Homework
Weekly homework submissions:
Week 1 HW: Principles and Practices
Here you will see a proposal in which I attempt to overstep my bounds in the field of microbiology with my identity as an artist and designer. If any of my statements are incorrect, incomplete, or biased, I would like to point out that this is due to my inexperience in the field, and I would gladly accept your support in correcting them. Q1. Describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
Week 2 HW: DNA Read, Write, & Edit
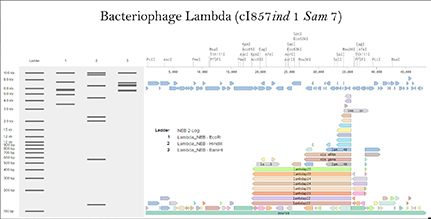
Part 1: Benchling & In-silico Gel Art First, I checked how to find Lambda through the database. I rewatched the Bootcamp recording by Adrian Filips and week 2 files of HTGAA2025 as well as the HTGAA2026 Recitation recordings on Benchling Basics provided by Cholpisit (Ice) Kiattisewee, and reviewed all the notes and presentations. NHI LAmbda webpage Biolabs Lambda webpage
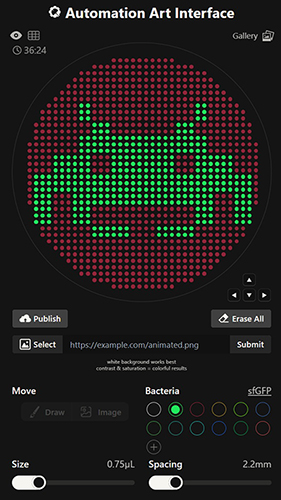
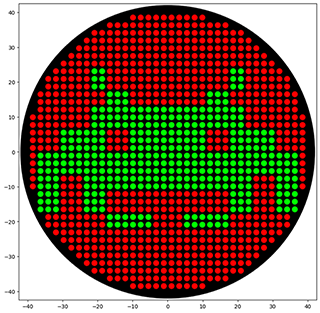
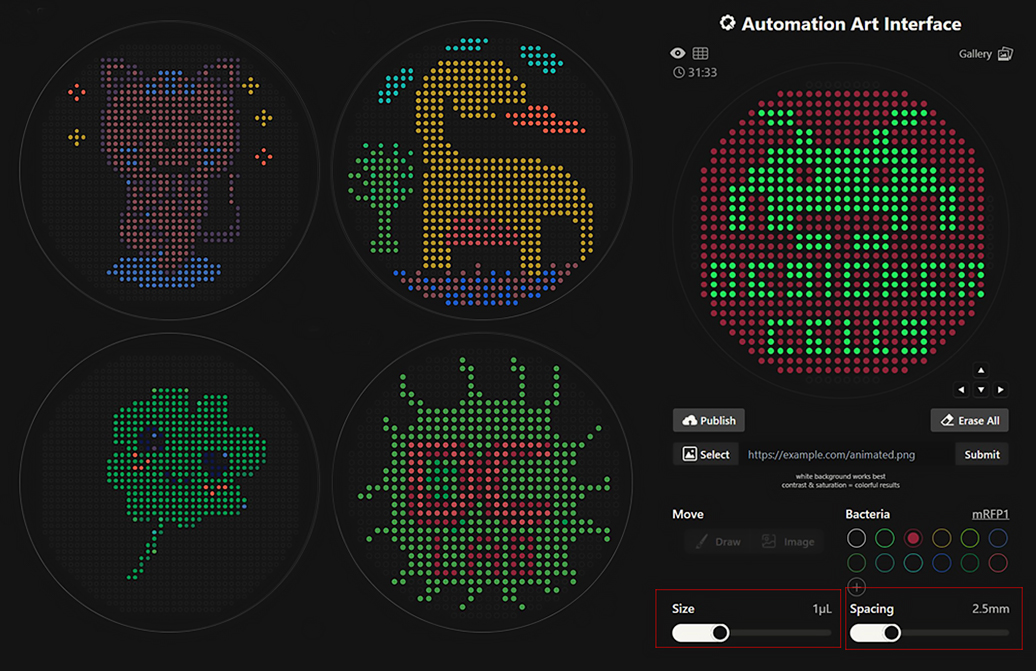
Python Script for Opentrons Artwork I have created Space Invaders with green and red because my node Designer Cells mentioned they have only red (mrfp1) and green (sfGFP) right now. I have evil plans to create also text “Designer Cells” down the Space Invaders logo:) - done!-
Part A: Conceptual Questions Answer any 9 of the following questions from Shuguang Zhang: (i.e. you can select two to skip) A.1 How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Approximately 6×10 23 amino acid molecules. 1 gram of amino acids would be 0.01 moles (1g/100g/mol). If 500g of meat is roughly 25% protein (about 125g), we’d have 1.25 moles. Multiplying by Avogadro’s number (6.022×10 23), we get approximately 7.5×10 23 molecules.
Part A: SOD1 Binder Peptide Design (From Pranam) Human Superoxide Dismutase 1 (SOD1, UniProt: P00441) is a cytosolic antioxidant enzyme responsible for detoxifying superoxide radicals. The A4V mutation (Alanine → Valine at position 4) destabilizes the N-terminal region, increases aggregation propensity, and is associated with a severe form of familial ALS. The goal of this assignment is to design short 12-mer peptides that bind preferentially to mutant SOD1 and evaluate their structural and therapeutic potential using PepMLM, AlphaFold3, and PeptiVerse.
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly Phusion High-Fidelity PCR Master Mix Components While the specific biochemical list of Phusion ingredients is not detailed in the excerpts, the sources confirm that PCR reactions are a core “DNA Skill” used to generate “linear fragments” or “gene fragments” for cloning. Typically, a high-fidelity master mix includes: DNA Polymerase: The enzyme responsible for synthesizing the new DNA strand; high-fidelity versions (like Phusion) have proofreading activity to minimize mutations. dNTPs (Deoxynucleotide Triphosphates): The chemical “LEGO bricks” (A, T, C, G) used to build the DNA chain. Buffer and Mg2+: Provides the optimal chemical environment and cofactors for the polymerase to function. Factors Determining Primer Annealing Temperature (Tm) The sources highlight Tmprediction as a critical computational filter in the protein design pipeline. The primary factors determining this temperature include: GC Content: The ratio of Guanine and Cytosine; higher GC content increases the Tmbecause G-C pairs have three hydrogen bonds compared to the two bonds in A-T pairs Primer Length: Longer primers generally have higher annealing temperatures. Salt Concentration: The concentration of ions in the PCR buffer affects the stability of the DNA duplex. PCR vs. Restriction Enzyme Digests The sources compare these as two methods for preparing DNA for assembly: Protocol: PCR uses primers and a polymerase to amplify a specific sequence into a linear fragment. Restriction digestion uses enzymes (like NdeI or XhoI) to cut a DNA backbone or insert at specific “cloning sites” to create sticky or blunt ends Preferable Use: PCR is preferred when you need to amplify a specific gene from a complex template or add “homology arms” for Gibson cloning. Restriction digestion (described as “Plan B” in the project) is often used for inserting fragments into standard backbones like pET-28a(+) but can add “1–2 weeks” to the timeline for additional cloning and screening steps Ensuring Appropriateness for Gibson CloningTo ensure DNA fragments are ready for Gibson Assembly, you must verify that the linear fragments (whether from PCR or digestion) have overlapping homology sequences at their ends. The sources recommend using Benchling for “in silico design” to check sticky-end orientation, digestion sites, and frame verification to ensure all parts will align correctly during the assembly reaction.
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
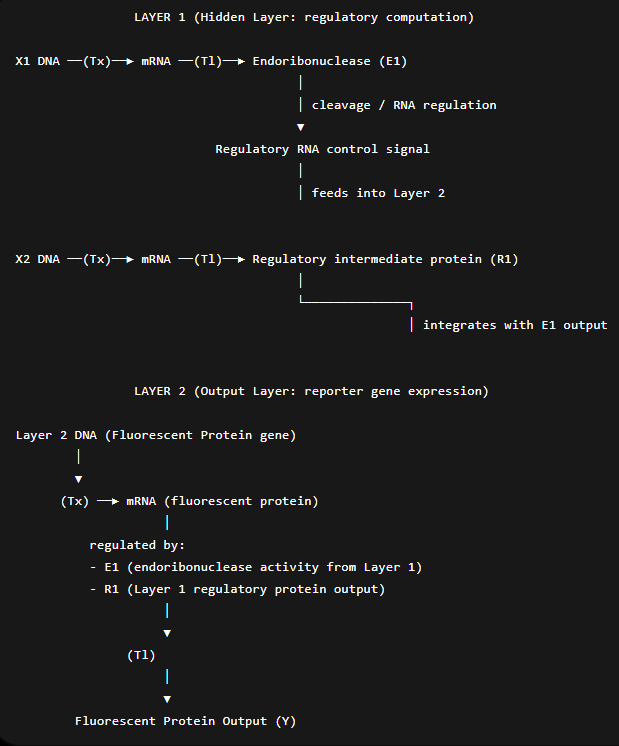
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) In-cell Artificial Neural Networks (IANNs) offer a major advantage over traditional Boolean genetic circuits by enabling synthetic systems to operate at the same level of abstraction as natural cellular signaling. Unlike Boolean circuits, which are restricted to discrete on/off logic, IANNs use continuous, analog signal processing with nonlinear activation functions, allowing them to represent smooth and highly complex input–output relationships such as band-pass or non-monotonic responses. This enables IANNs to approximate arbitrary continuous functions more efficiently and compactly than combinations of rigid logic gates. In addition, their continuous nature makes them better suited for modeling real biological systems, which are inherently noisy and graded rather than binary. As a result, IANNs are more robust for complex cellular decision-making tasks and pattern recognition in biological environments.
Homework Part A: General and Lecturer-Specific Questions General homework questions Cell-free protein synthesis offers much greater flexibility and experimental control than traditional in vivo systems because it removes the constraints of cell viability, membrane transport, and metabolic regulation. In CFPS, components can be precisely tuned (DNA template concentration, ions, chaperones, cofactors), and toxic or unstable proteins can be produced without affecting living cells. Two cases where CFPS is especially advantageous: Toxic proteins (e.g., membrane-disrupting peptides or nucleases) that would kill host cells in vivo. Rapid prototyping of genetic constructs, where many variants need to be tested quickly without cloning or cell line generation. A CFPS system typically contains: Cell extract (E. coli, wheat germ, or rabbit reticulocyte lysate): Provides ribosomes, tRNAs, aminoacyl-tRNA synthetases, and translation machinery. DNA or mRNA template: Encodes the target protein; serves as the blueprint for transcription/translation. Energy system (ATP regeneration components): Supplies ATP/GTP required for transcription and translation. Amino acids: Building blocks for protein synthesis. Salts and cofactors (Mg²⁺, K⁺, etc.): Stabilize ribosomes and enzymes. Nucleotides (NTPs): Required for transcription of mRNA from DNA. Optional additives (chaperones, membrane mimics, redox agents): Improve folding and functionality of expressed proteins. Importance of energy regeneration Protein synthesis is extremely energy-intensive; each peptide bond consumes multiple ATP/GTP equivalents. Without regeneration, ATP is rapidly depleted, stopping translation. To maintain continuous ATP supply, one method is:
Week 10 HW: Advanced Imaging & Measurement Technology
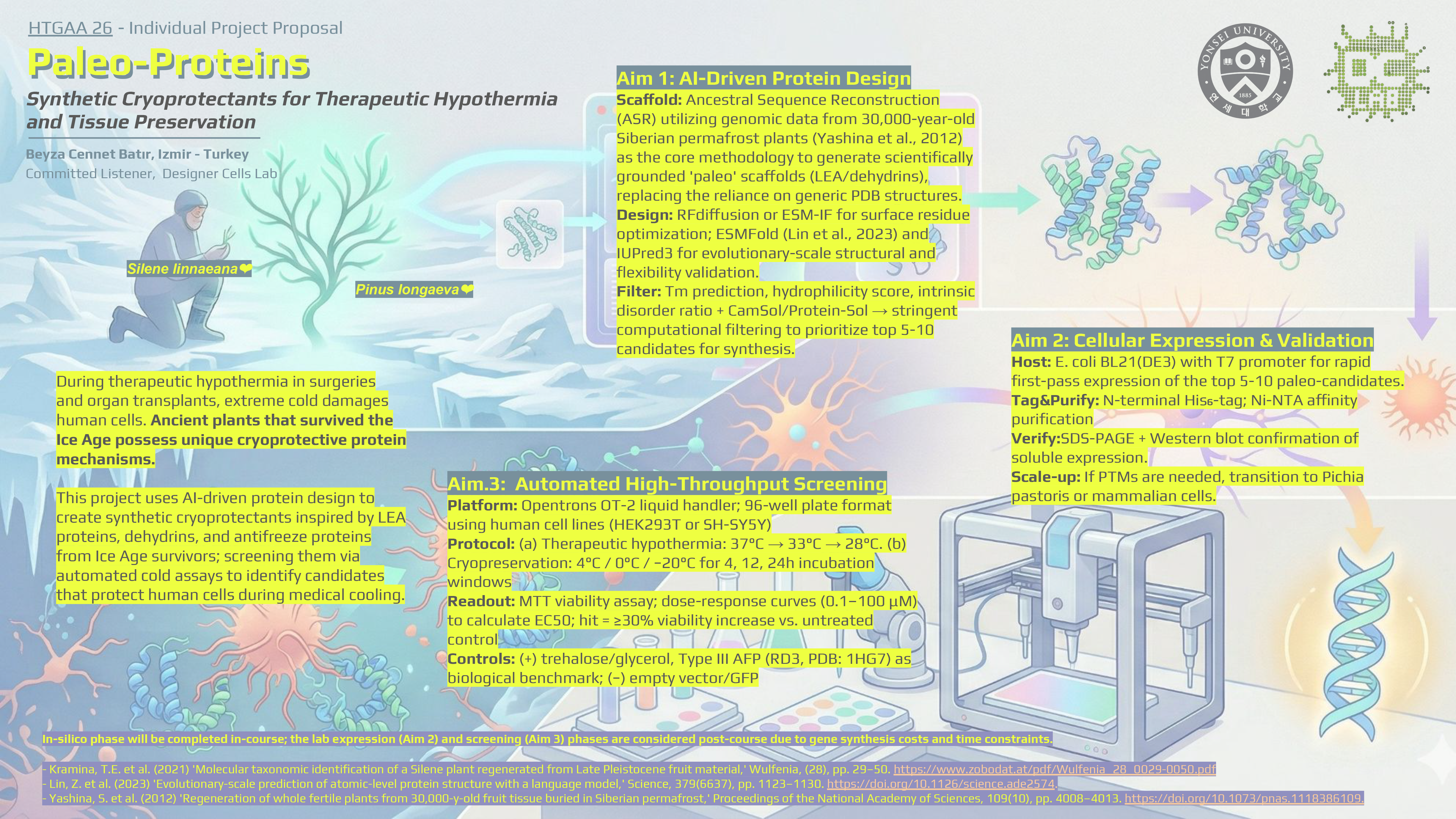
Homework: Final Project In my final project proposal, Paleo-Proteins project, success is measured through a multi-layered validation pipeline that spans from in silico sequence verification to automated functional assays in human cell lines. Below are the specific aspects being measured and the technologies employed to perform these measurements. Protein Identity and Structural Integrity The primary physical aspects to be measured are the molecular weight and immunological identity of the synthetic cryoprotectants (e.g., DHN-K2S). Measurement: I will confirm that the expressed protein matches the predicted molecular weight (e.g., ~11.4 kDa for DHN-K2S) and contains the intended N-terminal His₆-tag. Technologies: SDS-PAGE: A 12% precast gel will be used to provide gel-based confirmation of protein production and approximate size in less than 2 hours. Western Blot: Utilizing an anti-His₆-HRP antibody, this provides orthogonal identity confirmation, distinguishing the target protein from background cell-free synthesis (CFPS) components based on specific epitope recognition.
Week 11 HW: Bioproduction & Cloud Labs
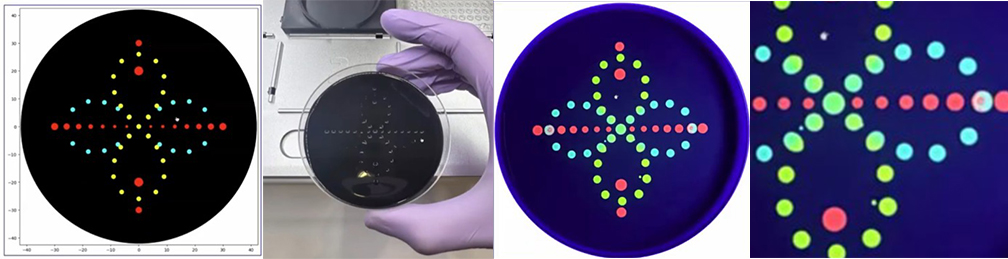
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Done :) global artwork experiment Make a note on your HTGAA webpages including: I was part of the community bioart project by supporting others design. I like collaborative artworks because of their inclusive atmosphere. I don’t have any recommendations. Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork 1. Roles of Each Cell-Free Reaction Component
Building Genomes (George Church, John Glass, Jef Boeke)
Lab: BioproductionWeek 13 HW: Biodesign & Engineered Living Material
Homework: Work on your Final Project Present it May 12 (MIT/Harvard) or May 13 (Committed Listeners) Done! :)
Week 14 HW: Bio Design & Bio Fabrication
Homework: Finish your Final Project Present it May 12 (MIT/Harvard) or May 13 (Committed Listeners) Done! :) https://docs.google.com/presentation/d/1vxVu8kgoHVHmmDpRqoX6xxGv62YjuYbUX5MRlUITn7I/edit?slide=id.g3e7d9f77350_242_2#slide=id.g3e7d9f77350_242_2



{kind=link}