Week 4 HW: Protein Design I
Week 4 Protein Design part I;
A. Conceptual questions answers
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
a Dalton is a unit of measure, equal to 1/12 of carbon-12 atom weight; approx. 1.66 x 10-27 Kg.
Type of meat is not specified, I like chicken so.. On average, 100g of chicken breast has 27g of protein; for 500g, that’s 135g protein;
1 gram =6.023 × 1023 atomic mass units => 135g are ~8.13 x 1025 Da;
Dividing the Daltons of protein in chicken by an a.a. weight, I should end up with roughly the number of molecules found in 500g of meat => ~8.12 x 1023 molecules.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Lmao. Animal based products, as well as plants’ are made up of fat (lipids), proteins, carbohydrates and DNA/RNA. Except from the genetic material, that just passes through the digestive system, each macromolecule from either category is essential for human health. Now, why doesn’t the DNA we ingest get translated and build up proteins to turn us into.. cows? Mainly because it gets denaturized by HCl from stomachs, and because only RNA can be read as instructions for proteins, and anyhow it is not enough to cause any problems. Even if some gets transcribed, a couple of proteins will not take over our body and transform it Ben10 style.
Honorable mention: Viruses; They can “manipulate” host genetic material to make up the few proteins needed for virus “replication”; even in this scenarios, humans don’t turn into viruses, just end up replicating them.
3. Why are there only 20 natural amino acids?
Firstly, because they’re enough- they have enough structural diversity to make up any needed complex protein (and provide balance between hydrophobic and hydrophilic; acid and alkaline; etc.). Secondly, the 20 essential a.a. have a low energy cost (metabolically, they do not require much energy to be synthesized in-body), and are chemically stable, unlike other a.a. existent in nature. Furthermore, scientific speculation also impacts answer- “Frozen accident” theory: early in evolution, this set was likely chosen, and the machinery became so specialized that changing it to include new amino acids would be disadvantageous.
4. Can you make other non-natural amino acids? Design some new amino acids.
Non-natural a.a. typically refer to non-proteinogenic a.a., they are still found in nature, as they can form spontaneously.
Chemically, an amino acid is an organic compound made up of an alpha carbon (central) bonded with a carboxylic group -COOH, an amino group -NH2, a hydrogen, and a variable chain -R, that determines what the a.a. is. So, practically, it is possible to make up “new” a.a.
So, for my proposed a.a. will contain a naphthalene group including a nitrogen atom, and 1, double bonded, new oxygen atom, along with other carbon-carbon chains:
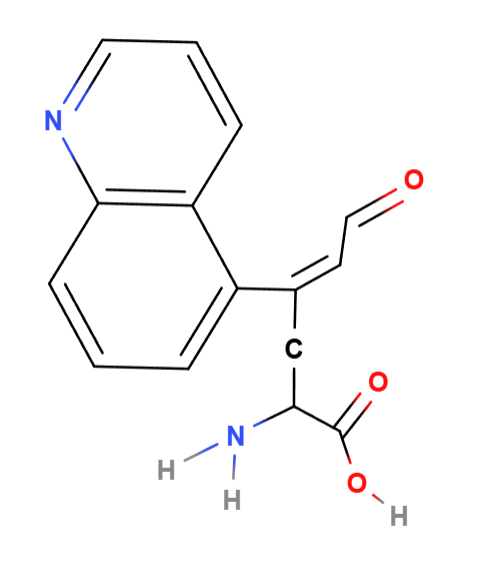
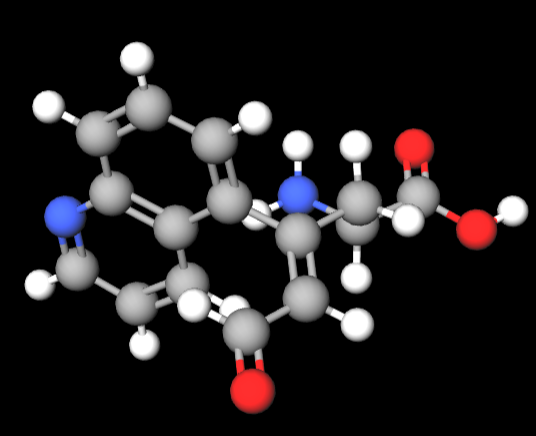
^ I used MolView.
5. Where did amino acids come from before enzymes that make them, and before life started? Chronologically, as in the Archean Eon, bacteria and archaea prevailed. Until then, bio-synthetical pathways for singular amino acids must have been already established;
It is believed life emerged from inorganic matter - Abiogenesis; So, that’s probably how amino acids even came to be, “before life started”. Others theories build upon extraterrestrial materials, as well as a.a., that could have come from a meteorite.
Personally, I resonate most with the Abiogenesis theory. And so, will advocate for it in present assignment.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
α-helices are usually right-handed, as their dense, tightly packed structure has a.a.’s R “poking out”- they’re made out of L-amino acids. So by using D-amino acids, and not having the structure fall apart, the whole stereochemistry ought to be reversed, thus resulting in a left-handed α-helix.
7. Can you discover additional helices in proteins?
Yes, besides α-helices, other helical structures are classified as protein structures; examples include π-helix and 3↓10-helix. It is also possible to design and synthesize foldamers with helical shapes that do not exist in nature.
8. Why are most molecular helices right-handed? Because almost all amino acids found in living organisms are left-handed. D-amino acids are scarce, and may not even serve building proteins as their purpose.
9. Why do β-sheets tend to aggregate?
β-sheets tend to aggregate because of intermolecular forces acting upon atoms from structural groups in a.a. (such as hydrogen bonds and hydrophobic interactions). This can lead to stacking of multiple sheets together- causing issues, specifically amyloidogenic as they lack structural integrity on their edges; thus forming β-sheets with neighboring strands.
• What is the driving force for β-sheet aggregation?
Non-covalent intermolecular forces, mostly non-polar hydrophobic ones.
10. Why do many amyloid diseases form β-sheets?
Because, thermodynamically speaking, β-sheets are more inert, and exhibit less energy needed- they’re more stable. In turn, making up cross- β structures. Hydrophobic forces cause parallel alignments, creating hydrogen bonds that are stable and protease-resistant.
• Can you use amyloid β-sheets as materials?
I figure that their increased stability and mechanical strength may facilitate amyloid β-sheets usage as building blocks for various materials. Speculatively speaking, they may be useful for in-human structures, but I think a protein plastic would be cool: poly amyloid β-sheets.
11. Design a β-sheet motif that forms a well-ordered structure
For a motif to be well-ordered it needs to be in the lowest energy state possible and be chemically and structurally stable (achievable by using the right amino acids); and that is usually the case for most proteins found in life organisms- depending still on the medium. Simply using β-sheets I could base the design on β hairpin model (3 antiparallel β-sheets), a Greek key motif with 4 or a β-barrel; moreover, I need to assure using the optimum a.a. interactions and proprieties (hydrophilic/phobic to let the protein “package itself” better and the polar residues outside to avoid internal protein disturbance). Also, protein motifs almost always depend on the rest of the protein, designing just the motif will be hard.
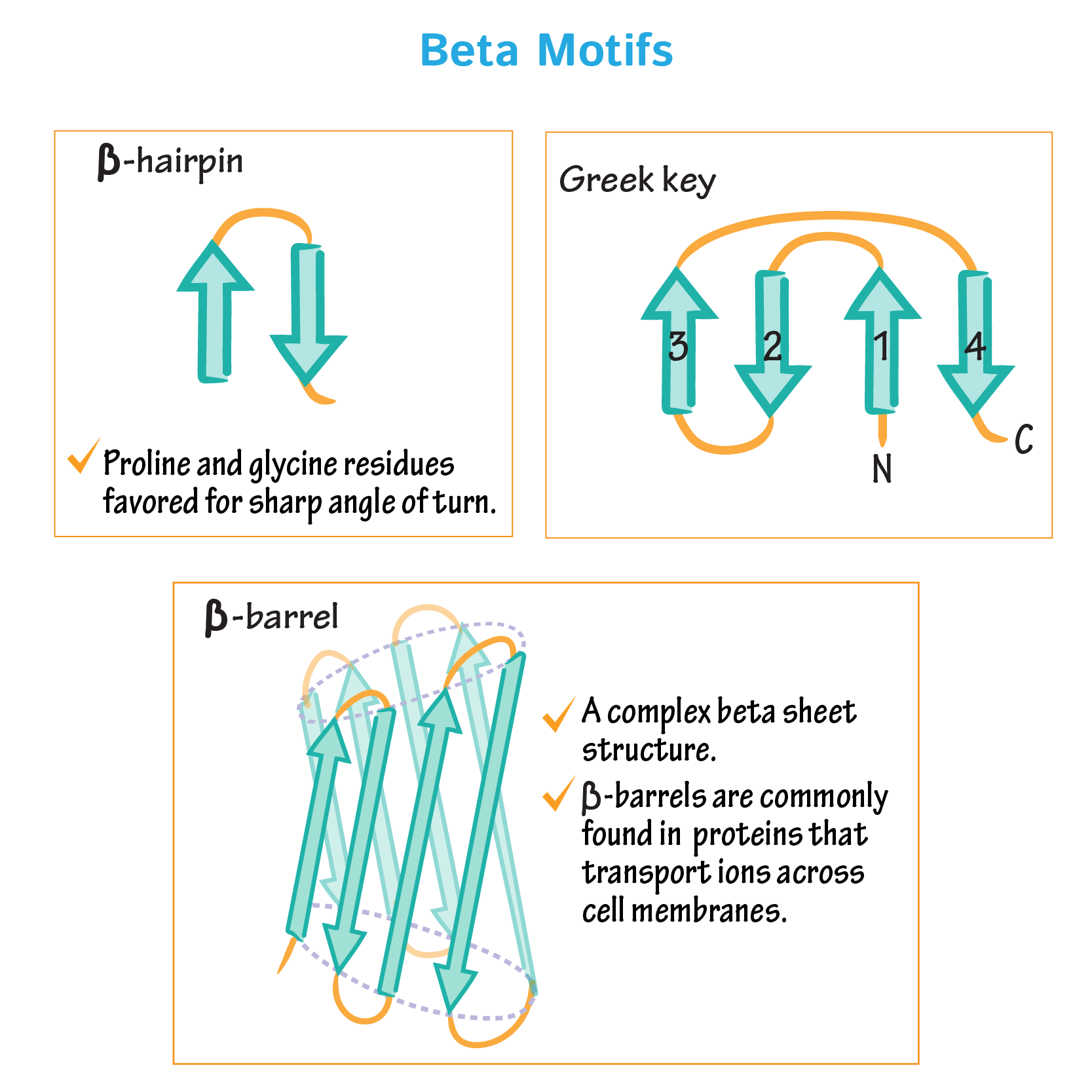
I based my design on GB1 domain Protein G
2 antiparallel β-sheets: MTYKLILN(GKTL)KGETTTEA
After changing some a.a. up for improved forces (C-C: disulfide bridges, ionic bonds, hydrophobic bonds) and reduced flexibility (a.a. -> P) and cutting some off, I ended up with a stable enough looking (I hope) motif:
MTPKLIKNGKTLKCETTT

B. Protein Analysis and Visualization
- Briefly describe the protein you selected and why you selected it.
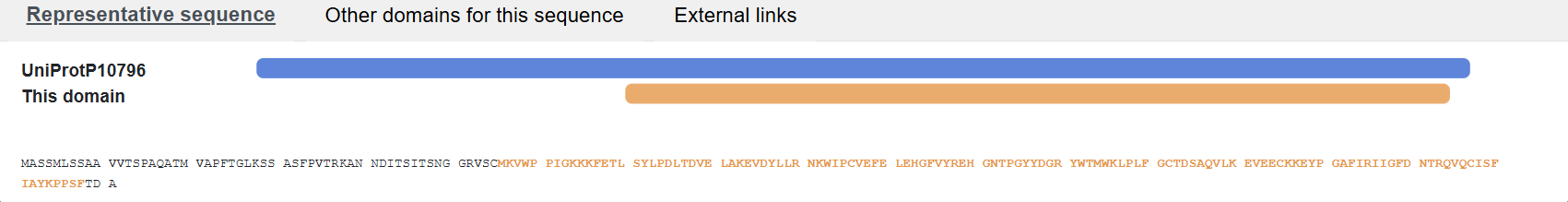
RuBisCO- Vegetal protein responsible for Carbon molecule morphism in Calvin cycle, fixing CO2 into PGA, prime step in making C6H12O6; I have chosen it because the other one didn’t work. Amino acid sequence used belongs to RuBisCO small subunit 1B (RBCS-1B);
sp|P10796|RBS1B_ARATH Ribulose bisphosphate carboxylase small subunit 1B, chloroplastic OS=Arabidopsis thaliana OX=3702 GN=RBCS-1B PE=1 SV=1
^ work protein isolated from Arabidopsis thaliana plant ( O03042 ).
- Amino acid sequence:
MASSMLSSAAVVTSPAQATMVAPFTGLKSSASFPVTRKANNDITSITSNGGRVSCMKVWPPIGKKKFETLSYLPDLTDVELAKEVDYLLRNKWIPCVEFELEHGFVYREHGNTPGYYDGRYWTMWKLPLFGCTDSAQVLKEVEECKKEYPGAFIRIIGFDNTRQVQCISFIAYKPPSFTDA
- using Amino acid- colab counter.:
- using UniProt’s BLAST tool:
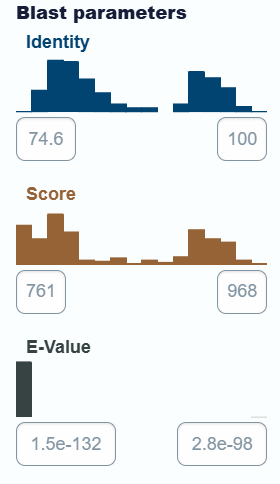
• Protein belongs to “Ribulose-bisphosphate carboxylase family”.
- using RCSB:
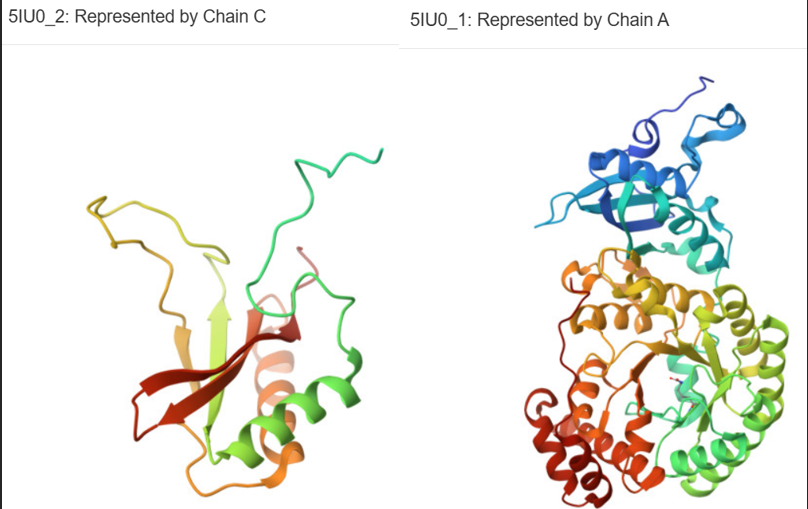
- Ribulose bisphosphate carboxylase small chain 1B, chloroplastic- correspondent to provided a.a. seq.
- Ribulose bisphosphate carboxylase large chain. Whole RuBisCO hexadecamer protein conists of 2 small (C, D | 181 a.a.) and 2 large subunits (A, B | 479 a.a.).
• Deposited: 2016-03-17; • Resolution: 1.50 Å (smaller than 2.70 => good quality).
• Molecules apart from protein in solved structure:

- using SCOP 2, I identified protein belogining to structure classification family:
Protein visualisation using Pymol software:
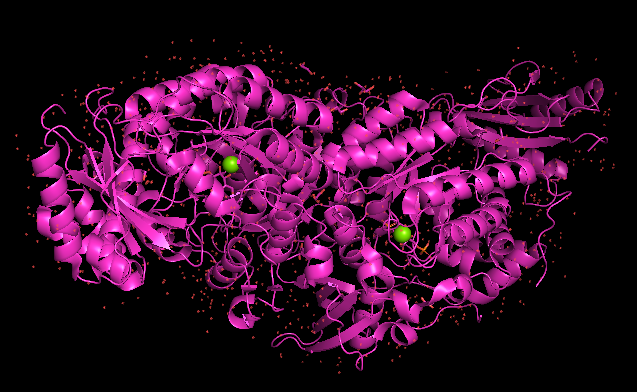
pdb_00005IU0 (whole complex)
CARTOON
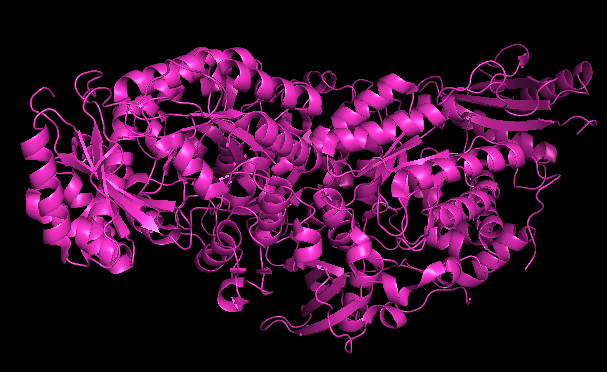
RIBBON

BALL AND STICK

COLORATION BY SECONDARY STRUCTURE
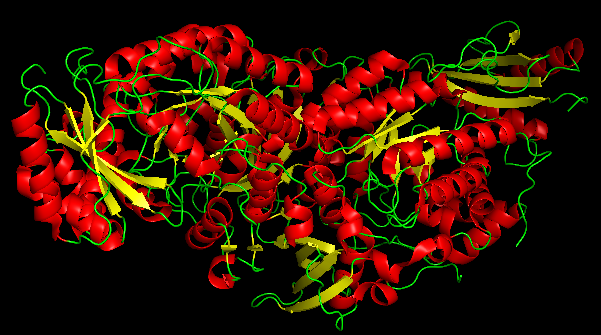
- It has more helices
PROTEIN SURFACE
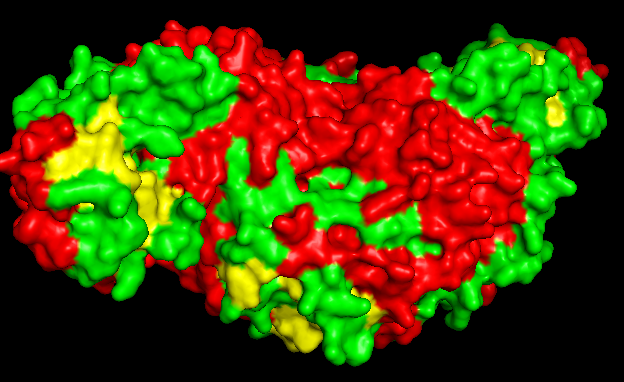 -it does have binding spots;
-it does have binding spots;
C. Using ML-Based Protein Design Tools
-using google colab on RuBisCO;
C1. Protein Language Modeling
• ESM2 link
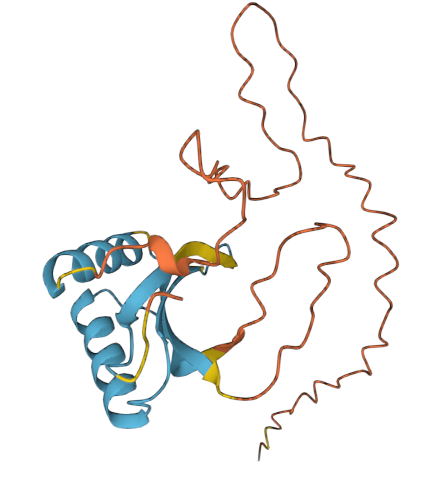
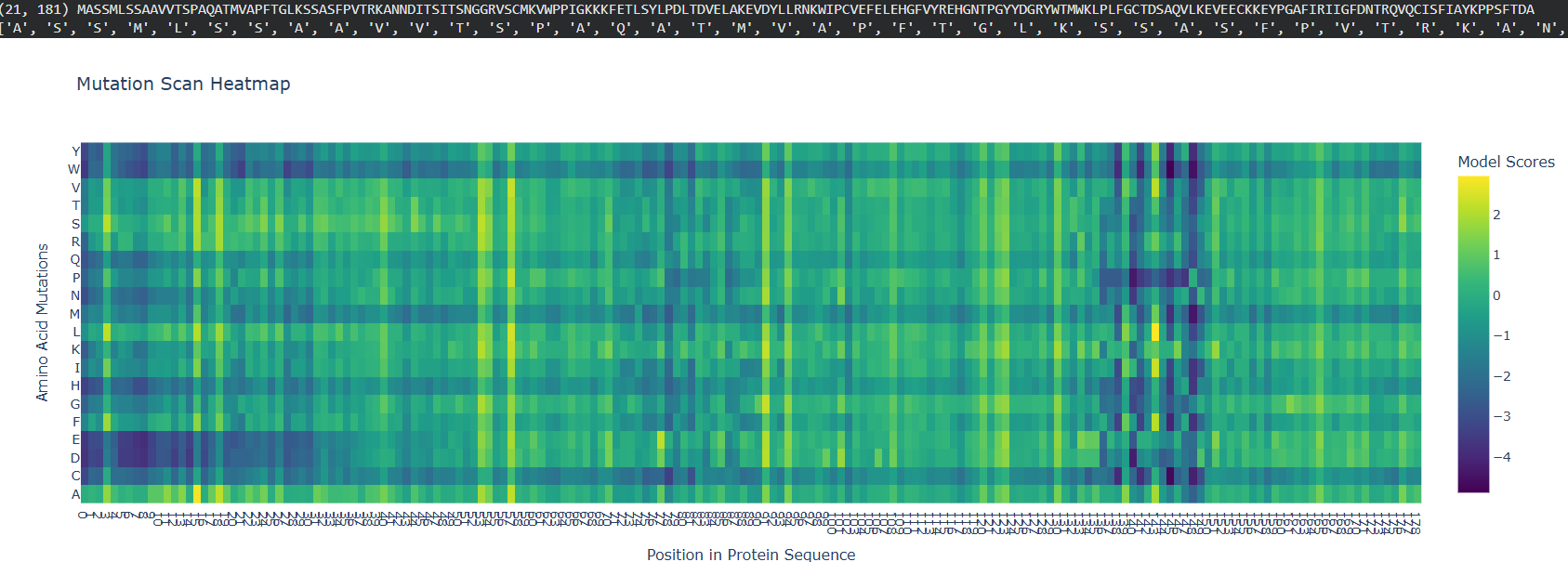
The initial segment of the protein shows high mutational tolerance, visible as a large dark blue block. After the transit peptide, map transitions into a region of high sensitivity- yellow and bright green.
Proline (P) and Glycine (G) amino acids often contain more “bright” spots. These are “helix breakers”; adding them to a stable helix typically disrupts the fold, leading to a high sensitivity score.
Bibliography
PyMol usage
COMMANDS:
bg_color [color] (e.g. I used: black/white);
fetch [pdb_id] (e.g. I used: 00005IU0);
show cartoon, spheres, sticks, surface, lines, ribbon;
as cartoon, spheres, etc -changes entire selection to chosen aspect, hides other styles;
color [color], resn [a.a.+a.a.+..] (e.g. color red, resn ASP+GLU for Acidic/Negative a.a.);
set [element_character], [measure] (e.g. set sphere_scale, 0.25, set stick_radius, 0.2);
dist [name], [selection1], [selection2] -measures distance between two atoms or groups;
png [filename.png] -saves current screen view;
INTERFACE:
- A (Actions): Major operations like renaming, deleting, or applying complex presets (e.g., “publication” mode).
- S (Show): Adds representations like cartoon, sticks, or surface.
- H (Hide): Removes specific representations (the opposite of Show).
- L (Label): Adds text labels for residues, atoms, or distances.
- C (Color): Changes color of the object using predefined palettes or residue-specific schemes]