As a Biology student, I have a strong determination to transform my life into one that’s more meaningful. I’m known as someone who easily connects with others, possesses self-confidence, and is a reliable, independent individual. I thrive on challenges and have a deep enthusiasm for new experiences. My ability to collaborate effectively and my strong work ethic ensure success in any endeavor. Furthermore, I take pride in being a great conversational companion, always ready to engage in meaningful discussions.
Description of the Bioengineering Application Rapid-Cycle Rice System The Rapid-Cycle Rice System is a bioengineering platform designed to shorten the rice harvest cycle to approximately 45–60 days by modulating flowering-time genetic pathways in combination with controlled growth environments. The system aims to enable high-frequency and rapid-response rice production, particularly under conditions of climate stress, such as extreme droughts, floods, or disruptions to food supply chains.
Part 1: Benchling & In-silico Gel Art 1. IMPORTING LAMBDA DNA 1.1 Creating an Account and Accessing Benchling Purpose: To access the platform for DNA simulation.
Post-Lab Question 1
A study by the Opentrons research community demonstrated how low-cost laboratory automation platforms can accelerate biological experiments. The researchers used the Opentrons OT-2 robot to automate liquid handling tasks such as pipetting, reagent mixing, and plate preparation.
In the study, the robot was programmed using Python scripts to perform repetitive experimental steps with high precision. Automation enabled researchers to run multiple biological assays simultaneously while reducing human error.
PART A — Conceptual Questions How many molecules of amino acids are in 500 g of meat? If we assume 500 g of protein: 500g÷100g/mol = 5 mol
Total of molecules: 5×6.022×10²³
≈ 3 × 10²⁴ amino acid molecules
Why do humans eat beef but do not become a cow, and eat fish but do not become fish? Proteins from food are not absorbed as intact proteins. Instead, they are broken down during digestion:
Part 1 — Generate Binders with PepMLM PepMLM was used to generate four 12-amino-acid peptides conditioned on the A4V mutant SOD1 sequence. The generated peptides showed perplexity values ranging from approximately 5.8 to 7.0, suggesting moderate model confidence in their plausibility as binders. Lower perplexity indicates that the peptide sequence better fits the model’s learned representation of protein–peptide interactions. The known SOD1-binding peptide FLYRWLPSRRGG was included as a reference for downstream structural and functional comparison.
The Rapid-Cycle Rice System is a bioengineering platform designed to shorten the rice harvest cycle to approximately 45–60 days by modulating flowering-time genetic pathways in combination with controlled growth environments. The system aims to enable high-frequency and rapid-response rice production, particularly under conditions of climate stress, such as extreme droughts, floods, or disruptions to food supply chains.
Problem Addressed: Food Security
Global food security is increasingly threatened by climate change, which has led to unpredictable growing seasons, reduced yields in conventional rice cultivation, more frequent crop failures, and continued dependence on long growth cycles (90 - 120 days) for staple crops.
From a scientific perspective, the Rapid-Cycle Rice System contributes to:
• Deeper understanding of the genetic regulation of flowering time and vegetative-to-reproductive transitions in cereal crops.
• Development of ultra-short life-cycle crop models, offering new experimental systems in plant biology.
• Integration of genome editing, high-throughput phenotyping, and controlled-environment agriculture, which may serve as a transferable framework for other staple crops.
Socially and ecologically, the system has the potential to:
• Enhance food resilience for vulnerable communities, particularly in post-disaster or climate-unstable regions.
• Reduce reliance on food imports during emergency conditions.
• Enable localized, high-frequency food production, thereby reducing pressure for agricultural land expansion.
• Lower the risk of food shortages by allowing rapid replanting following crop failure.
At the same time, the system introduces ecological considerations, including impacts on agroecosystem dynamics and genetic diversity, that will require careful governance and oversight.
The urgency of developing the Rapid-Cycle Rice System arises from the convergence of several trends:
• Accelerating climate impacts that exceed earlier predictive models.
• Geopolitical instability and global supply chain disruptions are increasing the likelihood of regional food crises.
• Rapid advances in genome editing technologies (e.g., CRISPR), phenotyping, and controlled-environment agriculture make this approach increasingly technically feasible.
In this context, the Rapid-Cycle Rice System represents not merely an agricultural innovation but a strategic intervention in global food system resilience.
2. Governance and Policy Goals
Ensuring the Ethical Development and Deployment of Rapid-Cycle Rice
Goal 1: Biosafety and Biosecurity
Ensure that the development and deployment of Rapid-Cycle Rice do not introduce biological risks or enable misuse.
Sub-goal 1.1 Prevent unintended ecological or biological harm. Implement risk assessment frameworks to evaluate gene flow, ecological disruption, and unintended phenotypic consequences before field deployment.
Sub-goal 1.2 Prevent misuse or unauthorized manipulation. Establish oversight mechanisms for genome editing workflows, seed distribution, and research access to reduce risks of dual-use or irresponsible deployment.
Goal 2: Environmental Sustainability and Ecosystem Protection
Ensure that rapid-cycle cultivation systems do not degrade agroecosystems or accelerate unsustainable agricultural practices.
Sub-goal 2.1 Maintain agro-biodiversity and genetic diversity. Promote conservation strategies and diversified cropping systems to avoid monoculture dominance and genetic homogenization.
Sub-goal 2.2 Minimize the ecological footprint of intensive production systems. Encourage resource-efficient controlled environments (energy, water, nutrients) and prevent excessive land-use or input intensification.
Goal 3: Equity and Fair Access
Ensure that the benefits of Rapid-Cycle Rice are distributed fairly, particularly among smallholder farmers and climate-vulnerable communities.
Sub-goal 3.1 Prevent technological monopolization and exclusion. Promote licensing models or public-sector partnerships that avoid concentration of control among a few corporations.
Sub-goal 3.2 Support equitable access for low-resource communities. Develop subsidy programs, open-access breeding resources, or public deployment strategies that enable adoption beyond high-income agricultural systems.
Goal 4: Farmer and Community Autonomy
Protect the rights and decision-making power of farmers and local communities in adopting or rejecting Rapid-Cycle Rice technologies.
Sub-goal 4.1 Ensure informed consent and transparency in deployment. Provide clear communication regarding genetic modifications, cultivation requirements, and potential ecological implications.
Sub-goal 4.2 Protect traditional agricultural practices and local knowledge systems. Ensure that the introduction of rapid-cycle systems does not undermine local seed sovereignty or culturally embedded farming methods.
3. Governance Actions
Governance Action 1: Mandatory Pre-Deployment Ecological and Biosafety Review
At present, risk assessment for genetically engineered crops is often fragmented, varies significantly across national contexts, and does not explicitly account for technologies characterized by ultra-short life cycles and high-frequency cultivation. To address this gap, this action proposes the introduction of a mandatory and standardized ecological and biosafety review specifically tailored to rapid-cycle or accelerated-growth crops prior to any field deployment or large-scale release. Such a review would systematically evaluate risks related to gene flow, ecological interactions, and the cumulative long-term effects of repeated high-frequency planting cycles.
This governance mechanism would involve national biosafety authorities, academic institutions, and independent ecological risk panels. Approval would be required before field trials, supported by inter-institutional data sharing and periodic reassessment following deployment to capture emergent risks. Funding would be provided through a combination of public research grants and regulatory fees. This approach assumes that regulatory bodies possess sufficient expertise to evaluate advanced plant bioengineering, that ecological risks can be meaningfully assessed before full deployment, and that developers are willing to comply with additional review requirements.
However, this action carries risks. Approval processes may become slow, potentially delaying deployment during food emergencies, and increased regulatory burdens could discourage public-sector or small-scale research initiatives. Even if successful, over-standardization may reduce sensitivity to local ecological contexts, and compliance costs could unintentionally exclude smaller research groups from participation.
Governance Action 2: Public-Interest Licensing and Seed Access Framework
Currently, advanced crop biotechnologies are frequently governed by restrictive intellectual property regimes, which can limit access for smallholder farmers and low-income regions. This governance action proposes the development of a public-interest licensing framework for Rapid-Cycle Rice, ensuring that essential genetic constructs and seed lines remain accessible for non-commercial, humanitarian, or climate-resilience applications.
This framework would be implemented through collaboration among universities, public research institutions, governments, international agricultural organizations, seed banks, and public breeding programs. It would rely on tiered licensing structures that distinguish commercial from humanitarian use, supported by public seed repositories and technology transfer initiatives. Incentives for participation would include public funding tied to access and equity commitments. This proposal assumes that public institutions retain some control over intellectual property, that open or semi-open licensing does not eliminate incentives for innovation, and that farmers can adopt the technology effectively when adequate support systems are in place.
Potential failures include reduced private-sector investment and weak enforcement of access conditions. Even successful implementation may generate unintended consequences, such as informal seed sharing that bypasses biosafety controls or increased tension between open access and the traceability of genetically modified seeds.
Governance Action 3: Technical Safeguards Embedded in Plant Design
Most existing governance approaches rely heavily on external regulation, with limited emphasis on embedding constraints directly within biological systems. This action proposes incorporating genetic or environmental containment mechanisms into Rapid-Cycle Rice varieties themselves, such as dependency on controlled environmental cues, reduced competitiveness outside managed systems, or conditional expression of rapid-flowering traits.
The development and oversight of these safeguards would involve bioengineers, plant geneticists, research institutions, and regulatory reviewers. Implementation would require design standards for built-in safeguards, validation during biosafety review processes, and continued monitoring after deployment. This approach reflects a “safety-by-design” governance model, emphasizing intrinsic risk reduction rather than post hoc control. It assumes that biological containment mechanisms are reliable and evolutionarily stable, that safeguards do not significantly compromise agronomic performance, and that researchers can accurately anticipate ecological interactions.
Nonetheless, safeguards may fail due to mutation or selective pressure, and their presence may create a false sense of security that weakens external oversight. If highly successful, over-engineered constraints could limit adaptability in real-world farming conditions, and reliance on controlled systems may exclude low-resource or smallholder settings.
4. Scoring Governance Actions Against Policy Goals
Based on the scoring matrix, Option 1 (Mandatory Pre-Deployment Ecological and Biosafety Review) is prioritized as the most rational governance approach for the Rapid-Cycle Rice System. This option consistently performs strongest in preventing biosecurity, biosafety, and environmental harms, which represent the most severe and least reversible risks associated with genome-edited staple crops deployed at high frequency. While this approach entails higher regulatory costs, potential delays in deployment, and unequal implementation capacity across regions, these trade-offs are justified given the long-term consequences of ecological disruption or biological misuse. Nevertheless, residual risks remain, including incomplete ecological foresight, procedural compliance without substantive review, and reduced flexibility during food emergencies.
This recommendation is primarily directed toward:
National Ministries of Agriculture and Environmental Protection, in coordination with public research institutions and biosafety authorities.
These actors are best positioned to:
• Establish standardized review frameworks,
• Coordinate scientific expertise across disciplines,
• Balance food security imperatives with long-term environmental stewardship.
At the international level, this approach could be harmonized through multilateral agricultural or biosafety agreements, ensuring consistency across borders for the governance of staple crops.
6. Ethical Reflection
Engaging with the Rapid-Cycle Rice concept raised ethical concerns that extend beyond traditional questions of crop safety and yield optimization. One key issue that emerged was the ethical tension between speed and responsibility: while rapid-cycle crops promise timely responses to food insecurity under climate stress, accelerating biological systems also compresses the time available for ecological learning, social deliberation, and institutional oversight. This highlighted a previously underappreciated risk that technologies designed for emergency resilience may normalize exception-based deployment, where urgency is repeatedly invoked to justify reduced scrutiny. Recognizing this risk reframed the ethical challenge not as whether rapid-cycle rice should exist, but under what conditions its deployment remains accountable and reversible.
Another ethical concern involved power asymmetries in technological control, particularly regarding who decides when and how such systems are used. Although rapid-cycle rice could enhance food security for vulnerable populations, it could also concentrate decision-making authority among governments, research institutions, or corporate actors, potentially undermining farmer autonomy and local knowledge systems. This raised questions about consent, transparency, and distributive justice that were not initially apparent when viewing the technology purely as a technical solution. In response, governance actions that emphasize participatory oversight, transparent risk communication, and public-interest deployment frameworks appear ethically necessary, not merely as safeguards against harm, but as mechanisms to preserve trust and legitimacy in climate-responsive agricultural innovation.
Homework Questions from Professor Jacobson
1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Biological DNA polymerase with proofreading has an error rate of approximately ~1 error per 10⁶ base additions (10⁻⁶), while the human genome size is shown in the scaling slides as approximately ~3.2 × 10⁹ base pairs (3.2 Gbp)
If polymerase made errors at 1 per 10⁶ bp, then copying the entire human genome would introduce ~3,200 errors per replication
This would be catastrophic if left uncorrected.
Biology resolves this through layered error correction, not polymerase alone:
Post-replication DNA repair. Mismatch repair systems (e.g. MutS/MutL, also shown in the lecture)
Cell-cycle checkpoints. Prevent propagation of heavily damaged DNA
2. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Average human protein length ≈ 1036 bp
Each nucleotide position can be A, T, G, or C, so the total number of possible DNA sequences is: 4^1036
This is an astronomically large number, far exceeding the number of atoms in the observable universe.
The most theoretically valid DNA sequences fail biologically or physically due to multiple constraints:
Secondary structure formation
Extreme GC or AT bias
Enzymatic and synthesis constraints
Homework Questions from Dr. LeProust
1. What’s the most commonly used method for oligo synthesis currently?
Solid-phase phosphoramidite chemical synthesis is the dominant method.
It uses stepwise nucleotide coupling on a solid support (e.g., CPG or silicon), repeating coupling–capping–oxidation–deprotection cycles.
2. Why is it difficult to make oligos longer than 200nt via direct synthesis?
Because error rates accumulate with each synthesis cycle. Even with ~99–99.5% coupling efficiency per base, long oligos suffer from truncations, deletions, and substitutions, causing a sharp drop in full-length product yield beyond ~200 nt.
3. Why can’t you make a 2000bp gene via direct oligo synthesis?
Direct chemical synthesis at that length would result in unacceptably high error rates and near-zero full-length yield. Instead, long genes are made by assembling many shorter oligos (e.g., 150–300 nt) using enzymatic assembly and error correction, followed by sequence verification.
Homework Question from George Church
1. [Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids in animals (cannot be synthesized de novo and must come from diet) are:
Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, Valine, and Arginine
Implication for the “Lysine Contingency”:
Because lysine is universally essential and metabolically costly, organisms cannot easily substitute away from lysine-rich proteins when lysine availability is low. This makes lysine a natural bottleneck linking nutrition, protein synthesis, and regulation (including PTMs like lysine acetylation). Rather than being replaceable, lysine’s indispensability amplifies selective pressure on regulatory systems to sense, conserve, and prioritize lysine usage, supporting the idea that the “lysine contingency” is a fundamental constraint, not a coincidence.
Week 2 HW: DNA Read, Write, and Edit
Part 1: Benchling & In-silico Gel Art
1. IMPORTING LAMBDA DNA
1.1 Creating an Account and Accessing Benchling
Purpose: To access the platform for DNA simulation.
Open benchling.com
Sign up
Log in
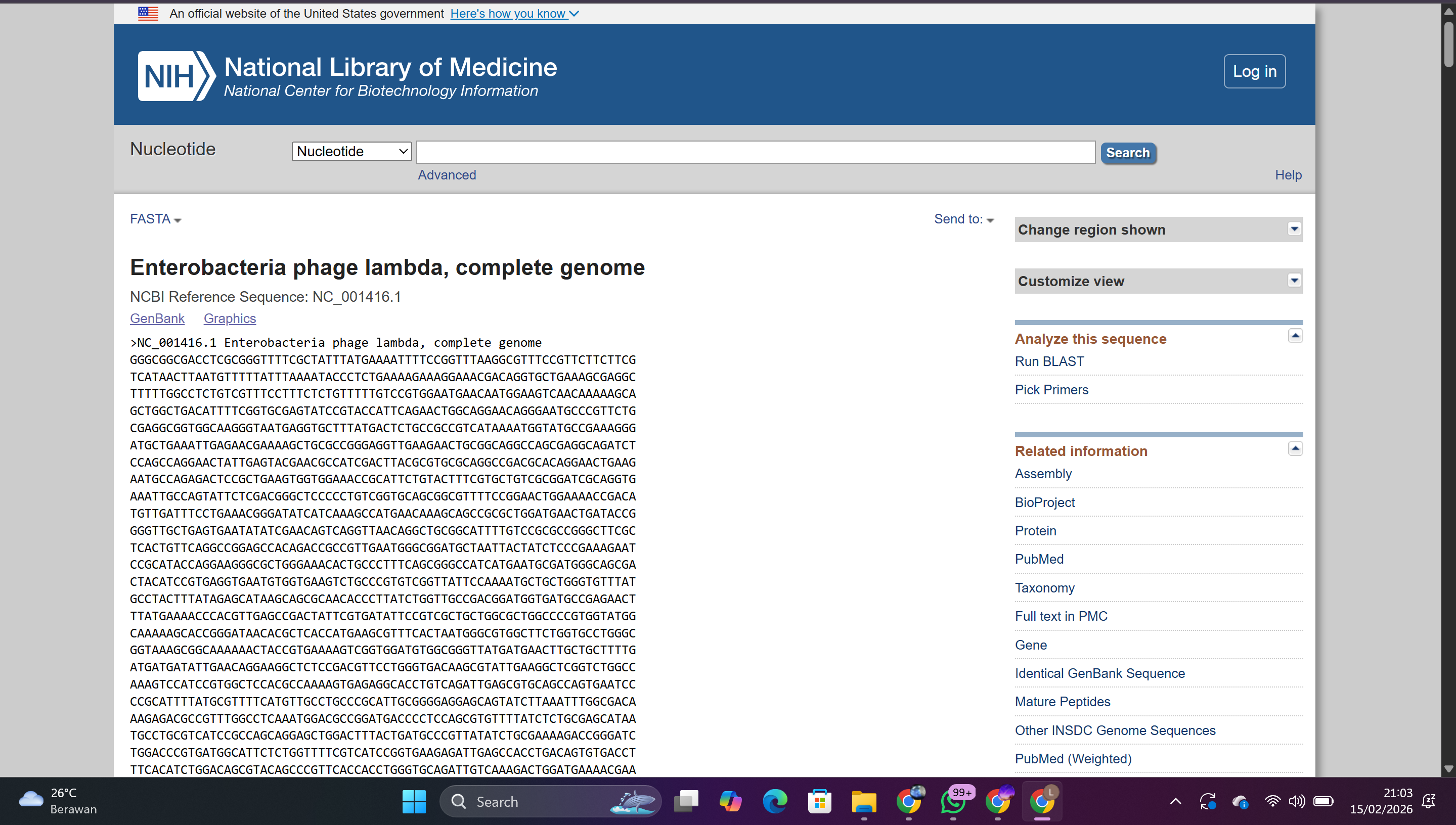
1.2 Importing Sequence from NCBI
Purpose: To obtain the DNA template for analysis.
Steps:
Search for Lambda DNA in the National Center for Biotechnology Information (NCBI) database
Copy the complete genome sequence
In Benchling:
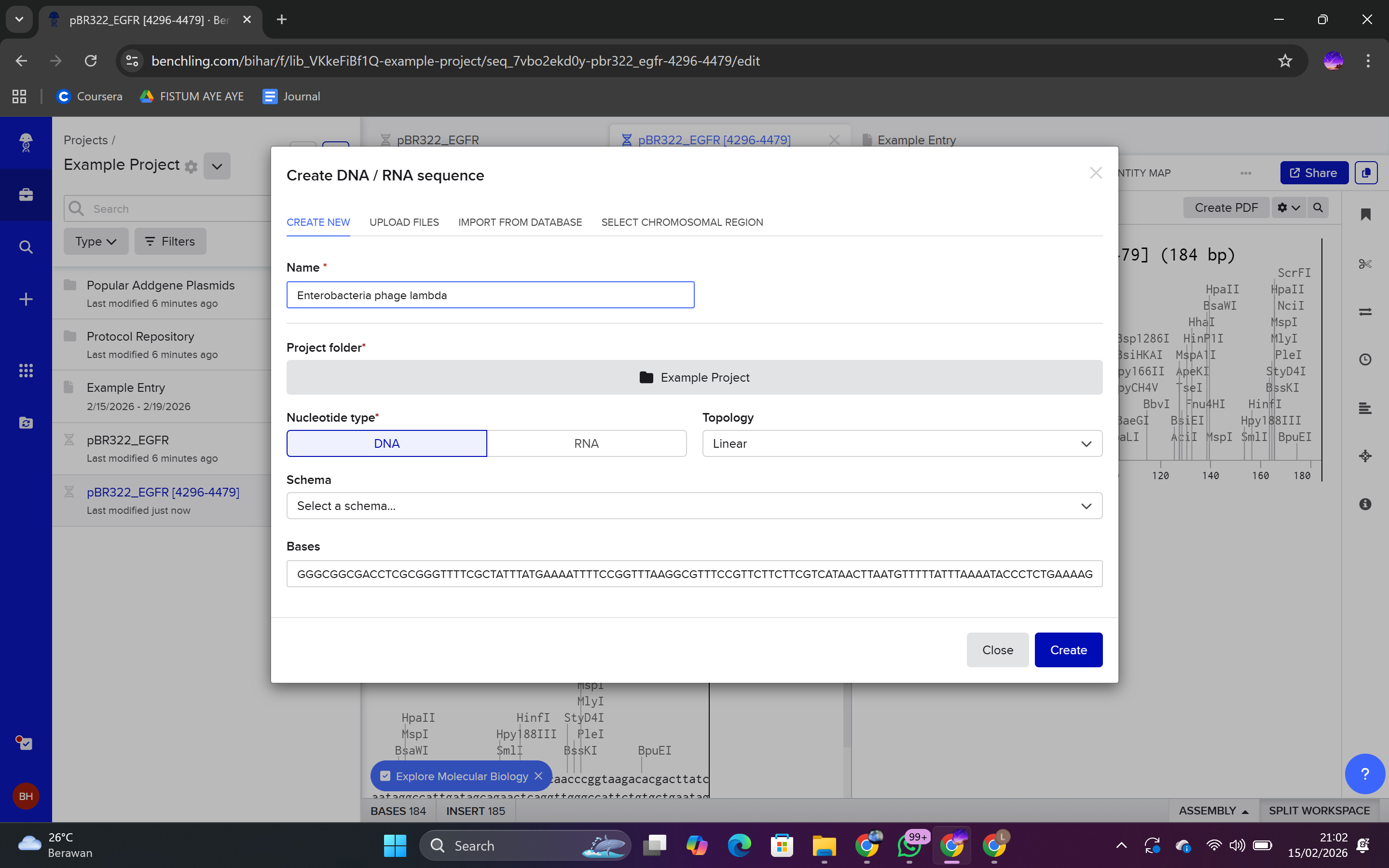
Click Create → DNA Sequence
Select Import
Paste the sequence
Save as Lambda DNA
Result: The ~48.5 kb Lambda DNA sequence was successfully imported into the workspace.
RESTRICTION DIGEST SIMULATION
2.1 Opening the Restriction Enzyme Tool
Purpose: To identify DNA cutting sites by specific enzymes.
Steps:
Open the Lambda DNA file
Click the scissors icon (Restriction Enzymes) on the right panel
Select the Digest tab
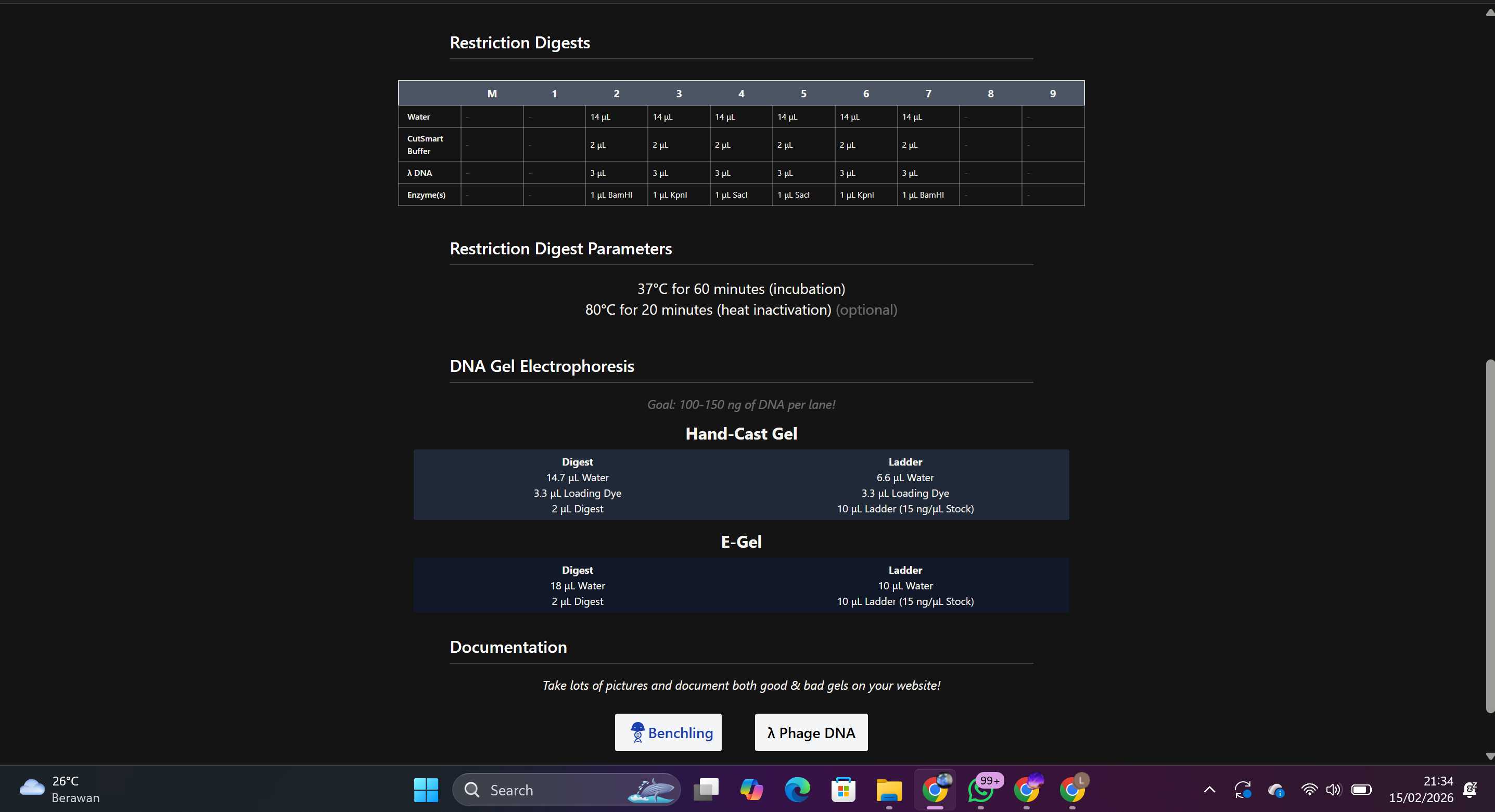
2.2 Adding Restriction Enzymes
Purpose: To generate DNA fragments with different cutting patterns.
Enzymes used:
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
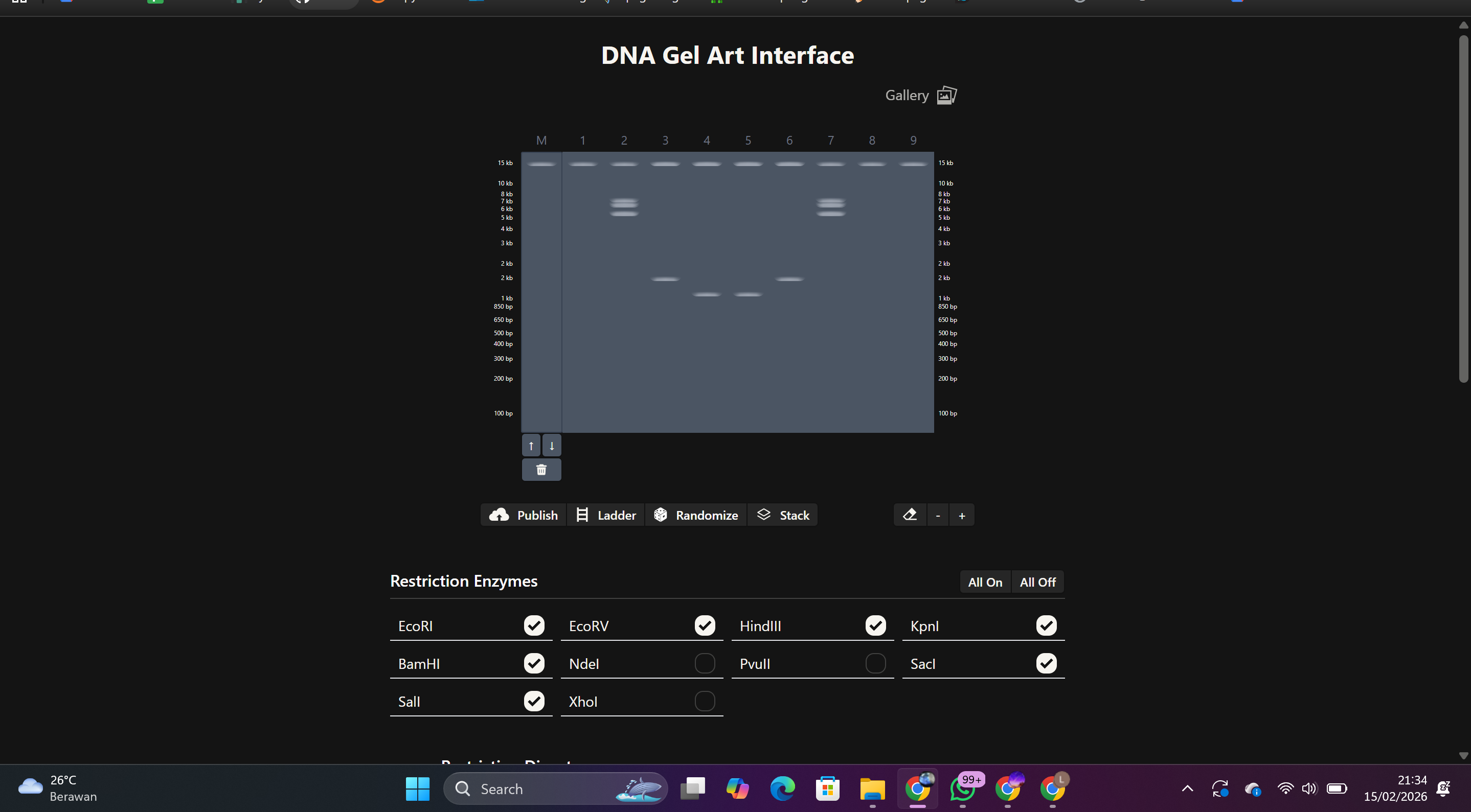
Each enzyme was entered individually to observe the number and size of resulting fragments.
Result:
DNA fragments of varying sizes were generated based on each enzyme’s recognition site.
DESIGNING A PATTERN IN THE STYLE OF LATENT FIGURE PROTOCOL
Purpose: To transform gel patterns into a bio-art visual composition.
Concept:
Each lane represents a visual column
Band positions act as visual points
Enzyme combinations create variation and detail
Steps:
Select digests with the most contrasting band patterns
Arrange multiple lanes side by side
Adjust enzyme combinations to form a silhouette
Capture a screenshot of the final gel
Final Result:
A gel pattern resembling a face or silhouette was created, inspired by Latent Figure Protocol, a bio-art approach that uses DNA visualization as a visual medium.
Part 3: DNA Design Challenge
3.1. Protein Selection
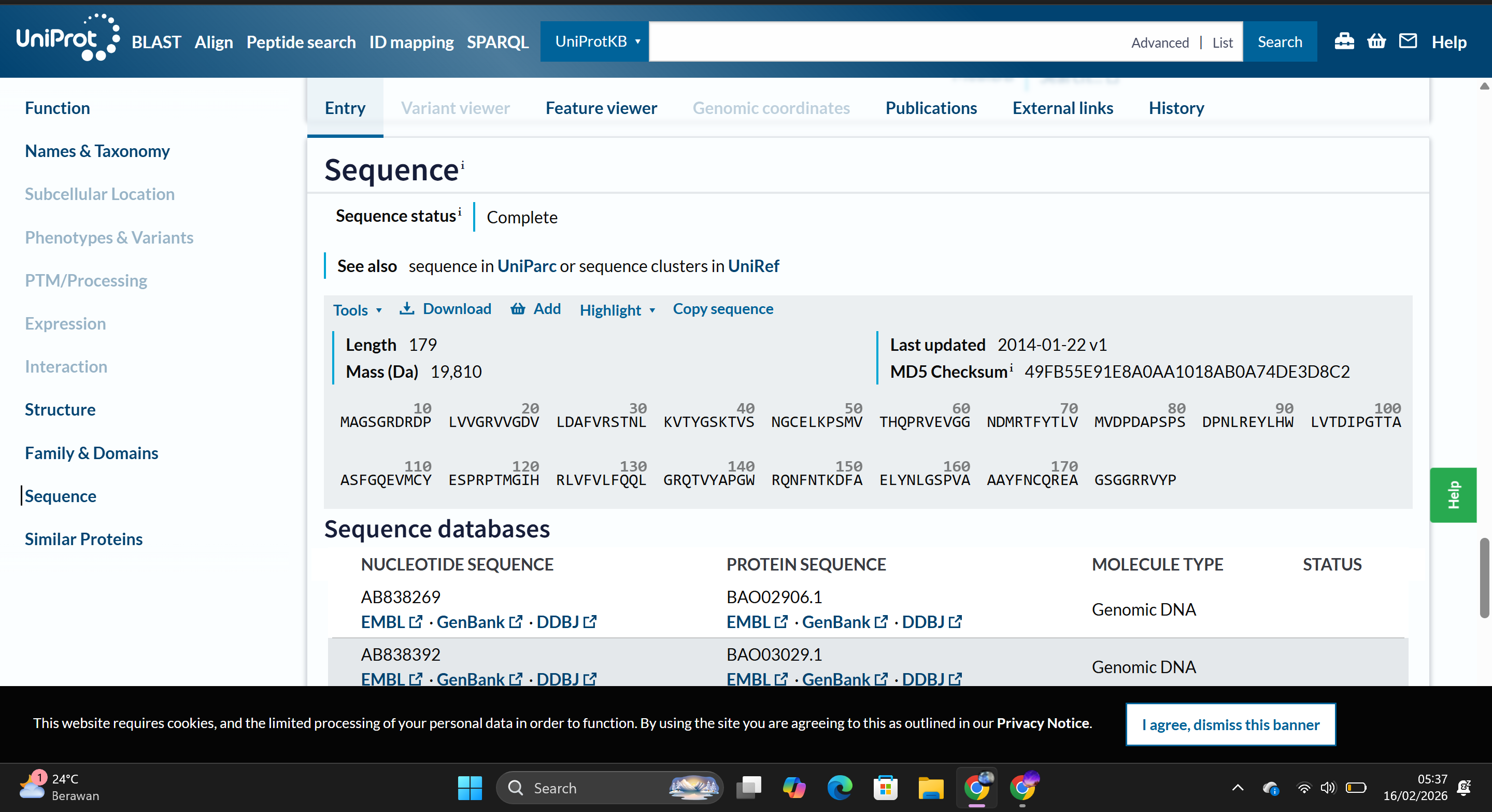
For this assignment, I selected the Hd3a (Heading date 3a) protein from Oryza sativa. Hd3a functions as a florigen protein, acting as a systemic flowering signal that induces the transition from vegetative to reproductive growth in rice. This protein is directly relevant to the “Rapid-Cycle Rice” concept, as modulating Hd3a expression can accelerate flowering and shorten the crop life cycle. Therefore, Hd3a represents a biologically rational target for bioengineering strategies aimed at improving food security through reduced cultivation time.
The amino acid sequence used in this assignment is:
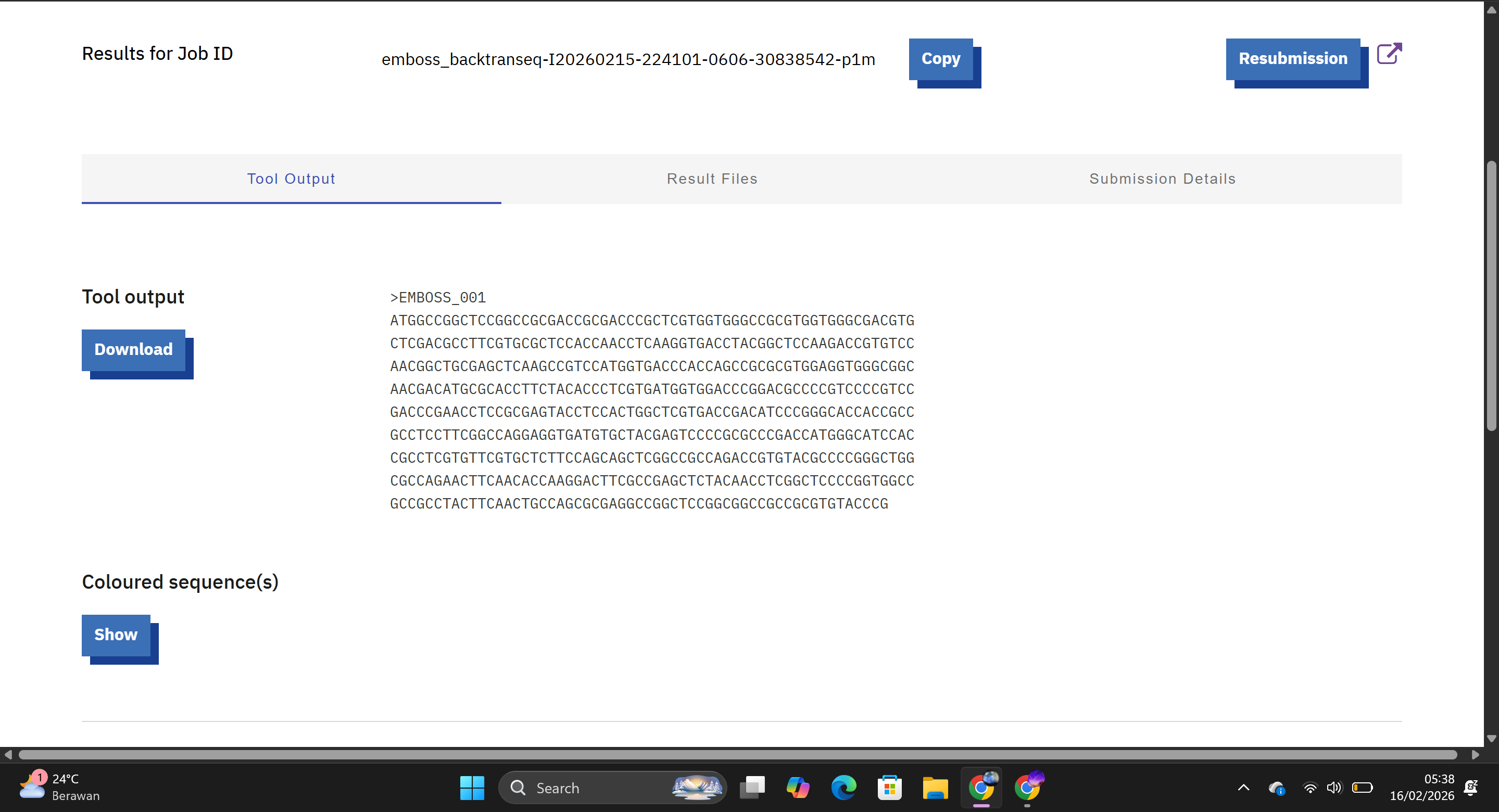
Based on the Central Dogma of molecular biology, a protein sequence can be used to infer a possible DNA sequence through reverse translation. Because the genetic code is degenerate (multiple codons can encode the same amino acid), a single protein sequence can correspond to many possible DNA sequences.
This tool generated a nucleotide sequence corresponding to the Hd3a protein sequence using the standard genetic code. The resulting DNA sequence theoretically encodes the Hd3a protein.
Although reverse translation produces a valid DNA sequence, the resulting sequence may not be optimal for expression in a chosen host organism. Different organisms exhibit codon bias, meaning they preferentially use certain codons over others for the same amino acid.
The reverse-translated DNA sequence was further optimized using the IDT Codon Optimization Tool for expression in Oryza sativa. The tool adjusts codon usage according to host codon bias, removes rare codons, and minimizes problematic motifs such as repeat regions or restriction sites.
An optimized DNA sequence improves protein yield, stability, and overall expression efficiency.
3.4. Protein Production from DNA
Once the optimized DNA sequence is obtained, the gene can be chemically synthesized and cloned into an expression vector. Protein production follows the Central Dogma:
DNA is transcribed into mRNA by RNA polymerase.
mRNA is translated by ribosomes into protein, where each three-nucleotide codon specifies one amino acid.
Protein production can be achieved using Cell-dependent expression systems
The gene is inserted into an expression plasmid and transformed into a host cell such as Escherichia coli or plant cells. A promoter within the plasmid initiates transcription, and the host’s ribosomes translate the mRNA into the Hd3a protein.
3.5. [Optional] How does it work in nature/biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level.
A single gene can produce multiple proteins at the transcriptional level primarily through alternative splicing. In eukaryotic organisms such as Oryza sativa, genes are composed of exons (coding regions) and introns (non-coding regions). After transcription, the pre-mRNA can be spliced in different ways, meaning certain exons may be included or skipped. This generates multiple mature mRNA isoforms from the same DNA sequence, each of which can be translated into distinct protein variants with different structures or functions. Additional mechanisms include alternative promoter usage and alternative polyadenylation, which also produce transcript diversity.
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Preparation of Hd3a Coding Sequence
The first step was preparing the coding sequence (CDS) of the Oryza sativa Hd3a gene.
The amino acid sequence of Hd3a was reverse translated into a DNA sequence using a sequence translation tool. The resulting nucleotide sequence was assumed to be codon-optimized for Escherichia coli expression. This optimized DNA sequence (537 bp) serves as the Coding Sequence (CDS) component in the expression cassette.
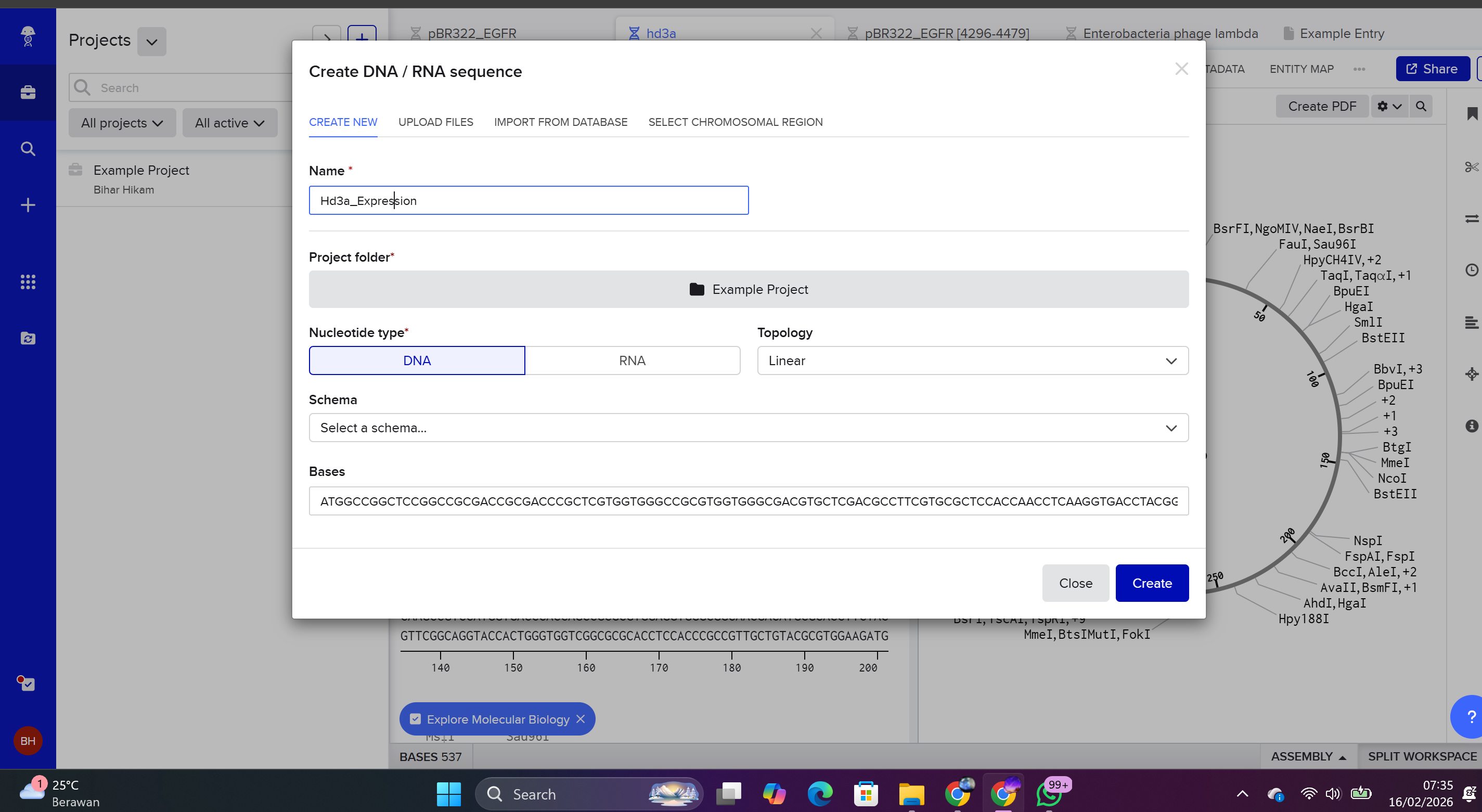
4.2. Build the DNA Insert Sequence (Expression Cassette) in Benchling
To construct the expression cassette:
Log in to Benchling.
Select New DNA/RNA Sequence.
Choose:
Molecule Type: DNA
Topology: Linear
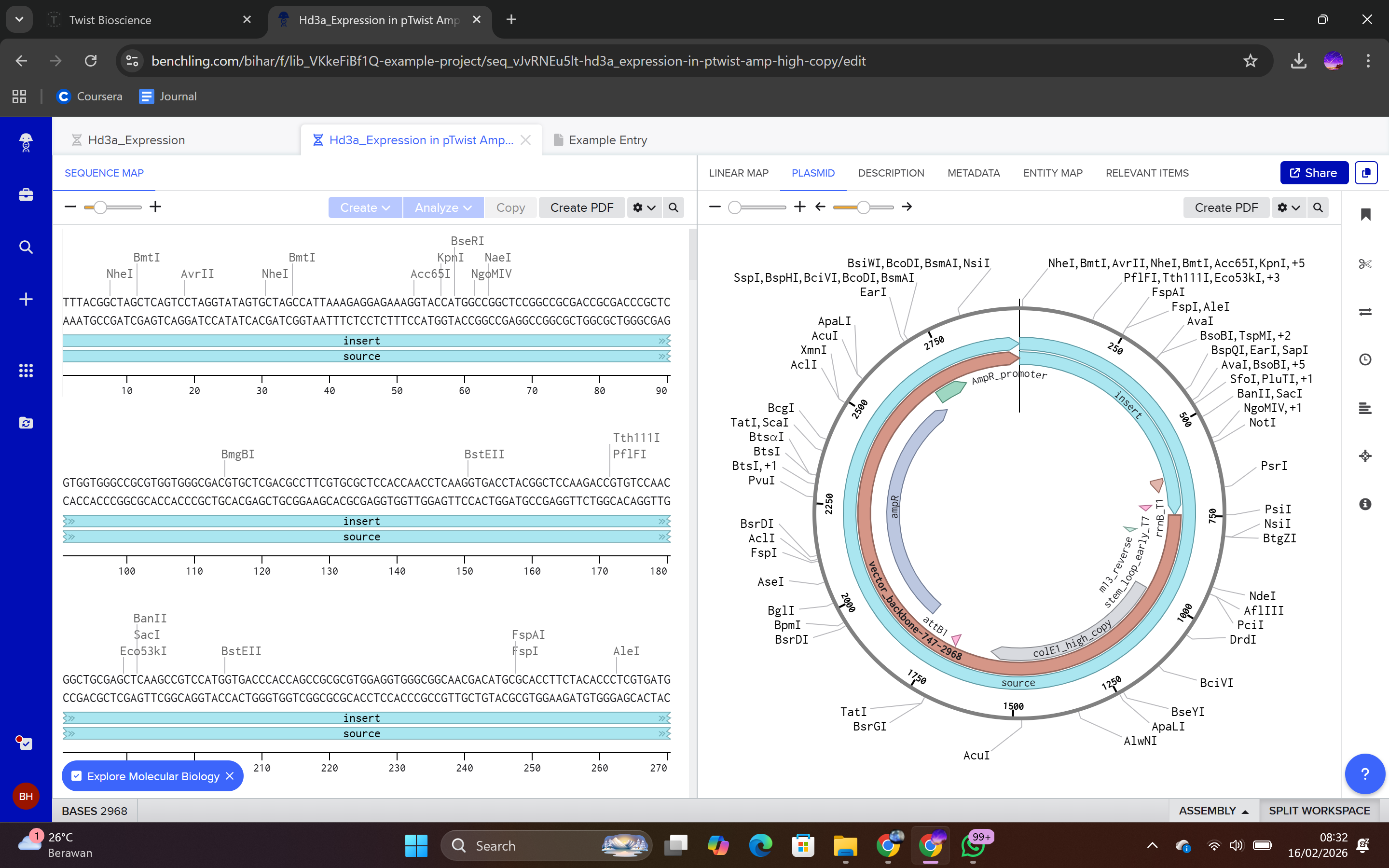
The insert was assembled in a 5’ → 3’ orientation by sequentially adding the following components:
Order of genetic elements:
Promoter: BBa_J23106 (35 bp)
Sequence: TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC
Function: Constitutive transcription initiation in E. coli.
Ribosome Binding Site: BBa_B0034 (21 bp)
Sequence: CATTAAAGAGGAGAAAGGTACC
Function: Facilitates ribosome recruitment for translation initiation.
Start Codon: ATG (3 bp)
Function: Initiates translation.
Hd3a Coding Sequence (537 bp)
Function: Encodes Hd3a protein.
7×His Tag: CATCACCATCACCATCATCAC (18 bp)
Function: Enables purification via Ni-NTA affinity chromatography.
Stop Codon: TAA (3 bp)
Function: Terminates translation.
Terminator: BBa_B0015 (127 bp)
Function: Terminates transcription.
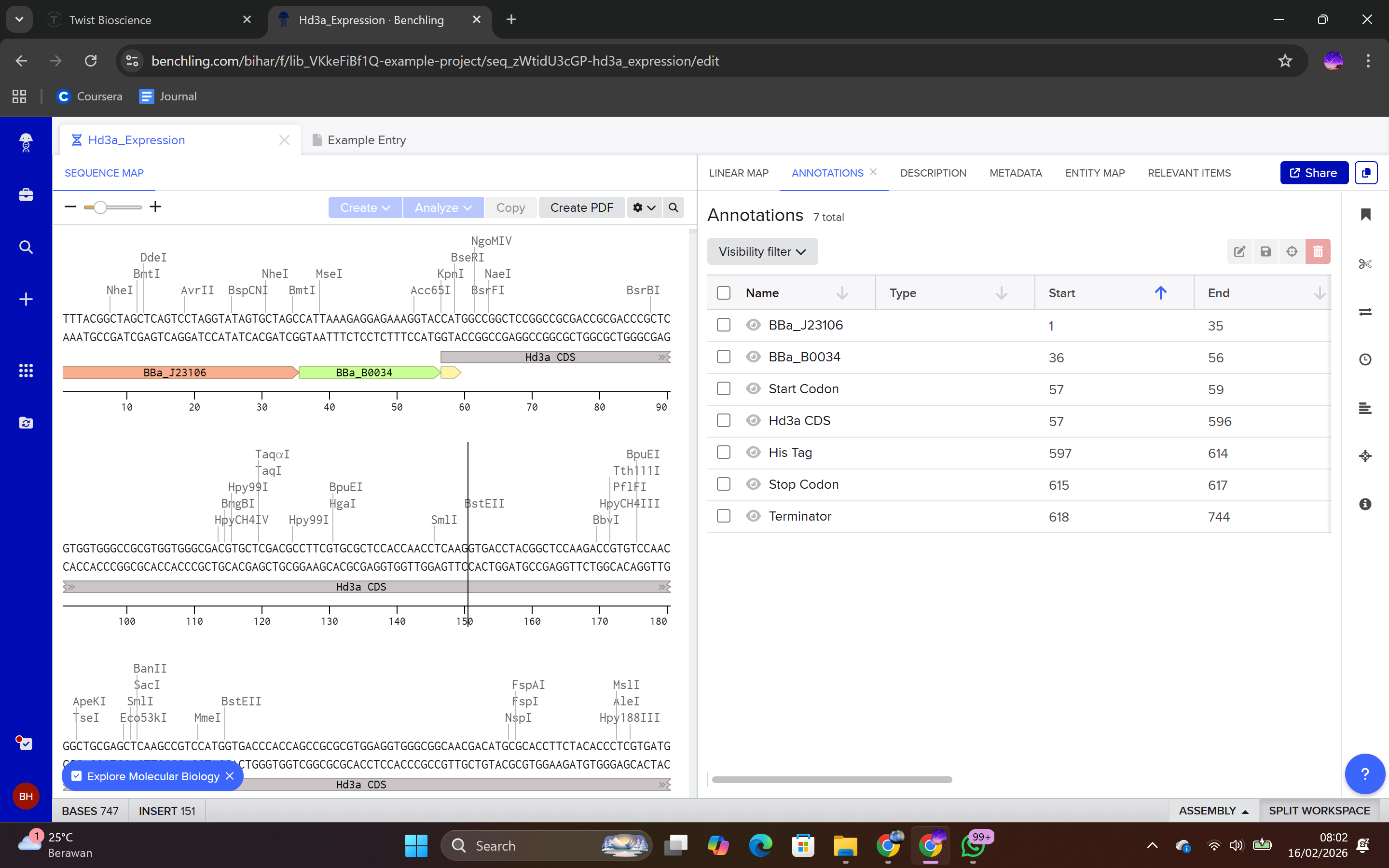
Each sequence component was annotated in Benchling to ensure clarity and proper documentation.
Annotation procedure:
Highlight the sequence segment.
Right-click.
Select Create Annotation.
Assign feature name (e.g., Promoter, RBS, CDS, His-tag, Terminator).
The completed sequence was then exported in FASTA format for submission to Twist Bioscience for gene synthesis.
4.3 Gene Synthesis via Twist Bioscience
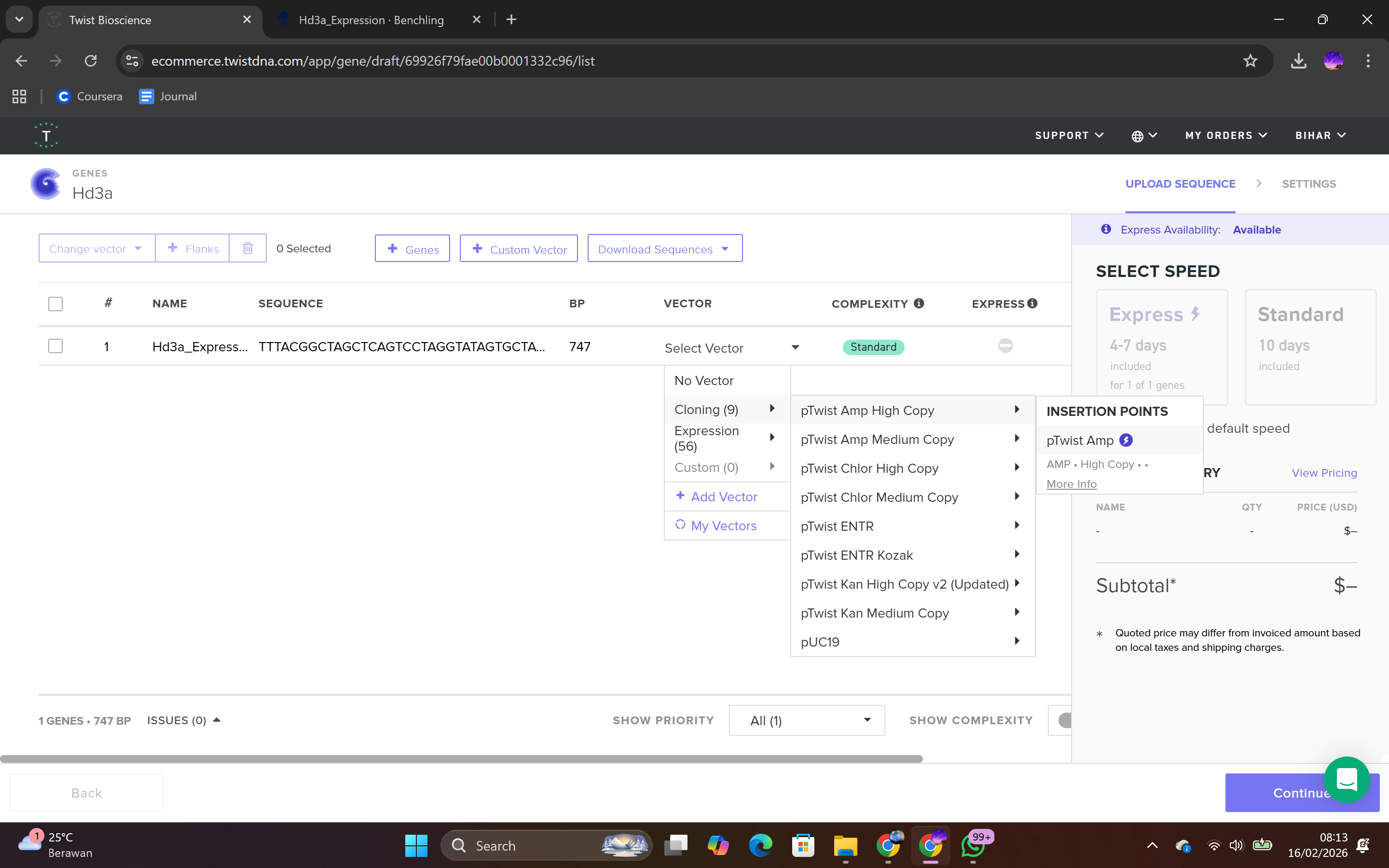
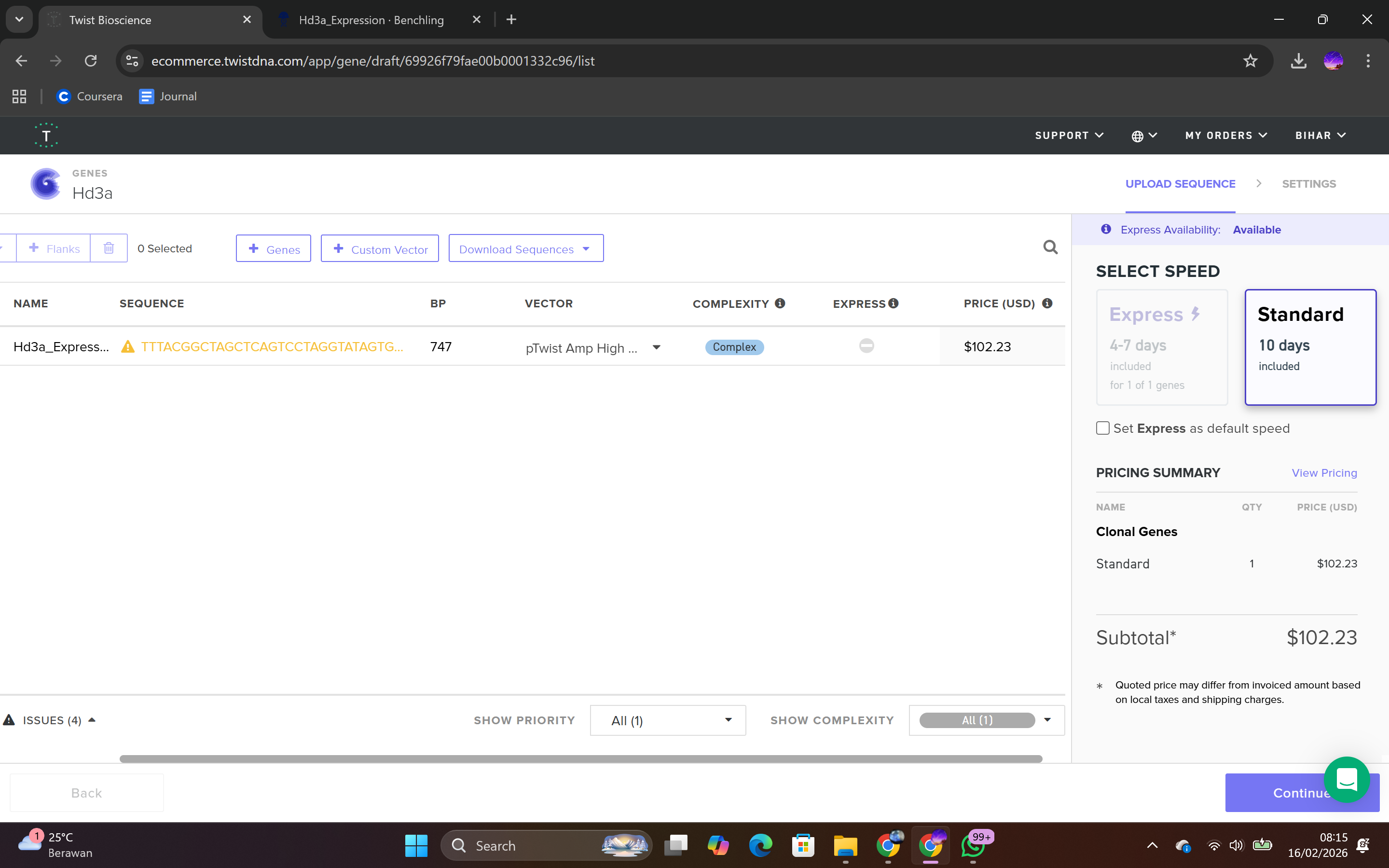
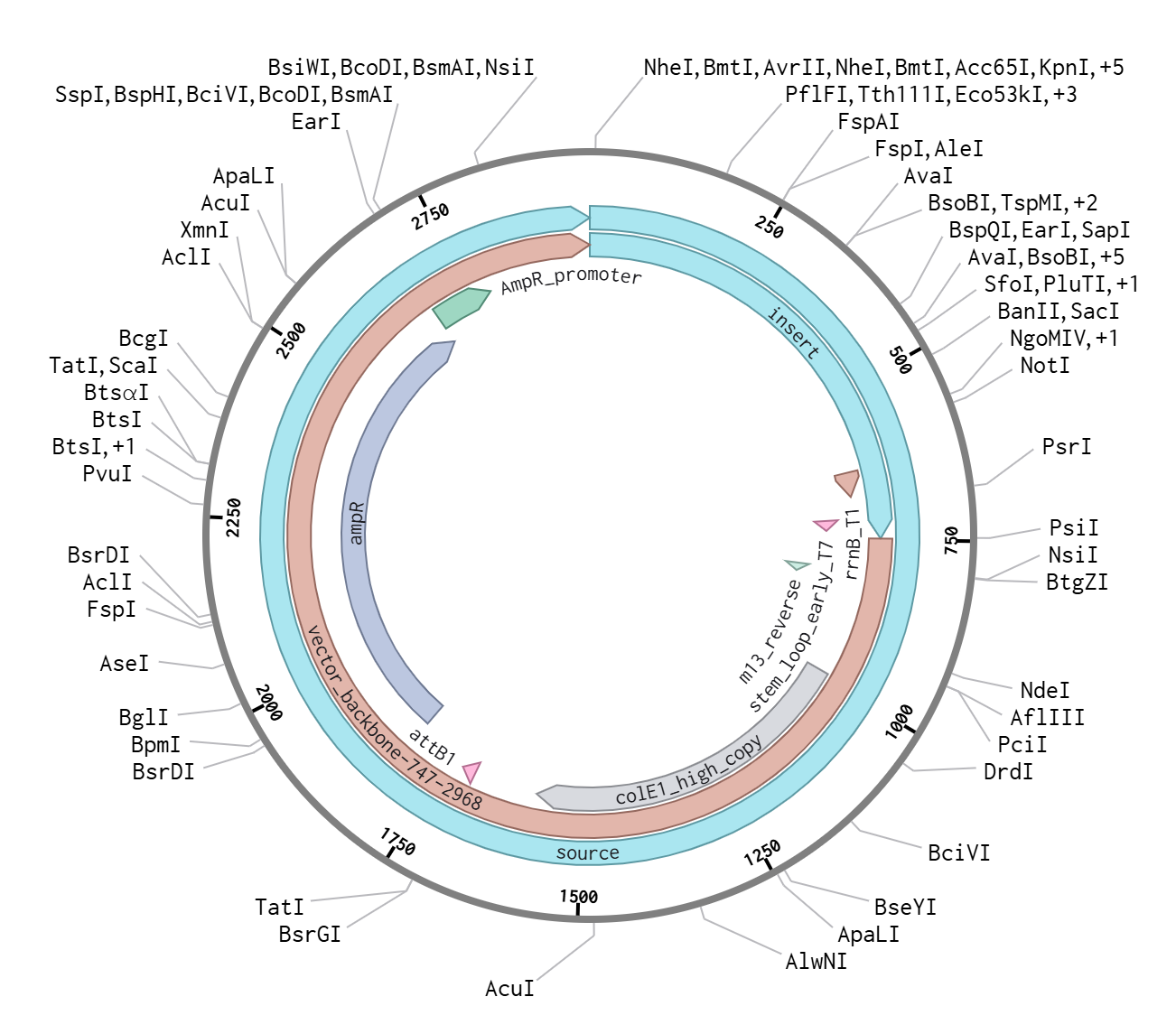
During the synthesis step, I selected the “Genes” category and chose the “Clonal Genes” option, which enables synthesis of the gene directly within a circular plasmid ready for transformation without additional cloning steps. The FASTA file was uploaded, and the plasmid backbone pTwist Amp High Copy was selected.
This backbone was chosen based on several biological considerations. It contains a high-copy-number origin of replication, allowing multiple plasmid copies per cell and thereby enhancing protein production in E. coli. It also carries an ampicillin resistance gene for selection, ensuring that only transformed cells survive under antibiotic pressure. The backbone provides the complete circular plasmid structure into which the Hd3a expression cassette is inserted.
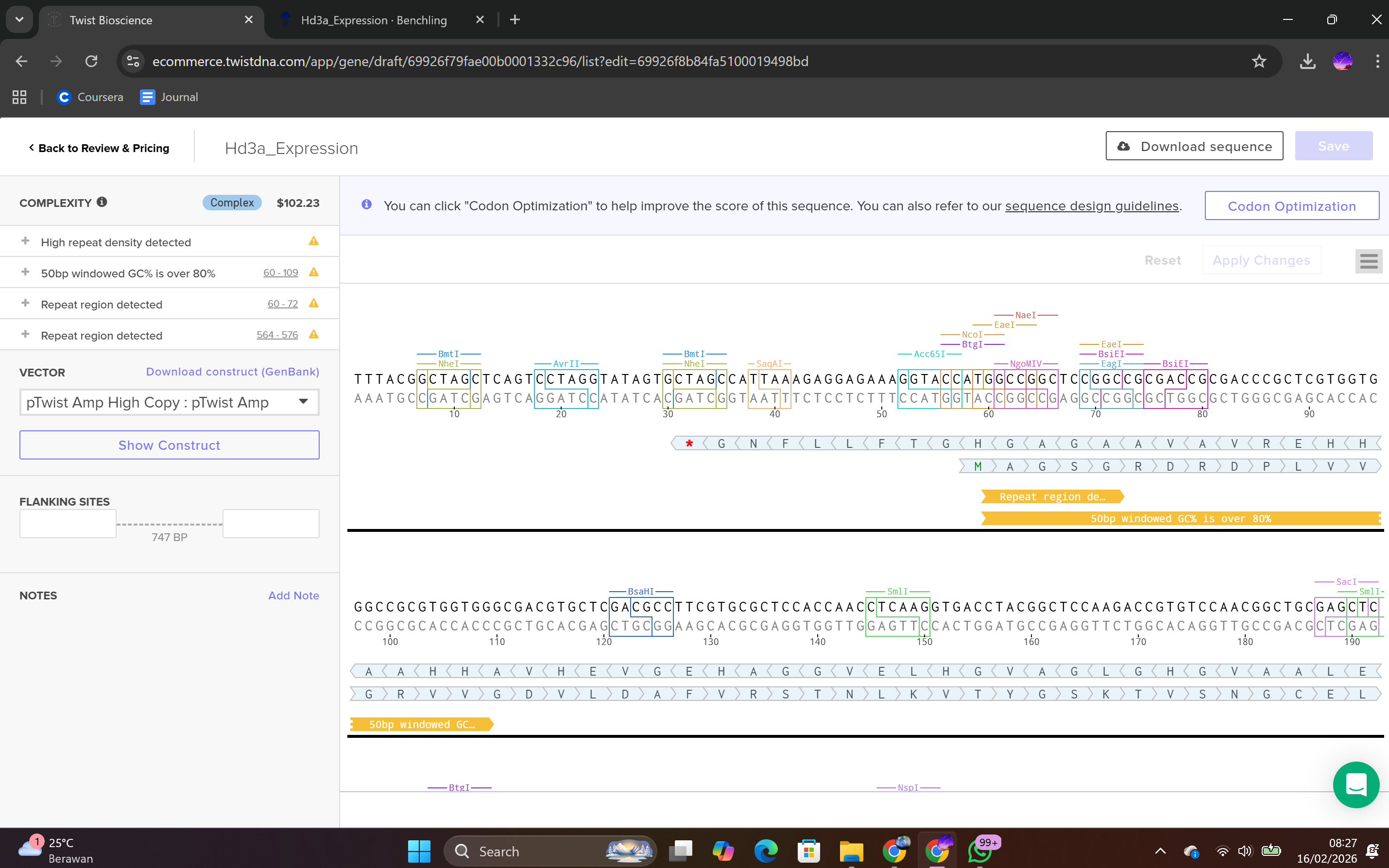
After selecting the vector backbone, the system provides a GenBank file containing the full plasmid sequence, including both the backbone and the Hd3a insert. I downloaded the complete construct as a GenBank file.
4.4 Verification of Final Plasmid
This file can be re-imported into Benchling to visualize the circular plasmid map, displaying the positions of the promoter, CDS, His-tag, terminator, origin of replication, and antibiotic resistance marker.
The process was halted at the vector selection stage, as final plasmid synthesis requires payment. However, theoretically, upon ordering and receiving the plasmid:
Transform the plasmid into competent E. coli cells.
Select transformants on ampicillin-containing agar plates.
Culture colonies.
Express Hd3a protein constitutively.
Purify protein using His-tag affinity purification.
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I would sequence genomic DNA and transcriptomic RNA (via cDNA) from rice varieties that exhibit variation in flowering time and growth rate, focusing on genes regulating developmental timing such as flowering-time regulators, photoperiod response genes, and growth hormone pathways (e.g., gibberellin signaling). The goal is to identify allelic variants, regulatory elements, and expression patterns associated with accelerated life cycles. By comparing fast- and normal-growing cultivars under different environmental conditions, we can pinpoint candidate loci for engineering rapid-cycle rice suited for climate stress and food security applications.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would focus on second-generation Illumina short-read sequencing. Illumina provides high accuracy and depth for detecting SNPs and quantifying gene expression (RNA-seq).
Is your method first-, second- or third-generation or other? How so?
Illumina is a second-generation sequencing technology because it relies on sequencing-by-synthesis with clonal amplification and parallel short-read generation.
What is your input? How do you prepare your input (e.g., fragmentation, adapter ligation, PCR)? List the essential steps.
The input is high-quality extracted rice genomic DNA. For Illumina, DNA is fragmented, adapters are ligated, fragments are amplified by PCR, and libraries are size-selected.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
In Illumina sequencing, fragmented DNA binds to a flow cell, undergoes bridge amplification to form clusters, and fluorescently labeled nucleotides are incorporated one base at a time; imaging detects fluorescence to determine each base (base calling).
What is the output of your chosen sequencing technology?
The output is digital sequence data in FASTQ format containing nucleotide sequences and quality scores. These reads can then be assembled into a reference-guided genome, used for variant calling, or analyzed for gene expression and regulatory differences associated with accelerated rice growth.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would synthesize a modular genetic construct to accelerate flowering and shorten the vegetative phase in rice. Specifically, I would design (1) a modified flowering activator gene such as an optimized Heading date 3a (Hd3a) coding sequence, (2) a tunable promoter responsive to environmental or chemical induction, and (3) a regulatory safeguard module. The construct would include a plant promoter, codon-optimized Hd3a CDS, terminator sequence, and optional CRISPR-based regulatory cassette targeting repressors of flowering. The purpose is to create a controllable rapid-cycle rice system that can reduce harvest time without permanently disrupting yield stability. The DNA synthesized would therefore be a modular plasmid insert containing promoter-gene-terminator architecture plus guide RNA cassette if CRISPR regulation is included.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use phosphoramidite-based chemical oligonucleotide synthesis combined with high-throughput gene assembly. This approach is currently the most scalable and accurate method for writing synthetic genes up to several kilobases. Short oligos (~150–200 nt) are synthesized chemically, then assembled enzymatically into full-length genes.
What are the essential steps of your chosen sequencing methods?
First, short oligonucleotides are synthesized using solid-phase phosphoramidite chemistry, where nucleotides are added sequentially with chemical protection/deprotection cycles. Second, oligos are cleaved, deprotected, and purified. Third, overlapping oligos are assembled into longer fragments via PCR-based assembly or Gibson Assembly. Fourth, the assembled construct is cloned into a plasmid vector and sequence-verified using NGS or Sanger sequencing to confirm accuracy.
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
Chemical synthesis accumulates errors as length increases, limiting direct synthesis to ~200 nt per oligo and requiring assembly for longer constructs. Error rates during synthesis and assembly necessitate sequence verification. While highly scalable and increasingly cost-effective, very large constructs (e.g., whole chromosomes) remain complex and time-consuming to assemble. Additionally, repetitive or high-GC regions can reduce synthesis efficiency.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would edit endogenous flowering-time regulatory genes in rice, particularly repressors or upstream regulators that delay the transition from vegetative to reproductive growth. The goal would be to fine-tune rather than eliminate gene function to shorten the life cycle while maintaining yield stability and stress resilience. For example, I would introduce targeted loss-of-function or promoter-weakening mutations in flowering repressors, or precise base edits in cis-regulatory regions to modulate expression levels.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-based genome editing, specifically CRISPR-Cas9, for targeted knockouts and CRISPR-based editors (e.g., cytosine or adenine base editors) for precise nucleotide substitutions. If fine transcriptional tuning is required, CRISPR interference or CRISPR activation systems could also be employed. These tools provide high specificity, programmability, and efficiency in plant genomes.
How does your technology of choice edit DNA? What are the essential steps?
CRISPR-Cas9 edits DNA by using a guide RNA to direct the Cas9 nuclease to a complementary genomic sequence adjacent to a PAM motif. Cas9 induces a double-strand break, which is repaired by the cell’s endogenous repair pathways, typically non-homologous end joining, generating small insertions or deletions, or homology-directed repair if a repair template is supplied. Base editors modify single nucleotides without generating double-strand breaks, improving precision.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
Preparation involves in silico design of guide RNAs targeting specific loci, off-target prediction analysis, cloning the gRNA into a plasmid expressing Cas9 or base editor machinery, and preparing plant transformation vectors. Inputs typically include the Cas9 or base editor protein, guide RNA construct, optional donor repair template, and rice cells or callus tissue for transformation via Agrobacterium-mediated delivery or particle bombardment.
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Limitations include variable editing efficiency depending on target locus and delivery method, off-target mutations, mosaicism in regenerated plants, and relatively low efficiency of HDR in plants. Additionally, complex traits like flowering time may involve polygenic networks, so editing a single locus may produce incomplete or context-dependent effects.
Week 3 HW: LAB Automation
Post-Lab Question 1
A study by the Opentrons research community demonstrated how low-cost laboratory automation platforms can accelerate biological experiments. The researchers used the Opentrons OT-2 robot to automate liquid handling tasks such as pipetting, reagent mixing, and plate preparation.
In the study, the robot was programmed using Python scripts to perform repetitive experimental steps with high precision. Automation enabled researchers to run multiple biological assays simultaneously while reducing human error.
The key innovation of this approach is that Opentrons is an open-source system. Scientists can modify the protocol, integrate custom labware, and automate workflows such as PCR setup, enzyme assays, and high-throughput screening. This significantly lowers the barrier to entry for laboratory automation and enables more scalable experimental design.
Post-Lab Question 2
Project Idea: Automated PCR Reaction Setup
For my final project, I plan to automate the preparation of PCR reactions for testing multiple DNA samples.
The workflow would involve:
The Opentrons robot distributing PCR master mix into all wells of a 96-well plate.
The robot adding different DNA templates into each well.
Mixing the reactions automatically.
Preparing the plate for thermocycling.
Automation would allow dozens of PCR reactions to be prepared quickly and reproducibly. This approach could be integrated with bioinformatics workflows where DNA sequences are designed computationally and then experimentally tested.
Week 4 HW: Protein Design part I
PART A — Conceptual Questions
How many molecules of amino acids are in 500 g of meat?
If we assume 500 g of protein: 500g÷100g/mol = 5 mol
Total of molecules: 5×6.022×10²³
≈ 3 × 10²⁴ amino acid molecules
Why do humans eat beef but do not become a cow, and eat fish but do not become fish?
Proteins from food are not absorbed as intact proteins. Instead, they are broken down during digestion:
Dietary proteins are digested into individual amino acids.
These amino acids enter the bloodstream.
Human cells use them as building blocks to synthesize human proteins according to human DNA instructions.
Why are there only 20 natural amino acids?
The genetic code evolved to encode 20 canonical amino acids through the translation machinery involving:
ribosomes
tRNA molecules
aminoacyl-tRNA synthetases
These 20 amino acids provide sufficient chemical diversity to build complex protein structures while maintaining translation accuracy and efficiency.
Can we make non-natural amino acids?
Yes. Scientists can design and synthesize non-natural amino acids.
Examples include:
Fluorinated amino acids –> These improve protein stability.
Where did amino acids come from before enzymes and life existed?
Several hypotheses explain the origin of amino acids:
Prebiotic chemistry
The Miller–Urey experiment demonstrated that amino acids can form from simple gases under conditions resembling early Earth.
Meteorites
Some meteorites contain organic molecules including amino acids.
If an α-helix were made from D-amino acids, what handedness would it have?
Natural proteins consist of L-amino acids, which form right-handed α-helices.
If the helix were made entirely from D-amino acids, the stereochemistry would reverse, producing a left-handed α-helix.
Why are most molecular helices right-handed?
Most biological helices are right-handed because proteins are composed almost exclusively of L-amino acids.
Why do β-sheets tend to aggregate?
β-sheets have a flat structure and strong backbone hydrogen bonding.
Because of this:
Multiple β-sheets can stack together
Intermolecular hydrogen bonds stabilize aggregates
Why do many amyloid diseases form β-sheets?
In diseases such as Alzheimer’s disease, proteins misfold into structures dominated by β-sheet stacking.
These structures form amyloid fibrils, which are highly stable and resistant to degradation, leading to toxic protein aggregates.
PART B — Protein Analysis and Visualization
Selected Protein
The selected protein is Green Fluorescent Protein (GFP) from the jellyfish Aequorea victoria.
This protein was chosen because:
It is widely used as a fluorescent reporter in molecular biology
Its structure is well characterized
It is commonly used in protein engineering studies
Amino Acid Sequence
GFP consists of 238 amino acids.
Frequently occurring residues include:
Glycine
Leucine
Serine
Glycine is common because it contributes flexibility to protein structures.
To obtain the amino acid sequence of the MS2 bacteriophage lysis protein that will serve as the reference sequence for downstream bioinformatic analyses.
Input
Protein sequence database entry of the MS2 phage lysis protein (FASTA format).
Output
The primary amino acid sequence of the MS2 lysis protein in FASTA format, which will be used as the query sequence for further analyses.
Homology Search
Tool used: BLAST
Objective
To identify homologous proteins from other bacteriophages or organisms in order to understand evolutionary relationships and detect conserved regions within the lysis protein family.
Input
Query amino acid sequence of the MS2 lysis protein (FASTA format).
Output
A list of homologous protein sequences with similarity scores, E-values, and alignment statistics.
Selected homologous sequences for further comparative analysis.
Multiple Sequence Alignment
Tool used: Clustal Omega
Objective
To compare multiple homologous protein sequences and identify conserved residues that may be functionally or structurally important for the lysis protein.
Input
A set of homologous protein sequences obtained from the BLAST search.
Output
Multiple sequence alignment showing conserved and variable residues.
Identification of highly conserved amino acid positions that may represent functional hotspots.
Protein Structure Prediction
Tool used: ESMFold
Objective
To predict the three-dimensional structure of the MS2 lysis protein, which provides structural context for understanding protein function and interaction sites.
Input
Amino acid sequence of the MS2 lysis protein in FASTA format.
Output
Predicted 3D protein structure in PDB format.
Confidence metrics indicating the reliability of the predicted structural regions.
Functional Site Prediction
Tool used: PeSTo
Objective
To identify residues within the protein structure that are likely involved in molecular interactions, such as membrane association or protein-protein interactions.
Input
Predicted 3D structure of the lysis protein in PDB format.
Output
Predicted interaction residues or functional sites within the protein structure.
Structural regions potentially involved in the lysis mechanism.
Structural Similarity Search
Tool used: FoldSeek
Objective
To identify proteins with similar three-dimensional structures, even if their sequences are not closely related, which may provide insights into structural conservation and potential functional analogs.
Input
Predicted protein structure (PDB format).
Output
A list of proteins with structurally similar folds.
Structural alignment statistics and similarity scores.
Sequence Optimization
Tool used: ProteinMPNN
Objective
To generate alternative amino acid sequences that are predicted to fold into the same backbone structure, potentially improving protein stability or functional properties.
Input
Protein backbone structure in PDB format.
Output
Optimized or alternative protein sequences predicted to maintain the same structural fold.
Candidate sequences for potential protein engineering or stability improvement.
Week 5 HW: Protein Design Part II
Part 1 — Generate Binders with PepMLM
PepMLM was used to generate four 12-amino-acid peptides conditioned on the A4V mutant SOD1 sequence. The generated peptides showed perplexity values ranging from approximately 5.8 to 7.0, suggesting moderate model confidence in their plausibility as binders. Lower perplexity indicates that the peptide sequence better fits the model’s learned representation of protein–peptide interactions. The known SOD1-binding peptide FLYRWLPSRRGG was included as a reference for downstream structural and functional comparison.
Part 2 — Evaluate Binders with AlphaFold3
AlphaFold3 modeling predicted moderate interaction confidence for the generated peptides, with ipTM values ranging from 0.55 to 0.68. Peptide P2 produced the highest ipTM score (0.68), slightly exceeding the known SOD1-binding peptide (0.65). Structural inspection suggested that several peptides localize near the N-terminal region where the A4V mutation occurs, while others bind the surface of the β-barrel or approach the dimer interface. These results indicate that PepMLM-generated peptides can achieve comparable structural binding confidence to the experimentally known binder.
Part 3 — Evaluate Peptides in PeptiVerse
PeptiVerse predictions revealed differences in therapeutic properties among the generated peptides. Peptide P2 showed the strongest predicted binding affinity, consistent with its high ipTM score from AlphaFold3. However, P4 demonstrated a more balanced profile, combining high predicted affinity with strong solubility and low hemolysis probability. Peptide P3 displayed lower solubility and a moderate hemolysis risk, which could limit therapeutic potential. Overall, peptides with higher structural confidence tended to show stronger predicted binding affinity, although favorable therapeutic properties were not always correlated with ipTM scores.
Part 4 — Optimized Peptides with moPPIt
Peptides generated with moPPIt differed from PepMLM peptides in that they were explicitly optimized for multiple objectives simultaneously, including binding affinity, motif targeting, solubility, and reduced hemolysis risk. As a result, moPPIt peptides tended to contain residue patterns consistent with known interaction motifs while maintaining physicochemical properties favorable for therapeutic development. Compared with the PepMLM peptides, which were generated through sequence-conditioned sampling, moPPIt designs appeared more targeted toward specific binding regions on SOD1, particularly near the A4V mutation site.
How to evaluate before clinical studies
Before advancing these peptides toward clinical studies, several additional evaluation steps would be necessary. First, structural validation using AlphaFold3 or molecular docking could confirm stable peptide–protein interfaces. Molecular dynamics simulations could then assess binding stability and conformational flexibility over time. In vitro experiments such as surface plasmon resonance or fluorescence binding assays would be required to measure actual binding affinity. Additionally, toxicity, protease stability, and aggregation assays would help determine whether the peptides are suitable therapeutic candidates.