Week 5 HW: Protein Design Part ii
- [] Homework — DUE BY START OF MAR 10 LECTURE
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc. Mechanis
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
- [] Task A
Your challenge:
Background: Design short peptides that bind mutant SOD1 and then decide which ones are worth advancing toward therapy. You will use three models developed in our lab:Part 1: Generate Binders with PepMLMHere is fully translated superoxide dimutase protein P00441 in uniprot with the initiator methionine included. We need to cleave that M off before we apply our requested mutation to progress with a mature enzyme. So not this…
But this..
To create our A4V SOD (love the rhyme) mutant…
The savvy student who fails to cleave the first methionine (M) can intuit the actual amino acid to change without thinking through any of the previous steps, but it’s nice to have a why in all things, since this is biology after all and we have evolution and ChatGPT. Please note that we will not want to use a protein sequence with any sort of truncation or wrapping on the sequence so here are my sequences for PPMLM-650M. To create our A4V SOD (love the rhyme) mutant…
Mutant A4V SOD for PepMLM-650
There are two options, full protein sequence and a 12-Sequence input which I settled on in later runs. MATKVVCVLKGD Within the PepMLM-650 codebase in Google Colab Notebook, there are sliders and input fields to parameterize individual runs. However, these parameters didn’t seem to encode, so I finally hard-coded changes, as I will show below as a series of excerpts pulled from the codebase. single_sequence = True #@param {type:"boolean"}
protein_seq = "MATKVVCVLKGD" #@param {type:"string"}
# Initial value for num_binders
num_binders = 4
# Initial values for top_k and peptide_length
top_k = 3
peptide_length = 12
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison. Scores above, I think. I feel like I’m in Gulliver’s Travels returning this exercise cold after 2 months so I am going to try and piece together memory using a peers homework. For the next phase I am now going to find the amino acid sequence for my SOD1 sequence. Now I’m not copying information because I just went to uniprot site myself and searched for SOD1 on the splash page and scrolling down on first page I found human sodc but I skipped over that one and found the sod for sheep
Record the perplexity scores that indicate PepMLM’s confidence in the binders. Given confusion perplexity scores are likely very high. The model confidence according to UniProt is spot on though, specifically model confidence is very high (pLDDT > 90). This is generated by AlphaFold as a per-residue confidence score (pLDDT) between 0 and 100. Now I am prepared to transition to part 2.
What about my mutants, though? I do not want to disrupt folding randomly, need an appropriate target region for mutation logic so I will leverage the MobiDB website. 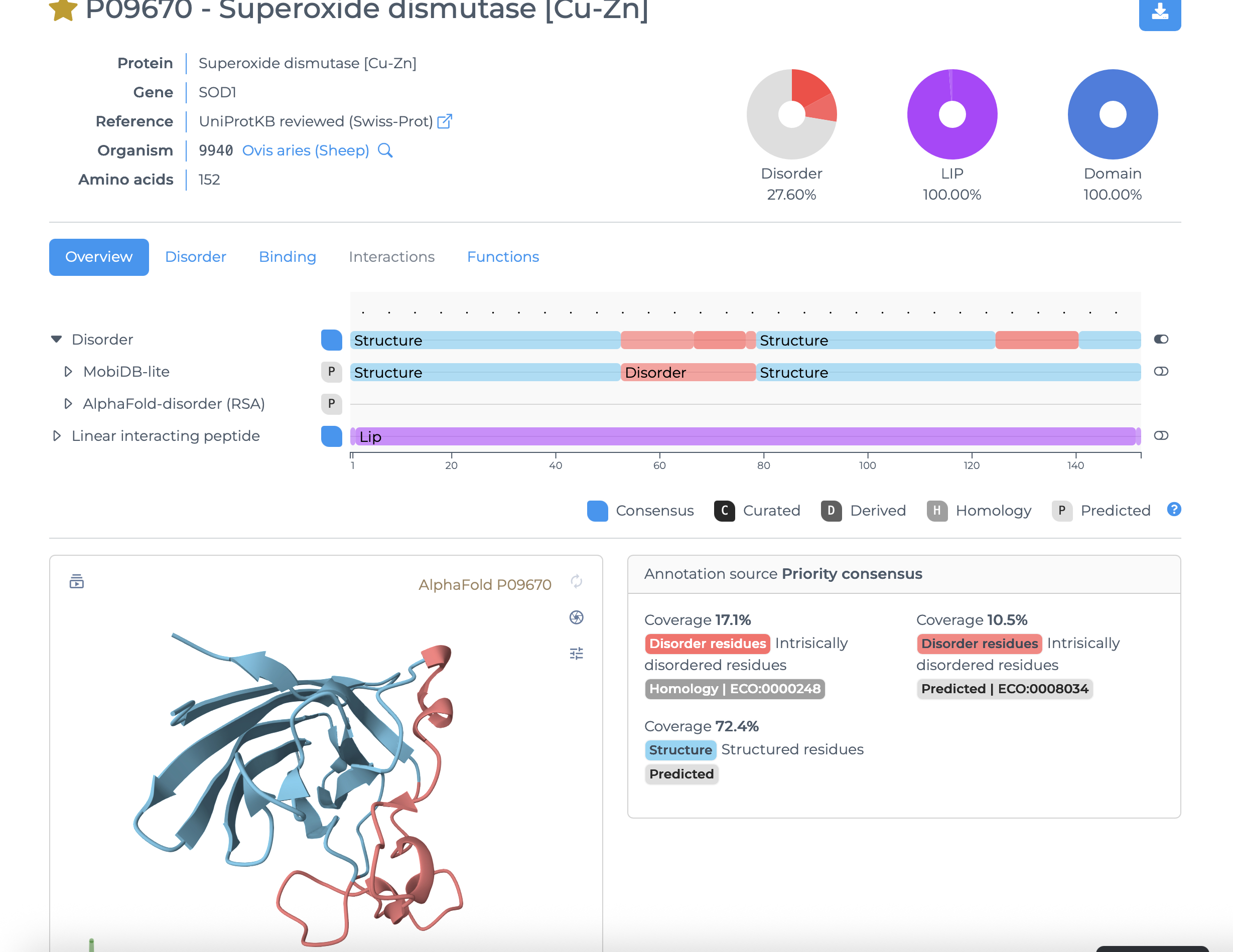
Enumerated Amino Acids with position and highlighted for subsequent mutagenesis based on encode segment flexibility, disruptability, and functional consequences
Amino acids selected for mutation
Final mutant sequence with three changes
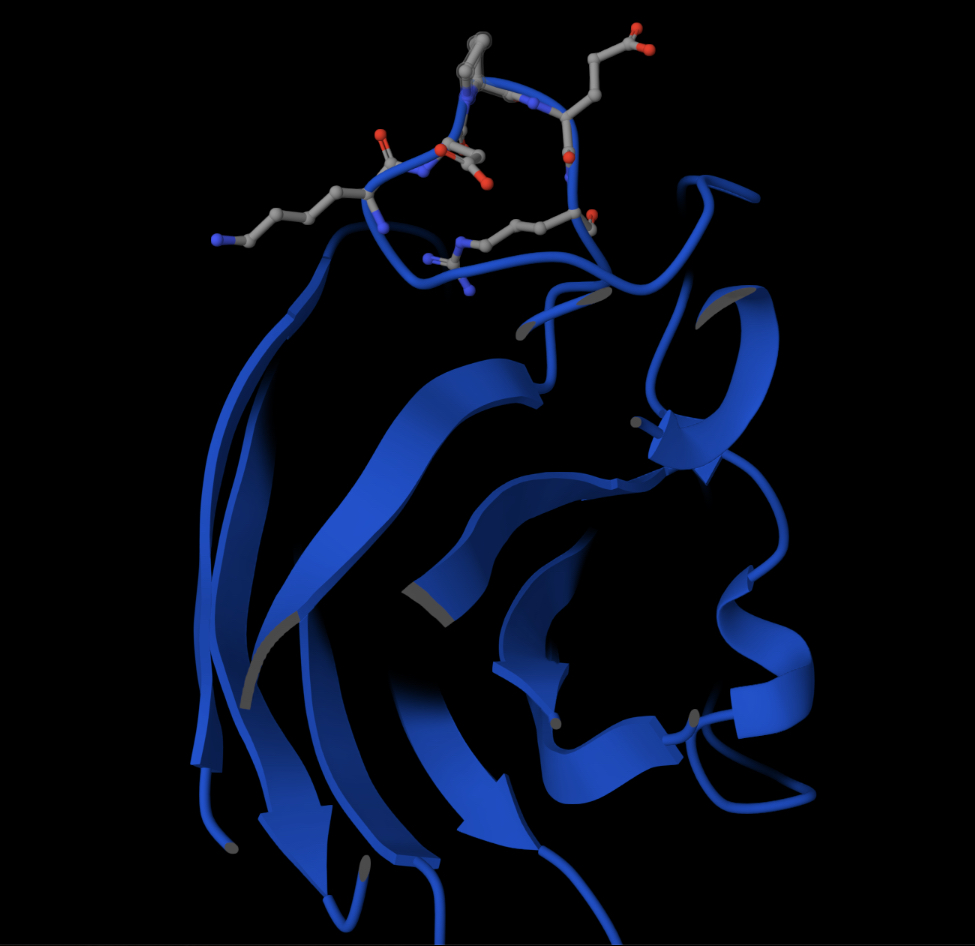
Part 2: Evaluate Binders with AlphaFold3Navigate to the AlphaFold Server: [alphafoldserver](https://alphafoldserver.com/welcome)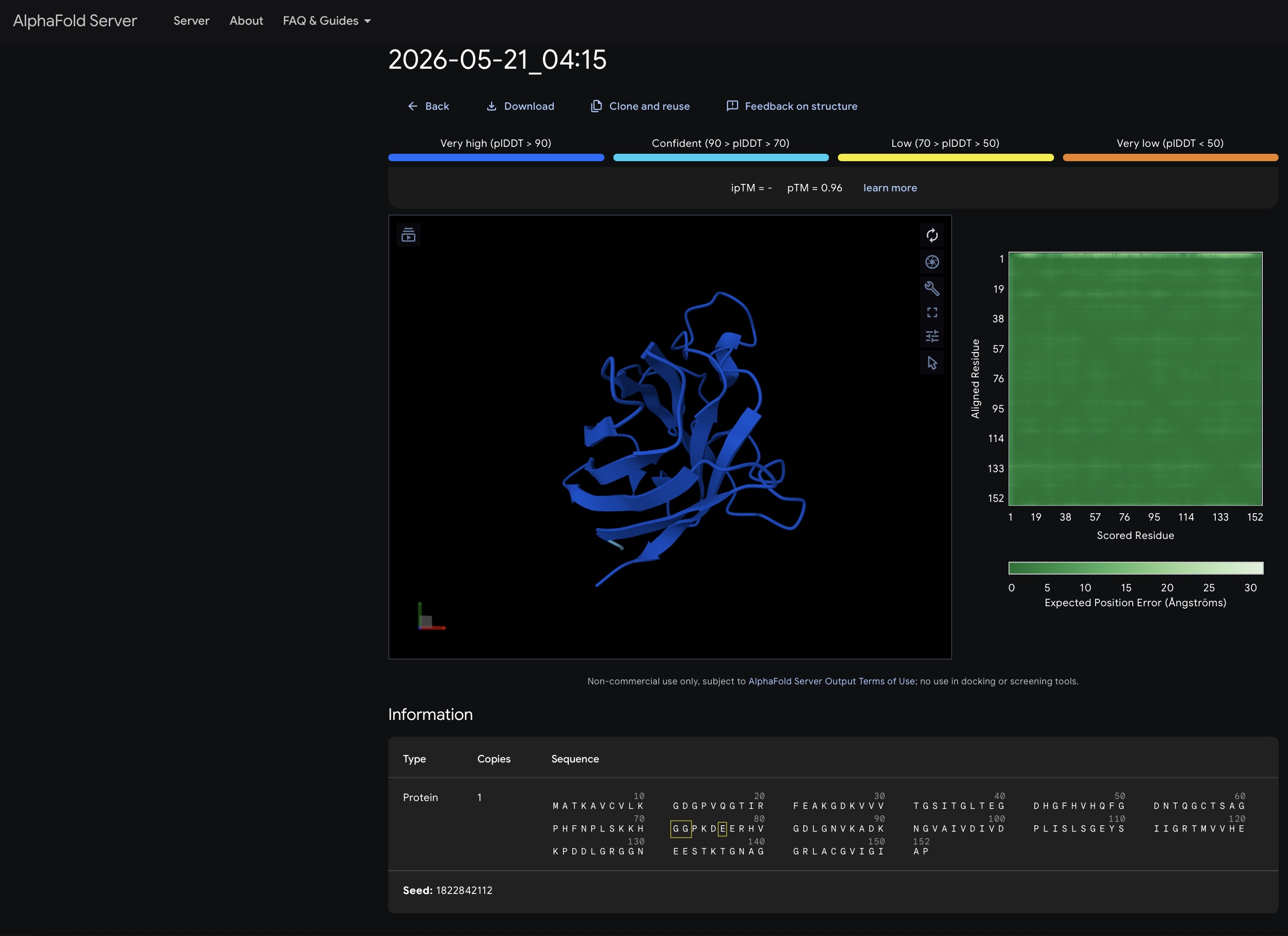 For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
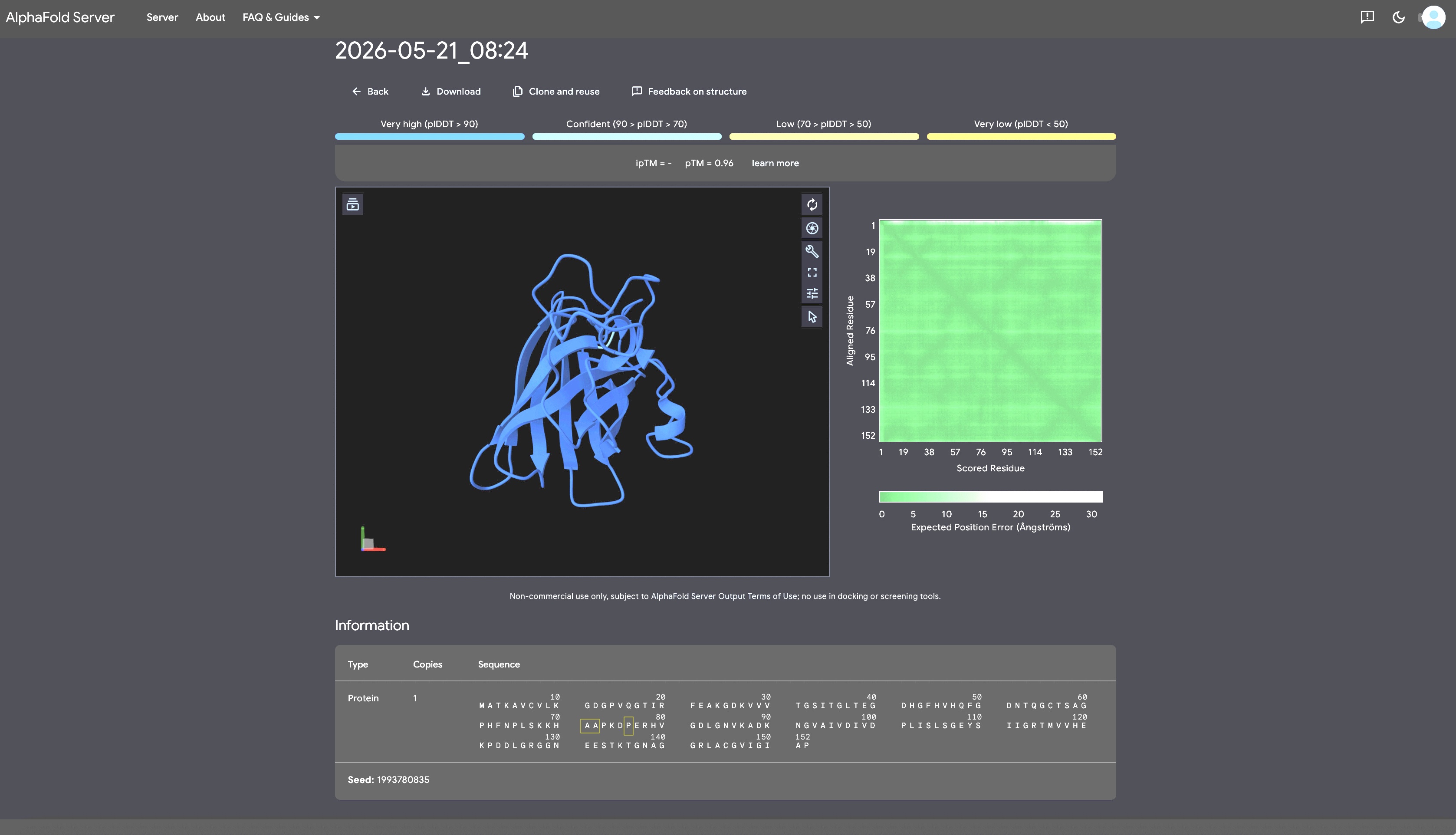 Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
 In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
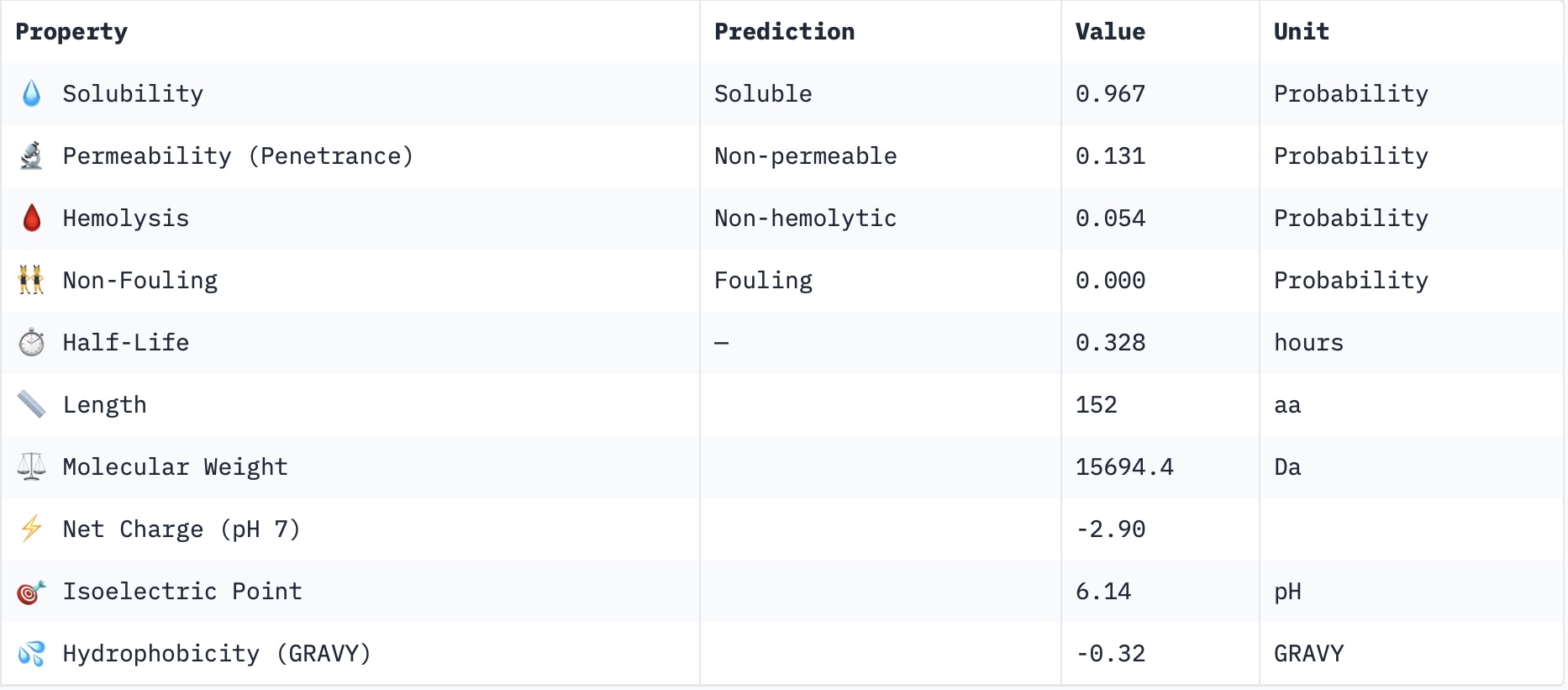
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerseStructural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide: 
Paste the peptide sequence.
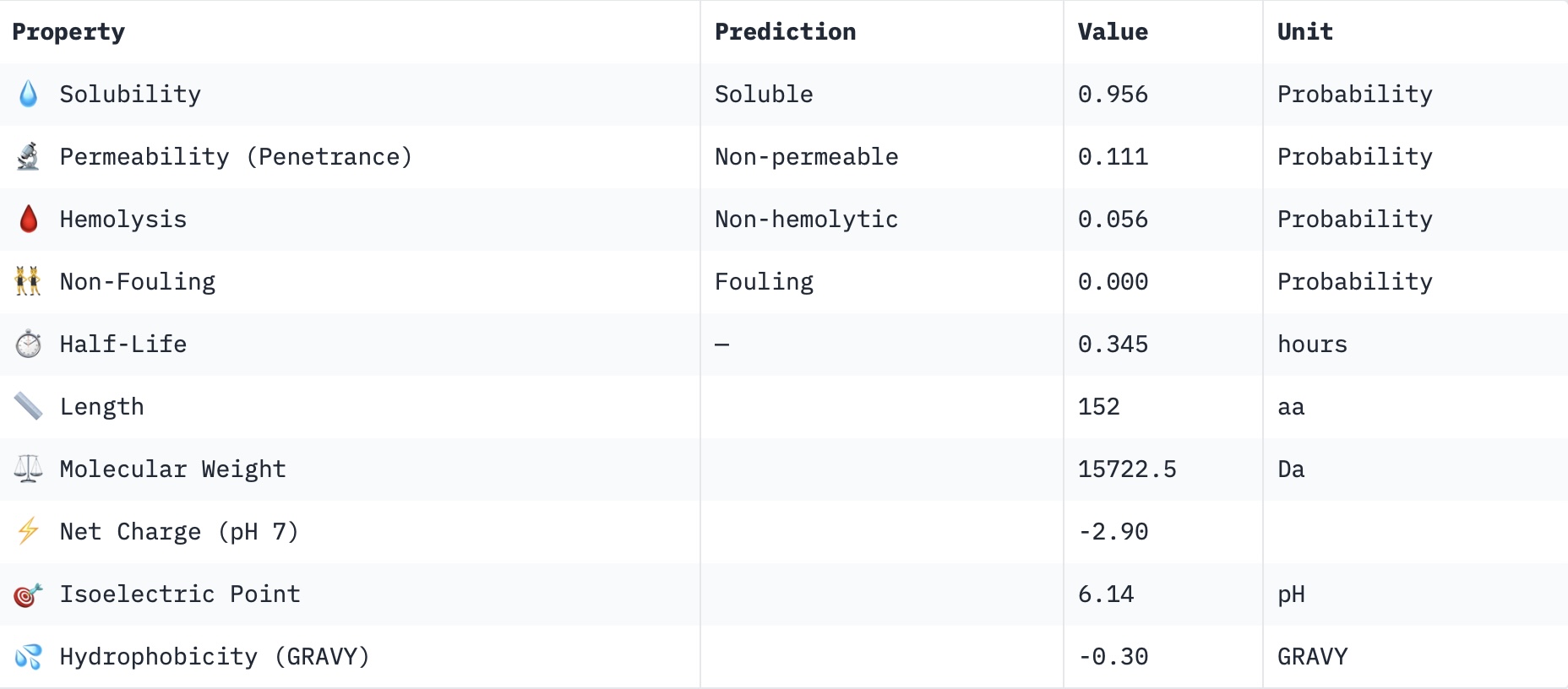
Check the boxes Predicted binding affinity Solubility Hemolysis probability Net charge (pH 7) Molecular weight Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties? Choose one peptide you would advance and justify your decision briefly.
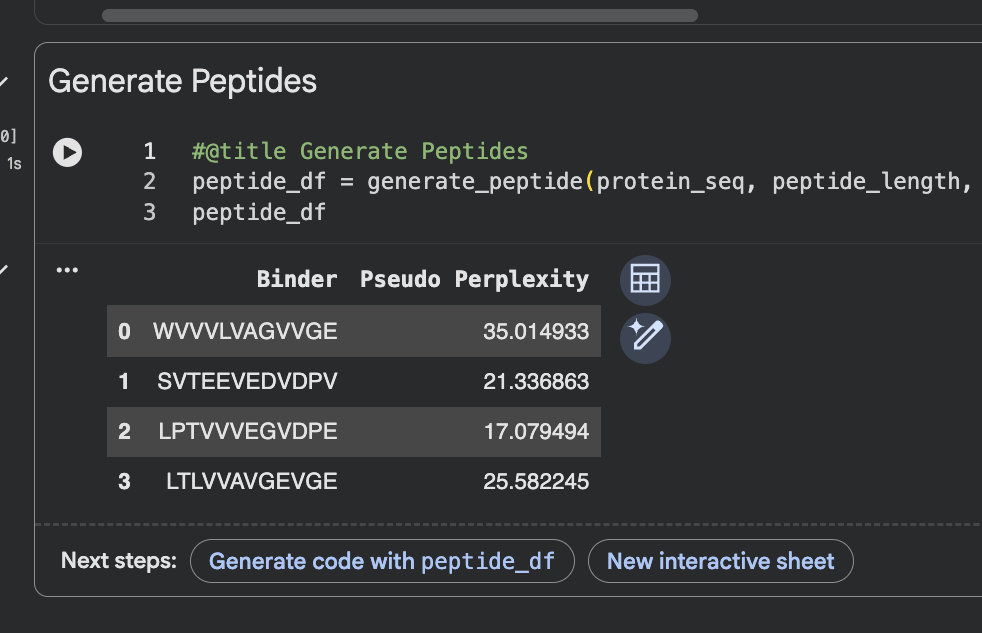
Part 4: Generate Optimized Peptides with moPPItNow, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once. Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
Paste your A4V mutant SOD1 sequence. Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch). Set peptide length to 12 amino acids. Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides. ~Set target for affinity to original WT: MATKAVCVLKGDGPVQGTIRFEAKGDKVVVTGSITGLTEGDHGFHVHQFGDNTQGCTSAGPHFNPLSKKHGGPKDEERHVGDLGNVKADKNGVAIVDIVDPLISLSGEYSIIGRTMVVHEKPDDLGRGGNEESTKTGNAGGRLACGVIGIAP After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies? It doesn’t stay connected to the necessary server to run. |

{kind=link}
{kind=link}
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
[] Task B - Boltz Document
https://docs.google.com/document/d/18Vd9TQL2FjpEU0QdlGCgHe1D0BDoMzcfPRiFEXQIAas/preview
Boltz Lab BRD4 Drug Discovery Platform Tutorial Introduction This exercise walks you through a real drug discovery workflow using the Boltz Lab platform - from predicting how known drugs bind a cancer target, all the way to running AI-generated molecule libraries and interpreting the results. You will work on BRD4 (Bromodomain-containing protein 4), an epigenetic reader protein and validated oncology target. BRD4 has been the subject of intense medicinal chemistry effort in recent years. What you will learn • How to use Boltz Lab to predict protein-ligand binding structures • How to interpret Binding Confidence and Optimization Score metrics • How to set up a virtual screening project in Boltz Lab • How to compare known drugs and AI-generated molecules in a single workflow • How to critically evaluate computational predictions from a drug discovery perspective
Background: BRD4 and BET Bromodomains
BRD4 is a member of the BET (Bromodomain and Extra-Terminal) family of epigenetic reader proteins. It recognises acetylated lysine residues on histone tails and recruits transcriptional machinery to gene promoters, driving expression of oncogenes including c-Myc. Dysregulated BRD4 activity is implicated in haematological malignancies, solid tumours, and inflammatory disease.
This exercise inspects the example of JQ1 - the landmark BRD4 inhibitor reported by Filippakopoulos et al. in Nature 2010. The three compounds below capture a hit-to-candidate optimisation journey, including a deliberately instructive stereochemical twist.
Stage
Compound
SMILES
Hit
Stripped
Back Core
CC1C2C(=C(SC=2NCCN=1)C)C
Lead
Triazole +
Acid
O=C(C[C@@H]1N=C(C)C2C(=C(SC=2N2C1=NN=C2C)C)C)O
Candidate
(+)-JQ1
O=C(C[C@H]1C2=NN=C(N2C3=C(C(C4=CC=C(C=C4)Cl)=N1)C(C)= C(S3)C)C)OC(C)(C)C
�� Note: Reference: Filippakopoulos P. et al. Selective inhibition of BET bromodomains. Nature 468, 1067-1073 (2010). Crystal structure PDB: 3MXF (BRD4 BD1 complexed with (+)-JQ1).
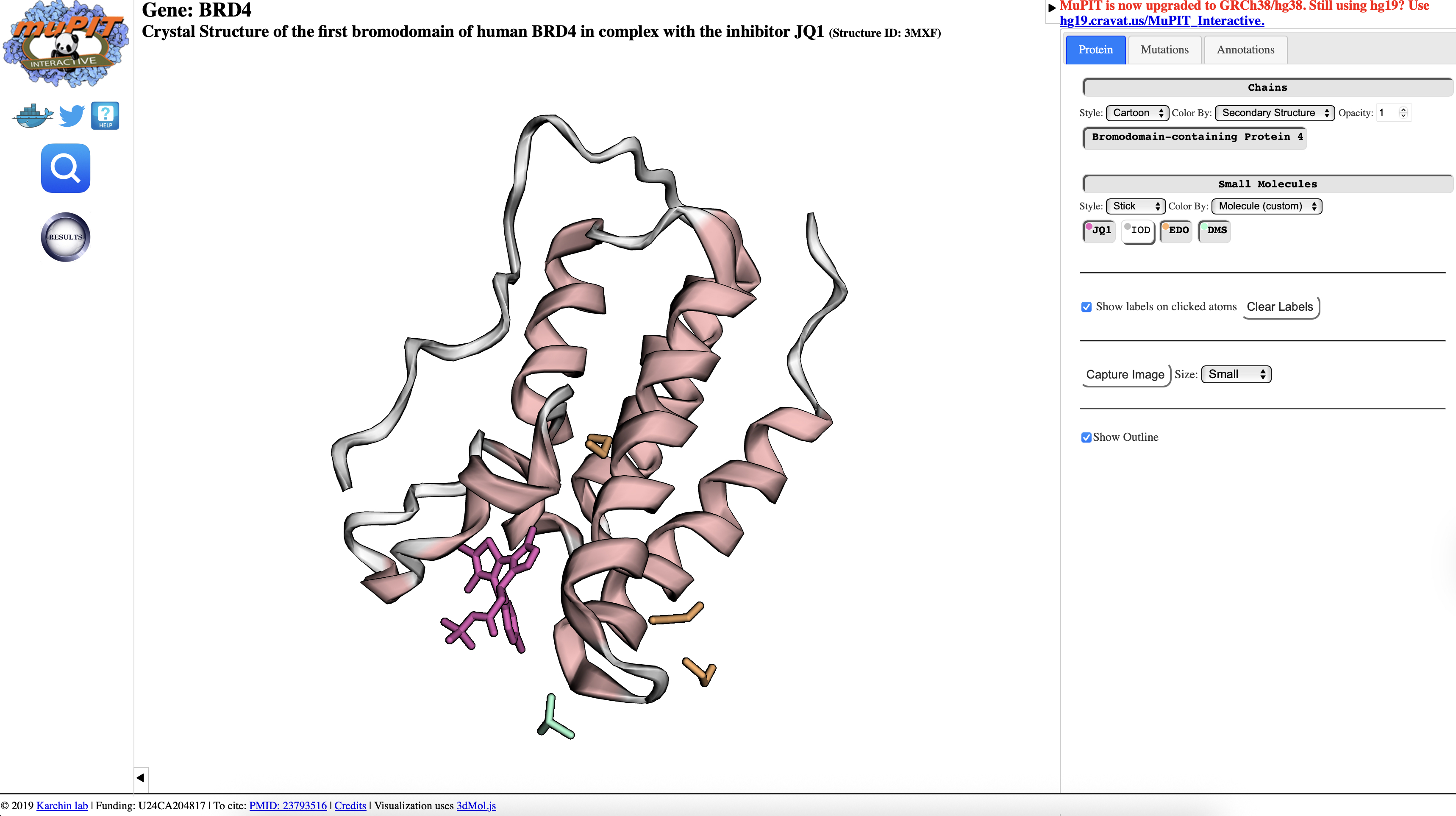 source
Tutorial designed by Geoffrey Smith
Boltz Lab | BRD4 Platform Tutorial — MIT Guest Lecture
Part 0: Sign-up to Boltz Lab
Go to lab.boltz.bio, click “Request Access”, add your name and email while specifying as organization name “HTGAA”, and click “Submit request”.
source
Tutorial designed by Geoffrey Smith
Boltz Lab | BRD4 Platform Tutorial — MIT Guest Lecture
Part 0: Sign-up to Boltz Lab
Go to lab.boltz.bio, click “Request Access”, add your name and email while specifying as organization name “HTGAA”, and click “Submit request”. We will try to make sure to approve your request within a day or two, giving you credits for both the exercise as well as further exploration. If you plan to use Boltz Lab for your final project and need more credits, please reach out to me at gabriele@boltz.bio.
We will try to make sure to approve your request within a day or two, giving you credits for both the exercise as well as further exploration. If you plan to use Boltz Lab for your final project and need more credits, please reach out to me at gabriele@boltz.bio.
Part 1: Structural Predictions in the Sandbox
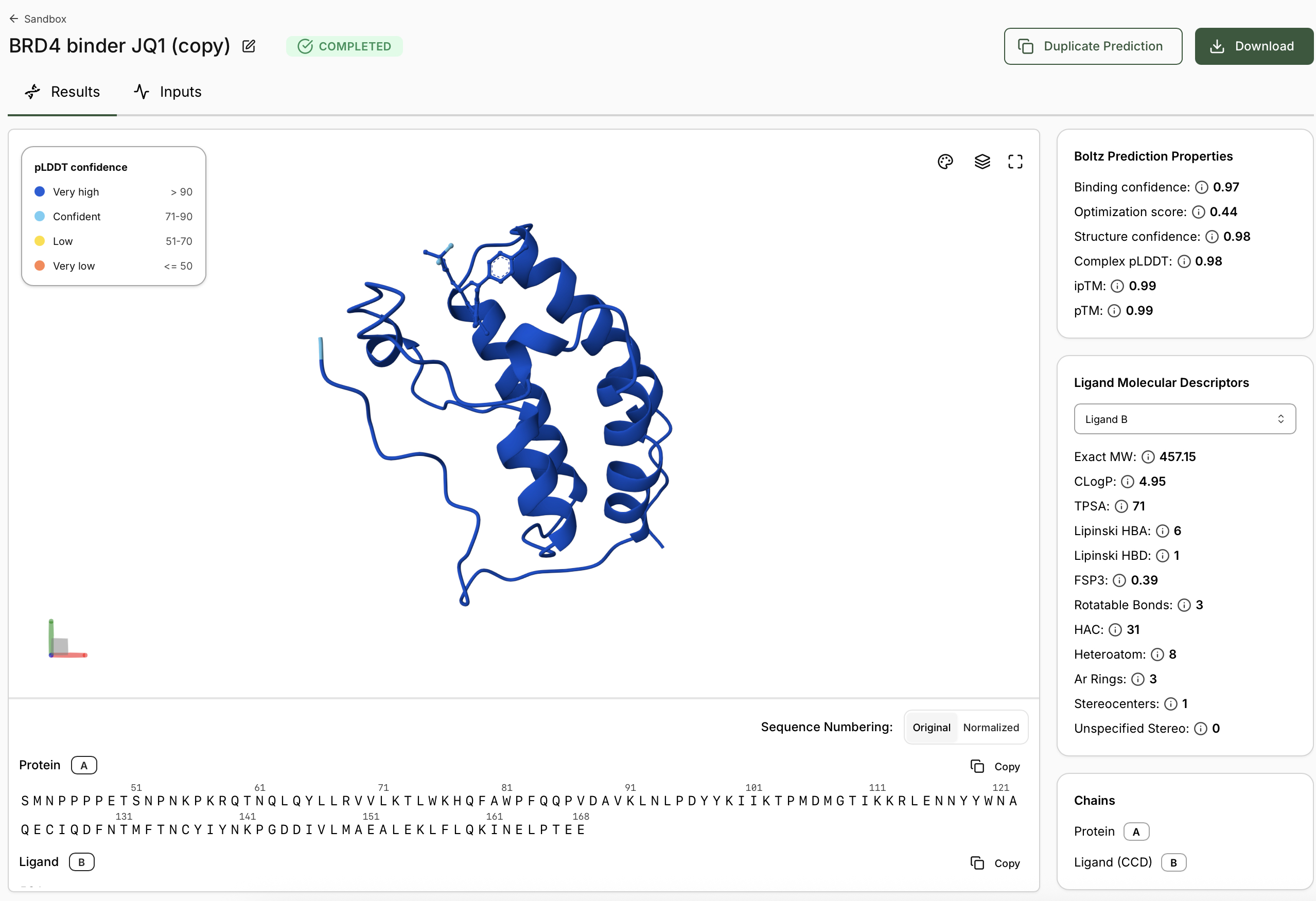
Start with three Boltz-2 predictions in the Sandbox to understand how the model scores protein– ligand interactions across a real drug discovery progression. 1.1 The Boltz-2 Metrics Explained Before you run your first prediction, understand these three key outputs: Metric Range What it means When to trust it Binding Confidence 0 - 1 How confidently Boltz-2 places the ligand in the binding site. Higher = predicted more likely to bind.
0.7 considered
reliable; > 0.8 high
confidence Optimization Score 0 - 1 A relative affinity for use in congeneric series, or between known binders. Higher = predicted to bind more tightly. Use for relative
ranking, Structure Confidence 0 - 1 Measures the confidence of the predicted structure Higher = more likely the structure predicted correctly. 0.8 considered high confidence.
You need all three to be high to trust a prediction. 1.2 Running Your Three Predictions Navigate to the Boltz Sandbox at lab.boltz.bio and log in to your account.
- Go to Sandbox → New Prediction
- Name this BRD4 binder JQ1
- Select ‘Complex’, add ‘Sequence from RCSB’, and add 3MXF
- Continue through Constraints (not needed for this example), and select Jq1 as the Binder for an affinity prediction.
- Submit the prediction.
- Use the ‘Duplicate Prediction’ in the results review, and remove the small molecule.
- Add in the SMILES for the Hit and Lead.
- When predictions complete, record your results in the table below
 Tutorial designed by Geoffrey Smith
Boltz Lab | BRD4 Platform Tutorial — MIT Guest Lecture
Compound
Binding Confidence
Optimization Score
Structure Confidence
Hit
Tutorial designed by Geoffrey Smith
Boltz Lab | BRD4 Platform Tutorial — MIT Guest Lecture
Compound
Binding Confidence
Optimization Score
Structure Confidence
Hit
| Compound | Binding Confidence | Optimization Score | Structure Confidence |
|---|---|---|---|
| Hit | |||
| Lead | |||
| JQI |
Discussion Questions
• Does Binding Confidence increase as you move from hit to clinical candidate? What would you expect, and why might it deviate?
• Inspect the predicted binding pose for JQ1. Can you identify potential key binding interactions.
• Compare the Optimization Scores. How do the scores compare for JQ1 vs the Lead. Part 2: Setting Up a BRD4 Design Project
Now you will create a small molecule Design Project - the Boltz Lab workflow for virtual screening and lead optimisation. We will set up BRD4 as a target using the clinical candidate as our structural reference.
2.1 Creating the Target
- From the dashboard, create a Design Projects via ‘New Project’
- Name your project: ‘BRD4 Workshop '
- Select ‘Small Molecule’
- Click Add Target and add the protein structure as in the Sandbox using PDB code 3MXF 5. Continue and let the apo structure complete. Continue if the structure looks good. 6. Leave binding residue selection blank, the platform will auto-detect the pocket 7. In the Molecular Probe field, paste the JQ1 SMILES.
- Predict Pocket Structure and complete the Target Set-Up �� Note: Why no binding residue selection? Boltz Lab uses the probe SMILES to identify the relevant binding pocket automatically. What the Probe Does The probe compound defines the active site geometry for the target. Boltz-2 uses the cofolded probe structure as an internal reference when scoring your library compounds. This is equivalent to providing a crystallographic template in traditional docking - except the model generates the structure on the fly.
Tutorial designed by Geoffrey Smith Boltz Lab | BRD4 Platform Tutorial — MIT Guest Lecture Part 3: Running Your Virtual Screen BRD4 is a well validated target, and therefore we will generate a small Library of 1K small molecule binders. For typical exploratory targets, Boltz recommends 20K as a minimum number of binders. 3.1: Run a Generative Design Campaign We will utilize the Boltz Lab small-molecule generative workflow. This generates novel molecules optimised for BRD4 binding using Boltz-2 as the scoring function.
- After creating the design project, Boltz Lab will prompt you to Generate binders with AI. 2. Name your experiment, provide a relevant hypothesis, and Create the Experiment. 3. The New Virtual Screen will be pre-configured with a Generative screen using the Enamine REAL space.
- Keep ‘Normal Filtering’ selected. This will ensure we only generate molecules acceptable to a medicinal chemist.
- Decide if you would like to apply any Molecule Filters. We recommend the ‘Drug-Like’ Preset.
- Select a custom number of Binders and enter 1K.
- Start the Virtual Screen.
- Allow binders to be generated, and View Results in Experiment �� Note: 1k molecules is a very small screen, for real applications where you plan to synthesize the molecule (e.g. your final project) we would recommend running at least 10-20k molecules. Part 4: Analysis and Discussion As your experiment completes, use the ‘Quick Add Candidates’ on the experiment screen to add JQ1 as a benchmark for generated designs. 4.1 Interpreting Your Results As your experiment completes, use the ‘Quick Add Candidates’ on the experiment screen to add JQ1 as a benchmark for generated designs. From your screen output, identify three categories of molecules: Category Criteria Likely interpretation High confidence binders Binding Confidence > 0.80 Opt. Score > 0.40 Strong predicted hits - inspect poses carefully Moderate confidence Binding Confidence 0.65–0.80 Opt. Score 0.25–0.40 Plausible binders - additional validation needed Low confidence / non-binders Binding Confidence < 0.65 Opt. Score < 0.25 Likely incorrect pose or non binding chemotype
Tutorial designed by Geoffrey Smith
Boltz Lab | BRD4 Platform Tutorial — MIT Guest Lecture
Discussion:
As the virtual screen completes, assess the following:
• How does JQ1 in the Design Project screen alongside the library. Does it score as the top compound?
• How do the top scoring binders compare in binding pose to JQ1?
• Try adding a second target to your project via the dropdown in the structure viewer, for example, BRD2 (PDB: 5UEN). Re-run the top scoring binders against BRD2 and compare which compounds score highly for BRD4 but not BRD2. This is a selectivity analysis - a key part of real BET inhibitor programs.
Resources and Further Reading
Resource
Link / Reference
Boltz Lab Platform
docs.boltz.bio
Key BRD4 Paper
Filippakopoulos P. et al. Nature 468, 1067–1073 (2010)
JQ1 PDB Structure
rcsb.org/structure/3MXF
Tutorial designed by Geoffrey Smith
Part C: Final Project: L-Protein Mutants
This homework requires computation that might take you a while to run, so please get started early.
Tools
See HTGAA Protein Engineering Tools spreadsheet