Week 2: DNA read write edit
for this week’s HW assignment I’ve chosen the protein mCherry. mCherry is a protein that expresses in red flourescent light emmitance. Fusing it to another protein will enable use to discern wheter the ‘other’ protein is expressed, by visibly observing the red flourescent light. essentially, mCherry functions as a global process visualisation tool across multiple SynBio applications.
Googling mCherry Protein I arrived at the uniprot.org database where I’ve obtained the mCherry sequence:
tr|A0A4D6FVK6|A0A4D6FVK6_ECOLI MCHERRY OS=Escherichia coli str. K-12 substr. MG1655 OX=511145 GN=mCherry PE=1 SV=1 MVSKGEEDNMAIIKEFMRFKVHMEGSVNGHEFEIEGEGEGRPYEGTQTAKLKVTKGGPLP FAWDILSPQFMYGSKAYVKHPADIPDYLKLSFPEGFKWERVMNFEDGGVVTVTQDSSLQD GEFIYKVKLRGTNFPSDGPVMQKKTMGWEASSERMYPEDGALKGEIKQRLKLKDGGHYDA EVKTTYKAKKPVQLPGAYNVNIKLDITSHNEDYTIVEQYERAEGRHSTGGMDELYK
Reverse translating amino sequence to dna codons using Biorinformatics.org’s Reverse Translate tool I got a 708 codon sequence:
atggtgagcaaaggcgaagaagataacatggcgattattaaagaatttatgcgctttaaa gtgcatatggaaggcagcgtgaacggccatgaatttgaaattgaaggcgaaggcgaaggc cgcccgtatgaaggcacccagaccgcgaaactgaaagtgaccaaaggcggcccgctgccg tttgcgtgggatattctgagcccgcagtttatgtatggcagcaaagcgtatgtgaaacat ccggcggatattccggattatctgaaactgagctttccggaaggctttaaatgggaacgc gtgatgaactttgaagatggcggcgtggtgaccgtgacccaggatagcagcctgcaggat ggcgaatttatttataaagtgaaactgcgcggcaccaactttccgagcgatggcccggtg atgcagaaaaaaaccatgggctgggaagcgagcagcgaacgcatgtatccggaagatggc gcgctgaaaggcgaaattaaacagcgcctgaaactgaaagatggcggccattatgatgcg gaagtgaaaaccacctataaagcgaaaaaaccggtgcagctgccgggcgcgtataacgtg aacattaaactggatattaccagccataacgaagattataccattgtggaacagtatgaa cgcgcggaaggccgccatagcaccggcggcatggatgaactgtataaa
Using the Gensmart Codon Opt Tool to optimise the above codon seq for E.coli. I chose to opt for E.coli because it is my understanding that this organism serves as a common platform for SynBio uses, and beacuse i’m new to the field, I rather stick to common practices to solidify my understanding when taking first steps in prot-design.
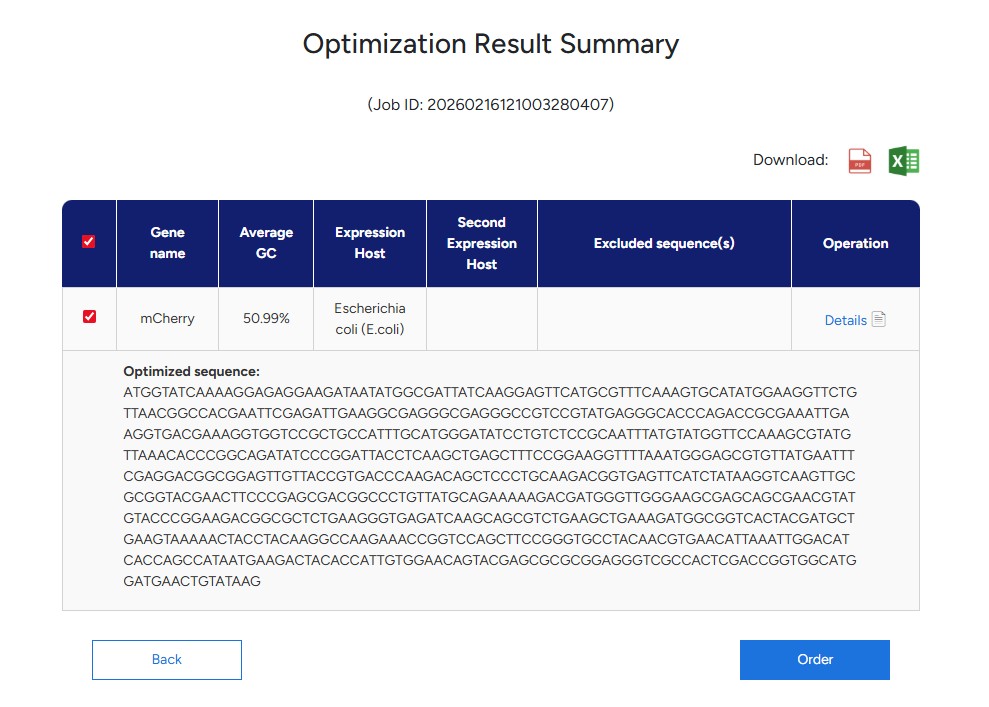
Resulting optimisation codon seq:
ATGGTATCAAAAGGAGAGGAAGATAATATGGCGATTATCAAGGAGTTCATGCGTTTCAAAGTGCATATGGAAGGTTCTGTTAACGGCCACGAATTCGAGATTGAAGGCGAGGGCGAGGGCCGTCCGTATGAGGGCACCCAGACCGCGAAATTGAAGGTGACGAAAGGTGGTCCGCTGCCATTTGCATGGGATATCCTGTCTCCGCAATTTATGTATGGTTCCAAAGCGTATGTTAAACACCCGGCAGATATCCCGGATTACCTCAAGCTGAGCTTTCCGGAAGGTTTTAAATGGGAGCGTGTTATGAATTTCGAGGACGGCGGAGTTGTTACCGTGACCCAAGACAGCTCCCTGCAAGACGGTGAGTTCATCTATAAGGTCAAGTTGCGCGGTACGAACTTCCCGAGCGACGGCCCTGTTATGCAGAAAAAGACGATGGGTTGGGAAGCGAGCAGCGAACGTATGTACCCGGAAGACGGCGCTCTGAAGGGTGAGATCAAGCAGCGTCTGAAGCTGAAAGATGGCGGTCACTACGATGCTGAAGTAAAAACTACCTACAAGGCCAAGAAACCGGTCCAGCTTCCGGGTGCCTACAACGTGAACATTAAATTGGACATCACCAGCCATAATGAAGACTACACCATTGTGGAACAGTACGAGCGCGCGGAGGGTCGCCACTCGACCGGTGGCATGGATGAACTGTATAAG
Releying on my understanding of ‘The Central Dogma’, one ’technology’ to produce the protein from the above sequence is the protein creation process that happens inside the Rybozome which is located in a cell’s nucleus. The Rybozome (which is also called R-RNA) is the site that ’takes-in’ M-RNA after it has transcribed a DNA sequence and undergone some editing to the original transcribing. The Rybozome has 2 parts. M-RNA sits in the small part, and gets read by the Rybozome which translates the codons it reads in the M-RNA, and ‘calls’ for T-RNAs to arrive at the big part of the Rybozome. Each T-RNA ‘holds’ and Amino Acid, and when ‘called’ by a respective codon sequence, it arrives at the Rybozome to ‘hand over’ that amino acid. Amino Acids bind to one another in the Rybozome’s bigger part and start forming a chain. This process repeats until the Rybozome ‘finishes’ reading the entire sequence, resulting in a chain of amino acids that will leave the Rybozome and continue to other processes that will eventually result in the aminos folding into a 3D structure that we call a protein.
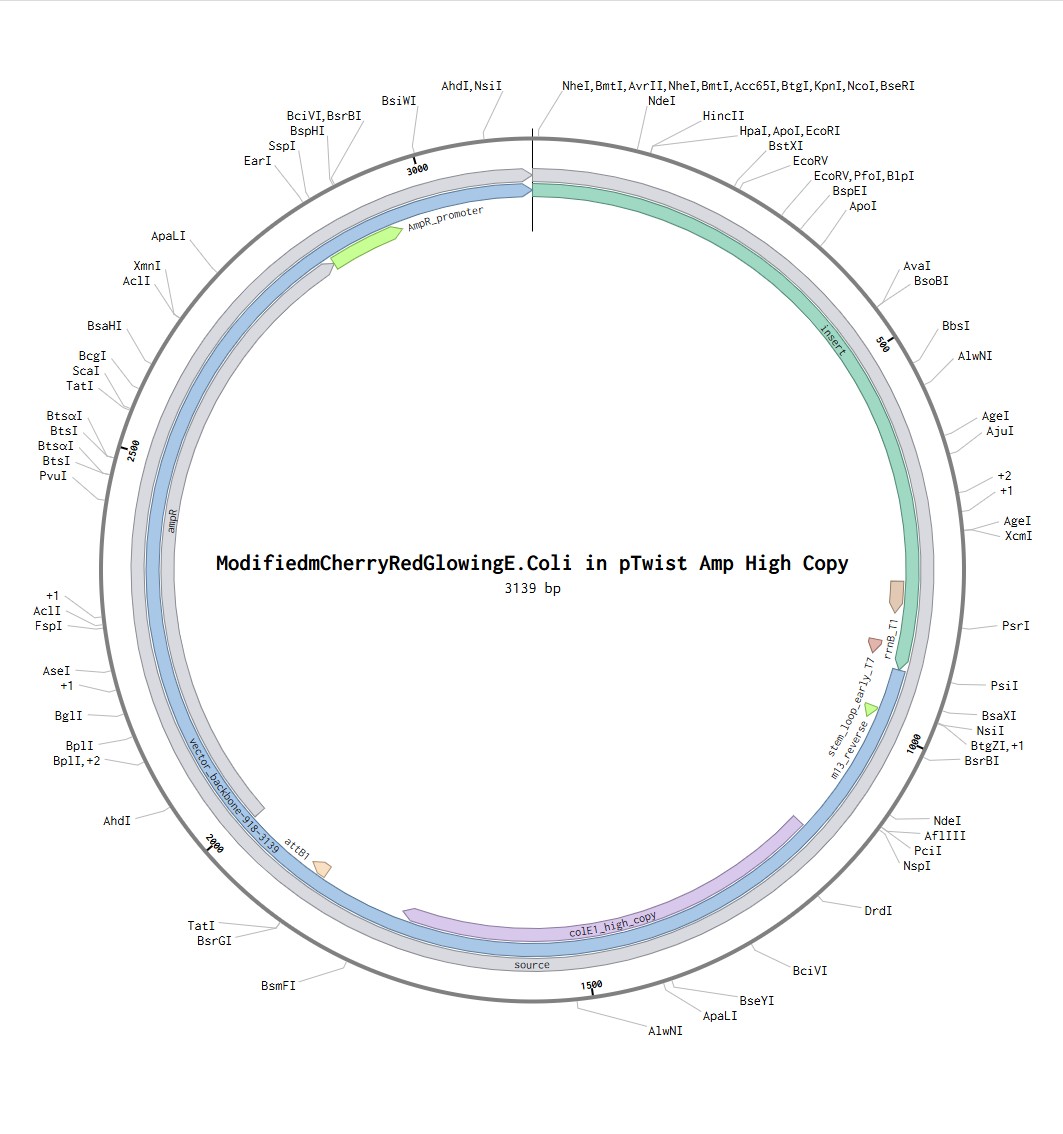
Putting my optimized codon seq into Benchling with the additional sequences provided, I have created a share link to the Benchling file.
Following the steps in the Twist instructions part, I was able to export my Benchling file in .fasta format, upload it to the Twist platform, choose pTwist Amp High Copy Vector and download the .gb file. reuploaded into benchling I can see the resulting expressions cassette! Whohoo (:
What DNA would you want to sequence (e.g., read) and why?
Within the given context, of being introduced to Synbio, I am inclined towards sequencing DNA of organisms and proteins that have something to do with processes of photosynthesis, CO2 sequestration and or flavor and aroma enhancement. The former two have versatile applications in the emerging field of bio-chemical energy production, while the latter have more focused applications in the field of cultivated lab meat, both of them I find highly impactful and worthwhile endeavors to pursue. In particular, I can propose to read the DNA of the RuBisCO enzyme, that binds a CO2 molecule to a RuBP molecule and creates two 3-carbon molecules. This is a part of the larger photosynthesis process where an organism is converting light energy into sugars and CO2 is being ‘stored’ in sugars. At the same time the photosynthetic process could also be tapped into, converting free electrons created in the process to electrical energy. Enhancing the processes underlying photosynthesis means potentially improving electrical energy yield and carbon sequestration, two much needed capabilities in our time.
In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
For reading RuBisCO genes, from my understanding, I would go for the Sanger reading process. It’s considered high accuracy, and fit for sequences that are on the order of ~700bp.
Is your method first-, second- or third-generation or other? How so?
Sanger is considered a first-gen method due to the fact that it reads the sequence one-by-one. ‘Next-Gen Methods’ tend to read arrays in parallel making them faster and more cost-effective.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
A Sanger reaction will typically require the reaction-medium, a template DNA (could be a purified PCR or a plasmid), and a primer. If using a Purified PCR, the primers used for the PCR process would work effectively as primers for the Sanger process as well. Each primer will read roughly ~700bp so for reading longer sequences you would need to tile primers one after the other. If the sequence is in the right size, you could sequence it in 2 ‘runs’ using a forward reading primer and a reverse reading primer and notice where they overlap.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
Lets say I am performing the Sanger reaction with a purified PCR. I would take the Sanger mix (the medium in which the Sanger reaction occurs), my purified RuBisCO PCRs, and the primer I used for the PCR creation itself. Based on the size of the PCR (in bp units) I will decide how many primers I need because Sanger reads around ~700bp. Preferably the best results are achieved either with a sequence length that is suitable for a single primer use or dual-primer use, in which case I will utilize both a forward reading primer and a reverse reading primer. ‘base calling’, or identification of ATCG nucleotides is done by attaching a fluorescent ddNTP after bases occasionally. The ddNTP stops the sequence’s extension, serving as a ‘cap’ of sorts. While copying the sequence many times, the RNA polymerase will attach the ddNTP label multiple times, effectively creating many copies of the DNA in all possible lengths. These ddNTP fluorescent caps will serve as a labels that could be seen after the entire batch had been separated by length in an electrophoresis process. A sorting algorithm is then deployed, identifying the different colors and lengths of each sequence that shows up, and translating the colors back to bases. so essentially cutting dna and attaching colored labels to cuts, ordering the cuts by size, translating color + size results back to bases sequence using software.
What is the output of your chosen sequencing technology?
The output of the Sanger process is the chromatogram, a color-coded graph that shows peaks where the respective base had been identified along the length of the sequence, and the base-called sequence that is derived from it.
What DNA would you want to synthesize (e.g., write) and why?
Going back to the first question in this assignment, in the given context, I am inclined towards modifying DNA of organisms and proteins that have something to do with processes of photosynthesis, CO2 sequestration and or flavor and aroma enhancement. The former two have versatile applications in the emerging field of bio-chemical energy production, while the latter have more focused applications in the field of cultivated lab meat, both of them I find highly impactful and worthwhile endeavors to pursue. In particular, I can propose to write the DNA of the RuBisCO enzyme, enhancing it’s ability to bind CO2 molecules to RuBP molecules more effectively. Enhancing the processes underlying photosynthesis means potentially improving electrical energy yield and carbon sequestration,.
What technology or technologies would you use to perform this DNA synthesis and why? How does your technology of choice edit DNA? What are the essential steps?
I believe that for modifying a DNA sequence I would order oligos of the chosen sequence, perform a fragment replacement on the specific point in the sequence I assume will result in enhancing the desired trait (In my case is improving the ability of RuBisCO enzyme to bind CO2 molecules to RuBP molecule), assemble all fragments together in assembly process (Gibson?), and then sequence the entire dna to verify that I’ve assembled it correctly.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
First I will need to make sure I have the proper assessment tools to determine success or failure. If the RuBisCO enzyme is binding CO2 molecules, it makes sense to creating a testing environment where I could measure rate of of CO2 dissappearance for example. I would then proceed to select sites for the mutation to happen (multiple sites or a specific target site, depending on available prior knowledge.). The next step would be to create primers that target these specific sites and change some bases, or if a larger chain needs to be replaced, I would create entire custome fragments. After that I would assemble the modifyied DNA, Sequence it to make sure I’ve assembled the intended sequence, and test it. Required inputs would include: template palsmids containing a PCR product of RuBisCO or the gene itself, primers or entire fragments for replacing, polymerase for duplicating the sequence, and assembly enzymes to ‘stich’ the new sequence together. If going with a plasmid, I will need the backbone itself, host cells to insert the plasmid to, and a sequencing technology to verify my work (Sanger was mentioned earlier but it depends on the actual planned sequence legnth in bp units)
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
The polymerase process may introduce errors in copying the sequence. This is also true for assembly of large fragments as well, they could introduce unintended changes during assembly, this tends to happen if you try to assemble a bigger fragment count, repetitive fragments, or very long ones.
References:
Adams, J. (2008) DNA sequencing technologies. Nature Education 1(1):193
https://www.nature.com/scitable/topicpage/dna-sequencing-technologies-690/
Blogpost: “Site Directed Mutagenesis by PCR”