Week 2 HW: DNA Read, Write, and Edit

Part 1: Benchling & In-silico Gel Art
1. IMPORTING LAMBDA DNA
1.1 Creating an Account and Accessing Benchling
Purpose: To access the platform for DNA simulation.
Open benchling.com
Sign up
Log in
1.2 Importing Sequence from NCBI
Purpose: To obtain the DNA template for analysis.
Steps:
Search for Lambda DNA in the National Center for Biotechnology Information (NCBI) database
Copy the complete genome sequence
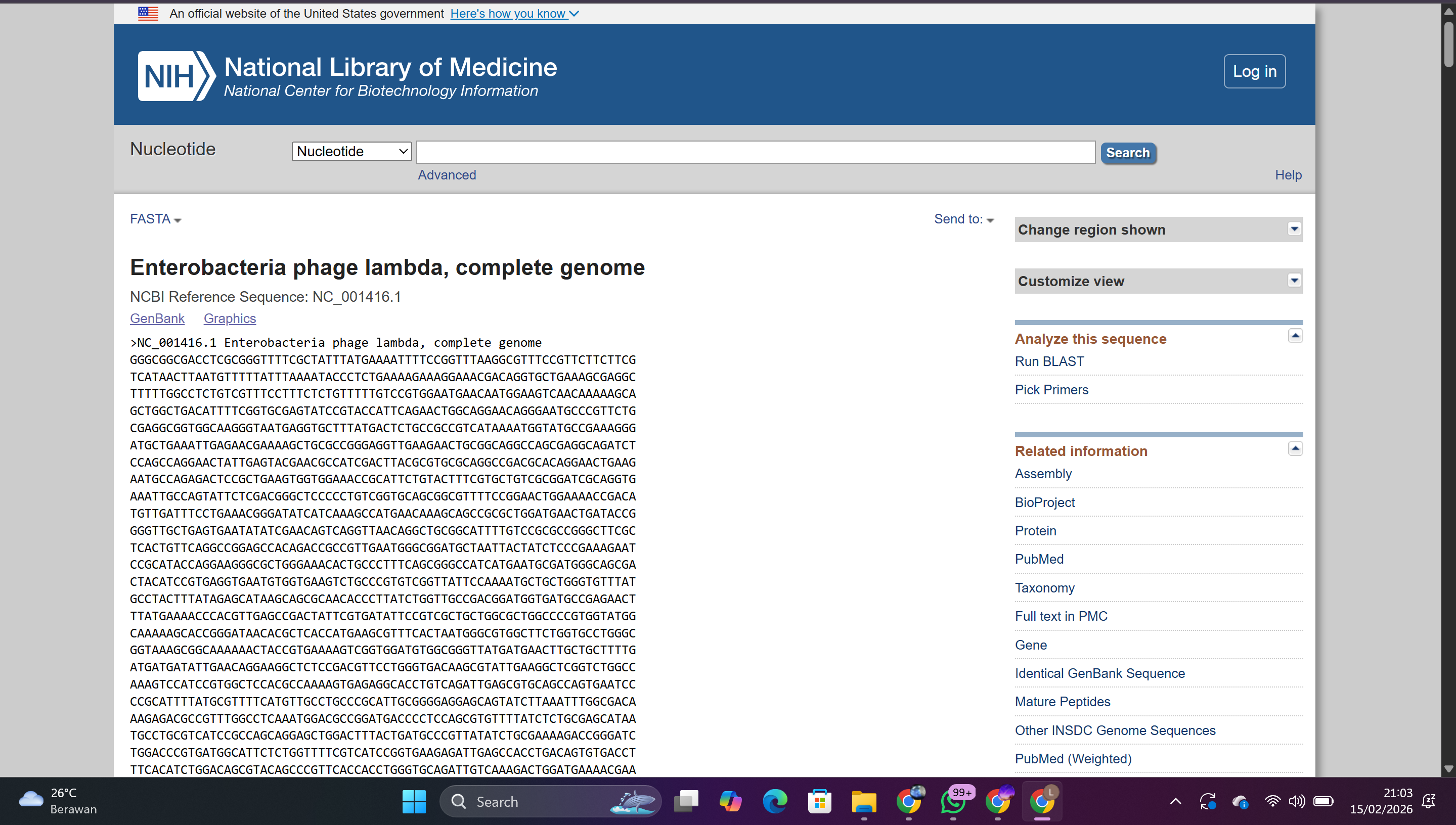
In Benchling:
Click Create → DNA Sequence
Select Import
Paste the sequence
Save as Lambda DNA
Result: The ~48.5 kb Lambda DNA sequence was successfully imported into the workspace.
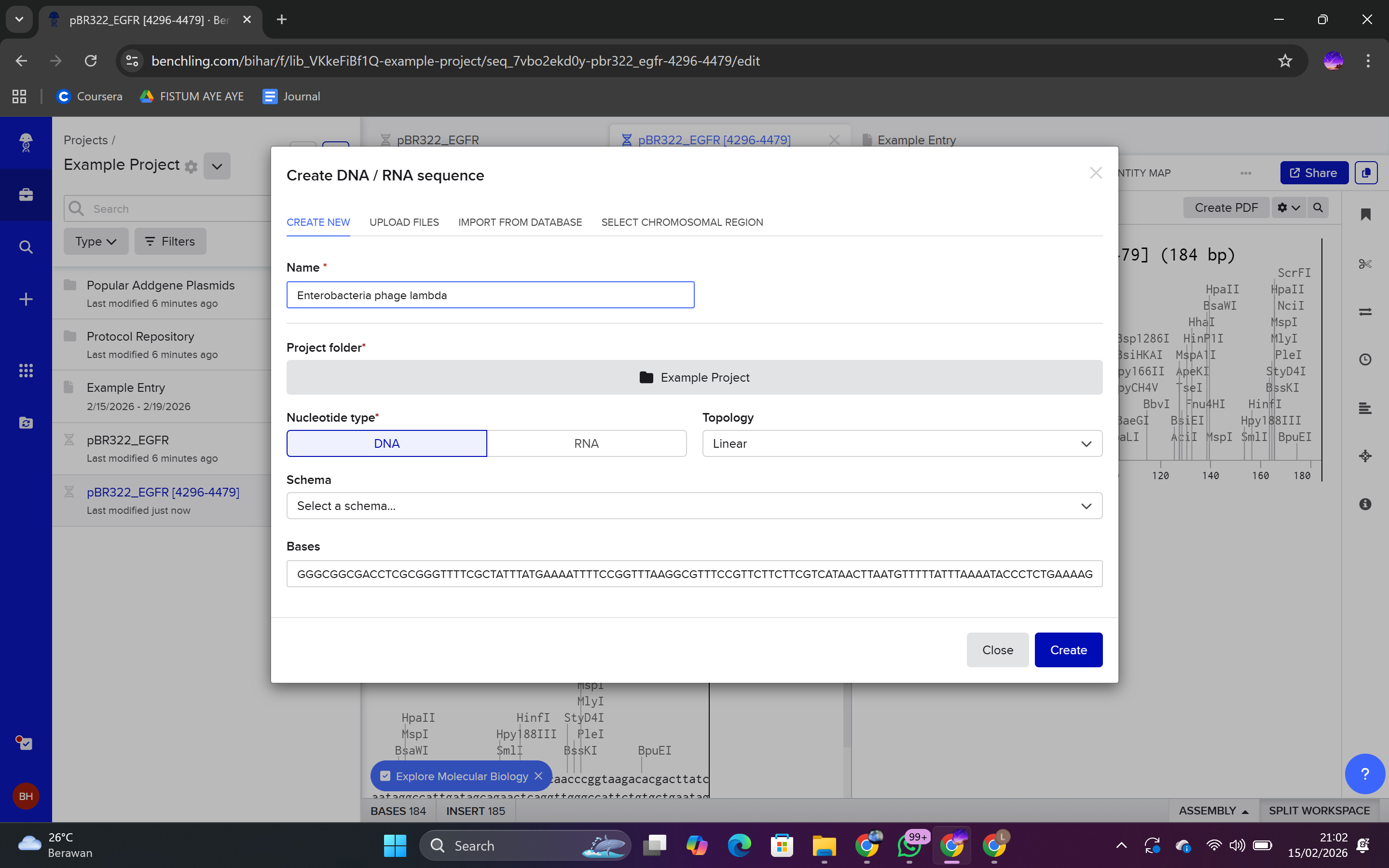
RESTRICTION DIGEST SIMULATION
2.1 Opening the Restriction Enzyme Tool
Purpose: To identify DNA cutting sites by specific enzymes.
Steps:
Open the Lambda DNA file
Click the scissors icon (Restriction Enzymes) on the right panel
Select the Digest tab
2.2 Adding Restriction Enzymes
Purpose: To generate DNA fragments with different cutting patterns.
Enzymes used:
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
Each enzyme was entered individually to observe the number and size of resulting fragments.
Result: DNA fragments of varying sizes were generated based on each enzyme’s recognition site.

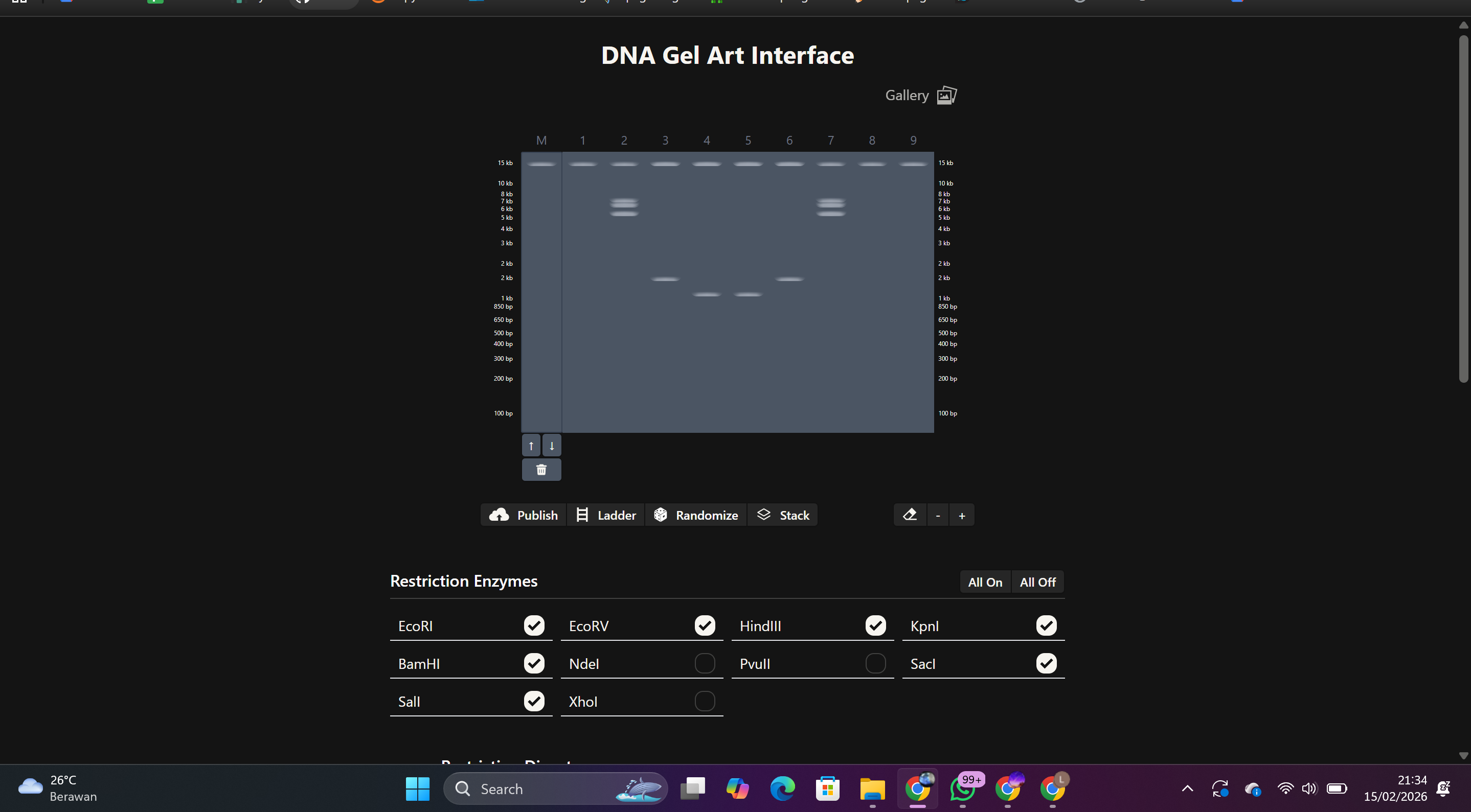
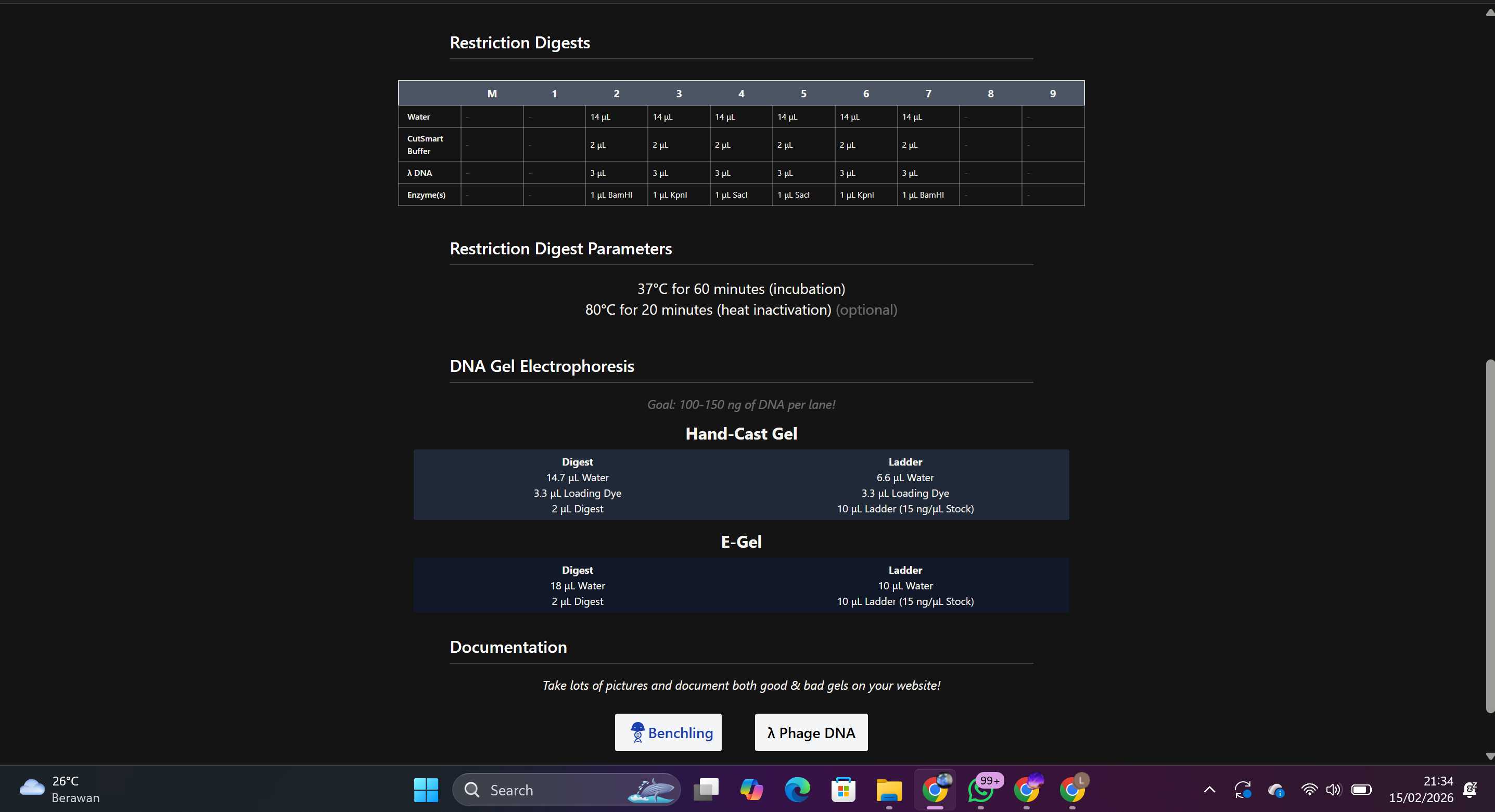
DESIGNING A PATTERN IN THE STYLE OF LATENT FIGURE PROTOCOL
Purpose: To transform gel patterns into a bio-art visual composition.
Concept:
Each lane represents a visual column
Band positions act as visual points
Enzyme combinations create variation and detail
Steps:
Select digests with the most contrasting band patterns
Arrange multiple lanes side by side
Adjust enzyme combinations to form a silhouette
Capture a screenshot of the final gel
Final Result: A gel pattern resembling a face or silhouette was created, inspired by Latent Figure Protocol, a bio-art approach that uses DNA visualization as a visual medium.
Part 3: DNA Design Challenge
3.1. Protein Selection
For this assignment, I selected the Hd3a (Heading date 3a) protein from Oryza sativa. Hd3a functions as a florigen protein, acting as a systemic flowering signal that induces the transition from vegetative to reproductive growth in rice. This protein is directly relevant to the “Rapid-Cycle Rice” concept, as modulating Hd3a expression can accelerate flowering and shorten the crop life cycle. Therefore, Hd3a represents a biologically rational target for bioengineering strategies aimed at improving food security through reduced cultivation time.
The amino acid sequence used in this assignment is:
MAGSGRDRDPLVVGRVVGDVLDAFVRSTNLKVTYGSKTVSNGCELKPSMVTHQPRVEVGGNDMRTFYTLVMVDPDAPSPSDPNLREYLHWLVTDIPGTTAASFGQEVMCYESPRPTMGIHRLVFVLFQQLGRQTVYAPGWRQNFNTKDFAELYNLGSPVAAAYFNCQREAGSGGRRVYP
3.2. Reverse Translation (Protein → DNA)
Based on the Central Dogma of molecular biology, a protein sequence can be used to infer a possible DNA sequence through reverse translation. Because the genetic code is degenerate (multiple codons can encode the same amino acid), a single protein sequence can correspond to many possible DNA sequences.
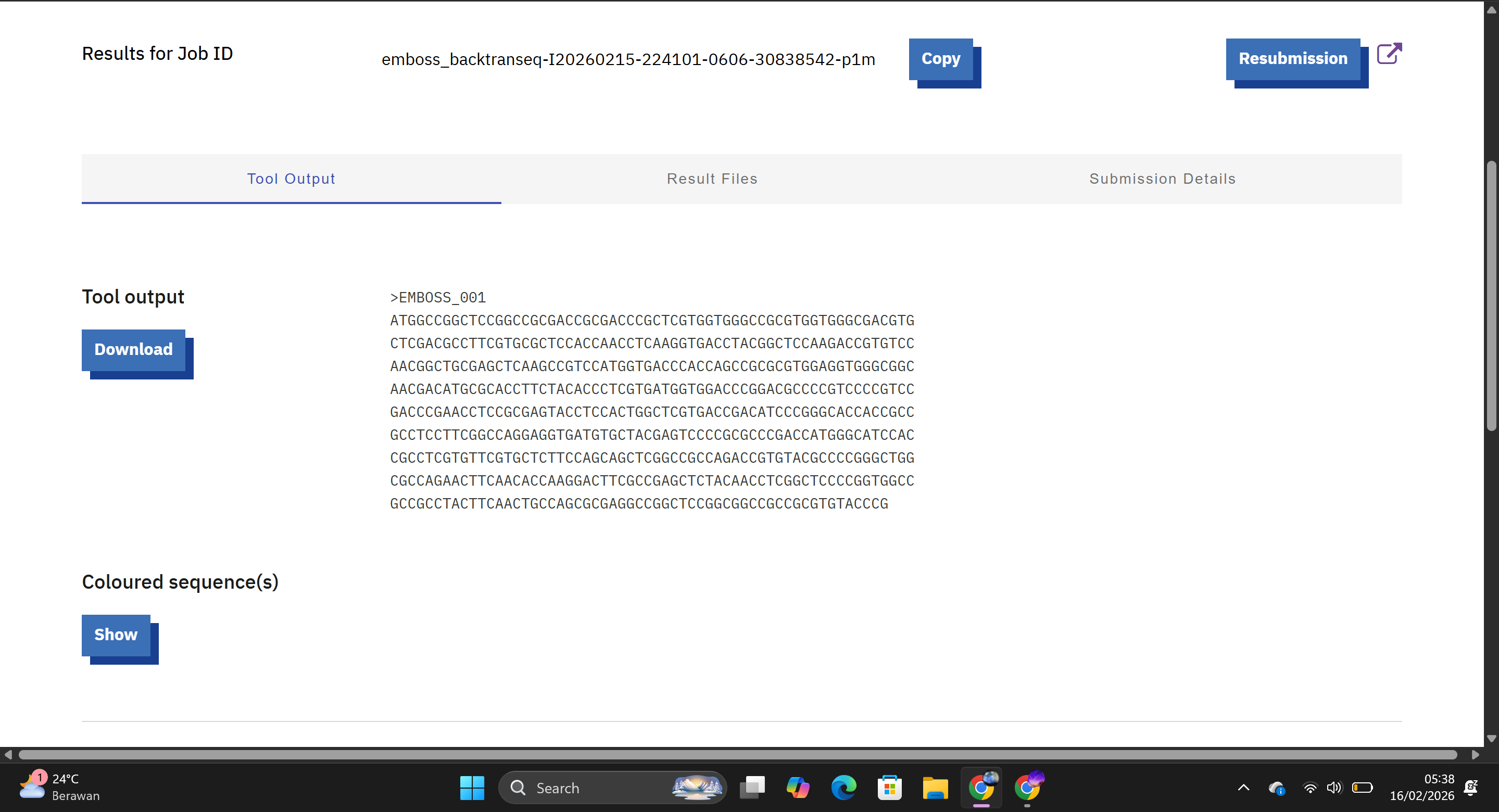
To perform reverse translation, I used the EMBOSS Backtranseq tool available through the European Bioinformatics Institute (EBI) at the following link: https://www.ebi.ac.uk/jdispatcher/st/emboss_backtranseq
This tool generated a nucleotide sequence corresponding to the Hd3a protein sequence using the standard genetic code. The resulting DNA sequence theoretically encodes the Hd3a protein.
Hd3a DNA sequence:
ATGGCCGGCTCCGGCCGCGACCGCGACCCGCTCGTGGTGGGCCGCGTGGTGGGCGACGTG CTCGACGCCTTCGTGCGCTCCACCAACCTCAAGGTGACCTACGGCTCCAAGACCGTGTCC AACGGCTGCGAGCTCAAGCCGTCCATGGTGACCCACCAGCCGCGCGTGGAGGTGGGCGGC AACGACATGCGCACCTTCTACACCCTCGTGATGGTGGACCCGGACGCCCCGTCCCCGTCC GACCCGAACCTCCGCGAGTACCTCCACTGGCTCGTGACCGACATCCCGGGCACCACCGCC GCCTCCTTCGGCCAGGAGGTGATGTGCTACGAGTCCCCGCGCCCGACCATGGGCATCCAC CGCCTCGTGTTCGTGCTCTTCCAGCAGCTCGGCCGCCAGACCGTGTACGCCCCGGGCTGG CGCCAGAACTTCAACACCAAGGACTTCGCCGAGCTCTACAACCTCGGCTCCCCGGTGGCC GCCGCCTACTTCAACTGCCAGCGCGAGGCCGGCTCCGGCGGCCGCCGCGTGTACCCG
3.3. Codon optimization
Although reverse translation produces a valid DNA sequence, the resulting sequence may not be optimal for expression in a chosen host organism. Different organisms exhibit codon bias, meaning they preferentially use certain codons over others for the same amino acid.
The reverse-translated DNA sequence was further optimized using the IDT Codon Optimization Tool for expression in Oryza sativa. The tool adjusts codon usage according to host codon bias, removes rare codons, and minimizes problematic motifs such as repeat regions or restriction sites.
Codon optimization is important because it:
Enhances translational efficiency
Reduces the presence of rare codons
Minimizes unfavorable mRNA secondary structures
Eliminates unwanted restriction enzyme recognition sites
An optimized DNA sequence improves protein yield, stability, and overall expression efficiency.
3.4. Protein Production from DNA
Once the optimized DNA sequence is obtained, the gene can be chemically synthesized and cloned into an expression vector. Protein production follows the Central Dogma:
DNA is transcribed into mRNA by RNA polymerase.
mRNA is translated by ribosomes into protein, where each three-nucleotide codon specifies one amino acid.
Protein production can be achieved using Cell-dependent expression systems
The gene is inserted into an expression plasmid and transformed into a host cell such as Escherichia coli or plant cells. A promoter within the plasmid initiates transcription, and the host’s ribosomes translate the mRNA into the Hd3a protein.
3.5. [Optional] How does it work in nature/biological systems?
- Describe how a single gene codes for multiple proteins at the transcriptional level.
A single gene can produce multiple proteins at the transcriptional level primarily through alternative splicing. In eukaryotic organisms such as Oryza sativa, genes are composed of exons (coding regions) and introns (non-coding regions). After transcription, the pre-mRNA can be spliced in different ways, meaning certain exons may be included or skipped. This generates multiple mature mRNA isoforms from the same DNA sequence, each of which can be translated into distinct protein variants with different structures or functions. Additional mechanisms include alternative promoter usage and alternative polyadenylation, which also produce transcript diversity.
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Preparation of Hd3a Coding Sequence
The first step was preparing the coding sequence (CDS) of the Oryza sativa Hd3a gene.
The amino acid sequence of Hd3a was reverse translated into a DNA sequence using a sequence translation tool. The resulting nucleotide sequence was assumed to be codon-optimized for Escherichia coli expression. This optimized DNA sequence (537 bp) serves as the Coding Sequence (CDS) component in the expression cassette.
4.2. Build the DNA Insert Sequence (Expression Cassette) in Benchling
To construct the expression cassette:
Log in to Benchling.
Select New DNA/RNA Sequence.
Choose:
Molecule Type: DNA
Topology: Linear
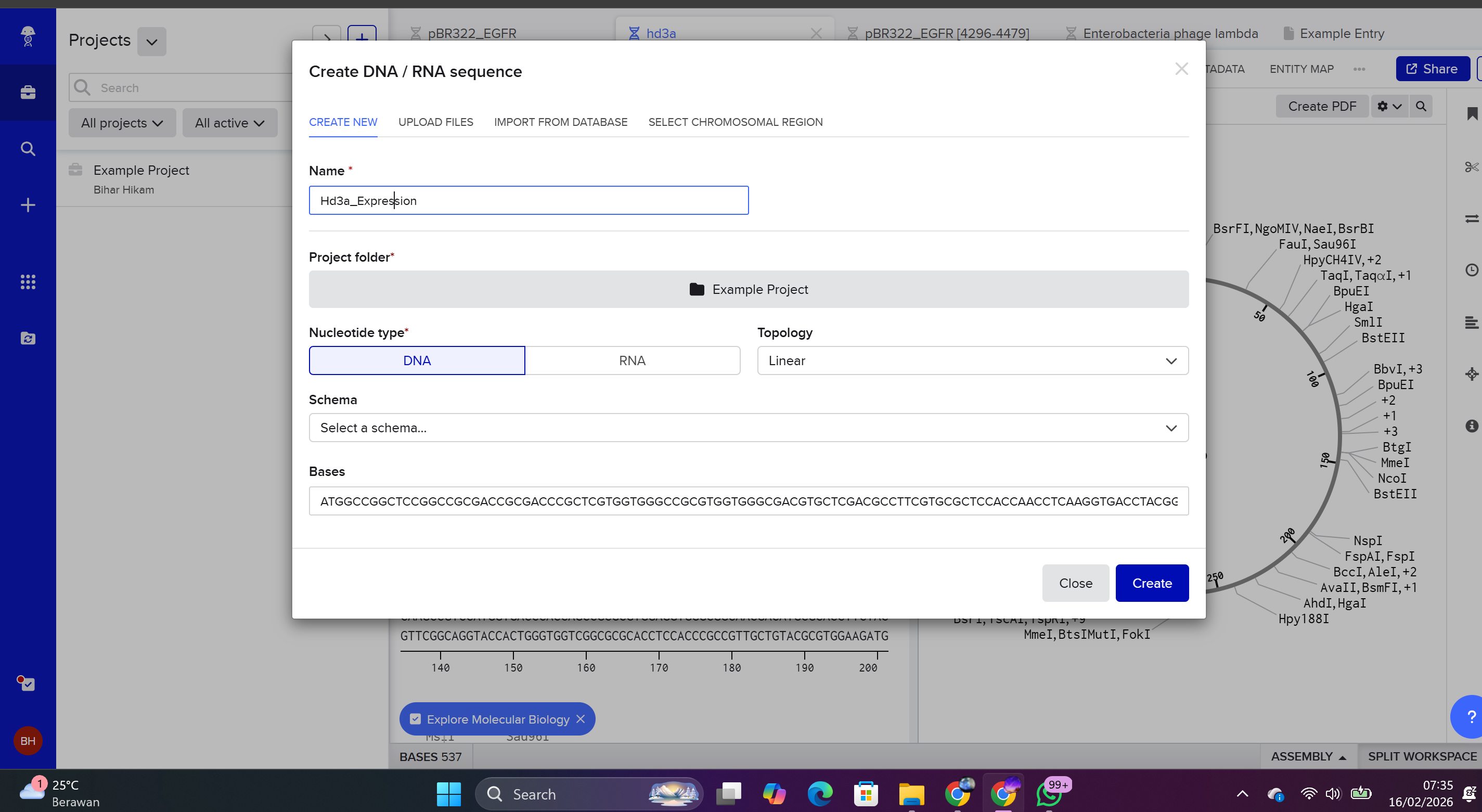
The insert was assembled in a 5’ → 3’ orientation by sequentially adding the following components:
Order of genetic elements:
Promoter: BBa_J23106 (35 bp)
Sequence: TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC
Function: Constitutive transcription initiation in E. coli.
Ribosome Binding Site: BBa_B0034 (21 bp)
Sequence: CATTAAAGAGGAGAAAGGTACC
Function: Facilitates ribosome recruitment for translation initiation.
Start Codon: ATG (3 bp)
Function: Initiates translation.
Hd3a Coding Sequence (537 bp)
Function: Encodes Hd3a protein.
7×His Tag: CATCACCATCACCATCATCAC (18 bp)
Function: Enables purification via Ni-NTA affinity chromatography.
Stop Codon: TAA (3 bp)
Function: Terminates translation.
Terminator: BBa_B0015 (127 bp)
Function: Terminates transcription.
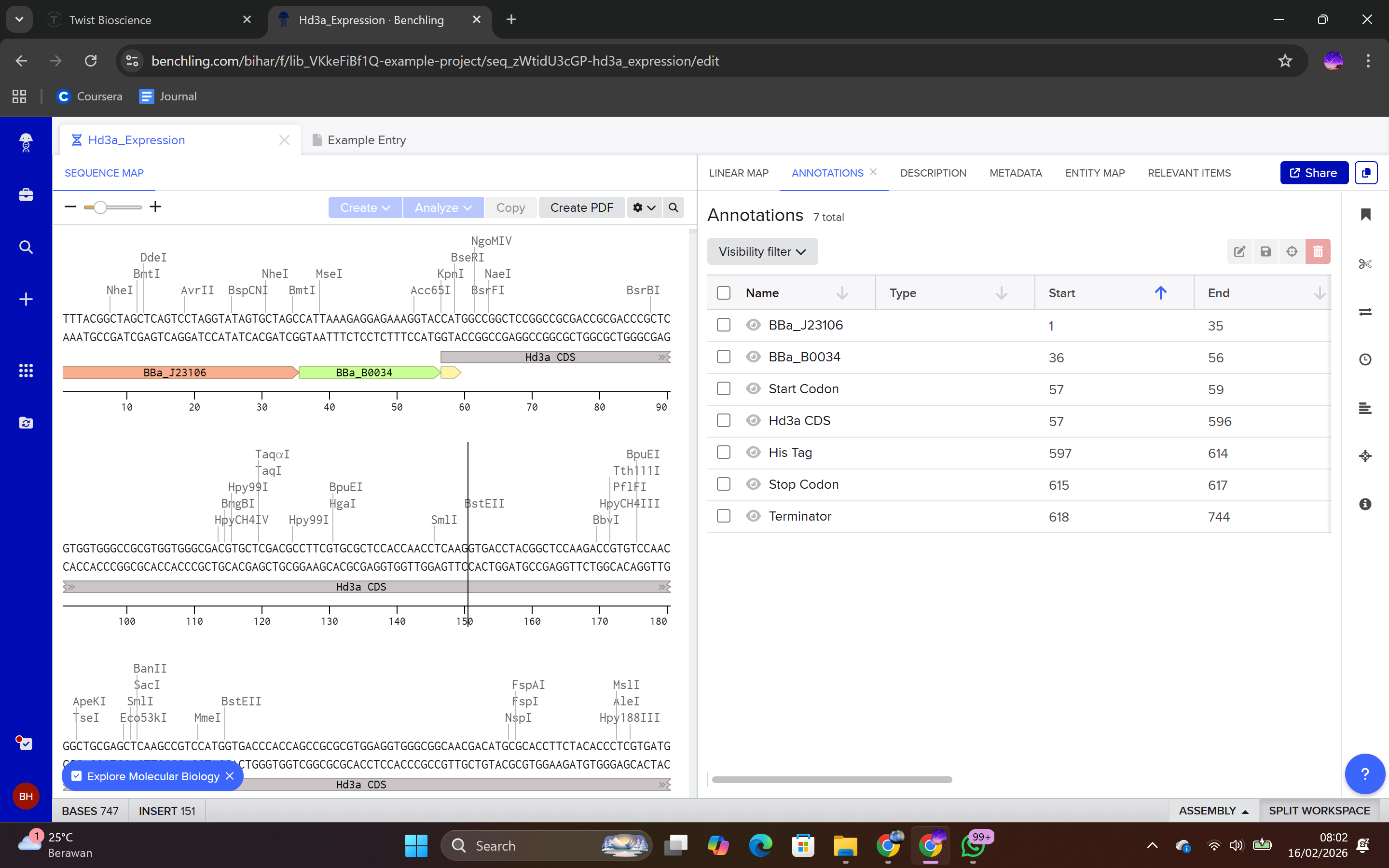
Each sequence component was annotated in Benchling to ensure clarity and proper documentation.
Annotation procedure:
Highlight the sequence segment.
Right-click.
Select Create Annotation.
Assign feature name (e.g., Promoter, RBS, CDS, His-tag, Terminator).

The completed sequence was then exported in FASTA format for submission to Twist Bioscience for gene synthesis.
4.3 Gene Synthesis via Twist Bioscience
During the synthesis step, I selected the “Genes” category and chose the “Clonal Genes” option, which enables synthesis of the gene directly within a circular plasmid ready for transformation without additional cloning steps. The FASTA file was uploaded, and the plasmid backbone pTwist Amp High Copy was selected.
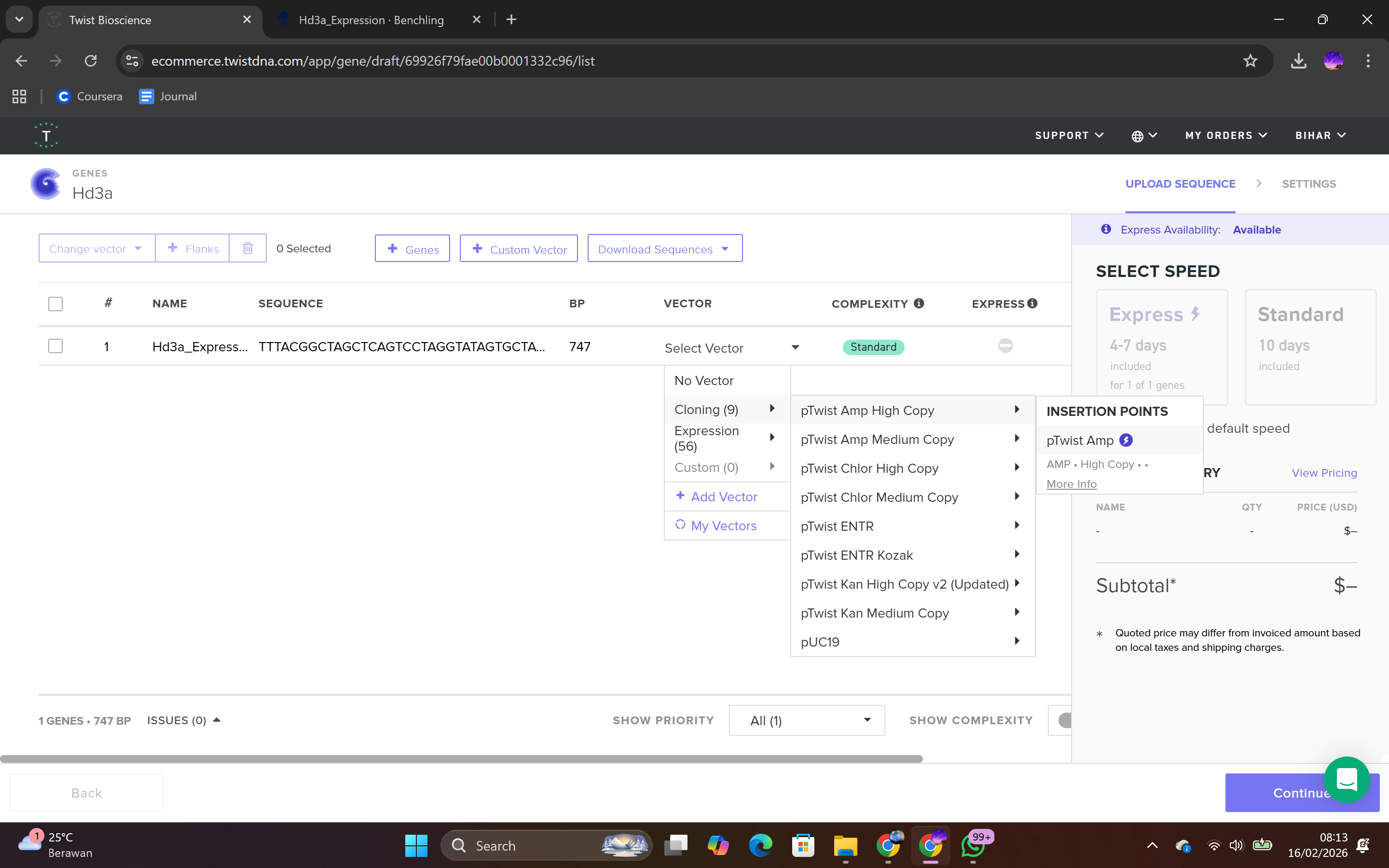
This backbone was chosen based on several biological considerations. It contains a high-copy-number origin of replication, allowing multiple plasmid copies per cell and thereby enhancing protein production in E. coli. It also carries an ampicillin resistance gene for selection, ensuring that only transformed cells survive under antibiotic pressure. The backbone provides the complete circular plasmid structure into which the Hd3a expression cassette is inserted.
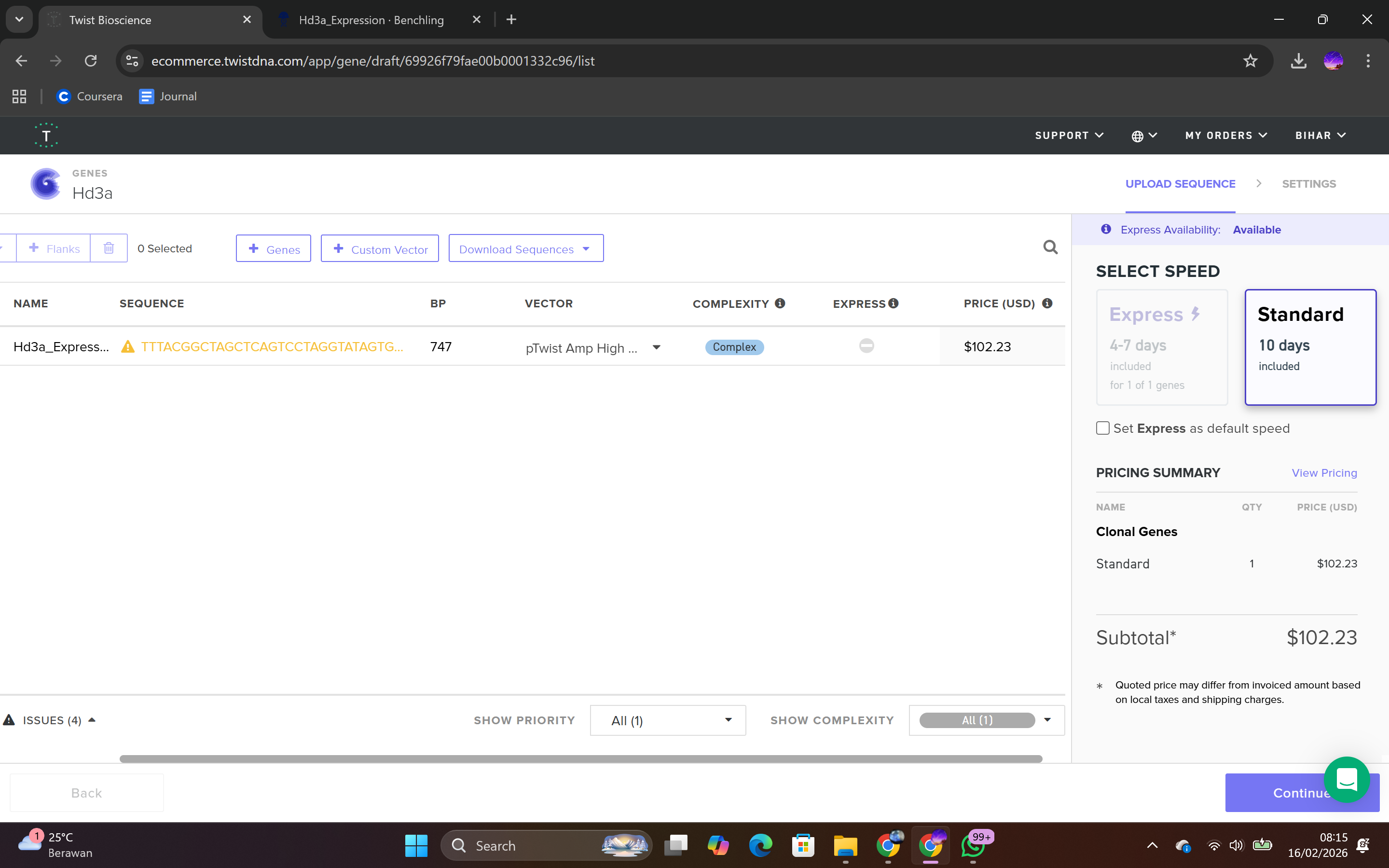
After selecting the vector backbone, the system provides a GenBank file containing the full plasmid sequence, including both the backbone and the Hd3a insert. I downloaded the complete construct as a GenBank file.
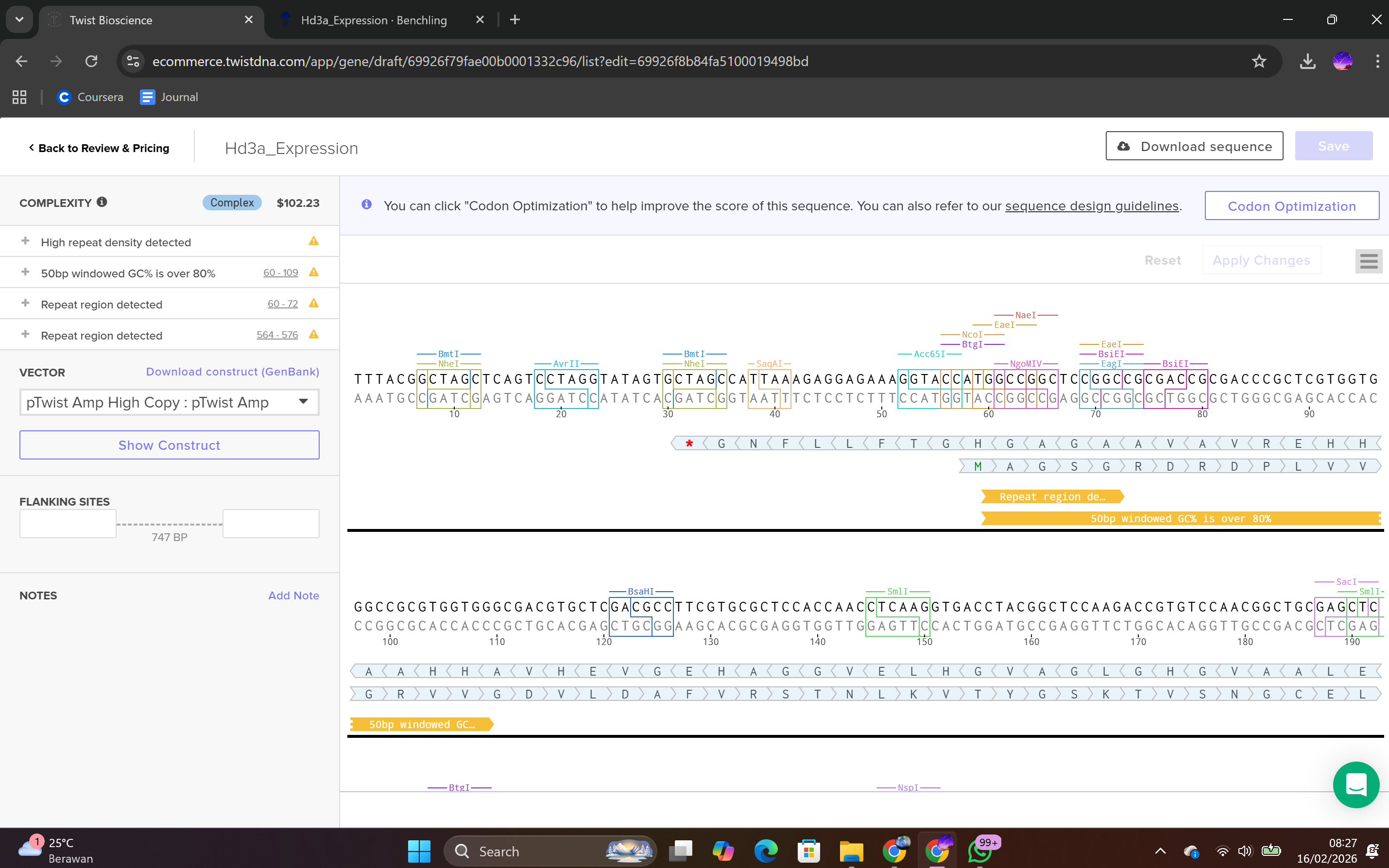
4.4 Verification of Final Plasmid
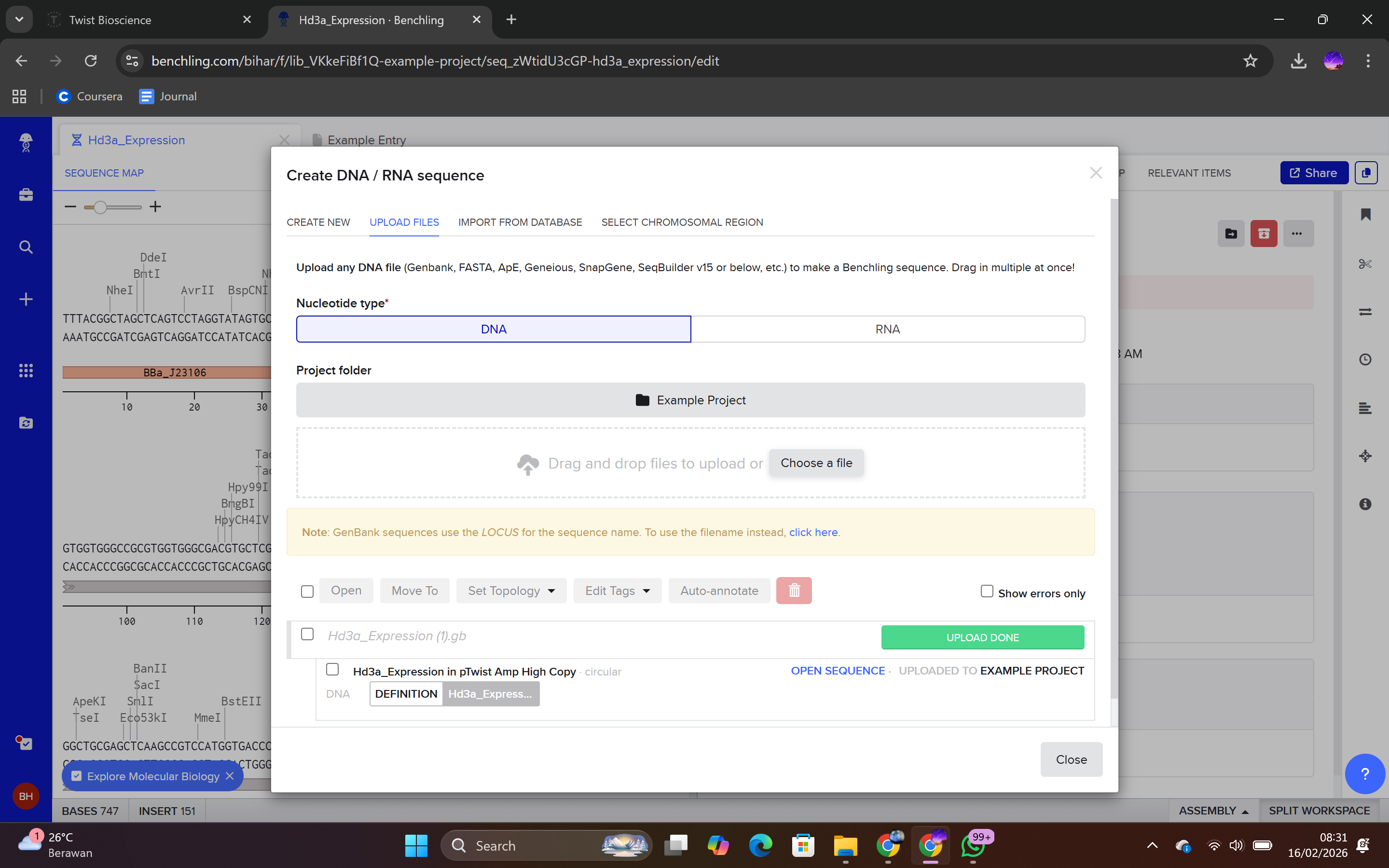
This file can be re-imported into Benchling to visualize the circular plasmid map, displaying the positions of the promoter, CDS, His-tag, terminator, origin of replication, and antibiotic resistance marker.
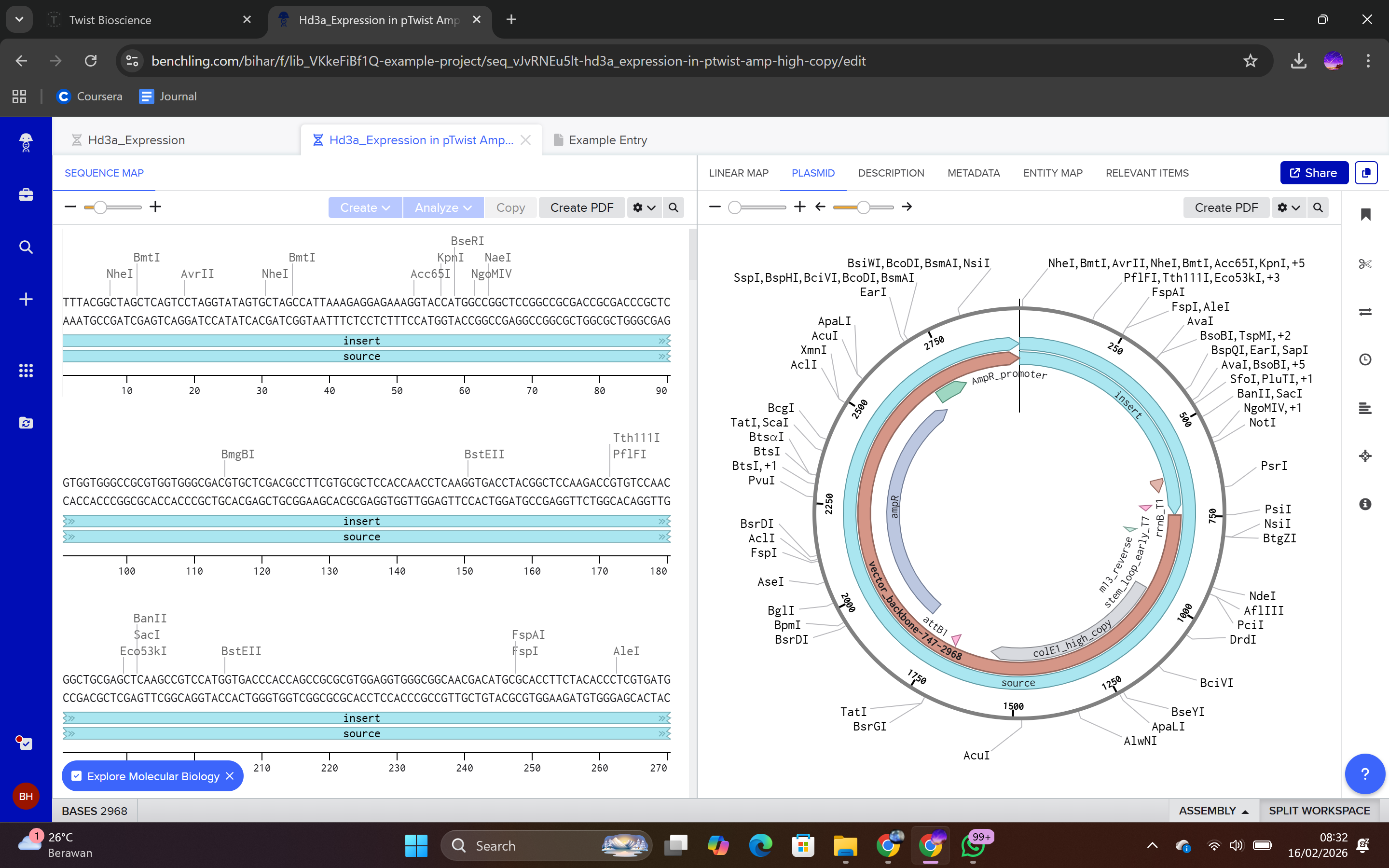
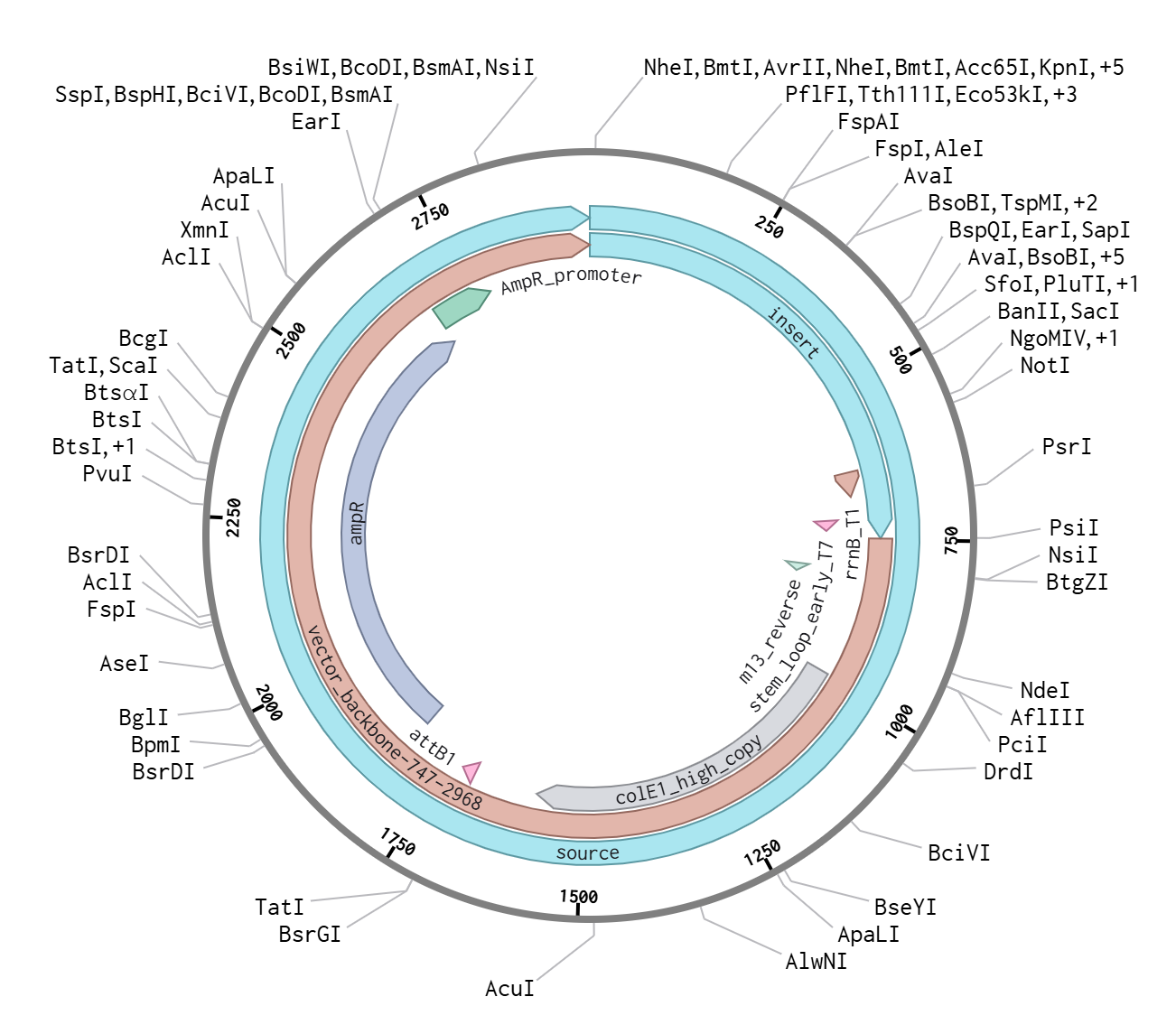
The process was halted at the vector selection stage, as final plasmid synthesis requires payment. However, theoretically, upon ordering and receiving the plasmid:
Transform the plasmid into competent E. coli cells.
Select transformants on ampicillin-containing agar plates.
Culture colonies.
Express Hd3a protein constitutively.
Purify protein using His-tag affinity purification.
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I would sequence genomic DNA and transcriptomic RNA (via cDNA) from rice varieties that exhibit variation in flowering time and growth rate, focusing on genes regulating developmental timing such as flowering-time regulators, photoperiod response genes, and growth hormone pathways (e.g., gibberellin signaling). The goal is to identify allelic variants, regulatory elements, and expression patterns associated with accelerated life cycles. By comparing fast- and normal-growing cultivars under different environmental conditions, we can pinpoint candidate loci for engineering rapid-cycle rice suited for climate stress and food security applications.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would focus on second-generation Illumina short-read sequencing. Illumina provides high accuracy and depth for detecting SNPs and quantifying gene expression (RNA-seq).
Is your method first-, second- or third-generation or other? How so?
Illumina is a second-generation sequencing technology because it relies on sequencing-by-synthesis with clonal amplification and parallel short-read generation.
What is your input? How do you prepare your input (e.g., fragmentation, adapter ligation, PCR)? List the essential steps.
The input is high-quality extracted rice genomic DNA. For Illumina, DNA is fragmented, adapters are ligated, fragments are amplified by PCR, and libraries are size-selected.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
In Illumina sequencing, fragmented DNA binds to a flow cell, undergoes bridge amplification to form clusters, and fluorescently labeled nucleotides are incorporated one base at a time; imaging detects fluorescence to determine each base (base calling).
What is the output of your chosen sequencing technology?
The output is digital sequence data in FASTQ format containing nucleotide sequences and quality scores. These reads can then be assembled into a reference-guided genome, used for variant calling, or analyzed for gene expression and regulatory differences associated with accelerated rice growth.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would synthesize a modular genetic construct to accelerate flowering and shorten the vegetative phase in rice. Specifically, I would design (1) a modified flowering activator gene such as an optimized Heading date 3a (Hd3a) coding sequence, (2) a tunable promoter responsive to environmental or chemical induction, and (3) a regulatory safeguard module. The construct would include a plant promoter, codon-optimized Hd3a CDS, terminator sequence, and optional CRISPR-based regulatory cassette targeting repressors of flowering. The purpose is to create a controllable rapid-cycle rice system that can reduce harvest time without permanently disrupting yield stability. The DNA synthesized would therefore be a modular plasmid insert containing promoter-gene-terminator architecture plus guide RNA cassette if CRISPR regulation is included.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use phosphoramidite-based chemical oligonucleotide synthesis combined with high-throughput gene assembly. This approach is currently the most scalable and accurate method for writing synthetic genes up to several kilobases. Short oligos (~150–200 nt) are synthesized chemically, then assembled enzymatically into full-length genes.
What are the essential steps of your chosen sequencing methods?
First, short oligonucleotides are synthesized using solid-phase phosphoramidite chemistry, where nucleotides are added sequentially with chemical protection/deprotection cycles. Second, oligos are cleaved, deprotected, and purified. Third, overlapping oligos are assembled into longer fragments via PCR-based assembly or Gibson Assembly. Fourth, the assembled construct is cloned into a plasmid vector and sequence-verified using NGS or Sanger sequencing to confirm accuracy.
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
Chemical synthesis accumulates errors as length increases, limiting direct synthesis to ~200 nt per oligo and requiring assembly for longer constructs. Error rates during synthesis and assembly necessitate sequence verification. While highly scalable and increasingly cost-effective, very large constructs (e.g., whole chromosomes) remain complex and time-consuming to assemble. Additionally, repetitive or high-GC regions can reduce synthesis efficiency.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would edit endogenous flowering-time regulatory genes in rice, particularly repressors or upstream regulators that delay the transition from vegetative to reproductive growth. The goal would be to fine-tune rather than eliminate gene function to shorten the life cycle while maintaining yield stability and stress resilience. For example, I would introduce targeted loss-of-function or promoter-weakening mutations in flowering repressors, or precise base edits in cis-regulatory regions to modulate expression levels.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-based genome editing, specifically CRISPR-Cas9, for targeted knockouts and CRISPR-based editors (e.g., cytosine or adenine base editors) for precise nucleotide substitutions. If fine transcriptional tuning is required, CRISPR interference or CRISPR activation systems could also be employed. These tools provide high specificity, programmability, and efficiency in plant genomes.
How does your technology of choice edit DNA? What are the essential steps?
CRISPR-Cas9 edits DNA by using a guide RNA to direct the Cas9 nuclease to a complementary genomic sequence adjacent to a PAM motif. Cas9 induces a double-strand break, which is repaired by the cell’s endogenous repair pathways, typically non-homologous end joining, generating small insertions or deletions, or homology-directed repair if a repair template is supplied. Base editors modify single nucleotides without generating double-strand breaks, improving precision.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
Preparation involves in silico design of guide RNAs targeting specific loci, off-target prediction analysis, cloning the gRNA into a plasmid expressing Cas9 or base editor machinery, and preparing plant transformation vectors. Inputs typically include the Cas9 or base editor protein, guide RNA construct, optional donor repair template, and rice cells or callus tissue for transformation via Agrobacterium-mediated delivery or particle bombardment.
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Limitations include variable editing efficiency depending on target locus and delivery method, off-target mutations, mosaicism in regenerated plants, and relatively low efficiency of HDR in plants. Additionally, complex traits like flowering time may involve polygenic networks, so editing a single locus may produce incomplete or context-dependent effects.