Subsections of Homework
Week 1 HW: Principles and Practices
![cover image]()
![cover image]()
1. First, describe a biological engineering application or tool you want to develop and why.
I am interested in developing materials that recover over time by mimicking fungal evolutionary conservatism and continuity. I am currently developing a pollen sensor intended to be placed in children’s playgrounds, while simultaneously reading Merlin Sheldrake’s Entangled Life. Together, these are inspiring me to think about design in a different way.
In Entangled Life, Sheldrake describes how certain fungi infect insects, such as ants, causing them to veer off the trajectory of their own evolutionary story and onto the evolutionary path of the fungus. This blurring of where one organism ends and another begins, prompted me to reflect on how can other objects operate in similar ways.
From this, I began to brainstorm how a pollen sensor, a public infrastructure where durability and reparability govern its sustainability and performance, could behave more like a fungus. How might it heal itself? How could it detect air quality and sense its environment without relying solely on electronic monitoring of pollen, carbon, dust, or humidity? What might be gained from infrastructure that is self-regenerative, adaptive, or even slowly evolving into another living organism?
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
In considering material that recovers especially public infrastructure, some governance that relates are environmental responsibility, ecological integration, public trust and safety standards regarding to public infrastucture utilities.
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
- Environmental responsibility
- Purpose: Self-recovering material used in public infrastructure does not introduce harm to surrounding environment and affect original biodiversity.
- Design: Require environmental impact assessments for all materials and define acceptable interactions between the infrastructure and local ecosystems.
- Assumptions: That materials designed to recover or regenerate will affect ecosystems and hence has to be carefully governed.
- Risks of Failure & “Success”:Failure is that the regenerative materials will alter local ecosystems in unpredictable or harmful ways. Success is that regenerative material will improve circularity in resources.
- Public safety and infrastructure standards compliance
- Purpose: Maintaining public trust by ensuring the material meets established safety stands especially as it begins to repair
- Design: Require regular inspection protocols that account for material change, aging, or regeneration over time. Establish clear thresholds for intervention.
- Assumptions: Public acceptance depends on reliability, when the infrastructure completely regnerates it is well integrated back into nature.
- Risks of Failure & “Success”: Failure is that the material recovery will compromise the structrual integrity and introduce new safety hazards. The success is that the infrastructures remains safe, reliable over a period of time until original functionality is overtaken by new identity.
- Ecological impact
- Purpose: The public infrastructure with regnerative material operates within ecological limits
- Design: Ensure ecological integration rather than disruption
- Assumptions: The rate of regeneration is influenced greatly and different location to location hence different types of material must be used, not a one size fits all.
- Risks of Failure & “Success”: Success is that the regenerative material will allow ecological compatibilty and creative positive interaction with its surroundings. Failutre is that failed integration between the organisms and their environment.
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
| Does the option: | Option 1 | Option 2 | Option 3 |
|---|
| Enhance Biosecurity | | | |
| • By preventing incidents | 3 | 3 | 3 |
| • By helping respond | 3 | 3 | 3 |
| Foster Lab Safety | 3 | 3 | 3 |
| • By preventing incident | 3 | 3 | 3 |
| • By helping respond | 3 | 3 | 3 |
| Protect the environment | | | |
| • By preventing incidents | 1 | 1 | 1 |
| • By helping respond | 1 | 1 | 1 |
| Other considerations | | | |
| • Minimizing costs and burdens to stakeholders | 1 | 2 | 2 |
| • Feasibility? | 2 | 2 | 1 |
| • Not impede research | 1 | 2 | `1 |
| • Promote constructive applications | 1 | 2 | 1 |
5.Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Drawing upon this scoring, I would prioritise priortise Public safety and infrastructure standards compliance because this goverance option addresses the risks associated with materials and installation, it ensures the regulations and compliances are met with industry standards which together contribute and build public trust. Some trades offs are safety may constrain certain material properties where it cannot completely replicate the conservatism or continuity like fungus. Key assumptions are that current safety standards do not yet provide a framework for selfregenerative materials, and there is uncertainty regarding how the materials will behave under rapdily changing conditions.
In preparation for Week 2’s lecture on “DNA Read, Write, and Edit,"
In addition, answer these questions in each faculty member’s section:
Homework Questions from Professor Jacobson:
- Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Error Rate:
1:1016
Throughput:
10 mS per
Base Addition
Compared to length of human genome
3 x 109 base pairs
Biology deal with that discrepency through error correcting gene sythesis.
- How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
10143 possible DNA sequences, and not all work to code for protein of interest because of codon usage bias and mRNA stability.
Homework Questions from Dr. LeProust:
- What’s the most commonly used method for oligo synthesis currently?
Most common use is each nucleotide addition has a small failure rate (~0.5–1%).
- Why is it difficult to make oligos longer than 200nt via direct synthesis?
It is difficult because loner sequences have more opportunities for incomplete coupling and other chemical damages.
- Why can’t you make a 2000bp gene via direct oligo synthesis?
Direct oligo synthesis only reliably produces ≤200 nt sequences
Homework Question from George Church:
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
- Arginine
- Histidine
- Isoleucine
- Leucine
- Lysine
- Methionine
- Phenylalanine
- Threonine
- Tryptophan
- Valine
It shows how the Lysine contingency is a noraml universal biologcal contstraint. All aniamsl need certain amino acids from food.
Week 2 HW: DNA Read, Write, Edit Life
Part 1: Benchling & In-silico Gel Art
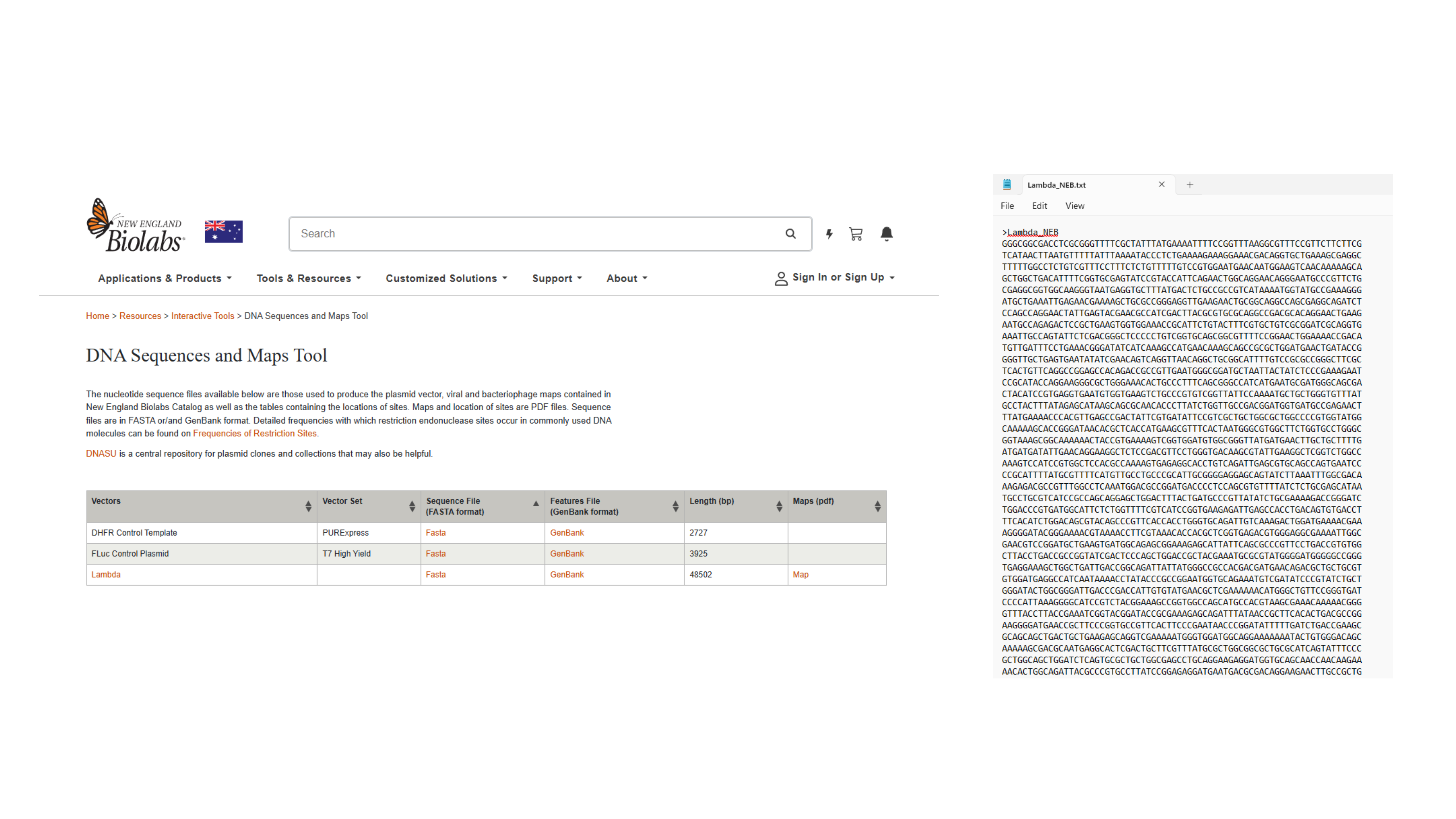
Navigating to Lambda sequence and saving as text. file

Importing Lambda file by saving FASTA Format of Sequence file
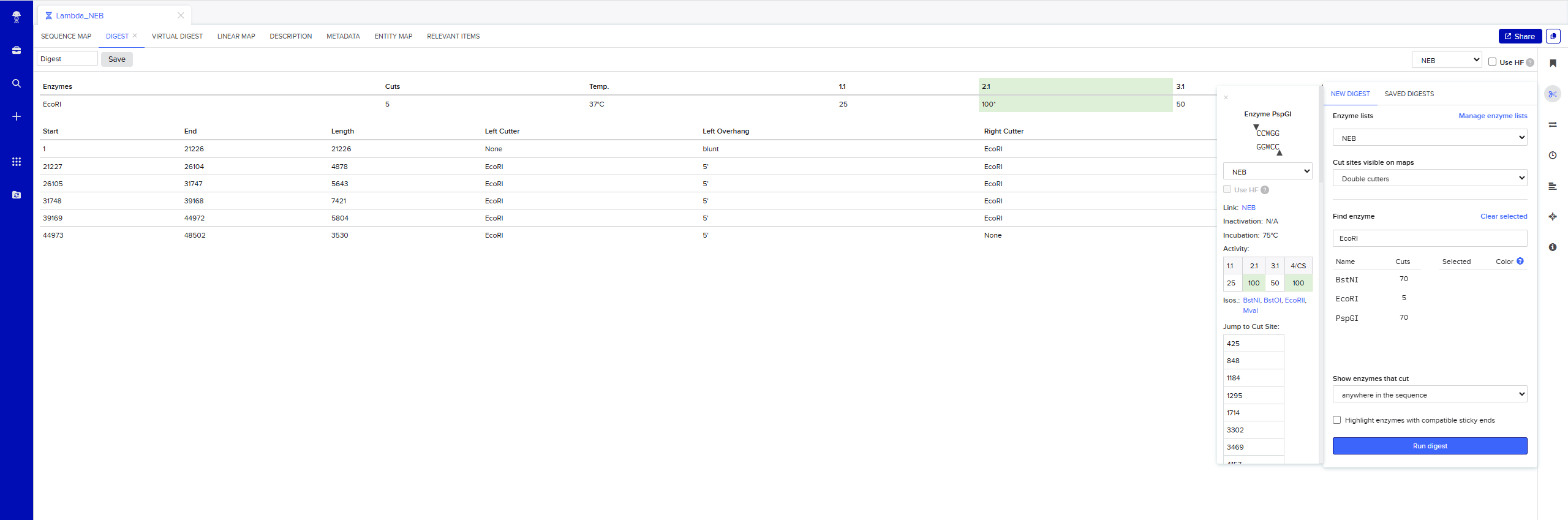
Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI
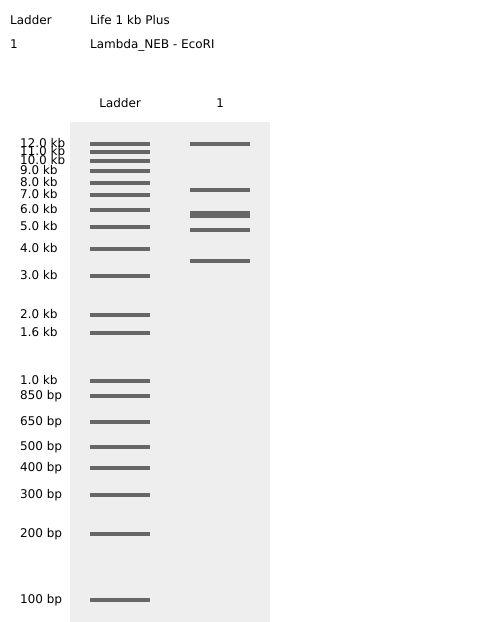
Testing Virtual digest example
Adding more restrcition enzymes

Simulate Restriction Enzyme Digestion with all the Enzymes
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks
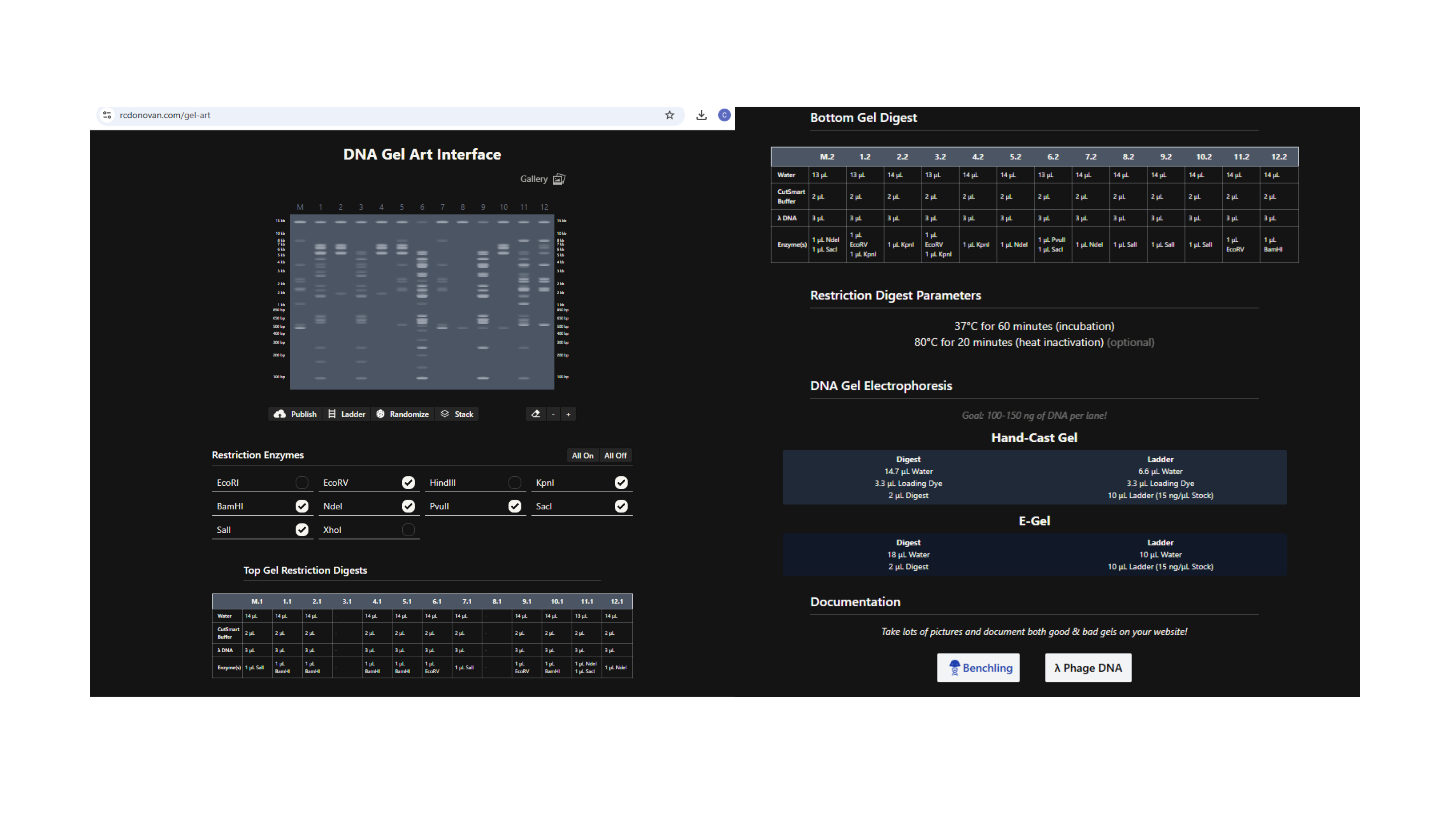
Using https://rcdonovan.com/gel-art to create randomised Gel Art
 Fireworks and Flower Gel Art Shape
Fireworks and Flower Gel Art Shape
Part 3: DNA Design Challenge
3.1 Choose your Protein
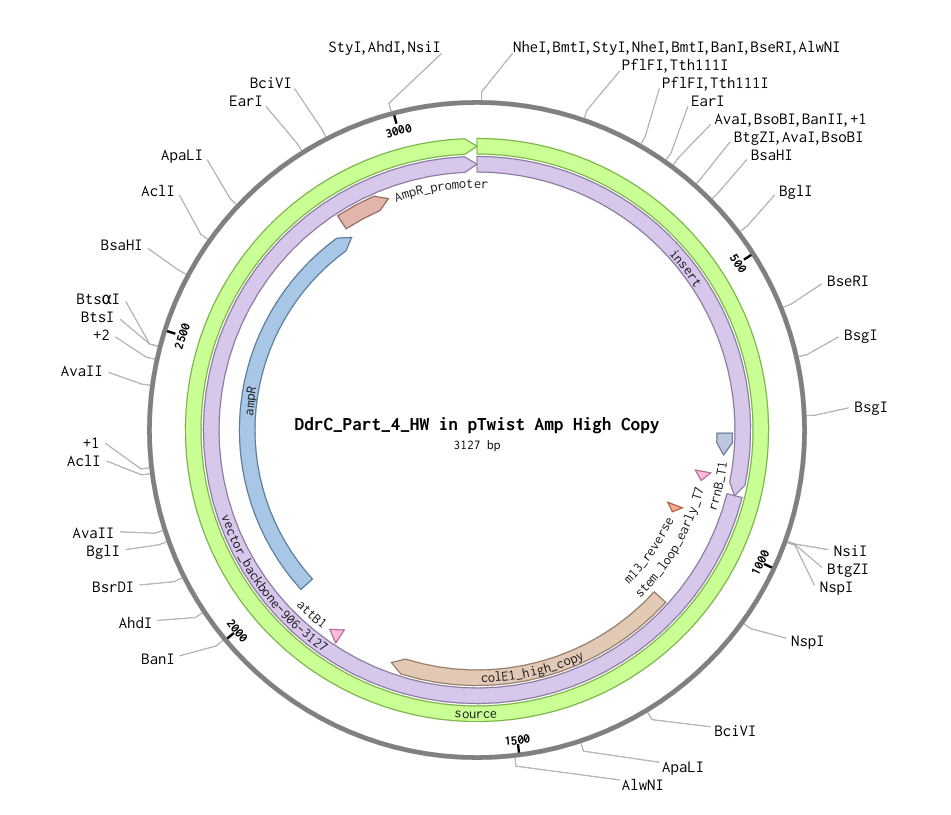
Figure 1
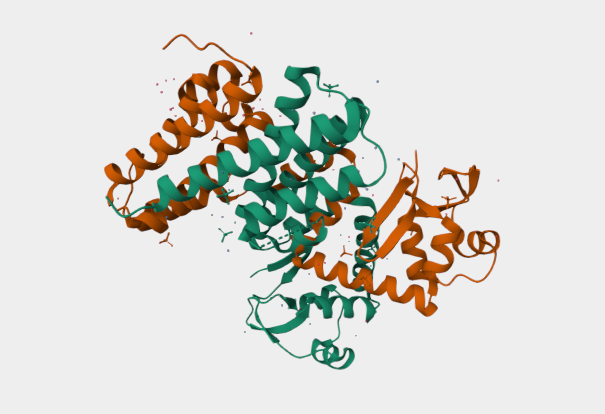
Figure 2
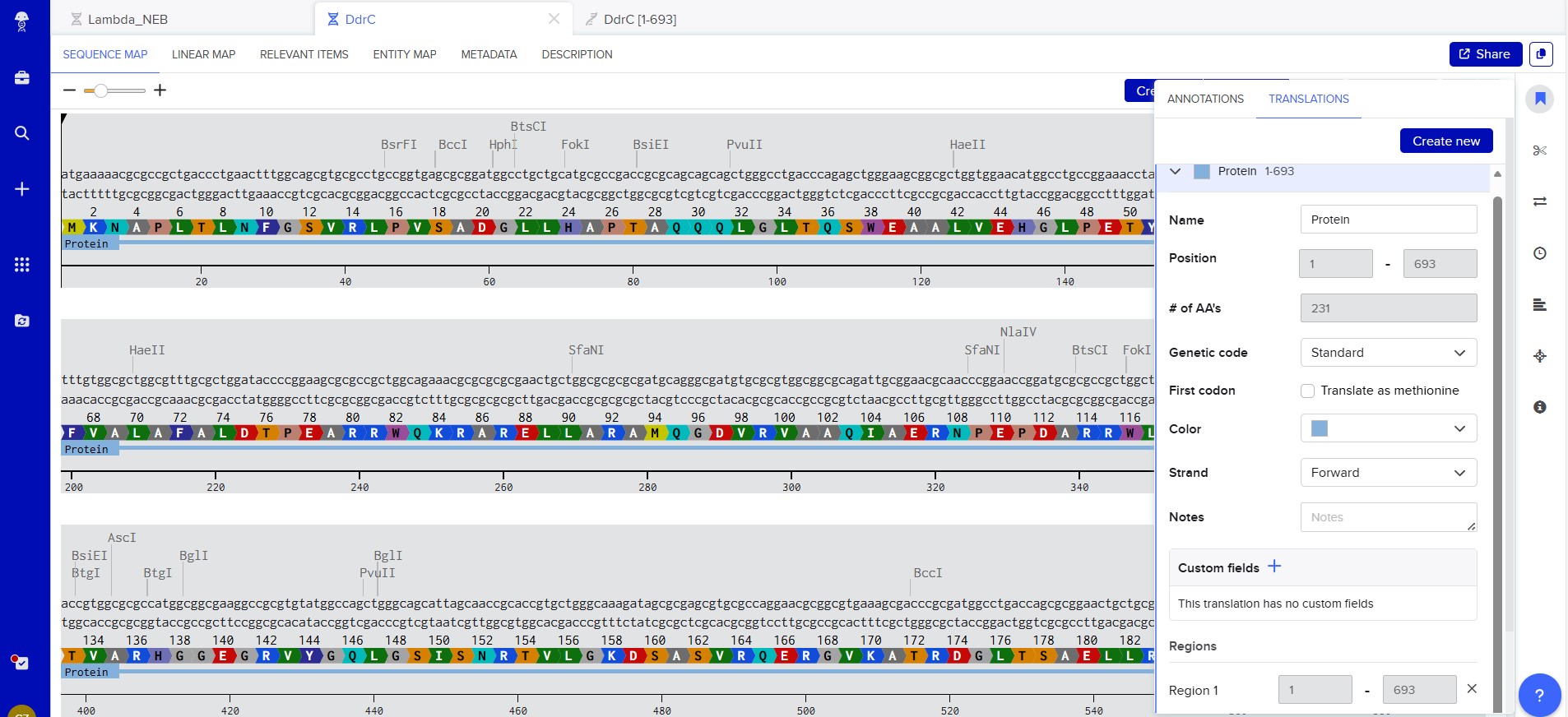
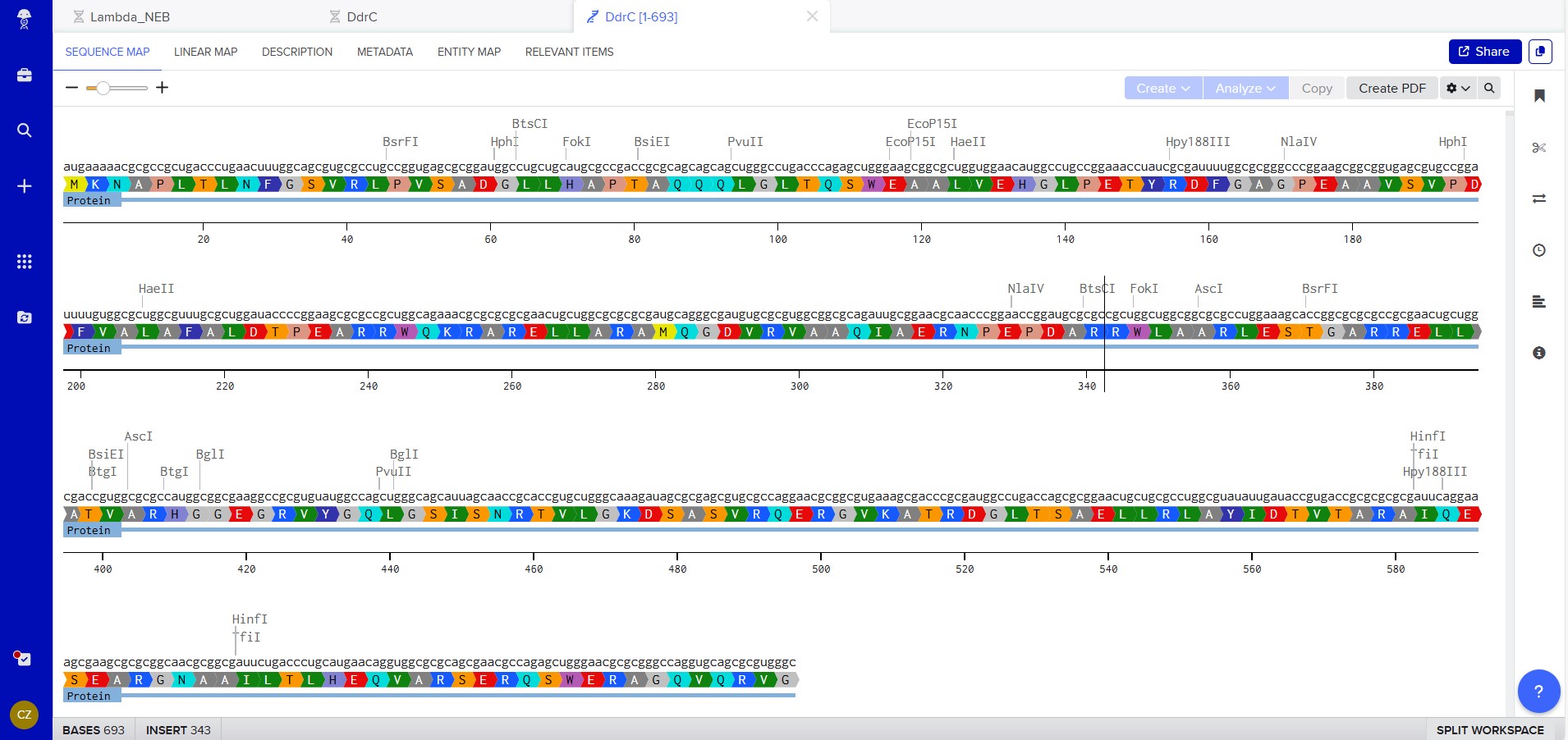
My chosen protein is DdrC Protein seen in figure 1 and 2 above, because of its ability to respond to DNA damage by putting a stop to the damage and alerting the cell to begin repair process. For my final project I would like to research into material that is able to self-repair and contain the deterioation. Hence I wanted to research about how DdrC from Deinococcus radioduran is able to stabilise DNA breaks by binding to both single and double stranded leisions. Something that I thought was quite interesting through the readings of this protein is its structural asymmetry allowing it precise lesion recognition (“The Unique Mechanism of DdrC in Enhancing DNA Stability,” 2024). It bears two asymmetric DNA binding sites located on either side of the dimer and can modulate the topology and level of compaction of circular DNA (Gueguen et al., 2022).
Protein Sequence
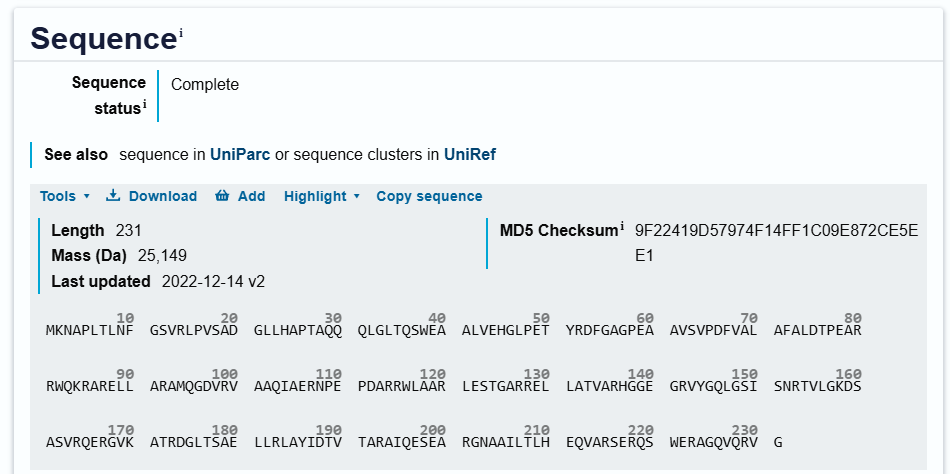
sp|Q9RYE6|DDRC_DEIRA DNA damage response protein C OS=Deinococcus radiodurans (strain ATCC 13939 / DSM 20539 / JCM 16871 / CCUG 27074 / LMG 4051 / NBRC 15346 / NCIMB 9279 / VKM B-1422 / R1) OX=243230 GN=ddrC PE=1 SV=2
MKNAPLTLNFGSVRLPVSADGLLHAPTAQQQLGLTQSWEAALVEHGLPETYRDFGAGPEA
AVSVPDFVALAFALDTPEARRWQKRARELLARAMQGDVRVAAQIAERNPEPDARRWLAAR
LESTGARRELLATVARHGGEGRVYGQLGSISNRTVLGKDSASVRQERGVKATRDGLTSAE
LLRLAYIDTVTARAIQESEARGNAAILTLHEQVARSERQSWERAGQVQRVG
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence
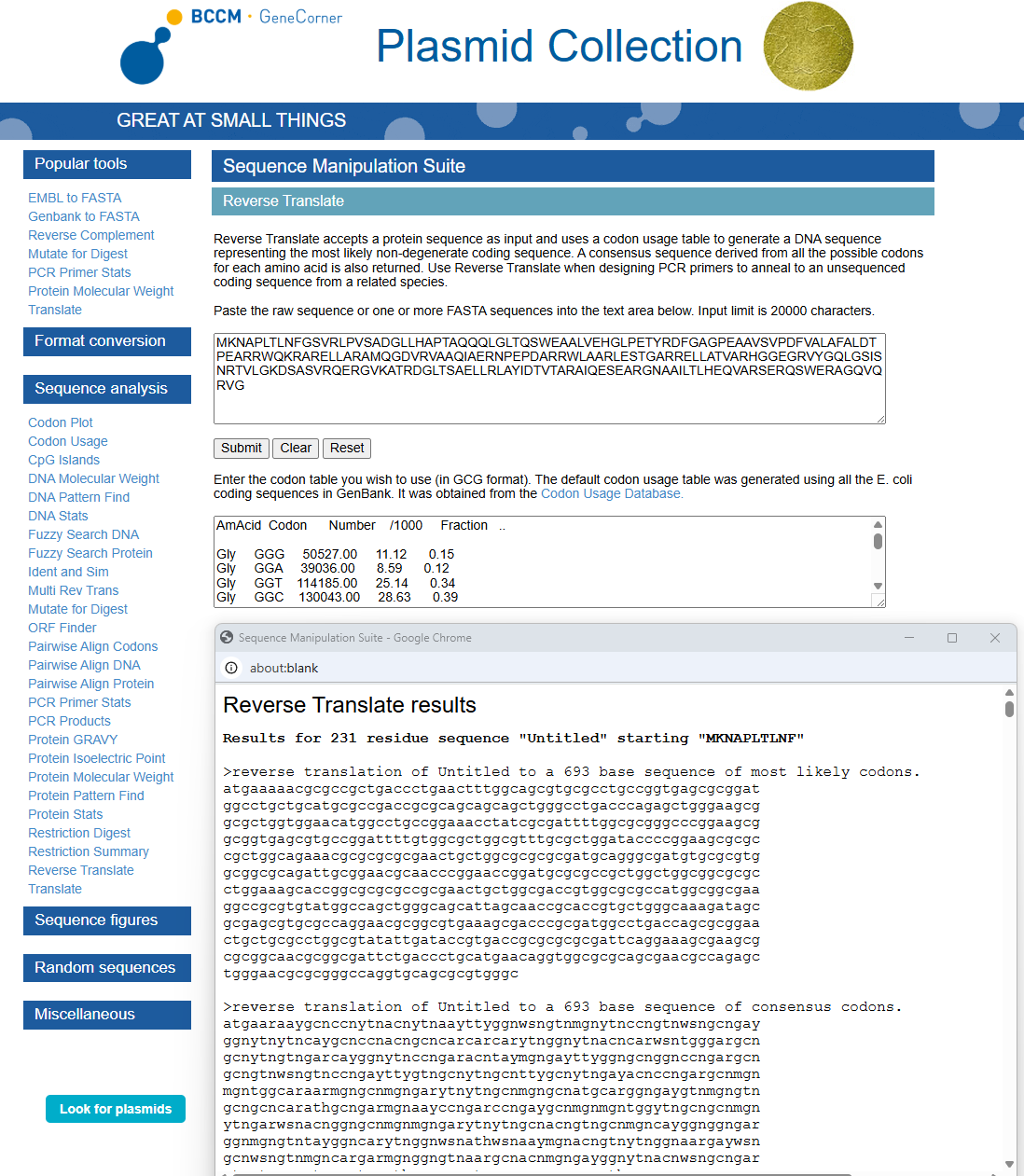
Using https://www.genecorner.ugent.be/rev_trans.html to create reverse translation of Protein DdrC
atgaaaaacgcgccgctgaccctgaactttggcagcgtgcgcctgccggtgagcgcggat
ggcctgctgcatgcgccgaccgcgcagcagcagctgggcctgacccagagctgggaagcg
gcgctggtggaacatggcctgccggaaacctatcgcgattttggcgcgggcccggaagcg
gcggtgagcgtgccggattttgtggcgctggcgtttgcgctggataccccggaagcgcgc
cgctggcagaaacgcgcgcgcgaactgctggcgcgcgcgatgcagggcgatgtgcgcgtg
gcggcgcagattgcggaacgcaacccggaaccggatgcgcgccgctggctggcggcgcgc
ctggaaagcaccggcgcgcgccgcgaactgctggcgaccgtggcgcgccatggcggcgaa
ggccgcgtgtatggccagctgggcagcattagcaaccgcaccgtgctgggcaaagatagc
gcgagcgtgcgccaggaacgcggcgtgaaagcgacccgcgatggcctgaccagcgcggaa
ctgctgcgcctggcgtatattgataccgtgaccgcgcgcgcgattcaggaaagcgaagcg
cgcggcaacgcggcgattctgaccctgcatgaacaggtggcgcgcagcgaacgccagagc
tgggaacgcgcgggccaggtgcagcgcgtgggc
3.3. Codon optimization
Using https://en.vectorbuilder.com/tool/codon-optimization.html to Condon Optimise DNA/RNA sequence
Selected Organism: Rat (Rattus Norvegicus)
In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Codon Optimization is required to improve the efficiency of translation of mRNA to Protein. It involves modifying the nucleotide sequence of a gene to replace rare or less-favoured codons with more frequently used codons in the host organsim. The organism I have chosen is a Rat and this is because the codon usuage differs from the orginal source of the DdrC gene. The codon optimisation will replace codons that are rare in rats with preferred rat codons allowing fro faster translation, higher protein expression and more efficient product of the DdrC protein.
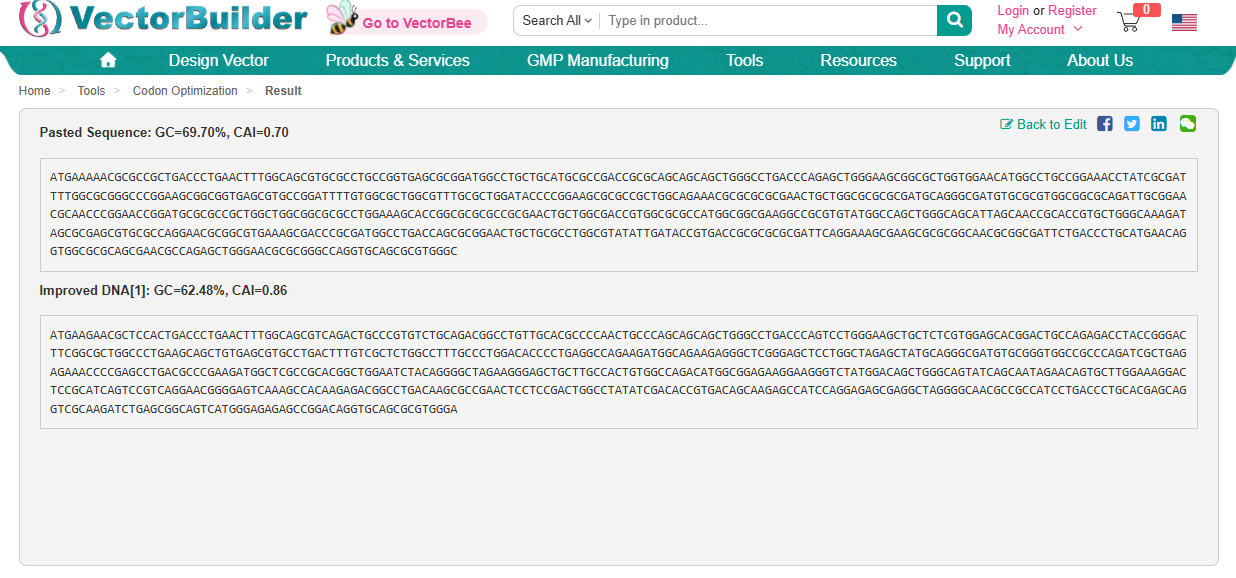
ATGAAGAACGCTCCACTGACCCTGAACTTTGGCAGCGTCAGACTGCCCGTGTCTGCAGACGGCCTGTTGCACGCCCCAACTGCCCAGCAGCAGCTGGGCCTGACCCAGTCCTGGGAAGCTGCTCTCGTGGAGCACGGACTGCCAGAGACCTACCGGGACTTCGGCGCTGGCCCTGAAGCAGCTGTGAGCGTGCCTGACTTTGTCGCTCTGGCCTTTGCCCTGGACACCCCTGAGGCCAGAAGATGGCAGAAGAGGGCTCGGGAGCTCCTGGCTAGAGCTATGCAGGGCGATGTGCGGGTGGCCGCCCAGATCGCTGAGAGAAACCCCGAGCCTGACGCCCGAAGATGGCTCGCCGCACGGCTGGAATCTACAGGGGCTAGAAGGGAGCTGCTTGCCACTGTGGCCAGACATGGCGGAGAAGGAAGGGTCTATGGACAGCTGGGCAGTATCAGCAATAGAACAGTGCTTGGAAAGGACTCCGCATCAGTCCGTCAGGAACGGGGAGTCAAAGCCACAAGAGACGGCCTGACAAGCGCCGAACTCCTCCGACTGGCCTATATCGACACCGTGACAGCAAGAGCCATCCAGGAGAGCGAGGCTAGGGGCAACGCCGCCATCCTGACCCTGCACGAGCAGGTCGCAAGATCTGAGCGGCAGTCATGGGAGAGAGCCGGACAGGTGCAGCGCGTGGGA
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
Cell-free protein synthesis - Using extracted cellular machinery
DNA is inserted into a reaction mixture and the sequence starts with ATG
Transcription machinery with RNA polymerase which binds ot the DNA and transcribes it into mRNA
mRNA is produced, containing codons
Translation of mRNA with reaction mixture containing: Ribosomes, tRNA molecules, 20 amino acids and ATP
Translation stars with Ribosomes binding to the mRNA, the translation starts at the codon AUG, and each codon is read in order. tRNA brings the correct amino acids and the amino acids are joined to form a polypetide chain.
Translation ends and the protein is released.
The protin then folds into the correct shape and becomes functional.
3.5. (Optional) How does it work in nature/biological systems?
- Describe how a single gene codes for multiple proteins at the transcriptional level.
A single gene can code for multiple proteins at the transcription level due to alternative splicing, it is a process where different sections of the a gene’s DNA can be selected and combined in different ways. This then results in the product of different proten variants with unqiue functions (Science of Bio Genetics, 2023).
- Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below.
Created new DNA sequence entry and translated to Protein
Created mRNA from DNA
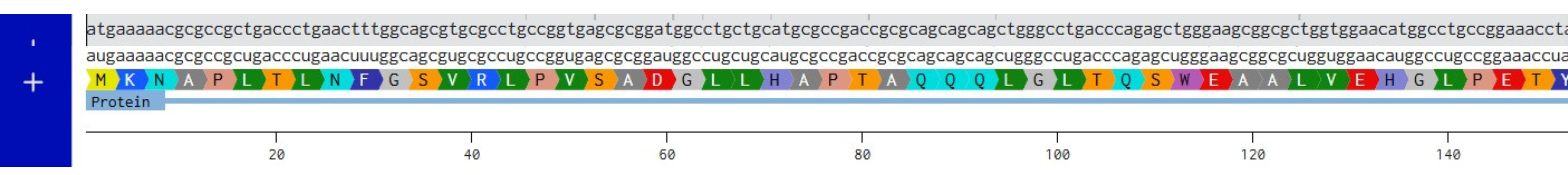
Combined DNA, mRNA and Protein
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account and a Benchling account
4.2. Build Your DNA Insert Sequence
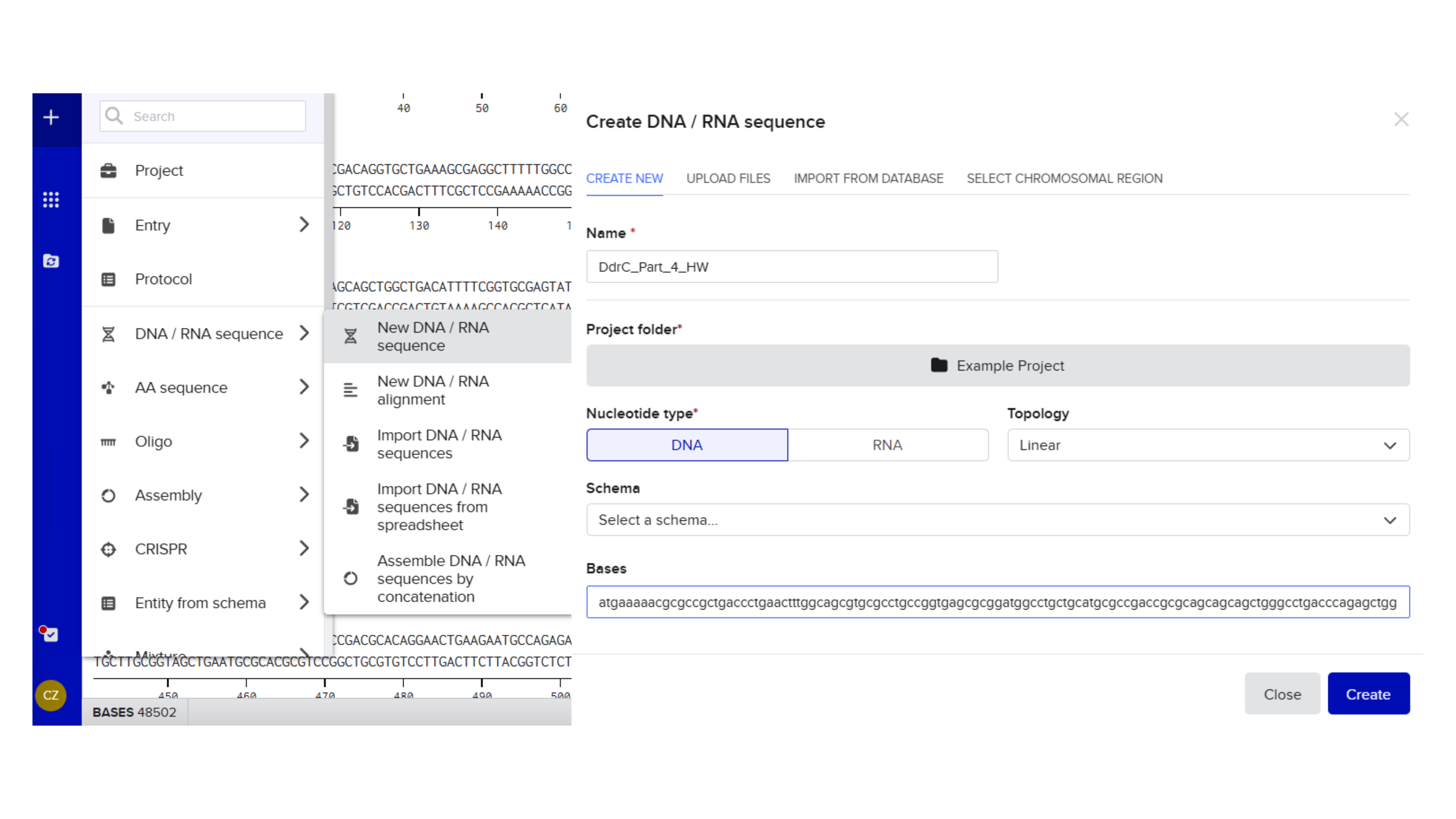
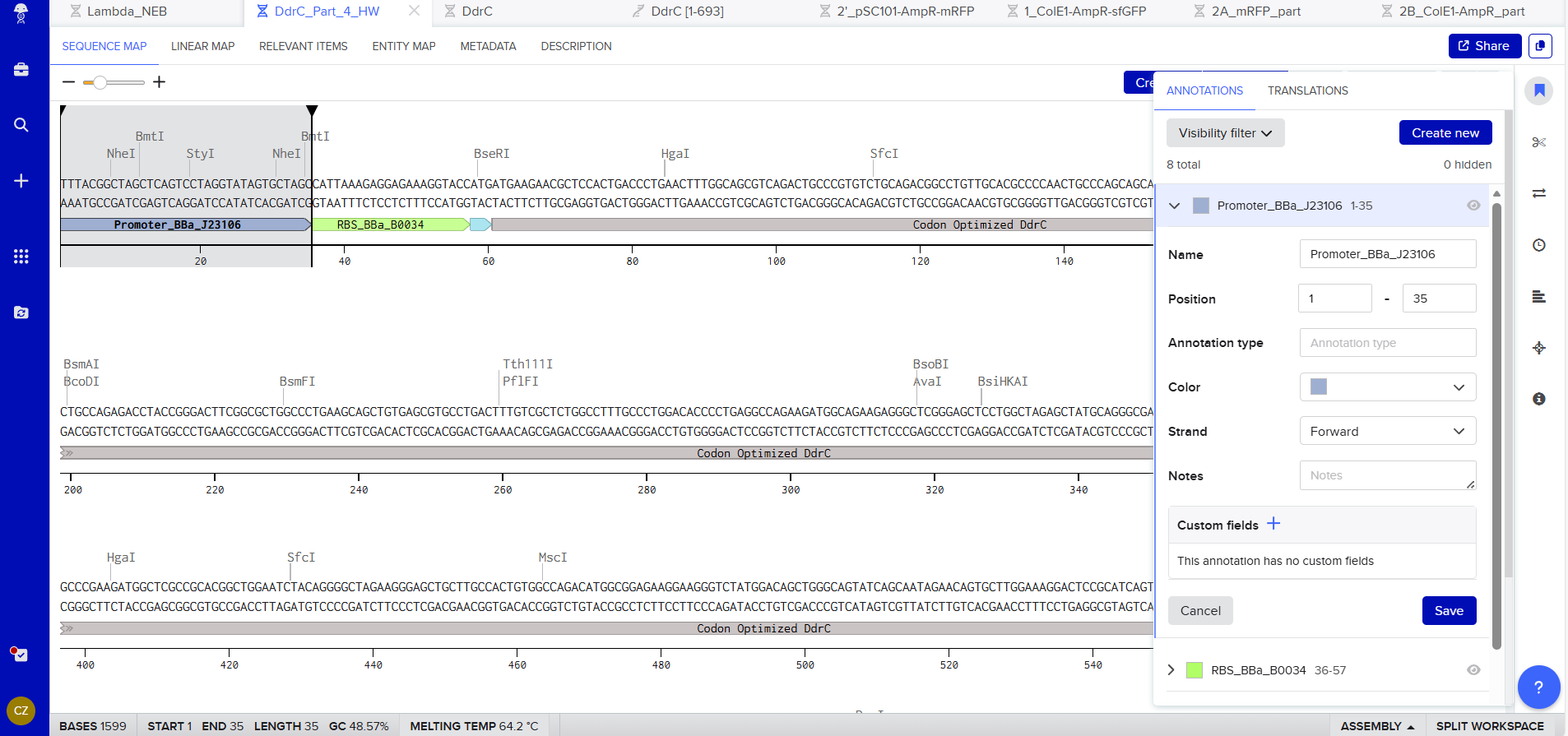
Promoter (e.g. BBa_J23106): TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC
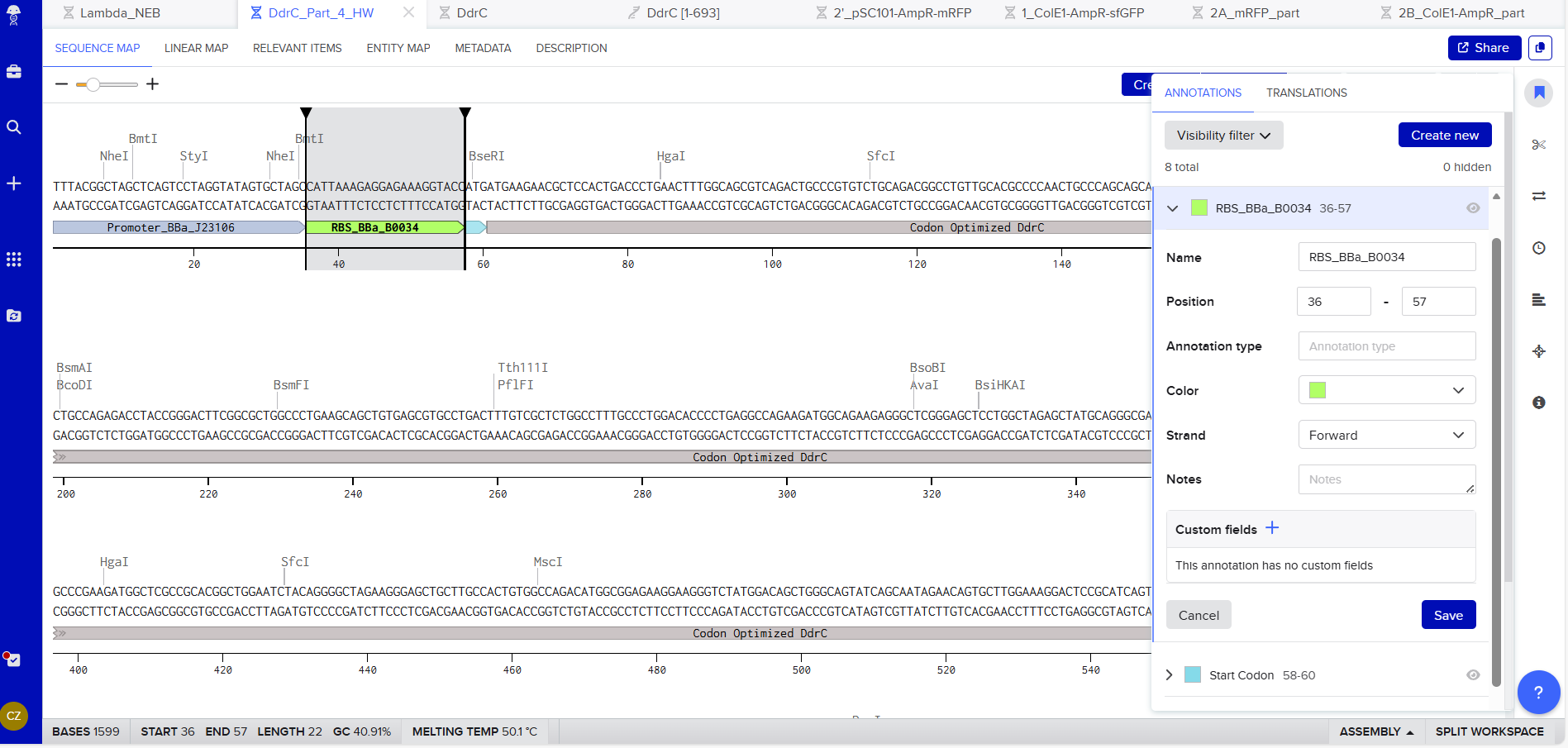
RBS (e.g. BBa_B0034 with spacers for optimal expression): CATTAAAGAGGAGAAAGGTACC
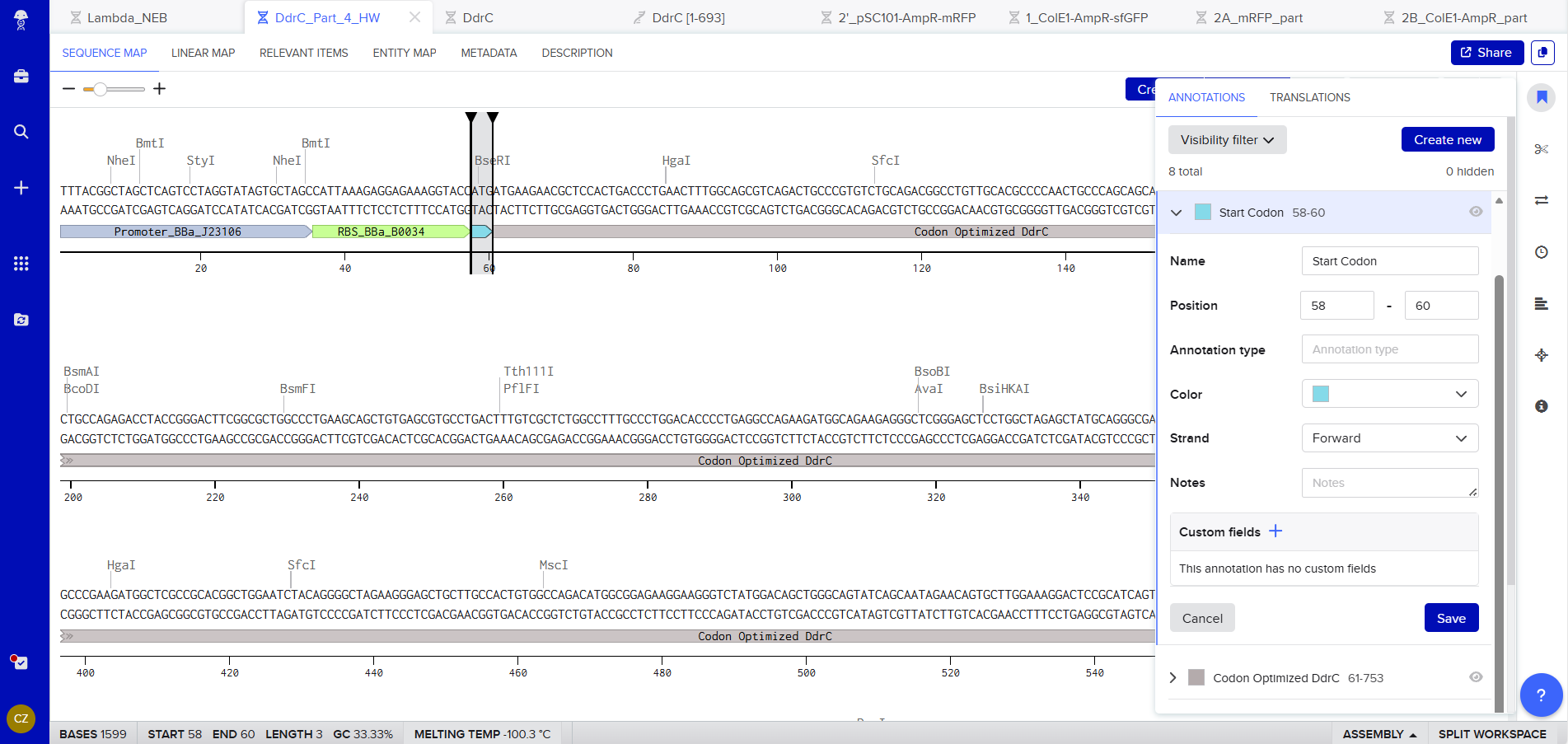
Start Codon: ATG
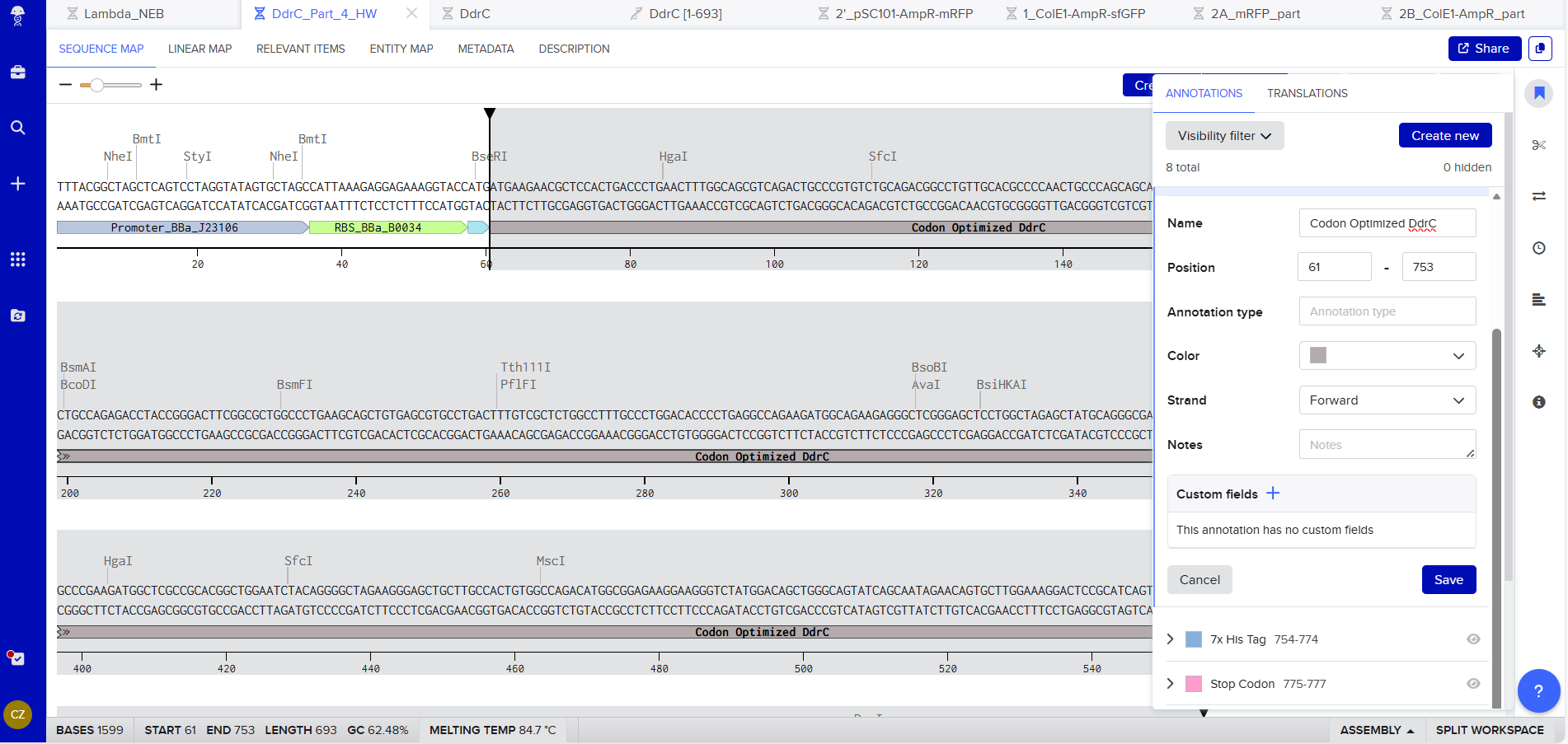
Coding Sequence codon optimized DNA
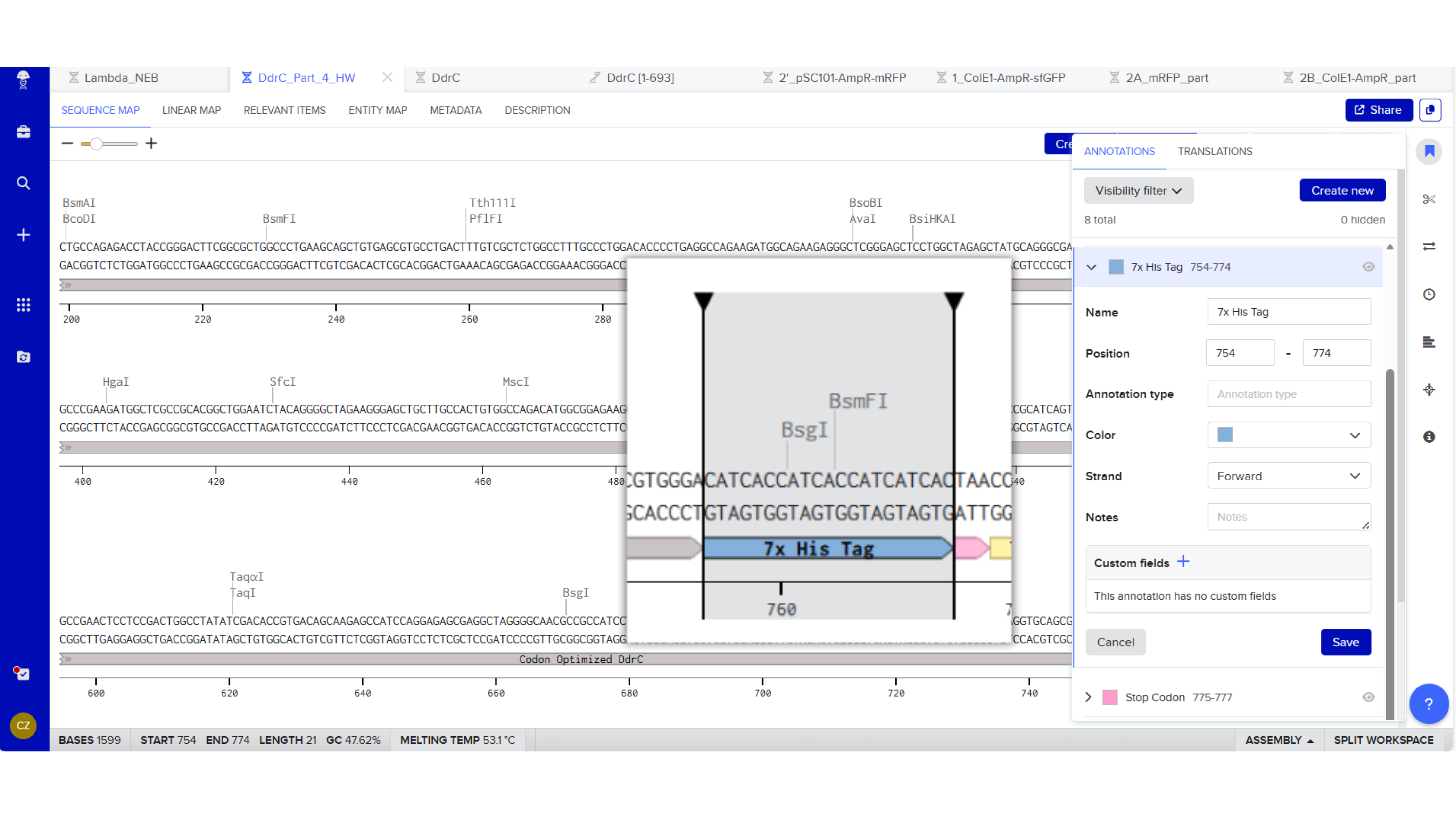
7x His Tag : CATCACCATCACCATCATCAC
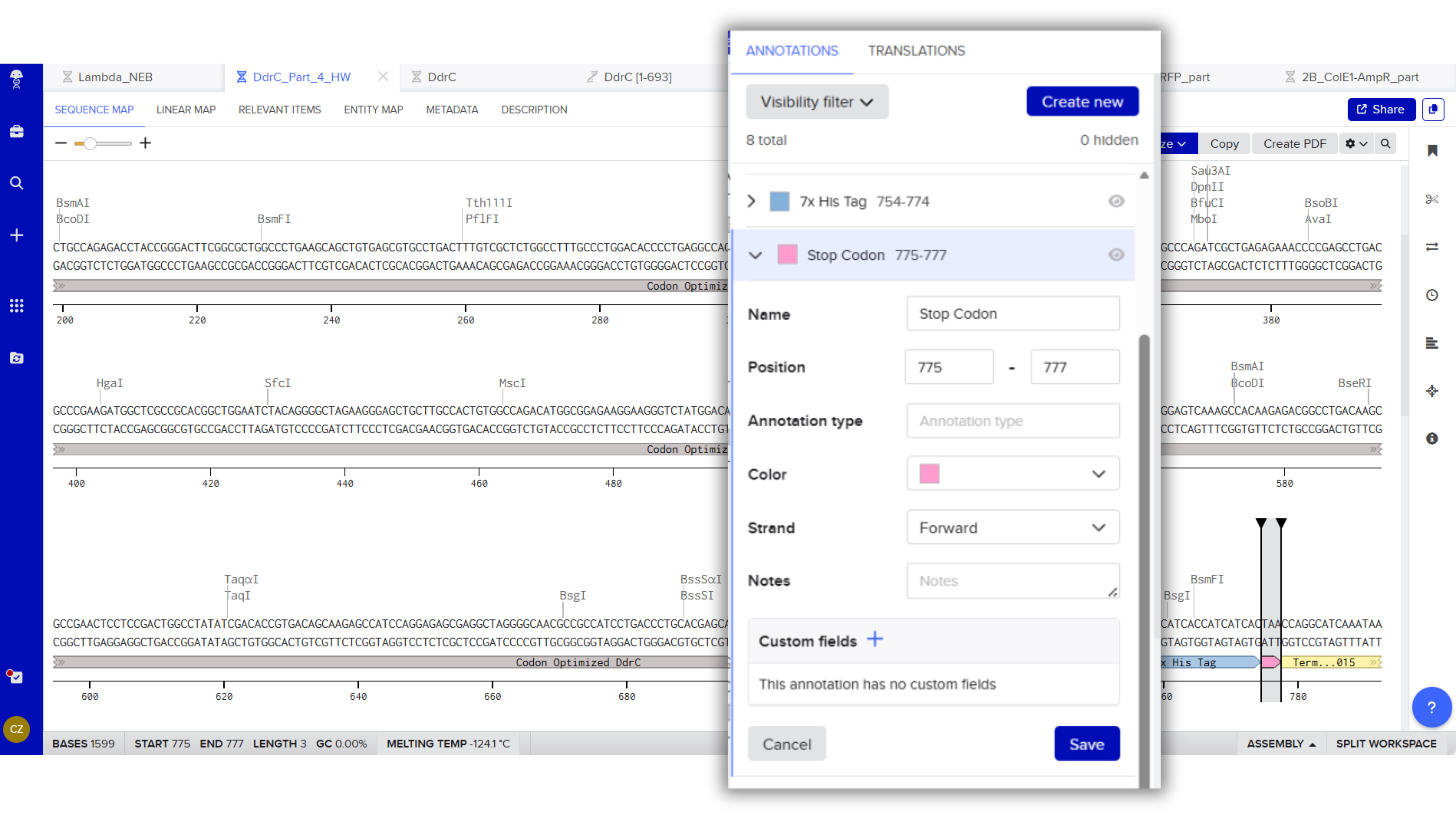
Stop Codon: TAA
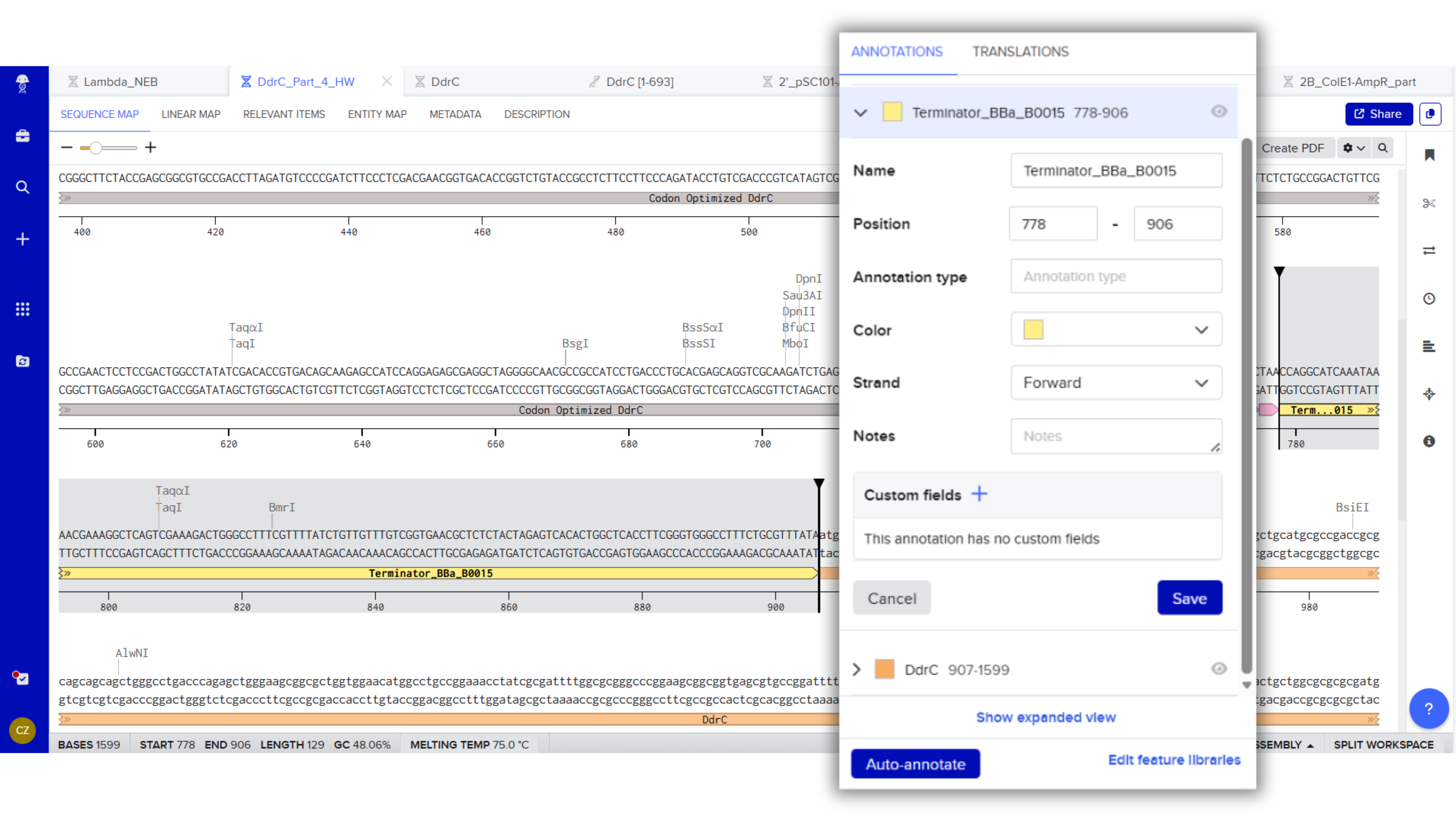
Terminator (e.g. BBa_B0015): CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
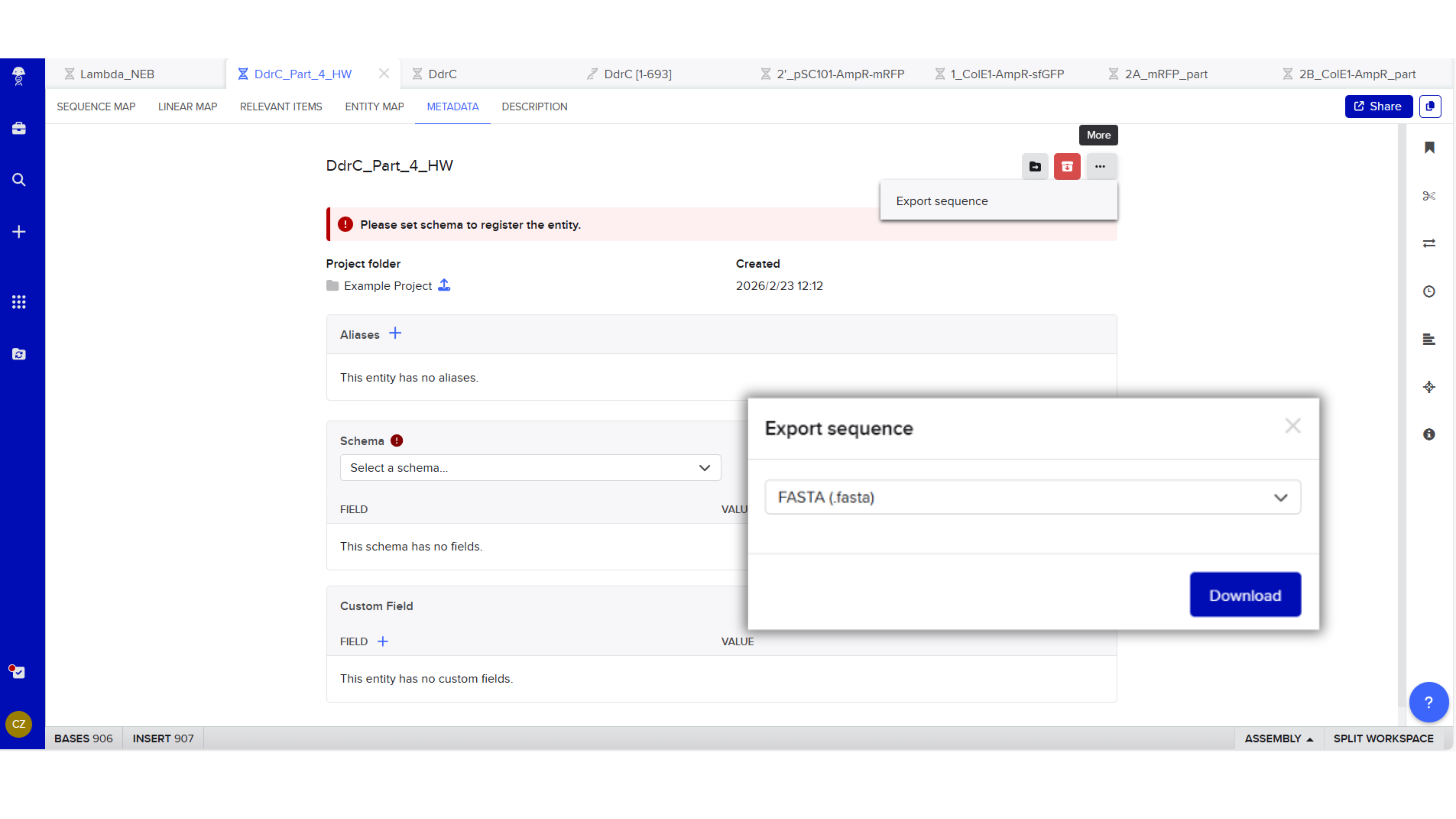
Export Sequence
4.3. On Twist, Select The “Genes” Option
4.4. Select “Clonal Genes” option
4.5. Import your sequence
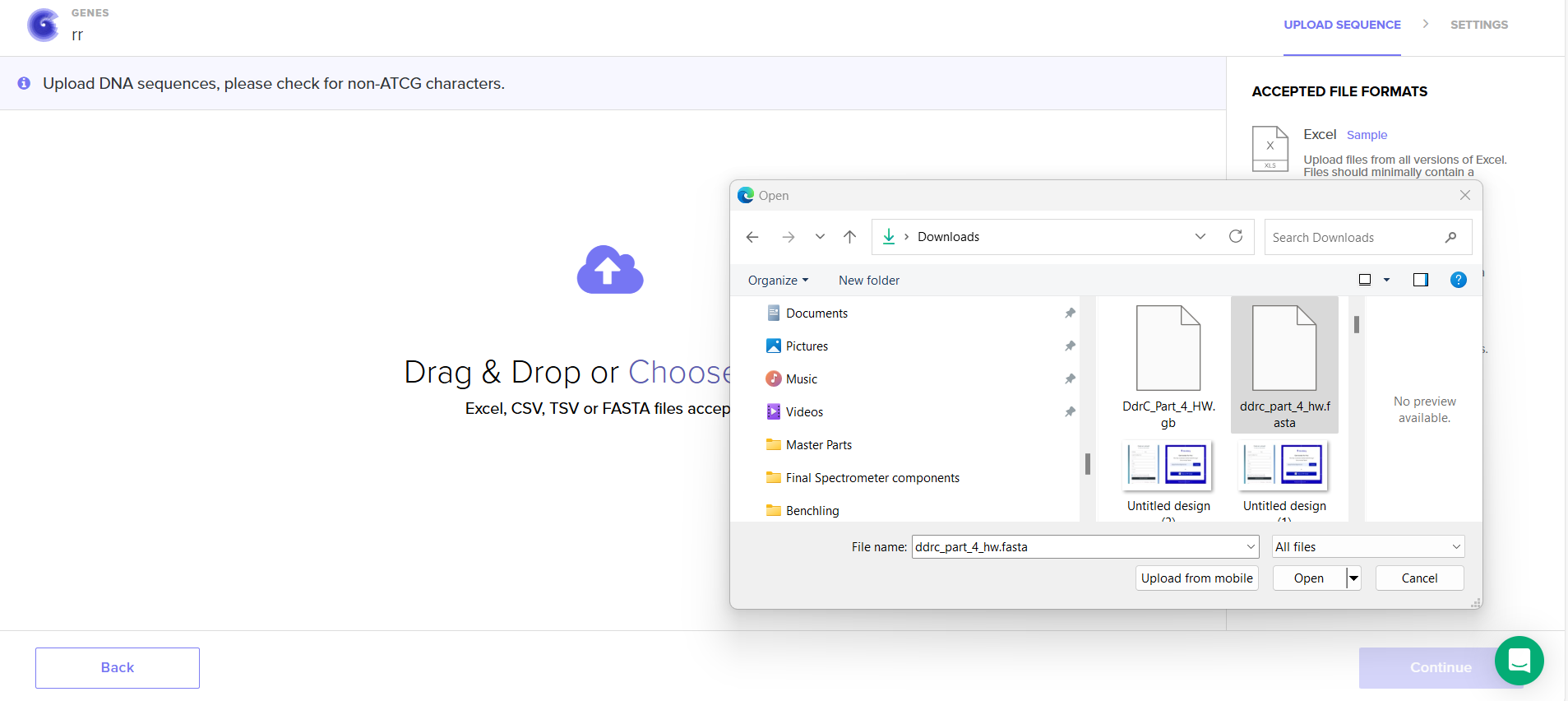
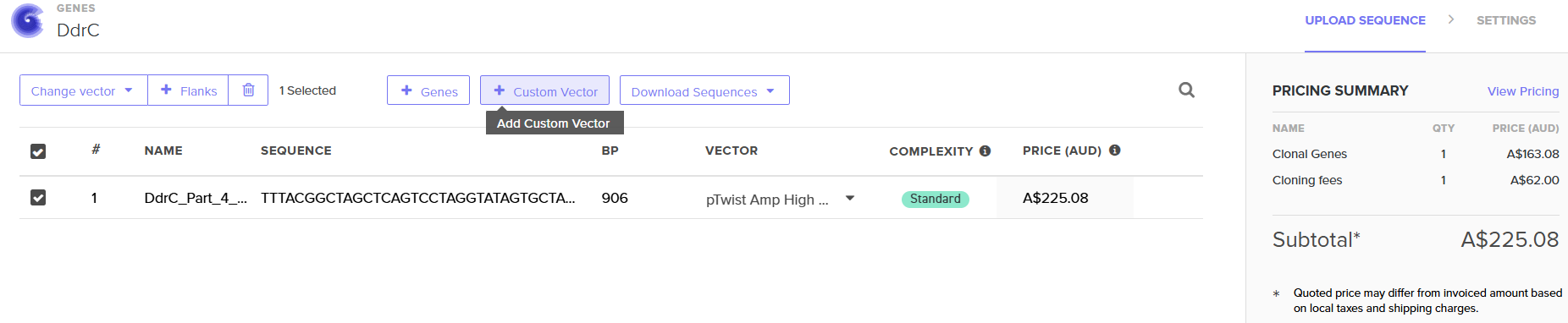
4.6. Choose Your Vector
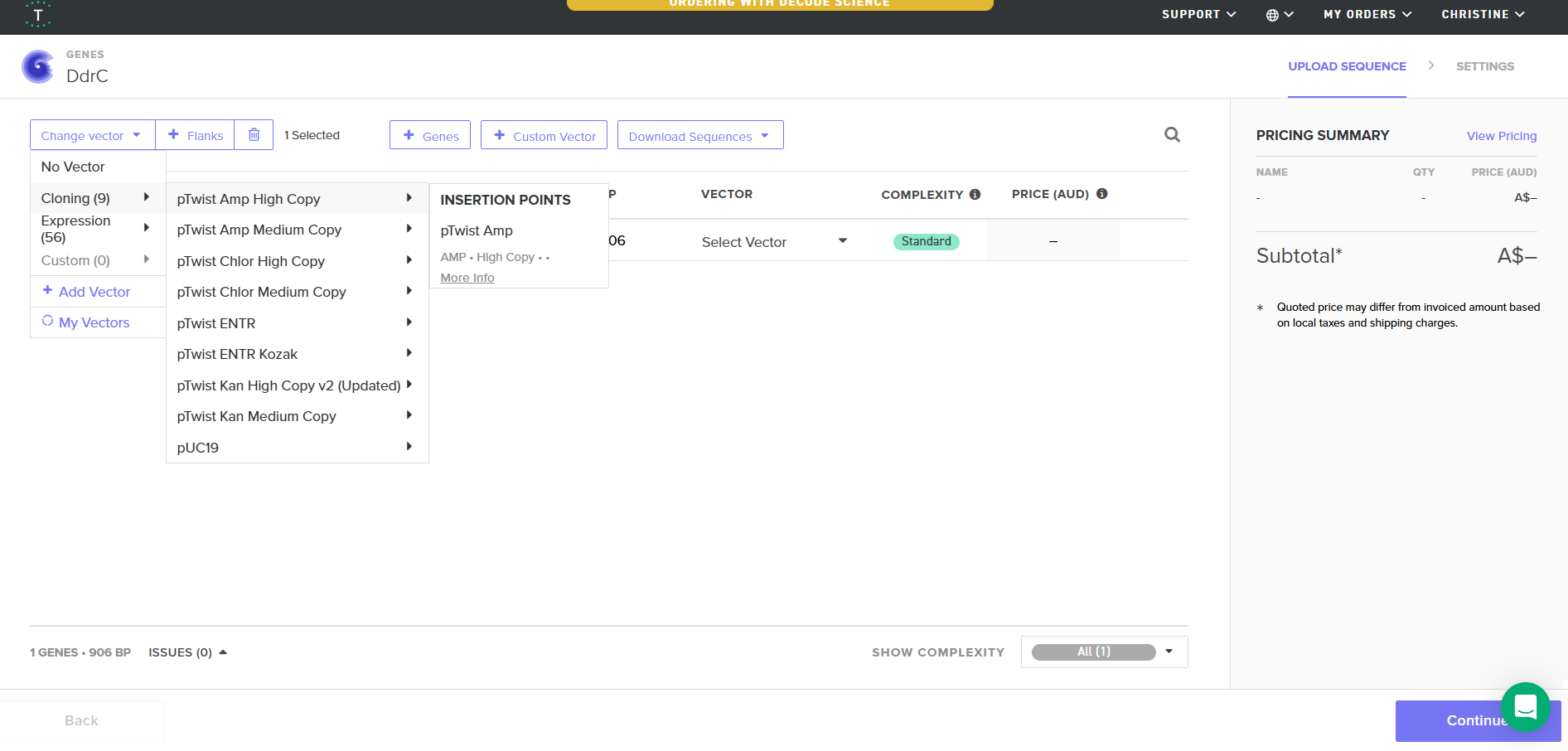
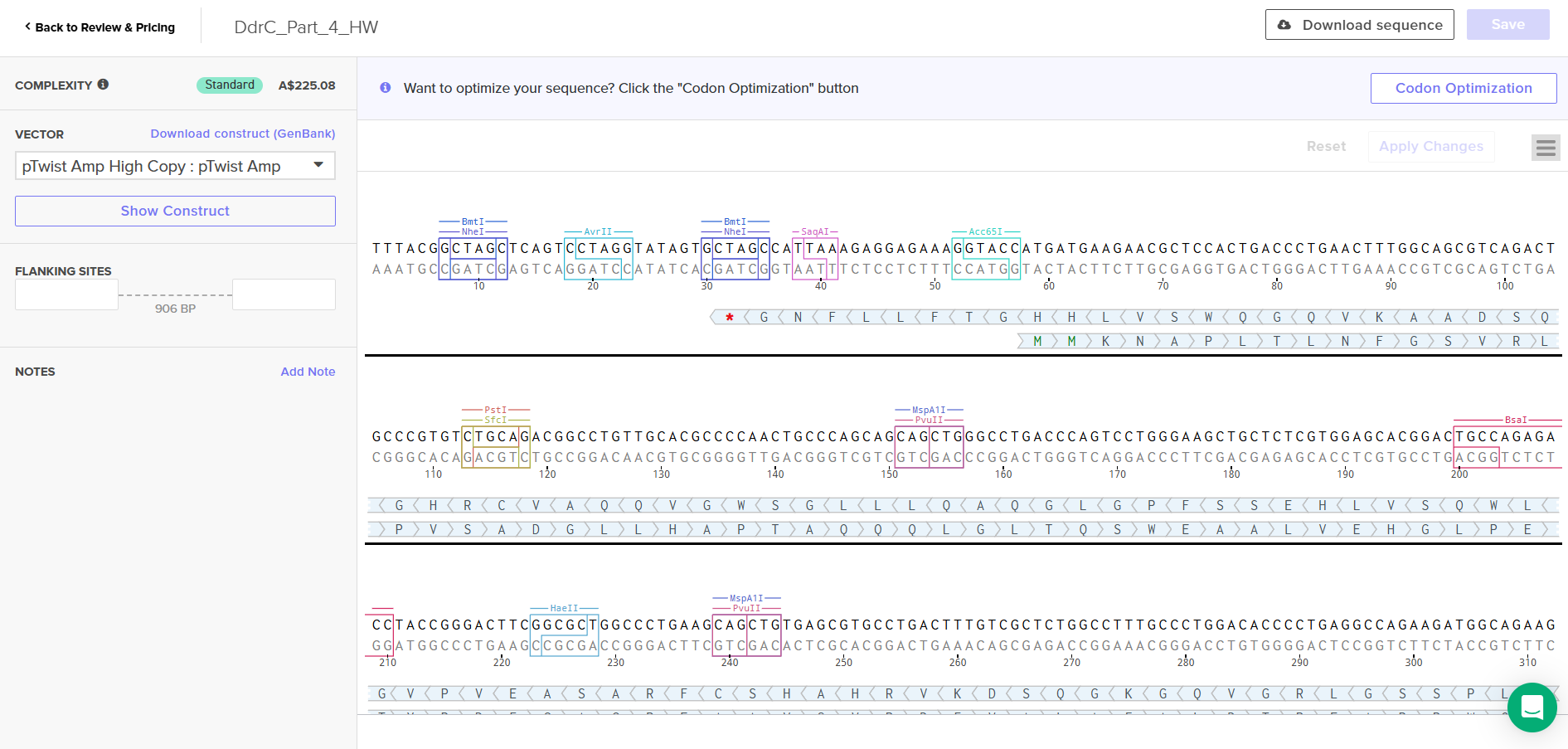
Download Construct (GenBank) and import to Benchling
Part 5: DNA Read/Write/Edit
a) What DNA would you want to sequence (e.g., read) and why?
I want to sequence the DNA (genome) of the bacterium called Paenibacillus Vortex seen in figure 1, because of these microorganism’s social motility and ability to employ cell-to-cell signaling to prompt acitvites such as attraction and repulsion under different environmental conditons (Myers, 2012). An example of this is when grown on hard surfaces, P.vortex will generate aggregates of dense bacteria that are pushed forward by repulsive chemotactic signals sensed from the cells at the back (Sirota-Madi et al., 2010). And when grown on soft surfaces these microorgranism can exhbit collective motility by forming foraging amrs that are sent out in search of food (Myers, 2012).
To achieve these cooperative ventures, P.Vortex has to be able to communicate with each other. The information exchanged are regarding its population size, myriad of indiviual environmental measurements at different locations, their internal states and their phenotypic and epigentic adjustments (Sirota-Madi et al., 2010). I want to know what sensors guides them to navigate the environment and what are the tranducers and responders that process the information into action. How do they communicate to each other?
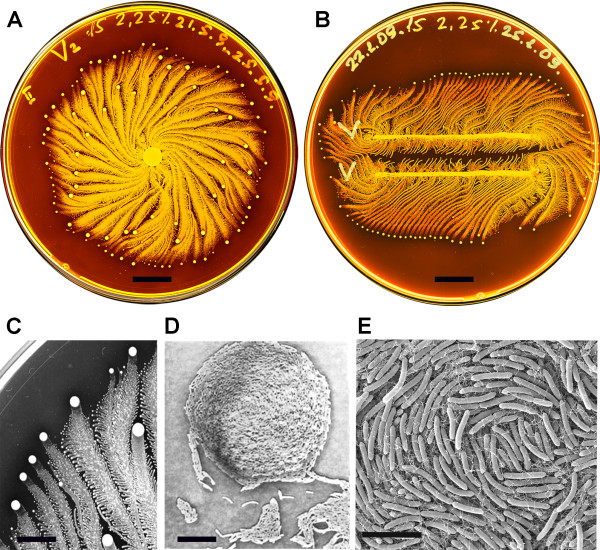
Figure 1, (Sirota-Madi et al., 2010)
Colony Organisation of the P.vortex bacteria
A. Whole colony view of P.vortex (grown on on 15 g/l peptone and 2.25% (w/v) agar for four days)
B. Two colonies of P.vortex (involuated in two parallel lines grown on on 15 g/l peptone and 2.25% (w/v) agar)
C. Colony pattern and vortex process (Magnification x20)
D. Mature indivdual Vortex (Magnification x500)
E. Each individual baterium has curvature (scans fro electron micropscope)
b) What technology or technologies would you use to perform this sequencing and why?
With reference to the research paper Genome sequence of the pattern forming Paenibacillus vortex bacterium reveals potential for thriving in complex environments by Sirota-Madi et al. (2010), the reserachers used a hybrid approach of 454 Life Sciences and Illumina, achieving a total of 289× coverage, with 99.8% sequence identity between the two methods (Sirota-Madi et al., 2010). This is because the reserachers claimed that p.Vortex has an exceptionally large number of signal transduction genes and a high combined regulatory score (TCS, TFs, transport, defense genes). In order to verify this, they would require a hihgly accurate genome, minimial assembly artifacts and reliable gene annotation. The hybrid approach can reduce the distortions that come from using one sequencting technology.
i) Is this your method first-, second-, or third-generation or other? How so?
Illumina and 454 Life Sciences are both second generation sequencing. 454 provides longer reads creating better contiguity
whilst Illumina provides massive depth hence providing higher accuracy. The researchers chose this combination of methods because of the De novo assembly problem (no reference genome). Short reads alone (especially early Illumina reads) are difficult to assemble reliably and the hybrid allowed more reliable identification of regulatory and signal transduction genes.
ii) What is your input? How do you prepare your input? List the essential steps.
Input:
Step 1: Bacteria growth
- P. vortex grown overnight in LB medium
- 37 °C, shaking at 200 rpm
- Ensures sufficient biomass
Step 2: Cell Harvesting
- DNA extracted from 2 ml culture (~10⁹ cells/ml)
Step 3: Cell Lysis
- Cells incubated with lysozyme for 45 minutes
- Breaks down bacterial cell wall (important for Gram-positive bacteria)
Step 4: DNA Extraction
- Qiagen DNeasy Blood & Tissue Kit
- Removes proteins, lipids, and other contaminants
Step 5: DNA Elution
- Eluted in 200 µl AE buffer (10 mM Tris-HCl, 0.5 mM EDTA pH 9.0)
iii) What are the essential steps of you chosen sequencing technology, how does it decode the bases of your DNA sample?
Sequencing approach incorporated 454 pyrosequencing with Illumina Genome Analyzer.
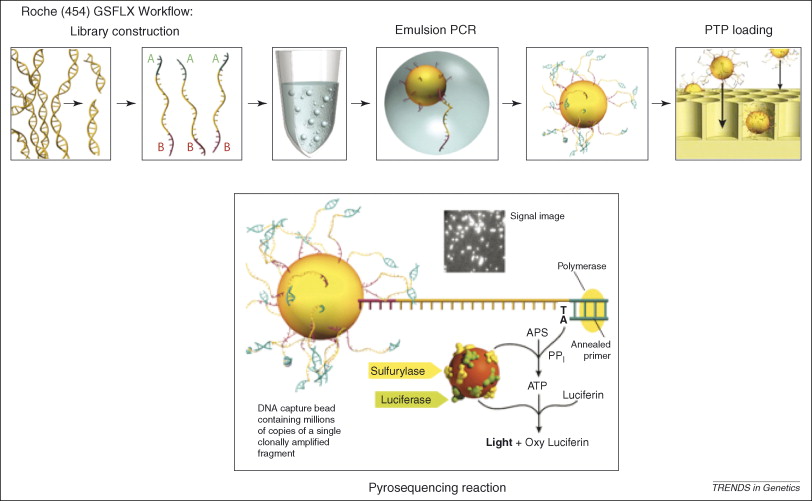 Figure 2, (MacLean, Jones, & Studholme, 2009)
Figure 2, (MacLean, Jones, & Studholme, 2009)
Referecin MacLean, Jones, and Studholme (2009) seen in figure 2, the essential steps of 454 pyrosequencing are:
DNA fragmentation and adapter ligation
Genomic DNA is broken into small fragments and short adapter sequences are attached.
Bead attachment and emulsion PCR
Each DNA fragment is attached to a microscopic bead and amplified inside an oil droplet.
Bead loading into wells
DNA-coated beads are placed into tiny wells, one bead per well.
Sequencing by nucleotide incorporation
DNA polymerase adds nucleotides to the growing DNA strand one type at a time.
Light generation (pyrosequencing)
When a nucleotide is incorporated, pyrophosphate is released, triggering a reaction that produces light.
Signal detection and base calling
The emitted light is recorded and used to determine the DNA sequence.
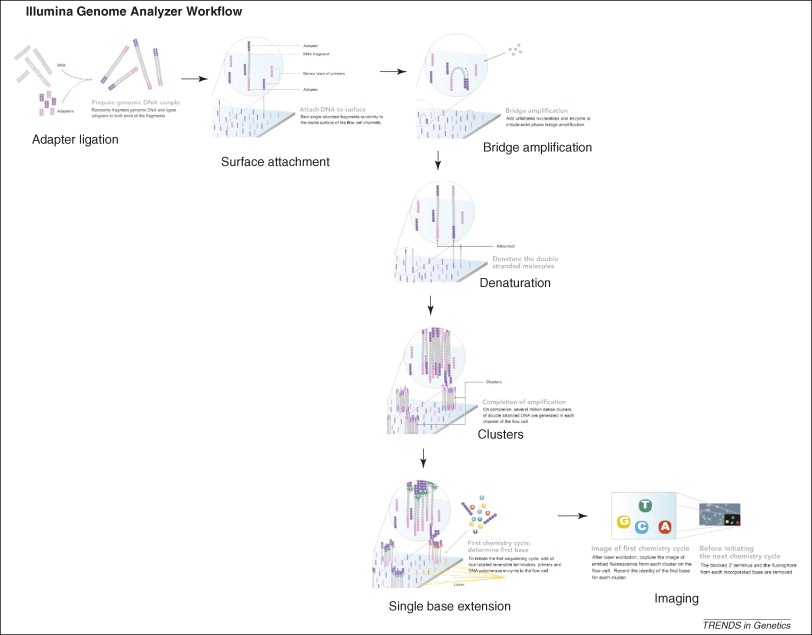 Figure 3, (MacLean, Jones, & Studholme, 2009)
Figure 3, (MacLean, Jones, & Studholme, 2009)
Referecin MacLean, Jones, and Studholme (2009) seen in figure 3, the essential steps of Illumina Genome Analyzer sequencing are:
DNA Fragmentation
Mixture of single-stranded, adaptor oligo-ligated DNA fragments
Attachment to the flow cell
Using a microfluidic cluster station to add these fragments to the surface of a glass flow cell.
Each flow cell is divided into eight separate lanes, and the interior surfaces have covalently attached oligos complementary to the specific adapters that are ligated onto the library fragments
- Cluster amplification (bridge PCR)
- Hybridization of these DNAs to the oligos on the flow cell occurs by an active heating and cooling step
- Subsequent incubation with reactants and an isothermal polymerase amplifies the fragments in a discrete area or ‘cluster’ on the flow cell surfaces
- Flow cell is placed into a fluidics cassette within the sequencer
- Each cluster is supplied with polymerase and four differentially labeled fluorescent nucleotides that have their 3′-OH chemically inactivated to ensure that only a single base is incorporated per cycle. (MacLean, Jones, & Studholme, 2009)
- Identify the incorporated nucleotide at each cluster
- Each base incorporation cycle is followed by an imaging step, and by a chemical step removes the fluorescent group and deblocks the 3′ end for the next base incorporation cycle. (MacLean, Jones, & Studholme, 2009)
- End of the sequencing run
- The sequence of each cluster is computed and subjected to quality filtering to eliminate low-quality reads of between 32 and 40bp. (MacLean, Jones, & Studholme, 2009)
iv) What is the output of your chosen sequencing technology?
Outputs are raw sequences, base quality scores and FASTQ files
DNA WRITE
a) What DNA would you want to synthesize (e.g., write) and why?

I want to synthesise the gene encoding the luminescent protein from Aequorea vicotria jellyfish (GFP Green Fluprescent Protein) with a Plasmid to emit light natrually without electricity.
b) What technology or technologies would you use to perfom this DNA synthesis and why?
The technologies I would use to perform this DNA synethesis are CRISPR-Cas9 becasue it is extremely precise and can insert GFP exactly in the desired location. It can work in bateria, yeast and plants and allows for stable genome integration.
DNA EDIT
a) What DNA would you want to edit and why?
- limonene synthase (Prem Kumar, Morehouse, Yu, & Oprian, 2019)*
The protein that I can to edit is limonene synthase (citrus sinensis) because I think it is interesting to edit smells, what would happen if you edit the smell of sweet orange and grow a fruit with the edited scent? Would it result in reduced fruit quality, the fruit will have no orange smell, how does smell evolve? What are some environmental factors that can change the DNA of certain smells.
b) What technology or technologies would you use to perfom these DNA edits and why?
I could utilise base editors where I can edit the DNA without creating double strand breaks, I can introduce specific point mutations in the Limonene synthase to tweak the enzyme’s function.
Bibliorgraphy
Gueguen, E., Bruto, M., Lemaire, D., Bertrand, E., Fichant, G., & Graille, M. (2022). Structural and functional characterization of DdrC, a novel DNA damage-induced nucleoid associated protein involved in DNA compaction. Nucleic Acids Research, 50(13). https://doi.org/10.1093/nar/gkac563
MacLean, D., Jones, J. D., & Studholme, D. J. (2009). Application of “next-generation” sequencing technologies to microbial genetics. Nature Reviews Microbiology, 7(4), 287–296. https://doi.org/10.1038/nrmicro2122
Morehouse, B.R., Kumar, R.P., Matos, J.O., Yu, Q., Bannister, A., Malik, K., Temme, J.S., Krauss, I.J. & Oprian, D.D. (2019) Direct Evidence of an Enzyme‑Generated LPP Intermediate in (+)-Limonene Synthase Using a Fluorinated GPP Substrate Analog. ACS Chemical Biology, 14, 2035‑2043. https://doi.org/10.1021/acschembio.9b00514
Myers, W. (2018). Bio design: Nature, science, creativity (Paola Antonelli, Foreword). Thames & Hudson.
Sirota-Madi, A., Olender, T., Helman, Y., Ingham, C., Brainis, I., Roth, D., Hagi, E., Brodsky, L., Leshkowitz, D., Galatenko, V., Nikolaev, V., Mugasimangalam, R. C., Bransburg-Zabary, S., Gutnick, D. L., Lancet, D., & Ben-Jacob, E. (2010). Genome sequence of the pattern forming Paenibacillus vortex bacterium reveals potential for thriving in complex environments. BMC Genomics, 11, 710. https://doi.org/10.1186/1471-2164-11-710
The unique mechanism of DdrC in enhancing DNA stability. (2024, December 30). Optimise.mfm.au. Retrieved from https://optimise.mfm.au/research/the-unique-mechanism-of-ddrc-in-enhancing-dna-stability/
Unknown author. (n.d.). JellyFish [Photograph]. The Pipette Pen. http://www.thepipettepen.com/wp-content/uploads/2016/04/7338555110_bc2ba135a5_z.jpg
Week 3 HW: Lab Automation
Generate an artistic design using the GUI at opentrons-art.rcdonovan.com
I wanted to draw Punch, the famous baby Japanese macaque from the zoo in Japan. The GUI created an approximate outline which I used as a base and added more details of different colour on top.
This was the end result
This was the settings with the coordinates.
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons
1. Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Urrutia Iturritza, M., Mlotshwa, P., Gantelius, J., Alfvén, T., Loh, E., Karlsson, J., Hadjineophytou, C., Langer, K., Mitsakakis, K., Russom, A., Jönsson, H. N., & Gaudenzi, G. (2024). An Automated Versatile Diagnostic Workflow for Infectious Disease Detection in Low-Resource Settings. Micromachines, 15(6), 708. https://doi.org/10.3390/mi15060708
detection in low-resource settings
The paper describes that having an automated diagnostic testing workflow in peripheral laboratories and in hospitals, clinic or epidemic control checkpoints is advantageous due to the simultaneous processing of multiple samples to provide rapid results to the patient and hence minimising the possibility of containing room or error during the sample handling or trasnportation, and also increases efficiency, however, most automation platforms are expensive and are not easily adaptable to new protocols. The paper proposes the need for a versatile, easy to use, rapid and reliable diagnostic testing workflow. They propose to combining open source modular automation such as Opentrons and automation compatible molecular biology protocols that can be easily adapted to a workflow for infectious diseases diagnosis.
The paper illustrates the feasibility of automation of the method with a low cost Neisseria meningitidis diagnostic test that utilises magnetic bends for pathogen DNA isolation, isothermal amplification and ferritin on a paper based microarray.
2. Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
This is an idea that was described by David in class but I thought can apply to my final projects. It is testing the biosensors at scale especially for my idea one. With the pollen sensor (can also be air quality biosensor) the automation tool comes in and does thousands of tests in parallel, to especially test as many different types of durations and data points in the design.
3. While your description/project idea doesn’t need to be set in stone, we would like to see core details of what you would automate. This is due at the start of lecture and does not need to be tested on the Opentrons yet.
Idea one

Context:
If fungi designed chairs?
- What if furniture was not designed by humans imposing geometry onto materials, but instead emerged from biological growth logic?
- What if chairs were a responsive organism rather than static furniture.
Inpsired by:
- The Algorithmic Beauty of Plants : Which models plant growth using mathematical rules (L-systems).
- Suzanne Simard : Who revealed underground fungal communication networks between trees.
- D’arcy Wentworth Thimpson : On growth and form 1917
Aim:
To simulate and materilaise a chain structure generated from fungal growth patterns, bio-processing and digital translation.
Method
Using eukaryotic cells cells as design tools.
Computational tools, scanning the cell and then conver it into a digital model,finding max and minium strength of the material, analysing the data. The equtions are then used to caluclate how the systems might create a chair.
Idea Two
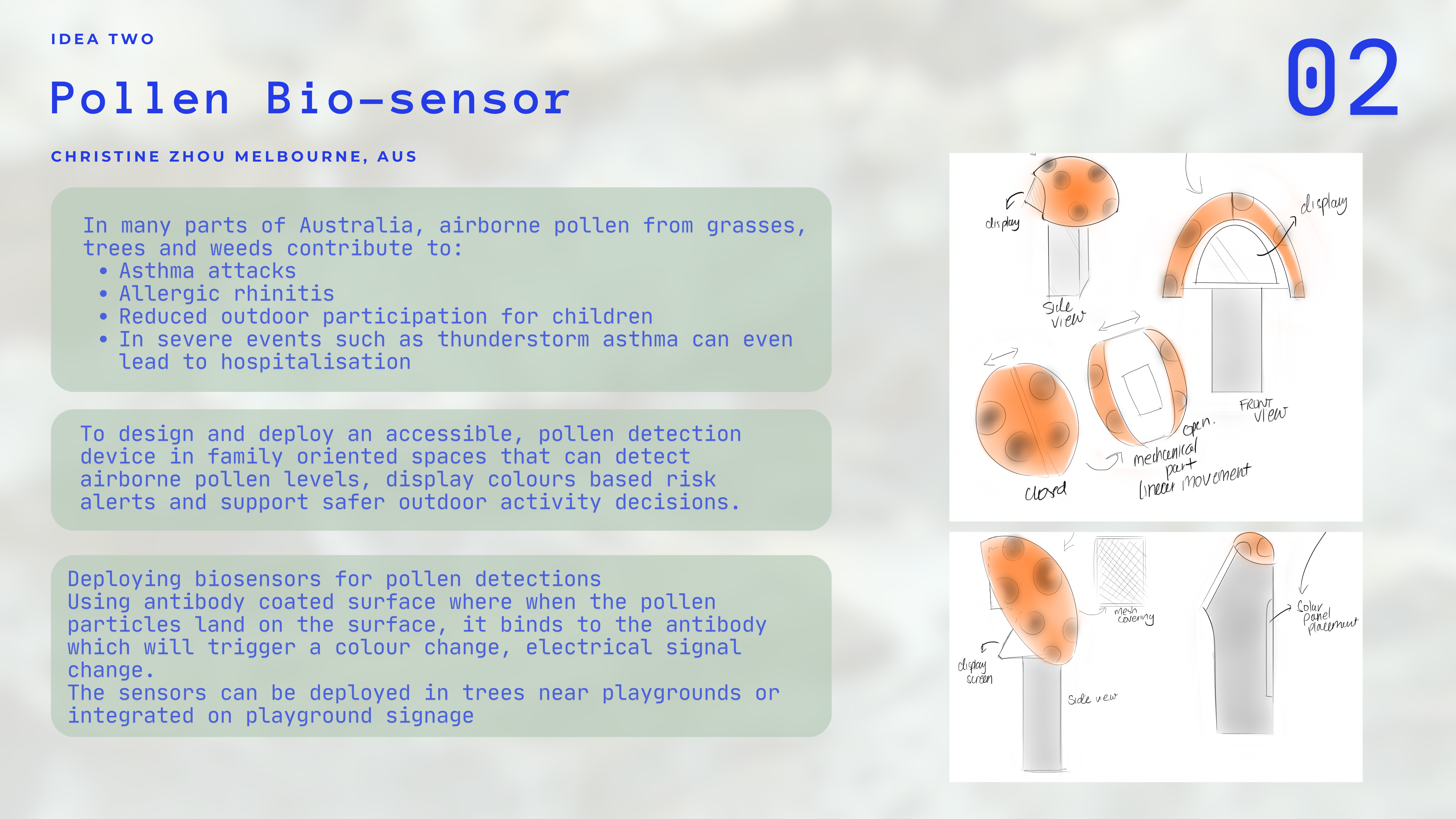
Context
In many parts of Australia, airborne pollen from grasses, trees and weeds contribute to
- Ashtma attacks
- Allergic rhinitis
- Reduced outfoor partcipation for children
- In severe events such as thunderstom asthma can event lead to death
Aim:
To design and deploy an accessible, pollen detection device in family oriented spaces that can detect airbourne pollen levels, display colours based risk alerts and super safer outdoor activity decisions.
Method:
Deploying biosensors for pollen detections
Using antibody coated surface where when the pollen particles land on the surface, it binds to the antibody which will trigger a colour change, electrical signal change.
The sensors can be Deployed in trees near playgrounds or integrated on playground signage
Idea Three
Storing music into bacteria (eschericha coli)
Context
By encoding music as DNA sequences, it can theoretically be stored inside living organisms, such as Escherichia coli, turning bacteria into living music libraries similar to CDs or Vinyls
Aim:
To encode a musical composition into the DNA of Escherichia coli cells, creating a biological storage system for music.
Theories to test:
How does sound age?
Can the sound mutate?
How does sunlight affect the sound stored?
Can external whethers activate the sound?
Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
meat is around 20-27 percent for beef.
500 x 0.20 =100 grams of protein
1 Dalton = 1.66054e-24 grams
1 amino acid = 110 Daltons
So protein / Dalton = 6.0221374e+25 grams
110 x 6.0221374e+25 = 6.0221374e+27 grams Of in 500 gram meat.
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When native mRNA is injected into the human body, it could trigger a series of heterologus immune responses and be degraded by the immune system, it is similar to how the human body resists viral unvarying.
DNA also needs to be in a nucleus, wrapped in histones and other proteins, with access to polymerase and other enzymes.
The immune systems in the human body identifies things that don’t match the own cells and attack or reject them.
Hou, X., Shi, J., & Xiao, Y. (2024). mRNA medicine: Recent progresses in chemical modification, design, and engineering. Nano research, 17(10), 9015–9030. https://doi.org/10.1007/s12274-024-6978-6
- Why are there only 20 natural amino acids?
The 20 standard amino acids are frozen evolutionary choices, solidified billions of years ago to balance protein functionality with metabolic efficiency.
Doig A. J. (2017). Frozen, but no accident - why the 20 standard amino acids were selected. The FEBS journal, 284(9), 1296–1305. https://doi.org/10.1111/febs.13982
Amino acids are also created through codons which are a chain of three bases stuck together. Nature has evoked to only produce 20 because of inherited redundancy, there are surveillance codons that encode for the same amino acids.
Why are there only 20 amino acids? | MyTutor. (n.d.). Www.mytutor.co.uk.https://www.mytutor.co.uk/answers/52609/Mentoring/Oxbridge-Preparation/Why-are-there-only-20-amino-acids/
Can you make other non-natural amino acids? Design some new amino acids.
Where did amino acids come from before enzymes that make them, and before life started?
In the origins of life on Earth, amino acids were synthesised chemically from a large mixture of organic compounds. After more complex forms of life arose, the ones that were able to synthesise their own amino acid survived and thats why, today, organisms are all able to synthesise their own aminos through enzymes and metabolic pathways.
Gutiérrez-Preciado, A., Romero, H., & Peimbert, M. (2010). Amino Acids, Evolution | Learn Science at Scitable. Nature.com. https://www.nature.com/scitable/topicpage/an-evolutionary-perspective-on-amino-acids-14568445/
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
An a-helix using D-amino acids will form a left-handed helix, while natural L-amino acids from right-handed helices, reversing the chirality of the amino acids to the D-form results in the exact mirror image, casting the polypeptide chain to coil in the opposite left handed direction.
Alpha Helix - an overview | ScienceDirect Topics. (2018). Sciencedirect.com. https://www.sciencedirect.com/topics/medicine-and-dentistry/alpha-helix
- Can you discover additional helices in proteins?
Right handed helices are energetically more stable, it has a lower state of energy due to having fewer steric clashes between the side chain and the main chain
Alpha Helix - an overview | ScienceDirect Topics. (2018). Sciencedirect.com. https://www.sciencedirect.com/topics/medicine-and-dentistry/alpha-helix
- Why are most molecular helices right-handed?
- Why do β-sheets tend to aggregate?
Part B: Protein Analysis and Visualization
Part B: Protein Analysis and Visualization
- Briefly describe the protein you selected and why you selected it.
Lysozyme (specifically C-type) is an enzyme that attacks the protective cell walls of bacteria by chewing through the peptidoglycan layer.
I chose this protein because it is a defence protein found in baterial enzyms of tears and saliva.
- Identify the amino acid sequence of your protein.
Amino Acid Sequence: KVFGRCELAA AMKRHGLDNY RGYSLGNWVC AAKFESNFNT QATNRNTDGS TDYGILQINS RWWCNDGRTP GSRNLCNIPC SALLSSDITA VVNCAKKIVS DGNGMNAWVA WRNRCKGTDV QAWIRGCRL
- How long is it? What is the most frequent amino acid?
129 amino acids
- You can use this Colab notebook to count the frequency of amino acids.
Most Frequent Amino Acid: Glycine (G) and Arginine (R) are highly prevalent, but Alanine (A) and Glycineoften tie for the lead depending on the specific species variant. In this sequence, Alanine and Glycine appear 12 times each.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
Does your protein belong to any protein family?
Lysozyme is ubiquitous across the animal kingdom (found in birds, mammals, and even some insects). It belongs to the Glycosyl hydrolase 22 family. These are enzymes that specifically hydrolyze the glycosidic bonds in complex sugars.
- Identify the structure page of your protein in RCSB
The classic high-resolution structure for this protein is found under PDB ID: 193L.
- When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The structure for 193L was published in 1995, though lysozyme was famously the first enzyme ever to have its structure solved via X-ray crystallography back in 1965.
It is an excellent quality structure. The resolution is 1.59 Å.
- Open the structure of your protein in any 3D molecule visualization software:
- PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
- Color the protein by secondary structure. Does it have more helices or sheets?
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Part C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
Choose your favorite protein from the PDB.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
- Deep Mutational Scans
a)Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
b)Can you explain any particular pattern? (choose a residue and a mutation that stands out)
c)(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
- Latent Space Analysis
a)Use the provided sequence dataset to embed proteins in reduced dimensionality.
b)Analyze the different formed neighborhoods: do they approximate similar proteins?
c)Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein Folding
- Folding a protein
a)Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
b)Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
C3. Protein Generation
- Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
a) Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
b)Input this sequence into ESMFold and compare the predicted structure to your original.
Part D. Group Brainstorm on Bacteriophage Engineering
Find a group of ~3–4 students
Read through the Phage Reading material listed under “Reading & Resources” below.
Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
Why do you think those tools might help solve your chosen sub-problem?
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
Include a schematic of your pipeline.
This resource may be useful: HTGAA Protein Engineering Tools
Each individually put your plan on your HTGAA website
Include your group’s short plan for engineering a bacteriophage
Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling
PeptiVerse: therapeutic property prediction
moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
Part 1: Generate Binders with PepMLM
- Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
- Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
- Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
- To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
- Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Part 2: Evaluate Binders with AlphaFold3
- Navigate to the AlphaFold Server: alphafoldserver.com
- For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
- Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
- In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
- Paste the peptide sequence.
- Paste the A4V mutant SOD1 sequence in the target field.
- Check the boxes
- Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
- Choose one peptide you would advance and justify your decision briefly.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
- Open the moPPit Colab linked from the HuggingFace moPPIt model card
- Make a copy and switch to a GPU runtime.
- In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Part C: Final Project: L-Protein Mutants
{kind=link}