Class Assignment — DUE BY START OF FEB 10 LECTURE Question 01 First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about. According to the well-regarded popular science writer Matthew Cobb (2022), since the Asilomar conference in 1975, molecular biologists have been the vanguard in self-regulating when playing God. This means we refrain from conducting our research irresponsibly by deploying unnecessarily hazardous experimental methods. Alas, this also means that some of the most exciting genetic engineering is no longer done. Consider Dr. Oswald Avery’s transforming principle experiment. Blindly take a population of virulent pneumonia bacteria and feed them harmless kin until they lose their aggressive function and magically adapt into weak and indifferent pneumonia. Since Asilomar, this is indeed one kind of experiment that trustworthy principal investigators must abstain from. I get it, and still I contemplate. Wasn’t Avery the best of us, though? Between Schrodinger and Watson, Crick, and Franklin – Dr. Avery intuited DNA into existence with his transforming principle and used it effectively. Surely I didn’t name my oldest son after this man for nothing?
Table of contents Software used: Terminal, git, xcode, hugo, benchling, rcdonovan website, twist website. Objective: This week explores the read–write–edit toolkit: sequencing and synthesis workflows, restriction digests and gel electrophoresis, and early genome-editing frameworks.
Homework for HTGAA 2026 (Week 03): Lab Automation Table of contents Software used: Terminal, git, Opentrons, rcdonovan website, Google Colab. Objective: This week we get hands-on (or at least code-on) with pipetting robots.
Homework: Protein Design I Assignment Objective: Learn basic concepts: amino acid structure, 3D protein visualization, and the variety of ML-based design tools. Brainstorm as a group how to apply these tools to engineer a better bacteriophage (setting the stage for the final project).
[] Homework — DUE BY START OF MAR 10 LECTURE Part A: SOD1 Binder Peptide Design (From Pranam) Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc. Mechanis
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
[]Homework — DUE BY START OF MAR 17 LECTURE
Week 6 HW: Genetic Circuits Part 01 Assignment: DNA Assembly Protocol and Study Questions What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? A proprietary gold standard heat-stable DNA polymerase alternative to Taq reagent synthesized and sold by Thermo Fisher Scientific. Unlike Taq which was isolated from thermophilic bacteria, Phusion emulates an archaea-based enzyme that evolved in the hydrothermal vents from extremeophile species. They function as DNA polymerases essentially in a form biomimickry with minimal replication error. The purpose of Phusion is to amplify target DNA sequences in the PCR protocol. Phusion PCR is more expensive but worth the investment to increase the accuracy of the run.
[]Homework — DUE BY START OF MAR 31 LECTURE at 2PM ET
Week 7 HW: Genetic Circuits Part 2 Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? IANNs are ideal for the continuous transcriptomic-driven change observed in cells that are constantly moving and communicating in their intracellular environment – through analog computations. In contrast, much of the early synbio genetic circuit engineering was digital, with discrete logic gate switch programming or perhaps even through gene knock out (present versus absent) if such a connection would be permitted.
[] Homework — DUE BY START OF Apr 7 LECTURE
Homework Part A: General and Lecturer-Specific Questions General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Describe the main components of a cell-free expression system and explain the role of each component. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment. Let it be noted, I really want to use George AI to engage this question, but I am running out of time, so I am just pitching shots up on the green to try and finish off with a putter and I also recognize metaphors are symptomatic of a weak mind, so there you have it. This is why we need AI too. Most people are susceptible to weak-minded syndrome. Therefore, since my overall understanding of what I am describing here is dulled, please do not try any of this at home. The bioenergetic cycles we wish to reconstruct in the cell-free environment literally resemble a water mill. I start with these two examples because one makes sense to me, and the other appears to be the same engineering concept but now there are some critical features missing that makes it more difficult to reconstruct the working order of things. I believe the same problem challenges us with cell-free systems. We have all the parts and experiments are clearly designed but what sustains them? There are clearly hidden variables that cannot be intuited at first glance. Now I am afraid if just put up my next image it will be swept away do to copyright infringement laws since I didn’t personally take a picture of this biochemical pathway. The irony of course is that the water mill and whatever mill above were invented when Newton was still alive if not before, likely long before, anyway the cellular aerobic respiration cycle was discovered inside of living organisms on Earth. In addition, we should note that the cellular cycle is part of many interacting open systems, and the other two mills are closed systems embedded in open living systems. Infact, is there anything sadder than a watermill without water for there in the bones of brick and iron is a functionless relic of a time before atomic energy had been harnessed. Perhaps a time we will return to in the end, but enough conjecture.
Week 10 HW: Imaging and Measurement Homework: Final Project Homework is partly based on data that will be generated in the Waters Immerse Lab in Cambridge, MA. Students will characterize green fluorescent protein (eGFP, a recombinant protein standard) structure (primary, secondary/tertiary) in the lab using liquid chromatography and mass spectrometry, as well as Keyhole Limpet Hemocyanin (KLH) oligomeric states using charge detection mass spectrometry (CDMS). Data generated in the lab needed to do the homework is included both within this document and in the Appendix of the laboratory protocol. Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements. There are many measurements that could be taken. The DNA of the mites and their larva is a target. In addition the bacteria and other microbiota and parasites that capitalize on the infestation damage to the host epidermis. This ofcourse brings up the geometry of the host tissue and biochemical molecules all of which can be measured quantiatively or qualitatively. Specifically, when it comes to host cells there are living and deceased keratinocytes and corneocytes and the odd hair follicle, especially in most heavily infested cases. There is superinfection residues full of bacterial cocci and rods or periods and semi colons as Dr. Betsy Dyer writes. There crushed and desciated erythrocytes, platelets, and leukocytes. In addition there will be scattering fluctuations of neturophils, eosinophils, macrophages, and lymphocytes. In addition, goats are always on the move grazing when they’re not cuddled up in hay or dirt. Therefore there will be pollen grains, plant frags, seed husks and hay chaff, and plant hairs in addition to other ecotoparasites. What I want to measure most though is stress.
Homework — DUE BY START OF APR 28 LECTURE
Week 11 HW Overview Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork [!info] Note that this homework is due a week later than it ordinarily would due to its release a week later than normal.
[x] Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
Week 14 HW Overview Assignment Part 1: Work on individual final project.
Reading & Resources
Subsections of Homework
Week 1 HW: Principles and Practices
Class Assignment — DUE BY START OF FEB 10 LECTURE
Question 01
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
According to the well-regarded popular science writer Matthew Cobb (2022), since the Asilomar conference in 1975, molecular biologists have been the vanguard in self-regulating when playing God. This means we refrain from conducting our research irresponsibly by deploying unnecessarily hazardous experimental methods. Alas, this also means that some of the most exciting genetic engineering is no longer done. Consider Dr. Oswald Avery’s transforming principle experiment. Blindly take a population of virulent pneumonia bacteria and feed them harmless kin until they lose their aggressive function and magically adapt into weak and indifferent pneumonia. Since Asilomar, this is indeed one kind of experiment that trustworthy principal investigators must abstain from. I get it, and still I contemplate. Wasn’t Avery the best of us, though? Between Schrodinger and Watson, Crick, and Franklin – Dr. Avery intuited DNA into existence with his transforming principle and used it effectively. Surely I didn’t name my oldest son after this man for nothing?
Unlike Dr. Avery, I am fortunate to be proposing my HTGAA 2026 project after the discovery of DNA and the Asilomar conference and the Once-in-a-Century Pandemic when mRNA Vaccines and CRISPR gene editing approaches were available. However, like Avery, with molecular biology, we can still take a population perspective to current wicked health problems. My first professional mentor, Dr. Paul Farmer, would often credit his milieu caring for the poorest of the poor as the center of his mission. Though I once aspired to that also, I now reflect that I am fortunate to just be an aspiring Molecular Biologist. In addition, when I marvel over everything that has been achieved since Avery, especially since the Human Genome Project and the advances of systems biology to synthetic biology, I see there are now viable alternatives in biological practice to help others and the living world.
This brings me to the project. I agree with Dr. Aubrey De Grey that biological aging is a vexing, immutable inequality in public health that must be solved. In fact, I am engaged in this research with my excellent Biology PHD mentors at North Carolina Agricultural and Technical State University (NCATSU), and one of them was on the team that first postponed senescence in Drosophila back when Star Wars movies were worthy of the hype.
Like Dr. De Grey, I believe exit velocity will be achieved in our lifetimes by engineering negligible senescence. The difference is that his model species are cohorts of robust, rejuvenated rodents centralized in a single laboratory, and I propose we develop many sites and open science approaches using goats instead. I also think that we will need to develop applied computational systems biology simulators (synthetic biology simulators too if they exist) and at the center of the approach needs to be the host-microbiome.
Why goats? They’re not even a monogastric species. Please hear me out.
According to ChatGPT, the oldest recorded goat in the Guinness World Records is McGinty (22 years, 5 months). The buck was a Brition, he was male, and from a Pygmy breed. My understanding is that Pygmy goats were originally bred to feed large cats. This record was set in 2003 and I assume it hasn’t been challenged since. Although I never had the pleasure of meeting McGinty, the general indifference evident by his nefarious name and the dusting of a few social media posts and overall absence of life-history information makes plain that likely society gave up on even understanding goat longevity decades ago. This means that despite living among Homo sapiens for more than 10,000 years and sustaining us in every challenging environment on Earth, we still know more about goats’ genomic diversity than life history. That’s not a bad thing, though, because goats’ genomes and immune systems are as infinitely fascinating as our own.
In addition to not being popular, goats live a life preoccupied by parasites, predators, and food insecurity that is only moderately improved by domestication, let’s be honest. I often reflect on the goats I met in the Galapagos Islands – the first example of extreme biological environments. Goats are not indigenous to the Galapagos. They are migrants. They didn’t migrate there on their own volition, though – instead they brought in rafts and boats a Century ago, and still to this few re-wilded stragglers refuse to go extinct. In fact it’s hard to find an island in the Galapagos that doesn’t have a pile of goat skulls on it. I understand the issue is complicated but either way you land on the issue, it’s hard to deny that goats are specialists in acclimating in extreme environments. Ironically, it’s Charles Darwin’s theory of Natural Selection that I would like to structure the computational systems biology goat longevity simulator around, particularly using Neo-Darwinian genetics and postponement of senescence work by Rose, Muller, Luckinbill, and Graves.
I propose a Long Term Experimental Evolution (LTEE) study that leverages synthetic biology and local animal husbandry to study the role of gut microbiomes on cellular senescence in goats. I hypothesize that understanding diversity and abundance in genetic circuitry constituting biological signaling pathways between adaptive, senescence-resistant microbes and Metazoan somatic tissues will yield the putative attractor switches we need to cure cellular senescence and put apoptosis on a toggle switch. Theoretically, though I certainly don’t plan to achieve this in 10 weeks or morally at all. The point is that once you understand that one contingent evolutionary endosymbiotic event transformed an alpha-proteobacteria into the power center for every Metazoan cell that came after, and then the effects of the mitochondria on oxidative stress accumulation and stabilization. Inevitably, we can trust that the solution to aging in somatic cells will never again be an if question.
Endpoints I will be investigating are biologically and statistically significant variation in “aging” host and microbe genes identified through differential gene expression. The study will be a multigenerational LTEE for Synthetic Biology 101, targeting the bidirectional interactions between living goat genes and pathways and the microbiota in their gut. My stakeholders are the American Milk Goat Breeding Association and Nanopore, and every isolated mountain village or homesteader that is still alive because of their goat herd.
Question 2
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
GPG01: Explore synthetic biology for goat life history for a putative Mitocarta or SASP gene and phenotypic pathways that may be useful in future studies to bioengineer negligible senescence in goats.
GPG02: Integrate aim 1 gene with OMICs data using computational model to explore molecule mediated bidirectional interactions between somatic host cells and microbes in goat microbiomes.
GPG03: Consider systems-level synthetic biology interventions for extreme environments that support goat metabolism and gut microbiome health.
Question 3
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design,
Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical
strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
Purpose: What is done now and what changes are you proposing?
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
CN Answer
Purpose: Humans have been artifically selecting phenotypes in goats to consume for our benefit for more than 10,000 years. I understand goat meat, milk, and fiber are necessary for human differential reproductive success and maintenance. I actualy have a working goat farm. My purpose is not adjudicate my species, or rescue another, my intention is to use this incredible opportunity to make ammends to another population of Metazoans by helping my peers use synthetic biology to help goats live longer, higher quality lives. I do not pursue this idea to make a more profitable goat commodity either. In summation my reasoning at HW 1 is: because I know postponing senescence in Metazoans is possible and I care about the welfare of all goats, I want to help others and myself advance negligible senesence in goat somatic cells safely and humanly too. The changes I am proposing for my HTGAA 2026 project though are only to expand fair, accurate, timely, accessible open science data about goat life history and genetics, so through synthetic biology we can help goats live longer, healthier lives.
Design: Based on what I know about Synthetic Biology today, which is far less than I care to admit without embarassment on a public website. What is needed to make it “work” is Dr. Aubrey De Grey brillance, vision, ability and a sincere heart for animal welfare. Eventually a sustainable research enterprise plan will be useful to achieve endpoints, quality benchmarks, and safety standards. Let me make a clear point first though, all I am proposing at this juncture is cast out net for data, reel it in and evaluate what I find. This review will require oversite from experts – can anyone put me in contact with Dr. George Church or Dr. Aubrey De Grey?
Assumptions: I love this question because I am a scientist and I think no other discipline is more pragmatic than us when it comes to how we manage uncertainty. This is the crux about assumptions. Uncertainty is dangerous. Case and point, because I care about goat welfare and recognize I do not understand enough about Synthetic Biology interventions to expect what I don’t understand about goat life-history, physiology, and genetics I would never do anything to disrupt in vivo what I am learning to explain and make predictions about – that being the bioengineering of negligible senesence in goats.
Risks of Failure & “Success”: Here risk communication and management are key. I was an Epidemiologist for 20 years before going back to school. In Epidemiology although all Pandemics are orphans, a breech in prevention is always the root cause. I say this to explain why I am so proactive about preventing failure, especially when it comes to public health. Another example, is part of my PhD training was working in a Molecular Microbiology lab on a LTEE for NASA. Here a significant portion of the job is monitor and improve protocols and practice to minimize contamination, especially on a 100 day LTEE study.
Stakeholders: Registry of Standard Biological Parts (RSBP), SAB Biotherapeutics (SABBio), World Health Organization (WHO), Rocky Hill Farm in WV (RHFWV)
Explore synthetic biology for goat life history for a putative MitoCarta or SASP gene and phenotypic pathways that may be useful in future studies to bioengineer negligible senescence in goats.
• By reducing uncertainty about the life history of goats.
♞
♞
♛
• By reducing uncertainty about synthetic biology interventions for negligible senescence in goats.
♞
Integrate aim 1 gene with OMICs data using computational model to explore molecule mediated bidirectional interactions between somatic host cells and microbes in goat microbiomes.
• By mapping major biological signaling pathways where communication goes from goat somatic cell to -> GIT microbiome
♞
• By mapping major biological signaling pathways where communication goes from GIT microbe in GIT microbiome to goat somatic cell or system
♞
Consider systems-level synthetic biology interventions for extreme environments that support goat metabolism and gut microbiome health.
• By cataloging goat metabolites and microbiota and their interactions
♟
♟
♟
• By modeling seed to goat food webs for diverse local environments.
♟
♟
♟
• By writing an aspirational study protocol.
♟
♟
♟
Other considerations
• Minimizing costs and burdens to stakeholders
♟
♟
♛
• Feasibility?
♟
♟
♟
• Not impede research
♛
♛
♛
• Promote constructive applications
♛
♛
♛
Assignment (Week 2 Lecture Prep) — DUE BY START OF FEB 10 LECTURE
In preparation for Week 2’s lecture on “DNA Read, Write, and Edit," please review these materials:
Lecture 2 slides.
The associated papers that are referenced in those slides.
In addition, answer these questions in each faculty member’s section:
Homework Questions from Professor Jacobson:
Question 1
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Based on the deck the error rate of polymerase is 1:10^6 or one error for every 1,000,000 base pairs. The size of the human genome according to the Molecular Biology of a Gene by Watson et al. (2007) the human genome is 3200 Mega base pairs in length which converts to 3,200,000,000 base pairs. Biology deals with the discrepancy through redundancy and replication forks moving from many different insertion sites at the same time. This way the redundancy offsets the discrepancy in the error rate. However errors still occur.
Question 2
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
There are two ways that I am aware of, Mass Spec and Edman Degredation. Both of these techniques identify amino residues that are synthesized as triple codons for a varity of lengths and structures. The total number of probable combinations is 3 codons multipled by 20 possible amino acids.
Homework Questions from Dr. LeProust: [Lecture 2 slides]
Question 1
What’s the most commonly used method for oligo synthesis currently?
Amplicon-Based Assays
Question 2
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Turn-around time on results due to added complexity from higher Chimera rate, drop out rate, and uniformity constraints above 100nt
Question 3
Why can’t you make a 2000bp gene via direct oligo synthesis?
I couldn’t find an exact answer in the deck, but an article by Yin et al (2024) cited below, which is relatively up-to-date, reports that the current length record for direct oligo synthesis is between 800 mer - 1728 mer. This alone is an accomplishment since authors explain that the rate of errors increases significantly above 100nt. The article also discusses the original 1000nt ceiling due to the steric hindrance of the substrate macromolecule. Please forgive my answer being a little choppy; I am still learning how to converse in this language.
Yin, Y., Arneson, R., Yuan, Y., and Fang, S. (2025). Long oligos: Direct chemical synthesis of
genes with up to 1728 nucleotides. Chemical Science, 16(4), 1966–1973. https://doi.org/10.1039/D4SC06958G
Homework Question from George Church: [Lecture 2 slides]
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
Question 1
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Question 2
[Given slides #2 & 4 (AA:NA and NA:NA codes)] What code would you suggest for AA:AA interactions?
Here I think you are asking me to provide an alternative code for the foundation of life? The irony is that is why I am here, I want you all to teach me how to rewrite code for AA:AA interactions. I know this because in preparing my answer I ran AA:AA interactions by AI for a second oppinion. My prompt was “what do you think Dr. George Church means by code for AA:AA interactions”. AI tells me that this code may need to demonstrate how sequences of amino acids influence physical interaction rules, which I interpret to be first order principles. Therefore what code would I suggest to influence AA:AA interactions that are able (if AI’s tip is correct) to perturb first order principles. My code as Dr. Nick Lane would say, should be efficient at turning the feedback loops of matter into biophysical waves of energy. In further prepartion I would turn to my greatest advisors in Natural Selection adaptation, bacteria and their metabolic motifs in Metazoans. I would need a coding system that respects phylogeny and doesn’t immagine I could ever devise a coding system more ingenious than the Krebs cycle or the molecularly machinery behind the deprotonation of hydrogen by Complex 5 in the Electron Transfer Chain. Still it’s a fascinating thought experiment if nothing else. To this end, I used your Acevodo-Rocha et al. (2016) paper and AI to find the Poliseno et al. (2024) paper and though I am not up-to-date on this team I do suspect there is an Epidemiologist among them, because their example of concordant and discordant pairing of coding and noncoding functions is what my coding system would be based on to optimize around the canonical rigidity of present AA:AA interaction.
Acevedo‐Rocha, C. G., & Budisa, N. (2016). Xenomicrobiology: A roadmap for genetic code engineering. Microbial Biotechnology, 9(5), 666–676. https://doi.org/10.1111/1751-7915.12398
Poliseno, L., Lanza, M., & Pandolfi, P. P. (2024). Coding, or non-coding, that is the question. Cell Research, 34(9), 609–629. https://doi.org/10.1038/s41422-024-00975-8
[(Advanced students)] Given the one paragraph abstracts for these real 2026 grant programs sketch a response to one of them or
Assignment (Your HTGAA Website) — DUE BY START OF FEB 10 LECTURE
Begin personalizing your HTGAA website in https://edit.htgaa.org/, starting with your homepage — fill in the template with
information about yourself, or remove what’s there and make it your own. Be creative! As with all assignments in HTGAA, be sure to
write up every part of this Homework on your HTGAA website in order to receive credit.
Important
For this week only, once your homework is complete and written up on your HTGAA website (and you’ve checked your published website at pages.htgaa.org and are happy with it), fill out the Homework 1 Completion form which David emailed out just after Lecture 1. This Google form expresses your interest in continuing with the course; without it you will not be accepted in HTGAA!
Week 2 HW: DNA Read Write and Edit
Table of contents
Software used:
Terminal,
git,
xcode,
hugo,
benchling,
rcdonovan website,
twist website.
Objective:
This week explores the read–write–edit toolkit: sequencing and synthesis workflows, restriction digests and gel electrophoresis, and early genome-editing frameworks.
Background:
DNA Read (George Church), Write (Joe Jacobson), & Edit (Emily Leproust). In addition to recitation and Tokyo Biohub node lab meetings
Methods:
Start with touchpoint of Design stage of SynBio DBTL cycle with In-silico Gel Art
Build DNA fragments in Benchling with restriction digests for Testing with Gel Electrophoresis
Learn from Benchling work & In-silico Gel Art
Start to Design or
Gel Electrophoresis
Obtain protein sequences
Plasmid digestion with restriction enzymes,
Preparing Twist DNA Synthesis Order
Tasks:
Documentation
Make sure to document every step of the in-silico and lab experiments. Make sketches, screenshots, notes, drawings… anything that helps you - and others - understand the experiment. Your documentation should help you - and others - to understand the topic. Don’t be afraid to add things that don’t work. Show your failures - and how you overcame them. Your Documentation should be a description of the amazing journey you are on!
Part 0: Basics of Gel Electrophoresis
Attend or watch all the lectures and recitation videos. Optionally watch bootcamp.
Part 1: Benchling & In-silico Gel Art
See the Gel Art: Restriction Digests and Gel Electrophoresis protocol for details.
Overview:
Make a free account at benchling.com
Import the Lambda DNA.
> All roads connect in SynBio. Here we have Bacteriophage Lambda (𝜆) which Lederberg discovered in 1950 targets E. coli strain K-12. This means we are on track with this phage if we focus on a specific gene in K-12 E. coli strain.
Simulate Restriction Enzyme (REs) Digestion with the following Type II Restriction Enzymes. These are one of the many bacterial adaptation strategies we have harvested in synbio to prepare plasmids. Bacteria use them to reduce the size of their genome to reduce mismatch with their environment. Each of these endonuclease agents below serve as tiny identification algorithms for directing cutting and splicing of plasmids in molecular biology experiments. Please note the “I” in REs represents roman numeral. Sticky means readily anneals to cut end or other strand cut by same enzyme.
The EcoRI RE is sourced from Escherichia coli> with palladrome cut at AATT
5’-GAATTC-3’
3’-CTTAAG-5’
leaving a 5’ sticky end.
The BamHI RE is sourced from Bacillus amyloliquefaciens and scans for
5’-GGATCC-3’
3’-CCTAGG-5’
to cut between G and G leaving a 5’ sticky end.
The HindIII RE is sourced from Haemophilus influenzae and scans for
5’-AAGCTT-3’
3’-TTCGAA-5’
leaving a 5’ sticky end.
The KpnI RE is sourced from Klebsiella pneumoniae, it requires small molecule cofactors including Mg and Ca ions to complete cut with fidelity; uses
5’-GGTACC-3’
3’-CCATGG-5’
and rather uniquely for this experimental RE set leaves a 3’ sticky end.
The EcoRV RE is sourced from Escherichia coli also and scans for
5’-GATATC-3’
3’-GTATAG-5’
and leaves the blunt end for this RE set.
The SacI RE is sourced from Streptomyces achromogenes and scans for
5’-GAGCTC-3’
3’-GTCGAG-5’
leaving a 5’ sticky end.
The SaII RE is sourced from Streptomyces albus and scans for
5’-GTCGAC-3’
3’-CAGCTG-5’
leaving a 5’ sticky end.
Source: Recognition sequences and cleavage patterns were verified using the REBASE database (Roberts et al., 2015).
Create a pattern/image w/style of Paul Vanouse’s Latent Figure Protocol artworks.
Use Ronan’s website as a helpful tool for quickly iterating on designs! Here is the link [https://rcdonovan.com/gel-art].
HW2 is structured purposefully to make us think like synbio engineers. For example, the reason we transition from Gel Electrophoresis to Restriction Digests is because we cannot move large strands of DNA and RNA through the GE matrix. We need small enough pieces of readable genetic material just to accomplish the lab assay. This makes RD a function necessary to achieve our design objectives. Benchling is a similar addition to the HW2 learning module, we need to see the restriction digests applied on our Lamba model and the computational ladder for converting the pieces of plasmid DNA in our GE matrix, it then helps that we can use Benchling in subsequent steps also.
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
Perform the lab experiment you designed in Part 1 and outlined in the Gel Art: Restriction Digests and Gel Electrophoresis protocol.
Now if your mind works like mine it might seem abrupt to leap from the movement of DNA through GE matrix to proteins but not if you understand the Central Dogma, sure, but even more the SynBio Design, Build, Test, Learn loop.
[https://doi.org/10.1371/journal.pbio.3002116]
Add a Bacterial chromosome and plasmid sequenced with Oxford Nanopore MiniON because I am annoyingly meticulous with discovery. In my HW2 discussion questions I am going to sing praises to Nanopore so also better to be consistent in DNA read inputs. I will download chromosome and plasmid DNA and load into Benchling. Please note the Genbank files do not play nicely with Benchling, so I will need to shift to FASTAs.
Escherichia coli DSM 30083 = JCM 1649 = ATCC 11775
> Chromosome GenBank: https://www.ncbi.nlm.nih.gov/nuccore/CP033092.2/
> CP033092.2 Escherichia coli DSM 30083 = JCM 1649 = ATCC 11775 chromosome, complete genome
> Plasmid GenBank: https://www.ncbi.nlm.nih.gov/nuccore/CP033091.2/
> CP033091.2 Escherichia coli DSM 30083 = JCM 1649 = ATCC 11775 plasmid unnamed, complete sequence
Part 3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
[Example from our group homework, you may notice the particular format — The example below came from UniProt]
sp|P03609|LYS_BPMS2 Lysis protein OS=Escherichia phage MS2 OX=12022 PE=2 SV=1 METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLL EAVIRTVTTLQQLLT
Considering: RpoS in E. coli K-12 will download Amino Acids for protein below and convert backwards to genome if I do not find an online reference that isn’t deleted.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
[Example: Get to the original sequence of phage MS2 L-protein from its genome phage MS2 genome - Nucleotide - NCBI]
Lysis protein DNA sequence
atggaaacccgattccctcagcaatcgcagcaaactccggcatctactaatagacgccggccattcaaacatgaggattacccatgtcgaagacaacaaagaagttcaactctttatgtattgatcttcctcgcgatctttctctcgaaatttaccaatcaattgcttctgtcgctactggaagcggtgatccgcacagtgacgactttacagcaattgcttacttaa
Lysis protein DNA sequence with Codon-Optimization
ATGGAAACCCGCTTTCCGCAGCAGAGCCAGCAGACCCCGGCGAGCACCAACCGCCGCCGCCCGTTCAAACATGAAGATTATCCGTGCCGTCGTCAGCAGCGCAGCAGCACCCTGTATGTGCTGATTTTTCTGGCGATTTTTCTGAGCAAATTCACCAACCAGCTGCTGCTGAGCCTGCTGGAAGCGGTGATTCGCACAGTGACGACCCTGCAGCAGCTGCTGACCTAA
3.4. You have a sequence! Now what?
3.5. [Optional] How does it work in nature/biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level.
Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein like a provided example at [https://2026a.htgaa.org/2026a/course-pages/weeks/week-02/index.html]
Reading DNA
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account, and Benchling account…
create Twist and Benchling accounts
Pick our protein! I will pick a protein related to aging for final project, I am just trying to keep my head above water on HW2 so the protein I pick is the example provided. See below in codeblock but what sort of nucleotides are “M E T…”? Clearly those aren’t nucleotides they are single letter representatives of amino acids, known as codons, constructed from 3 nucleotides. Here we are given in a top-down Build of a protein, which we must run the Central Dogma in reverse to translate back to RNA and then transcribe back to DNA.
Here is an example of what running backwards looks like crudely. In this instance we go all the way back (1996) to the original sequence of phage MS2 L-protein from its genome. This is an excerpt from the GenBank file: representing a “phage MS2 genome” GenBank record [https://www.ncbi.nlm.nih.gov/nuccore/V00642].
Please note this sequence doesn’t come from the bottom of the GenBank file instead
the selected region is required which must be further trimmed to match the code provided below from the HW2 blog. With correct NCBI links we can now confirm this code from the blog actually came from this GenBank record [ https://www.ncbi.nlm.nih.gov/nuccore/NC_001417.2?from=1678&to=1905&report=genbank]. I will also move this GenBank file into Benchling instead of previous file.
A closer match of genome nucleotides is obtainted through another NCBI lookup [https://www.ncbi.nlm.nih.gov/nuccore/NC_001417.2?report=fasta&from=1643&to=1938] though even here the resulting gene fragment must be further trimmed
Reflecting, since we need the gene that codes for the LYS_BPMS2 Lysis protein in the Escherichia phage MS2 we go back to a GenBank file from 1996 when virology was the approach in molecular biology for engineering tag segments of RNA strand with stems looped in the translation phase of the Central Dogma of molecular biology. Based on the orignal RNA virus from which MS2 was derived.
select Genes on the page with prompt “what can twist build for you?” for HW2
name the project “L protein” with “L” for “Lysis” for HW2.
select Clonal Genes order card and press “Order Now” when prompted to select gene type for HW2.
avoid my mistake, this next page is going to take us to an “Excel Like” worksheet that we will develop our request with. The old school way was to download and upload meticulously formatted Excel spreadsheets; we are advanced humans capable of using web forms. Before we enter the DNA we require into this order form we have to work through the DNA we were given to read in HW2
Completing the optimization process on Twist Website we now have a Codon-Optimized Lysis protein DNA sequence.
Optional: If we were going to synthesize more of this protein we now have a set of genetic instructions to read to grow those proteins. However there are different methods from which we can Build those proteins. We can consider cell-dependent or cell-free approaches. Explain more about these when I pick my protein.
In preparation for next steps remember that my Codon-Optimized Lysis protein DNA sequence
In Benchling instructions to transcribe gene to RNA
“Highlight the DNA sequence of interest.”
“Right-click and select Copy Special.”
“Choose the Reverse Complement option to get the anti-sense strand (RNA equivalent).”
“Create a New DNA/RNA Sequence and paste the sequence, ensuring the type is set to “RNA”.”
4.2. Build Your DNA Insert Sequence
Let’s first organize our directories in Benchling for the assembly line
Create folder for Registry of Standard Biological Parts [https://parts.igem.org/Part:BBa_J23106]
In that folder create the following folders:
> A_Promoter
> B_RBS
> C_Start Codon
> D_Coding Sequence
> E_7x His Tag
> F_Stop Codon
> G_Terminator
HW2 Objective of assembly: make a sequence that will make E. coli glow fluorescent green under UV light by constitutively (always) expressing sfGFP (a green fluorescent protein).
In Benchling, select New DNA/RNA sequence
Give your insert sequence a name and select DNA with a Linear topology (this is a linear sequence that will be inserted into a circular backbone vector of our choosing).
Go through each piece of the given DNA sequences highlighted below (Promoter, RBS, Start Codon, Coding Sequence, His Tag, Stop Codon, Terminator) and paste the sequences into the Benchling file one after the other (replacing the coding sequence with your codon optimized DNA sequence of interest!). Each time you add a new piece of the sequence, make sure to annotate by right clicking over the sequence and creating an annotation that describes what each piece (e.g., Promoter, RBS, etc.) is (see image below).
RBS (e.g. BBa_B0034 with spacers for optimal expression)
CATTAAAGAGGAGAAAGGTACC
Start Codon
ATG
Coding Sequence (your codon optimized DNA for a protein of interest, sfGFP for example)
AGCAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCAATTCTTGTTGAATTAGATGGTGATGTTAATGGGCACAAATTTTCTGTCCGTGGAGAGGGTGAAGGTGATGCTACAAACGGAAAACTCACCCTTAAATTTATTTGCACTACTGGAAAACTACCTGTTCCGTGGCCAACACTTGTCACTACTCTGACCTATGGTGTTCAATGCTTTTCCCGTTATCCGGATCACATGAAACGGCATGACTTTTTCAAGAGTGCCATGCCCGAAGGTTATGTACAGGAACGCACTATATCTTTCAAAGATGACGGGACCTACAAGACGCGTGCTGAAGTCAAGTTTGAAGGTGATACCCTTGTTAATCGTATCGAGTTAAAGGGTATTGATTTTAAAGAAGATGGAAACATTCTTGGACACAAACTCGAGTACAACTTTAACTCACACAATGTATACATCACGGCAGACAAACAAAAGAATGGAATCAAAGCTAACTTCAAAATTCGCCACAACGTTGAAGATGGTTCCGTTCAACTAGCAGACCATTATCAACAAAATACTCCAATTGGCGATGGCCCTGTCCTTTTACCAGACAACCATTACCTGTCGACACAATCTGTCCTTTCGAAAGATCCCAACGAAAAGCGTGACCACATGGTCCTTCTTGAGTTTGTAACTGCTGCTGGGATTACACATGGCATGGATGAGCTCTACAAA
7x His Tag (Let’s add a 7×His tag at the C-terminus of the protein to enable protein purification from E. coli)
CATCACCATCACCATCATCAC
Once you’ve completed this, click on Linear Map to preview the entire sequence. If you intend to have a TA review a sequence in the future, this is a good way to verify that all sections are annotated!
This is not required for this exercise, but to share your design with others, please ensure that link sharing is turned on! (Optional) Share your final sequence link with a TA for review!
This insert sequence you built is commonly referred to as an expression cassette in molecular biology (a sequence you can drop into any vector and it’ll perform its function). Go ahead and download the FASTA file for the sequence you made.
Here is my practice assembled copy of the HW2 gene fragment I will import in Twist. However, I will not submit an actual order to Twist because this is just my demonstration Clonal Gene fragment copy. I will repeat these steps with my own functional gene for official purchase order.
For this demonstration, we’ll choose Clonal Genes. You’ll select clonal genes or gene fragments depending on your final project.
Historically, HTGAA projects using clonal genes (circular DNA) have reached experimental results 1-2 weeks quicker because they can be transformed directly into E. coli without additional assembly.
Gene fragments (linear DNA) offer greater design flexibility but typically require an assembly or cloning step prior to transformation. An advantage is If designed with the appropriate exonuclease protection, gene fragments can be used directly in cell-free expression.
4.4. Import your sequence
You just took an amino acid sequence of interest and converted it into DNA, codon optimized it, and built an expression cassette around it! Choose the Nucleotide Sequence option and Upload Sequence File to upload your FASTA file.
4.5. Choose Your Vector
Since we’re ordering a clonal gene, you will need to refer to Twist’s Vector Catalog to choose your circular backbone. You can think of this as taking your linear expression cassette for your protein of interest, and completing the rest of the circle!
The backbone confers many special properties like antibiotic resistance, an origin of replication, and more. Discuss with your node to decide on appropriate antibiotic options. At MIT/Harvard, you can use Ampicillin, Chloramphenicol, or Kanamycin resistance.
Twist vectors do not contain restriction sites near the insert fragment, so make sure to flank your design with cut sites if you are intending to extract this DNA insert fragment later.
For this demonstration, choose a Twist cloning vectors like pTwist Amp High Copy.
Click into your sequence and select download construct (GenBank) to get the full plasmid sequence:
Go back to your Benchling account. Inside of a folder, click the import DNA/RNA sequence button and upload the GenBank file you just downloaded.
This is the plasmid you just built with your expression cassette included. Congratulations on building your first plasmid!
Important
For your final projects, remember to include:
Fully annotated Benchling insert fragment
Desired Twist cloning vector
Part 5: DNA Read/Write/Edit
It’s helpful to visualize DNA designs using SBOL Canvas (Synthetic Biology Open Language) to convey your designs. Here’s an example of what you just annotated in Benchling:
4.3. On Twist, Select The “Genes” Option
4.4. Select “Clonal Genes” option
For this demonstration, we’ll choose Clonal Genes. You’ll select clonal genes or gene fragments depending on your final project.
Historically, HTGAA projects using clonal genes (circular DNA) have reached experimental results 1-2 weeks quicker because they can be transformed directly into E. coli without additional assembly.
Gene fragments (linear DNA) offer greater design flexibility but typically require an assembly or cloning step prior to transformation. An advantage is If designed with the appropriate exonuclease protection, gene fragments can be used directly in cell-free expression.
4.5. Import your sequence
You just took an amino acid sequence of interest and converted it into DNA, codon optimized it, and built an expression cassette around it! Choose the Nucleotide Sequence option and Upload Sequence File to upload your FASTA file.
4.6. Choose Your Vector
Since we’re ordering a clonal gene, you will need to refer to Twist’s Vector Catalog to choose your circular backbone. You can think of this as taking your linear expression cassette for your protein of interest, and completing the rest of the circle!
The backbone confers many special properties like antibiotic resistance, an origin of replication, and more. Discuss with your node to decide on appropriate antibiotic options. At MIT/Harvard, you can use Ampicillin, Chloramphenicol, or Kanamycin resistance.
Twist vectors do not contain restriction sites near the insert fragment, so make sure to flank your design with cut sites if you are intending to extract this DNA insert fragment later.
For this demonstration, choose a Twist cloning vectors like pTwist Amp High Copy.
Click into your sequence and select download construct (GenBank) to get the full plasmid sequence:
Go back to your Benchling account. Inside of a folder, click the import DNA/RNA sequence button and upload the GenBank file you just downloaded.
This is the plasmid you just built with your expression cassette included. Congratulations on building your first plasmid!
Important
For your final projects, remember to include:
Fully annotated Benchling insert fragment
Desired Twist cloning vector
Part 5: DNA Read/Write/Edit
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would like to use directed evolution in outbred goats to select my DNA to sequence. Therefore my plan is to stick with the HTGAA method until I have a Nanopore sequencer and reagents for genomic surveillance of my herd. My argument for why is still developing but essentially I have anecdotal observations to support a hypothesis. An example of the type of genes I would like to sequence is the second vector I uploaded – the RpoS gene in the K-12 strain of E. coli. The gene was sequenced with a Nanopore sequencer.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
There is no substitute in my opinion for a Nanopore sequencer, with a distant second being a PacBio. Ofcourse, Nanopore sequencers are far less popular than Illumina, and despite the fact that I am a big fan of Craig Venter, I still prefer the scientific opportunities available with Nanopore. In fact a significant reason why I went back to school post reproductive fitness equals zero, is because when I graduated from college they still hadn’t completed the Human Genome Project. I learned about Nanopores during the COVID-19 Pandemic when I started one of the first wastewater surveillance programs in the U.S. I believe the accuracy, speed, and flexibility of pore facilitated single base sequencing reads in parallel multithreaded readings fits my future research goals exactly and I am on the cusp of becoming a Nanopore super user.
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
What is the output of your chosen sequencing technology?
I am focused on deep time series sequencing data that is broad enough to include changes in diversity of microbiome and host metatranscriptomic, epigenetic, and metabolomic signals as well as metagenomic changes. I also want to develop pipelines that I always have the opportunity to contribute to but never want to own or primarily benefit from. I believe paywall science is an etiologic mechanism that favors contagion.
The first benefit of 3rd and 4th generation sequencers, particularly the Nanopore machines, is that they do not even require PCR amplification. Don’t get me wrong, I love PCR as a flexible assay, but as an Epidemiologist I have never been comfortable with making more copies of pathogens on principle. I realize this a bit of a semantic argument and there are plenty of bio safety measures in place. At the same time the same biosecurity measures are drivers of inequality in applied Molecular Biology capabilities. What does it mean when the technology itself becomes a driver of inequality to scientific techniques everyone in a generation should have access to? I believe it means it’s time to keep innovating.
In addition I think there is wisdom in sequencing the actual shoddy molecules collected from the field, particularly for my applications. This is a Biosecurity advantage and better fit to the Epi Triangle anyway. However I am not saying there are not scenarios where higher level Biosecurity reference labs with PCR pipelines are not necessary. I just think some sequencers should be managed and maintained by governments and smaller non-PCR-based Nanopores should be prioritized by individual field researchers, like I intend to be.
Now there is an elephant in the room, thoug,h and that’s data storage. I have been wrestling with data storage my entire career, and I know my interest in Nanopore sequencing isn’t going to make these challenges go away anytime soon. Therefore, I am all for DNA storage of genomic sequencing information about animals in plant DNA ideally. If a safe method is already available, storage in animal subjects would be incredible as well. What DNA storage is maintained in goat horns or sheep’s wool? I need to investigate the methodology further to see if this is even possible. I am ashamed to admit that until I read Dr. Church’s Epilogue in Regeneration, I had never even thought about this before.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I am dazzled by DNA origamis for synthetic materials, but the complexity of the methods to achieve static outputs is not necessarily a tradeoff I would invest time in right now. Genetic circuits are different though, I am fully attentive to this revolution. Particularily like we see in the examples provided by the Elowitz Lab [https://www.elowitz.caltech.edu/research#!computationandsyntheticcircuits].
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
I would like to learn more about the CHOMP (circuits of hacked orthogonal modular proteases) method to integrate binary logic into functional programming modules in biological circuitry. The CHOMP method can then be used to control regulatory cascades and even more exciting to me binary logic gates. My research interests are nonlinear models in aging and cellular senescence that utiilizes elucidated insights to improve areas of stasis with the potentional for rejuvination. Waves of molecular decisions all of which with decipherable underlying binary logic gates based on Boolean logic. The engineering methods focus on viruses and bacteria. The programming motif they target is incoherent feed-forward loops. The amino acid they interact with is the Nitrogen end of Tyrosine which they expand to a four protein circuit. They image their single-transcript adaptive pulse circuits using time-lapse images. The result of the engineering is a rachet to control intrinsic nonlierarity of input and output biological systems. The scalabity and accuracy are tunable by the application. The speed is slow to design and as fast as biological circuits once implemented.
Another method I would be interested in investigating further is the Asish et al. (2026) lab’s noninvasive biosensor application using live-cell diffusion-weighted imaging to investigate the effect of Gly-Ser spacers in transcription.
Source: Xiaojing J. Gao et al. ,Programmable protein circuits in living cells.Science361,1252-1258(2018).DOI:10.1126/science.aat5062
Asish N. Chacko et al. ,A programmable genetic platform for engineering noninvasive biosensors.Sci. Adv.12,eaec1211(2026).DOI:10.1126/sciadv.aec1211
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I am for animals. I want to contribute to dextinction and life extinction of all endangered and vulnerable organisms I can serve. I will contribute to human longevity as an afterthought to Natural diversity and sustainability. I am not beholden to humans though. I believe in the sanctity of all life.
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions: How does your technology of choice edit DNA? What are the essential steps? What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
The synthesis of DNA and OMIC ontologies starts with phylogeny, small molecules, and phenotypes like disease. Therefore my career plan is to build the throughput for molecule mediate bidrectional interactions between Host physiology and the microbes in the microbiome and metagenome around the host. This is the big tent vision. Now how do get there. Imagine if I lay it all out here step-by-step. How could I do what I aspire to do if 1000 people do it first before me. Still, I come to HTGAA a pleb at the stairs to the Temple of Zeus, with my goats. Not to sacrifice, though. I will not be a culling scientist. There is no scientific discovery in the text of life that is worth sacrificing a living thing. I am a builder by nature anyway, I want to observe life without intervention, well some intervention is necessary, but not like it’s currently done. Therefore what to do?
Here is what I can share at HW2. Everywhere an organism lives, say a goat, is a DNA and RNA wake of material. Much of it is waste material, residues from competing metabolic systems stacked and ready to be interpreted and transformed into data. With data comes constraints, especially OMIC data, it’s endless and massive in scale, randomized and chaotic. I like the idea of applied systems biology pipelines built around dead biological material. You can catalogue and reconstruct living systems from waste chemistry. Do you need an Almond in it’s shell to understand the life history of that nut, not really, a fragment of husk in a pile on the ground will tell you about the almond and the animal that consumed it.
COVID-19, as a front-line Epidemiologist, in the center of the maelstrom did not equivocate in its lessons. First and foremost, public health apparatuses like mRNA vaccination research and deployment infrastructure is useful when it’s available, accessible, and appropriately matched to the agent. The rest is wastewater. Especially, where the infrastructure of sewers is insufficient to remove waste from a community fast enough, can be used to trace outbreaks in near real time. Wastewater surveillance is harder where the water is plentiful, deep, and fast-flowing. The great news for wastewater epidemic surveillance is that the structural inequalities above the sewers, exist within the sewers, and drive disease transmission in Outbreaks. This isn’t a hunch; the data support it. This is why I will continue to be interested in wastewater surveillance also when I enter the workforce.
However, I will focus on much broader networks of waste than wastewater, which is what makes the intersection of gut microbiomes, microbes, and host physiology the biological nexus for me. Thus, applications, many options here – especially in agriculture. I like agriculture because soil is the ultimate biological pile of waste. I have watched animal waste turn into dirt for several years now, and from that waste, plants grow. The animals eat those plants and turn it into animal tissues using systems of heredity and variability that have nothing to do with anything I did. I just get the animal in front of a plant and they complete their reproductive and maintenance programs. If I keep the animals water clean and their housing dry they do not get sick. These animals and the environment are an engine that I can run passively – they make the world a better place.
At the same time, though, this natural experiment produces a lot of opportunities to study molecule-mediated bidirectional relationships between animal hosts and the microbes in their microbiome and metagenome. Fortunately, for my experimental milieu, my species is driving Earth to its extreme of the boundary conditions for habitability, which certainly makes science more interesting – especially when local interventions can be developed to support sustainability, health, and longevity.
The last sentence is key for the edits I would dare to make. Never blindly though. This is why I will structure my lab within evolution directed sythesis.
CRISPR/Cas9
Short tutorial for designing gRNAs: https://blog.addgene.org/how-to-design-your-grna-for-crispr-genome-editing
Benchling specific tutorial for designing gRNAs: https://www.benchling.com/blog/how-to-design-grnas-to-target-your-favorite-gene
List of Cas editors and their PAM sites: https://www.synthego.com/guide/how-to-use-crispr/pam-sequence
Base Editors
Base editors contain a nicking or dead Cas9 enzyme fused to a deaminase. a.) PAM requirement: Base editors contain a nicking or dead Cas9 enzyme fused to a deaminase. For designing your guide RNA for base editing you will therefore have a PAM requirement like you would have for any Cas9 experiment. b.) Deamination window: An additional design constraint is that the sequence window in which deamination occurs is only a few base pairs long. You can find information on the deamination windows in the review below (even though some new editors are not included).
BE4 and ABE7.10 are good starting points and both use SpCas9 with NGG Pam requirement. Base editors with other PAM sites have been constructed too.
Review of base editors (2018) including a list of all base editors, their editing window and PAM requirement: https://www.nature.com/articles/s41576-018-0059-1?WT.feed_name=subjects_animal-biotechnology
Other editors:
Prime editor https://www.nature.com/articles/s41586-019-1711-4
Tutorials/tools:
https://primeedit.nygenome.org/https://www.nature.com/articles/s41551-020-00622-8http://pegfinder.sidichenlab.org/
TALEN For TALENs, you can assume no sequence restrictions – One of the technology’s previous restrictions was a T starting base, but this has since been overcome. In contrast to the CRISPR/Cas technologies above, your DNA sequence is recognized through interactions between the DNA and the TALEN: each TAL in the array recognizes one base. (Note: In order to introduce a double strand break, you will need to design to TALENs targeting the opposing strands.)
Short guide: https://www.addgene.org/talen/guide/
One of the available design resources: https://tale-nt.cac.cornell.edu/node/add/talen
Directed evolution for overcoming starting base restriction:https://academic.oup.com/nar/article/41/21/9779/1276340
Additional Resources:
Gel Purification of DNA: after DNA gel electrophoresis, cutting a band of DNA out of the agarose gel allows isolation and purification of a specific DNA fragment:
Addgene: Protocol - How to Purify DNA from an Agarose Gel
Overview of synthetic, unnatural organisms using recoding:
Synthetic genomes with altered genetic codes (2020)
DNA recorders, Sense+Read+Write:
Lineage tracing and analog recording in mammalian cells by single-site DNA writing (2021)
Molecular electronics, integrating single molecules into electronic chips:
Molecular electronics sensors on a scalable semiconductor chip: A platform for single-molecule measurement of binding kinetics and enzyme activity (2022)
Review of genome editors (zinc finger nucleases, TALENs, CRISPR) at the time CRISPR was emerging as editing technology: https://www.cell.com/trends/biotechnology/pdf/S0167-7799(13)00087-5.pdf
Clinical trials of genome-editing therapies: https://www.nature.com/articles/d41573-020-00096-y
This week we get hands-on (or at least code-on) with pipetting robots.
Background:
No lecture. Recitation and Tokyo Biohub node lab meetings. Submit three slides with ideas to our node by 24Feb2026.
Ideas for Tokyo Biohub Deck
GPG01: Identify transcription indicators in post reproductive goat life history indicative of alterations to NAD(H), ROS signaling, tissue specific oxidative stress and inflammation.
GPG02: Explore application of G-protein coupled receptors (GPCRs) in goats a method Chen et al. (2019) proposes more broadly to monitor bioactive microbial metabolites with associations to physiology.
GPG03: Consider systems-level synthetic biology agricultural interventions to improve yield of metabolite specific food-stuffs to support molecule mediated bidirectional interactions between goat hosts and microbiota.
Questions:
For this week, we’d like for you to do the following:
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
While your description/project idea doesn’t need to be set in stone, we would like to see core details of what you would automate. This is due at the start of lecture and does not need to be tested on the Opentrons yet.
I propose using a cloud laboratory and automation tools to process environmental metagenomics samples with and by Oxford nanopore sequencers. Here is the problem I am attempting to address. I am only one person. My time is always constrained profoundly. This means I always am burning and undercooking items with all my pans in the fire. Still, personally and professionally I am comitted red tooth and claw to environmental protection of biodiversity and abundance of natural ecologies and agricultural middle corridors. In addition I am personally offended by inequality, especially when it comes to the allocation of scientific discovery capacity and supply lines. The most diverse places on Earth are the most imperiled and at the same time least equipped with tooling to achieve the scientific advacements they need to protect their habitats and communities. Allow me to also preface that HTGAA is a small example that the bottlenecks of which I speak are not in human capacity, it’s techology, energy, infrastructure, brick and mortar. I believe that cloud computing and automation tools are a stopgap measure urgently needed to fill the breech and provide platforms to the facilitate the synergies of natural and unnatural selection required to advance sustainability and biodiversity. However engineering these partnerships are going to be just as important as the technological capabilities. The great thing about HTGAA is that we are doing this work from the bottom-up by participating in these cohorts.
Aside from HTGAA, my work with goats actually comes from the same engineering aspiration. I never saw a goat in the U.S. until I became a community health worker and started working outside of my country. Once I left the U.S. goats were much more plentiful, especially in rural mountainous regions. I am raising goats now to learn animal husbandry of these critical animals so that I can better understand how to help raise goats anywhere, in any locale, with any resource constraint. Goats in my opinion are the first automation tool that humans partnered with to survive in extreme environments. Through this partnership goats and humans expanded their gene and environment match with the physical constraints they were encountering in their struggle to ensure their families thrived. Goats and humans share many strengths and weaknesses, mainly their dedication to their families and security of FDR’s essential freedom from uninhabitable temperature and violence, hunger, thirst.
Now to the assignment, but from this perspective, the paper I reviewed from Childs et al. (2025) compared manual and automated metagenomic workflows using Oxford nanopore sequencers and found minimal differences in outcomes assessed. The first reason I chose this paper is because it starts with a fundamental truth, long-read sequencing has transformed our understanding of the microbiome. In fact, metagenomic and microbiome catalogues were not even attempted reliably until these machines entered the OMICs revolution. Enter the pipetter. I can attest, this is monastic work. The challenge is not the tool, it’s the lab space, and the sheer magnitude of the wells that the pipetter must span. Experimental protocols require percise allocation of minute quanties of fluids over and over again. From a personal vantage I quite enjoy the process, for there are few activities more zen in my day but then I am also hyper-privleged. Again inequality rears its head into the hallways of science. Who enters the cloister of the dwindling lab spaces in the world to the shelter of the bench and how many minuites do they have to spend to achieve their objectives. Here too is another ineqality though, because let’s be honest, not their objectives but the objectives of their research supervisors–because labs are also part of the caste system.
How do we untangle all of these knots to do the do the critical work. Could it be automation is answer? This will depend on who has access to automation. Are we talking about robotic workflows that are accessible to anyone with curiosity about microbiomes and metagenomes. Likely not anytime soon. I guess it will be more about the workflows done by students with professors. This is where the revolution of OMICs and Next-Generation Sequencers must be fought. What about private start-ups, I don’t know enough to speculate here. I can ponder the task of expanding the paradigm so any student with want of bench exposure using sequencers can have it. Honestly, I think HTGAA is pursuing this admirably. The cause is certainly just. If students and professors with and without wetlab spaces can both access cloud platforms and automation labs then we can realize the type of contingent niche environment that theoretically at least could be scaled-up and that is far better than not having a foothold at all. The Childs lab (2026) certainly seems to understand this charge when they explain that automation is a game changer fit to improve throughput, reproducibility, and accuracy. What is less clear is if the solution is the automated workflow or the Oxford nanopore sequencers that true read the sample one base pair at a time very quickly and then write that information into a cloud library for template recognition against other long-read sequences with annotation.
I didn’t really leave myself enough time to do this properly, ironically because this is lambing season, but Child’s et al. (2025) do make some very interesting observations in their side-by-side comparison of manual and automated workflows. I will apply these to my project now as well.
Childs et al. (2025) explain that many of the current studies they reviewed for their article only contain high throughput amplicon from the COVID-19 Pandemic. I do not see this as a challenge at all. Instead, when I think about the COVID-19 Pandemic as front-line warrior for Metazoans I see the good we accomplished when political will was aligned with scientific aspirations, and trust that the only reason naysayers have any leeway now to gripe about the deluge of SARS-CoV-2 data and genetic contamination, is because they are alive because of mRNA vaccines and wastewater surveillance, which Oxford nanopore significantly supported.
The liquid handling robot arm of the Childs et al. (2025) study was a Bravo Automated Liquid Handing Platform. I want one. Is it worth the cost though. Apparently, the findings are not sufficient to justify a purchase, based on read length alone. In the study the manual and Bravo study arms both analyzed the same 24 samples from a range of environments across a 96-well plate. Except for read length, which was on average longer in the manual arm than the automated. We can assume, if we have ever pipetted, that the automated arm would be more consistent in the allocation of microfluidics but confounding from variation in diverse soil samples appears to have made this distinction difficult to show. Meanwhile, the manual arm included eludication of DNA samples that the automata didn’t replicate, that doesn’t seem fair to me. However, if the automated workflow literally is not able to do all of the workflow steps than that is a strong point for manual over automated arms until the landscape is level.
Here’s the big takeaway though for my project. Childs et al. (2025) did find that improved automated libraries reduced PCR artefacts and increased sensitivity provide a more accurate snapshot of the ecological taxa of the microbiota – in other words more families, species, sub-species in the samples of less abundant organisms. This is what I want to hear, because if this process was applied to five studies instead of one then we would have 5x’s the power in detection of rare organisms that contribute to the diversity of the soil ecosystems, which is what I aspire most to understand and preserve.
Final Project Ideas — DUE BY START OF FEB 24 LECTURE
Methods:
Cloud Computing
Tasks:
Assignment: Python Script for Opentrons Artwork — DUE BY YOUR LAB TIME!
Your task this week is to Create a Python file to run on an Opentrons liquid handling robot.
Review this week’s recitation and this week’s lab for details on the Opentrons and programming it.
Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept.
If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead.
[!warning] Ask for help early! If you are having any trouble with scripting, contact your TAs as soon as possible for help.
Do not wait until your scheduled robot time slot or you may not be able to complete this assignment!
If the Python component is proving too problematic even with AI and human assistance, download the full Python script from the GUI website and submit that:
Use the download icon pointed to by the red arrow in this diagram.
If you use AI to help complete this homework or lab, document how you used AI and which models made contributions.
Sign up for a robot time slot if you are at MIT/Harvard/Wellesley or at a Node offering Opentrons automation. The Python script you created will be run on the robot to produce your work of art!
At MIT/Harvard? Lab times are on Thursday Feb.19 between 10AM and 6PM.
At other Nodes? Please coordinate with your Node.
Submit your Python file via this form.
Post-Lab Questions — DUE BY START OF FEB 24 LECTURE
One of the great parts about having an automated robot is being able to precisely mix, deposit, and run reactions without much intervention, and design and deploy experiments remotely.
For this week, we’d like for you to do the following:
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
While your description/project idea doesn’t need to be set in stone, we would like to see core details of what you would automate. This is due at the start of lecture and does not need to be tested on the Opentrons yet.
Example 1: You are creating a custom fabric, and want to deposit art onto specific parts that need to be intertwined in odd ways. You can design a 3D printed holder to attach this fabric to it, and be able to deposit bio art on top. Check out the Opentrons 3D Printing Directory.
Example 2: You are using the cloud laboratory to screen an array of biosensor constructs that you design, synthesize, and express using cell-free protein synthesis.
Echo transfer biosensor constructs and any required cofactors into specified wells.
Bravo stamp in CPFS reagent master mix into all wells of a 96-well / 384-well plate.
Multiflo dispense the CFPS lysate to all wells to start protein expression.
PlateLoc seal the plate.
Inheco incubate the plate at 37°C while the biosensor proteins are synthesized.
XPeel remove the seal.
PHERAstar measure fluorescence to compare biosensor responses.
Final Project Ideas — DUE BY START OF FEB 24 LECTURE
As explained in this week’s recitation, add a slide in your Node’s section of this slide deck with an idea you have for an Individual Final Project. Be sure to put your name on your slide!
By Eyal Perry, Laura Maria Gonzalez, Dominika Wawrzyniak, Alex Hadik, Suvin Sundararajan, Ronan Donovan
This notebook contains a few examples that demonstrates how the Opentrons OT-2 can be used to draw arbitrary patterns using the the Python Opentrons API. These examples can and should be used as your template as you try to pattern your own colorful, synthetically engineered bacteria.
To use this, make your own copy of this Colab, and in that copy you can run and edit the last section (and your work will be saved in your copy!).
Note: After learning about how to program designs using colab and python, you may choose to print more designs with automated tools like Opentrons Art Interface.
Each example consists of two blocks of code:
The first code block is where the pattern is drawn using .aspirate(), .move_to(), and dispense_and_detach() (as a wrapper around .dispense()) commands (similar to G-code). This block will typically generate no output, as it’s just loading the code (but doesn’t run it yet). This block of code can later be copied as-is and saved as a .py file to be executed on a real Opentrons machine.
The second code block runs a simple simulation that visualizes the pipetted pattern by executing your code in the simulator in this colab. This block will draw the state of the plate after running the robot code.
At the end is a section for you to code your design in, with the same two code blocks. Make your own copy of this Colab notebook and work in your copy. When ready, upload the link to your first block in this section to the linked google form a day before your lab date! Don’t edit the second block in this section, as only the first block will be run on the robot.
Several important notes:
All units are in mm
Never go beyond a radius of 40mm from (0,0). If you do, you might hit the walls of the petri dish and all hell breaks loose, or you might dispense onto the wall of or even outside the petri dish. (Some common “90mm” or “100mm” petri dishes only have an inner diameter of 84mm in the bottom plate, and the tip occupies a radius of a couple mm.)
For the Black Agar Plates, dispense 1 uL drops by default. (If you are trying for a particular effect, going slightly higher in some places may be acceptable.) While that may sound like a small quantity, the E.coli will still be visible (especially after growing) and small “pixel” sizes can produce more detailed patterns.
Be careful of dispensing samples too close to each other! They will move around slightly depending on the size of the drop. 1uL drops 2mm apart may sometimes run together or may stay mostly distinct even after incubation; 1uL drops 5mm apart will almost always stay distinct, but give you less than half the “resolution” for your art. Midway between those - 3.5mm separation - may be a happy medium. (See past year photos here and in the Lab Protocol and count dots along one axis; these of course show the ones which were lucky enough to mostly not run together…)
On the robot if you dispense and immediately move the tip 1cm to the left it will create a streak of bio-ink (shaped according to the viscosity of the liquid). The simulator accounts for this basic effect, and you will see spurious lines between your dots or to random locations in the visualization. We have provided a routine dispense_and_detach() that dispenses and moves the tip slightly up & down to fully clear the droplet; you can use this in your code both for the simulator and the robot to avoid streaking.
We have defined standard configuration for the robot deck for this lab, and our template code follows it. We plan to have Red-, Yellow-, Green-, Cyan-, and Blue-fluorescing bacteria (but no others) at all sites in the robot for your use, and have provided a routine location_of_color() you can use to retrieve our standardized configuration’s location of a named color (which you can pass to an aspirate() call).
Pay attention to any text output from the simulator (typically just above the plate image) - it can give useful diagnostics and statistics. Don’t get so focused on your beautiful drawing that you forget to check this every once and a while.
Remember not to waste any resources (here tips & reagents, as explained in the Lab Protocol – you can confirm via the “Volume Totals by Color” and “Tip Count” summaries shown after every successful run – but don’t cross-contaminate your color wells.
The visualization is not 100% accurate. We don’t model any flud dynamics, so any streaking if you don’t use dispense_and_detach(), any effects of dispensing from z>0, and even the droplet sizes in all cases are not physically realistic; and the simulator doesn’t have an awareness of the 3D positions of labware. (Feel free to contribute improvements to the simulator!)
The simulation is not even close to a 100% complete reimplementation of the Opetrons API. Some commands will work on the OT-2 but will cause errors in the simulation (Feel free to contribute!).
After your code is done, to submit it to be run on a robot:
Make sure your code is accessible to us: in your colab click the “Share” icon in the upper right, set “General access” so that “Anyone with the link” can be a “Viewer”.
Copy to the clipboard a link to your code: right-click in the first code block (which has the metadata = {...} section near the top and your code at the end) and choose “Copy link to cell”
Paste this URL into the Google form for submitting to the OT-2 and submit at least a day before your robot time slot.
The following block of code contains required installations and the simulation/visualization code. It only needs to be run once per runtime.
When run, it will output errors declaring “ERROR: pip’s dependency resolver does not currently take into account all the packages that are installed.” and list some package incompatibilities; that is expected, and is a result of the Opentrons API requiring an old version of some libraries. (No other errors are expected.)
This block can be re-run in a runtime without ill effect (but will show the same errors every time).
Run this block once per runtime to set up your environment
#@title Run this block once per runtime to set up your environment
The colab now comes with too new a version of numpy; opentrons still needs an older one.
So set up venv-like isolation of my pip installs (separated from colab packages) for all subsequent cells.
(Without doing this, colab would require restarting the runtime right after installing a different numpy version.)
import sys, os
py = f"{sys.version_info.major}.{sys.version_info.minor}"
PKG = f"/content/venv/lib/python{py}/site-packages"
os.makedirs(PKG, exist_ok=True)
if PKG not in sys.path: sys.path.insert(0, PKG)
os.environ[“PIP_TARGET”] = PKG # routes !pip / %pip installs into the venv
os.environ[“PYTHONNOUSERSITE”] = “1”
Install opentrons into the venv (and all its dependencies!) BEFORE any import numpy etc.
%pip install -q –upgrade –target “$PKG” opentrons
Now opentrons has been cleanly installed in its own venv-like environment with
versions of packages it likes; proceed to use it “normally” from here.
from opentrons import types
import matplotlib.pyplot as plt
plt.rcParams[“figure.figsize”] = (10,10)
Petri dish size constants
PETRI_INNER_DIAMETER = 84 # 84mm is hopefully a tight lower bound on inner diameter of “90mm” & “100mm” petri dishes
MAX_DRAW_RADIUS = PETRI_INNER_DIAMETER/2 - 2 # leave 2mm margin for the tip size, drop size, miscalibration, etc.
Define some classes for our custom HTGAA Opentrons simulator/visualizer
def same2DLocation(loc1, loc2): # ignores z (=> tests x, y, and labware)
return loc1.point.x == loc2.point.x and loc1.point.y == loc2.point.y and loc1.labware == loc2.labware
def mock_print(str):
#print("…\n" + str)
pass
each PipetteSim instance tracks what it’s dispensed; if you have multiple, need to call visualize() on each.
(can’t unify multiple by making the instance variables into class variables; note this colab has at least
one instance per example, and we don’t want those sharing dispense states.)
class PipetteSim: # modeled after InstrumentContext in the opentrons api
def init(self, instrument_official_name, mount_LR, tip_rack_list, well_colors):
if instrument_official_name != “p20_single_gen2”:
raise ValueError(f"Unsupported pipette {instrument_official_name} – should be p20_single_gen2")
self.max_volume = 20
self.instrument_official_name = instrument_official_name
if mount_LR != "right":
raise ValueError(f"Unsupported pipette mount {mount_LR} -- should be right")
self.mount_LR = mount_LR
if tip_rack_list[0].labware_official_name != "opentrons_96_tiprack_20ul":
raise ValueError(f"Unsupported tip rack {tip_rack_list[0].labware_official_name} -- should be opentrons_96_tiprack_20ul")
self.tip_rack_list = tip_rack_list
self.well_colors = well_colors
self.droplets_x = []
self.droplets_y = []
self.droplets_size = []
self.droplets_color = []
self.smears = [] # list of 3-tuples: (xlist, ylist, color)
self.location = nullLocation # used by dispense_and_detach()
self.justDispensedAt = None
self.current_volume = 0
self.aspirated_loc = None
self.totalAspirated = {} # 'color' : total
self.totalDispensed = {} # 'color' : total
self.curr_color = 'orange'
self.has_tip = False # (in the opentrons api!)
self.tip_count = 0
def del(self):
if self.has_tip:
raise Exception("### ERROR: Run completed without dropping the tip!") # python prints but ignores exceptions in destructors
used by our dispense_and_detach() routine
def _get_last_location_by_api_version(self): # (in the opentrons api!)
return self.location
use the well id to make up a location on the petri dish diagram:
D6 in the center, A1 lower left, H12 upper right (assuming 96-well, but will work for any)
def smearIfJustDispensed(self, loc): # (NOT in opentrons api)
assert(isinstance(loc, (types.Location, WellMock)))
if self.justDispensedAt is not None:
newloc = loc if isinstance(loc, types.Location) else self.petriLocOfWell(loc)
if not same2DLocation(self.justDispensedAt, newloc):
line_end = self.justDispensedAt.move(0.5 * (newloc.point - self.justDispensedAt.point))
self.smears.append(([self.justDispensedAt.point.x, line_end.point.x],
[self.justDispensedAt.point.y, line_end.point.y],
self.curr_color))
self.justDispensedAt = None
def dispense(self, volume, location): # (in opentrons api)
assert(isinstance(location, types.Location)) # not allowing dispensing into well or trashbin/wastechute for this lab – petri only!
assert(isinstance(volume, (int, float)))
if (location.point.x2 + location.point.y2 > MAX_DRAW_RADIUS**2):
raise ValueError(f’Dispensing outside “safe” area: Point ({location.point.x}, {location.point.y}) is more than’ +
f" {MAX_DRAW_RADIUS}mm away from the petri dish’s center.")
if not self.has_tip:
raise RuntimeError(“dispense() called when no tip was being held”)
if self.current_volume < volume:
raise ValueError(f"You dispensed {volume}uL, which is more than was in the pipette ({self.current_volume}uL).")
if volume <= 0:
raise ValueError(f"Dispensing {volume}uL – you should dispense a positive amount.")
if location.point.z < 0:
raise ValueError(f"dispense() passed a location with z={location.point.z} – do not go below z=0!")
if location.point.z >= 10:
print(f"Dispensing from a location with z={location.point.z} – do you really want to dispense from that high?")
self.smearIfJustDispensed(location)
self.current_volume -= volume
self.droplets_x.append(location.point.x)
self.droplets_y.append(location.point.y)
self.droplets_size.append(volume * 100) # unprincipled scale factor (1uL->100 sq.pt), but it works
self.droplets_color.append(’lime’ if self.curr_color.lower()==‘green’ else self.curr_color) # map green -> lime (looks more like GFP)
self.totalDispensed.setdefault(self.curr_color, 0)
self.totalDispensed[self.curr_color] += volume
self.location = location
self.justDispensedAt = location
def aspirate(self, volume, location): # (in opentrons api)
assert(isinstance(volume, (int, float)))
assert(isinstance(location, (types.Location, WellMock)))
if not self.has_tip:
raise RuntimeError(“aspirate() called when no tip was being held”)
if volume + self.current_volume > self.max_volume:
raise ValueError(f"Aspirating {volume}uL + {self.current_volume}uL already in pipette = {volume + self.current_volume}uL,"
f" which is more than the pipette can hold ({self.max_volume}uL).")
if volume <= 0:
raise ValueError(f"Aspirating {volume}uL – you should aspirate a positive amount.")
if self.aspirated_loc is not None and self.aspirated_loc != location:
raise RuntimeError(f"Cross-contaminating wells {self.aspirated_loc} and {location} with a single pipette")
self.aspirated_loc = location
self.smearIfJustDispensed(location)
self.current_volume += volume
if isinstance(location, WellMock):
if location.well_id.upper() not in (id.upper() for id in self.well_colors.keys()):
raise ValueError(f"aspirate() was passed well location {location} which hasn’t been configured to have a color.")
color = location.color()
newloc = location
else: # legal for aspirate() but we should probably treat this as an error for this lab? right now marking it white…
print(f"WARNING – aspirate() passed a Location rather than a well – are you sure you know what you’re doing?")
if location.point.z < 0:
raise ValueError(f"aspirate() passed a location with z={location.point.z} – do not go below z=0!")
color = ‘white’ # we don’t know where they’re asiprateing from… use an unusual color to mark it.
newloc = self.petriLocOfWell(location)
self.curr_color = color
self.totalAspirated.setdefault(color, 0)
self.totalAspirated[color] += volume
self.location = newloc
def pick_up_tip(self): # (in opentrons api)
loc = types.Location(types.Point(x=-MAX_DRAW_RADIUS, y=MAX_DRAW_RADIUS, z=0), ‘Pickup Tip’)
self.smearIfJustDispensed(loc)
if self.has_tip:
raise RuntimeError(“pick_up_tip() called when already holding a tip”)
self.has_tip = True
assert(self.aspirated_loc is None)
self.tip_count += 1
self.current_volume = 0
self.location = loc
def drop_tip(self): # (in opentrons api)
loc = types.Location(types.Point(x=MAX_DRAW_RADIUS, y=MAX_DRAW_RADIUS, z=0), ‘Drop Tip’)
self.smearIfJustDispensed(loc)
if not self.has_tip:
raise RuntimeError(“drop_tip() called when no tip was being held”)
self.has_tip = False
self.aspirated_loc = None
self.current_volume = 0
self.location = loc
def move_to(self, location): # (in opentrons api)
if location.point.z < 0:
raise ValueError(f"move_to() passed a location with z={location.point.z} – do not go below z=0!")
self.smearIfJustDispensed(location)
self.location = location
def visualize(self): # (NOT in opentrons api)
print("\n=== VOLUME TOTALS BY COLOR ===")
for color in self.totalAspirated.keys() | self.totalDispensed.keys():
comment = ’’
if self.totalAspirated.setdefault(color, 0) != self.totalDispensed.setdefault(color, 0):
comment = “\t\t##### WASTING BIO-INK : more aspirated than dispensed!”
print(f"\t{color}:\t\t aspirated {self.totalAspirated[color]}\t dispensed {self.totalDispensed[color]}{comment}")
print(f"\t[all colors]:\t[aspirated {sum(self.totalAspirated.values())}]\t[dispensed {sum(self.totalDispensed.values())}]")
print(f"\n=== TIP COUNT ===\n\t Used {self.tip_count} tip(s) (ideally exactly one per unique color)")
print("\n") # plus prints its own newline
## uncomment (only) one of these corresponding to the background medium you're printing on
plt.gca().add_patch(plt.Circle((0, 0), radius=PETRI_INNER_DIAMETER/2, color='#000000', fill=True)) # petri dish - 84mm inner diam, black agar plate
#plt.gca().add_patch(plt.Circle((0, 0), radius=PETRI_INNER_DIAMETER/2, color='#000000', fill=False)) # petri dish - 84mm inner diam, paper insert
#plt.gca().add_patch(plt.Circle((0, 0), radius=PETRI_INNER_DIAMETER/2, color='#d7ca95', fill=True)) # petri dish - 84mm inner diam, agar plate
plt.scatter(self.droplets_x, self.droplets_y, self.droplets_size, c=self.droplets_color)
for xlist,ylist,color in self.smears:
plt.gca().plot(xlist, ylist, color=color, linewidth=4, solid_capstyle='round')
plt.xlim((-(PETRI_INNER_DIAMETER/2 + 0.5), PETRI_INNER_DIAMETER/2 + 0.5))
plt.ylim((-(PETRI_INNER_DIAMETER/2 + 0.5), PETRI_INNER_DIAMETER/2 + 0.5))
plt.show()
class WellMock:
def init(self, well_id, well_color, labware_official_name):
self.well_id = well_id
self.labware_official_name = labware_official_name
self.well_color = well_color if well_color else ‘purple’
def get_row_col(self): # (NOT in opentrons api)
row = ord(self.well_id[0].upper())
col = int(self.well_id[1:])
return (row, col)
def set_row_col(self, row, col):# (NOT in opentrons api)
self.well_id = chr(row) + str(col)
def color(self): # (NOT in opentrons api)
return self.well_color
def bottom(self, z): # (in opentrons api)
assert z >= 0
return self
def center(self): # (in opentrons api)
return self
def top(self, z=0): # (in opentrons api)
assert(isinstance(z, (int, float)))
return types.Location(types.Point(x=0, y=0, z=z), 'Well')
# return self
def move(self, location): # (NOT in opentrons api) -- why do we have this here? what do we think it should do, move a well?
assert(isinstance(location, types.Location))
return self
def __eq__(self, other):
return self.__class__ == other.__class__ and self.__dict__ == other.__dict__
def __repr__(self):
return self.well_id
if "p20" in instrument_official_name:
self.display_name = "P20"
self.vol_range = (1, 20)
elif "p300" in instrument_official_name:
self.display_name = "P300"
self.vol_range = (20, 300)
elif "p1000" in instrument_official_name:
self.display_name = "P1000"
self.vol_range = (100, 1000)
else:
mock_print("WARNING: UNSUPPORTED PIPETTE")
assert false
def advance_tip(self):
row, col = self.starting_tip.get_row_col()
row += 1
if row > ord('H'):
row = ord('A')
col += 1
if col > 12:
mock_print("WARNING: OUT OF TIPS!!!")
assert false
self.starting_tip.set_row_col(row, col)
def pick_up_tip(self):
row, col = self.starting_tip.get_row_col()
assert(row >= ord('A') and row <= ord('H'))
assert(col >= 1 and col <= 12)
mock_print(self.display_name + " is picking up a tip from " + str(self.starting_tip))
self.advance_tip()
def drop_tip(self):
mock_print(self.display_name + " is dropping a tip");
def aspirate(self, volume, well):
assert(isinstance(volume, (int, float)))
assert(isinstance(well, WellMock))
assert volume >= self.vol_range[0] and volume <= self.vol_range[1]
mock_print("##### " + str(well.labware_official_name) + " [" + str(well.well_id) + "] ---> (" + str(volume) + "uL)")
def dispense(self, volume, well):
assert(isinstance(volume, (int, float)))
assert(isinstance(well, WellMock))
assert volume >= self.vol_range[0] and volume <= self.vol_range[1]
mock_print("##### " + str(well.labware_official_name) + " [" + str(well.well_id) + "] <--- (" + str(volume) + "uL)")
def blow_out(self):
mock_print(self.display_name + " blow out")
def mix(self, repetitions, volume, well):
assert(isinstance(repetitions, int))
assert(isinstance(volume, (int, float)))
assert(isinstance(well, WellMock))
assert volume >= self.vol_range[0] and volume <= self.vol_range[1]
mock_print("##### " + str(well.labware_official_name) + " [" + str(well.well_id) + "] - Mixing - " + str(repetitions) + " times, volume " + str(volume) + "uL")
def move_to(self, location, force_direct=False):
assert(isinstance(force_direct, bool))
assert(isinstance(location, WellMock))
mock_print(self.display_name + " is moving");
class OpentronsMock:
def init(self, well_colors):
self.well_colors = well_colors
self.pipette = None
#self.location_cache = None # unimplemented: opentrons api’s more canonical way to get last_location, but these protocols don’t need it
def home(self):
mock_print("Going home!")
# the opentrons api names these arguments: self, load_name, location, label
def load_labware(self, labware_official_name, deck_slot, display_name):
mock_print("Loaded " + str(labware_official_name) + " in deck slot " + str(deck_slot))
return LabwareMock(labware_official_name, deck_slot, display_name, self.well_colors)
# the opentrons api names these arguments: self, module_name, location
def load_module(self, module_official_name, deck_slot=0):
mock_print("Loaded module " + str(module_official_name) + " in deck slot " + str(deck_slot))
return ModuleMock(module_official_name, deck_slot, self.well_colors)
# the opentrons api names these arguments: self, instrument_name, mount, tip_racks
def load_instrument(self, instrument_official_name, mount_LR, tip_rack_list):
self.pipette = PipetteSim(instrument_official_name, mount_LR, tip_rack_list, self.well_colors)
return self.pipette
def pause(self):
mock_print("Robot pause")
def visualize(self):
self.pipette.visualize()
Put your name in the ‘author’ field of the metadata near the top of the first block, give your protocol a ‘protocolName’ there, and fill in the ‘description’ of what the protocol will do
Write code to create your design at the very end of the first block
DEVELOPMENT TIP: Write your code in short runnable chunks, and after you’ve written each one run both of your clode blocks (running the first one loads your code, running the second one executes it on the simulator) to see that it’s doing what you expect. Simulate often!
Learn basic concepts: amino acid structure, 3D protein visualization, and the variety of ML-based design tools.
Brainstorm as a group how to apply these tools to engineer a better bacteriophage (setting the stage for the final project).
Key Links:
HTGAA Protein Engineering Tools, HTGAA Protein Engineering Feedback
Part A. Conceptual Questions
Answer any of the following questions by Shuguang Zhang:
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Used George AI on this question. First, we begin by equating 1 Dalton (Da) to 1.66 x 10⁻²⁴ grams so that we could compute 100 Da amino acid weighs 100 grams. We decided to go with Fermi estimation instead of foundational biochemistry. Since skeletal muscle is roughly 20-25% protein by mass we decided 500g x 0.25 = 125 g protein. Then Georgeai explained that in polypeptide chains every amino acid residue loses one H₂O during peptide bond formation which is why the average residue weight is closer to 110 Da than the free amino acid weight of 128 Da. average molecular weight of approximately 110 Da. Next 125 g ÷ 110 g/mol ≈ 1.14 mol. We then assume that the average amino acid has a molecular formula of C₃H₇NO₂ has 13 atoms per residue (3C + 7H + 1N + 2O = 13), and multiplying by the number of residues gives you total atoms: 7.53 × 10²³ residues × 13 atoms/residue = 9.789 × 10²⁴ atoms.
2. Why humans eat beef but do not become a cow, eat fish but do not become fish?
Although the saying goes we are what we eat, our genomes disagree. The genes preserve function against infinite intrinsic and extrinsic stresses or other organisms’ genes. Vertically inherited animal genomes from ancestors, parents, and offspring are protected in the germ line but there also exists the functional genes that create proteins in specialized tissue mosaics that also selfishly persist as long as constituent cells survive – all cells are selfish like that. These specialized cells are sustained, though by new macromolecules which is likely why the human eat the cow in the first place. Here though Natural Selection has again preserved the reproductive programs of each species genome – starting with the taste buds and olfactory centers of the human brain. A carnivore has a digestive tract that can disassemble and denature raw muscle protein very efficiently, largely because they have different enzymes. Are lions susceptible to prion diseases? That question will make more sense in a moment, but a quick search of the internet indicates they are highly resistant (particularily to Chronic Wasting Disease). Again the enzymes in their gut protect them against 97-98% of CWDs they are exposed to. The risk to humans is different, but then so is the type of prion disease. Human also have digestive tracts though that denature the proteins and lipids of the cow before it even enters the digestive tract. Unfortunately humans also have large scale industrial agriculture and rampid inequality in access to cuts and quality of cow meat. This means that economics largely determines what parts of cows humans will be exposed and how concentrated the cows on the feed lots will be along with the condition of slaughter floor. It’s hard to know today because enzymes outside of niches are fragile things, but once humans started cooking their cow meat before shredding and emulsification in their digestive systems maybe they had more of the lion’s enzymes. The loss of those enzymes is no more felt than in the epidemiological triangle between prion in cow and the prion in man, here not gene-to-gene but protein-to-protein interaction between these two animals and the zoonotic pathogen adds a new layer of complexity.
3. Why there are only 20 natural amino acids?
> This is such a great question. Likely it was because Earth is a special planet, at least the most exceptional planet we have any knowledge about, thus far, for forming these 20 amino acids. Foundational to this question is what I love most about the study of Earth life. Evolution is change in matter in response to energetic landscapes in constant motion. Everything in biology is nested in this evolutionary onion through which cosmic evolution begets planetary evolution all the way down to the sub-atomic particles of atoms and back up to cells-within-cells. All this evolutionary change, as my astrobiologist friends tell me, started with the creation of our Universe 13.8 billion years ago. Therefore, as I learned from Dr. Graves to Dr. Lane, single-celled organisms evolved into multicellular organisms through Natural Selection and biochemical pathways that can be traced back from the 20 natural amino acids and other macromolecules that then formed single-celled organisms and multicelled organisms. We can trace origin of individual atoms in those micromolecules to macromolecules, one element at a time or in families. Throughout all evolutionary nesting, from the Cosmos to the Microcosmos, there is the unifying story of energy flowing and matter cycling. This is the root cause of the first tradeoff in Natural Selection – the struggle for existence of living organisms constrained by dynamic environmental conditions. Life, or the 20 natural amino acids, thus formed from elemental particles flowing from energy sink to energy sink over the vast horizon of evolutionary time in one long continuous chain reaction. If anyone doubts this, break every natural amino acid down to elemental atoms, starting with Hydrogen the most abundant element in the Universe.
6. Can you make other non-natural amino acids? Design some new amino acids.
We sure can but we have to be heretical and leave the canonical 2o natural amino acids behind. The process then becomes a matter of what your trajectory will be through the process. Are you going to design new amino acids within cells or externally and then from the bottom up or top down? You can make cell-free systems that include canonical and novel amino acids. However, without a hydrothermal vent underlying your invention for evolutionary scales of time, it’s hard to harness those molecules in the engine of living organisms and environmental niches through Natural Selection. Of course, we are pursuing a cell-free life, which will put the onus of selection on the creator. Do you want Elon Musk designing your child’s spouse? I digress, though. We are long from that point, RIP Dolly.
7. Where did amino acids come from before enzymes that make them, and before life started.
> Earth was formed from the same cosmic evolutionary process that formed the other planets and their moons over 4 billion years ago. Cells are an administrative variable today. What is less understood is the abiotic chemistry of the primordial oceans, could the hydrothermal vents Infact we know that orginally, when the Earth was first formed, it didn’t even have a moon. Then there was movement of Jupiter and Mars was forever changed and a giant piece of rock hit Earth which led to the formation of our Moon. Again, all this was over 4 billion years ago, and there were no amino acids or enzymes at this time. Now, I am going to throw a bunch of science at you, but just remember when I do – the Earth and Moon are a twin study. The simple answer is that when Amino Acids are synthesized in a lab, outside of a cell, they form a racemic mixture of both L-amino acids and D-amino acids. However, when they are synthesized within cells between genes and proteins for specific functions, they are almost exclusively L-amino acids. This is because cells use enzymes to speed up biological synthesis, and this contributes to the preponderance of L-amino acids. Samples of the moon’s surface from the Apollo missions contained glycine, alanine, glutamic acid, aspartic acid, serine, and threoine. This indicates that there was life generating amino acids on the moon, but the process stopped. Coincidentally, that over 4 billion years ago when the Earth was forming, a huge rock collided with it, leading to the formation of the orbiting Moon body. This allows us to deduce that some simple R-group amino acids left Earth and entered the vacuum of space where they essentially were frozen in time. Meanwhile, more natural L-amino acids continued to evolve on Earth. Where did they evolve? In bacterial and archaeal cells, the central dogma of molecular biology tells us that genes cannot transcribe and translate proteins without enzymes. Now knowing that ontogeny recapitulates phylogeny the question now becomes, who is the index bacterium, Earth life’s original synthesizer? Several months later, nearing end of course, predictions about amino acids must be made using their structural properties. There is jurisprudence in their design. At the center of every essential amino acid is an alpha carbon that holds the pivot between a reducer and an oxidizer. On the reducer end we start with a single H in the smallest AA (I dare not recall it now in case I say the wrong name, guess is leucine). On the other end of the scale is the oxidizing COO-. We call the complete molecule a zwitterion, I think. Each end of each amino acid is signal of pka roughly between 2 and 9 and in the middle the pH balances at around 7. Simple numbers have significant consequences in the most eloquent manner. Because of the Krebs Cycle, and the metabolism of living organisms that produce amino acids their growth remains balanced in advance and retreat. The balance of amino acid design maintains the goverance of chirality that ensures the assembly of living macromolecules through the geometry underlying the Central Dogma of Molecular Biology is conserved at the foundation and divergent at the periphery. Both ends of all of these continuums maintain a clockwork universe regardless of your faith system.
8. If you make an alpha-helix using D-amino acids, what handedness (right or left) would you expect?
The 20 primary amino acids are all L-amino acids, as are most protein building blocks of cells. Alpha-helices here will be the B-DNA, favoring right-handedness. Thus by the power of deduction that leaves D-amino acids, the exceptions, to the way of left-handedness. Now, the cool science here is that B-DNA favors right-handedness on Earth, but because of complementary strand synthesis, the pattern of right-handedness carries over to the A-form RNA during transcription, but not exclusively! This means that our converse hypothesis was correct, and there is another handedness in RNA, also known as Z-RNA. Furthermore, there are specific conditions that generate the Z-RNA form.
9. Can you discover additional helices in proteins?
Yes, there are primary, secondary, tertiary, and quaternary forms in proteins based on the structural chemistry of the bonding molecules. The ⍺-helix is the most ordinary secondary protein structure but in biology there are already exceptions. Examples of these exceptional helices include the pi helix or coiled coil.
10. Why most molecular helices are right-handed?
Most life on Earth is evolutionary rooted in B-DNA helices with a right-handed confirmation due to origin in saltwater oceans, passed on to self-replicating cells synthesized from macromolecules shaped by complementarities in form dominated by non-covalent weak interactions. source,
11. Why do beta-sheets tend to aggregate?
Two ways to answer this question: Hysteresis and Natural Selection. This is a why question after all, and that’s what evolution shines light on, the why behind biological structure. If you dig to the bottom of the fossil pile or record you find energetic attractor conditions for the clumping of polypeptides. Let’s start with the canonical amino acid ingredients of beta-sheet secondary structures. Every amino acid from the natural 20 is capable of contributing to beta-sheets but some are very improbable to be represented, including: Proline, Glycine, Asparatic acid, Glutamic acid, and Lysine. Conversely, some frequent amino acid suspects and properties favor beta-sheet formation. For example, the alternating chain pattern of hydrophobicity and polarity. Therefore, high probability beta-sheet producers include: Valine, Isoleucine, Phenylalanine, Serine, and Tyrosine.
12. What is the driving force for b-sheet aggregation?
β-sheet aggregation is driven by a symphony of free-energy minimization building from a backbone hydrogen-bond stability, hydrophobic desolvation, solvent entropy crescendo, and percussion of highly repetitive intermolecular packing tempo characteristic of extended peptide conformations.
13. Why many amyloid diseases form b-sheet?
first, we must understand the protein continuum. At the foundation, amino acids are connected with peptide bonds, hence peptides ennumerate from the stringing together of amino acids in chains with specific amide donor and carbonyl acceptors adding the structure. The first line of proteins are primary, and then there are the secondary proteins that have two main branches the alpha-helices and the beta-sheets or beta-pleated sheets with beta strands on the end. The b-sheets form parallel and antiparallel patterns that lead to superstructures or globular proteins, also known as tertiary and quaternary proteins. Anyway, the beta-sheets are the predominate stabilizing structure in amyloid diseases and there are many include progeriod diseases like Huntington’s disease, or neural-degenerative diseases like Alzheimer’s disease, or Parkinson’s disease, or even Amyotropic Lateral Sclerosis (ALS) as well as the growing threat through confined area agriculture of prion diseases. The even deeper dive, or physics of the amyloid beta-sheet formation, is the bioenergetic stabilization of backbones with so much hydrogen-bond stability networking, hydrophobic desolvation, steric zipper packing, and conformational self-templating, which provides an easily exploitable kinetic trap for the native protein state that amyloid formation can co-opt.
14. Can you use amyloid b-sheets as materials?
A good scientist can use anything to make the world a better place, and amyloid b-sheets are no exception. For example, like nucleotide bases, these molecular scaffolds enforce a planar geometry, which any Minecraft artist knows makes an excellent array of possible forms with a bit of imagination. Geometry is highly underrated these days. Beta sheets are uniformly stackable and scalable with the functionality that follows such capabilities in material.
15. Design a b-sheet motif that forms a well-ordered structure.
Copied from Wikipedia
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins.
I summon mTOR for my protein (including 4JSV, 4JSN, 4DRH, 4DRI, 3ML9, 5WBH, 5GPG, 5H64, 5FLC, 6BCX, 6BCU, 6SB0, 6SB2, 6ZWM, 7PE7, 7PEC, 8ERA, 9ED4, 9ED6, 9ED7, 9ED8).
Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions.
Briefly describe the protein you selected and why you selected it. The protein I am now selecting now that I better understand the exercise and tooling is the mighty mTORTOR mechanistic target of Rapamycin protein kinase. I pick this protein because just trying to write the name is challenging. In add there is conserved and then there is mTOR conserved, it’s as common as Cytochrome C. Also I like the puzzle of navigating all of the codes just to plug it into the codebase – the combinations are amazing as my son’s Pokomon characters would say. Lastly, it has a lot of wiring for considering circuitry as a quintessential regulator of Natural Selection programs for growth, maintenance, and metabolism under stress.
Identify the amino acid sequence of your protein.
My protein is human mTOR, which like everything in biology, must be extremely complicated. Thus the one entity has many partial constructs shown below in the table. The full human mTOR sequence from Uniprot with accession number
P42345
There are 2549 amino acids in human mTOR. I am going to use the revert back to the surfeit locus protein 2 for the remaining questions and answers.
The sequence for surfeit locus protein 2 is:
MDEPPSDVLAFLRQHPSLRLLPNTRKVRCSLTGHELPCRLPELQEYTRGKKYQRLSSSFSNFDYAAFEPHIVPSTKNRHQLFCKLTLRHINKSPEHVLRHTQGRRYQRALHQYEECQKQGVEYVPACLLHKRKKREDQTNSDELPGQRTGFWEPASSDEEDALSDDSMTDLYPPELFTKRELGKPKNDDTPEDFLTDQQDEKPEHSEEKSFREREEARVGHKRGRKLRKKQLTSLTKKFKSYHHKPKNFSSFKQLGR
My old protein show below in current analyses is surfeit locus protein 2 protein is: 257 amino acids. The most common amino acid is: L, which appears 27 times. All Amino Acid Frequencies:
Total Sequence Length: 257
Amino Acid | Count | Frequency (%)
L | 27 | 10.51%
K | 25 | 9.73%
E | 24 | 9.34%
R | 24 | 9.34%
S | 20 | 7.78%
P | 18 | 7.00%
D | 15 | 5.84%
Q | 15 | 5.84%
T | 14 | 5.45%
H | 13 | 5.06%
F | 12 | 4.67%
G | 10 | 3.89%
A | 8 | 3.11%
Y | 8 | 3.11%
V | 7 | 2.72%
N | 7 | 2.72%
C | 5 | 1.95%
M | 2 | 0.78%
I | 2 | 0.78%
W | 1 | 0.39%
6. How many protein sequence homologs are there for your protein? Hint: Use the pBLAST tool to search for homologs and ClustalOmega to align and visualize them. Tutorial Here
Does your protein belong to any protein family?
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
Are there any other molecules in the solved structure apart from protein?
Does your protein belong to any structure classification family? SURF2
Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Part C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein. Copy the notebook below and set up a colab instance with GPU for this section:
HTGAA_ProteinDesign2026.ipynb
Choose your favorite protein from the PDB. We will now try multiple things, report each of those results in your homework page:
Protein Language Models:
Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality
Analyze the different formed neighborhoods: do they approximate similar proteins?
Place your protein in the resulting map and explain its position and similarity to its neighbors
Attention Maps
Analyze the attention maps of ESM2. Investigate if its layers correlate to the 2D map of residue distances of your protein
Protein Folding:
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Protein Generation:
Inverse-Folding a protein
Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one
Input this sequence into ESMFold and compare the predicted structure to your original
Last step in my copy of HTGAA_ProteinDesign2026.ipynb script
New Sequence:MPPLPPEVVAFLAQHPHLVALPGQPLVRCTLTGEELPAELPVLRAHVATPRHQALAAREKNFDFSKYEPHIVPSRWDPDKLFCRLCLKEIPKTPEAVEAHVNSKEHQEALKEYEEAKKRGKRYIPKRLRKRRRRRRRRRRRRRGRRKRRKRRPPPPRRRPRRKRRRRRRRLVPREWLRRRRRRRRRRRRRRRPRRRRRPPRRVVGAAPEPAVAALAEAPAPPAPPAPPPPPEERPEPPPPPPERREPPPEELEEEEE
Part D. Group Brainstorm on Bacteriophage Engineering
Find a group of ~3–4 students
Review the Bacteriophage Final Project Goals:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
Why you think those tools might help solve your chosen sub-problem.
One or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
Include a schematic of your pipeline
This resource may be useful: HTGAA Protein Engineering Tools
Individually put your plan on your website page
Each group’s short plan for engineering a bacteriophage
Schedule time ( HTGAA Protein Engineering Feedback) to get feedback/discuss your ideas, and put the feedback on your website
[Optional] Part E. Find a drug for an oncology target
Week 5 HW: Protein Design Part ii
[] Homework — DUE BY START OF MAR 10 LECTURE
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc. Mechanis
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
[] Task A
Your challenge:
Background: Design short peptides that bind mutant SOD1 and then decide which ones are worth advancing toward therapy. You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling.
Here is fully translated superoxide dimutase protein P00441 in uniprot with the initiator methionine included. We need to cleave that M off before we apply our requested mutation to progress with a mature enzyme.
So not this…
1 2 3 4
M A T K
But this..
1 2 3 4
A T K A
To create our A4V SOD (love the rhyme) mutant…
1 2 3 4
A T K V
The savvy student who fails to cleave the first methionine (M) can intuit the actual amino acid to change without thinking through any of the previous steps, but it’s nice to have a why in all things, since this is biology after all and we have evolution and ChatGPT. Please note that we will not want to use a protein sequence with any sort of truncation or wrapping on the sequence so here are my sequences for PPMLM-650M.
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
To create our A4V SOD (love the rhyme) mutant…
1 2 3 4
A T K V
Mutant A4V SOD for PepMLM-650
There are two options, full protein sequence and a 12-Sequence input which I settled on in later runs. MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Within the PepMLM-650 codebase in Google Colab Notebook, there are sliders and input fields to parameterize individual runs. However, these parameters didn’t seem to encode, so I finally hard-coded changes, as I will show below as a series of excerpts pulled from the codebase.
single_sequence = True #@param {type:"boolean"}
protein_seq = "MATKVVCVLKGD" #@param {type:"string"}
# Initial value for num_binders
num_binders = 4
# Initial values for top_k and peptide_length
top_k = 3
peptide_length = 12
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Scores above, I think. I feel like I’m in Gulliver’s Travels returning this exercise cold after 2 months so I am going to try and piece together memory using a peers homework. For the next phase I am now going to find the amino acid sequence for my SOD1 sequence. Now I’m not copying information because I just went to uniprot site myself and searched for SOD1 on the splash page and scrolling down on first page I found human sodc but I skipped over that one and found the sod for sheep
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Given confusion perplexity scores are likely very high. The model confidence according to UniProt is spot on though, specifically model confidence is very high (pLDDT > 90). This is generated by AlphaFold as a per-residue confidence score (pLDDT) between 0 and 100. Now I am prepared to transition to part 2.
What about my mutants, though? I do not want to disrupt folding randomly, need an appropriate target region for mutation logic so I will leverage the MobiDB website.
Enumerated Amino Acids with position and highlighted for subsequent mutagenesis based on encode segment flexibility, disruptability, and functional consequences
Navigate to the AlphaFold Server: [alphafoldserver](https://alphafoldserver.com/welcome)
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
There were two mutation pathways considered in this section the A4V change resulted in a 0.1 ipTM drop. I can comment on the mutation I engineered above, which introduces a three-residue change (G71A/G72A + E76P). The predicted template modeling (pTM) score and interface predicted template modeling (ipTM) scores are based on the template modeling (TM) score which are all metrics available in the AlphaFold Server visualization. The TM was originally proposed by Zang_&_Skolnick based on the Global Distance Test (GDT) and MaxSub. The scores are evaluated using a statistical association, measured by a correlation coefficient, after adjusting for differences in protein size. An interesting observation in Abramson et al. (2024) methods article they do not resport statistical tests of association due to small n populations paper. The paper describes pTM and ipTM as global ranking variables that can increase rates of disorder in model. In addition chain ranking can be performed with a variation of the pTM metric and pLDDT can be averaged for putative residues.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
There can be alignment between the computational prediction scores from AlphaFold Server, specifically ipTM rank score, which are both “significant” because they are larger than 0.80 but the wild type ipTM score is 0.01 larger than the mutant ipTM. In regards to the thermodynamics expressed in the PeptiVerse datasheets. Solubility and penetrance increase in the WT as hydrophobicity declines, compared to the mutant, which has greater hydrophobicity and lower solubility and penetrance. The explanation for the thermodynamic differences between the wild type and the mutants is the exposure of hydrophobic bases to the solution, leading to more water cages forming around them in the mutant than in the wild type.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
~Make sure to switch De Novo to enter manual sequence
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
~Set target for affinity to original WT: MATKAVCVLKGDGPVQGTIRFEAKGDKVVVTGSITGLTEGDHGFHVHQFGDNTQGCTSAGPHFNPLSKKHGGPKDEERHVGDLGNVKADKNGVAIVDIVDPLISLSGEYSIIGRTMVVHEKPDDLGRGGNEESTKTGNAGGRLACGVIGIAP
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies? It doesn’t stay connected to the necessary server to run.
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
Boltz Lab
BRD4 Drug Discovery Platform Tutorial
Introduction
This exercise walks you through a real drug discovery workflow using the Boltz Lab platform - from predicting how known drugs bind a cancer target, all the way to running AI-generated molecule libraries and interpreting the results.
You will work on BRD4 (Bromodomain-containing protein 4), an epigenetic reader protein and validated oncology target. BRD4 has been the subject of intense medicinal chemistry effort in recent years.
What you will learn
• How to use Boltz Lab to predict protein-ligand binding structures
• How to interpret Binding Confidence and Optimization Score metrics
• How to set up a virtual screening project in Boltz Lab
• How to compare known drugs and AI-generated molecules in a single workflow • How to critically evaluate computational predictions from a drug discovery perspective
Background: BRD4 and BET Bromodomains
BRD4 is a member of the BET (Bromodomain and Extra-Terminal) family of epigenetic reader proteins. It recognises acetylated lysine residues on histone tails and recruits transcriptional machinery to gene promoters, driving expression of oncogenes including c-Myc. Dysregulated BRD4 activity is implicated in haematological malignancies, solid tumours, and inflammatory disease.
This exercise inspects the example of JQ1 - the landmark BRD4 inhibitor reported by Filippakopoulos et al. in Nature 2010. The three compounds below capture a hit-to-candidate optimisation journey, including a deliberately instructive stereochemical twist.
Stage
Compound
SMILES
Hit
Stripped Back Core
CC1C2C(=C(SC=2NCCN=1)C)C
Lead
Triazole + Acid
O=C(C[C@@H]1N=C(C)C2C(=C(SC=2N2C1=NN=C2C)C)C)O
Candidate
(+)-JQ1
O=C(C[C@H]1C2=NN=C(N2C3=C(C(C4=CC=C(C=C4)Cl)=N1)C(C)= C(S3)C)C)OC(C)(C)C
�� Note: Reference: Filippakopoulos P. et al. Selective inhibition of BET bromodomains. Nature 468, 1067-1073 (2010). Crystal structure PDB: 3MXF (BRD4 BD1 complexed with (+)-JQ1).
source
Tutorial designed by Geoffrey Smith
Boltz Lab | BRD4 Platform Tutorial — MIT Guest Lecture
Part 0: Sign-up to Boltz Lab
Go to lab.boltz.bio, click “Request Access”, add your name and email while specifying as organization name “HTGAA”, and click “Submit request”.
We will try to make sure to approve your request within a day or two, giving you credits for both the exercise as well as further exploration. If you plan to use Boltz Lab for your final project and need more credits, please reach out to me at gabriele@boltz.bio. Part 1: Structural Predictions in the Sandbox
Start with three Boltz-2 predictions in the Sandbox to understand how the model scores protein– ligand interactions across a real drug discovery progression.
1.1 The Boltz-2 Metrics Explained
Before you run your first prediction, understand these three key outputs:
Metric
Range
What it means
When to trust it
Binding Confidence
0 - 1
How confidently Boltz-2 places the ligand in the binding site.
Higher = predicted more likely to bind.
0.7 considered reliable; > 0.8 high confidence
Optimization Score
0 - 1
A relative affinity for use in congeneric series, or between known binders. Higher = predicted to bind more tightly.
Use for relative ranking,
Structure Confidence
0 - 1
Measures the confidence of the predicted structure
Higher = more likely the structure predicted correctly.
0.8 considered high confidence.
You need all three to be high to trust a prediction.
1.2 Running Your Three Predictions
Navigate to the Boltz Sandbox at lab.boltz.bio and log in to your account.
Go to Sandbox → New Prediction
Name this BRD4 binder JQ1
Select ‘Complex’, add ‘Sequence from RCSB’, and add 3MXF
Continue through Constraints (not needed for this example), and select Jq1 as the Binder for an affinity prediction.
Submit the prediction.
Use the ‘Duplicate Prediction’ in the results review, and remove the small molecule.
Add in the SMILES for the Hit and Lead.
When predictions complete, record your results in the table below
Tutorial designed by Geoffrey Smith
Boltz Lab | BRD4 Platform Tutorial — MIT Guest Lecture
Compound
Binding Confidence
Optimization Score
Structure Confidence
Hit
Compound
Binding Confidence
Optimization Score
Structure Confidence
Hit
Lead
JQI
Discussion Questions • Does Binding Confidence increase as you move from hit to clinical candidate? What would you expect, and why might it deviate?
• Inspect the predicted binding pose for JQ1. Can you identify potential key binding interactions.
• Compare the Optimization Scores. How do the scores compare for JQ1 vs the Lead. Part 2: Setting Up a BRD4 Design Project
Now you will create a small molecule Design Project - the Boltz Lab workflow for virtual screening and lead optimisation. We will set up BRD4 as a target using the clinical candidate as our structural reference.
2.1 Creating the Target
From the dashboard, create a Design Projects via ‘New Project’
Name your project: ‘BRD4 Workshop '
Select ‘Small Molecule’
Click Add Target and add the protein structure as in the Sandbox using PDB code 3MXF 5. Continue and let the apo structure complete. Continue if the structure looks good. 6. Leave binding residue selection blank, the platform will auto-detect the pocket 7. In the Molecular Probe field, paste the JQ1 SMILES.
Predict Pocket Structure and complete the Target Set-Up
�� Note: Why no binding residue selection? Boltz Lab uses the probe SMILES to identify the relevant binding pocket automatically.
What the Probe Does
The probe compound defines the active site geometry for the target. Boltz-2 uses the cofolded probe structure as an internal reference when scoring your library compounds. This is equivalent to providing a crystallographic template in traditional docking - except the model generates the structure on the fly.
Tutorial designed by Geoffrey Smith
Boltz Lab | BRD4 Platform Tutorial — MIT Guest Lecture
Part 3: Running Your Virtual Screen
BRD4 is a well validated target, and therefore we will generate a small Library of 1K small molecule binders. For typical exploratory targets, Boltz recommends 20K as a minimum number of binders.
3.1: Run a Generative Design Campaign
We will utilize the Boltz Lab small-molecule generative workflow. This generates novel molecules optimised for BRD4 binding using Boltz-2 as the scoring function.
After creating the design project, Boltz Lab will prompt you to Generate binders with AI. 2. Name your experiment, provide a relevant hypothesis, and Create the Experiment. 3. The New Virtual Screen will be pre-configured with a Generative screen using the Enamine REAL space.
Keep ‘Normal Filtering’ selected. This will ensure we only generate molecules acceptable to a medicinal chemist.
Decide if you would like to apply any Molecule Filters. We recommend the ‘Drug-Like’ Preset.
Select a custom number of Binders and enter 1K.
Start the Virtual Screen.
Allow binders to be generated, and View Results in Experiment
�� Note: 1k molecules is a very small screen, for real applications where you plan to synthesize the molecule (e.g. your final project) we would recommend running at least 10-20k molecules.
Part 4: Analysis and Discussion
As your experiment completes, use the ‘Quick Add Candidates’ on the experiment screen to add JQ1 as a benchmark for generated designs.
4.1 Interpreting Your Results
As your experiment completes, use the ‘Quick Add Candidates’ on the experiment screen to add JQ1 as a benchmark for generated designs.
From your screen output, identify three categories of molecules:
Category
Criteria
Likely interpretation
High confidence binders
Binding Confidence > 0.80 Opt. Score > 0.40
Strong predicted hits - inspect poses carefully
Moderate confidence
Binding Confidence 0.65–0.80 Opt. Score 0.25–0.40
Plausible binders - additional validation needed
Low confidence / non-binders
Binding Confidence < 0.65 Opt. Score < 0.25
Likely incorrect pose or non binding chemotype
Tutorial designed by Geoffrey Smith
Boltz Lab | BRD4 Platform Tutorial — MIT Guest Lecture
Discussion:
As the virtual screen completes, assess the following:
• How does JQ1 in the Design Project screen alongside the library. Does it score as the top compound? • How do the top scoring binders compare in binding pose to JQ1?
• Try adding a second target to your project via the dropdown in the structure viewer, for example, BRD2 (PDB: 5UEN). Re-run the top scoring binders against BRD2 and compare which compounds score highly for BRD4 but not BRD2. This is a selectivity analysis - a key part of real BET inhibitor programs.
Resources and Further Reading
Resource
Link / Reference
Boltz Lab Platform
docs.boltz.bio
Key BRD4 Paper
Filippakopoulos P. et al. Nature 468, 1067–1073 (2010)
JQ1 PDB Structure
rcsb.org/structure/3MXF
Tutorial designed by Geoffrey Smith
Part C: Final Project: L-Protein Mutants
This homework requires computation that might take you a while to run, so please get started early.
Tools
See HTGAA Protein Engineering Tools spreadsheet
Week 6 HW: Genetic Circuits Part i
[]Homework — DUE BY START OF MAR 17 LECTURE
Week 6 HW: Genetic Circuits Part 01
Assignment: DNA Assembly
Protocol and Study Questions
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
A proprietary gold standard heat-stable DNA polymerase alternative to Taq reagent synthesized and sold by Thermo Fisher Scientific. Unlike Taq which was isolated from thermophilic bacteria, Phusion emulates an archaea-based enzyme that evolved in the hydrothermal vents from extremeophile species. They function as DNA polymerases essentially in a form biomimickry with minimal replication error. The purpose of Phusion is to amplify target DNA sequences in the PCR protocol. Phusion PCR is more expensive but worth the investment to increase the accuracy of the run.
What are some factors that determine primer annealing temperature during PCR?
I don’t know if a question will formally cover this, but PCR methods include initialization, annealing, and extension. Heat is first applied in the initialization step of a hot start PCR protocol. There are two temperature modalities with a typical run: 205 °F or 208 °F. Phusion polymerase would be a proper reagent for a hot start PCR run. The next phase, denaturation, again includes a 201-208 °F step to separate double-stranded DNA templates by breaking hydrogen bonds. The next temperature cycle is the annealing step where temperature drops to 122–149 °F. A key factor with temperature annealing is to be exact with temperature and time to avoid an off-target reaction mixture.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Fundamentally, the first difference between restriction enzymes and PCR is the inventor. Bacteria invented restriction enzymes to decrease the size of their individual single chromosome genome through Natural Selection to adapt to environmental niches faster. A professor once explained it to me like the traveller who embarks into the desert. Why would they carry junk DNA they do not need when they can prioritize every ounce of storage space for genes they will need to survive in the desert? PCR is an entirely different angle. Now we, the scientists, are using a laboratory machine to initialize, anneal, and extend sections of DNA we are interested in replicating experimentally. In fact, it is the exact opposite mechanism, like the continuum between divestment and investment of DNA. In terms of protocols, restriction enzymes are more of a puzzle based on the actual information available genetically, and PCR can be applied to any segment of DNA anywhere on the genome that can be extracted. Additionally, there is a quality differential between approaches in the input DNA.
How does the plasmid DNA enter the E. coli cells during transformation?
Bacteria naturally use three methods to transfer genetic material, including conjugation, transduction, and transformation. Conjugation requires direct contact. Transduction uses phages as intermediates. Transformation occurs in nature when bacteria incorporate genetic material from dead bacteria in the environment. Scientists have learned how to leverage bacterial transformation using heat shock and electroporation.
Describe another assembly method in detail (such as Golden Gate Assembly (GGA))
Golden Gate is one of my favorite parks in Cali and GGA is a new (circa 1996) assembly method detailed by New England Biolabs. Key components are Type IIS restriction enzymes, T4 DNA Ligase, the “backbone”, and Transcription Activator Like Effectors (TALEs). Conceptually, GGA is revolutionary because it unites restriction enzymes and PCR amplicon assembly in an expedited way. The workflow for GGA has a two-modality kit for either BsaI or BsmBI directed assemblies, both are restriction enzymes with recognition sites that generate 4-bp overhangs when cut. The they have different recognition sites, BsaI is for standard assembly and uses GGTCTC and BsmBI is a hierarchical system mod that uses CGTCTC.
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
An alternative to GGA is Gibson Assembly. In this approach:
Step01: The homologous DNA assembly fragments must overlap by 20-40 bps.
Step02: The reaction mix with 5’ exonuclease is applied which chews back 5’ ends of both fragments.
Step03: The matching x overlapping ends then anneal or base pair spontaneously which aligns the joined fragments in the correct order.
Step04: The addition of a high-fidelity DNA polymerase (i.e., Phusion) then extends the missing bases to produce the 3’ ends.
Step05: The Taq DNA ligase is then applied to fill in the gaps, and with that all fragmentary assembles continuous double-stranded DNA plasmid or linear construct.
Model this assembly method with Benchling or a similar tool!
Repeating a similar Benchling workflow to BioClub Japan teammate Nourelden Rihan's notebook. Not only is Dr. Rihan a natural leader of excellence in the work but his formatting makes me wish I were Egyptian.
In the workflow is to stich a GFP Protein to a plasmid. I tried the ENA route with a different vector and GFP ideal for mitochondrial superfluorescence imaging. I then took a different course.
First, I instead start with a eGFP protein without any additional fragments attached to it– shown here as U55761_EGFP_CDS translated into AA sequence for better artistic effect.
U55761_EGFP_CDS
Amino acid Count
Ala A 8 3.3%
Arg R 6 2.5%
Asn N 13 5.4%
Asp D 18 7.5%
Cys C 2 0.8%
Gln Q 8 3.3%
Glu E 16 6.7%
Gly G 22 9.2%
His H 9 3.8%
Ile I 12 5.0%
Leu L 21 8.8%
Lys K 20 8.4%
Met M 6 2.5%
Phe F 12 5.0%
Pro P 10 4.2%
Ser S 10 4.2%
Thr T 16 6.7%
Trp W 1 0.4%
Tyr Y 11 4.6%
Val V 18 7.5%
Pyl O 0 0.0%
Sec U 0 0.0%
Net Charge
After wrestling with my pre-loaded mito_mGFP plasmid, I used ChatGPT to find the simplest, vanilla, mammalian expression vector with CMV promoter and selectable marker possible. Mission Accomplished – not quite, many more steps to follow after this. Also shown in this screenshot from the Benchling account is the primers that I had to create to guide the Gibson Assembly to follow.
pcDNA31_plasmidwREIIandPCRPrimers
Assignment: Asimov Kernel
Protocol and Study Questions
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
Resources
Primer Design: HTGAA’s Supplement to Gibson Assembly Recitation
NEB’s (New England Biolabs) video Introduction to Gibson Assembly
NEB’s (New England Biolabs) explanation & protocols for Gibson Assembly®
Week 7 HW: Genetic Circuits Part 2
[]Homework — DUE BY START OF MAR 31 LECTURE at 2PM ET
Week 7 HW: Genetic Circuits Part 2
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs are ideal for the continuous transcriptomic-driven change observed in cells that are constantly moving and communicating in their intracellular environment – through analog computations. In contrast, much of the early synbio genetic circuit engineering was digital, with discrete logic gate switch programming or perhaps even through gene knock out (present versus absent) if such a connection would be permitted.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Perhaps it’s still a bit conceptual at this time, but the redox lesion-boundary in my personal HTGAA project may be a useful application for framing an intracellular artificial neural network (IANN). My goal would be to create a perceptron-esque intracellular circuit used to classify tissue sample sites by integrating continuous biochemical inputs into a weighted threshold graded output that identifies lesion boundaries in host animals. This data will be dependent on a time-series with continuous expression and variation that is spatially distributed across host goats. The input layer in this model might not even include a mite or mite eggs as is traditionally used but instead models inputs like ROS, hypoxia, inflamatory damage that spreads across hosts in similar phenotypic patterns. The weights for this model would be the expected promoter strength, affinity, repression, similarity. Activation would be evaluated using threshold nonlinear gene response (it’s biology afterall) and the output would be fluorescent markeing (highlighting) of lesion bounardy cellular expression.
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
With a minimal understanding of this week’s lab but help of the world wide web search and protcol droid Chat-GPT, I am now going to try to adapt the Cortassa lab’s (2004) computational mitochondrial oscillator on the reactive oxygen species model to the diagram challenge for the intracellular multilayer perceptron challenge. Why? I am endlessly fascinated by both. The Cortassa model hypotheses target spontaneous metabolic oscillations in heart cells, and is built using sound biology and stone-cold math. I also respect that Cortassa was modelling the mitochondria in 2003; we didn’t know anything about mitochondria back then, compared to what we know now. Another great feature of the Cortassa model is the scalability across time. Often, when we discuss the interstellar nature of scale in biology, we focus on space, but time is just as perplexing when it comes to the bioenergetics of metabolic redox reactions, as Cortassa et al (2004) describe, with timescales from milliseconds to hours. Lastly, the Cortassa model even considers Fluorescent probes, so as a side note, if this lab is looking for a PostDoc.
Cortassa Lab Dynamic Mitochondrial Oscillator Model
The dynamics are nonlinear
Each modelled mitochondrion is assumed to possess an inner membrane anion channel abbreviated as IMAC.
An IMAC is activated
An IMAC is modulated by MG2+ and PH
An IMAC is inhibited by amphiphilic molecules. The model has two state changes:
There is the relaxation mitochondrial oscillator state with slow and fast spaces. Over the slow space ROS builds up in the mitochondrial matrix
There is the stable mitochondrial oscillatory state
Source:
Cortassa S, Aon MA, Winslow RL, O’Rourke B. A mitochondrial oscillator dependent on reactive oxygen species. Biophys J. 2004 Sep;87(3):2060-73. doi: 10.1529/biophysj.104.041749. PMID: 15345581; PMCID: PMC1304608.
Cortassa S, Aon MA, Marbán E, Winslow RL, O’Rourke B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J. 2003 Apr;84(4):2734-55. doi: 10.1016/S0006-3495(03)75079-6. PMID: 12668482; PMCID: PMC1201507.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for?
Fungal melanin is used for radiation shielding in spaceflight. Fungal chitan and chitosan hydrogels are being developed medical grade purification and regenerative tissue scaffolds. Mycelium foam as a new line of sustainable nanoscale fiber networks for textiles, upholstery, and shoe support materials. Fungal materials in form of mycelium composites with straw are also being used to engineer sustainable biobuilding materials. I found this nest in a pile of holly branches my wife had trimmed – I was opposed to it exactly for this reason. Nonetheless, holding this nest and thinking about existing fungal materials I can’t help but wonder why the Robin who built this wasn’t contemplating this question as well. Certainly in the evolutionary history of animal housing and nest building there are many unknown libraries of fungal materials integrated in the selection logic in say a bird’s intuition about different materials in different states of decay for each segment of the nest. I wonder how many generations it took for the American Robin to master the architecture for this nest. I think that fungal materials could also be designed using variations of the approach above to help more birds build their own nests – like the HTGAA Habitat for Humanity in a time when so many local nest-building species are increasingly losing their habitats and habits to parking lots and rental properties.
What are their advantages and disadvantages over their traditional counterparts?
I think it’s only upside for the applications I am envisioning. Except for biofilms, which pose a whole new set of challenges that I must investigate further. Mostly I am thinking of applications for housing animals and supporting animals in their construction projects. For example, birds will abandon a nest if a human touches it after they build it but set the material aside in a pile and the birds will help themselves to any innovation that meets their design needs. Same for dogs as well, I recently learned from one of our farm animal companions - Baku’s new preferred housing.
What might you want to genetically engineer fungi to do and why?
There are so many things that we could genetically engineer with Fungi, so why is it so confounding just to pick one? Ironically, it’s easier to imagine what a bird would genetically engineer with fungi. Shelter is the first demand after all for survival; a bird’s nest could be a noble odyssey for scientific discovery. To start, what do birds build nests with? On this farm, birds use sticks, hay, and straight wool. What do all of these substrates have in common? For the most part, they are straight and bendable, but there are other notable attributes. Hay and Wool for example, are resistant to most environmental antagonists: water, wind, bugs, and fire. I also think they use composites because they are structurally reliable with a predictable function. For example, all three are tough enough and not heavy. A bird can pick up a piece with its beak, fly back to the nest, and drop it where they want it. Does this make it a tool? Anyway, once they drop a piece of stick, wool, or hay they then just have two more steps: tuck in side A and then tuck in side B – done. The problem with all three of these materials is that they exist independently of the task of providing birds with material for nests. Enter a fungi farm for bird nest materials. All we need are yeast plasmids, a PCR machine, a gel electrophoresis device, reagents, electricity, computers, and revolutionary institutions to teach us how. Fortified with our why and how, we commence…
What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
There are several ways to approach this question. We choose bacteria because of their remarkable ability to adapt. While it’s important to acknowledge the bacterium’s collaboration with natural selection, it took over a billion years for the kingdom to develop this trait. Traditionally, we prioritize bacteria in synthetic biology because bacteria occupy the niche first. Why is this significant? Well, it can be summarized in one sentence: the niche is where matter and energy intersect. Fungi eventually occupy the niche also and when they do, they provide structure and in a less specific way. Perhaps their universality is because fungi possess the genes of plants and animals. Scale, is another reason Fungi. Bacteria leaned millions of years ago to the illusion of invisibility to multicellular ocular adaptations, which was advantageous to their race for niches against multicellular competitors. Not to downplay humans too much, we figured this out too, and we can even watch bacteria at their little spinning wheels and transform them from the inside out. However, be it tangential or not, I still wonder where the evolutionary studies are following the synbio bacteria prospectively as they reintegrate with nature. Therefore, in summary, fungi over bacteria because they replicate so slowly for microbiota, which for example better adjusts to human chronological understanding of space and time.
Installation Guide and Walk-through of the Neuromorphic Wizard Software.
Open link for the installation file: https://drive.google.com/drive/folders/10_gEzYV2J5hVOdKt6sNBeSH8cEMMGX8O
Click the three dots.
Click Download.
Move the folder to somewhere you can access it from Terminal with cd command.
If the downloaded file is zipped, then unzip it.
In terminal run cd into NeuromorphicWizard and ls contents
In terminal run conda create -n neuro_wiz python==3.10
In terminal run conda activate neuro_wiz
In terminal run pip install -r requirements.txt
In terminal run python main.py
Assignment Part 3: First DNA Twist Order
Review the Individual Final Project documentation guidelines.
Submit this Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs. DUE MARCH 20 FOR MIT/HARVARD/WELLESLEY STUDENTS
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
Homework Part A: General and Lecturer-Specific Questions
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Describe the main components of a cell-free expression system and explain the role of each component.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Let it be noted, I really want to use George AI to engage this question, but I am running out of time, so I am just pitching shots up on the green to try and finish off with a putter and I also recognize metaphors are symptomatic of a weak mind, so there you have it. This is why we need AI too. Most people are susceptible to weak-minded syndrome. Therefore, since my overall understanding of what I am describing here is dulled, please do not try any of this at home. The bioenergetic cycles we wish to reconstruct in the cell-free environment literally resemble a water mill. I start with these two examples because one makes sense to me, and the other appears to be the same engineering concept but now there are some critical features missing that makes it more difficult to reconstruct the working order of things. I believe the same problem challenges us with cell-free systems. We have all the parts and experiments are clearly designed but what sustains them? There are clearly hidden variables that cannot be intuited at first glance.
Now I am afraid if just put up my next image it will be swept away do to copyright infringement laws since I didn’t personally take a picture of this biochemical pathway.
The irony of course is that the water mill and whatever mill above were invented when Newton was still alive if not before, likely long before, anyway the cellular aerobic respiration cycle was discovered inside of living organisms on Earth. In addition, we should note that the cellular cycle is part of many interacting open systems, and the other two mills are closed systems embedded in open living systems. Infact, is there anything sadder than a watermill without water for there in the bones of brick and iron is a functionless relic of a time before atomic energy had been harnessed. Perhaps a time we will return to in the end, but enough conjecture.
A cell-free system picks up where the watermill ends, beginning from the assumption that what we are doing here will be as difficult as designing an engine powered by a river that is no longer there. Thus, the cell-free system must build the mill and the river and then let go, with the prediction being that if we are successful, this new cell-free system will keep running independently of our intervention.
Now, in full disclosure, I might not be resilient enough to imagine a cell-free system world because I have been so conditioned by the Earth-assisted invention cycle, and I am overly sentimental about the importance of existing Earth Systems, as we inherited them from previous generations. That being said, I am not fully naive either and recognize where we are headed. Cell-Free systems are acceptable when the alternative is extinction.
Certainly, if we have a mill already built, there are many less promising alternatives than finding a way to regenerate the river that was and fire that mill back up, and sustain them both.
Function is clearly the keystone in the design of any cell-free form. Consider the aerobic respiration cycle in our cells, moving from left to right. Let’s categorize two functional buckets in the first cut. The first bucket is for the river, or the input systems, or energy sinks that are milled to power the functions in the other bucket, the cellular motion. The Glucose goes into the river bucket; it is derived as an independent input. Fortunately, in this model, at least Glucose is initially derived from external foodstuffs and converted into a crude energy source, regulated by dosage and intake. However, Glucose will also be an output of the internal system and will later be stored. Another key takeaway about the Glucose it’s one of the only initial inputs (aside from Odd-Numbered-Fatty-Acids) that is not an essential Amino Acid. This is why, when I lived in Boston and worked for the Boston Public Health Commission (BPHC), I survived on bargain fruit from the grocery store– no one was taking Glucose away from me just because I was a poor Epidemiologist.
Another key feature of the aerobic respiration cycle is the neighborhoods in the overall metabolic map that different essential amino acids contribute to synthesizing enzymes required by the cell organelles. A similar process is observed in the waterwheel-powered grain mill. If you go inside the structure of a working watermill you will see an array of different shaking and turning gears and swinging rods all in concert to grind up harvested grains and kernels. However, today the water does not flow directly beside the mill, it has been rerouted and piped for pressurization, and then it falls onto the wheel to turn the primary shaft. The great thing about CFS is they too can utilize different pipes or pipette arrangements for additive components of the expression. For example the first reaction in the Glycolysis cascade involves the addition of Hydroxy-proline to Pyruvate as well as conversion to Glyoxylate. In addition, there are four other critical Amino acids, including Alanine, Glycine, Serine, and Cysteine, and then the addition of Threonine, which is also added at the same time to Acetyl-CoA, so it’s ready as a key enzyme for OXPHOS in the Mitochondria or CFS mitochondrial equivalent. However before the Acetyl-CoA can even be completely synthesized the Pyruvate must also be prepared as well as the Lucine.
In fact, Acetyl-CoA is actually a much more nuanced synthesis step because although some Leucine will directly lead to the final Acetyl-CoA most will come through the build-up or synthesis of Aceto-Acetate along with Phenylalanine and tyrosine. Only then can the Aceto-Acetyl-CoA be synthesize with Even Numbered Fatty Acids and Lyine and Tryptophan to further increase the supply of Acetyl-CoA.
Now what’s extraordinary about all these steps so far is that they only get us to the actual Kreb’s Cycle, which is the essential design element. In living cells, almost all of these reactions take place in the mitochondria. That said, there is a transition-state relationship between the mitochondria and the nucleus of the living cell that varies across phylogeny. Specifically, the variation here is the level of completeness in the transition of mitochondrial genetic information and associated functions to nuclear genome transcription and translation. This transition could potentially by supported by CFS designs that could identify artificial selection acclimations and adaptations that could further facilitate what many believe will be an inevitable combination of mitochondrion and nuclear genomes.
Once inside the Krebs Wheel there are many advances to consider. For example unlike a waterwheel it matters which direction the Kreb’s Cycle turns. Temporality is also a requirement. There are many molecules and reactions that must already be working in the Krebs’ Cycle when the Glucose starts flowing. For example, there must be a balanced exchange between Aspartate and Oxalo-Acetate. Infact it is explainable here to see why Oxalo-Acetate has direct pathway back to Glucose almost like a signal that the cycle is ready to commence. However before Oxalo-Acetate can send that signal it must be synthesized by Phenylalanine and Tyrosine through Fumerate. However then some Oxalo-acetate must synthesize Citate which will then result in 2-Oxo-Glutarate which is also maintained through a titration with Glutamate that is synthesize from Proline and Histidine. Then a secondary loop exists between here and Succinyl-CoA that is also independently maintained by influx of Methyl-Malonyl-CoA synthesized by Valine and Propionyl-CoA synthesized from Isoleucine, Methionine, and other Odd-Numbered Fatty acids. In addition Succinyl-CoA combines with Glycine to form Porphyrins.
CFS must maintain all of these inflows and outflows if CFS is also going to be dependent on aerobic respiration.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
The classic chassis for a prokaryotic cell-free protein synthesis (CFPS) system is derived from bacterial cells, most commonly Escherichia coli lysates. These systems leverage many of the same evolutionary advantages that have allowed bacteria to dominate nearly every ecological niche on Earth: rapid growth, extraordinary metabolic efficiency, and immense adaptive capacity. Bacterial populations replicate clonally at remarkable speed, generating extensive genetic and phenotypic variation over short timescales. While this adaptability contributes to the robustness and productivity of bacterial systems, it also presents challenges for CFPS reproducibility. Even within the same culture lineage, significant divergence can emerge between generations as cells continuously adapt to environmental conditions, metabolic pressures, and selective constraints. As a result, maintaining consistency in CFPS preparations requires careful control of bacterial growth state, timing, and storage conditions. Researchers must decide which physiological state of the culture is most appropriate for lysate preparation, since a population harvested at one time point may differ substantially from the same culture only hours later. Delayed harvesting can lead to nutrient depletion, stress responses, population collapse, or contamination by competing microorganisms, all of which can alter lysate composition and downstream protein synthesis performance. To preserve experimental reproducibility, ancestral stocks are typically cryopreserved and periodically compared against actively growing cultures. Maintaining these reference populations and monitoring culture integrity introduces additional cost and labor, but it is essential for ensuring consistent inference about the CFPS system being modeled. Once stable growth conditions and colony maintenance strategies are established, however, bacterial CFPS platforms can produce specific proteins rapidly and at exceptionally high yield, making them powerful tools for synthetic biology, biosensing, metabolic engineering, and biologics production.
The classic chassis for a cell-free eukaryotic system (CFES) uses yeast cells. Even at the smallest scale of CFES, there is more control over folding machinery, enzymatic dynamics, and chaperone mechanisms. CFES also offers additional functional categories that can be compared with those of larger Metazoans, including sophisticated immune systems, complex multiprotein structures, signaling networks, and specialized membrane structures. CFES takes more resources and steps to prepare for growth, and their cell lines are almost always more fragile than their CFPS counterparts. The machinery supporting CFES growth is also more expensive, which increases inequality in the research landscape between scientists. Inequality anywhere corrodes the quality of the science. For example, there are institutions where only engineers have access to even basic CFES technology, and entire departments of professors and their students see the machines only in conference pamphlets. This results in specialization of science based on having and not having facilities and technologies, which means considerable gaps form between curiosity, ability, and the type of research questions being pursued. This means that scientific progress in CFES systems, as opposed to CFPS, becomes like Moneyball, and this is certainly not just in the USA. Entire countries may face a CFPS ceiling, in which the only way they can pursue scientific questions about multicellular organisms is to leave their country and join another research institution. This ensures that inequalities in CF research breakthroughs persist, which is detrimental to science as a whole. For example, most of my knowledge about CFES comes from HTGAA; indeed, I didn’t even know what I didn’t know before this course, and there is still much to learn before I can fully articulate an answer to many of these questions.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
In CFPS, this is a much easier problem to solve. This is because in CFPS, if the protein we are targeting is synthesized when the match between the environment and genotypic expression prioritizes an expression of a target phenotype, for example, as we might see in V. Cholera that adapts proteins to form a Type Six Secretion System for competition with other bacteria in their colony. In CFES, the specificity of protein function outpaces the abundance of protein across a population. Variation that becomes an obstacle in CFES as individual organisms in a population develop differences in their gene networks that produce target proteins and the reason for the abundance of particular proteins can be counterintuitive to CFES scientists planning experiments, as anyone who has ever tried to grow a multicellular organism for resource harvesting. For example, the blueberry bushes that never produce fruit, or a milk goat that never produces enough milk.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
I am going to develop a stress inflammatory signaling system in the Ste20 kinase family from the ground up. I will use MAP4K2 as my model.
What would your synthetic cell do? What is the input and what is the output?
I choose MAP4K2 because it is so complex and high up in the chain and stress signaling molecules. I realize this violates the minimal cell part of the instruction but actually another way of looking at simplicity is just repeating the same word over and over again and in that sense this might be the simplest gene of all. The full name is mitogen-activated protein kinase kinase kinase kinase kinase 2. My version of a MAP4K2 cell will be an upstream stress-responsibe kinase that will direct amplification of inflammatory and oxidative stress signaling cascades. The inputs will be TNF-α signaling, TRAF adaptor complexes, stress receptor activation, small GTPase signaling, Oxidative stress, and environmental stress. Outputs will be MAP3Ks, JNK pathway activation, p38 pathway modulation, c-Jun activation, and Cytoskeletal signaling.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Unfortunately not, I could express some protein, demonstrate kinase activity in vitro, and exhibit some substrate phosphorylation but I will not be able to recreate the full biological function of mitogen-activated protein kinase kinase kinase kinase kinase 2 without membrane-based signaling geometry, localization, and scaffold interactions.
Could this function be realized by genetically modified natural cell?
The good news is mitogen-activated protein kinase kinase kinase kinase 2 would be more effective as a modified natural cell than actual minimal synthetic cell which would be more complicated if that is even possible to comprehend. This is because MAP4K2 would benefit from genetically modified cell’s existing membrane organization, protein scaffolding, standing kinase networks, existing ATP homeostasis parameters, as well as the natural cells cytoskeleton, signaling receptors, phoshorylation machinery, and organization of spatial compartments. Furthermore in addition to the existing infrastructure in the natural cell I could add through the genetic modification flourescent reporters to beter visualize pathway activiation associated with my MAP4K2.
Describe the desired outcome of your synthetic cell operation.
My desired outcome of this simplified synthetic system to investigate which components are minimally required for MAP4K2 function.
Design all components that would need to be part of your synthetic cell.
Although I wish I had written my dissertation on this topic, now I’d better keep it short for this question. A minimal mapping of all of the components that will need to be part of my MAP4K2 synthetic cell are a Liposome, TX/TL, MAP4K2 DNA and mRNA, ATP and Mg²⁺, phosphorylation substrate, and fluorescent readout. Additional modules would include my base MAP4K2 platform and some simple peptide substrates, MAP3K10, scaffold/recruitment system, liposome membrane anchoring, and JNK/p38 pathway.
What would be the membrane made of?
I would use a liposome membrane made from phospholipids. Ingredients would include: POPC, POPG, Cholesterol, and Ni-NTA lipids.
What would you encapsulate inside? Enzymes, small molecules.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
How will you measure the function of your system?
I will measure the function of my mitogen-activated protein kinase kinase kinase kinase 2 synthetic cell by detecting phosphorylation of an internal substrate. Successful function would be demonstrated by increased phosphorylation signal compared to no-kinase and kinase-dead controls, either using fluorescent phorphorylation reporter or Phos-tag gel shift assay.
Example solution
Based on: Lentini, R. et al., 2014. Nat comm, 5, p.4012.
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output?
Expand the sensing capacity of bacteria. Input: theophylline (inert to bacteria). Output of the SMC: IPTG. Output of the whole system: GFP produced in bacteria. (Theophyline aptamer reference: Martini, L. & Mansy, S.S., 2011. Cell-like systems with riboswitch controlled gene expression. Chemical Communications, 47(38), p.10734.)
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
No. If the IPTG were not encapsulated, it would go into the bacteria without the need of theophylline-induced membrane channel synthesis, thus the synthetic cell actuator would not exist.
Could this function be realized by genetically modified natural cell?
Yes, in this particular case: the theophylline aptamer could be incorporated into a transformed gene. This lacks generality though – it is easier to make SMC than modify bacteria, so in this system a single bacteria reporter can be used to detect various small molecules.
Describe the desired outcome of your synthetic cell operation.
In the presence of SMC, bacteria sense theophylline.
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
Phospholipids + cholesterol.
What would you encapsulate inside? Enzymes, small molecules.
cell-free Tx/Tl system, IPTG, gene for membrane transporter under the control of theophylline aptamer.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
Bacterial, because of the theophylline riboswitch used as SMC input.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The membrane is permeable to the input molecule (theophylline), the output is IPTG that will cross the membrane via the membrane pore created after theophyline-initiated gene expression.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids: POPC, cholesterol
Enzymes: bacterial cell-free Tx/Tl
Genes: a-hemolysin (aHL) to encapsulate in SMC
Biological cells: E.coli transformed with GFP under T7 promoter and a lac operator
How will you measure the function of your system?
Measure GFP output of the cells via flow cytometry. Alternatively, use enzymatic reporter, like luciferase, and measure bulk output of the enzyme.
(a) In the absence of artificial cells (circles), E. coli (oblong) cannot sense theophylline.
(b) Artificial cells can be engineered to detect theophylline and in response release IPTG, a chemical signal that induces a response in E. coli.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
How will the idea work, in more detail? Write 3-4 sentences or more.
What societal challenge or market need will this address?
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Homework Part B: Individual Final Project
We’d like students to start exploring their final project in depth this week! Of your three Aims, for this week you should have at least Aim 1 decided and written down.
Put your chosen final project slide in the appropriate slide deck following the instructions on slide 1:
MIT/Harvard/Wellesley ONE FINAL PROJECT IDEA
Committed Listener ONE FINAL PROJECT IDEA
Submit this Final Project selection form if you have not already.
Begin planning how you will write your final project documentation based on these guidelines
Prepare your first DNA order and put it in the “Twist (MIT)” or “Twist (Nodes)” tab of the 2026 HTGAA Ordering: DNA, Reagents, Consumables spreadsheet, as appropriate.
First Twist order deadline for MIT/Harvard/Wellesley students is Friday, April 3 at 11PM ET
First Twist order deadline for Committed Listeners is Friday, April 10 at 11PM ET. (Your Node Lead will place the Twist order, so please work with them to finalize your constructs and ordering decisions.)
Reading & Resources
Cell-free protein synthesis (explanation by minipcr's DNAdots)
Validation of Cell-Free Protein Synthesis Aboard the International Space Station (ACS Synthetic Biology paper by Ally Huang et al.)
Week 10 HW: Imaging and Measurement
Week 10 HW: Imaging and Measurement
Homework: Final Project
Homework is partly based on data that will be generated in the Waters Immerse Lab in Cambridge, MA. Students will characterize green fluorescent protein (eGFP, a recombinant protein standard) structure (primary, secondary/tertiary) in the lab using liquid chromatography and mass spectrometry, as well as Keyhole Limpet Hemocyanin (KLH) oligomeric states using charge detection mass spectrometry (CDMS). Data generated in the lab needed to do the homework is included both within this document and in the Appendix of the laboratory protocol.
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
There are many measurements that could be taken. The DNA of the mites and their larva is a target. In addition the bacteria and other microbiota and parasites that capitalize on the infestation damage to the host epidermis. This ofcourse brings up the geometry of the host tissue and biochemical molecules all of which can be measured quantiatively or qualitatively. Specifically, when it comes to host cells there are living and deceased keratinocytes and corneocytes and the odd hair follicle, especially in most heavily infested cases. There is superinfection residues full of bacterial cocci and rods or periods and semi colons as Dr. Betsy Dyer writes. There crushed and desciated erythrocytes, platelets, and leukocytes. In addition there will be scattering fluctuations of neturophils, eosinophils, macrophages, and lymphocytes. In addition, goats are always on the move grazing when they’re not cuddled up in hay or dirt. Therefore there will be pollen grains, plant frags, seed husks and hay chaff, and plant hairs in addition to other ecotoparasites. What I want to measure most though is stress.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
What does the farmer’s molecular lab contain. This will certainly increase in the coming decades as the struggle to draw nutrients from the Earth is increasingly a necessity and struggle. The same for the labor of keeping herds alive admist the global conflagurations and increasing heat and parasites. I predict by the time I retire to the dirt every farmer worth their salt will have a ready supply of gel electrophoresis, DNA sequencing, and mass spectrometry equipment and proper freezer in their shed lest they have a lab in Boston at the ready to ring.
Homework: Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at
The high MW estimate from Benchling is 28006.50 Da for 1-247 AA. The low MW estimate with BLAST search and selection of P42212 · GFP_AEQVI combined with the ExPASy calculator 1-238 AA link resulted in Compute pI/MW - Results
GFP_AEQVI (P42212)
Description:
Green fluorescent protein
Organism:
Aequorea victoria (Water jellyfish) (Mesonema victoria)
The parameters have been computed for the following feature:
FT CHAIN 1 238 Green fluorescent protein
The computation has been carried out on the complete sequence (238 amino acids).
Molecular weight (Da):
26886.32 (average mass)
26869.36 (monoisotopic mass)
Theoretical pI:
5.67
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Q
Determine z for each adjacent pair of peaks (n, n+1).
A
[ z = (m/zn+1) / ((m/zn) − (m/zn+1)) ]
Q
Determine z for adjacent peaks at m/z 875.4421 and 903.7148.1
A
Formula:
z = (m/zn+1) / ((m/zn) − (m/zn+1))
Substitute values:
z = 903.7148 / (875.4421 − 903.7148)
Simplify denominator:
z = 903.7148 / (−28.2727)
Solve:
z ≈ −31.96
Take absolute value:
z ≈ 32
Therefore, the charge state is +32.
1 Peak values adapted from Ade Larsen homework data. Formatting of LaTeX blocks for math inside Git using the AI protocol droid Chat-GPT.
Q
Determine the MW of the protein using the relationship between m/zn, MW, and z.
A
[ MW = z · (m/zn) − z · H ]
Q
Determine the molecular weight (MW) of the protein using the relationship between m/zn, MW, and z.1
A
Relationship between ion mass and charge:
m/z = (MW + zH) / z
where:
MW = neutral molecular weight of the protein
z = charge state
H = mass of a proton
Proton mass:
H = 1.0073 Da
Rearrange equation to solve for MW:
MW = z · (m/zn) − z · H
Substitute values:
MW = 32 · (875.4421) − 32 · (1.0073)
Simplify:
MW = 28014.1472 − 32.2336
Solve:
MW ≈ 27981.91 Da
Therefore, the molecular weight of the protein is
~27.98 kDa.
1 Peak values adapted from Ade Larsen homework data. Theoretical GFP molecular weight from Aequorea victoria GFP sequence analysis. Formatting of LaTeX blocks for math inside Git using the AI protocol droid Chat-GPT.
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
Calculate the accuracy of the measurement using the deconvoluted MW from Section 2.2 and the predicted molecular weight from Section 2.1.1
A
Accuracy relationship:
Accuracy =
| MWexperiment − MWtheory |
/ MWtheory
Values:
MWexperiment = 27981.91 Da
MWtheory = 26886.32 Da
Substitute values:
Accuracy =
|27981.91 − 26886.32|
/ 26886.32
Simplify numerator:
Accuracy =
1095.59 / 26886.32
Solve:
Accuracy ≈ 0.0407
Therefore, the experimental measurement differs from the theoretical molecular weight by
~0.0407.
1 Experimental molecular weight determined from ESI-MS deconvolution using adjacent charge-state analysis. Theoretical molecular weight derived from the Aequorea victoria GFP amino acid sequence.
Homework: Waters Part II — Secondary/Tertiary structure
We will analyze eGFP in its native, folded state and compare it to its denatured, unfolded state on a quadrupole time-of-flight MS. We will be doing MS-only analysis (no liquid chromatography, also known as “direct infusion” experiments) on the Waters Xevo G3-QToF MS.
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
Figure 2. Comparison of the mass spectra between denatured (top) and native (bottom) eGFP standard on the Waters Xevo G3 QTof MS.
When a protein unfolds because of an environmental exposure, be it from acid or heat or other denaturant, the surface area of that protein is more exposed to protons from the environment that can bond to the proton during ionization. The mass spectrometer then uses electrospray ionization (ESI) to further shift the charge state of the protein. The way this works is the protein is ejected from a charged needle and the droplets formed evaporate. The time of flight is then the movement of these charged droplets as they transition into a gaseous phase.
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800 $\frac{m}{z}$? What is the charge state? How can you tell?
Figure 3. Native eGFP mass spectrum from the Waters Xevo G3 Q-Tof MS. The inset is a zoomed-in view of the charge state at ~2800 $\frac{m}{z}$ on a mass spectrometer with 30,000 resolution.
The provided peak for charge state 10+ is ~2,800 m/z. The predicted MW of eGFP is ascertained to be ~ 26,886.32 Da. To calculate m/z for ionized protein droplet m/z = (MW + zH) / z which is the protein ion relationship where MW is molecular weight and z is charge state, and H is the mass of one proton. Now we plugin values. The mass of one proton H = 1.0073 Da. Now we rearrange the relationship equation with our available information to solve for z, so that z = MW / (m/z) - H, and m/z = is the given peak near 2,800 m/z or slighly less at m/z = 2799.42. Now we find the deconvoluted MW from before which is MW = 27,981.91 Da. Then we substitute values into our final forumula, z = 27.981.91 / (2,799.42 - 1.0073). Next z = 27,981.91 / 2,798.41. Next z ≈ 9.99. Then we round to the whole number, so z ≈ 10, which means are estimated charge state of 10 carries 10 positive charges for the native eGFP protein ion of ~ 2,800 mz.
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
We can make an educated guess. Using zoom view in figure 1 there are multiple possible peaks around m/z = 1,473.7 and they all share the same charge state. To measure distribution, we take a min and max for example 1,473.63 and 1,473.67. We then subtract the min from the max to compute our peak spacing which amounts to about 0.04. We then use the isotope spacing relationship formula where z = 1 / 0.04 which equals approximately 24.94 which we round to z = 25. This produces our back of the envelope approximation of +25 charge state.
Homework: Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
Amino Acid Frequencies from Benchling
AA3
AA1
Count
Percent
Ala
A
8
3.2%
Arg
R
6
2.4%
Asn
N
13
5.3%
Asp
D
18
7.3%
Cys
C
2
0.8%
Gln
Q
8
3.2%
Glu
E
17
6.9%
Gly
G
22
8.9%
His
H
15
6.1%
Ile
I
12
4.9%
Leu
L
22
8.9%
Lys
K
20
8.1%
Met
M
6
2.4%
Phe
F
12
4.9%
Pro
P
10
4.0%
Ser
S
10
4.0%
Thr
T
16
6.5%
Trp
W
1
0.4%
Tyr
Y
11
4.5%
Val
V
18
7.3%
Pyl
O
0
0.0%
Sec
U
0
0.0%
How many peptides will be generated from tryptic digestion of eGFP?
Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
mass
position
#MC
modifications
peptide sequence
4472.1752
170-210
0
HNIEDGSVQLADHYQQNTPI GDGPVLLPDNHYLSTQSALS K
2566.2931
217-239
0
DHMVLLEFVTAAGITLGMDE LYK
2437.2608
5-27
0
GEELFTGVVPILVELDGDVN GHK
2378.2577
54-74
0
LPVPWPTLVTTLTYGVQCFS R
1973.9062
142-157
0
LEYNYNSHNVYIMADK
1503.6597
28-42
0
FSVSGEGEGDATYGK
1266.5783
87-97
0
SAMPEGYVQER
1083.4979
240-247
0
LEHHHHHH
1050.5214
115-123
0
FEGDTLVNR
982.4952
133-141
0
EDGNILGHK
821.3940
81-86
0
QHDFFK
790.3552
75-80
0
YPDHMK
769.3913
47-53
0
FICTTGK
711.2944
103-108
0
DDGNYK
655.3813
98-102
0
TIFFK
602.2780
211-215
0
DPNEK
579.3137
128-132
0
GIDFK
507.2925
164-167
0
VNFK
502.3235
124-127
0
IELK
Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
Figure 4. Example conditions for predicting the number of tryptic peptides from the eGFP standard. Please replicate all parameters shown above.
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
Figure 5a. Total ion chromatogram (TIC) of the eGFP peptide map. The peak at 2.78 minutes is circled, and its MS data is shown in the mass spectrum in Figure 5b, below.
Depending on the peaks we select as GTE to 10% relative, about 26 peaks.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
Yes they are approximately the same.
Identify the mass-to-charge ($\frac{m}{z}$) of the peptide shown in Figure 5b. What is the charge ($z$) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ($\small{[M\!\!+\!\!H]^+}$) based on its $\frac{m}{z}$ and $z$.
Figure 5b. Mass spectrum figure to show $\frac{m}{z}$ for the chromatographic peak at 2.78 min from Figure 5a above. The inset is a zoom-in of the peak at $\frac{m}{z}$ 525.76, to discern the isotope peaks.
Figure 5c. Fragmentation spectrum of the peptide eluting at retention time 2.78 minutes in Figure 5a (above).
Start with the calculation of mass of the singly charged form of peptide mass from Figure 5b
[M + H]+experiment ≈ 1050.52694 Da
Then compare to the expected PeptideMass value, with the closest expected peptide being FEGDTLVNR
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
(Recall that $ \text{Accuracy} = \frac{|MW_{\text{experiment}} - MW_{\text{theory}}|}{MW_{\text{theory}}} $ )
Now we determine mass accuracy, here is the formula
Accuracy =|MWexperiment − MWtheory|
/ MWtheory
Then we substitute in our values from the previous problem
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
88% of Amino Acids in eGFP in Figure 6
Bonus Peptide Map Questions
8. Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c? (HINT: Use your results from Question 2 above to match the peptide molecular weight that is closest to that shown in Figure 5b. Copy and paste its sequence into this tool online to predict the fragmentation pattern based on its amino acid sequence: http://db.systemsbiology.net/proteomicsToolkit/FragIonServlet.html. What is the sequence of the eGFP peptide that best matches the fragmentation spectrum in Figure 5c?
Does the peptide map data make sense, i.e. do the results indicate the protein is the eGFP standard? Why or why not? Consult with Figure 6, which depicts the % amino acid coverage of peptides positively identified using their calculated mass and fragmentation pattern.
Homework: Waters Part IV — Oligomers
Homework: Waters Part V — Did I make GFP?
*Part V copied from Ade Larsen page because I did not attend Waters Lab as CL
Reading & Resources
Fundamentals of peptide and protein mass spectrometry (Steve Carr, the Broad Institute of MIT and Harvard): https://www.youtube.com/watch?v=PFOodSbH9IY
This link has 2 tutorial video presentations on some of the basics of mass analyzers and different information you can learn from “Tandem” MS (also called MS/MS): https://www.asms.org/about-mass-spec/fundamentals-hardware-instrumentation
History of LC and MS, a video presentation by Professor James Jorgenson: https://player.vimeo.com/video/53604465
Nature Methods perspectives article on “Best Practices for intact protein analysis for top-down mass spectrometry: https://www.nature.com/articles/s41592-019-0457-0
Principles of Intact Protein Analysis: https://www.youtube.com/watch?v=ySql2iKRN6U
What is Mass Spectrometry?: https://www.asms.org/docs/default-source/what-is-ms-booklet/whatisms-ppt_201243e71d0ea09c6d75a448ff000066efb8.pdf?sfvrsn=627b70c3_0
Basics of Reverse Phase Liquid Chromatography: https://www.ionsource.com/tutorial/chromatography/rphplc.htm
Peptide and protein for Bioanalysis using LC-MS: https://www.youtube.com/watch?v=vsQ-Kr4Gdoo
Article - Native vs Denatured : An in Depth Investigation of Charge State and Isotope Distributions: https://pmc.ncbi.nlm.nih.gov/articles/PMC7539638/
Week 11 HW: Building Genomes
Homework — DUE BY START OF APR 28 LECTURE
Week 11 HW Overview
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
[!info] Note that this homework is due a week later than it ordinarily would due to its release a week later than normal.
[x] Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse.
[x] If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
Make a note on your HTGAA webpages including: what you contributed to the community bioart project
(e.g., “I made part of the DNA on the bottom right plate”)?
What you liked about the project?
My favorite part about the project was when different people started collaborating or interacting on a design. I liked the timer and not knowing if someone else recognized the pattern and the opportunity to scale up quickly on a design when different contributors stepped up and worked on the same art. Then at some point in the process, I started to see the maker space as a giant multi-threaded game of GO in color, and it even seemed like some folks started picking up on that and making better shapes. Initially, the competitive streak triggered and I thought game on. Then, I remembered, wait, we are supposed to be working together here. Now, there was a point when the exercise was not as cool. Specifically, when it was first unveiled, everyone was talking about an Egyptian flag, for example, and I was thinking I didn’t see a flag and there were these sections where it was like someone just painted over everything else. However, it was then unveiled (maybe I was just slow on all these details, but it was all a surprise to me:) that there was this hysteresis process built into the application which I thought was brilliant. Then I was right as rain about the whole exercise and better appreciated all of the evolutions.
What about this collaborative art experiment could be improved next year?
Well, here’s the thing, now that I know about the hysteresis slider, I’m just going to wait until the end of the lab to start plotting. Seriously, though, one idea might be to make it like an archaeological dig or a puzzle. No, I’ve got it. I think we need to make it like a massive mind-sweeper puzzle, and add a component so a portal square can be detected, revealing a new chamber that leads to another massive mind-sweeper puzzle. Alternatively, if we don’t want to just digress into a video game, the next iteration could be a 96-well plate or a gel or a bacterial plasmid,and we could all receive an invitation to make contributions (even redesign experiments), almost like an interactive collaborative Wiki. They also have chemistry and microbe simulators. For example, there is the BEAKER application by THIX (https://thix.co/beaker). Of course, the dream would be an HTGAA BEAKER collaboration with a history slider, so we could scroll backward through experimental changes, including the lab notebook. The great thing about BEAKER is that it also lets you auto-document reaction steps, which could scroll as well. Then, if we could add more organic and biochemical molecules and run an electric pulse through water, we could even run Miller-Laurie (sp?) experiments. I also thought there were a number of lab assignments and demonstrations from the lecture that could have been run in a similar collaborative sandbox. I could also the promise in benchling to support this process. For example we could all be given a link to set-up our own lab notebooks based on a template which was already there this semester but we could just build on that.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Getting It Done Note:
Ronan provides a link in lab slide deck to a simulation tool
We are transported to a virtual engineering space where we can design a floor plan, an assembly model, and in the assembly model variables. In addition, radio buttons were included to set the preferences for the simulation space. We can even adjust camera views to observe the cloud lab setup we design.
Ronan provides a link in lab slide deck to Benchling directory
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)Karim_&_Jewett_paper
The role will involve protein overexpression and the preparation of extracts.
Agrawal_et_al_paper
BL21 (DE3) is a distinct and mildly adapted strain of E. coli bacteria. In this experiment, the bacteria’s chromosome is a chassis for the T7 (bacteriophage) RNA Polymerase gene. What really makes this particular bacterium useful for us as well is its likelihood to exhibit leaky expression Hayat et al_paper.
Salts/Buffer A necessary component of cell-free protein synthesis, according to Bartsch et al (2024) is salts and buffers Bartsch_et_al_2024. For example,
Potassium Glutamate Following protocol in Karim and Jewett paper (2016) 60mM (note our concentration too) was added to cold wash before centrifugation to prepare E. coli cell for extraction
HEPES-KOH pH 7.5 Just like in a saltwater aquarium, included in a buffer which is used to maintain a pH neutral at 7.5, external to internal environment, during cellular transcription, translation, and other metabolic biochemical pathways.
Magnesium Glutamate In Karim and Jewett paper (2016), 8mM was added (note our concentration too) to a CFME reaction assay mixture that contained 5 extracts – each with its own unique twist in the form of an enzyme overexpressed to represent a step in a bio-synthetic enzymatic pathway for n-butanol production before and after CFME assay enhancement.
Potassium phosphate monobasic is also introduced in Karim and Jewett paper (2016) as an additive in the 2 YTPG growth media for E. coli BL21(DE3) cells.
Potassium phosphate dibasic s also introduced in Karim and Jewett paper (2016) as an additive in the 2 YTPG growth media for E. coli BL21(DE3) cells.
Energy / Nucleotide System
Ribose Serves a salvage role as precursor for nucleotide biosynthesis and ATP generation.
Glucose First energy substrate to generate ATP from pyruvate without any additional enzymes following reactions with nicotinamide adenine dinucleotide (NAD) and coenzyme A (CoA) Hunt_et_al_2025.
AMP Another salvage metabolism functional small molecule. Adenosine Monophosphate (AMP) needed to generate Adenosine Diphosphate (ADP) which required to synthesize Adenosine Triphosphate (ATP) through endogenous kinase mechanism.
CMP Supports transcription-like synthesis of mRNA intermediates where Cytidine Monophosphate (CMP) is a precursor to Cytidine Diphosphate (CDP) and then Cytidine Triphosphate (CTP).
GMP Guanosine Monophosphate (GMP) Using Bio-MOD (biologically derived medicines on demand) platform can be produced (Hunt et al, 2025). Synthesized from Guanine to form Guanosine Diphosphate (GDP) and then Guanosine Triphosphate (GTP) which is necessary for ribosome translocation and support of translation.
UMP Uridine Monophosphate (UMP) is another salvage metabolism precusor essential for synthesis of Uridine Diphosphate (UDP) and then Uridine Triphosphate (UTP). Elongates RNA and supports the transcription phase of CFS.
Guanine On a basic level is the Nucleotide used to synthesize GMP, then GDP, and GTP.
Translation Mix (Amino Acids)17 Amino Acid Mix Standard mixture for translation of amino acids to proteins in CFS, where a sufficient quantity of nonreactive essential amino acids is thus bioavailable. This mix only includes 1mM of tyrosine and cysteine.
Tyrosine One of two nucleotides, along with
Cysteine that are synthesized separately because of degradation concerns and their contribution to oxidation.
Additives
Nicotinamide Supplement added to glucose to simulate glycolysis to generate initial ATP in the cell-free synthesis technique (Hunt et al, 2025).
Backfill
Nuclease Free Water Both deep eutectic solvents (DES) and polyethylene glycol (PEG) are utilized in pre-delution for pipettablity according to Bartsch et al (2024).
2. Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
In addition to the ratio of time 1:20 hour master process
3. Bonus question: How can transcription occur if GMP is not included but Guanine is?
I am going to guess looking at the biochemical synthesis pathway and say it’s because there appears to be a DNA-Directed and RNA-Directed Synthesis loop…in progress
C: Planning the Global Experiment | Cell-Free Master Mix Design
1. Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP
mRFP1
mKO2
mTurquoise2
mScarlet_I
Electra2
The amino acid sequences are shown in the HTGAA Cell-Free Benchling folder.
I am going to use a markdown table to organize my answer:
~ Source: FBASE (https://www.fpbase.org), all other information comes from the HTGAA Cell-Free Benchling folder and Chat-GPT used for answers that could not be found on FBASE.
2. Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
3. The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions
[X] In order to be eligible for this, make sure that your final project slide is in the “2026 Committed Listener ONE FINAL PROJECT IDEA” slide deck.
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
Use this simulation tool to create an interesting looking cloud lab out of the Ginkgo Reconfigurable Automation Carts. This is just a minimal implementation so far, but I would love to see some fun designs!
Tip
[x] Note from Ronan: If you are interested in helping me build out future HTGAA cloud lab software, please fill out this form!
Reading:
Recitation slides from week 3
Nebula RACs TA Onboarding slides from HTGAA Summer Research
Using a GPT-5-driven autonomous lab to optimize the cost and titer of cell-free protein synthesis
Design-driven optimization of low-cost reagent formulations for reproducible and high-yielding cell-free gene expression
Common Nebula protocols & their parameters

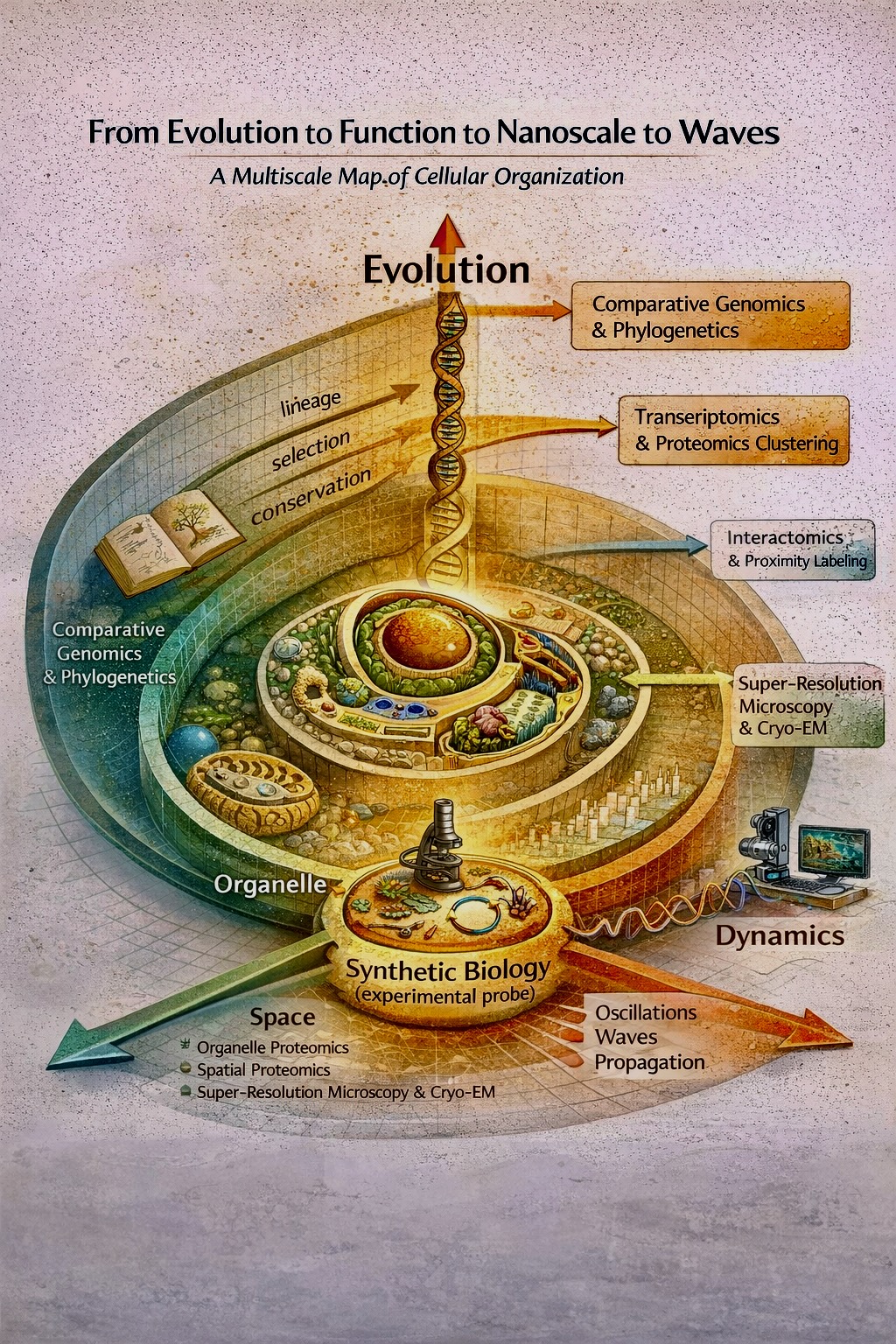




![DNA-based digital data storage technology. Source: Archives in DNA: Workshop Exploring Implications of an Emerging Bio-Digital Technology through Design Fiction - Scientific Figure on ResearchGate. Available from: https://www.researchgate.net/figure/DNA-based-digital-data-storage-technology_fig1_353128454 [accessed 11 Feb 2025]](image4.png)



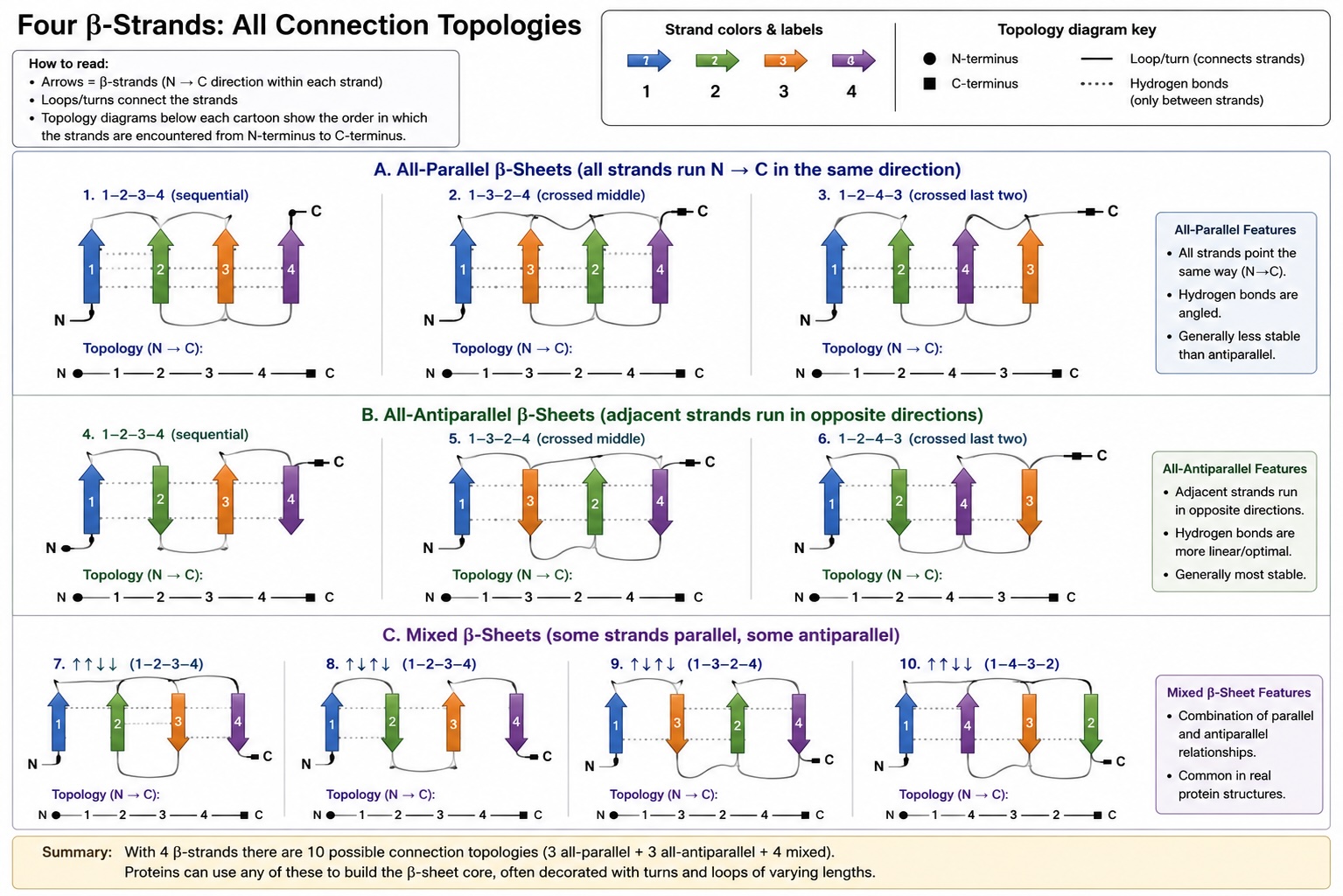

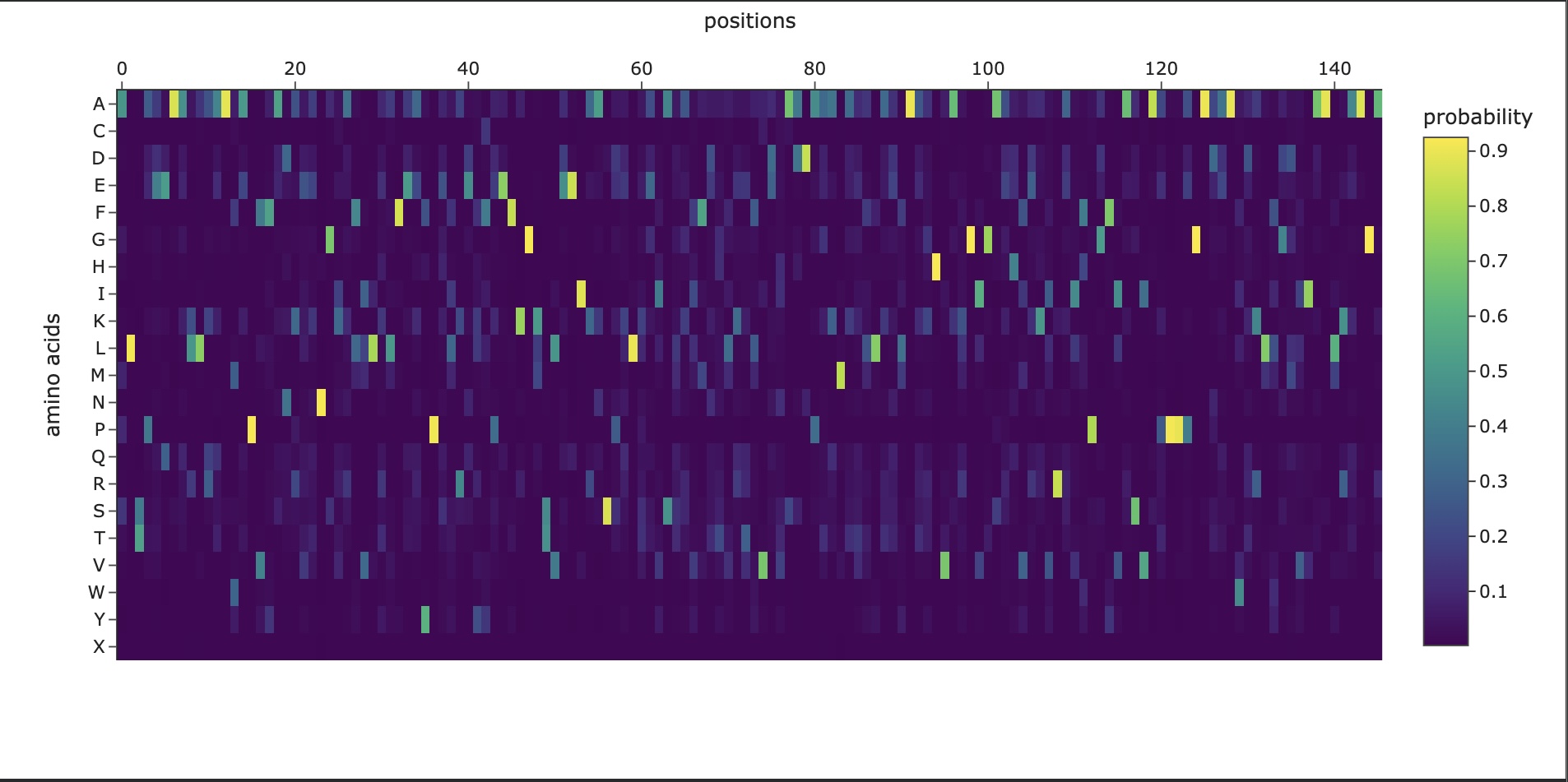
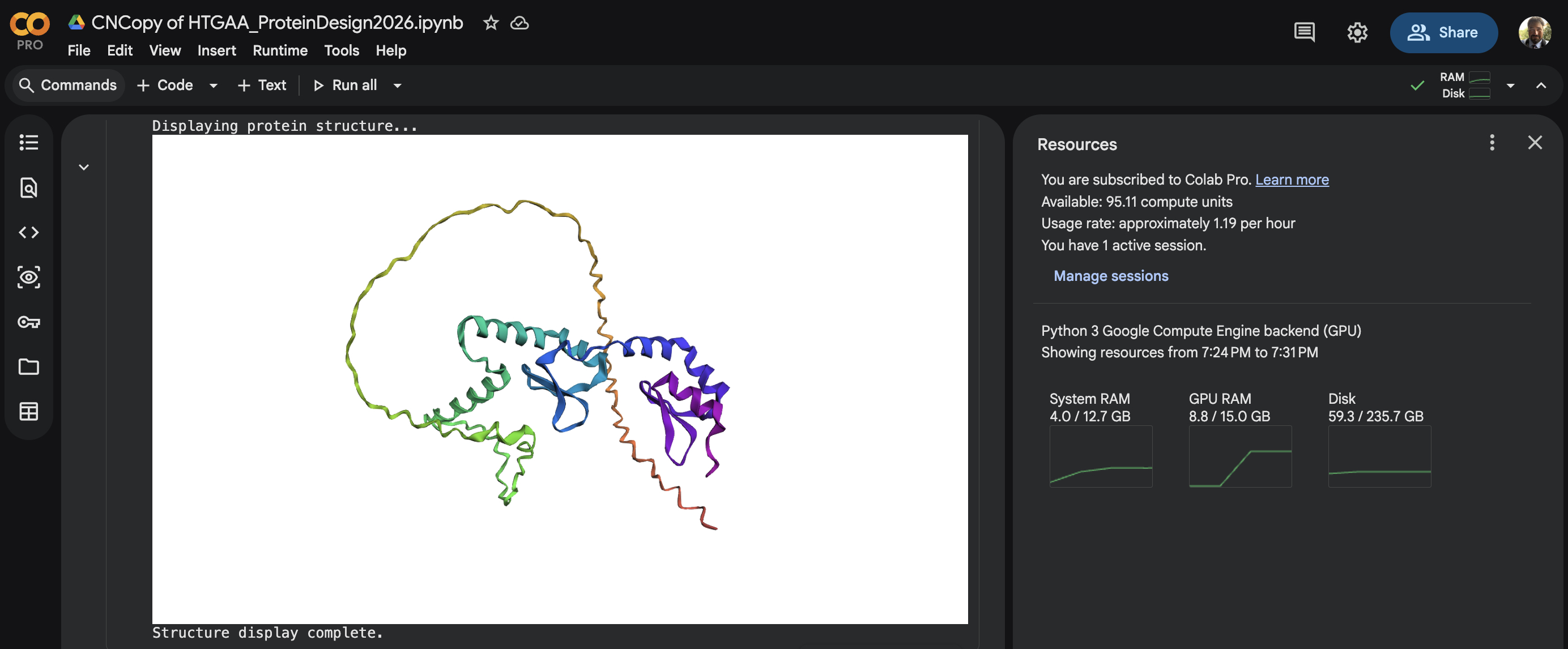
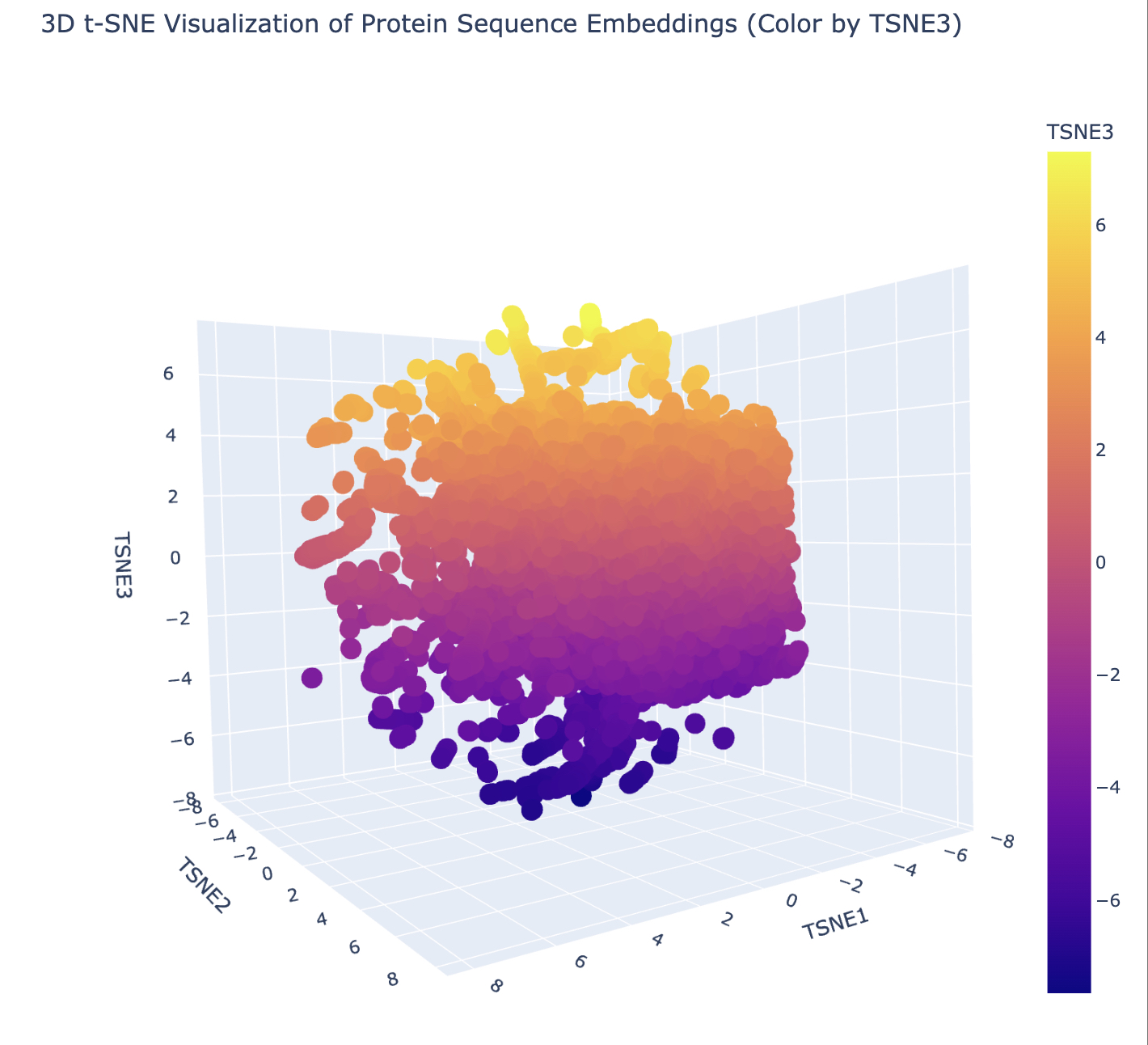
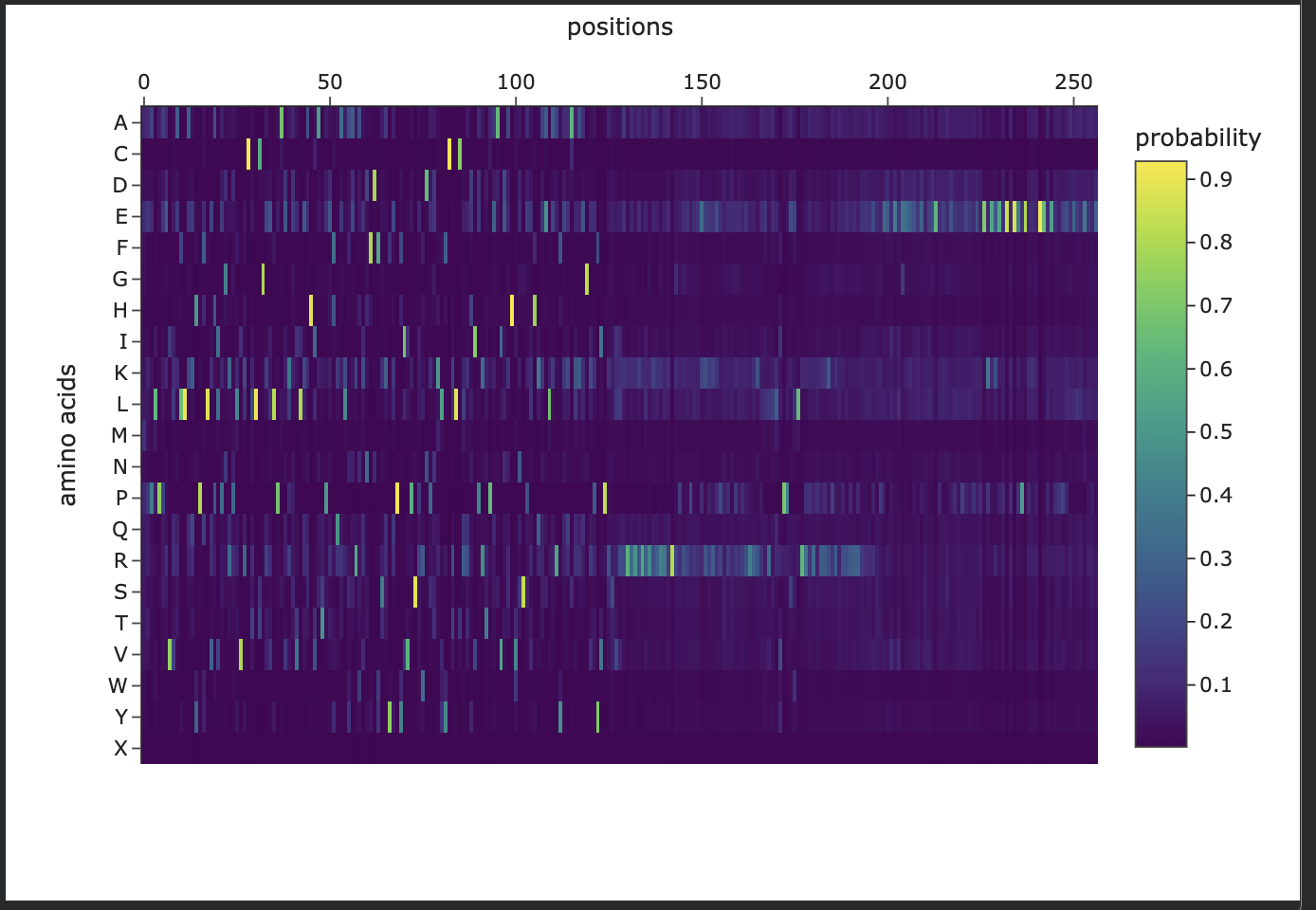
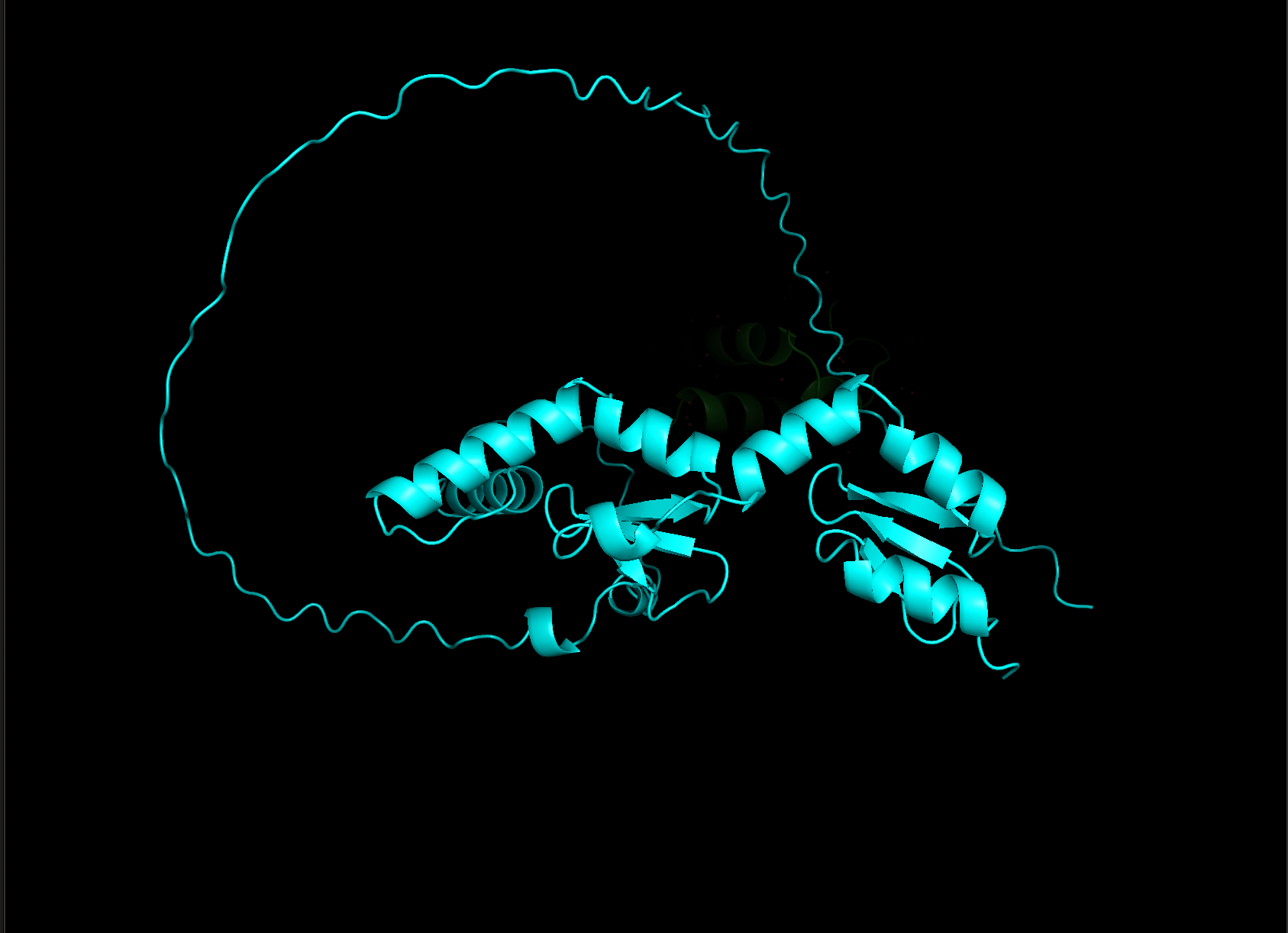

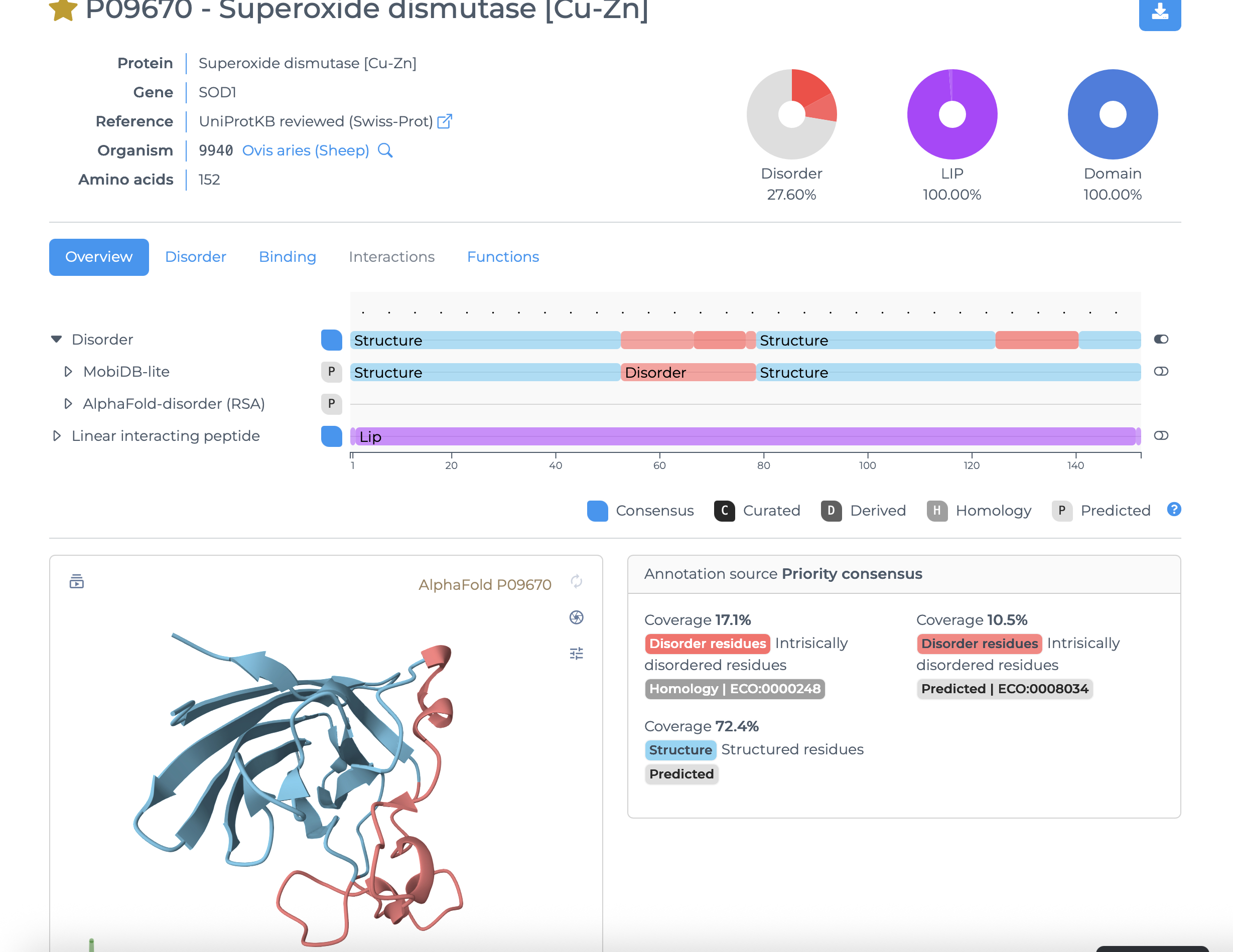
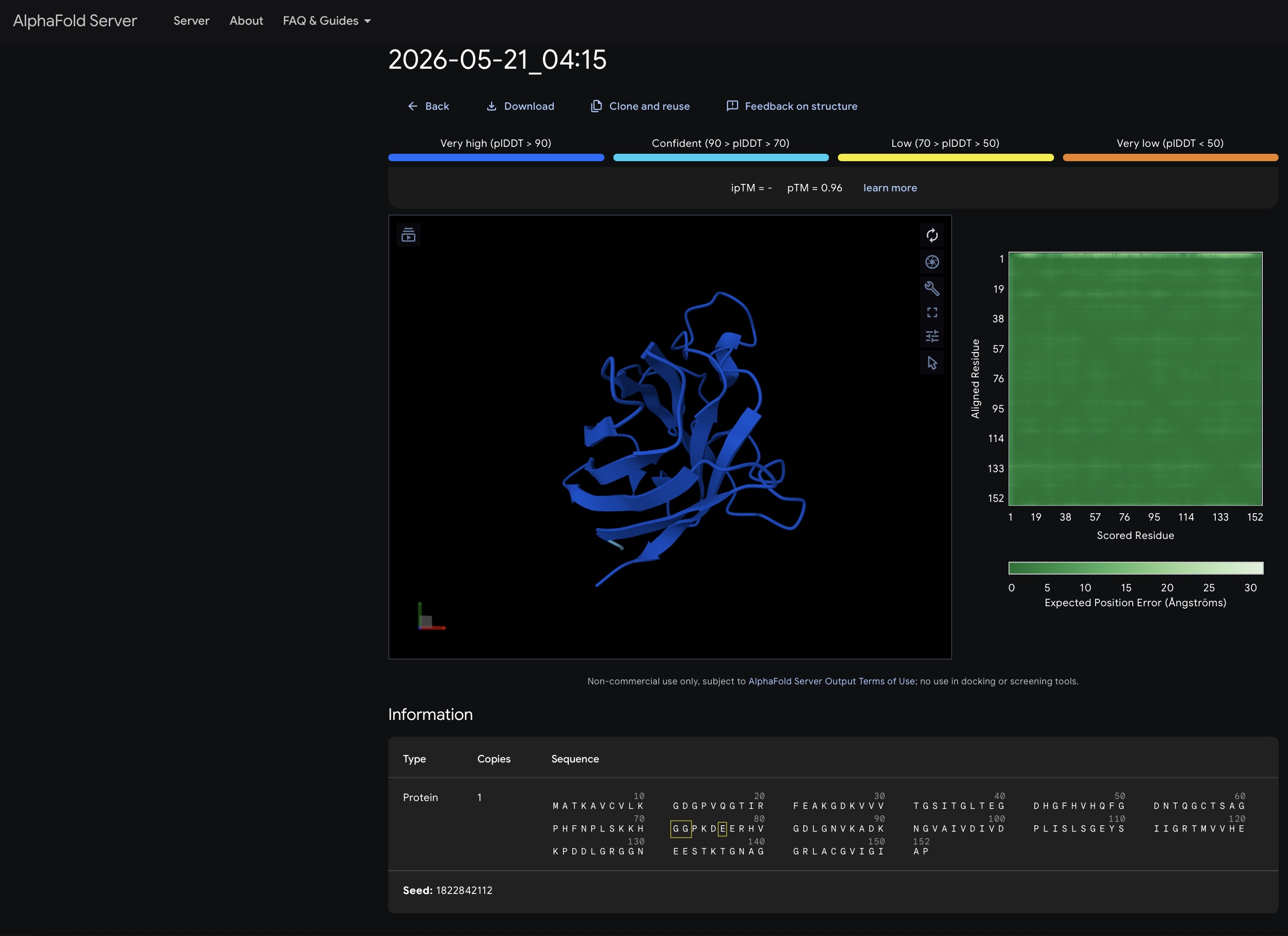
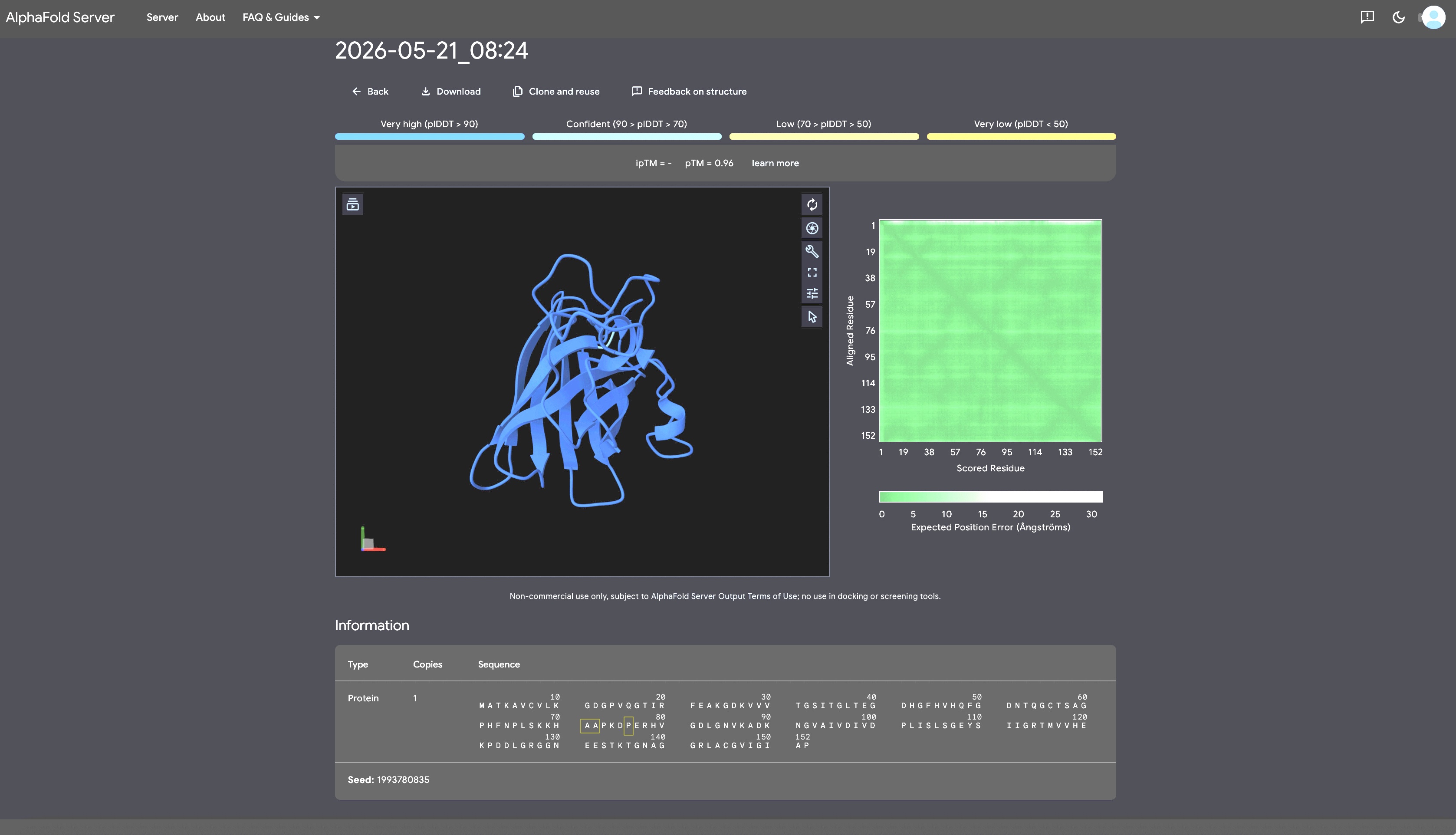

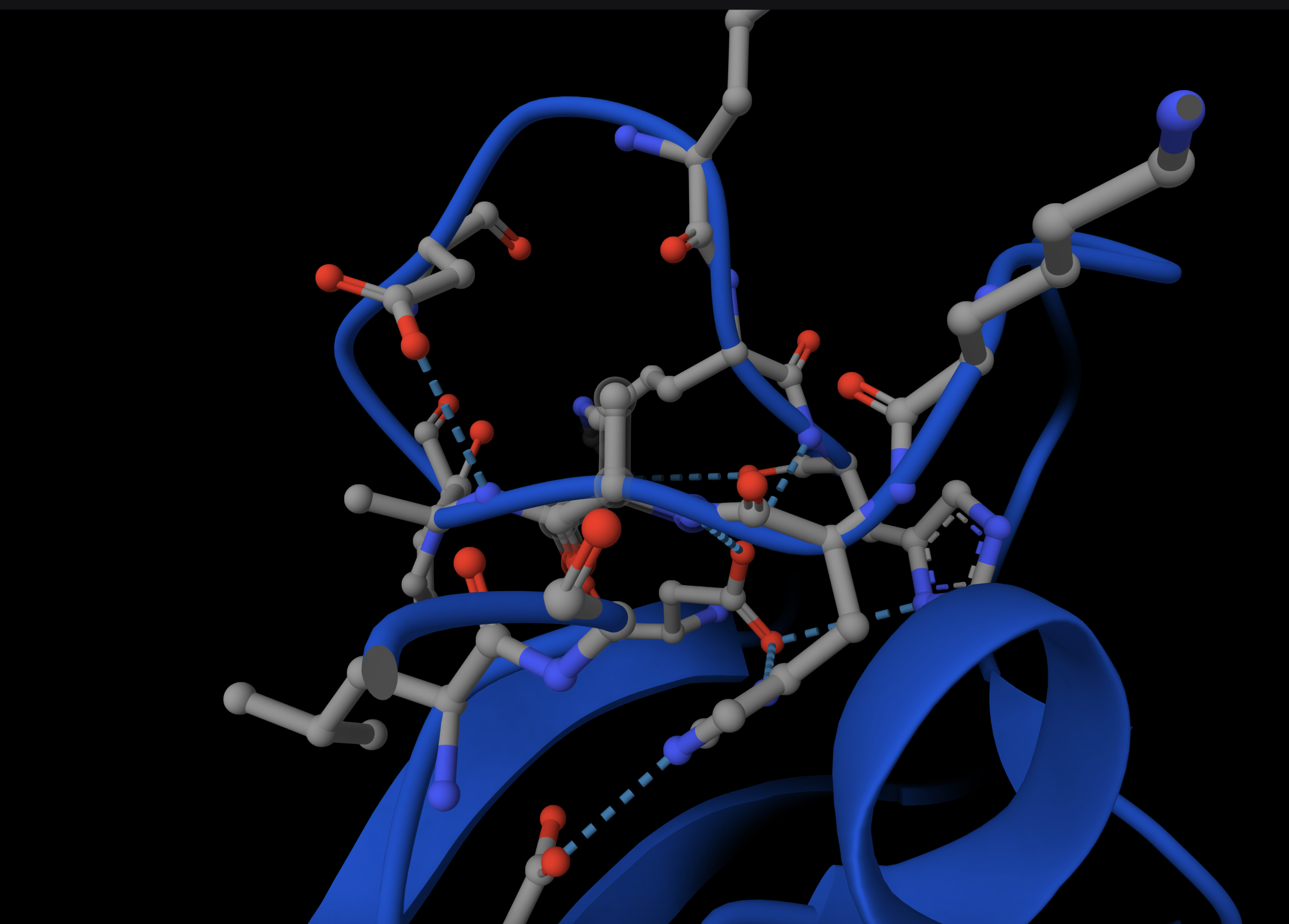
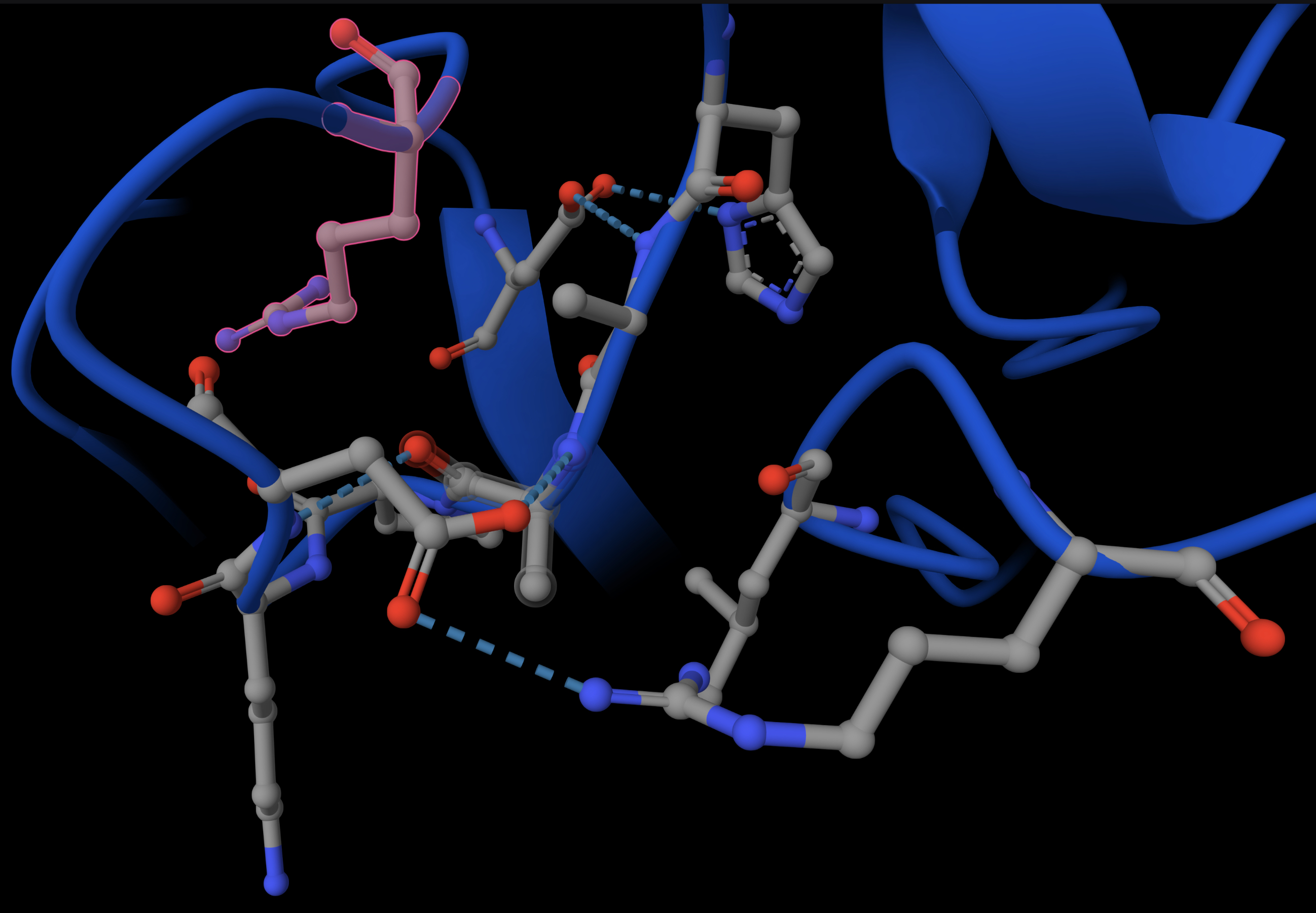
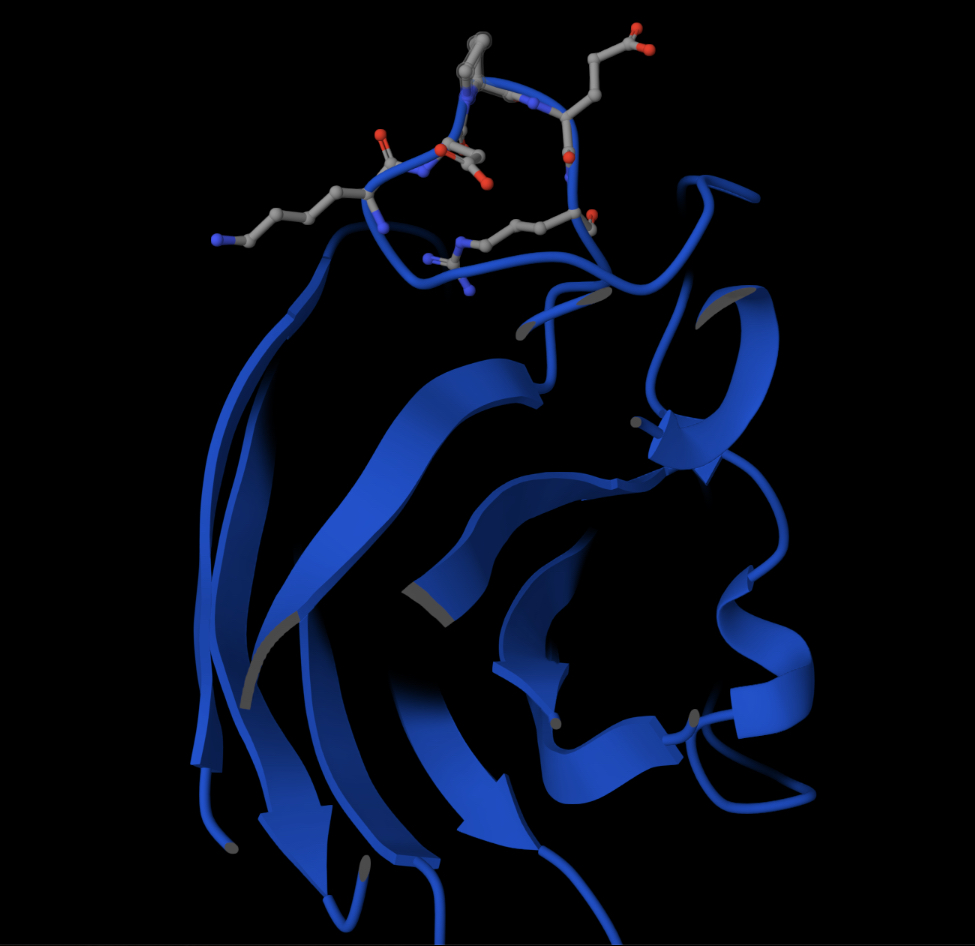
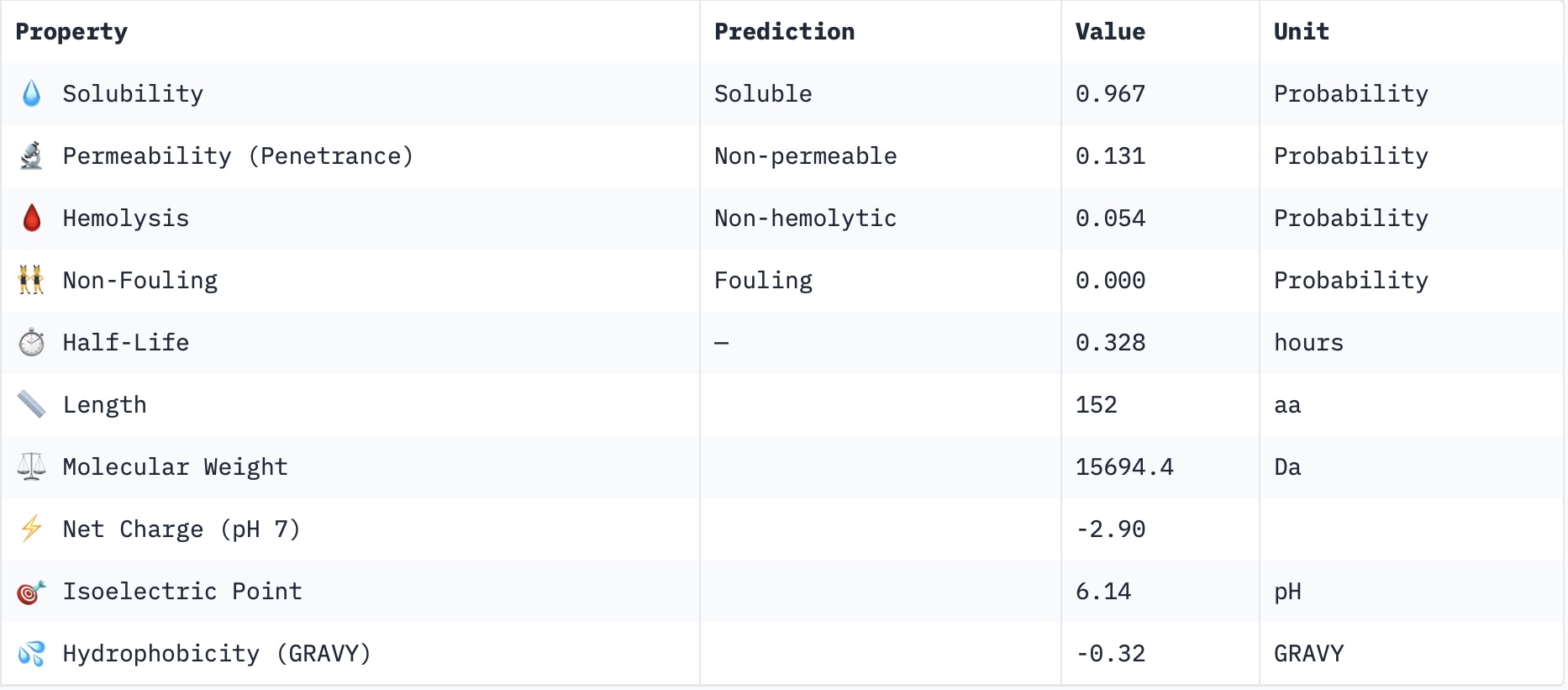
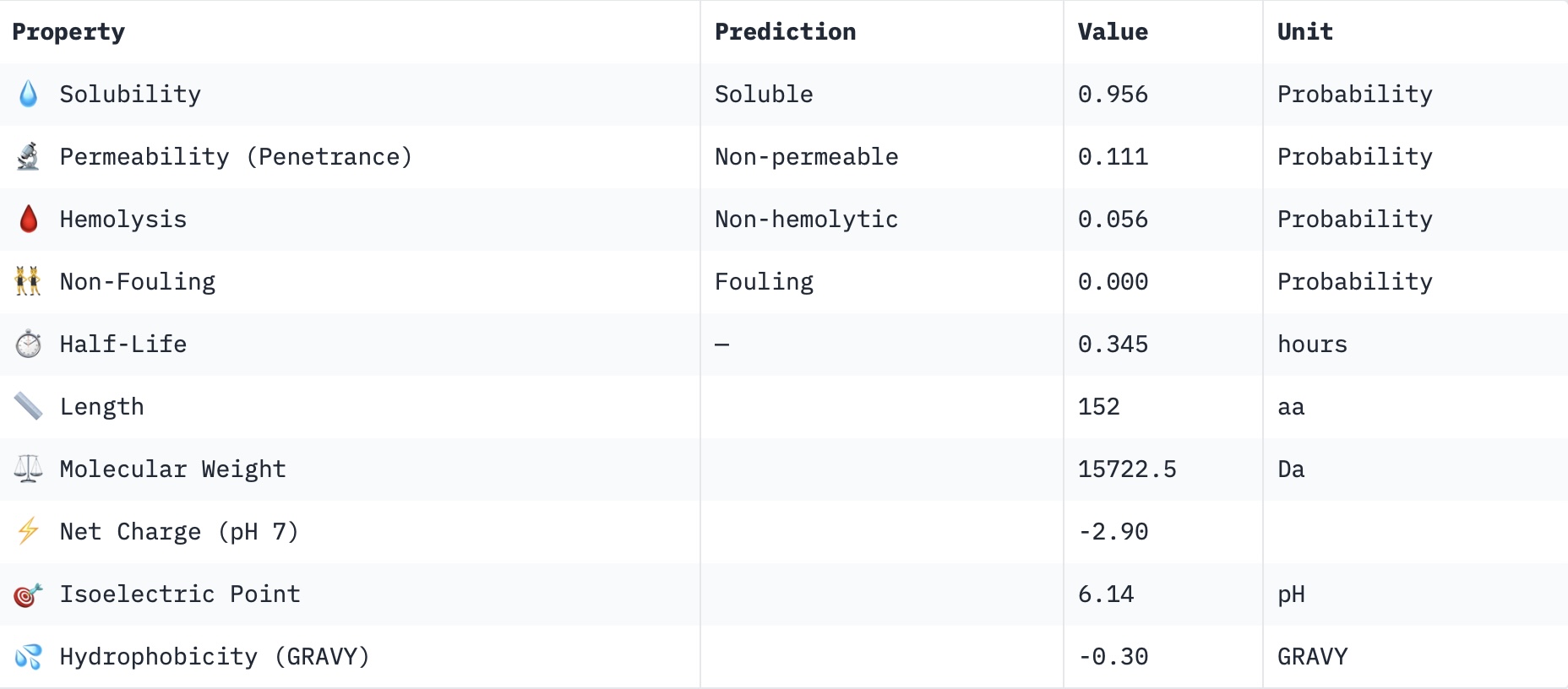
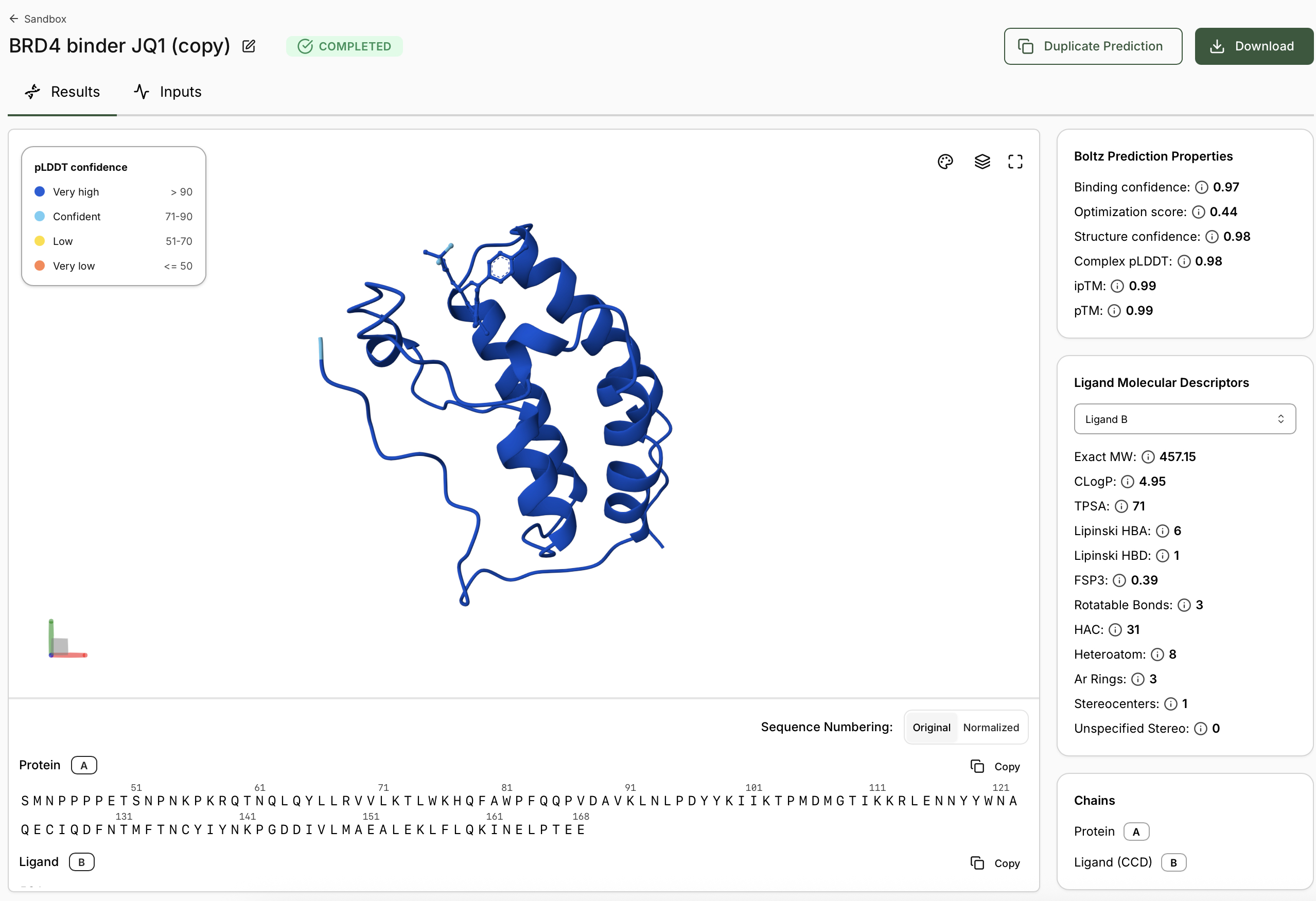



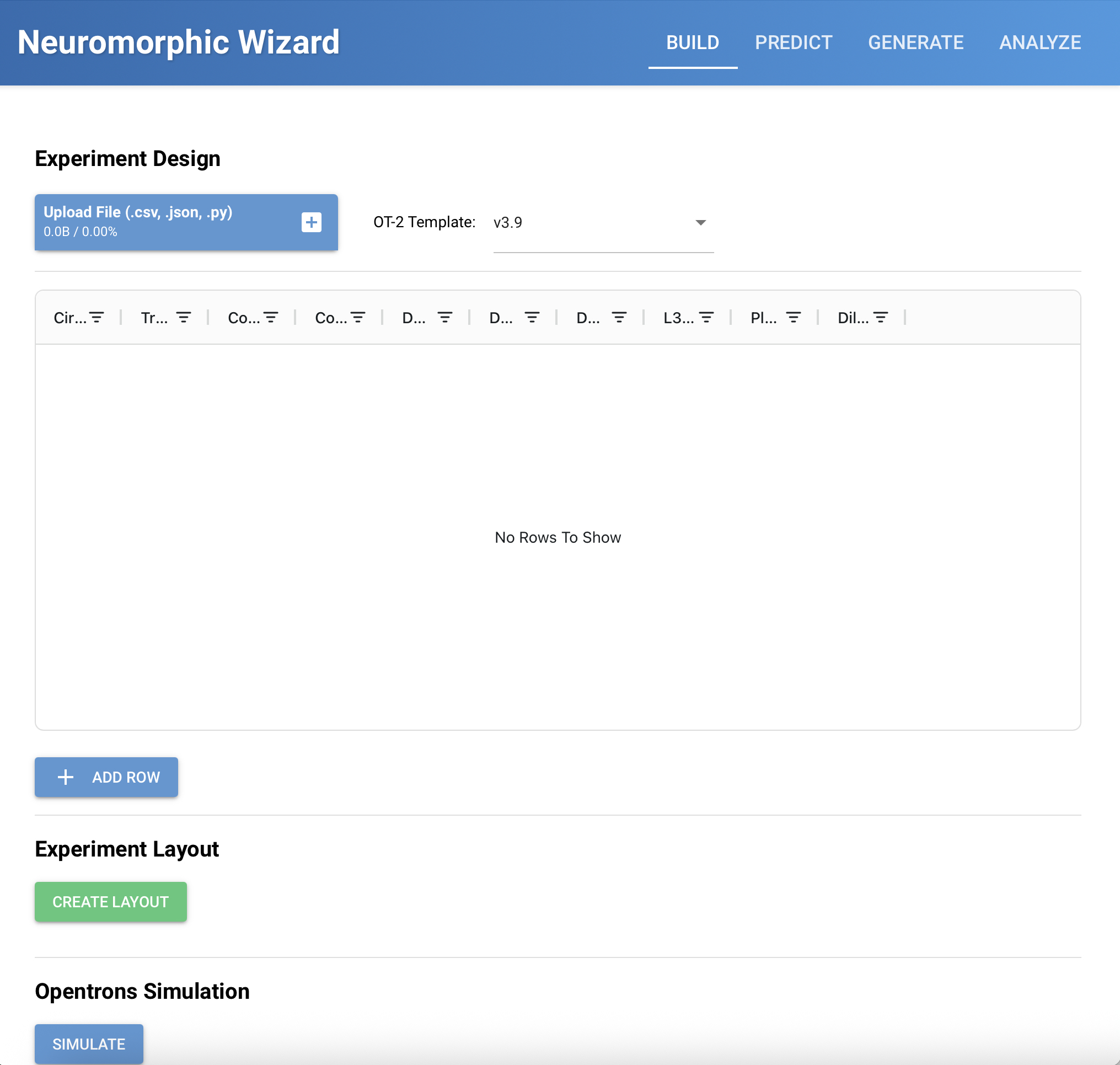


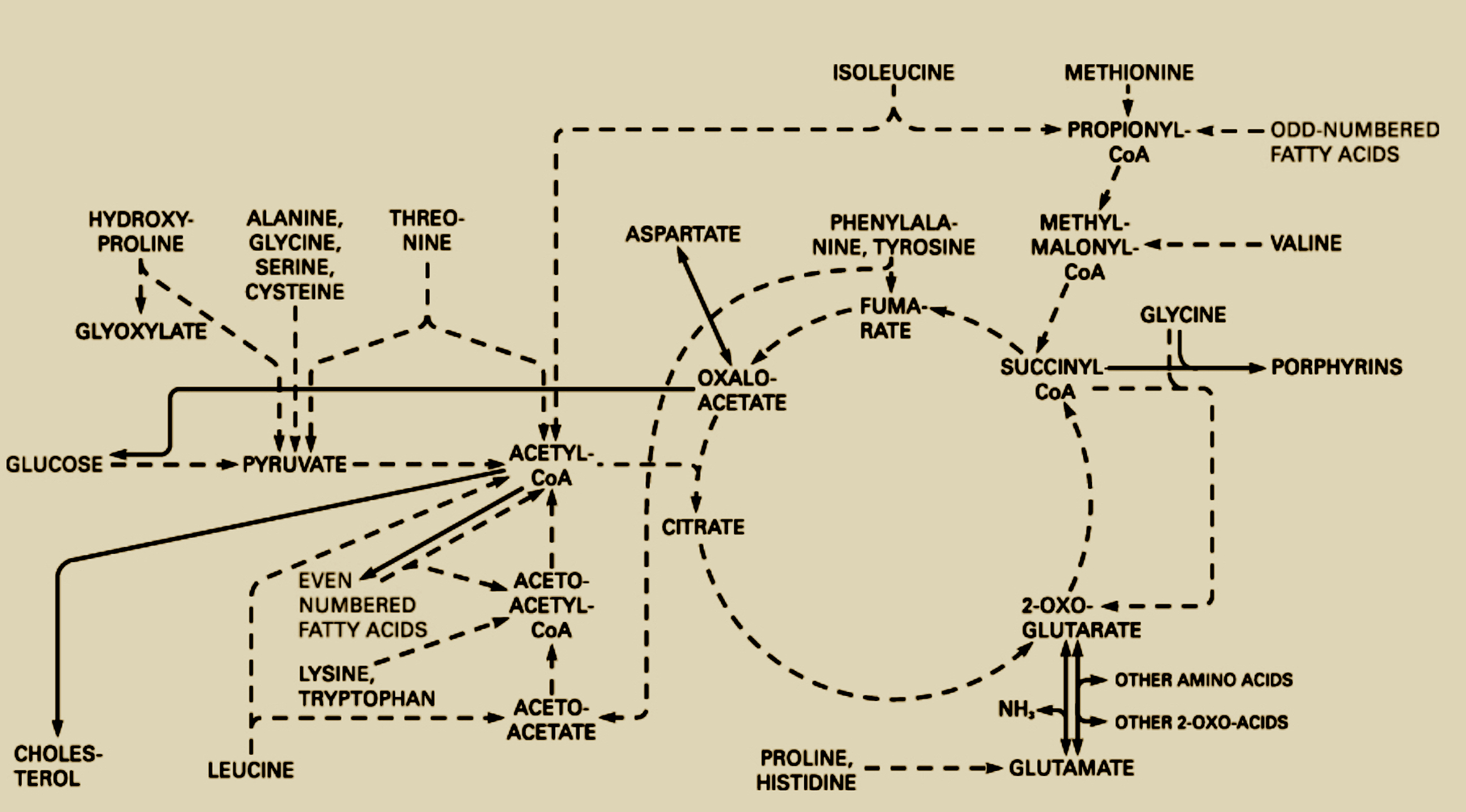

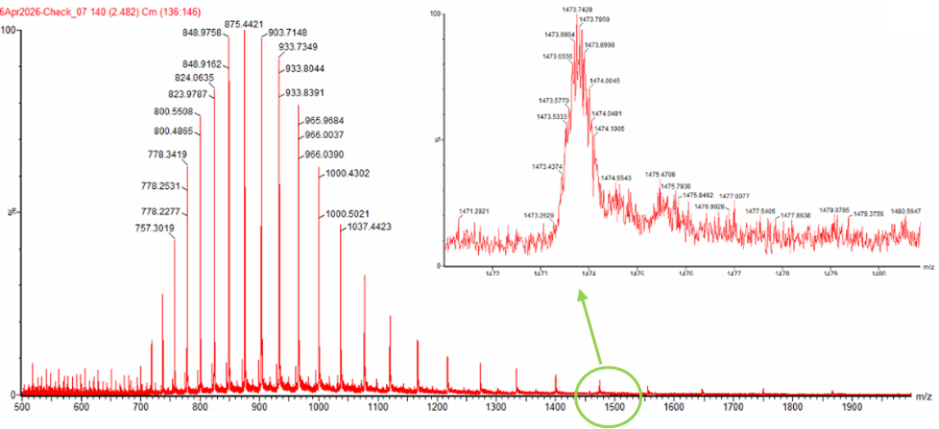
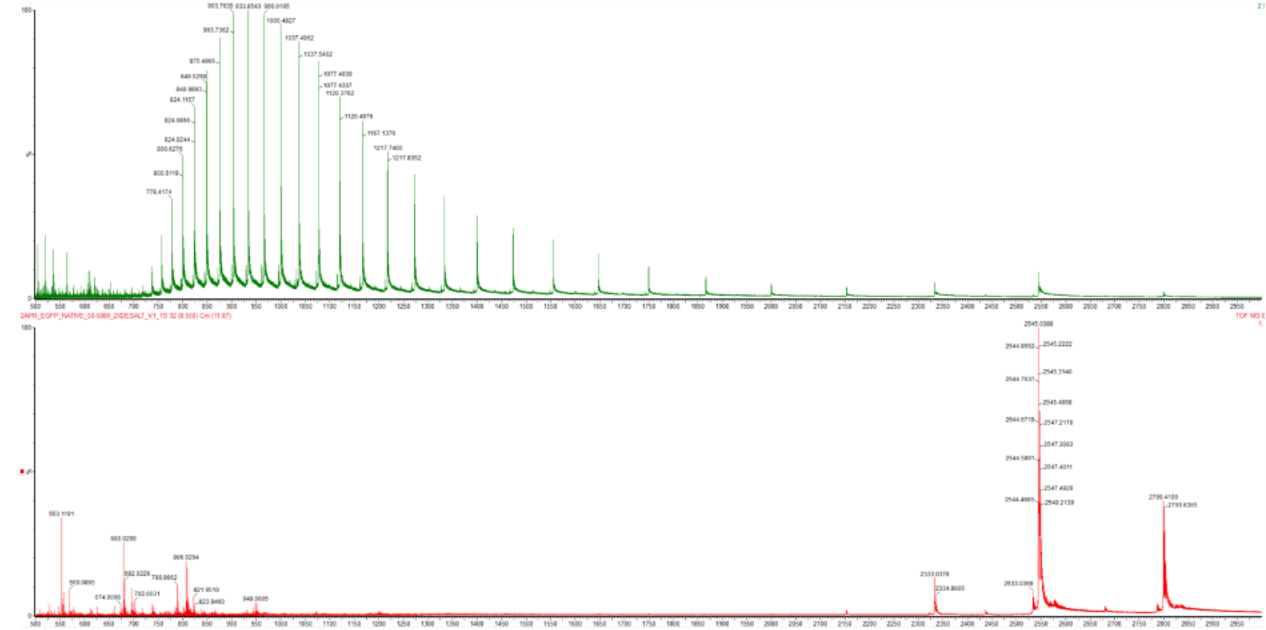
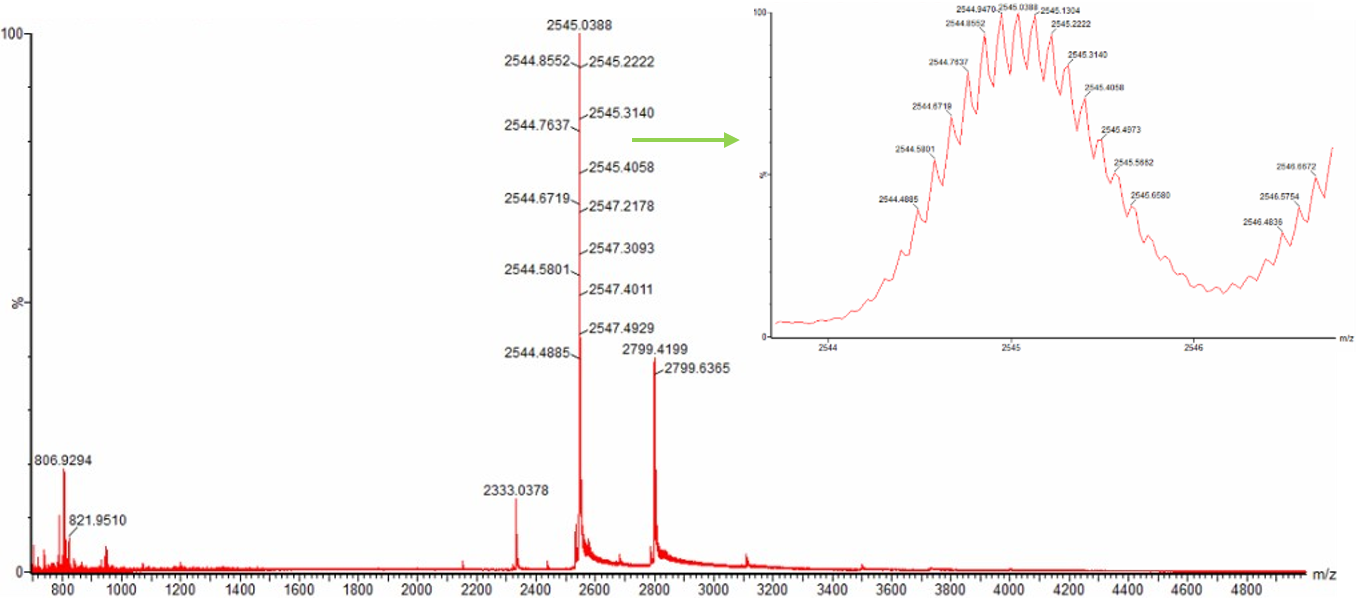

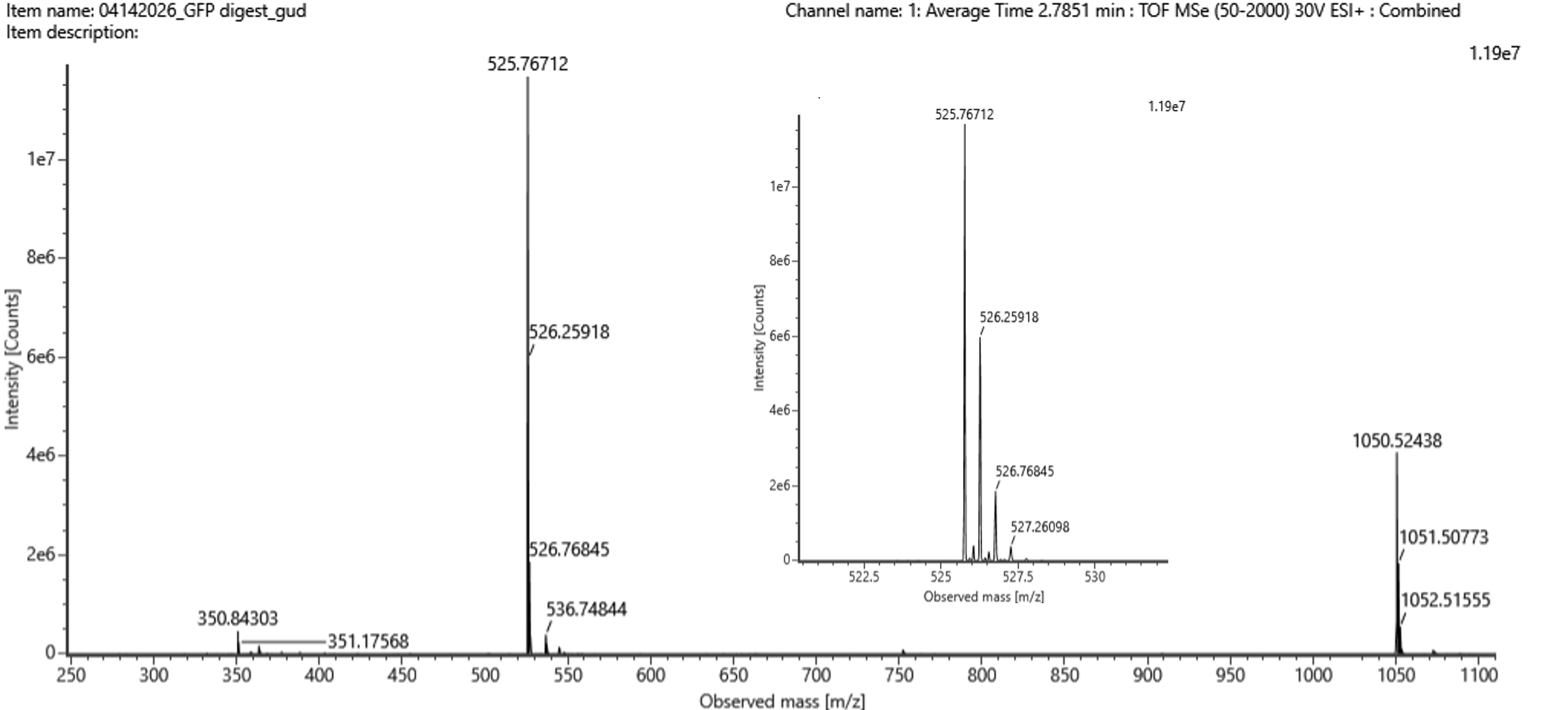


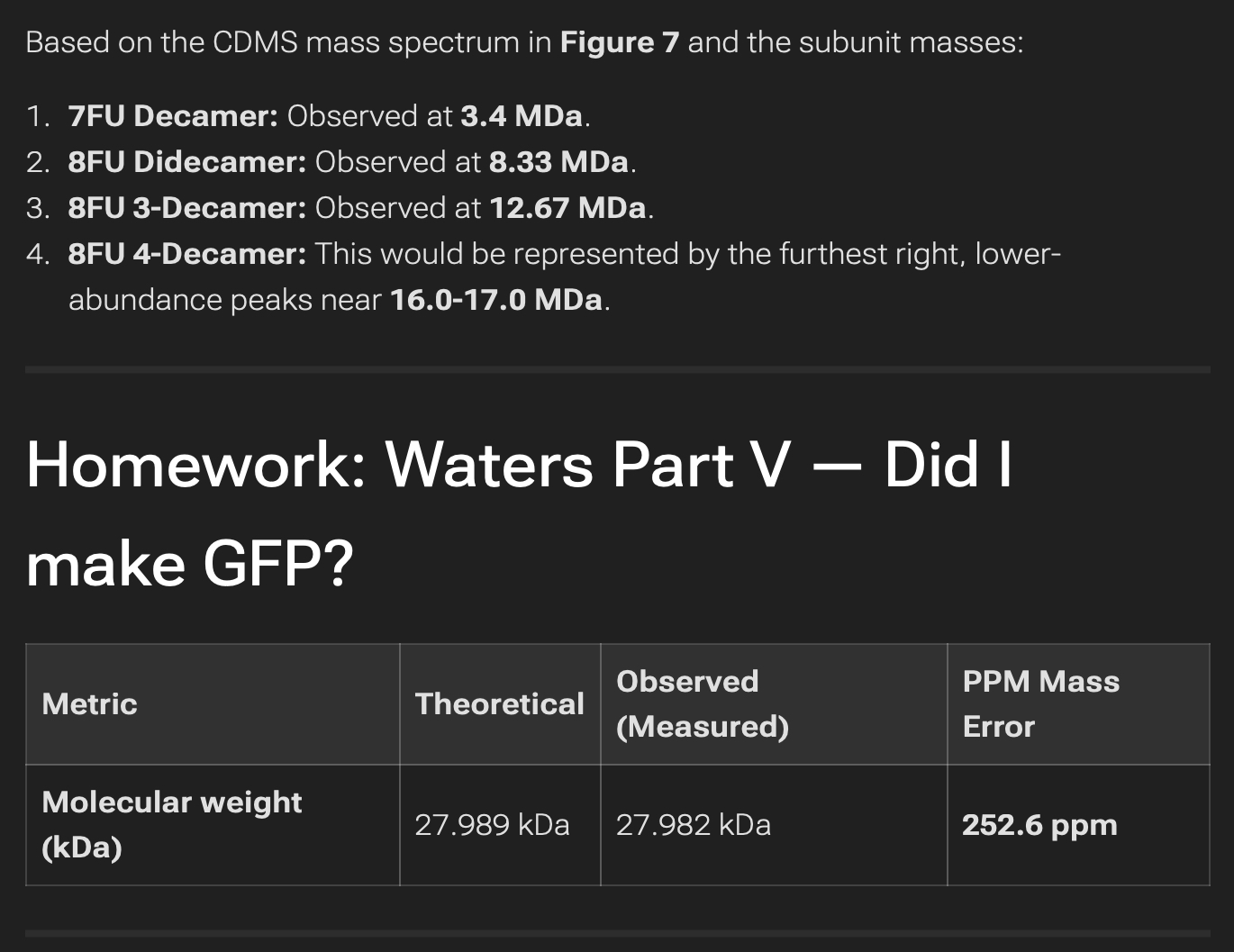

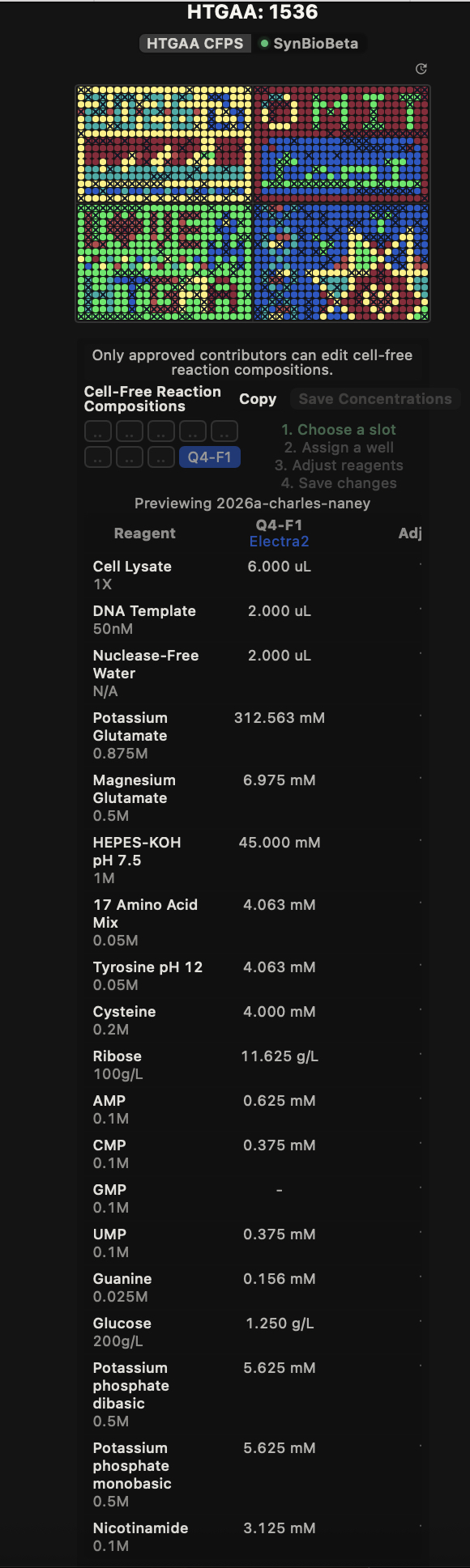
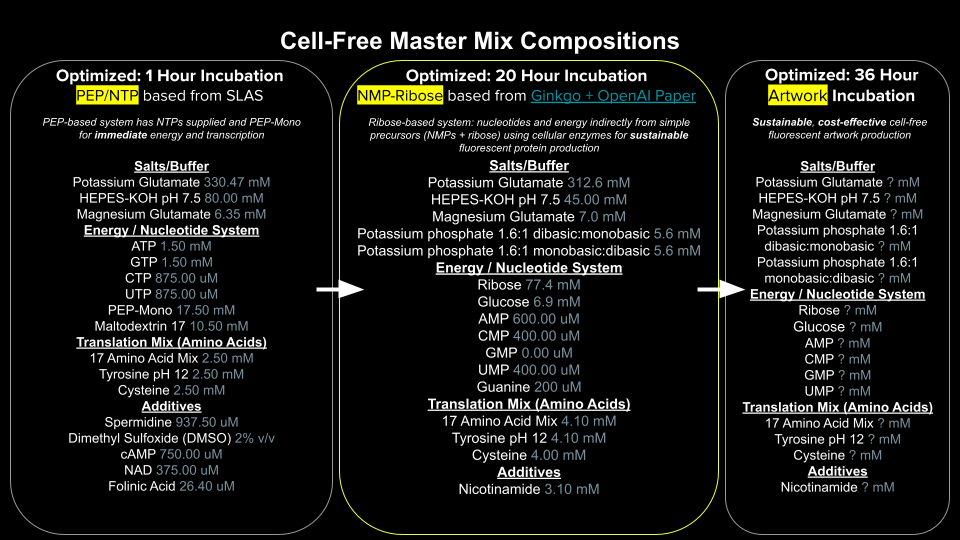


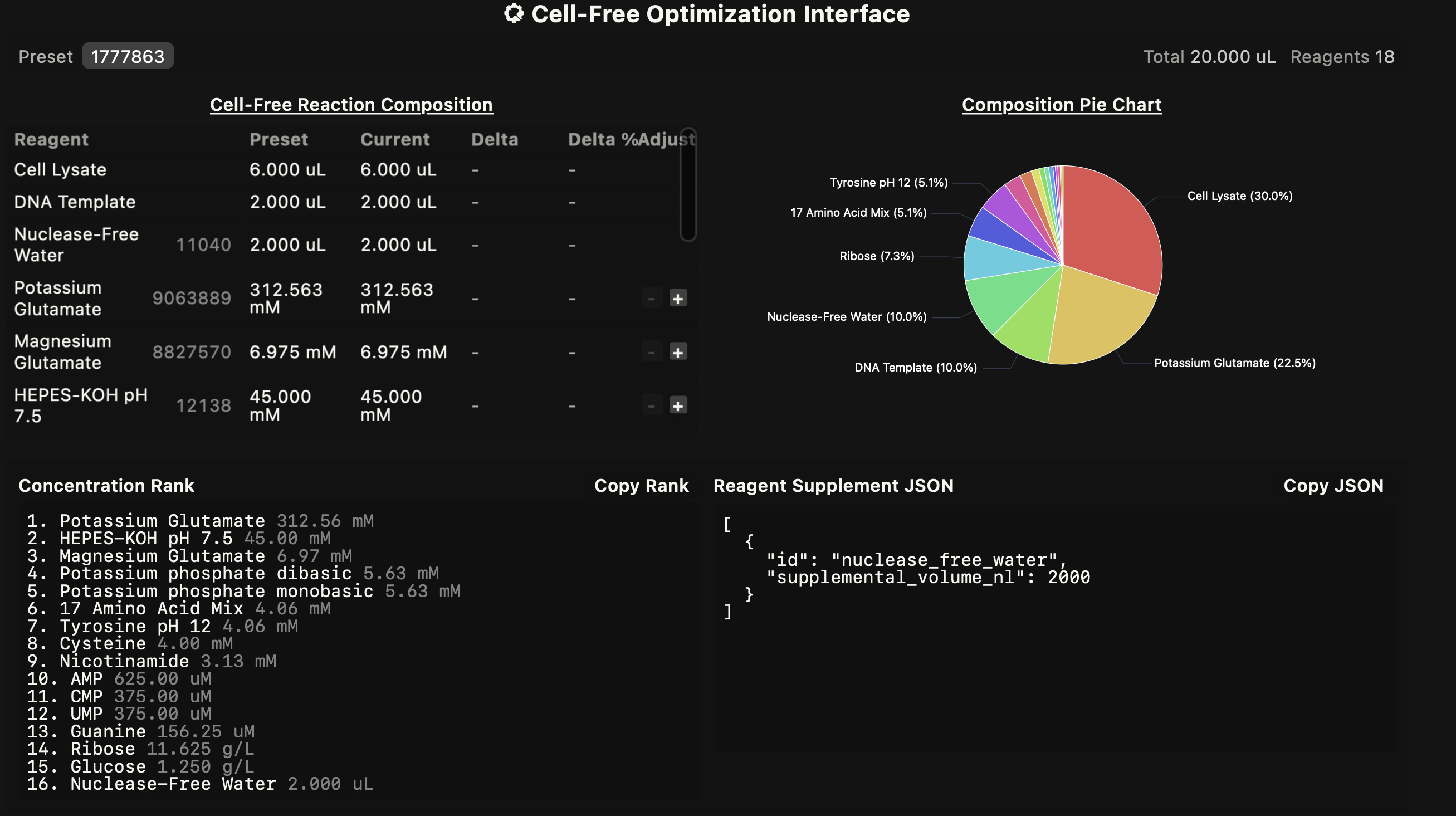

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}