Week 10 Lab: Mass Spectrometry at Waters
 In this week’s lab, we had the pleasure of visiting Waters Immerse Cambridge and learning about mass spectrometry and advanced imaging up close.
In this week’s lab, we had the pleasure of visiting Waters Immerse Cambridge and learning about mass spectrometry and advanced imaging up close.
The team for today:

During our visit, we explored several advanced mass spectrometry workflows used for modern protein characterization and biochemical analysis. We learned how LC-MS can be used to determine molecular weight, probe protein folding and structure, and even reconstruct amino acid sequences from peptide fragments. Throughout the lab, we had the opportunity to work closely with cutting-edge instrumentation and gain hands-on exposure to techniques commonly used in both research and industry.
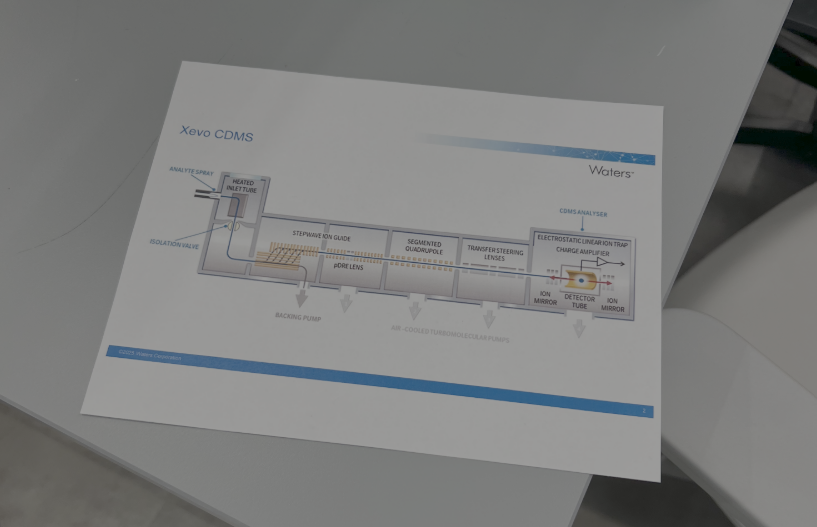
1. Mass Measurements of Megadalton-Sized Protein Complexes with Charge Detection Mass Spectrometry (CDMS)
Our first station focused on Charge Detection Mass Spectrometry (CDMS), a specialized technique designed for analyzing extremely large biomolecules that are difficult to study using conventional mass spectrometry methods. We investigated different oligomeric states of Keyhole Limpet Hemocyanin (KLH), a massive protein complex in the megadalton range. It was fascinating to see how CDMS can directly measure both the mass-to-charge ratio and the charge of individual ions, enabling accurate mass determination of huge biological assemblies such as protein complexes and viruses.
2. Protein Structure and Shape – Native versus Denatured Protein Measurement on the Xevo G3 QTof
In this station, we explored how protein folding influences mass spectrometry measurements. Using eGFP samples, we compared the spectra of proteins in their native folded state and their denatured, unfolded state. We learned that folded proteins generally exhibit fewer charge states because their compact structure limits protonation sites, while denatured proteins unfold and expose more sites for ionization. This gave us a very intuitive demonstration of how mass spectrometry can provide insight not only into molecular weight, but also into higher-order protein structure.

3. Primary Amino Acid Sequence – Peptide Mapping on the Waters BioAccord LC-MS
Our final station focused on peptide mapping and protein sequencing using the Waters BioAccord LC-MS system. We enzymatically digested eGFP with trypsin to generate smaller peptide fragments, which were then separated and analyzed by LC-MS. By fragmenting these peptides further inside the mass spectrometer, we could reconstruct parts of the protein’s amino acid sequence and better understand how peptide mapping is used for protein identification and characterization in modern biochemistry workflows.

Thank you so much for hosting us - it was an amazing experience to learn more about these advanced instruments and the incredible capabilities of modern mass spectrometry and biochemical analysis!