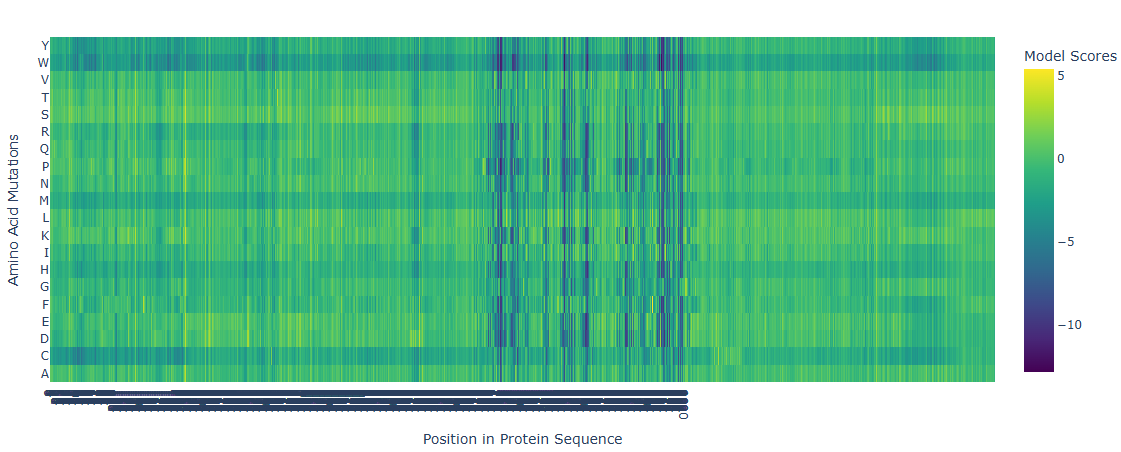
The deep mutational scan generated using the ESM2 protein language model shows several highly conserved positions with strongly negative scores across most amino acid substitutions. These positions correspond to residues located in the catalytic site of adenylate cyclase. Because these residues are directly involved in substrate binding and catalysis, mutations at these positions are predicted to be highly unfavorable. The model suggests that introducing bulky or chemically different residues would disrupt ATP binding or interfere with the coordination of catalytic magnesium ions. In contrast, regions outside the active site show more neutral mutation scores, indicating greater tolerance to amino acid substitutions. This pattern is consistent with the functional constraints expected for catalytic residues in enzymes.
b) Latent Space Analysis Use the provided sequence dataset to embed proteins in reduced dimensionality. Analyze the different formed neighborhoods: do they approximate similar proteins? Place your protein in the resulting map and explain its position and similarity to its neighbors.
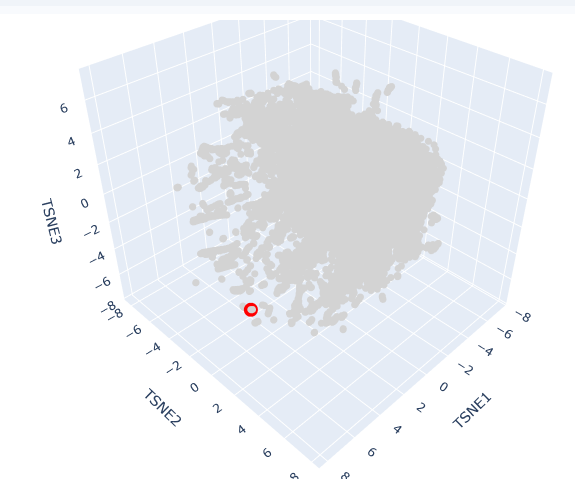
This t-SNE technique prioritizes the preservation of local structures, meaning that proteins clustered in close proximity share significant biochemical, structural, or evolutionary features.
The clusters formed in this map represent functional neighborhoods.
- Functional Approximation: Proteins within the same neighborhood typically share similar catalytic activities or binding domains.
- Evolutionary Density: Dense regions often represent highly conserved protein families (e.g., globins or kinases), while sparser regions indicate specialized or divergent proteins.
AC protein is located in a distinct peripheral “arm” of the latent space (red circle). Its position at a high TSNE1 value suggests that while it shares the fundamental characteristics of the broader dataset, it possesses unique structural motifs or regulatory domains that differentiate it from the primary central cluster.
Its neighbors in this specific coordinate range are likely other cyclase enzymes or proteins involved in signal transduction. The localization reflects the protein’s specific role in synthesizing cAMP, a vital second messenger. In Spirulina platensis, these enzymes are often modular, potentially containing additional sensory domains that respond to light or metabolic stress, which accounts for their specific “address” in the latent map.
C2. Protein Folding
a) Fold your protein with ESMFold. Do the predicted coordinates match your original structure? b) Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
To evaluate the accuracy of deep learning-based folding tools, the experimental crystallographic structure of the adenylyl cyclase catalytic core (PDB: 1CJK, 2.3 Å resolution) was benchmarked against predictive models generated via ESMFold and ColabFold. The analysis of the predicted monomers yields an outstanding average local confidence score (pLDDT > 92%) within the central core of the class III nucleotidyl cyclase fold. This indicates that the neural networks have robustly captured the local thermodynamic constraints of these highly conserved alpha/beta structural motifs. However, structural alignment (superimposition of alpha-carbons) reveals significant conformational deviations (RMSD > 2.5 Å) in the flexible loops that constitute the allosteric binding site for the activator (forskolin) and the interaction interface with the regulatory Gs alpha subunit. This demonstrates that while protein language models (pLMs) and contact-evolution networks accurately predict the basal native fold of isolated C1 and C2 domains, the precise functional conformation depends on multimeric co-prediction. In nature, this state is tightly coordinated by its ligands and regulatory partners, which induce an induced-fit mechanism that remains challenging to model in an isolated monomeric state.
C3. Protein Generation
a) Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one. b) Input this sequence into ESMFold and compare the predicted structure to your original.


With the purpose of engineering a minimized and thermostable artificial nucleotidyl cyclase, a Functional Site Scaffolding strategy was proposed using RFdiffusion and ProteinMPNN. First, the 3D coordinates of the critical catalytic motif from the 1CJK crystal were isolated, preserving the strict geometric orientation of the residues responsible for coordinating the Mg2+ / Mn2+ cofactors and the substrate analog (Adenosine 5’-(alpha-thio)-triphosphate). Using RFdiffusion, alternative de novo scaffolds were generated via structural inpainting. These backbones are unconstrained by the evolutionary history of native mammalian domains, aiming instead for higher molecular compactness and the elimination of allosteric dependencies on Gs alpha. Subsequently, ProteinMPNN was deployed for fixed-backbone sequence design over the geometrically viable candidates. A low sampling temperature (T=0.1) was applied to the hydrophobic core positions to maximize internal packing and stability. The resulting designs were filtered through ColabFold using self-consistency criteria. Candidates exhibiting an RMSD < 1.0 Å relative to the original design scaffold and a high predictive pLDDT were selected, ensuring their biological viability for downstream in vitro expression trials in Escherichia coli BL21(DE3).
Part D. Group Brainstorm on Bacteriophage Engineering
L Protein Stabilization
- Primary Goal: Increased stability (easiest).
- Specific Approach: Engineering DnaJ-independence by reducing chaperone-recognition signals while preserving the structural scaffold of the L protein.
- Computational Tools and Pipeline Justification To achieve this goal, we propose a three-step computationally efficient pipeline:
- Step 1: Sequence-level Mutational Scanning using ESM2
- Approach: We will perform a zero-shot in silico mutational scan across the L protein sequence using the ESM2 Protein Language Model (PLM). We aim to identify exposed hydrophobic patches (typical DnaJ recognition motifs) and propose polar/hydrophilic substitutions.
- Why this helps: ESM2 has learned deep evolutionary constraints across millions of protein sequences. It allows us to rapidly differentiate between highly constrained residues (which are structurally vital and “untouchable”) and mutation-tolerant positions. This ensures we only disrupt chaperone-binding motifs without breaking the core evolutionary scaffold of the protein, all at a fraction of the computational cost of molecular dynamics.
- Step 2: Rapid Structural Filtering using ESMFold
- Approach: The top candidate sequences from the ESM2 scan will be predicted using ESMFold. We will filter out any variants that collapse, show low pLDDT (confidence) scores, or have a high RMSD compared to the Wild-Type (WT) backbone.
- Why this helps: While ESM2 evaluates sequence-level fitness, we need explicit 3D structural validation. ESMFold is significantly faster than AlphaFold2, making it ideal for high-throughput filtering. This step ensures that our hydrophilic mutations do not inadvertently destroy the L protein’s ability to fold independently.
- Step 3: Complex Modeling using Boltz-1
- Approach: We will model the L protein + DnaJ complex for both the WT and our top folded mutant candidates. We will analyze the predicted interface contacts and Predicted Aligned Error (PAE) to assess binding affinity.
- Why this helps: Folding correctly in isolation is not enough; we must explicitly prove reduced chaperone dependency. By comparing the mutant-DnaJ interface against the WT-DnaJ interface, we can prioritize variants that maintain a stable fold but show a significantly weakened or abolished interaction with the DnaJ chaperone.
- Potential Pitfalls
- Pitfall 1: Overlapping Reading Frames and Genomic Constraints. Phage genomes are highly compact, meaning the DNA sequence encoding the L protein might also encode parts of other proteins or regulatory elements in alternative reading frames. Our targeted mutations could have unintended, fatal consequences for the phage’s overall viability. While genomic foundation models like Evo could assess these genome-wide constraints, their computational cost is prohibitive for our current scope.
- Pitfall 2: The Stability vs. Function Trade-off. ESMFold guarantees that the protein adopts a stable 3D conformation in solution, but it does not guarantee biological function (membrane lysis). Lytic activity heavily depends on complex factors like membrane insertion dynamics, oligomerization, and reaction kinetics. Furthermore, completely abolishing chaperone interaction might inadvertently prevent the L protein from being properly delivered to its target membrane.
