Subsections of Homework
Week 1: Principles and practices
1. First, describe a biological engineering application or tool you want to develop and why.
I’m curious about cellular agriculture and lab-grown meat. in this project im proposing to develop a living, light-activated scaffold that produces and spatially distributes oxygen inside a growing tissue. this is one of the needed early steps toward making a thick, steak-like cut rather than thin sheets or ground meat. Today, animal cells grown for food struggle beyond a few millimeters because oxygen and nutrients don’t diffuse well. the interior becomes starved and dies unless you add complex and expensive hardware. My project reframes that bottleneck as a biological engineering opportunity: a biofabricated “breathing” matrix that couples geometry + metabolism so that illumination drives localized oxygen generation and makes it visible and tunable. In this course’s context it would be explored as a living installation: a translucent scaffold whose oxygen field can be visualized in real time under light/dark cycles, producing both data and an intuitive, aesthetic demonstration of how engineered living materials might reduce reliance on expensive hardware in future cultivated-meat systems.
2. Describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
Governance policy goal #1: ensuring Biosafety and non-malfeasance
Make sure the system can’t cause harm to people, ecosystems, or lab staff through accidental release, contamination, or unsafe handling.
- Making sure every engineered organism used in the project is not viable outside of the controlled lab conditions.
- Contamination monitoring and incident reporting standards for all project related activities in the wet lab.
- Use ‘Low Risk’ Chassis Organisms and avoid incorporating traits that increase survivability of harmful actions.
Governance policy goal #2: maximize public benefit
directing this tool toward clear societal value—lowering barriers to safer, more resource-efficient cellular agriculture research and accelerating pathways to scalable cultivated meat.
- Define ‘Constructive Use’ criteria and require study to explicitly show qualitative improvement over one of the following criteria : lower resource use, improving oxygen diffusion limits, reducing complexity of tissue cultivation, reducing cost of tissue cultivation.
- Encourage standardized open-source documentation for non-sensitive aspects like negative results and measurement methods.
Governance policy goal #3: promote equity & autonomy
Ensure this tool’s benefits are broadly shared rather than concentrated, and that people retain meaningful choice and informed consent.
- cultural and livelihood impacts: include early stakeholder perspectives (food cultures, labor/farming communities) to reduce the risk that “technical success” drives social harm or displacement.
- if such a risk is assesed as high, co-develope a slow transition plan.
3. Describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Action 1: Project specified containnment regime
Purpose: Replace generic lab norms with a project-specific containment regime so accidental release and unsafe handling are structurally less likely.
Design: A mandatory SOP covering labeling, storage, transport, and validated inactivation/disposal for every culture/run. chain-of-custody log for strains and materials.
Assumptions: Containment does not undermine biological performance.
Risks: Failure - checkbox compliance or incomplete logs, resulting in bad lab norms and culture. Success risk - issue of a compliance-overhead that will evemtually become a barrier for smaller teams unless tooling/templates reduce burden.
Action 2: Pre-registered public-benefit targets
Purpose: Ensure the work advances constructive uses by tying it to explicit, testable public-benefit goals rather than novelty.
Design: Before experiments, declare 1–2 primary public-benefit targets (reduced process complexity/reduced resource use) and define how they will be evaluated. after, conduct a brief impact evaluation including tradeoffs and limits.
Assumptions: Labs track real constraints; teams won’t optimize for non-beneficial metrics.
Risks: Failure - metric gaming or proxies that don’t translate. Success risk - the encourageing or incentivizing a ‘follower’ culture where the first metrics to have consenseus are repeated as defaults.
Action 3: stakeholder review & benefit-sharing
Purpose: Ensure “success” does not override cultural values, informed consent, or fairness in who benefits from the technology.
Design: A stakeholder checkpoint with at least two external perspectives (labor/farming and food culture/ethics) plus explicit benefit-sharing commitments (open standards, non-exclusive licensing norms, accessible documentation, and clear communication of uncertainties to support informed choice).
Assumptions: Early stakeholder engagement surfaces blind spots. benefit-sharing commitments foster trust between community and research/venture.
Risks: Failure risk -tokenism or performative consultation. Success risk - added friction slows iteration—but that is an intentional tradeoff to protect autonomy and prevent concentrated capture.
Action 4: responsible release of documentation
Purpose: Maximize reproducibility and shared learning while reducing misuse risk.
Design: Two publication layers: Open (concept, results, non-sensitive documentation) and Restricted (step-by-step replication details and other speceficities). an external mechanism decides classification of research documents. access to the Restricted layer will be granted by same mechanism.
Assumptions: Sensitive details can be identified; restriction won’t destroy scientific value.
Risks: Failure risk - misassuming the layer definitions as too open (misuse) or too closed (no benefit). Success risk - restricted knowledge becomes a chokepoint that concentrates power and limits equitable access.
Score (from 1-3 with, 1 as the best) each of your governance actions against your rubric of policy goals.

Drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
I would prioritize a combined governance package aimed at two audiences: First would be an institutional biosafety governance body (such as an IBC) and the second would focus on field-facing actors (research funders, journals, and cultivated-meat research networks). The core package is Action 1 (Containment Regime), Action 2 (Pre-registered public-benefit targets), and Action 3 (Stakeholder review & benefit sharing). I would start with Action 1 because it most directly reduces non-malfeasance risks (accidental release, unsafe handling, or unmanaged contamination). Action 2 ensures the project is oriented toward constructive use by requiring explicit evidence of benefit rather than novelty alone. Action 3 is prioritized early because success in this domain can create downstream social impacts—such as concentration of ownership or displacement pressures—so stakeholder input and benefit-sharing commitments are necessary to protect autonomy and legitimacy.
The primary trade-off is speed and ease of iteration vs. safety, accountability, and equity. These actions add overhead (documentation, evaluation, review), and if implemented too rigidly they could become barriers for smaller teams. the assumption is that this can be mitigated with lightweight templates and clear defaults. A key uncertainty is whether lab-scale proxies for oxygen distribution translate to real thick-tissue outcomes. Action 2 should make a clear and defined outline through central—pre-registering of what counts as improvement, to structured and goal-oriented claims rather than post-hoc storytelling. Overall, this prioritized set aims to make the project safe by default, oriented toward public benefit, and socially accountable if it succeeds.
References
Rouwkema, J. & Khademhosseini, A. (2008). Vascularization in tissue engineering.
https://www.sciencedirect.com/science/article/abs/pii/S0167779908001583Lovett, M., Lee, K., Edwards, A. & Kaplan, D. L. (2009). Vascularization Strategies for Tissue Engineering.
https://pmc.ncbi.nlm.nih.gov/articles/PMC2817665/The Good Food Institute (2024). Trends in cultivated meat scale-up and bioprocessing.
https://gfi.org/wp-content/uploads/2024/04/Trends-in-cultivated-meat-scale-up-and-bioprocessing.pdf
Prof. Jacobson’s Questions:
- Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Polymerase makes about 1 error per 106 bases. The human genome is about 3.2×109 bp, so a genome-length copy would imply roughly ~3,200 errors. Biology closes that gap by layering proofreading and post-replication mismatch repair on top of polymerase.
- How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Because many amino acids have multiple equivalent codons, the number of possible DNA sequences encoding the same protein is huge. In practice, many variants fail because the codon choice will change translation efficiency and because sequence composition changes mRNA structure.
Dr. LeProust’s Questions:
- What’s the most commonly used method for oligo synthesis currently?
The most common method is solid-phase phosphoramidite chemical synthesis. It builds DNA one nucleotide at a time on a solid support through repeated cycles (coupling, capping, oxidation, deblock).
- Why is it difficult to make oligos longer than 200nt via direct synthesis?
Because each added base requires another chemical cycle, small inefficiencies and side reactions add-up over hundreds of cycles.
- Why can’t you make a 2000bp gene via direct oligo synthesis?
A 2000 bp gene would require ~2000 sequential synthesis cycles, making correct full-length yield very low because errors and truncations will become significant along the process.
Prof. Church’s Question:
Using Google & Prof. Church’s slide #4 : What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The EAAs in all animals are Arginine, Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, and Valine. To my understanding ‘The Lysine Contingency’ was a strategy in the ‘Jurassic Park’ fiction implemented by Henry Wu to disable dinosaurs’ ability to create Lysine by themselves, thus forcing them to obtain it through supplements provided by the park, or die. Since I now know no known-animal has the ability to self-produce the EAA Lysine, this renders as complete nonsense - because the dinosaurs did not have the ability to create the Lysine in the first place. The concept of a kill-switch however, still stands valid as a bio-safety measure.
References:
- Yongqing Hou, Guoyao Wu (2016) Nutritionally Essential Amino Acids https://www.sciencedirect.com/science/article/pii/S216183132201273X
- Lysine Contingency https://jurassicpark.fandom.com/wiki/Lysine_contingency
Week 10: Week 10 — Advanced Imaging & Measurement Technology
Final Project Homework:
Identify at least one aspect of your project that you will measure.
One aspect I will measure is the spatial response of receiver cells to a signaling source, by quantifying reporter intensity as a function of distance from sender cells. Another aspect I will measure, especially for Aim 0.5, is the activation of the fat-related proxy, using eGFP fluorescence as a readout for co-expression of ‘TesA in the receiver cells. Together, these give me two measurable outputs: whether the system forms a real gradient in space, and whether that gradient is successfully coupled to a fat-related expression program.
Describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements. What are the technologies you will use?
I would like to measure the following metrics:
a. I will confirm fragment sizes during cloning and validation steps to validate and check whether the expected DNA products are present using gel electrophoresis.
b. Receiver activation by tracking eGFP fluorescence intensity over time and, when possible, as a function of distance from sender cells using video timelapse. This would let me see whether the circuit is simply on or off, and whether it produces any spatial pattern rather than a uniform response. I would compare this against controls such as receiver-only culture.
c. ‘TesA expression, since the conceptual point of Aim 0.5 is to link fluorescence to a fat-related expression program. I would ideally use a protein-level assay kit such as an 4–20% Mini-PROTEAN® TGX™ Precast Gel to check whether a protein of the expected size is being produced. In principle, mass spectrometry could also be used to confirm protein identity more precisely, although for the scope of Aim 0.5 it would likely be a more advanced method than I strictly need.
d. Whether expression of ‘TesA corresponds to any increase in free fatty acid production (FFA). Even a coarse measurement here would strengthen the experiment, because it would move the result toward actual biological activity. For this I could use a free fatty acid assay kit / colorimetric assay such as this MAK466 Sigma-Aldrich Free Fatty Acid assay kit for a crude evaluation.
e. I would monitor culture performance: growth, viability, and environmental parameters like media conditions, temperature, and possibly pH. Since both the sender and receiver rely on engineered expression, these background conditions matter because weak growth or excessive burden could distort the interpretation of fluorescence or proxy expression. I plan to use standard microbiology measurements such as optical density, growth observations, and controlled culture conditions.
Week 11: Bioproduction & Cloud Labs

Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
What I contributed to the community bioart project
I looked up the final artwork, but I could only find the second version, which unfortunatley I could not contribute to in time. Even so, I found the project very compelling. Since I missed this round’s contributions, and respecting the week 11 homework description would be very happy to join HTGAA as a TA next semester!
What I liked about the project
What I liked most was its collaborative r/place-like quality and the nod to an original cultural phenomenon shaped by online communities. In the context of this course, that idea became even more meaningful, because it was extended through real distributed participation: people in different locations around the world were all taking part in the same collective experiment. I appreciated how the project made that network visible through a shared visual output.
What could be improved for next year
One possible improvement would be to expand the expressive range of the system by introducing more colors, more wells, or even a custom color-mixing interface. That could make the final artifact richer and allow participants to contribute with more nuance and variation.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Roles of each component in the cell-free reaction
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
The lysate provides the core molecular machinery needed for cell-free transcription and translation, including ribosomes, tRNAs, metabolic enzymes, and translation factors. Because this lysate comes from BL21 (DE3) Star, it also includes T7 RNA polymerase, which transcribes genes placed under a T7 promoter.
Salts / Buffer
Potassium Glutamate
Potassium glutamate helps recreate an intracellular-like ionic environment and supports ribosome and enzyme function during transcription and translation. It is often used as a major salt in bacterial cell-free systems because it better mimics cytoplasmic conditions than simple chloride salts.
HEPES-KOH pH 7.5
HEPES-KOH is the buffering system that keeps the reaction near a stable physiological pH. Maintaining pH is important because both transcription and translation enzymes are sensitive to acid/base changes over the course of the reaction.
Magnesium Glutamate
Magnesium is an essential cofactor for many enzymes in the reaction, especially RNA polymerase, ribosomes, and enzymes involved in nucleotide handling. If magnesium is too low or too high, the reaction can fail or become inefficient, so it is one of the most critical tuning parameters in CFPS.
Potassium phosphate monobasic
This phosphate salt helps contribute to the buffering and ionic balance of the reaction. Together with the dibasic form, it helps stabilize pH and phosphate availability in the system.
Potassium phosphate dibasic
This works with monobasic phosphate as part of a conjugate buffer pair. It helps maintain pH stability and contributes to the chemical environment needed for efficient enzyme activity.
Energy / Nucleotide System
Ribose
Ribose serves as a carbon source that can support nucleotide and energy metabolism in longer-running cell-free reactions. It is especially relevant in systems that regenerate resources over time rather than relying only on a single high-energy phosphate donor.
Glucose
Glucose provides an additional metabolic energy source that can feed endogenous enzymatic pathways in the lysate. In longer reactions, it helps sustain ATP regeneration indirectly and support continued protein production.
AMP
AMP is a nucleotide monophosphate precursor that can be recycled into higher-energy nucleotide forms in extended energy-regeneration systems. In this setup it supports rebuilding the nucleotide pool rather than supplying ATP directly.
CMP
CMP is a precursor for cytidine nucleotide regeneration and helps replenish the RNA-building pool needed for transcription. It is part of the lower-energy nucleotide set used in longer-duration reactions.
GMP
GMP supports regeneration of guanosine nucleotide pools used in RNA synthesis and other reaction processes. Like the other monophosphates, it is part of a resource-efficient long-duration system.
UMP
UMP is the uridine nucleotide precursor used to support RNA synthesis after regeneration into higher-energy forms. Its inclusion helps sustain transcription over longer reaction times.
Guanine
Guanine is a free nucleobase that can feed salvage pathways in the lysate to help replenish guanine nucleotide pools. It supports longer-term reaction economy by contributing to nucleotide regeneration.
Translation Mix (Amino Acids)
17 Amino Acid Mix
This mixture provides most of the amino acids needed as building blocks for protein synthesis. They are consumed directly by the ribosome as the target protein is translated.
Tyrosine
Tyrosine is often added separately because of solubility or stability issues in concentrated amino acid mixes. It serves the same role as the others: supplying a required amino acid for protein synthesis.
Cysteine
Cysteine is also commonly handled separately because it is chemically more reactive and less stable in stock solutions. It is required for translation of proteins containing cysteine residues and can also influence redox-sensitive folding contexts.
Additives
Nicotinamide
Nicotinamide supports metabolic cofactor balance because it is related to NAD-dependent biochemical pathways. In cell-free reactions, additives like this can help maintain metabolic activity and improve reaction longevity.
Backfill
Nuclease Free Water
Nuclease-free water is used to bring the reaction to its final volume without introducing DNases or RNases that could degrade templates or transcripts. It acts as the clean solvent base for the reaction mixture.
Main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix
The main difference is that the 1-hour optimized PEP-NTP mix is built for fast, high-output expression using directly supplied high-energy nucleotide triphosphates and a strong phosphate-based energy donor such as PEP. By contrast, the 20-hour NMP-Ribose-Glucose mix is designed for longer-duration reactions and relies on a more gradual metabolic regeneration strategy, using nucleotide monophosphates plus carbon sources like ribose and glucose to sustain the reaction over time.
In other words, the PEP-NTP system prioritizes short-term speed and strong expression, while the NMP-Ribose-Glucose system prioritizes resource efficiency and longer reaction lifetime. The second mix is generally more metabolically distributed and slower, but better suited for extended incubation.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
sfGFP
sfGFP is useful in cell-free systems because it is engineered for robust folding and is also a fast-maturing GFP variant, which helps fluorescence appear quickly and reliably even when expression conditions are not ideal. Like other GFP-like proteins, however, chromophore maturation still depends on oxygen, so fluorescence can lag if oxygen availability is limited.
mRFP1
mRFP1 is a slowly maturing red fluorescent protein, so the protein may be present before the fluorescence is fully visible, which can make short cell-free reactions underestimate expression. It is reported to have relatively low acid sensitivity, which can help preserve signal if the reaction drifts slightly in pH over long incubations.
mKO2
mKO2 is generally valued because it is a relatively fast-maturing orange fluorescent protein, which is helpful when comparing fluorescence over limited reaction times. It also has moderate acid sensitivity, so pH drift in a long cell-free incubation could reduce its apparent brightness.
mTurquoise2
mTurquoise2 is known for its high brightness and photostability, which makes it a strong reporter when repeated imaging or long observation windows are needed. As a GFP-family fluorophore, it still requires oxygen-dependent chromophore maturation, so final fluorescence depends not only on translation but also on post-translational maturation conditions.
mScarlet-I
mScarlet-I is especially attractive in cell-free systems because it was engineered for accelerated maturation relative to mScarlet, which helps red signal appear more quickly in practical experiments. This is useful in long but finite reactions such as 20–36 hour incubations, where maturation speed strongly affects how much fluorescence is visible by the endpoint.
Electra2
Electra2 is a newer blue fluorescent protein, so one relevant consideration is that its performance may be more context-dependent and less broadly benchmarked than older standards like sfGFP or mTurquoise2. As with other fluorescent proteins, usable signal still depends on proper folding and chromophore maturation, so suboptimal reaction chemistry could reduce apparent output even if the protein is translated.
Hypothesis for improving fluorescence over a 36-hour incubation
Hypothesis: Increasing the buffering capacity of the cell-free mastermix and carefully re-optimizing magnesium concentration will improve the 36-hour fluorescence endpoint of mKO2, because stronger pH stability should reduce acid-related signal loss while optimized Mg²⁺ should support translation and folding efficiency.
I would therefore test a condition with slightly higher or more stable buffer capacity together with a small Mg²⁺ titration series. The expected effect is that mKO2 would retain more of its fluorescence over long incubation, rather than losing apparent brightness because of pH drift or suboptimal folding conditions.
Week 2: DNA read write edit
for this week’s HW assignment I’ve chosen the protein mCherry. mCherry is a protein that expresses in red flourescent light emmitance. Fusing it to another protein will enable use to discern wheter the ‘other’ protein is expressed, by visibly observing the red flourescent light. essentially, mCherry functions as a global process visualisation tool across multiple SynBio applications.
Googling mCherry Protein I arrived at the uniprot.org database where I’ve obtained the mCherry sequence:
tr|A0A4D6FVK6|A0A4D6FVK6_ECOLI MCHERRY OS=Escherichia coli str. K-12 substr. MG1655 OX=511145 GN=mCherry PE=1 SV=1 MVSKGEEDNMAIIKEFMRFKVHMEGSVNGHEFEIEGEGEGRPYEGTQTAKLKVTKGGPLP FAWDILSPQFMYGSKAYVKHPADIPDYLKLSFPEGFKWERVMNFEDGGVVTVTQDSSLQD GEFIYKVKLRGTNFPSDGPVMQKKTMGWEASSERMYPEDGALKGEIKQRLKLKDGGHYDA EVKTTYKAKKPVQLPGAYNVNIKLDITSHNEDYTIVEQYERAEGRHSTGGMDELYK
Reverse translating amino sequence to dna codons using Biorinformatics.org’s Reverse Translate tool I got a 708 codon sequence:
atggtgagcaaaggcgaagaagataacatggcgattattaaagaatttatgcgctttaaa gtgcatatggaaggcagcgtgaacggccatgaatttgaaattgaaggcgaaggcgaaggc cgcccgtatgaaggcacccagaccgcgaaactgaaagtgaccaaaggcggcccgctgccg tttgcgtgggatattctgagcccgcagtttatgtatggcagcaaagcgtatgtgaaacat ccggcggatattccggattatctgaaactgagctttccggaaggctttaaatgggaacgc gtgatgaactttgaagatggcggcgtggtgaccgtgacccaggatagcagcctgcaggat ggcgaatttatttataaagtgaaactgcgcggcaccaactttccgagcgatggcccggtg atgcagaaaaaaaccatgggctgggaagcgagcagcgaacgcatgtatccggaagatggc gcgctgaaaggcgaaattaaacagcgcctgaaactgaaagatggcggccattatgatgcg gaagtgaaaaccacctataaagcgaaaaaaccggtgcagctgccgggcgcgtataacgtg aacattaaactggatattaccagccataacgaagattataccattgtggaacagtatgaa cgcgcggaaggccgccatagcaccggcggcatggatgaactgtataaa
Using the Gensmart Codon Opt Tool to optimise the above codon seq for E.coli. I chose to opt for E.coli because it is my understanding that this organism serves as a common platform for SynBio uses, and beacuse i’m new to the field, I rather stick to common practices to solidify my understanding when taking first steps in prot-design.
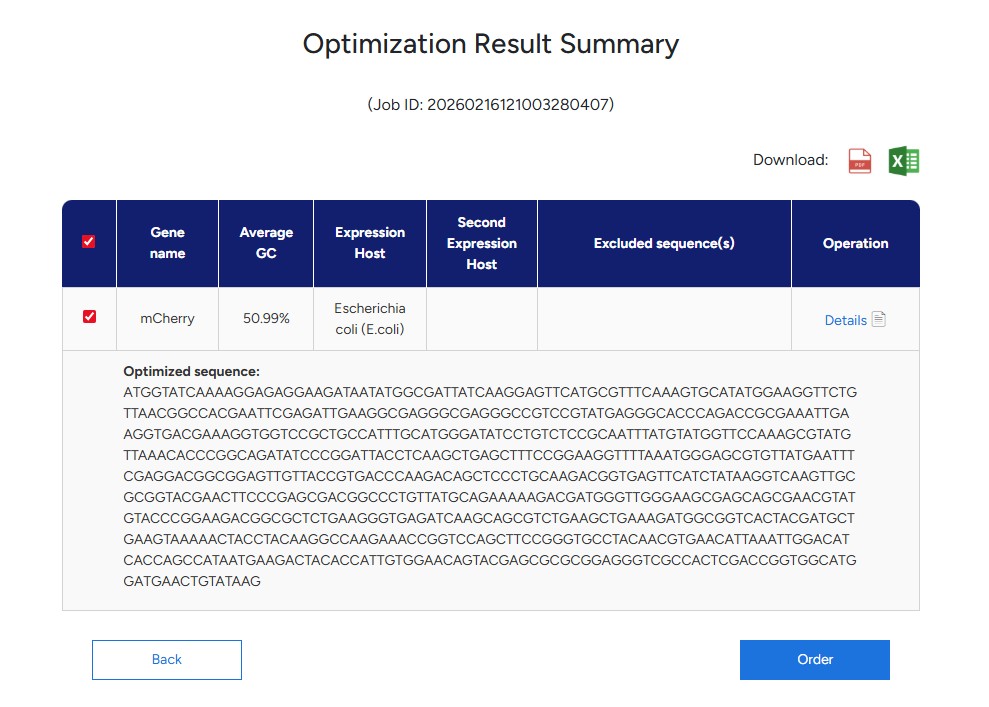
Resulting optimisation codon seq:
ATGGTATCAAAAGGAGAGGAAGATAATATGGCGATTATCAAGGAGTTCATGCGTTTCAAAGTGCATATGGAAGGTTCTGTTAACGGCCACGAATTCGAGATTGAAGGCGAGGGCGAGGGCCGTCCGTATGAGGGCACCCAGACCGCGAAATTGAAGGTGACGAAAGGTGGTCCGCTGCCATTTGCATGGGATATCCTGTCTCCGCAATTTATGTATGGTTCCAAAGCGTATGTTAAACACCCGGCAGATATCCCGGATTACCTCAAGCTGAGCTTTCCGGAAGGTTTTAAATGGGAGCGTGTTATGAATTTCGAGGACGGCGGAGTTGTTACCGTGACCCAAGACAGCTCCCTGCAAGACGGTGAGTTCATCTATAAGGTCAAGTTGCGCGGTACGAACTTCCCGAGCGACGGCCCTGTTATGCAGAAAAAGACGATGGGTTGGGAAGCGAGCAGCGAACGTATGTACCCGGAAGACGGCGCTCTGAAGGGTGAGATCAAGCAGCGTCTGAAGCTGAAAGATGGCGGTCACTACGATGCTGAAGTAAAAACTACCTACAAGGCCAAGAAACCGGTCCAGCTTCCGGGTGCCTACAACGTGAACATTAAATTGGACATCACCAGCCATAATGAAGACTACACCATTGTGGAACAGTACGAGCGCGCGGAGGGTCGCCACTCGACCGGTGGCATGGATGAACTGTATAAG
Releying on my understanding of ‘The Central Dogma’, one ’technology’ to produce the protein from the above sequence is the protein creation process that happens inside the Rybozome which is located in a cell’s nucleus. The Rybozome (which is also called R-RNA) is the site that ’takes-in’ M-RNA after it has transcribed a DNA sequence and undergone some editing to the original transcribing. The Rybozome has 2 parts. M-RNA sits in the small part, and gets read by the Rybozome which translates the codons it reads in the M-RNA, and ‘calls’ for T-RNAs to arrive at the big part of the Rybozome. Each T-RNA ‘holds’ and Amino Acid, and when ‘called’ by a respective codon sequence, it arrives at the Rybozome to ‘hand over’ that amino acid. Amino Acids bind to one another in the Rybozome’s bigger part and start forming a chain. This process repeats until the Rybozome ‘finishes’ reading the entire sequence, resulting in a chain of amino acids that will leave the Rybozome and continue to other processes that will eventually result in the aminos folding into a 3D structure that we call a protein.
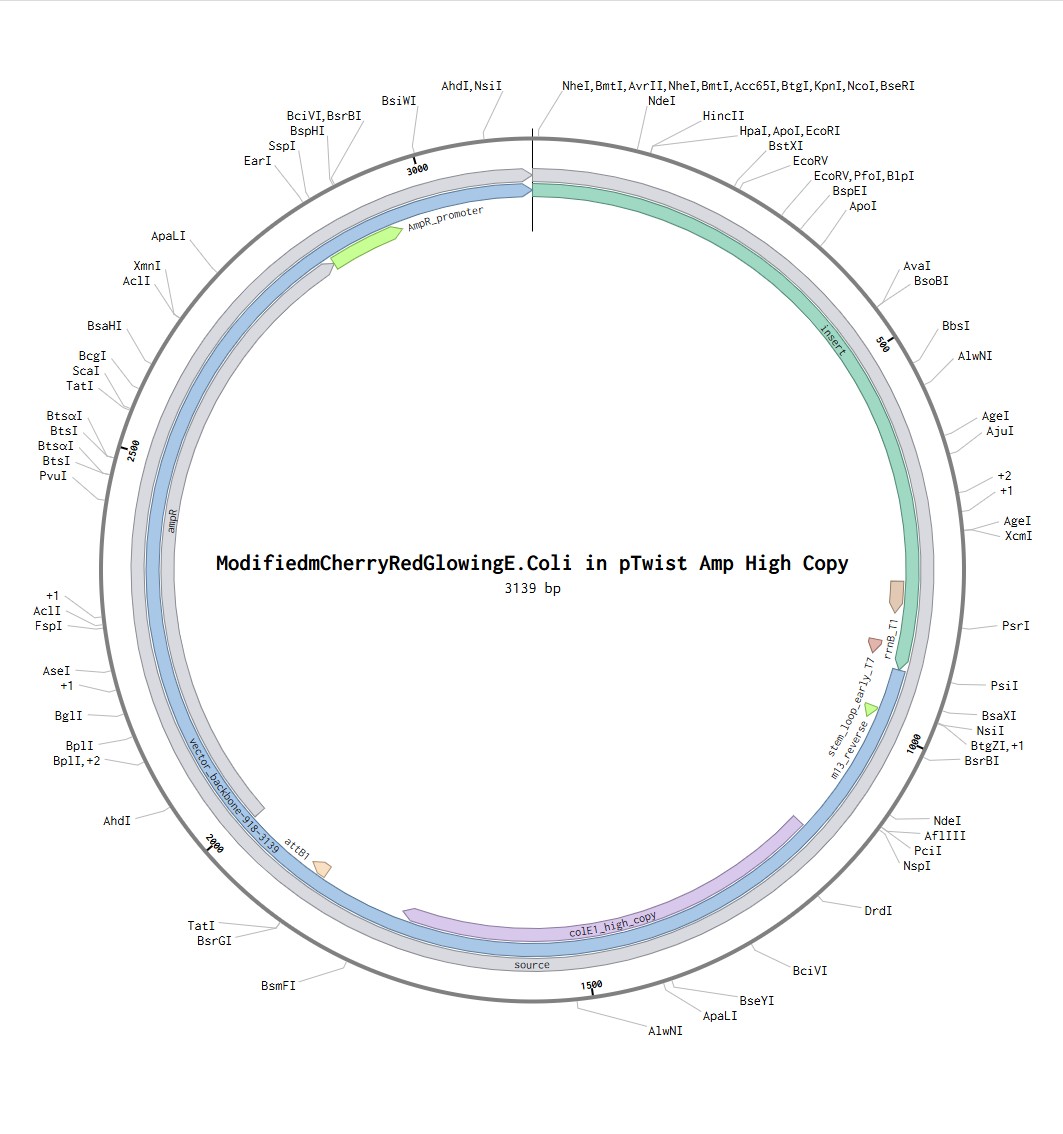
Putting my optimized codon seq into Benchling with the additional sequences provided, I have created a share link to the Benchling file.
Following the steps in the Twist instructions part, I was able to export my Benchling file in .fasta format, upload it to the Twist platform, choose pTwist Amp High Copy Vector and download the .gb file. reuploaded into benchling I can see the resulting expressions cassette! Whohoo (:
What DNA would you want to sequence (e.g., read) and why?
Within the given context, of being introduced to Synbio, I am inclined towards sequencing DNA of organisms and proteins that have something to do with processes of photosynthesis, CO2 sequestration and or flavor and aroma enhancement. The former two have versatile applications in the emerging field of bio-chemical energy production, while the latter have more focused applications in the field of cultivated lab meat, both of them I find highly impactful and worthwhile endeavors to pursue. In particular, I can propose to read the DNA of the RuBisCO enzyme, that binds a CO2 molecule to a RuBP molecule and creates two 3-carbon molecules. This is a part of the larger photosynthesis process where an organism is converting light energy into sugars and CO2 is being ‘stored’ in sugars. At the same time the photosynthetic process could also be tapped into, converting free electrons created in the process to electrical energy. Enhancing the processes underlying photosynthesis means potentially improving electrical energy yield and carbon sequestration, two much needed capabilities in our time.
In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
For reading RuBisCO genes, from my understanding, I would go for the Sanger reading process. It’s considered high accuracy, and fit for sequences that are on the order of ~700bp.
Is your method first-, second- or third-generation or other? How so?
Sanger is considered a first-gen method due to the fact that it reads the sequence one-by-one. ‘Next-Gen Methods’ tend to read arrays in parallel making them faster and more cost-effective.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
A Sanger reaction will typically require the reaction-medium, a template DNA (could be a purified PCR or a plasmid), and a primer. If using a Purified PCR, the primers used for the PCR process would work effectively as primers for the Sanger process as well. Each primer will read roughly ~700bp so for reading longer sequences you would need to tile primers one after the other. If the sequence is in the right size, you could sequence it in 2 ‘runs’ using a forward reading primer and a reverse reading primer and notice where they overlap.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
Lets say I am performing the Sanger reaction with a purified PCR. I would take the Sanger mix (the medium in which the Sanger reaction occurs), my purified RuBisCO PCRs, and the primer I used for the PCR creation itself. Based on the size of the PCR (in bp units) I will decide how many primers I need because Sanger reads around ~700bp. Preferably the best results are achieved either with a sequence length that is suitable for a single primer use or dual-primer use, in which case I will utilize both a forward reading primer and a reverse reading primer. ‘base calling’, or identification of ATCG nucleotides is done by attaching a fluorescent ddNTP after bases occasionally. The ddNTP stops the sequence’s extension, serving as a ‘cap’ of sorts. While copying the sequence many times, the RNA polymerase will attach the ddNTP label multiple times, effectively creating many copies of the DNA in all possible lengths. These ddNTP fluorescent caps will serve as a labels that could be seen after the entire batch had been separated by length in an electrophoresis process. A sorting algorithm is then deployed, identifying the different colors and lengths of each sequence that shows up, and translating the colors back to bases. so essentially cutting dna and attaching colored labels to cuts, ordering the cuts by size, translating color + size results back to bases sequence using software.
What is the output of your chosen sequencing technology?
The output of the Sanger process is the chromatogram, a color-coded graph that shows peaks where the respective base had been identified along the length of the sequence, and the base-called sequence that is derived from it.
What DNA would you want to synthesize (e.g., write) and why?
Going back to the first question in this assignment, in the given context, I am inclined towards modifying DNA of organisms and proteins that have something to do with processes of photosynthesis, CO2 sequestration and or flavor and aroma enhancement. The former two have versatile applications in the emerging field of bio-chemical energy production, while the latter have more focused applications in the field of cultivated lab meat, both of them I find highly impactful and worthwhile endeavors to pursue. In particular, I can propose to write the DNA of the RuBisCO enzyme, enhancing it’s ability to bind CO2 molecules to RuBP molecules more effectively. Enhancing the processes underlying photosynthesis means potentially improving electrical energy yield and carbon sequestration,.
What technology or technologies would you use to perform this DNA synthesis and why? How does your technology of choice edit DNA? What are the essential steps?
I believe that for modifying a DNA sequence I would order oligos of the chosen sequence, perform a fragment replacement on the specific point in the sequence I assume will result in enhancing the desired trait (In my case is improving the ability of RuBisCO enzyme to bind CO2 molecules to RuBP molecule), assemble all fragments together in assembly process (Gibson?), and then sequence the entire dna to verify that I’ve assembled it correctly.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
First I will need to make sure I have the proper assessment tools to determine success or failure. If the RuBisCO enzyme is binding CO2 molecules, it makes sense to creating a testing environment where I could measure rate of of CO2 dissappearance for example. I would then proceed to select sites for the mutation to happen (multiple sites or a specific target site, depending on available prior knowledge.). The next step would be to create primers that target these specific sites and change some bases, or if a larger chain needs to be replaced, I would create entire custome fragments. After that I would assemble the modifyied DNA, Sequence it to make sure I’ve assembled the intended sequence, and test it. Required inputs would include: template palsmids containing a PCR product of RuBisCO or the gene itself, primers or entire fragments for replacing, polymerase for duplicating the sequence, and assembly enzymes to ‘stich’ the new sequence together. If going with a plasmid, I will need the backbone itself, host cells to insert the plasmid to, and a sequencing technology to verify my work (Sanger was mentioned earlier but it depends on the actual planned sequence legnth in bp units)
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
The polymerase process may introduce errors in copying the sequence. This is also true for assembly of large fragments as well, they could introduce unintended changes during assembly, this tends to happen if you try to assemble a bigger fragment count, repetitive fragments, or very long ones.
References:
Adams, J. (2008) DNA sequencing technologies. Nature Education 1(1):193
https://www.nature.com/scitable/topicpage/dna-sequencing-technologies-690/
Blogpost: “Site Directed Mutagenesis by PCR”
Week 3: Opentrons
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
The paper I’ve chosen is AssemblyTron: flexible automation of DNA assembly with Opentrons OT-2 lab robots by John A. Bryant Jr., Mason Kellinger, Cameron Longmire, Ryan Miller, and R. Clay Wright.
Published in Synthetic Biology (Volume 8, Issue 1, 2023), the paper presents a new open-source script for the Opentrons OT-2 robot called “AssemblyTron.” The paper overviews automation in the context of synthetic biology’s repeating workflow (the DBTL cycle) and argues that experimental progress is often constrained by the labor and tacit expertise required to carry out repetitive, error-prone bench work. These standards are crucial for reliable experimentation, but the paper suggests automation can help by:
Reducing opportunities for human pipetting error and improving consistency
Lowering the hands-on training burden for repetitive liquid-handling steps
Minimizing time, cost, and waste
The authors describe using AssemblyTron to streamline DNA assembly/cloning workflows on the OT-2, including automating PCR fragment preparation and the setup of multipart DNA assembly reactions from designed DNA parts/fragments. The discussion concludes by emphasizing the potential for lowering barriers and increasing accessibility in synthetic biology experimentation, along with future directions for improving and extending the script.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
One idea that I could explore with automation regarding the Living Scaffold Final Project I suggested in Week 1 would be automated “parameter sweeps” for mapping purposes. Automation would let me treat the living scaffold as a controllable, testable system: running repeatable parameter sweeps, collecting data, and potentially implementing closed-loop control of light and media conditions to map oxygen distribution. Basically what I’m suggesting here is to run the same experiment across many conditions without human variability, to see how oxygen generation/distribution changes with different inputs:
Different light cycles (intensity, pulsing, duty cycle)
Different media compositions (nutrients, buffering, additives that affect scaffold properties)
Different co-culture ratios (photosynthetic layer density vs scaffold thickness)
Different geometry variants (channels/porosity patterns)
Week 4: Protein Design 1
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Some definitions regarding measurement units:
A Dalton (Da) is a unit used to describe molecular mass. A useful heuristic is:
1 Da ≈ 1 g/mol
A mole (mol) is a counting unit in chemistry: 1 mol = 6.022 × 10^23 molecules (Avogadro’s number).
If we assume an amino acid (AA) is ~100 Da, this corresponds to ~100 g/mol. Meat is not entirely protein; if we assume protein is ~20% of meat by mass, then 500 g meat ≈ 100 g protein.
100 g ÷ (100 g/mol) = 1 mol ≈ 6.022 × 10^23 AA units.
So, 500 g of meat contains on the order of ~6 × 10^23 amino-acid “units” worth of protein mass, given the assumptions above. (OpenStax, n.d.)
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When we digest proteins, our body breaks them down into smaller peptides and then into amino acids. Protein breakdown in digestion can be described in three stages:
- Stomach: Hydrochloric acid (HCl) lowers pH and helps denature proteins (unfolding them). The enzyme pepsin begins cutting long polypeptides into shorter peptides. (OpenStax, n.d.)
- Small intestine: Pancreatic proteases (e.g., trypsin and chymotrypsin) continue cutting peptide bonds into smaller peptides and some free amino acids. (OpenStax, n.d.)
- Brush border: Enzymes on the intestinal “brush border” complete digestion into amino acids and very short peptides that can be absorbed into the bloodstream and distributed throughout the body for building human proteins. (OpenStax, n.d.)
Why are there only 20 natural amino acids?
The standard genetic code encodes 20 amino acids, with two rare genetically encoded additions: selenocysteine (including in humans) and pyrrolysine (in some microbes). A key point is that amino acids are not only “letters” for variety; they are the building blocks that enable proteins to reliably fold into stable, soluble, close-packed 3D structures with functional binding pockets. (Doig, 2016)
Evolution likely began with a smaller set of amino acids, then expanded the “vocabulary” as biological complexity increased. However, beyond a certain point, adding new amino acids yields diminishing returns and increases system-level costs: folding stability becomes harder to maintain, translation errors become more costly, and codon assignments become harder to manage without confusion. This challenges the idea that the set is purely a “frozen accident” and supports the view that the 20-amino-acid set is close to an optimal balance between chemical diversity and translational reliability. (Doig, 2016)
Where did amino acids come from before enzymes that make them, and before life started?
This answer splits into two domains: extra-terrestrial and terrestrial sources. Extra-terrestrially, amino acids have been detected in carbon-rich meteorites, suggesting that early Earth could have received a chemically diverse mixture of amino acids from space. (Kirschning, 2022)
Terrestrially, multiple plausible prebiotic routes could generate amino acids through abiotic chemistry in different environments—so it’s unlikely there was a single origin location. Examples include Strecker-type synthesis and Miller–Urey style discharge experiments, as well as hydrothermal and iron–sulfur mineral settings that can promote reaction networks from simple precursors. Wet–dry cycles (e.g., in hydrothermal fields) are important because they can repeatedly concentrate reactants and support peptide formation, bridging from free amino acids toward short peptides. (Kirschning, 2022)
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
An α-helix made from D-amino acids is expected to be left-handed (the mirror-image of the common right-handed α-helix formed by L-amino acids). This follows from stereochemistry: switching from L to D reverses the preferred helical handedness. (Doig, 2016)
Can you discover additional helices in proteins?
Yes. Besides the α-helix, proteins can also contain 3_10 helices and π-helices. A 3_10 helix is typically tighter than an α-helix (fewer residues per turn), often appearing as short segments. A π-helix is rarer and is often described as an insertional “bulge” within α-helices that can contribute to function (e.g., shaping binding pockets). (Cooley et al., 2010)
Why are most molecular helices right-handed?
In proteins built from L-amino acids, the right-handed α-helix is strongly favored because it minimizes steric clashes and supports favorable backbone geometry and hydrogen bonding. Left-handed α-helices in L-amino-acid proteins tend to be destabilized by unfavorable backbone/side-chain interactions. (Doig, 2016)
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets tend to aggregate because exposed β-sheet edges can “zipper” together through backbone hydrogen bonds (edge-to-edge pairing). In water, aggregation is often further stabilized when hydrophobic side chains pack together and exclude water, promoting larger assemblies. (Nowick, 2008)
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Many amyloid diseases involve proteins that misfold and assemble into highly stable β-sheet-rich fibrils (amyloid), which can accumulate in tissues and disrupt function. β-sheet structures can expose edges that promote repeated “zippering” and stacking into fibrils, making them unusually stable. (Nowick, 2008)
Because amyloid fibrils can be rigid, chemically stable, and programmable by sequence, they are also being explored as functional biomaterials (with careful design and safety considerations). As one review summarizes: “The rigidity, chemical stability, high aspect ratio, and sequence programmability of amyloid fibrils have made them attractive candidates for functional materials…” (Li & Zhang, 2021)
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
Myoglobin is an oxygen-binding protein in muscle that stores and shuttles O₂ inside cells using a heme group, and its oxygen/oxidation state is a major reason meat looks red vs brown. In the cultivated-meat “marbling/structure” project, it’s directly tied to the sensory realism problem: getting cultured tissue to develop the same color cues people associate with meat. It interests me because it links a visible outcome (color) to a real underlying biological variable (oxygen handling and tissue state), which is a legible bioengineering lever I can explore in my final project proposition.
Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid?
Downloaded the AA sequence for Myoglobin (one of five variations in humans) from the Uniprot website
sp|P02144|MYG_HUMAN Myoglobin OS=Homo sapiens OX=9606 GN=MB PE=1 SV=2 MGLSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETLEKFDKFKHLKSEDEMKASE DLKKHGATVLTALGGILKKKGHHEAEIKPLAQSHATKHKIPVKYLEFISECIIQVLQSKH PGDFGADAQGAMNKALELFRKDMASNYKELGFQG
it is 154 Amino Acids long, and the most frequent Amino Acid within the sequence is Lysine (coded in the sequence as K), and it appears 20 times in the chain.
How many protein sequence homologs are there for your protein? Does your protein belongs to any protein family?
Using the Uniprot BLAST tool, I analysied the Myoglobin sequence and found that there are 250 homologs for this protein across the swiss-prot and trEMBL datasets.
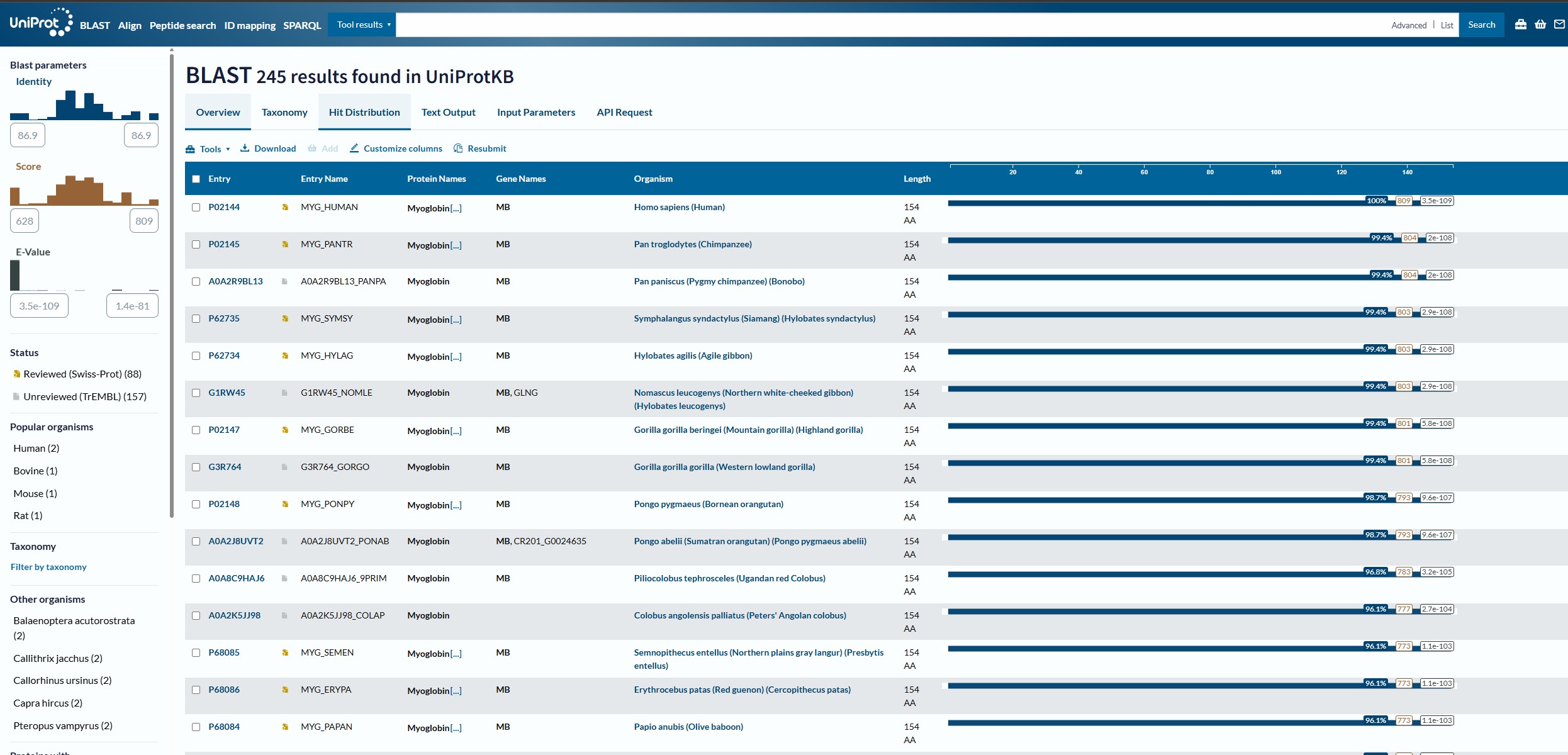
looking up Myoglobin I arrived at the Interpro webpage where Myoglobin was identified to be a member of the globin domain (Pfam category PF00042)
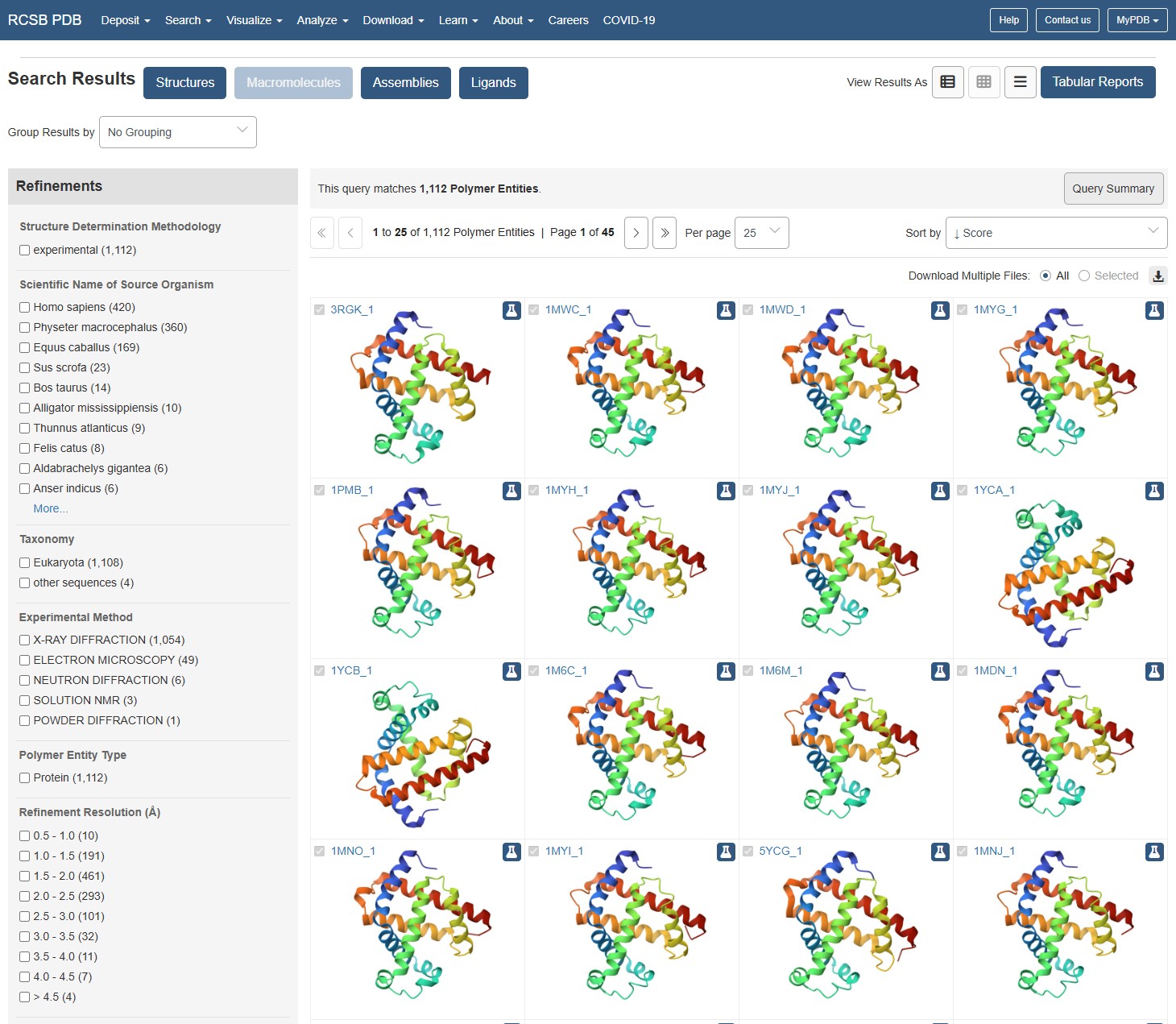
Identify the structure page of your protein in RCSB When was the structure solved? Is it a good quality structure?
using the RCSB website to identify Myoglobin structure, I can see the structure has been documented many times, in increasing resolutions. The earliest documented structure on the website dates to 1975. The best resolutions available (10 entities) are within the 0.1-0.5A range, and these are dated to be between 2005-2024. I would say for the latest entities, they populate the best resolution category available on the webpage so they should be considered as excellent quality.
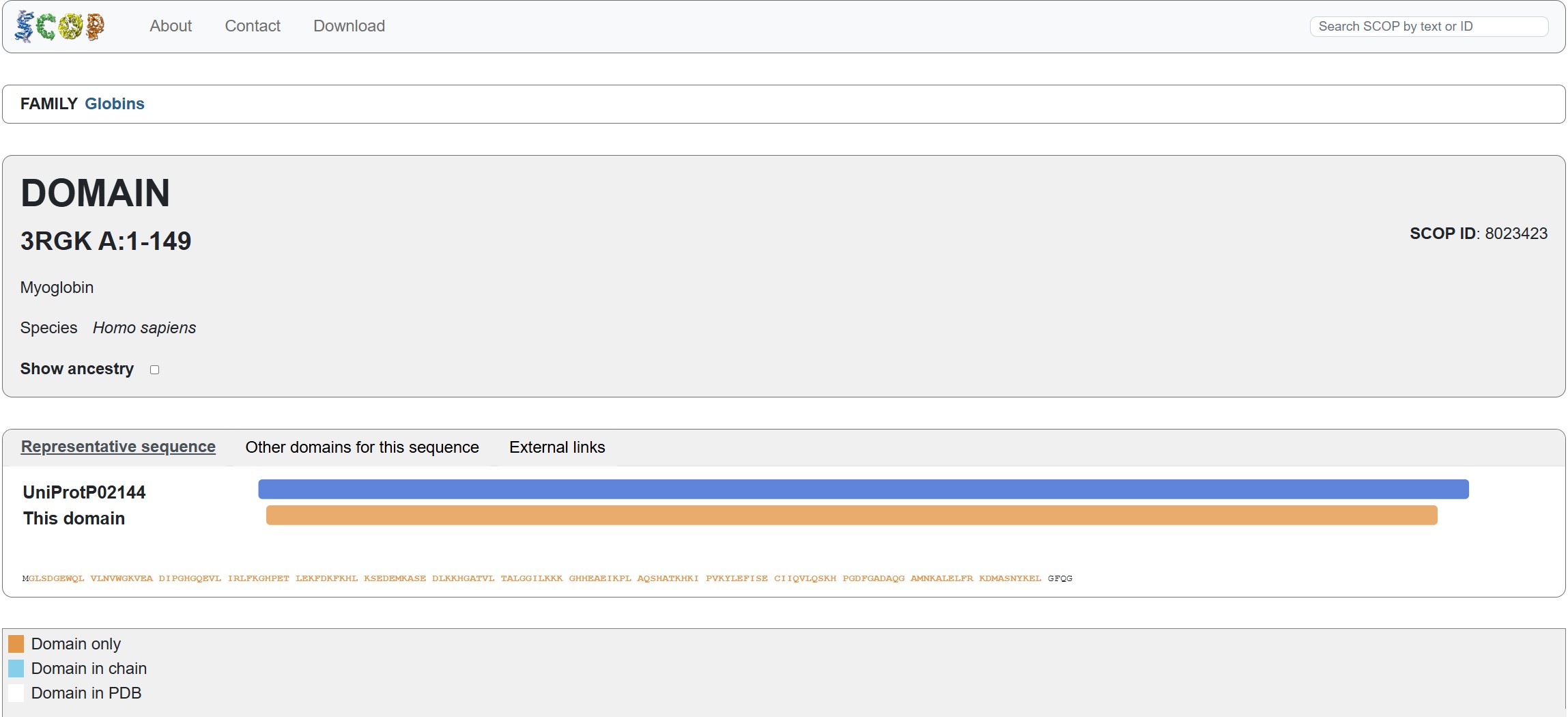
Are there any other molecules in the solved structure apart from protein? Does your protein belong to any structure classification family?
Using the 3D render and data provided for 6KA9 (the first RCSB search result: Crosslinked alpha(Fe-CO)-beta(Ni) human hemoglobin A), I can see there are O molecules scattered across the 3D render. This seems to be due to the protein modeled while being in an aqueos solution. Besides the water, only Polymer and Ligand groups are present in the render, which make the entire protein quaternary structure.
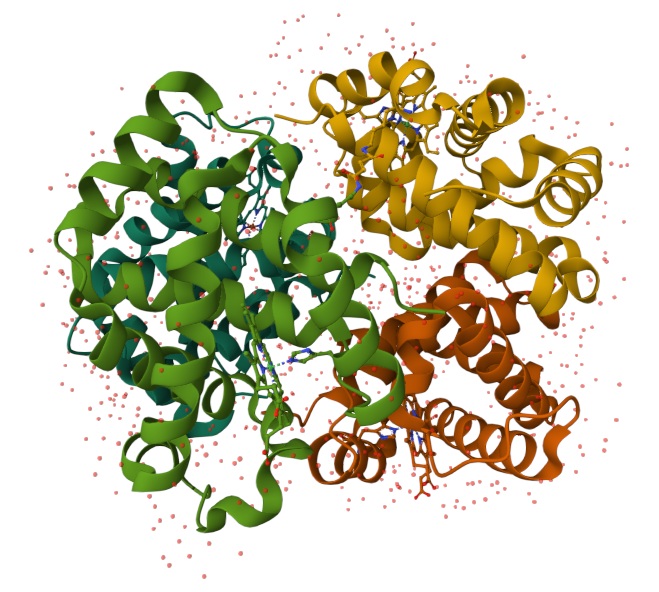
Does your protein belong to any structure classification family?
Using the SCOP tool shows that Myoglobin is a member of the 3RGK A:1-149 domain (SCOP ID 8023423). it also notes that it is a member of the Globins family, which matches the protein family we say Myoglobin associated with in the earlier question.
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
For this assignment, I used the RCSB 3D visualisation webapp that runs Mol* (WebGL) for 3D renderings. Because the “cartoon” representation for the ligand groups did not show anything, I assigned them a “line” representation so they will be visible.
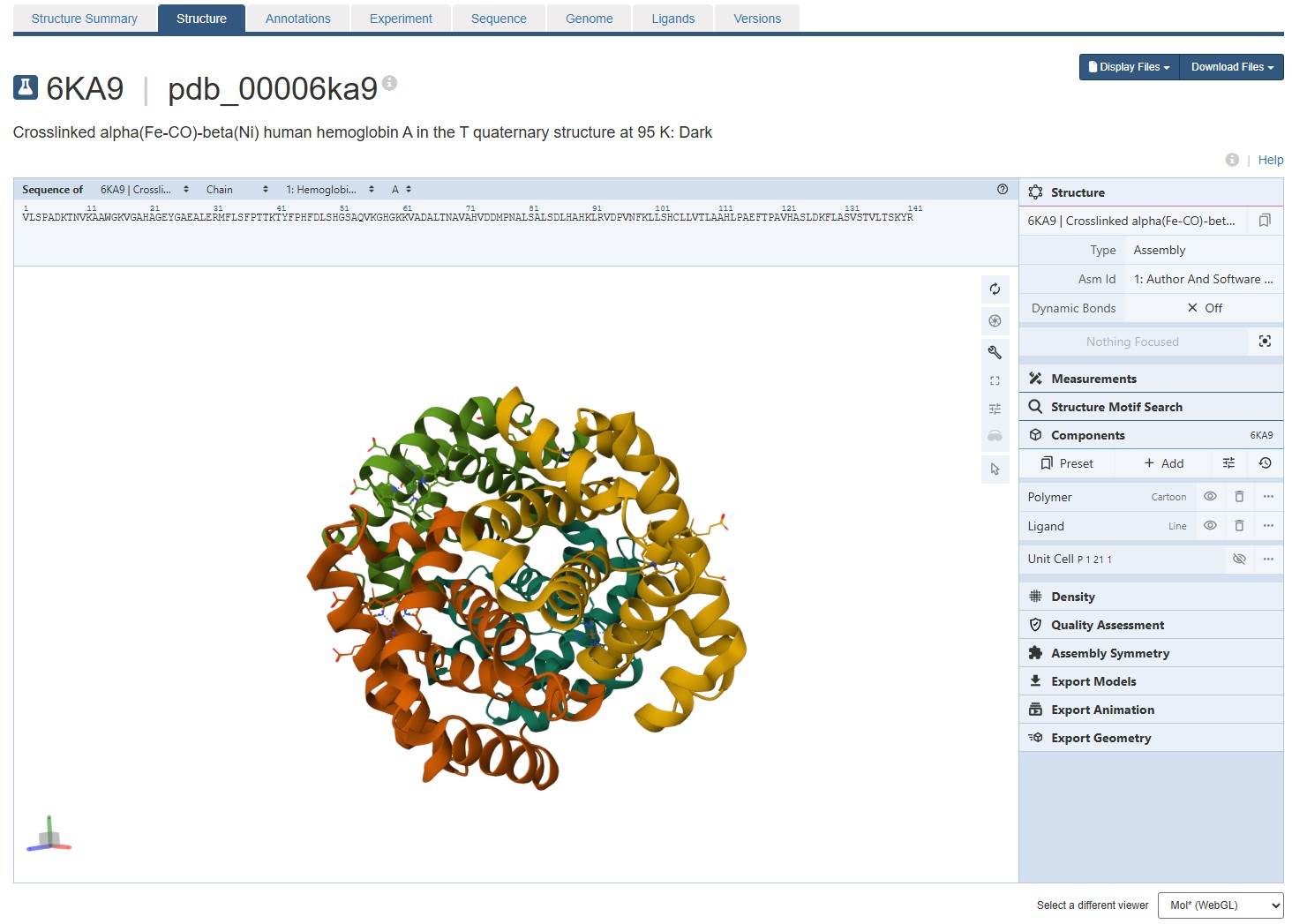
Mol* does not seems to have a “ribbon” representation. I switch to JSMol in the RCSB 3D Visualisation webapp, and chose a ribbon representation.
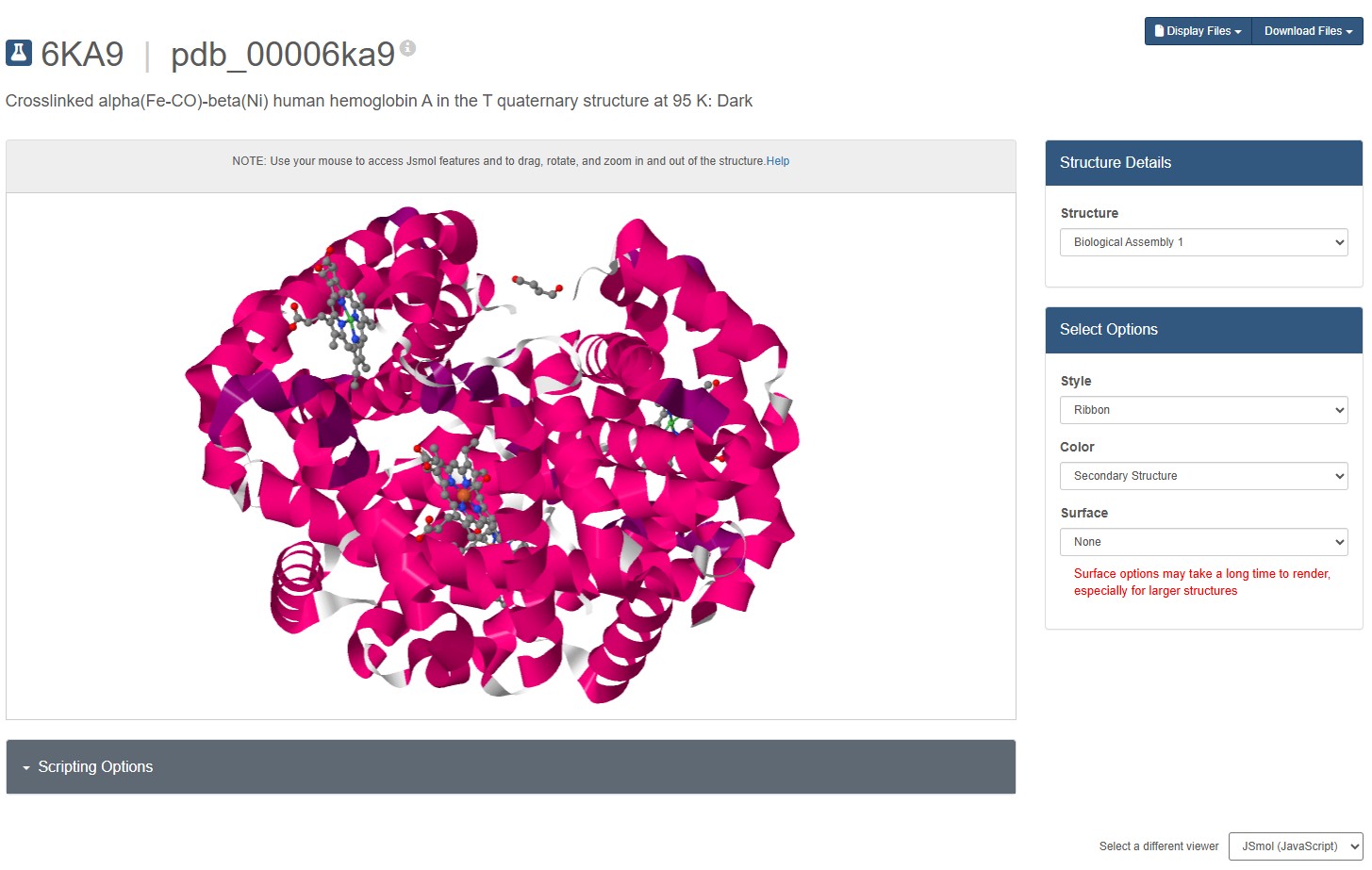
Going back to Mol* (because I think it creates nicer renderings), we get the “ball and stick” representation.
Coloring secondary structures (and honestly we could even determine this before, but to be on the safe side), we can see the Myoglobin protein variant I’m visualising consists only of α-helices as Polymer structures (They are all colored the same).
Coloring the protein for hydrophobicity, we discover the molecules vary in their hydrophobic charecteristics, varying across a scale of 11 different degrees. The spread is a bit confusing and I’m not sure I can draw a solid conclusion from it. It seems the protein is mostly hydrophillic, but certain molecules demonstrate a strong hydrophobic attribute.
Representing the protein using surfaces, we can immidiatly tell there is a central pocket. Another interesting observation available through the surface render is that the Ligand groups have their own ‘mini pockets’ where they are tucked into the Polymer structure.
Surface representation
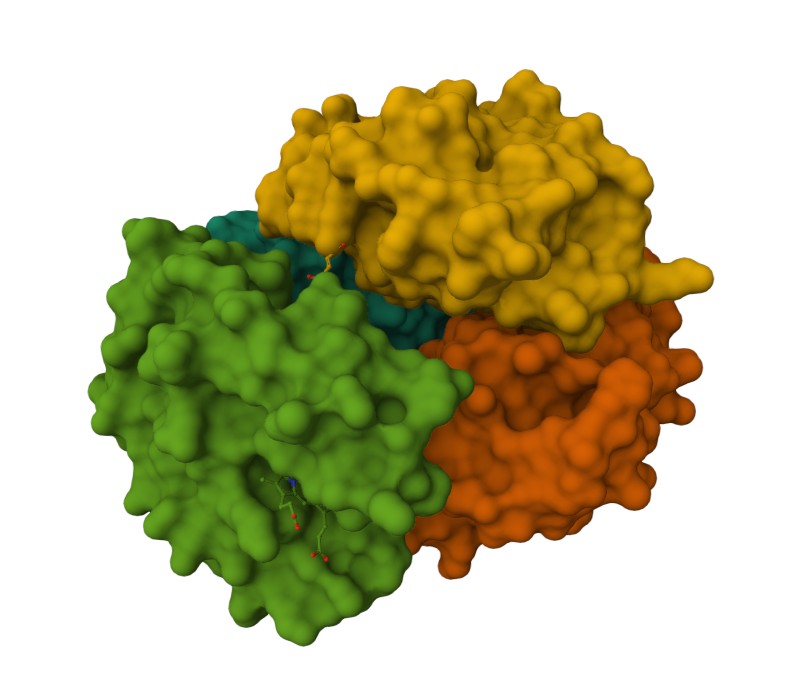
Observing the central pocket
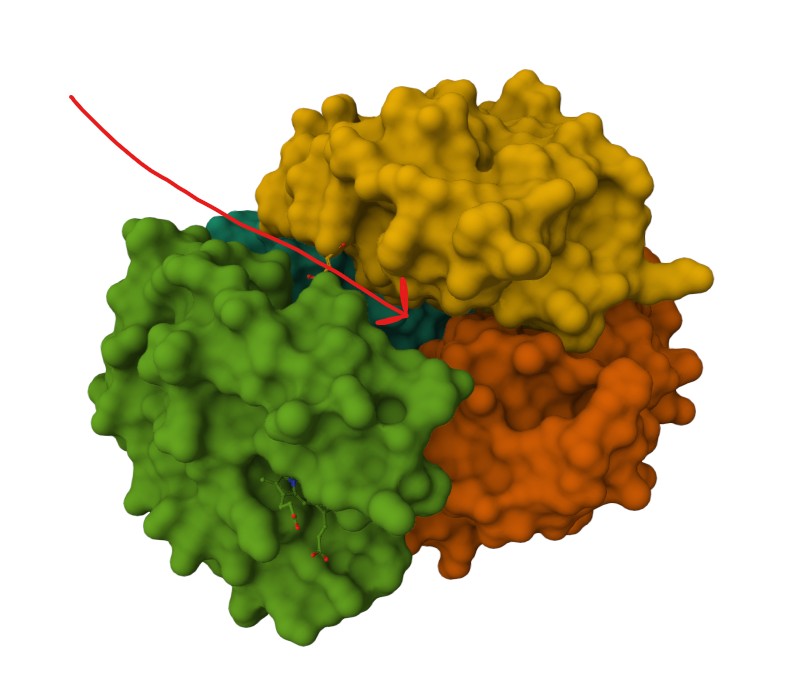
Tucked Ligand groups
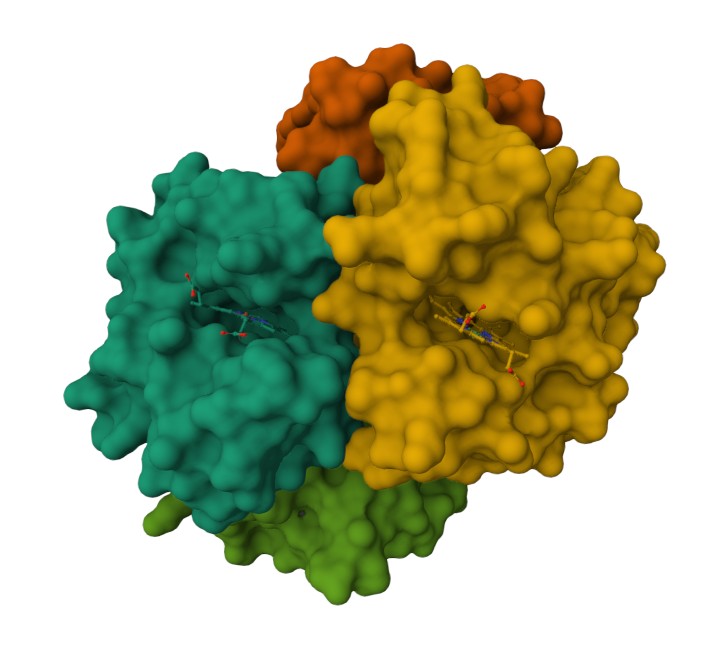
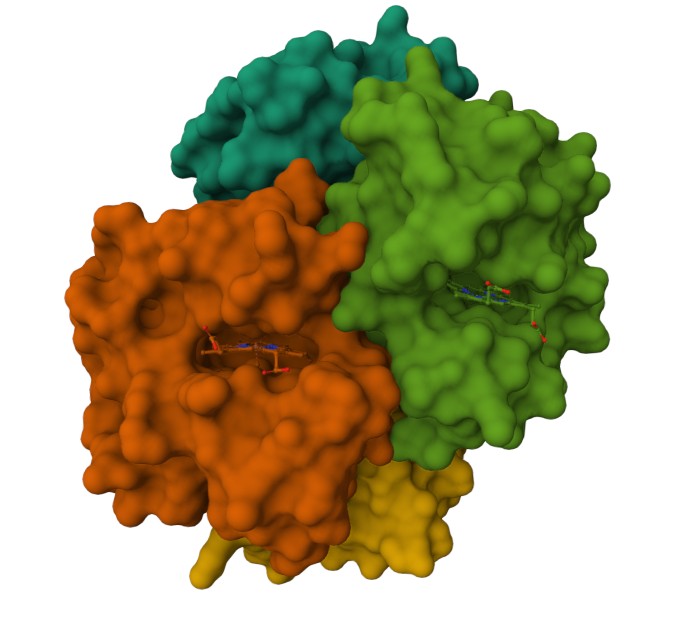
C1. Protein Language Modeling
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods. Can you explain any particular pattern?
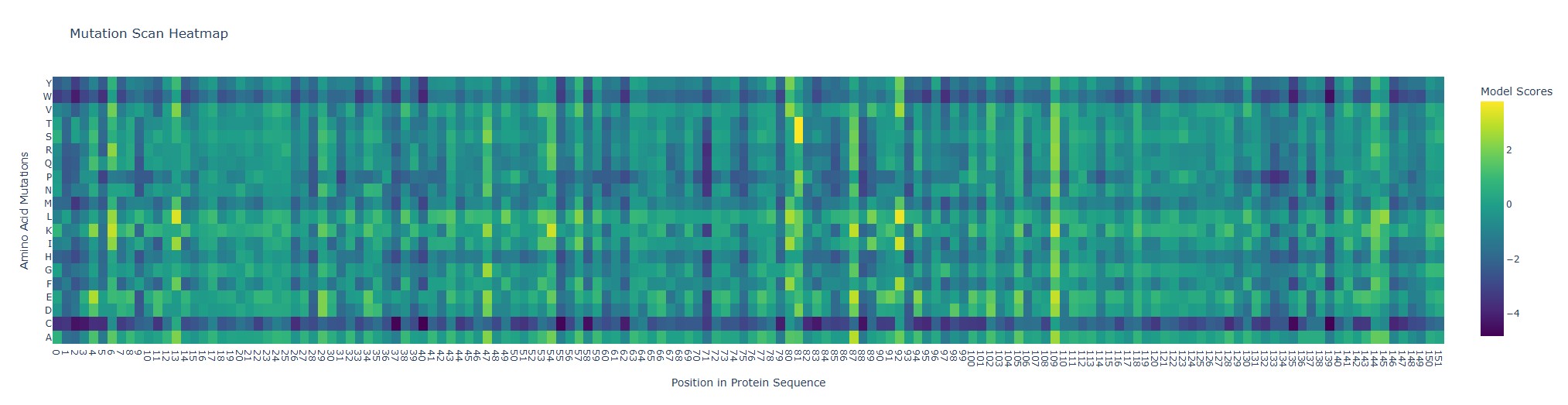
Looking at the heatmap, position 92 (wild-type Q, glutamine) stands out because the substitutions Q→L and Q→I get unusually high model scores (bright yellow). In ESM2 terms, that means the model thinks Leu or Ile “fit” well at that position in a myoglobin-like sequence.
Latent Space Analysis: Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
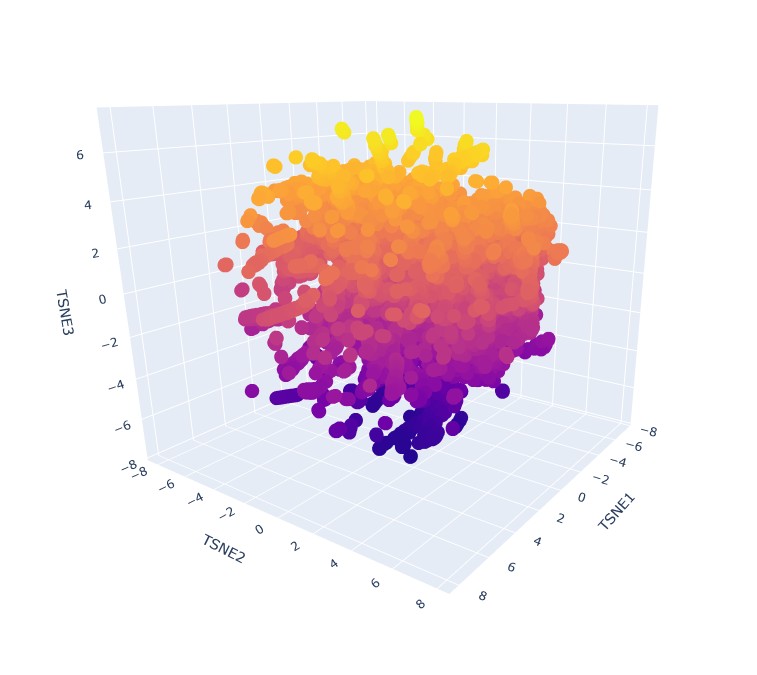
Yes, it seems that when zooming in, specific proteins tend to group in neighbourhoods based on their 3 t-SNE coordinates. like this group:
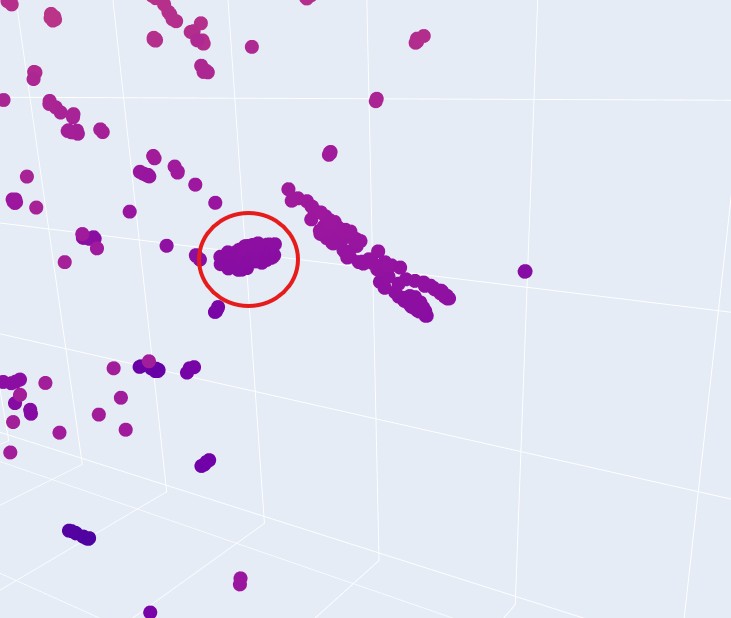
This implies that they have very similar sequence, domain or group, and therefore can be expected to have similar fold structures. They do not, however share the same taxonomy. That is understood immidiatly when you hover over some group of proteins and see that their taxonomy is varied across multiple species.
References
Cooley, R. B., Arp, D. J., & Karplus, P. A. (2010). Evolutionary origin of a secondary structure: π-helices as cryptic but widespread insertional variations of α-helices enhancing protein functionality. Proceedings of the National Academy of Sciences, 107(25), 11285–11290. https://pmc.ncbi.nlm.nih.gov/articles/PMC2981643/
Doig, A. J. (2016). Frozen, but no accident – why the 20 standard amino acids were selected. FEBS Journal. https://febs.onlinelibrary.wiley.com/doi/10.1111/febs.13982
Kirschning, A. (2022). On the evolutionary history of the twenty encoded amino acids. Chemistry–A European Journal, 28, e202201419. https://pmc.ncbi.nlm.nih.gov/articles/PMC9796705/
Li, C., & Zhang, X. (2021). Amyloids as building blocks for macroscopic functional materials: Designs, applications and challenges. International Journal of Molecular Sciences, 22(5), 1–28. https://pmc.ncbi.nlm.nih.gov/articles/PMC8508955/
Nowick, J. S. (2008). Exploring β-sheet structure and interactions with chemical model systems. Accounts of Chemical Research, 41(10), 1319–1330. https://pmc.ncbi.nlm.nih.gov/articles/PMC2728010/
OpenStax. (n.d.). Chemical digestion and absorption: A closer look. In Anatomy and Physiology 2e. https://openstax.org/books/anatomy-and-physiology-2e/pages/23-7-chemical-digestion-and-absorption-a-closer-look
Week 5: Protein Design 2
This week, we need to find a favorable peptide to bind into the mutated (A4V) version of a SOD1 enzyme.
Part 1: Generate Binders with PepMLM
This is the original sequence of SOD1 as it appears here.
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Changing the 4th residue (In much of the SOD1/ALS literature, that first methionine (M) is considered removed in the mature protein, so residue numbers shift by minus 1.)
wild type: MATKAVCV… mutant: MATKVVCV…
Running the sequence of the A4V mutated SOD1 in the PepMLM Collab, asking for 4 different peptides at the length of 12 AA’s with K = 5.

I want to note that I do observe X to appear in 3 out of 4 sequences, more specifically the top 2 scoring sequences end with an X. This may be an issue downstream.

Noting that the Alphafold server does not allow to input sequences containing an X AA, I return to the PepMLM notebook to continue generating peptides under the same parameters until I have 4 valid candidates for Alphafold.

It took a couple more iterations but finally I got 4 valid, generated, peptide predictions.

Running the SOD1 V4A mutation alone first in the Alphafold server to get familiar with it’s shape before I model more peptides in the scene.

For clarity, highlighted in pink is the mutated residue (I manually replaced the V residue in the 4th position with an A residue before inputting the sequence to the Alphafold server)

I can also verify this seems to be a good representation of the modeled SOD1 mutated protein using it’s Alignment Residue graph and high pTm score of 0.96. Below is the provided explantation for the scores from the Alphafold server
How can I interpret confidence metrics to check the accuracy of structures?
pTM and ipTM scores: the predicted template modeling (pTM) score and the interface predicted template modeling (ipTM) score are both derived from a measure called the template modeling (TM) score. This measures the accuracy of the entire structure (Zhang and Skolnick, 2004; Xu and Zhang, 2010). A pTM score above 0.5 means the overall predicted fold for the complex might be similar to the true structure. ipTM measures the accuracy of the predicted relative positions of the subunits within the complex. Values higher than 0.8 represent confident high-quality predictions, while values below 0.6 suggest likely a failed prediction. ipTM values between 0.6 and 0.8 are a gray zone where predictions could be correct or incorrect. TM score is very strict for small structures or short chains, so pTM assigns values less than 0.05 when fewer than 20 tokens are involved; for these cases PAE or pLDDT may be more indicative of prediction quality.

Moving on to model the interactions of the generated peptides from PepMLM with the SOD1 V4A For the first peptide on our list, the WRYYAAQAAWKE variant, the model shows a proximity to the N-terminus where the mutation sits, and in the illustrated view we can see it is actually binding. According to ipTM and pTM scores, we learn the folded-form prediction has high confidence grade from the model, while the spatial predicition (how these two models actually interact in the simulated space) is very low, making this first simulation unreliable.


For the second peptide, the WLYPYVAVALAA variant, the model shows again there is no binding between the peptide and the SOD1 V4A protein. This time, the model provides a high confidence score for the fold(pTM), but the ipTM scores read as a ‘failed prediction’ (“values below 0.6 suggest likely a failed prediction.”) and results seems even further away from the target site (Residue 4 near the N-Terminuus). to conclude, the peptide does not serve us well.


For the third peptide, the WRVSVVGVVHGG, results look more promising. ipTM and pTM scores both look strong. the Cartoon view shows that the peptide binds to the SOD1 protein, and it does sit relatively closer to the N-terminus. Getting more comforable with the Alphafold UI I saw an option to switch the illustration mode, where it seems to actually bind to the SOD1 (went ahead and updated all the other simulations as well). I’m unsure if this counts as binding ‘on the target site’, as it’s not precisely touching it, but it is very close. another observation worth mentioning is that it seems this interaction has completley changed the SOD1’s spatial configuration. I think this peptide is a good candidate to proceed.


For the fourth peptide the KVNGAYAGRWLE, which also had the worst psuedo-perplexity score from the PepMLM model (27.7), we see a failed ipTM score, making this simulation irrelevant. A strong pTM results gives confidence about the folding structures, noting this is the first generated-peptide to demonstrate a secondary structure of beta sheets when folded. However, the low ipTM score does not allow me to proceed with this one.


Lastly, simulating the reference peptide FLYRWLPSRRGG also yields failed ipTM scores. I find it surprising, as I was expecting the reference peptide to demonstrate the desired behavior to serve as a reference. while this peptide seems to be bind to the protein for the most part, I cannot trust the outcome due to the very low confidence of the model in the spatial predicition between the two elements. Kind of happy to know one of my generated peptides (WRVSVVGVVHGG) seems more promising than the reference one.


Moving onward to the Peptiverse model to analyise the different metrics we care about regarding our generated peptides: 💧 Solubility 🔬 Permeability (Penetrance) 🩸 Hemolysis 👯 Non-Fouling ⏱️ Half-Life 🔗 Binding Affinity (in context of our mutated SOD1-V4A sequence) 📏 Length ⚖️ Molecular Weight ⚡ Net Charge (pH 7.4) 🎯 Isoelectric Point 💦 Hydrophobicity (GRAVY)
Peptide 1 — WRYYAAQAAWKE AlphaFold3 gives this complex the lowest ipTM (0.17), and structurally the peptide looks mostly extended and only lightly attached, with limited contact against the SOD1 surface. That visually suggests a weak or unstable interface. PeptiVerse agrees: it predicts weak binding (6.13 pKd/pKi). On the therapeutic side it is still attractive in that it is soluble, permeable, and non-hemolytic, but the structural model does not make it look like a strong binder.

Peptide 2 — WLYPYVAVALAA This one looks much better structurally: the peptide appears to sit along one face of SOD1 with a broader contact patch, and the ipTM of 0.56 is clearly stronger than pep1, pep4, or the reference. PeptiVerse also gives it the best affinity score of the set (7.367, medium binding). It is predicted to be soluble, permeable, and non-hemolytic, so this is the clearest case where the structure and property model are both favorable. The main caution is that it is fairly hydrophobic and not predicted as “non-folding,” but overall it still looks like the best-balanced lead.

Peptide 3 — WRVSVVGVVHGG Structurally, this is the strongest-looking AlphaFold3 hit: it has the highest ipTM (0.71) and the peptide seems to make a long, continuous interface across the protein surface, which is exactly the kind of pose you would hope for in a binder. But PeptiVerse does not rank it as the best binder; it is still only weak binding (6.653) and is also predicted to be non-permeable. So this is the clearest mismatch between the two models: best structural interface confidence, but not best therapeutic profile.

Peptide 4 — KVNGAYAGRWLE This complex is intermediate-to-weak by structure: ipTM is 0.43, and the peptide looks more like a surface appendage on one side rather than a strongly buried binder. PeptiVerse is consistent with that and predicts weak binding (6.095). It is still soluble and non-hemolytic, but also non-permeable, so there is not a strong reason to prioritize it over pep2 or even pep3.

Reference peptide — FLYRWLPSRRGG The reference peptide gives a low ipTM (0.32) and looks only partially engaged, with the chain remaining fairly extended and not deeply wrapped into the SOD1 surface. PeptiVerse also predicts weak binding (5.968), the weakest of the five by affinity score. Its upside is that it is soluble, highly permeable, non-hemolytic, and the only one predicted as non-folding, which is a nice therapeutic feature, but its binding looks less compelling than the better generated candidates.

Across the whole set, higher ipTM does not map perfectly onto stronger predicted affinity. The clearest example is peptide 3, which has the highest ipTM and pTM scores (which made me favor it moving onward earlier) but only weak predicted affinity, while peptide 2 has the best PeptiVerse affinity despite a lower ipTM than pep3. Also, none of the stronger candidates are predicted to be hemolytic or poorly soluble; all five are predicted soluble and non-hemolytic. The best overall balance of predicted binding plus therapeutic properties is peptide 2 (WLYPYVAVALAA). I would choose peptide 2 because it gives the best combined picture across both models: the strongest predicted affinity in PeptiVerse, a reasonably strong AlphaFold3 interface (ipTM 0.56), and the model indicates signals of being soluble, permeable, and non-hemolytic.
In the next step we are tasken with running the moPPIt model on the google collab notebook provided. Following instructions I inputted all required data points. However, the final step of retreiveing a csv file with the scored results failed. I tried to change the Runtime modes to CPU, and V5E-1 TPU mode, (originally ran it on the T4 GPU) both have also failed. error code reads “RuntimeError: moo.py failed with code 2”.
Week 6: Genetic Circuits
Week 6 Homework: Genetic Circuits
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix contains several key components required for accurate DNA amplification. One central component is the Phusion DNA polymerase, a high-fidelity enzyme with proofreading activity that synthesizes new DNA strands and reduces the error rate compared to standard Taq polymerase. The mix also contains dNTPs (deoxynucleotide triphosphates), which serve as the molecular building blocks used to construct the new DNA strand during amplification.
Another important component is the reaction buffer, which maintains the correct chemical environment for the polymerase to function efficiently. This buffer includes salts and magnesium ions (Mg²⁺), which are essential cofactors for polymerase activity and influence primer binding and enzyme performance. The mix may also include stabilizing agents that help preserve enzyme activity during thermal cycling. Because it is provided as a master mix, many of these ingredients are already pre-balanced, which reduces pipetting error and improves consistency across reactions.
2. What are some factors that determine primer annealing temperature during PCR?
Primer annealing temperature during PCR is determined primarily by the melting temperature (Tm) of the primers. The Tm depends on several sequence features, especially primer length, GC content, and base composition, since G–C pairs form three hydrogen bonds and are therefore more thermally stable than A–T pairs. Primers with higher GC content or longer length typically have a higher Tm and therefore require a higher annealing temperature.
A second factor is the degree of complementarity between the primer and the target sequence. If the primer sequence has mismatches with the target, annealing may be weaker and require optimization at a lower temperature, though this may reduce specificity. The salt concentration and reaction conditions in the PCR mix also affect hybridization behavior, because ionic conditions influence DNA duplex stability. In practice, the annealing temperature is usually chosen a few degrees below the primer Tm in order to balance efficient binding with high specificity.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR and restriction enzyme digestion can both generate linear DNA fragments, but they do so through very different mechanisms. PCR creates a linear fragment by enzymatically amplifying a specific region of DNA using primers, polymerase, dNTPs, and thermal cycling. The protocol involves repeated cycles of denaturation, primer annealing, and extension, which selectively amplify the region bounded by the primer pair. PCR is especially useful when a fragment must be isolated precisely, modified through primer design, or generated from a small starting amount of template DNA.
By contrast, a restriction enzyme digest generates linear DNA by cutting an existing DNA molecule at specific recognition sequences. The protocol is generally simpler: plasmid or DNA template is mixed with one or more restriction enzymes, appropriate buffer, and water, then incubated at the temperature optimal for enzyme activity. Restriction digestion is often preferable when the desired fragment is already flanked by known restriction sites and does not require amplification or sequence redesign. It is also useful for opening plasmids or excising inserts when convenient restriction sites are available.
PCR is more flexible because primer design allows the user to define fragment boundaries and add overlaps or other sequence features. However, it can introduce amplification errors or nonspecific bands if poorly optimized. Restriction digests are often cleaner and simpler when the sequence already contains the required cut sites, but they are limited by the availability and placement of those sites. In practice, PCR is often preferred when custom design flexibility is needed, whereas restriction digestion is preferred when a pre-existing construct already contains a convenient cloning architecture.
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure that digested and PCR-generated DNA fragments are appropriate for Gibson cloning, the most important requirement is that adjacent fragments contain overlapping homologous ends, typically around 20–40 base pairs long. These overlaps allow the Gibson Assembly enzymes to join fragments in the correct order. When PCR is used, these overlaps are usually added directly through primer design. When restriction digestion is involved, the resulting fragment must still be checked to make sure it contains the correct overlap regions relative to the other assembly partners.
It is also important to confirm that the fragments correspond to the intended sequence and orientation. This can be checked computationally by mapping the PCR primers and restriction enzyme cut sites in sequence software such as Benchling. In addition, the fragments should ideally be clean and specific, meaning the PCR gives a single product of the expected size and the digest yields the correct linearized or excised band. Gel electrophoresis and gel extraction are commonly used to confirm fragment size and purify the correct product before assembly.
A final precaution is to verify that the fragment ends do not include unwanted features that would interfere with assembly, such as incorrect overlaps, missing bases, or incompatible sequence order. In short, appropriate Gibson-ready fragments are confirmed by sequence design, overlap design, size verification, and purification before the actual assembly reaction is performed.
5. How does the plasmid DNA enter the E. coli cells during transformation?
During transformation, plasmid DNA enters E. coli cells after the cells have been made competent, meaning temporarily capable of taking up external DNA. In a standard chemical transformation protocol, the cells are treated with salts such as calcium chloride, which helps neutralize the negative charges on both the DNA and the bacterial cell membrane. This reduces electrostatic repulsion and allows the plasmid DNA to associate more closely with the cell surface.
A brief heat shock then creates a transient physical imbalance across the membrane, which helps drive the plasmid DNA into the cell. The exact molecular mechanism is still described somewhat operationally rather than as a single perfectly resolved pathway, but the key idea is that heat shock temporarily increases membrane permeability and promotes uptake of the plasmid. In electroporation, a different method, a short electrical pulse creates temporary pores in the membrane through which DNA can enter.
Once inside the cell, the plasmid is maintained and replicated if it contains an origin of replication compatible with the host. Cells that successfully took up the plasmid can then be selected on antibiotic plates if the plasmid also carries an antibiotic resistance marker.
6. Describe another assembly method in detail (such as Golden Gate Assembly)
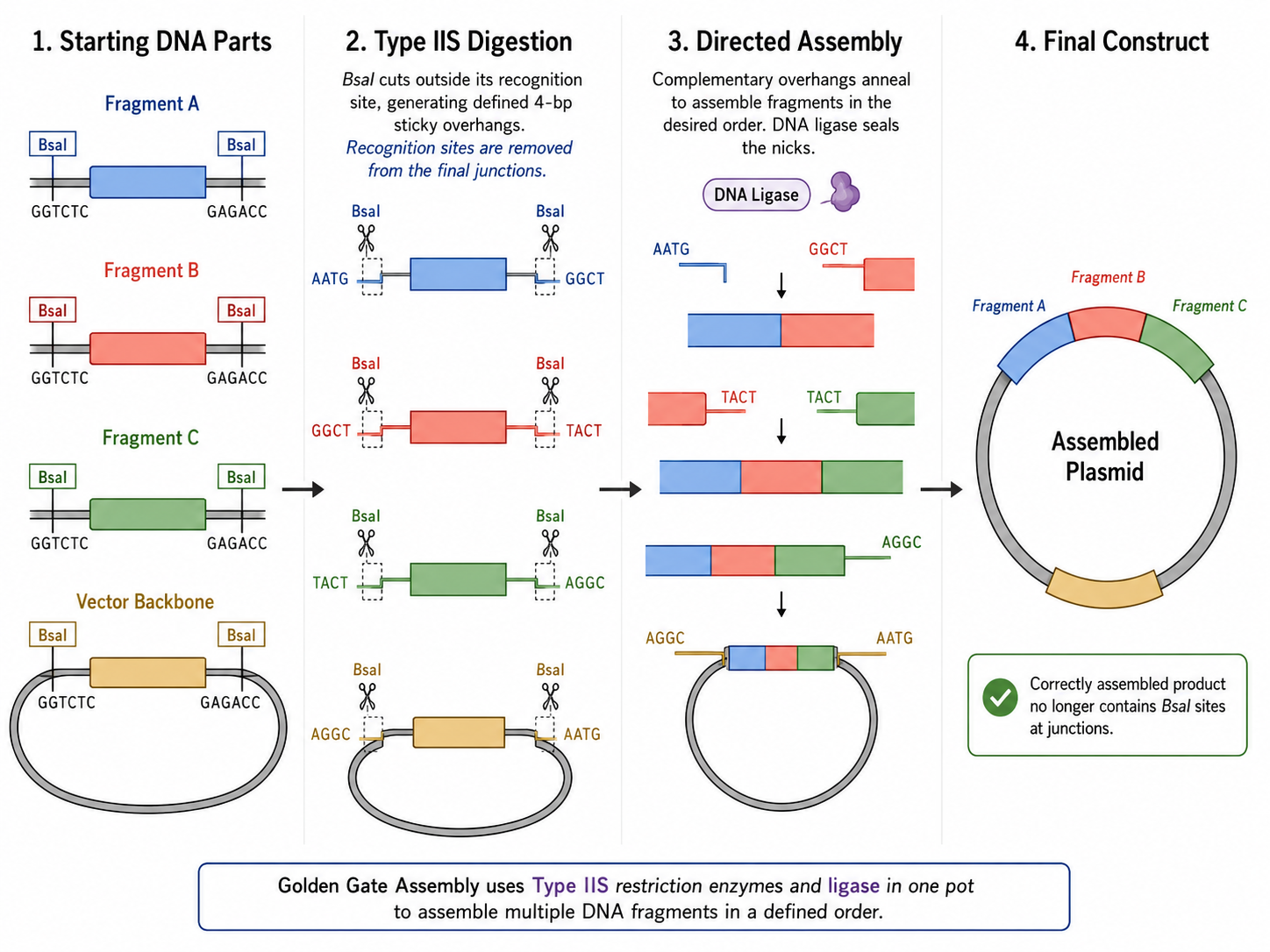
Golden Gate Assembly is a DNA assembly method that uses Type IIS restriction enzymes together with DNA ligase to join multiple DNA fragments in a defined order within a single reaction. Unlike standard restriction enzymes, Type IIS enzymes such as BsaI cut outside of their recognition sequence, which allows the user to design custom 4-base overhangs on each fragment. These overhangs determine exactly which fragments ligate to each other, enabling seamless and directional assembly without leaving unwanted restriction-site scars between parts. In practice, the DNA fragments and destination vector are designed so that digestion creates complementary overhangs, and the reaction is cycled between temperatures that favor digestion and ligation. Because correctly assembled products no longer contain the original Type IIS recognition sites, they are not re-cut, which enriches the desired final construct over time. Golden Gate Assembly is especially useful for modular cloning, multi-part assemblies, and workflows where many parts must be assembled rapidly and in a predefined order. It is often preferable when standardized part architecture is available, whereas Gibson Assembly may be more flexible when custom overlaps are easier to design than restriction-site-based overhangs.
Week 7: Genetic Circuits 2
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
The main advvantage I see of an IANN over a traditional genetic circuit (Boolean in nature) is that the IANN seems better suited for biological situations that require a more sophisticated estimation than a sharp TRUE/FALSE. We would need more ‘sensitive tools’ if we are to deal with many inputs, gradients, dynamic thresholds and intermidiate states - for all mentioned, binary logic is insufficient. I’m interested in cultivated meat, and in this course I will try to demonstrate control over marbeling of fat and meat tissues. This concept matters here because fat distribution is not a binary problem but a morphogentic spatial multi-input issue. IANN’s can seem more relevant when the goal is to account for several factors and generate a site-specific nuanced output.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
A useful application for an IANN, in the context of my meat-fat marbling catalogue direction, would be a system that helps decide where cells in a growing tissue should become more fat-like, more muscle-like, or remain in an intermediate state based on several inputs at once. The inputs could include things like oxygen level, nutrient availability, local signaling molecules, and maybe cell density or position within the scaffold, and the output would be a graded patterning decision rather than a single differentiation switch. What interests me is that this could eventually help produce different marbling motifs or classes instead of just maximizing fat everywhere. A limitation, though, is that a system like this would probably be very sensitive to diffusion, timing, signal decay, and spatial arrangement, so even if the logic works conceptually, getting a stable and precise pattern in real tissue would be difficult. Some of the papers I’ve been reading make that feel plausible as a direction, but also make clear how quickly these systems become messy once spatial biology is involved.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.

Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
One example of an existing fungal material is mycelium-based packaging or building material, where the fungal network grows through agricultural waste and binds it into a lightweight solid. I find that interesting because it can replace things like foam packaging or some synthetic insulation materials while using low-value waste as feedstock. The main advantages are that it is more biodegradable, potentially lower-emission, and can be grown rather than heavily manufactured. At the same time, the disadvantages are that it is usually less standardized and sometimes less strong, water-resistant, or durable than conventional materials, so it can be harder to use in situations where reliability and repeatability matter a lot.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
If I were engineering fungi, I would be interested in improving the mechanical properties of fungal materials, for example making them stronger or more consistent so they become more useful as design or construction materials. That feels meaningful to me because a lot of the promise of mycelium materials depends not just on being sustainable, but on actually performing well enough to compete with existing options. An advantage of working in fungi instead of bacteria is that fungi already naturally grow as large material-forming networks, so they are better suited for building structural biomaterials rather than just producing molecules in liquid culture. The downside is that fungi are usually slower and more complex to engineer than bacteria, but for material applications they may still be the more relevant organism.
References:
Basu, S., Gerchman, Y., Collins, C. H., Arnold, F. H., & Weiss, R. (2005). A synthetic multicellular system for programmed pattern formation. Nature, 434(7037), 1130–1134.
Mordvintsev, A., Randazzo, E., Niklasson, E., & Levin, M. (2020, February 11). Growing neural cellular automata. Distill, 5(2), e23. https://distill.pub/2020/growing-ca/
GeeksforGeeks. (n.d.). The perceptron, the basis of artificial neural networks. https://www.geeksforgeeks.org/deep-learning/what-is-perceptron-the-simplest-artificial-neural-network
Vasle, A. H., & Moškon, M. (2024). Synthetic biological neural networks: From current implementations to future perspectives. BioSystems, 245, 105164. https://doi.org/10.1016/j.biosystems.2024.105164
Week 9: Cell-Free Systems
Cell-Free Systems Homework
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis (CFPS) offers major advantages over traditional in vivo expression because it removes the constraints imposed by maintaining living cells. Since the reaction occurs in vitro, the experimenter has much tighter control over variables such as DNA concentration, energy source, salts, cofactors, reaction timing, and additives. This makes CFPS especially useful for rapid prototyping, because gene circuits or expression constructs can be tested directly without cloning into cells and waiting for growth. It is also easier to study toxic proteins or unstable pathways in CFPS, since there is no living host whose growth is harmed by the product. In addition, freeze-dried cell-free systems are portable, low-maintenance, and can be deployed with minimal equipment, making them well suited for low-resource settings and space applications. This was demonstrated in the BioBits study aboard the ISS, where freeze-dried cell-free reactions were rehydrated and used to express aptamers and fluorescent proteins under microgravity conditions.
Two cases where cell-free expression is more beneficial than cell production are:
- Point-of-need biosensing or diagnostics, where portability and rapid response are more important than long-term cell growth.
- Expression of toxic or burdensome proteins, where the product would damage or slow the growth of living cells.
2. Describe the main components of a cell-free expression system and explain the role of each component.
A cell-free expression system contains the molecular machinery needed for transcription and translation outside of living cells. One major component is the cell extract, which provides ribosomes, tRNAs, translation factors, metabolic enzymes, and in some cases RNA polymerase. Another component is the DNA template, which encodes the gene or circuit to be expressed. The system also includes nucleotides for transcription, amino acids for translation, and salts/cofactors such as magnesium and potassium that support enzyme activity and ribosome function.
A critical part of the mixture is the energy system, which regenerates ATP and other high-energy molecules needed to power transcription and translation. Many systems also include supplements such as folinic acid, tRNA mixtures, cofactors, and reducing agents to improve expression efficiency. In the BioBits ISS paper, the reaction mixture included ATP, GTP/UTP/CTP, amino acids, potassium glutamate, ammonium glutamate, magnesium glutamate, NAD, CoA, spermidine, putrescine, phosphoenolpyruvate, and cell extract, all of which helped support robust gene expression in vitro.
3. Why is energy provision/regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is critical in cell-free systems because transcription and translation consume large amounts of ATP and GTP. Unlike living cells, CFPS reactions do not have intact metabolism to continuously replenish these molecules, so without an energy-regeneration strategy the reaction would stop quickly. Sustained protein synthesis therefore depends on including a substrate that can support ATP regeneration over time.
One common method is to include phosphoenolpyruvate (PEP) as an energy source. In crude extract systems, endogenous enzymes can use PEP to regenerate ATP and maintain the reaction for longer periods. The BioBits formulation used aboard the ISS included 33 mM phosphoenolpyruvate, showing exactly this kind of strategy for sustaining cell-free transcription and translation. In my own experiment, I could use a PEP-based energy system and compare protein yield over time to confirm that the reaction remains active.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
A prokaryotic cell-free system is usually faster, cheaper, and easier to optimize than a eukaryotic system. It is well suited for expressing bacterial proteins, fluorescent reporters, enzymes, or genetic circuits that do not require complex post-translational modifications. For example, eGFP is a good protein to produce in a prokaryotic CFPS system because it folds relatively well, is easy to detect, and does not require glycosylation or other advanced processing.
A eukaryotic cell-free system is better for proteins that depend on more complex folding environments or post-translational modifications such as disulfide bonding, glycosylation, or membrane insertion. For example, a human cytokine or secreted antibody fragment would be a better candidate for a eukaryotic system, because bacterial extracts often cannot reproduce the same maturation steps needed for proper activity. In short, prokaryotic systems are excellent for speed and simplicity, while eukaryotic systems are preferable when the target protein requires more biologically complex processing.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Membrane proteins are difficult to express in cell-free systems because they tend to misfold, aggregate, or precipitate when no membrane-like environment is available. To optimize expression, I would design a screen in which the same membrane-protein DNA template is tested across multiple reaction conditions that vary membrane mimics, detergent concentration, magnesium concentration, and temperature. A key addition would be some form of membrane support, such as liposomes, nanodiscs, or mild detergents, so that the newly synthesized protein has a hydrophobic environment into which it can insert.
I would also monitor both total expression and soluble/functional expression, since a high total yield is not useful if the protein is aggregated. Suitable readouts might include SDS-PAGE, fluorescence tagging, or an activity assay if the membrane protein has measurable function. A lower reaction temperature and codon-optimized DNA could also help improve folding. Overall, the main challenge is not just producing the protein, but producing it in a membrane-compatible state that preserves structure and activity.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
One possible reason is that the DNA template concentration or quality is poor. If the template is degraded, contaminated, or present at too low a concentration, transcription may be inefficient. A troubleshooting strategy would be to verify the DNA on a gel, re-purify it, and test a range of template concentrations.
A second possible reason is that the reaction chemistry is suboptimal, for example incorrect magnesium concentration, depleted energy source, or poor buffer balance. Since CFPS is very sensitive to reaction composition, I would troubleshoot by running a small matrix of conditions and adjusting magnesium, potassium, and energy-substrate levels systematically.
A third possible reason is that the target protein itself is difficult to express or fold. This is especially likely for large, toxic, disulfide-rich, or membrane proteins. In that case, I would try lowering the temperature, changing the reaction time, adding chaperones or membrane mimics, or switching to a different cell-free system better suited to the protein class.
Homework Question from Kate Adamala
Design an example of a useful synthetic minimal cell
Pick a function and describe it.
My synthetic minimal cell would function as a localized ammonia detoxification sensor-actuator for aquatic environments. Its purpose would be to detect elevated ammonia in contaminated water and respond by producing an enzyme-based output that helps convert ammonia into a less harmful form.
What would your synthetic cell do? What is the input and what is the output?
The input would be elevated environmental ammonia. The output would be expression of a reporter such as GFP together with an enzymatic detoxification module, for example glutamine synthetase activity coupled to ATP-dependent ammonia assimilation.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Not as effectively. Without encapsulation, the reaction would simply mix into the surrounding environment and lose the separation between sensing, response, and controlled release. Encapsulation makes the system behave more like an artificial cell rather than just a biochemical mixture.
Could this function be realized by genetically modified natural cell?
Yes, it could, but that would introduce issues of survival, biocontainment, ecological competition, and environmental release. A synthetic minimal cell would reduce those concerns by remaining non-living and function-limited.
Describe the desired outcome of your synthetic cell operation.
In the presence of elevated ammonia, the synthetic cell should produce a measurable fluorescent signal and activate a detoxification pathway, demonstrating both environmental sensing and a functional biochemical response.
Design all components that would need to be part of your synthetic cell.
The system would need:
- a lipid vesicle membrane
- a cell-free transcription/translation system
- an ammonia-responsive genetic control element
- a reporter gene such as GFP
- an output enzyme system relevant to ammonia capture or conversion
- ATP and metabolic cofactors
- permeable access for ammonia or a transport mechanism if needed
What would the membrane be made of?
The membrane could be made of POPC + cholesterol, similar to common synthetic-cell vesicle formulations, because this provides a reasonably stable phospholipid bilayer.
What would you encapsulate inside? Enzymes, small molecules.
Inside I would encapsulate:
- bacterial cell-free Tx/Tl extract
- amino acids, nucleotides, salts, and energy mix
- ATP-regeneration substrate
- plasmid DNA for the reporter and detoxification module
- glutamine synthetase or related enzymatic module if not produced in situ
Which organism would your Tx/Tl system come from?
A bacterial system would likely be sufficient, because the sensing and output logic do not require mammalian transcription factors or mammalian-specific signaling pathways. A bacterial extract would also be simpler and cheaper.
How will your synthetic cell communicate with the environment?
Ammonia is a small molecule and can cross membranes to some extent, so the input may not require a dedicated channel. If permeability is insufficient, I could incorporate a membrane pore such as alpha-hemolysin (aHL) to facilitate exchange.
Experimental details
Lipids: POPC, cholesterol
Genes: GFP reporter, alpha-hemolysin (aHL) if needed for permeability, and a detoxification-related gene such as glnA (glutamine synthetase)
How will you measure function?
I would measure:
- GFP fluorescence as the sensing readout
- ammonia concentration before and after treatment as the functional output
- optionally, vesicle integrity by microscopy or dye retention
Homework Question from Peter Nguyen
Write a one-sentence summary pitch sentence describing your concept.
I propose a freeze-dried cell-free textile patch that activates on contact with sweat and reports dehydration-related electrolyte imbalance through a visible fluorescent color change.
How will the idea work, in more detail?
The concept is a wearable textile patch containing freeze-dried cell-free reactions embedded into a layered fabric substrate. When activated by sweat, the patch rehydrates and the cell-free biosensor responds to a target chemical signature correlated with electrolyte loss or dehydration stress. The output could be a fluorescent or colorimetric signal visible to the wearer or captured by a phone camera. Because the system is cell-free, it avoids the maintenance and containment problems associated with living engineered cells while remaining lightweight and low-cost. The broader idea follows the same logic seen in portable cell-free diagnostics: on-demand activation, minimal equipment, and visible readout in low-resource conditions.
What societal challenge or market need will this address?
This could address the need for simple, wearable, low-cost monitoring tools for athletes, outdoor workers, military personnel, or people exposed to heat stress. A textile-integrated indicator could help users detect physiologically dangerous conditions earlier without needing batteries or complex electronics.
How do you envision addressing the limitation of cell-free reactions?
The main limitation is that freeze-dried cell-free systems are typically single-use and require rehydration. I would address this by designing the patch as a replaceable disposable insert within a reusable textile holder. Stability could be improved through lyophilization and protective packaging, and the system would be intentionally framed as an intermittent-use sensor rather than a permanent continuous monitor. The ISS BioBits paper is relevant here because it shows that freeze-dried cell-free systems can remain portable, stable, and functional in constrained environments while producing visually trackable fluorescent outputs.
Homework Question from Ally Huang
Background information
A major challenge in long-duration spaceflight is the need for lightweight, low-maintenance biological tools that can function without extensive laboratory infrastructure. Cell-free systems are promising because they do not require living cells, can be freeze-dried for storage, and can be activated on demand. This makes them relevant for space biology, where crew time, mass, stability, and biocontainment are all constrained. The ISS validation of BioBits showed that freeze-dried cell-free transcription, translation, and biosensing can function robustly in microgravity, making this platform scientifically interesting both for astronaut health monitoring and for biological analysis in future space missions.
Molecular or genetic target
A stress-responsive RNA or DNA target associated with microbial contamination in spacecraft water systems, detected using a sequence-specific toehold-switch or aptamer-based BioBits sensor.
Describe how your target relates to the challenge
A nucleic-acid target related to microbial contamination is directly relevant to astronaut safety because spacecraft currently rely heavily on delayed or ground-based monitoring of potentially contaminated samples. A cell-free biosensor that detects a specific microbial sequence would provide a simpler and more immediate onboard screening tool. This is scientifically interesting because it combines portable synthetic biology with spaceflight constraints, and it builds directly on the demonstrated ability of BioBits to express toehold-switch and aptamer-based biosensors aboard the ISS.
Hypothesis or research goal
My hypothesis is that a freeze-dried BioBits cell-free reaction can be used in space to detect a specific microbial nucleic-acid sequence relevant to spacecraft environmental monitoring and produce a visible fluorescence readout using the Genes in Space toolkit. The reasoning is that BioBits has already been shown to support transcription, translation, and biosensor function aboard the ISS, including toehold-switch-based detection of RNA targets and direct fluorescence readout with the Genes in Space Fluorescence Viewer. If a custom target sequence associated with a spacecraft waterborne microbe were inserted into a toehold-switch detection scheme, the same general architecture could be repurposed into a practical onboard monitoring assay. This would support faster environmental testing in space and reduce dependence on returning samples to Earth for analysis.
Experimental plan
I would prepare freeze-dried BioBits reactions containing a toehold-switch plasmid designed against a target microbial RNA sequence. Samples tested would include: correct target RNA, mismatched RNA control, no-target negative control, and blank reaction control. Reactions would be rehydrated and incubated using the BioBits workflow, then monitored with the P51 / Genes in Space fluorescence viewer. The main data collected would be fluorescence intensity over time and endpoint comparison between target and control conditions. A successful result would show fluorescence only in the presence of the correct target sequence, similar to the toehold-switch logic validated aboard the ISS.
References
- Kocalar, S., Miller, B. M., Huang, A., Gleason, E., Martin, K., Foley, K., Copeland, D. S., Jewett, M. C., Alvarez Saavedra, E., & Kraves, S. (2024). Validation of cell-free protein synthesis aboard the International Space Station. ACS Synthetic Biology, 13, 942–950. https://doi.org/10.1021/acssynbio.3c00733