Week 4: Protein Design 1
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Some definitions regarding measurement units:
A Dalton (Da) is a unit used to describe molecular mass. A useful heuristic is:
1 Da ≈ 1 g/mol
A mole (mol) is a counting unit in chemistry: 1 mol = 6.022 × 10^23 molecules (Avogadro’s number).
If we assume an amino acid (AA) is ~100 Da, this corresponds to ~100 g/mol. Meat is not entirely protein; if we assume protein is ~20% of meat by mass, then 500 g meat ≈ 100 g protein.
100 g ÷ (100 g/mol) = 1 mol ≈ 6.022 × 10^23 AA units.
So, 500 g of meat contains on the order of ~6 × 10^23 amino-acid “units” worth of protein mass, given the assumptions above. (OpenStax, n.d.)
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When we digest proteins, our body breaks them down into smaller peptides and then into amino acids. Protein breakdown in digestion can be described in three stages:
- Stomach: Hydrochloric acid (HCl) lowers pH and helps denature proteins (unfolding them). The enzyme pepsin begins cutting long polypeptides into shorter peptides. (OpenStax, n.d.)
- Small intestine: Pancreatic proteases (e.g., trypsin and chymotrypsin) continue cutting peptide bonds into smaller peptides and some free amino acids. (OpenStax, n.d.)
- Brush border: Enzymes on the intestinal “brush border” complete digestion into amino acids and very short peptides that can be absorbed into the bloodstream and distributed throughout the body for building human proteins. (OpenStax, n.d.)
Why are there only 20 natural amino acids?
The standard genetic code encodes 20 amino acids, with two rare genetically encoded additions: selenocysteine (including in humans) and pyrrolysine (in some microbes). A key point is that amino acids are not only “letters” for variety; they are the building blocks that enable proteins to reliably fold into stable, soluble, close-packed 3D structures with functional binding pockets. (Doig, 2016)
Evolution likely began with a smaller set of amino acids, then expanded the “vocabulary” as biological complexity increased. However, beyond a certain point, adding new amino acids yields diminishing returns and increases system-level costs: folding stability becomes harder to maintain, translation errors become more costly, and codon assignments become harder to manage without confusion. This challenges the idea that the set is purely a “frozen accident” and supports the view that the 20-amino-acid set is close to an optimal balance between chemical diversity and translational reliability. (Doig, 2016)
Where did amino acids come from before enzymes that make them, and before life started?
This answer splits into two domains: extra-terrestrial and terrestrial sources. Extra-terrestrially, amino acids have been detected in carbon-rich meteorites, suggesting that early Earth could have received a chemically diverse mixture of amino acids from space. (Kirschning, 2022)
Terrestrially, multiple plausible prebiotic routes could generate amino acids through abiotic chemistry in different environments—so it’s unlikely there was a single origin location. Examples include Strecker-type synthesis and Miller–Urey style discharge experiments, as well as hydrothermal and iron–sulfur mineral settings that can promote reaction networks from simple precursors. Wet–dry cycles (e.g., in hydrothermal fields) are important because they can repeatedly concentrate reactants and support peptide formation, bridging from free amino acids toward short peptides. (Kirschning, 2022)
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
An α-helix made from D-amino acids is expected to be left-handed (the mirror-image of the common right-handed α-helix formed by L-amino acids). This follows from stereochemistry: switching from L to D reverses the preferred helical handedness. (Doig, 2016)
Can you discover additional helices in proteins?
Yes. Besides the α-helix, proteins can also contain 3_10 helices and π-helices. A 3_10 helix is typically tighter than an α-helix (fewer residues per turn), often appearing as short segments. A π-helix is rarer and is often described as an insertional “bulge” within α-helices that can contribute to function (e.g., shaping binding pockets). (Cooley et al., 2010)
Why are most molecular helices right-handed?
In proteins built from L-amino acids, the right-handed α-helix is strongly favored because it minimizes steric clashes and supports favorable backbone geometry and hydrogen bonding. Left-handed α-helices in L-amino-acid proteins tend to be destabilized by unfavorable backbone/side-chain interactions. (Doig, 2016)
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets tend to aggregate because exposed β-sheet edges can “zipper” together through backbone hydrogen bonds (edge-to-edge pairing). In water, aggregation is often further stabilized when hydrophobic side chains pack together and exclude water, promoting larger assemblies. (Nowick, 2008)
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Many amyloid diseases involve proteins that misfold and assemble into highly stable β-sheet-rich fibrils (amyloid), which can accumulate in tissues and disrupt function. β-sheet structures can expose edges that promote repeated “zippering” and stacking into fibrils, making them unusually stable. (Nowick, 2008)
Because amyloid fibrils can be rigid, chemically stable, and programmable by sequence, they are also being explored as functional biomaterials (with careful design and safety considerations). As one review summarizes: “The rigidity, chemical stability, high aspect ratio, and sequence programmability of amyloid fibrils have made them attractive candidates for functional materials…” (Li & Zhang, 2021)
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
Myoglobin is an oxygen-binding protein in muscle that stores and shuttles O₂ inside cells using a heme group, and its oxygen/oxidation state is a major reason meat looks red vs brown. In the cultivated-meat “marbling/structure” project, it’s directly tied to the sensory realism problem: getting cultured tissue to develop the same color cues people associate with meat. It interests me because it links a visible outcome (color) to a real underlying biological variable (oxygen handling and tissue state), which is a legible bioengineering lever I can explore in my final project proposition.
Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid?
Downloaded the AA sequence for Myoglobin (one of five variations in humans) from the Uniprot website
sp|P02144|MYG_HUMAN Myoglobin OS=Homo sapiens OX=9606 GN=MB PE=1 SV=2 MGLSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETLEKFDKFKHLKSEDEMKASE DLKKHGATVLTALGGILKKKGHHEAEIKPLAQSHATKHKIPVKYLEFISECIIQVLQSKH PGDFGADAQGAMNKALELFRKDMASNYKELGFQG
it is 154 Amino Acids long, and the most frequent Amino Acid within the sequence is Lysine (coded in the sequence as K), and it appears 20 times in the chain.
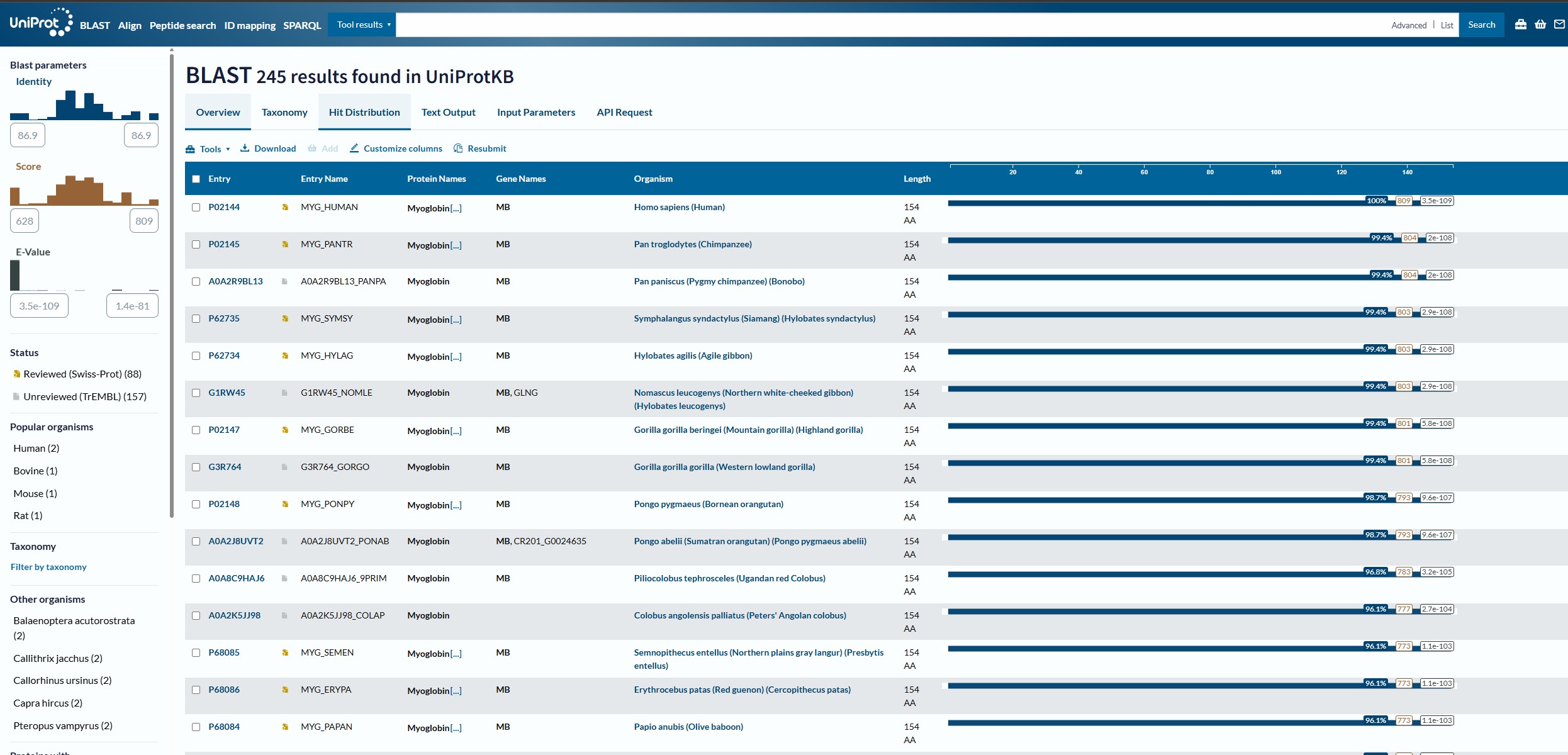
How many protein sequence homologs are there for your protein? Does your protein belongs to any protein family?
Using the Uniprot BLAST tool, I analysied the Myoglobin sequence and found that there are 250 homologs for this protein across the swiss-prot and trEMBL datasets.
looking up Myoglobin I arrived at the Interpro webpage where Myoglobin was identified to be a member of the globin domain (Pfam category PF00042)
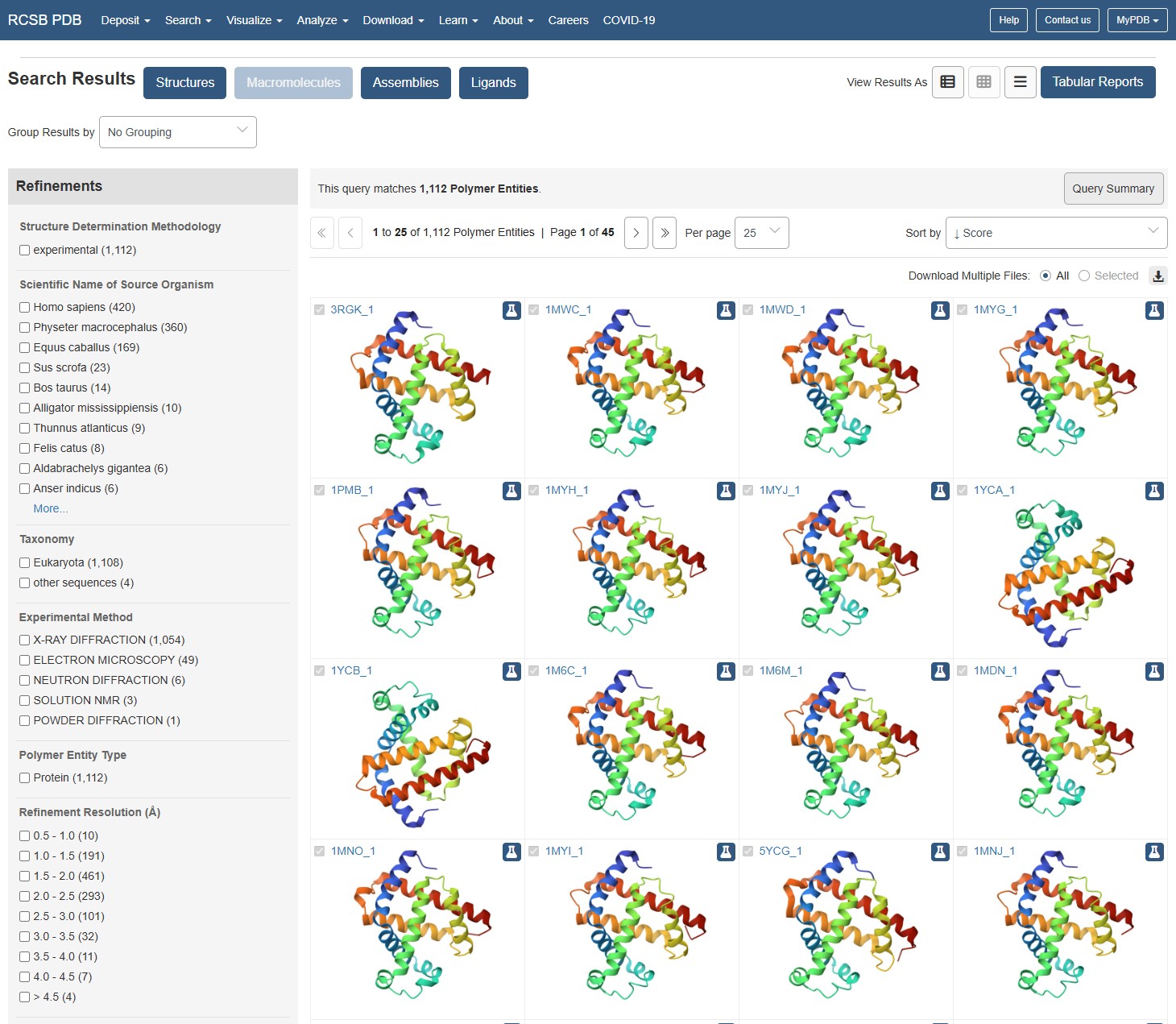
Identify the structure page of your protein in RCSB When was the structure solved? Is it a good quality structure?
using the RCSB website to identify Myoglobin structure, I can see the structure has been documented many times, in increasing resolutions. The earliest documented structure on the website dates to 1975. The best resolutions available (10 entities) are within the 0.1-0.5A range, and these are dated to be between 2005-2024. I would say for the latest entities, they populate the best resolution category available on the webpage so they should be considered as excellent quality.
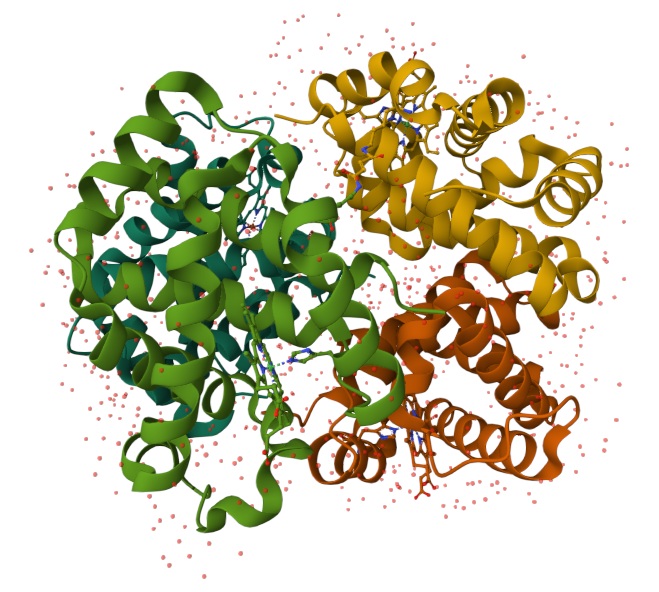
Are there any other molecules in the solved structure apart from protein? Does your protein belong to any structure classification family?
Using the 3D render and data provided for 6KA9 (the first RCSB search result: Crosslinked alpha(Fe-CO)-beta(Ni) human hemoglobin A), I can see there are O molecules scattered across the 3D render. This seems to be due to the protein modeled while being in an aqueos solution. Besides the water, only Polymer and Ligand groups are present in the render, which make the entire protein quaternary structure.
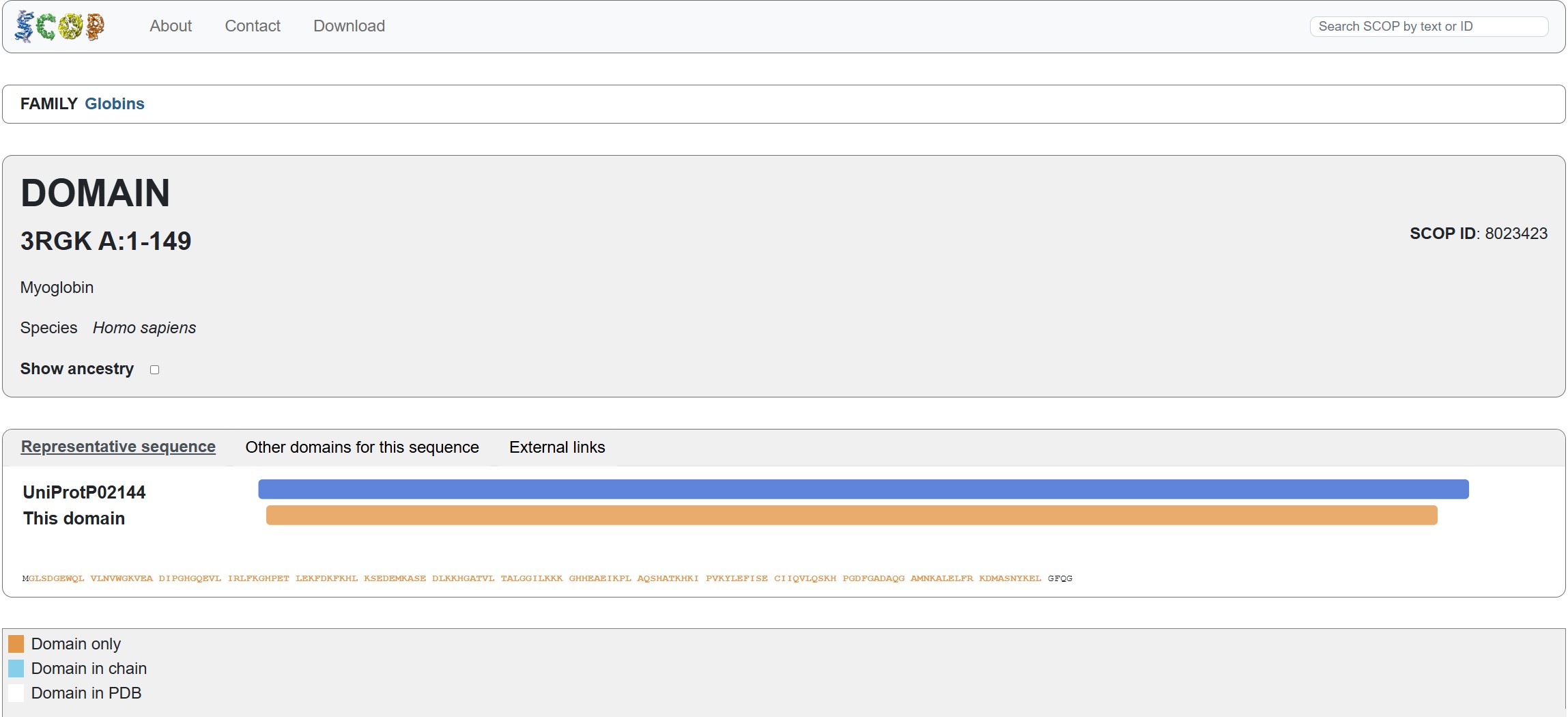
Does your protein belong to any structure classification family?
Using the SCOP tool shows that Myoglobin is a member of the 3RGK A:1-149 domain (SCOP ID 8023423). it also notes that it is a member of the Globins family, which matches the protein family we say Myoglobin associated with in the earlier question.
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
For this assignment, I used the RCSB 3D visualisation webapp that runs Mol* (WebGL) for 3D renderings. Because the “cartoon” representation for the ligand groups did not show anything, I assigned them a “line” representation so they will be visible.
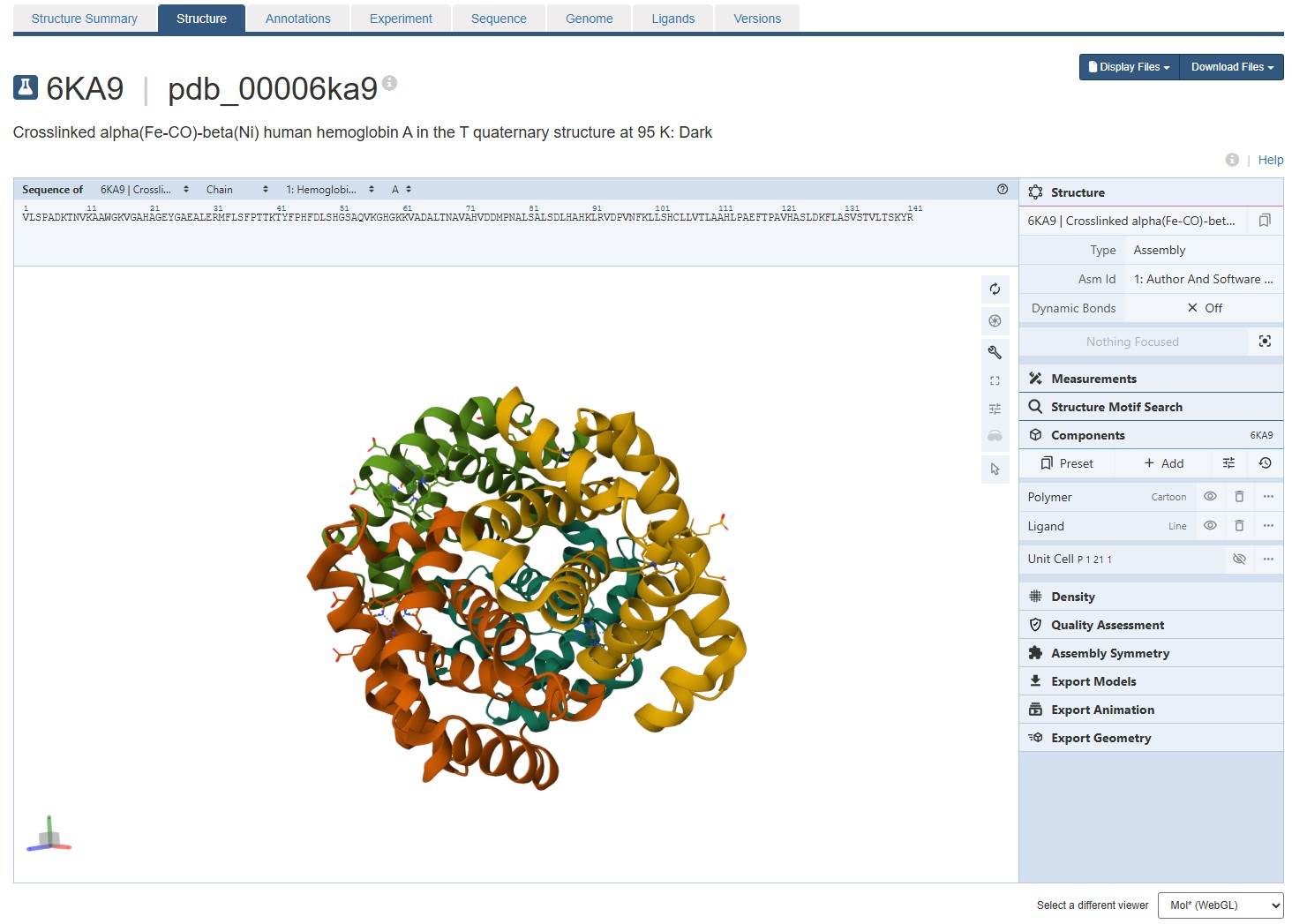
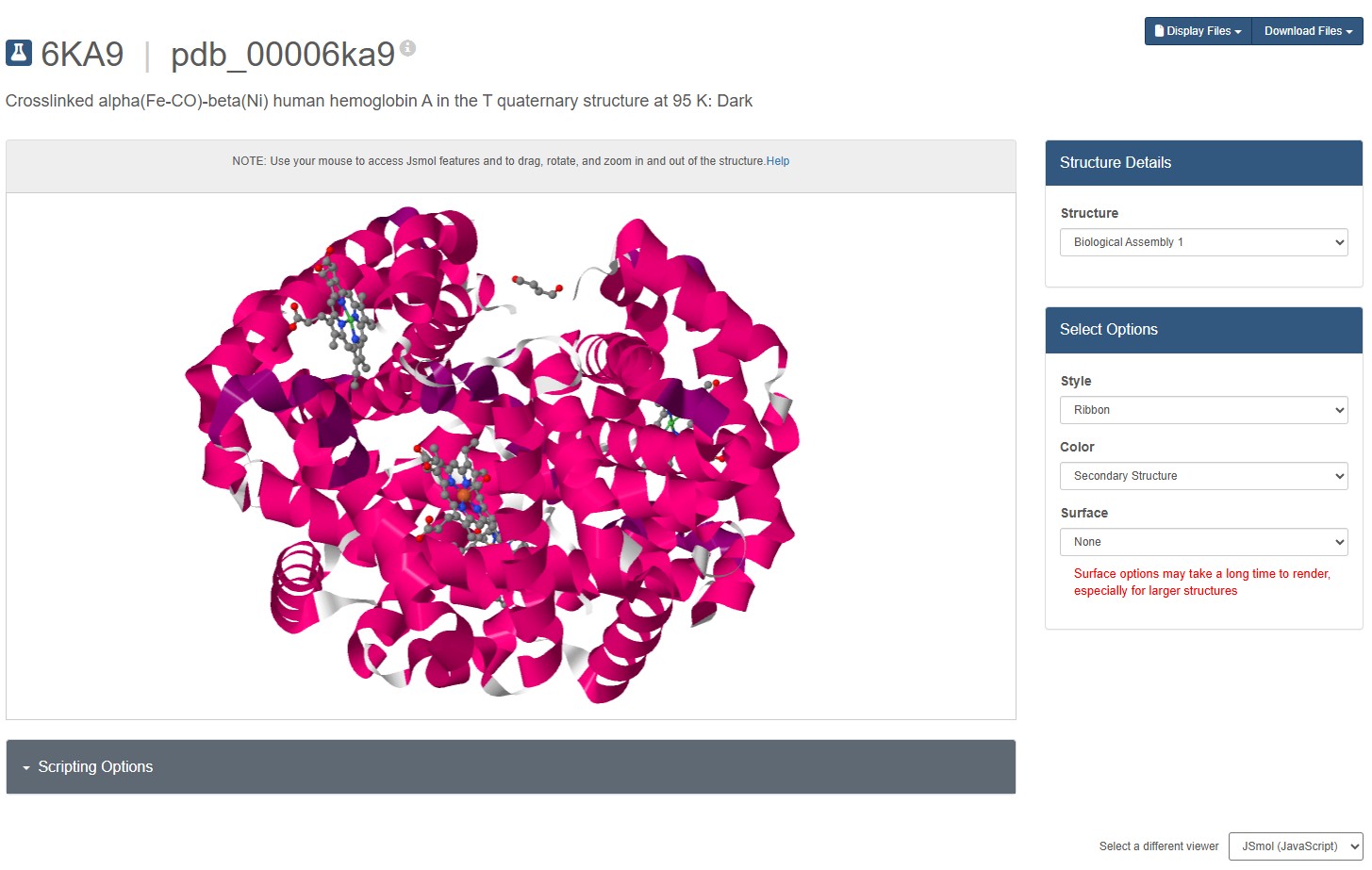
Mol* does not seems to have a “ribbon” representation. I switch to JSMol in the RCSB 3D Visualisation webapp, and chose a ribbon representation.
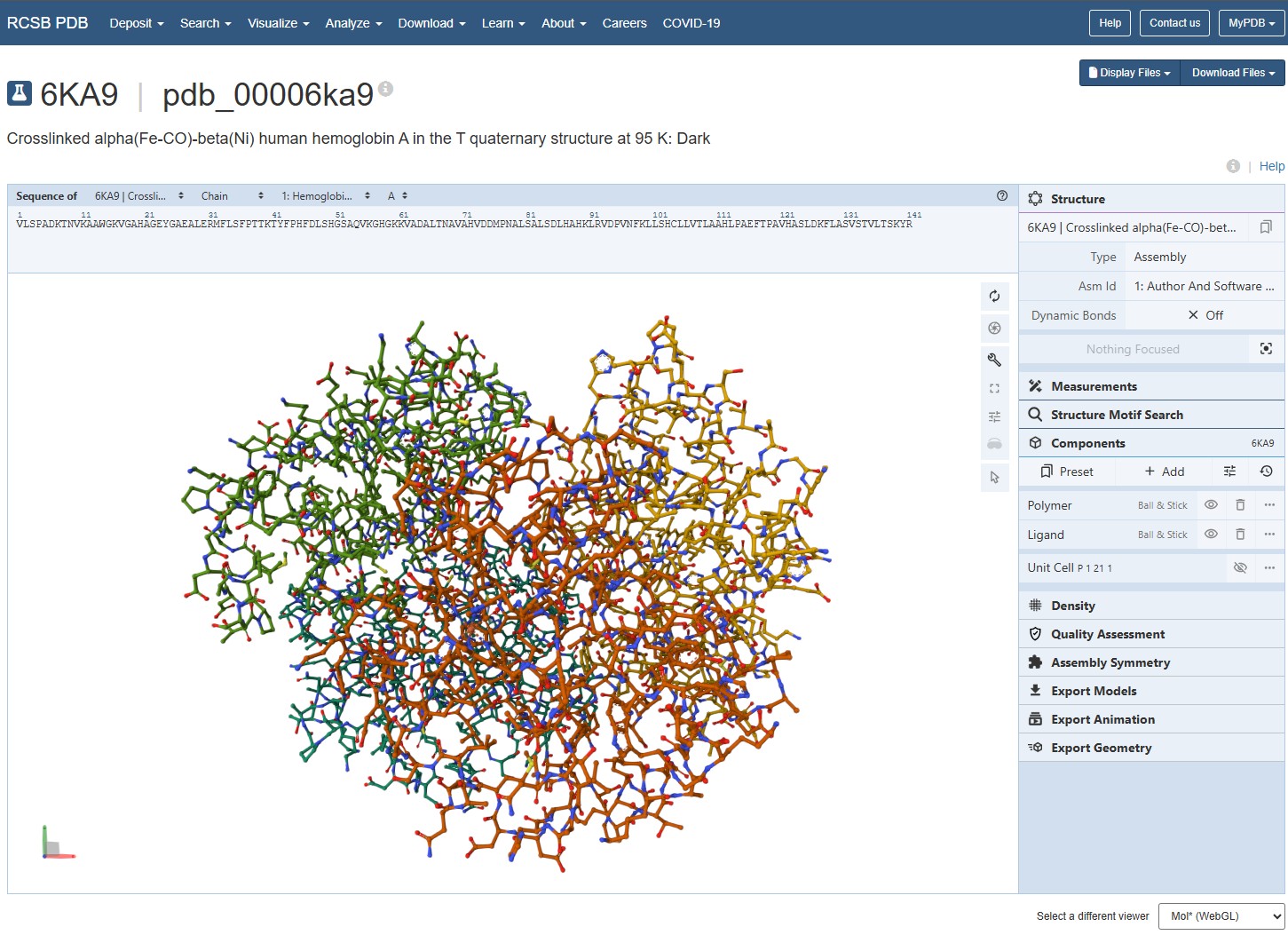
Going back to Mol* (because I think it creates nicer renderings), we get the “ball and stick” representation.
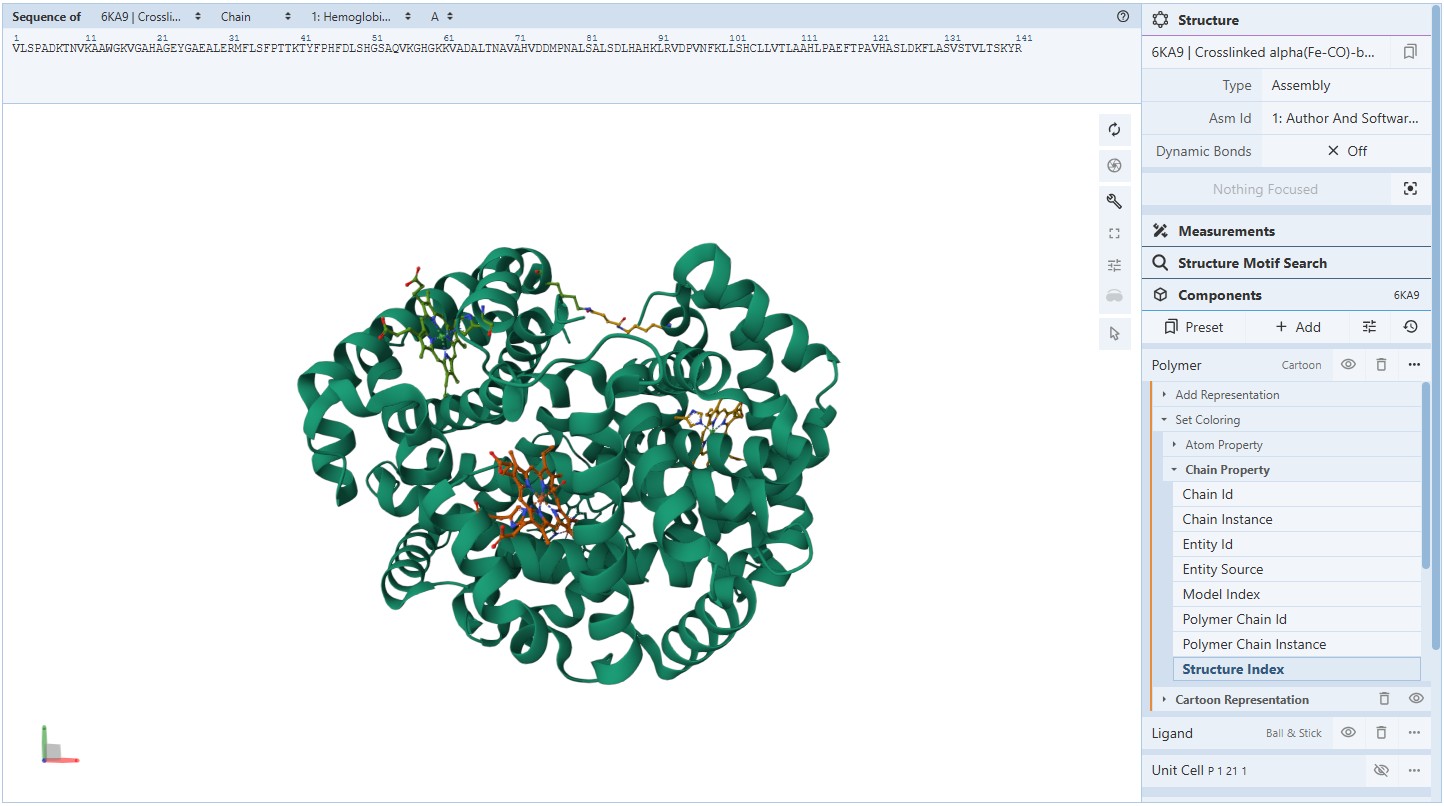
Coloring secondary structures (and honestly we could even determine this before, but to be on the safe side), we can see the Myoglobin protein variant I’m visualising consists only of α-helices as Polymer structures (They are all colored the same).
Coloring the protein for hydrophobicity, we discover the molecules vary in their hydrophobic charecteristics, varying across a scale of 11 different degrees. The spread is a bit confusing and I’m not sure I can draw a solid conclusion from it. It seems the protein is mostly hydrophillic, but certain molecules demonstrate a strong hydrophobic attribute.
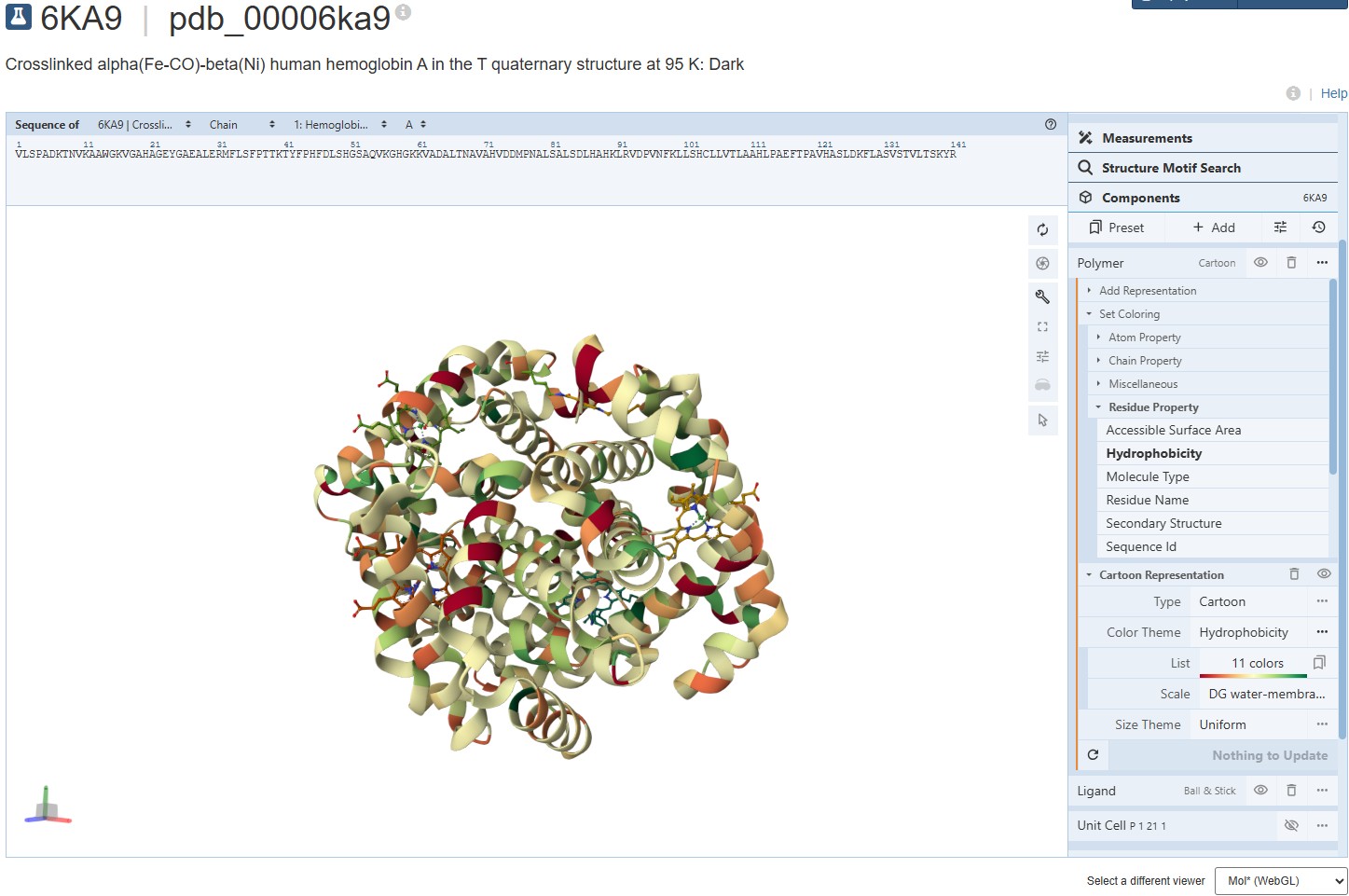
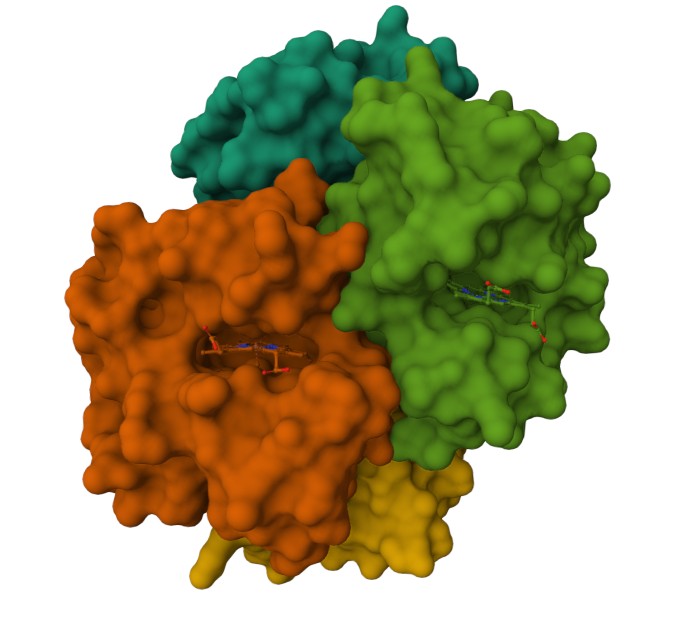
Representing the protein using surfaces, we can immidiatly tell there is a central pocket. Another interesting observation available through the surface render is that the Ligand groups have their own ‘mini pockets’ where they are tucked into the Polymer structure.
Surface representation
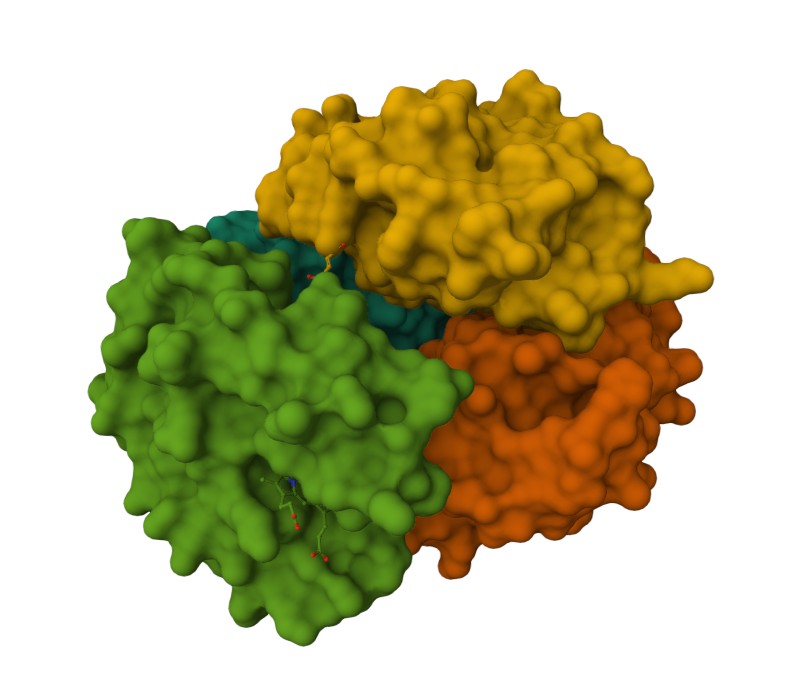
Observing the central pocket
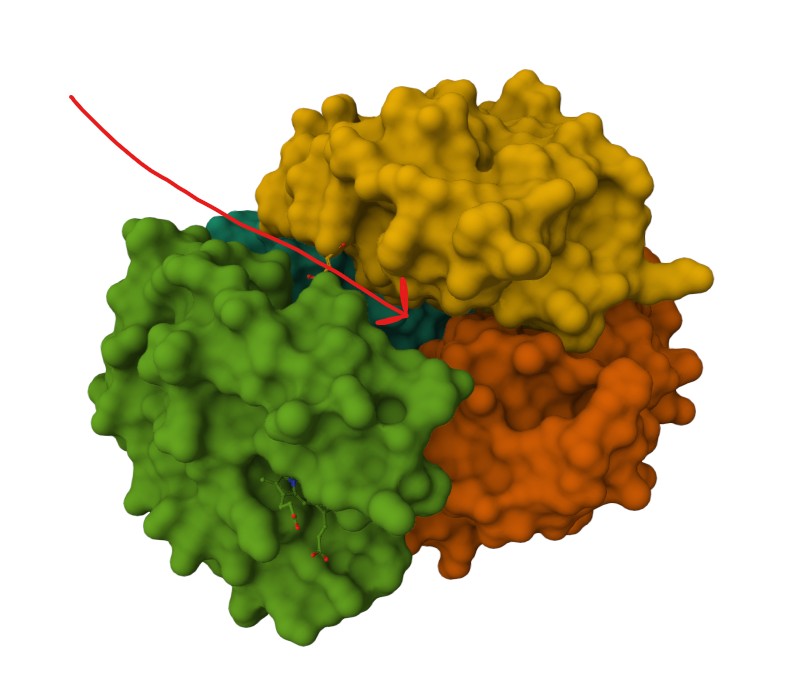
Tucked Ligand groups
C1. Protein Language Modeling
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods. Can you explain any particular pattern?
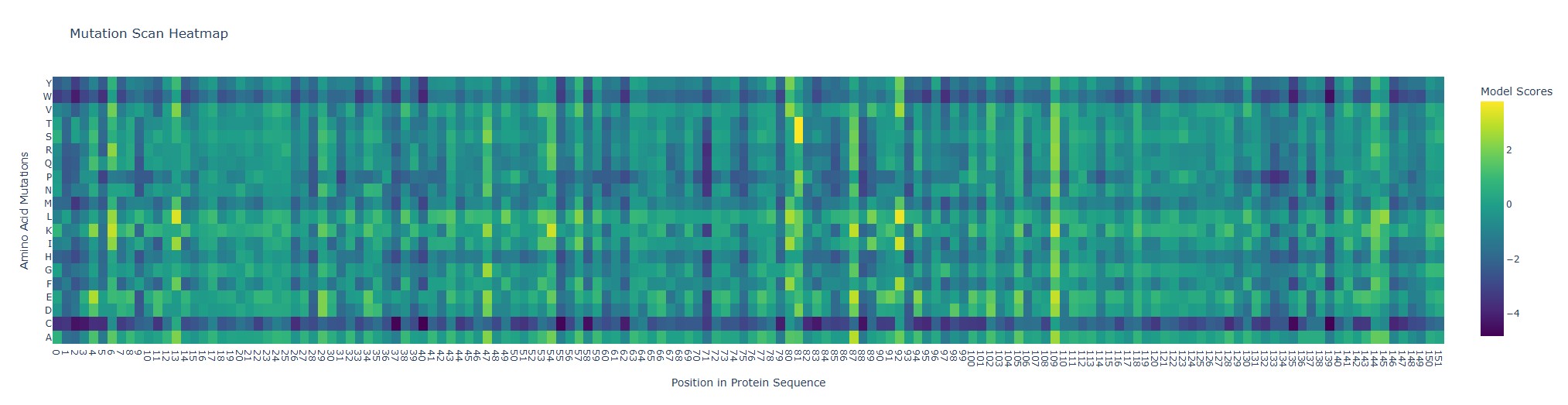
Looking at the heatmap, position 92 (wild-type Q, glutamine) stands out because the substitutions Q→L and Q→I get unusually high model scores (bright yellow). In ESM2 terms, that means the model thinks Leu or Ile “fit” well at that position in a myoglobin-like sequence.
Latent Space Analysis: Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
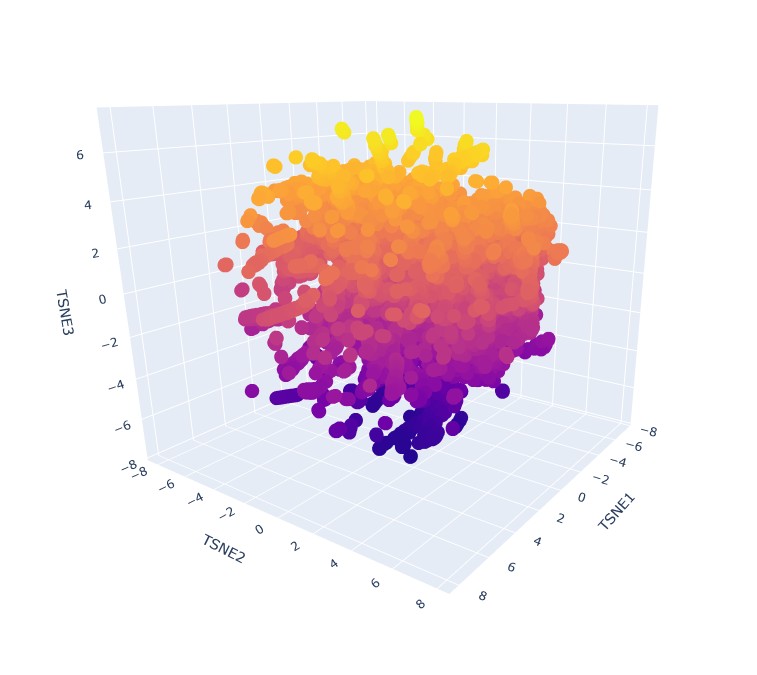
Yes, it seems that when zooming in, specific proteins tend to group in neighbourhoods based on their 3 t-SNE coordinates. like this group:
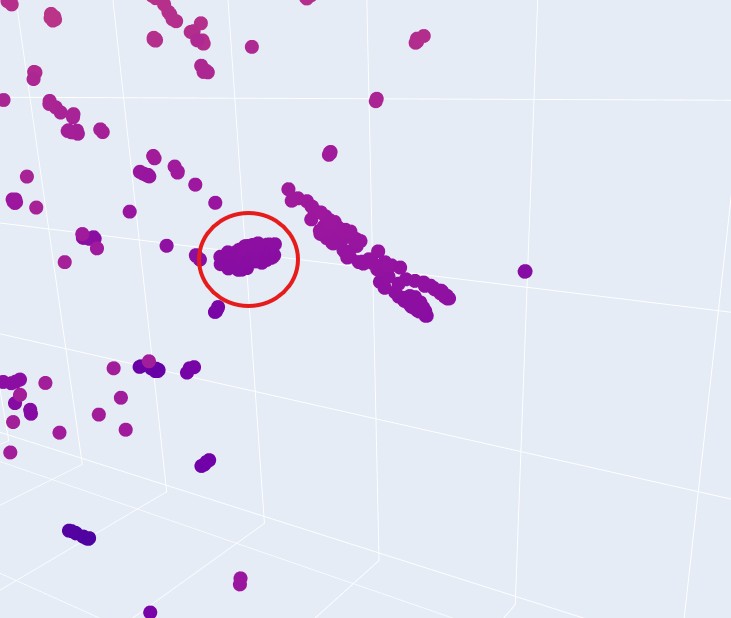
This implies that they have very similar sequence, domain or group, and therefore can be expected to have similar fold structures. They do not, however share the same taxonomy. That is understood immidiatly when you hover over some group of proteins and see that their taxonomy is varied across multiple species.
References
Cooley, R. B., Arp, D. J., & Karplus, P. A. (2010). Evolutionary origin of a secondary structure: π-helices as cryptic but widespread insertional variations of α-helices enhancing protein functionality. Proceedings of the National Academy of Sciences, 107(25), 11285–11290. https://pmc.ncbi.nlm.nih.gov/articles/PMC2981643/
Doig, A. J. (2016). Frozen, but no accident – why the 20 standard amino acids were selected. FEBS Journal. https://febs.onlinelibrary.wiley.com/doi/10.1111/febs.13982
Kirschning, A. (2022). On the evolutionary history of the twenty encoded amino acids. Chemistry–A European Journal, 28, e202201419. https://pmc.ncbi.nlm.nih.gov/articles/PMC9796705/
Li, C., & Zhang, X. (2021). Amyloids as building blocks for macroscopic functional materials: Designs, applications and challenges. International Journal of Molecular Sciences, 22(5), 1–28. https://pmc.ncbi.nlm.nih.gov/articles/PMC8508955/
Nowick, J. S. (2008). Exploring β-sheet structure and interactions with chemical model systems. Accounts of Chemical Research, 41(10), 1319–1330. https://pmc.ncbi.nlm.nih.gov/articles/PMC2728010/
OpenStax. (n.d.). Chemical digestion and absorption: A closer look. In Anatomy and Physiology 2e. https://openstax.org/books/anatomy-and-physiology-2e/pages/23-7-chemical-digestion-and-absorption-a-closer-look