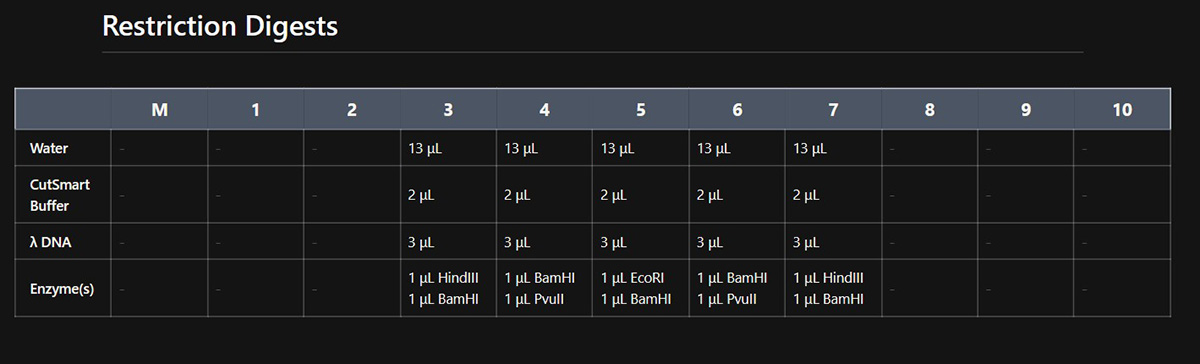
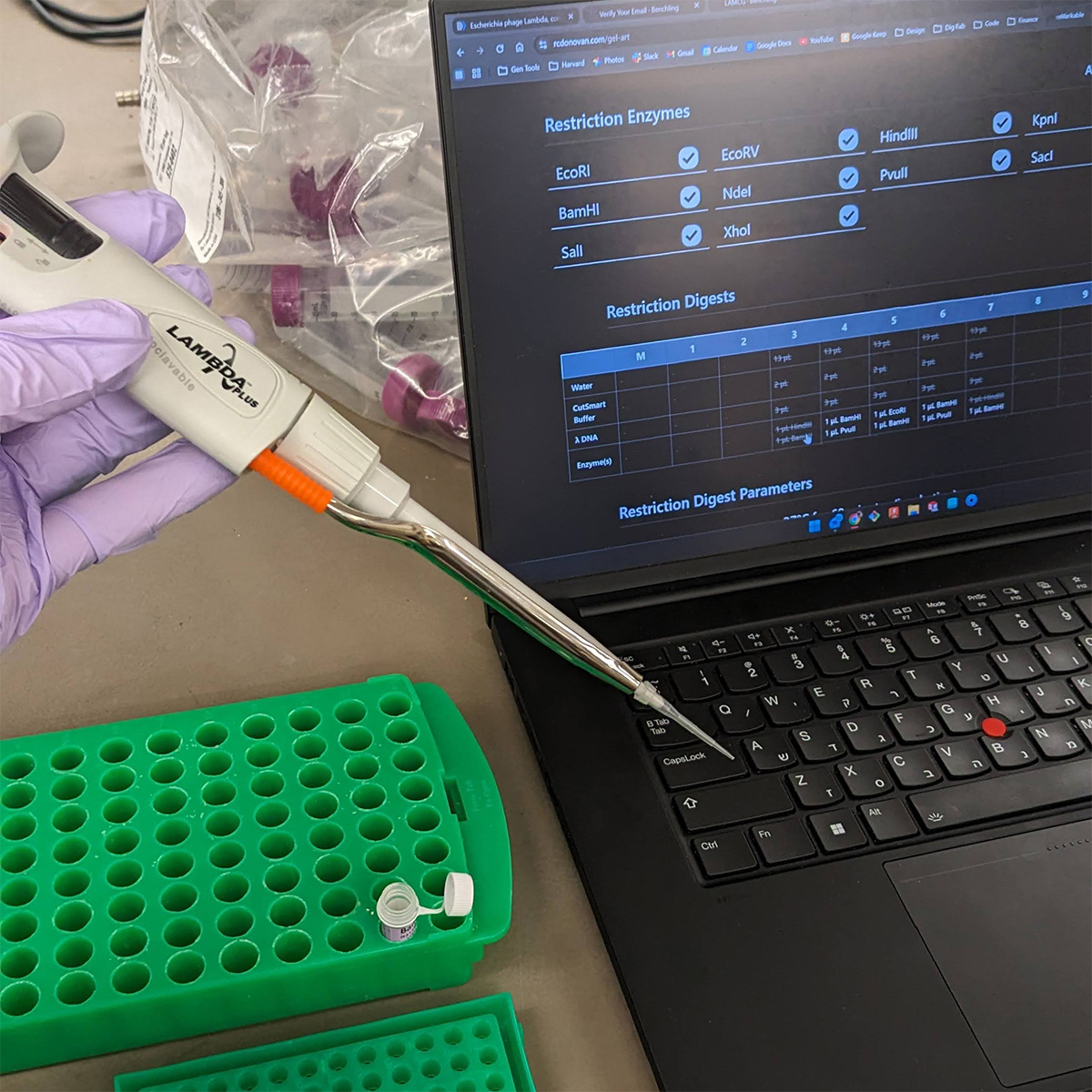
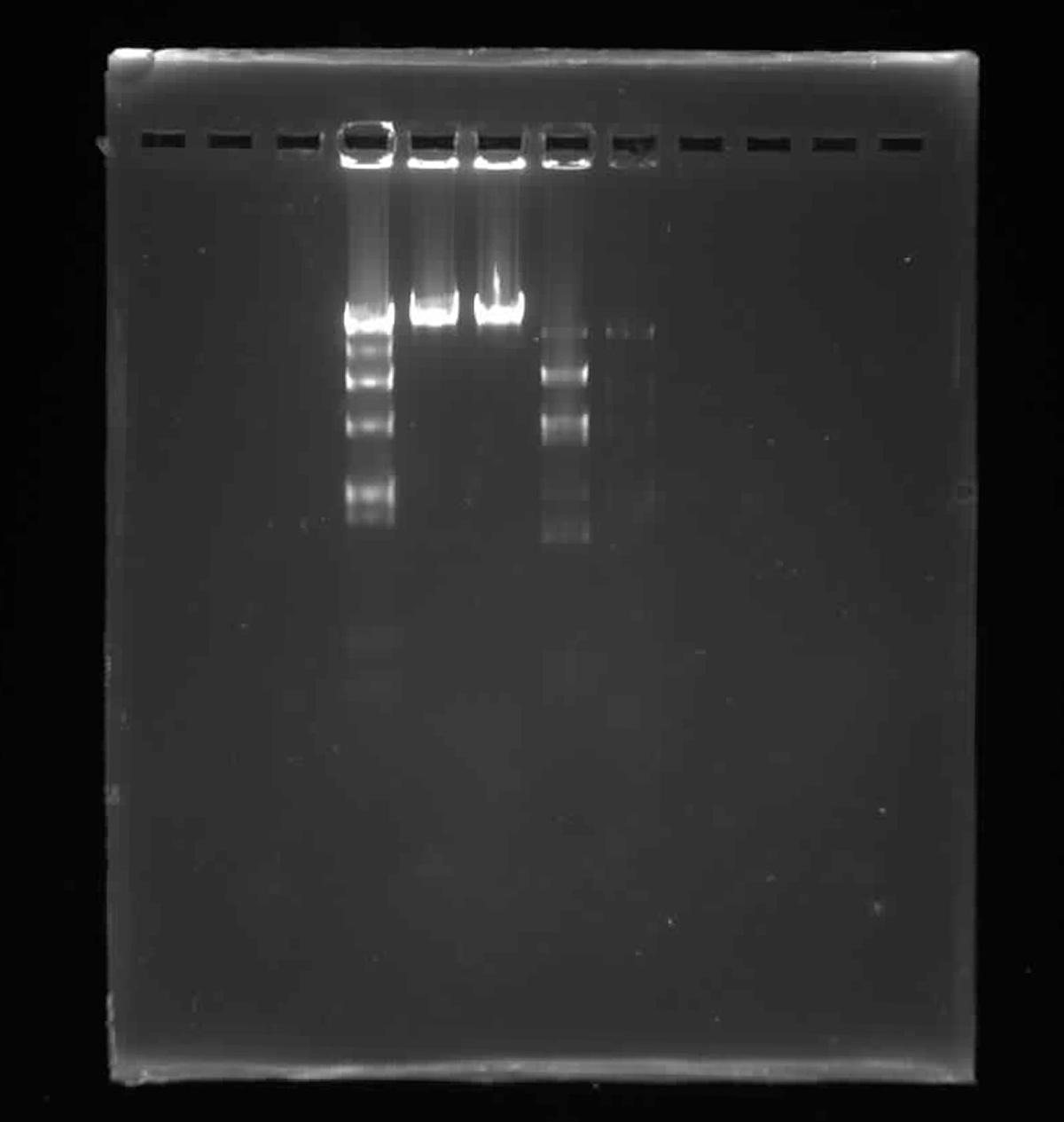
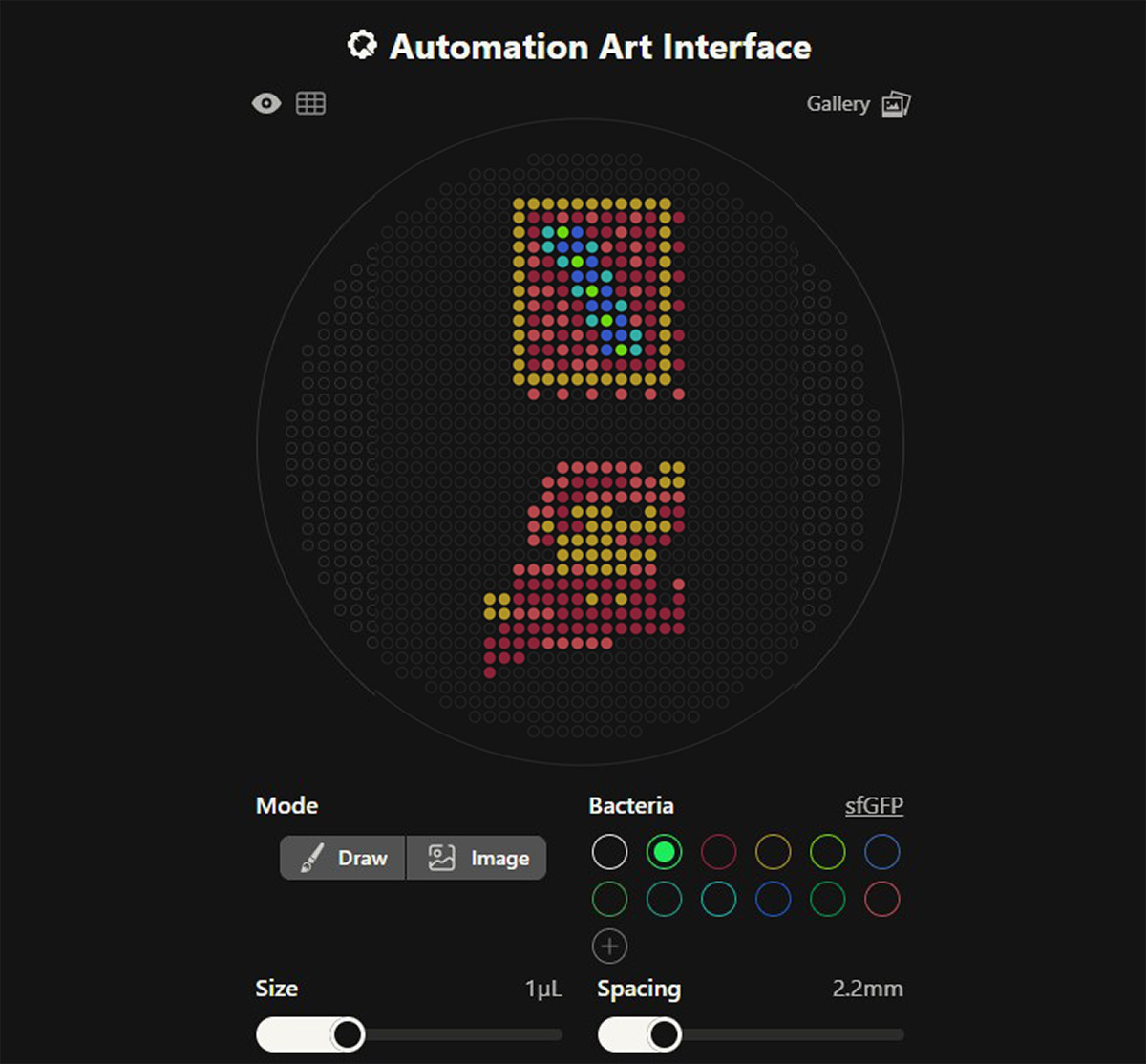
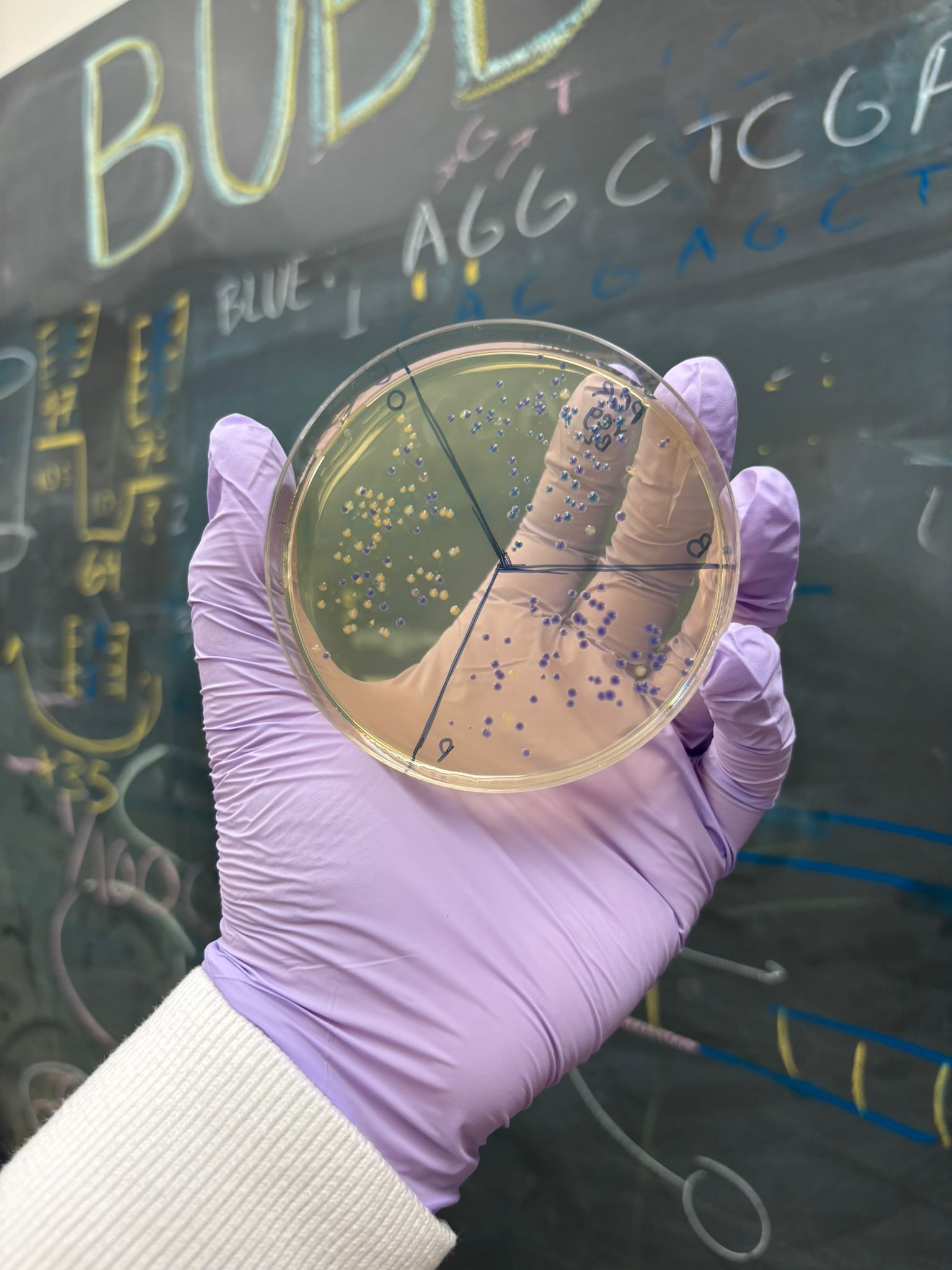
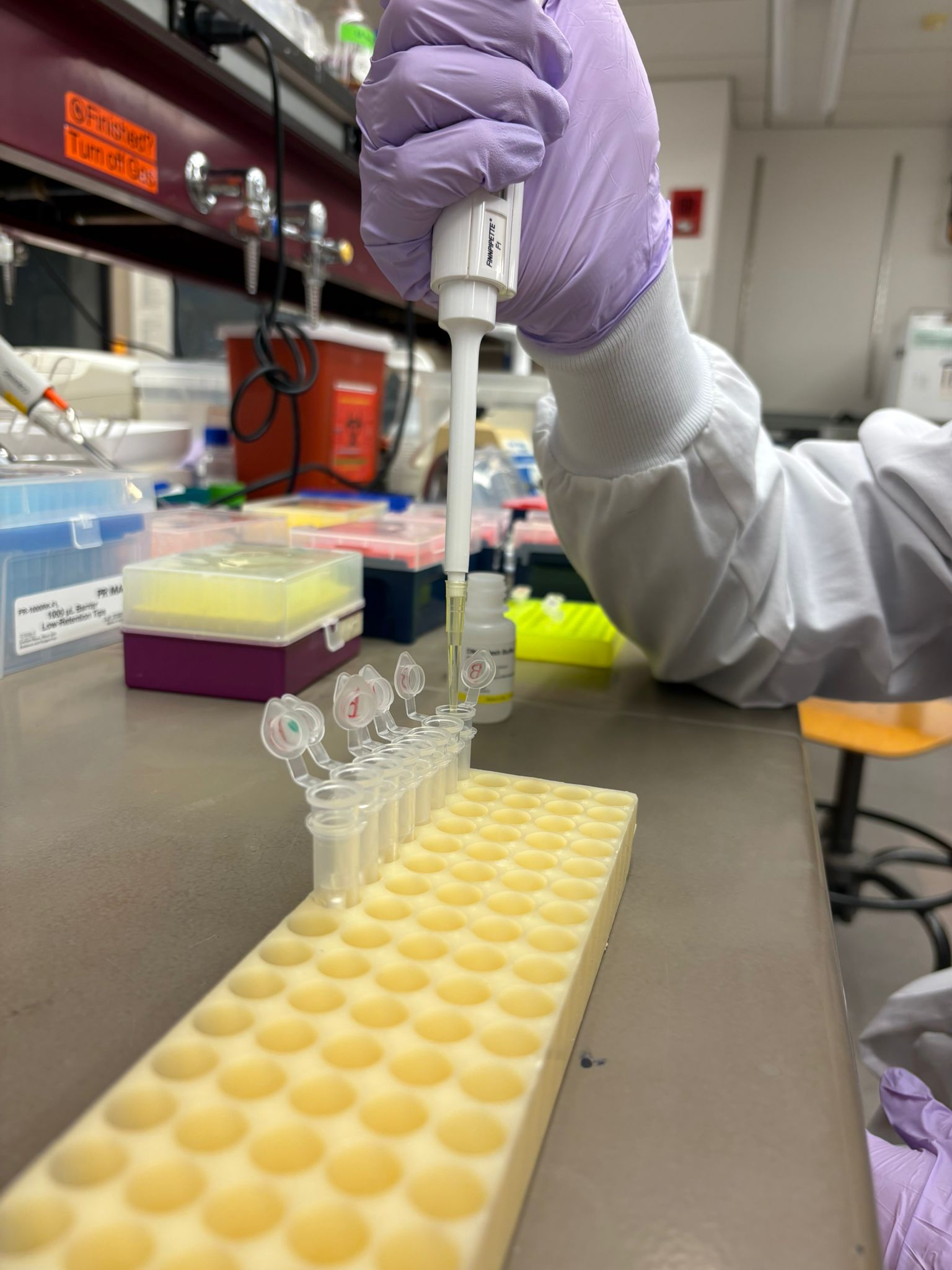
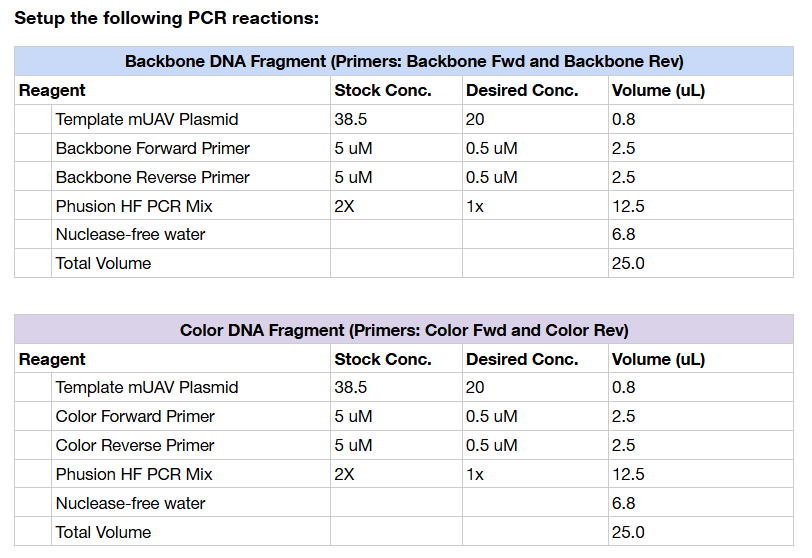
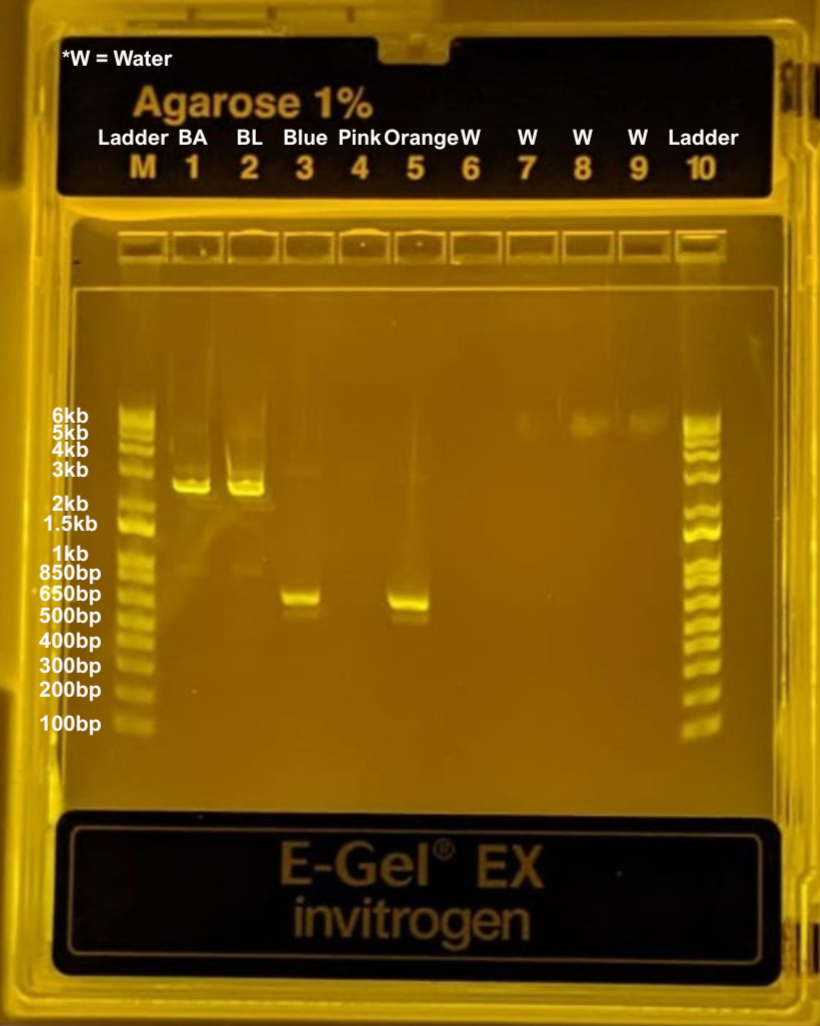
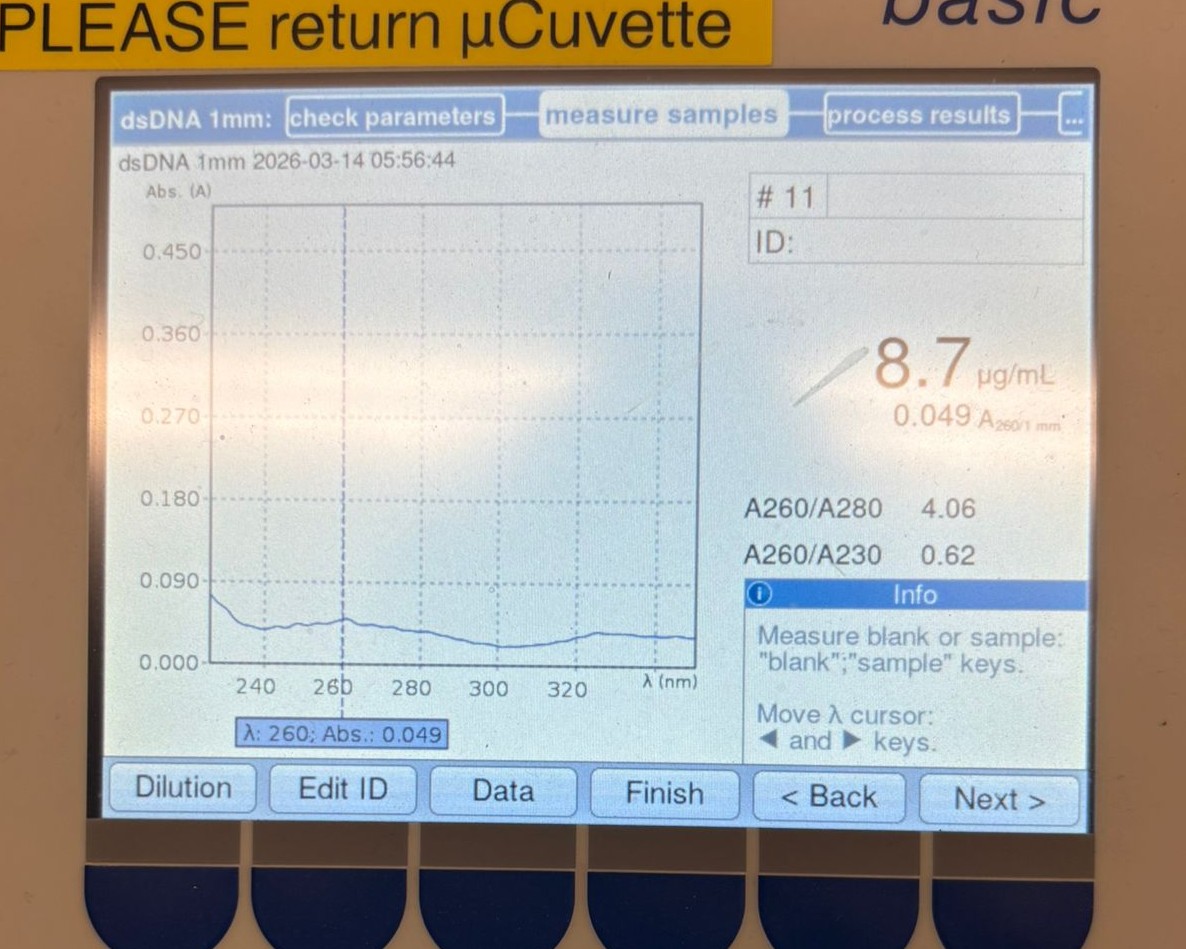
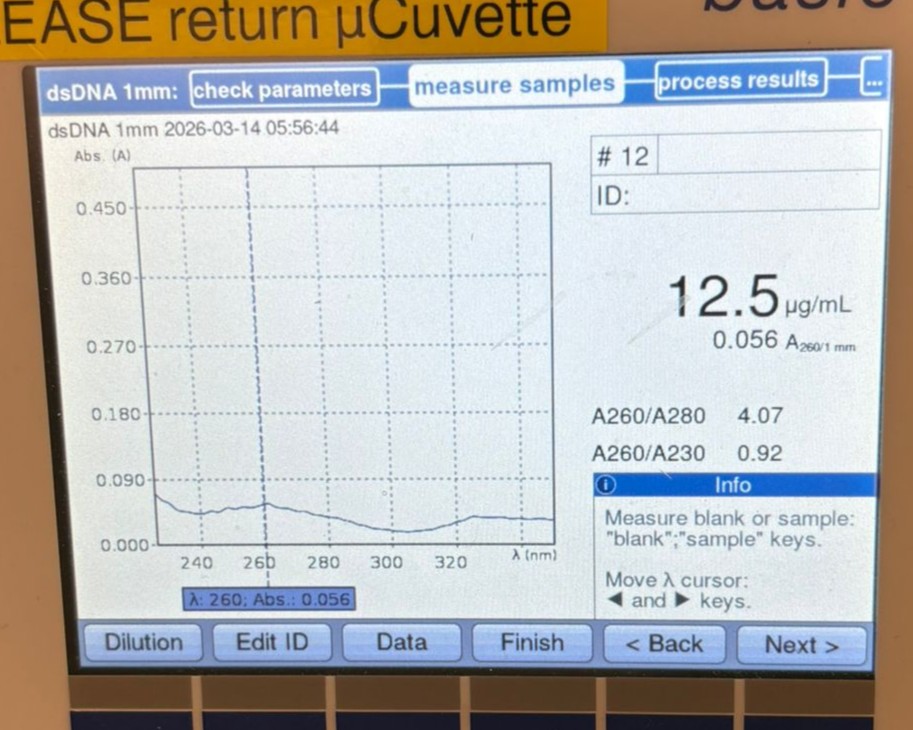
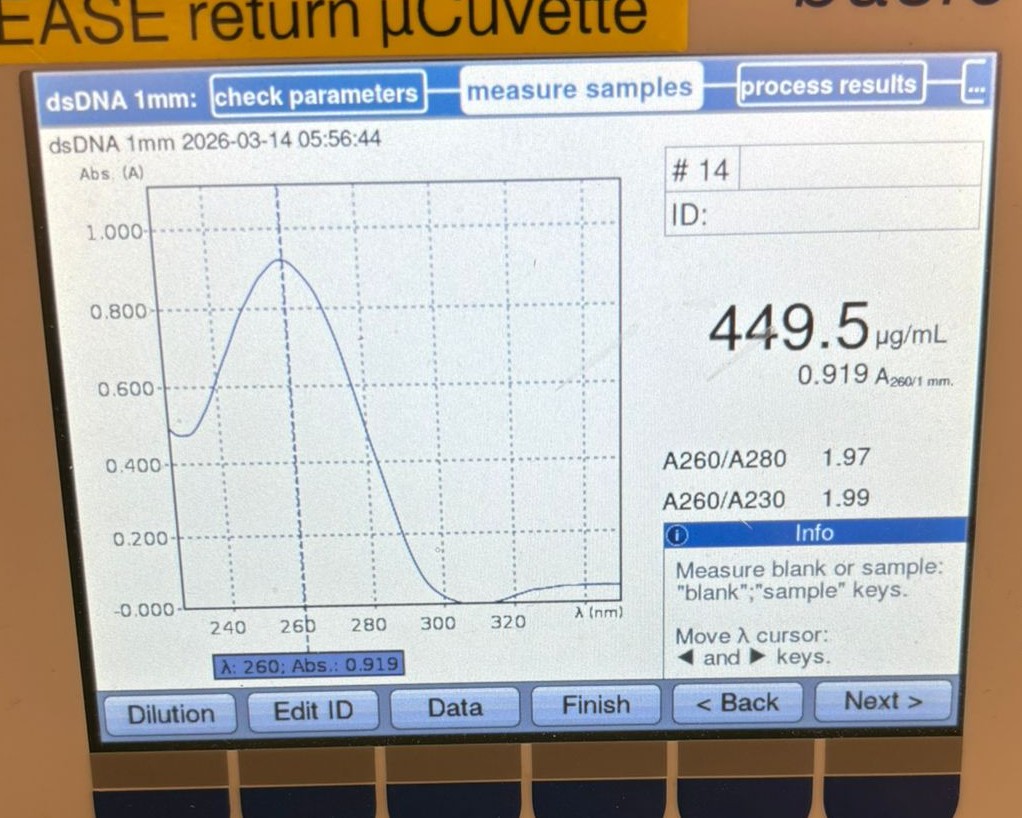
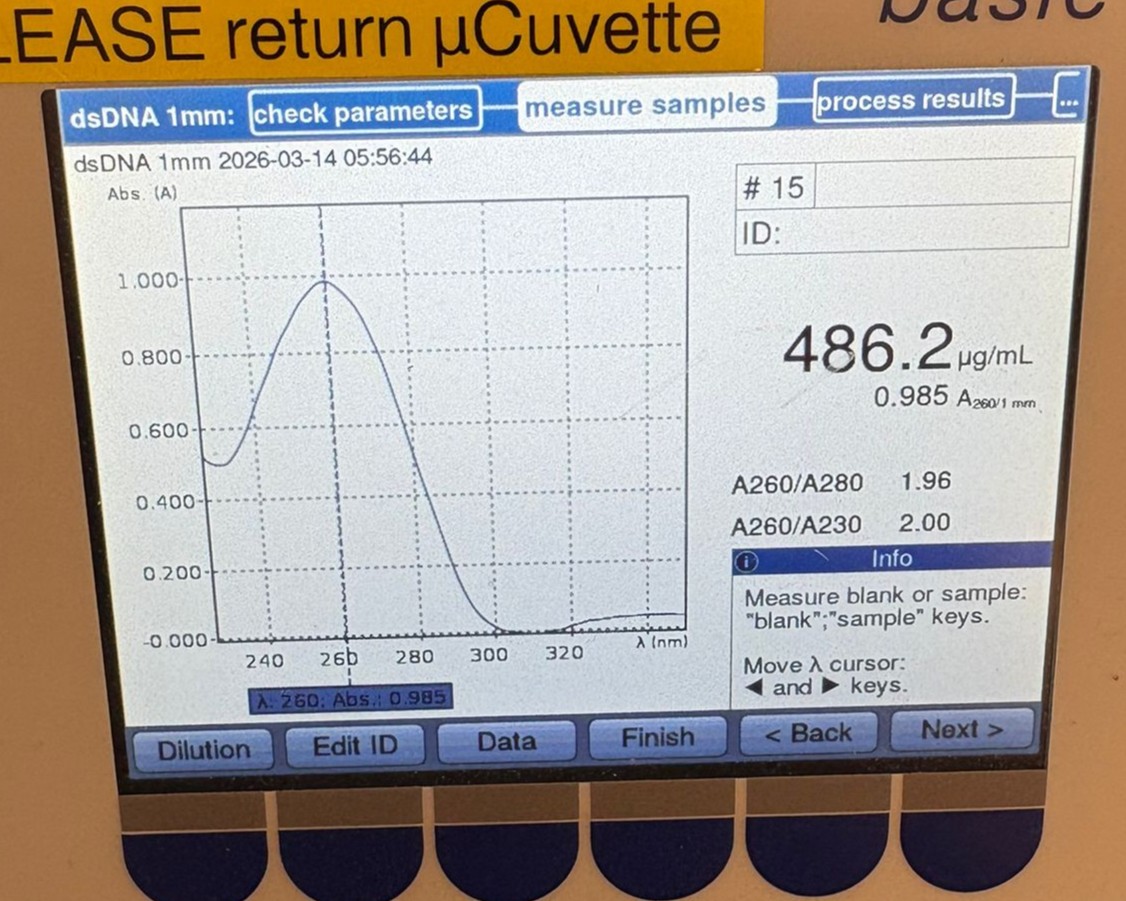
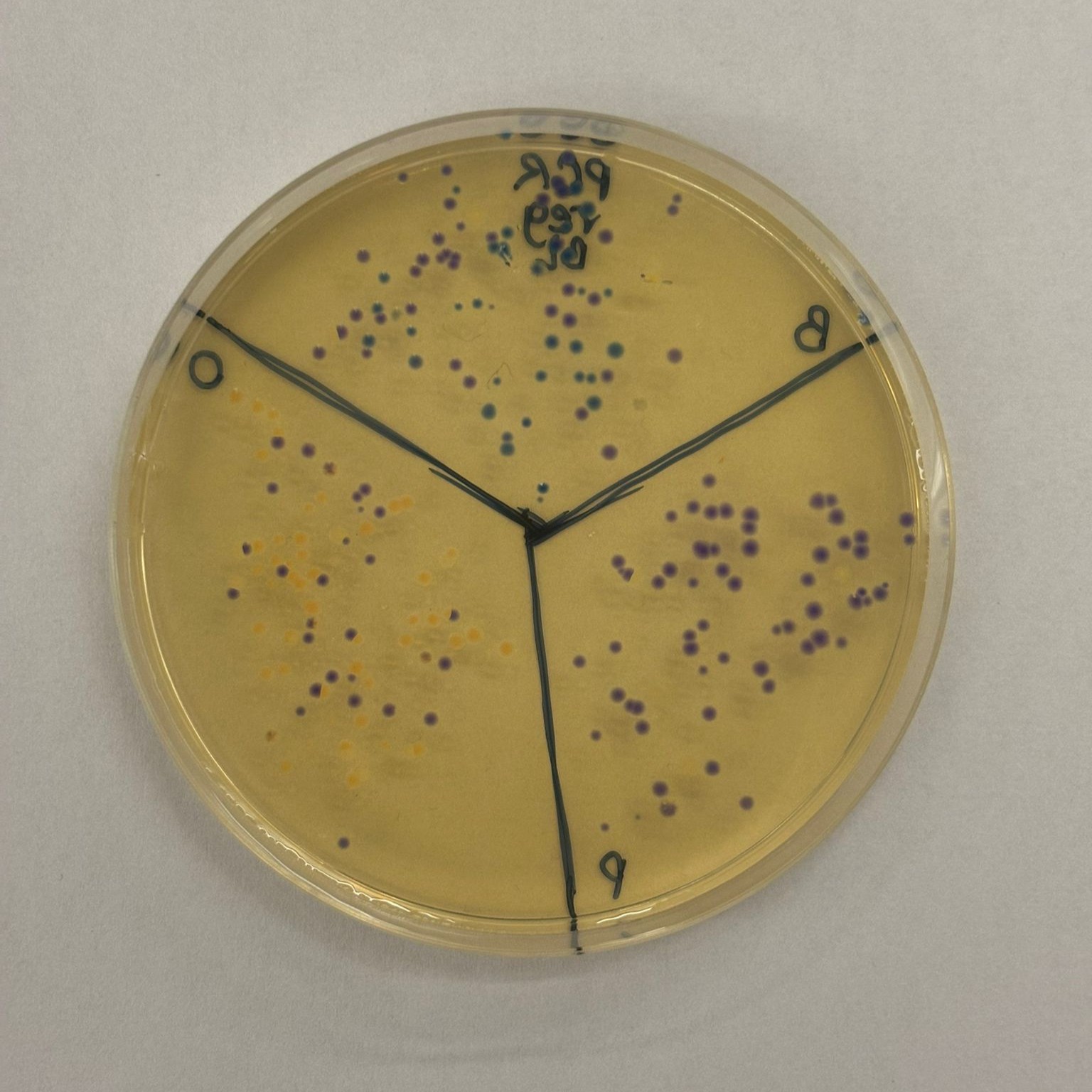
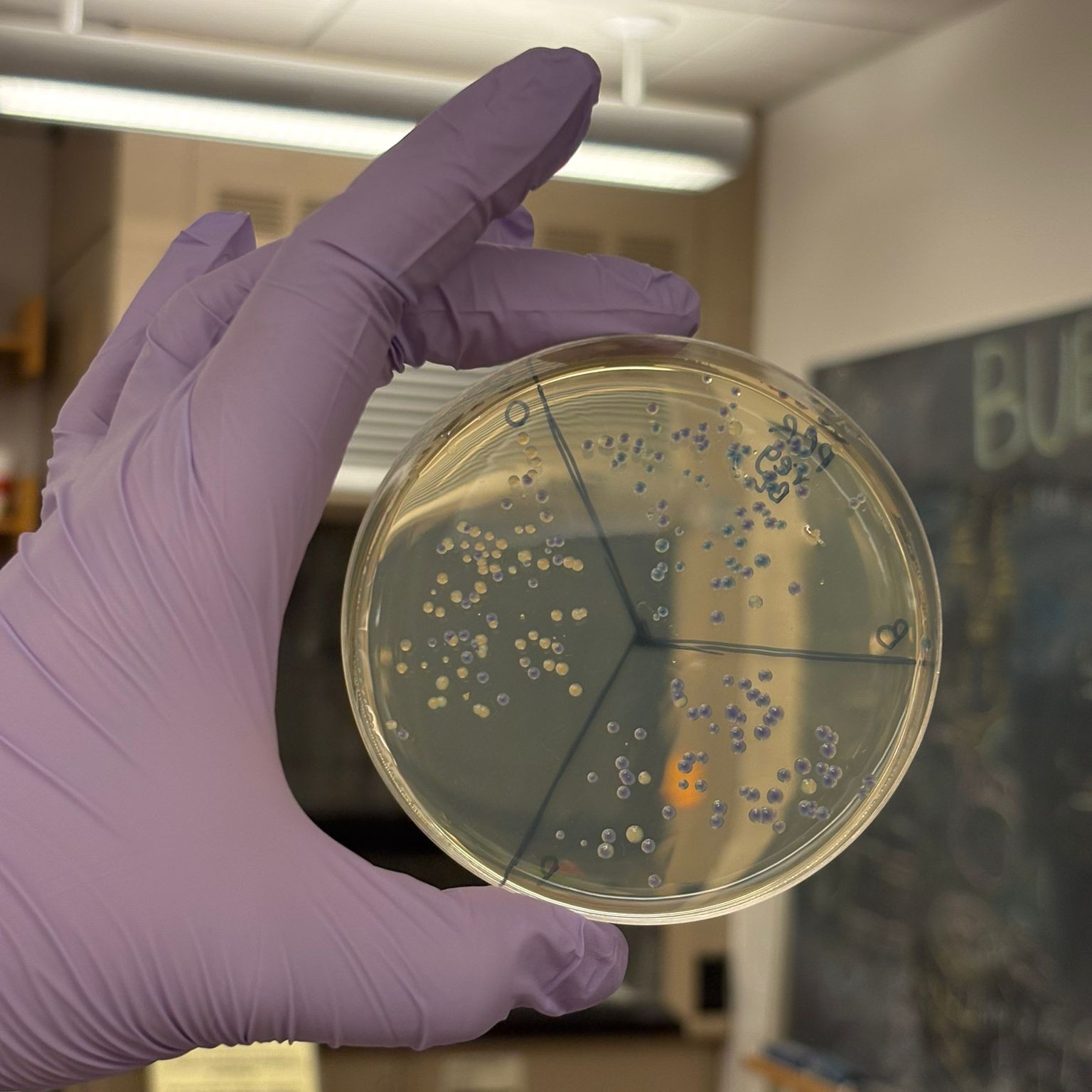
This week’s lab was about getting familiar with cuttind-edge lab automation tools.
We were introduced to the Opentron, which to me was a close relative to 3d printing hardware and other gantry based fabrication method.
It runs on a python script indicating coordinates for the working head to go-to, and has a pump and a motor where you would imagine the filament extruder motor and the heating element to be in a standard FDM 3d printer. Really enjoyed using this cool device.
Thanks to Ronan Donovan’s awesome online tool, we could generate python scripts very quickly using a simple UI that translated our graphics into the code itself, very similar to what a slicer software does for an STL file to a 3D printer or what a CAM engine does for a STEP file for CNC operations.
from opentrons import types
import string
metadata = {
'protocolName': '{YOUR NAME} - Opentrons Art - HTGAA',
'author': 'HTGAA',
'source': 'HTGAA 2026',
'apiLevel': '2.20'
}
Z_VALUE_AGAR = 2.0
POINT_SIZE = 1
mrfp1_points = [(-7,31), (-5,31), (-1,31), (3,31), (5,31), (9,31), (13,31), (-7,29), (1,29), (3,29), (7,29), (9,29), (5,27), (9,27), (13,27), (-5,25), (3,25), (5,25), (9,25), (-7,23), (-5,23), (-3,23), (5,23), (7,23), (9,23), (13,23), (-3,21), (5,21), (9,21), (-5,19), (-1,19), (7,19), (9,19), (13,19), (-7,17), (-1,17), (9,17), (-3,15), (1,15), (9,15), (13,15), (-7,13), (-5,13), (-1,13), (9,13), (-7,11), (-3,11), (3,11), (5,11), (7,11), (9,11), (13,11), (-1,-5), (1,-5), (3,-5), (5,-5), (-3,-7), (-1,-7), (-3,-9), (11,-9), (13,-9), (-3,-11), (-1,-11), (13,-11), (-5,-13), (7,-13), (9,-13), (11,-13), (-9,-19), (-7,-19), (-5,-19), (-3,-19), (-1,-19), (1,-19), (3,-19), (5,-19), (7,-19), (9,-19), (-9,-21), (-7,-21), (-5,-21), (-3,-21), (-1,-21), (3,-21), (7,-21), (9,-21), (13,-21), (-3,-23), (-1,-23), (1,-23), (3,-23), (5,-23), (7,-23), (9,-23), (11,-23), (13,-23), (-11,-25), (-9,-25), (-7,-25), (-5,-25), (-3,-25), (-1,-25), (1,-25), (3,-25), (5,-25), (7,-25), (9,-25), (11,-25), (13,-25), (-13,-27), (-11,-27), (-9,-27), (-7,-27), (-13,-29), (-11,-29), (-9,-29), (-13,-31)]
mko2_points = [(-9,33), (-7,33), (-5,33), (-3,33), (-1,33), (1,33), (3,33), (5,33), (7,33), (9,33), (11,33), (-9,31), (11,31), (-9,29), (11,29), (-9,27), (11,27), (-9,25), (11,25), (-9,23), (11,23), (-9,21), (11,21), (-9,19), (11,19), (-9,17), (11,17), (-9,15), (11,15), (-9,13), (11,13), (-9,11), (11,11), (-9,9), (-7,9), (-5,9), (-3,9), (-1,9), (1,9), (3,9), (5,9), (7,9), (9,9), (11,9), (11,-3), (13,-3), (11,-5), (13,-5), (-1,-9), (1,-9), (3,-9), (9,-9), (-5,-11), (1,-11), (3,-11), (5,-11), (7,-11), (9,-11), (11,-11), (-3,-13), (-1,-13), (1,-13), (3,-13), (-3,-15), (-1,-15), (1,-15), (3,-15), (5,-15), (7,-15), (9,-15), (-3,-17), (1,-17), (3,-17), (5,-17), (-13,-21), (-11,-21), (1,-21), (5,-21), (-13,-23), (-11,-23)]
mscarlet_i_points = [(-3,31), (1,31), (7,31), (5,29), (-7,27), (3,27), (7,27), (-7,25), (7,25), (-7,21), (-5,21), (7,21), (-7,19), (-3,19), (-5,17), (-3,17), (7,17), (-7,15), (-5,15), (-1,15), (-3,13), (1,13), (-5,11), (-1,11), (1,11), (-7,7), (-3,7), (1,7), (5,7), (9,7), (13,7), (-3,-3), (-1,-3), (1,-3), (3,-3), (5,-3), (7,-3), (-5,-5), (-3,-5), (7,-5), (9,-5), (-5,-7), (1,-7), (3,-7), (5,-7), (7,-7), (9,-7), (11,-7), (13,-7), (-7,-9), (-5,-9), (-7,-11), (-7,-13), (5,-13), (-9,-17), (-7,-17), (-5,-17), (-1,-17), (7,-17), (9,-17), (13,-19), (-9,-23), (-7,-23), (-5,-23), (-5,-27), (-3,-27), (-1,-27), (1,-27), (3,-27)]
electra2_points = [(-1,29), (-3,27), (-1,27), (1,25), (-1,23), (1,23), (3,21), (1,19), (3,19), (5,17), (3,15), (5,15), (3,13)]
mturquoise2_points = [(-5,29), (-5,27), (1,27), (-3,25), (3,23), (-1,21), (5,19), (1,17), (7,15), (7,13)]
venus_points = [(-3,29), (-1,25), (1,21), (3,17), (5,13)]
point_name_pairing = [("mrfp1", mrfp1_points),("mko2", mko2_points),("mscarlet_i", mscarlet_i_points),("electra2", electra2_points),("mturquoise2", mturquoise2_points),("venus", venus_points)]
# Robot deck setup constants
TIP_RACK_DECK_SLOT = 9
COLORS_DECK_SLOT = 6
AGAR_DECK_SLOT = 5
PIPETTE_STARTING_TIP_WELL = 'A1'
# Place the PCR tubes in this order
well_colors = {
'A1': 'sfGFP',
'A2': 'mRFP1',
'A3': 'mKO2',
'A4': 'Venus',
'A5': 'mKate2_TF',
'A6': 'Azurite',
'A7': 'mCerulean3',
'A8': 'mClover3',
'A9': 'mJuniper',
'A10': 'mTurquoise2',
'A11': 'mBanana',
'A12': 'mPlum',
'B1': 'Electra2',
'B2': 'mWasabi',
'B3': 'mScarlet_I',
'B4': 'mPapaya',
'B5': 'eqFP578',
'B6': 'tdTomato',
'B7': 'DsRed',
'B8': 'mKate2',
'B9': 'EGFP',
'B10': 'mRuby2',
'B11': 'TagBFP',
'B12': 'mChartreuse_TF',
'C1': 'mLychee_TF',
'C2': 'mTagBFP2',
'C3': 'mEGFP',
'C4': 'mNeonGreen',
'C5': 'mAzamiGreen',
'C6': 'mWatermelon',
'C7': 'avGFP',
'C8': 'mCitrine',
'C9': 'mVenus',
'C10': 'mCherry',
'C11': 'mHoneydew',
'C12': 'TagRFP',
'D1': 'mTFP1',
'D2': 'Ultramarine',
'D3': 'ZsGreen1',
'D4': 'mMiCy',
'D5': 'mStayGold2',
'D6': 'PA_GFP'
}
volume_used = {
'mrfp1': 0,
'mko2': 0,
'mscarlet_i': 0,
'electra2': 0,
'mturquoise2': 0,
'venus': 0
}
def update_volume_remaining(current_color, quantity_to_aspirate):
rows = string.ascii_uppercase
for well, color in list(well_colors.items()):
if color == current_color:
if (volume_used[current_color] + quantity_to_aspirate) > 250:
# Move to next well horizontally by advancing row letter, keeping column number
row = well[0]
col = well[1:]
# Find next row letter
next_row = rows[rows.index(row) + 1]
next_well = f"{next_row}{col}"
del well_colors[well]
well_colors[next_well] = current_color
volume_used[current_color] = quantity_to_aspirate
else:
volume_used[current_color] += quantity_to_aspirate
break
def run(protocol):
# Load labware, modules and pipettes
protocol.home()
# Tips
tips_20ul = protocol.load_labware('opentrons_96_tiprack_20ul', TIP_RACK_DECK_SLOT, 'Opentrons 20uL Tips')
# Pipettes
pipette_20ul = protocol.load_instrument("p20_single_gen2", "right", [tips_20ul])
# PCR Plate
temperature_plate = protocol.load_labware('opentrons_96_aluminumblock_generic_pcr_strip_200ul', 6)
# Agar Plate
agar_plate = protocol.load_labware('htgaa_agar_plate', AGAR_DECK_SLOT, 'Agar Plate')
agar_plate.set_offset(x=0.00, y=0.00, z=Z_VALUE_AGAR)
# Get the top-center of the plate, make sure the plate was calibrated before running this
center_location = agar_plate['A1'].top()
pipette_20ul.starting_tip = tips_20ul.well(PIPETTE_STARTING_TIP_WELL)
# Helper function (dispensing)
def dispense_and_jog(pipette, volume, location):
assert(isinstance(volume, (int, float)))
# Go above the location
above_location = location.move(types.Point(z=location.point.z + 2))
pipette.move_to(above_location)
# Go downwards and dispense
pipette.dispense(volume, location)
# Go upwards to avoid smearing
pipette.move_to(above_location)
# Helper function (color location)
def location_of_color(color_string):
for well,color in well_colors.items():
if color.lower() == color_string.lower():
return temperature_plate[well]
raise ValueError(f"No well found with color {color_string}")
# Print pattern by iterating over lists
for i, (current_color, point_list) in enumerate(point_name_pairing):
# Skip the rest of the loop if the list is empty
if not point_list:
continue
# Get the tip for this run, set the bacteria color, and the aspirate bacteria of choice
pipette_20ul.pick_up_tip()
max_aspirate = int(18 // POINT_SIZE) * POINT_SIZE
quantity_to_aspirate = min(len(point_list)*POINT_SIZE, max_aspirate)
update_volume_remaining(current_color, quantity_to_aspirate)
pipette_20ul.aspirate(quantity_to_aspirate, location_of_color(current_color))
# Iterate over the current points list and dispense them, refilling along the way
for i in range(len(point_list)):
x, y = point_list[i]
adjusted_location = center_location.move(types.Point(x, y))
dispense_and_jog(pipette_20ul, POINT_SIZE, adjusted_location)
if pipette_20ul.current_volume == 0 and len(point_list[i+1:]) > 0:
quantity_to_aspirate = min(len(point_list[i:])*POINT_SIZE, max_aspirate)
update_volume_remaining(current_color, quantity_to_aspirate)
pipette_20ul.aspirate(quantity_to_aspirate, location_of_color(current_color))
# Drop tip between each color
pipette_20ul.drop_tip()