HTGAA Week 6 Genetic circuit part 1 DNA Assembly
1.What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix is a concentrated DNA polymerase solution containing a high fidelity reaction buffer, MgCI (Magnesium chloride) and dNTPs ( deoxynucleoside triphosphates). It is used in PCR as polymerase solution can help fill the gaps in the sequence cloned during Gibson or HiFi assembly. The reaction buffer serves as a stabilizer and MgCI and dNTPs serve as building blocks for PCR. Reference list BioChain Institute Inc. (2024). Biochain Institute Inc. [online] Biochain Institute Inc. Available at: https://www.biochain.com/blog/using-dntp-in-polymerase-chain-reaction-pcr/. New England Biolabs (2026). [online] Neb.com. Available at: https://www.neb.com/en-gb/products/m0531-phusion-high-fidelity-pcr-master-mix-with-hf-buffer [Accessed 23 Mar. 2026].
HTGAA Week 7 Genetic circuits part 2 Part 1: Intracellular Artificial Neural Networks
1.What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? The advantage of IANNs over Boolean genetic circuits is their analog sensing and nonlinear processing capabilities, while traditional genetic circuits function as an On and Off system the IANNs can work in a continuous way due to its broader and stronger range of nonlinear inputs. Additionally, the IANNs have better pattern recognition and generalization technology, they can detect more complex patterns in data and can organise them better than the Boolean can.The IANNs are also less likely to fail and have a better error tolerance than the Boolean which is very susceptible to fail if there is a single issue with a gate. Moreover, the IANNs have a better scalable capacity due to the protein splicing mechanism allowing to create a multiple input-output circuit. The IANN technology can also better adapt and evolve to its environment compared to the Boolean which has a fixed function in its environment. Finally, IANNs have a strong memory quality that can be passed through to subsequent generations within the cell due stoichiometric cleavage and splicing which is irreversible.
HTGAA Week 9 Cell Free Systems Part A General and Lecture specific questions
General Homework Questions
1.Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Time : CFPS can be executed in very little time, a couple of hours however in vivo methods will take a few days to a few weeks. Time is a key part of efficient research as less time is wasted waiting to synthesize proteins to experiment on. System : CFPS is an open access system as it has no membrane and gives direct access to the molecule which one wants to work on, it offers more flexibility. It also allows for better control over the synthesis as one can specifically choose which components to input in a protein and better predict protein folding, this simplifies monitoring as well. Versus an in vivo method which has a membrane and therefore a closed system, this means the host and its other components will also react to any modification making it harder to control and direct, the cell’s functioning can often get in the way of the synthesis or create unpredicted issues. Tolerance: CFPS can tolerate high rates of toxic and difficult proteins as it is non living, however, in vivo technologies are more sensitive to toxins as it is likely to harm the host. CFPS also works using non-natural components offering a wider spectrum of possibilities. Complexity : CFPS is a simple PCR based procedure while in vivo requires more complex cloning and transformation steps. Reference List Silverman, A. D., et al. (2020). Quarto: A User’s Guide to Cell-Free Protein Synthesis. Nature Reviews Genetics. Gregorio, N. E., et al. (2019). Cell-free microbial synthesis of proteins. Frontiers in Bioengineering and Biotechnology.
HTGAA Week 10 Advanced imaging & measurement technology Final Project
1.Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
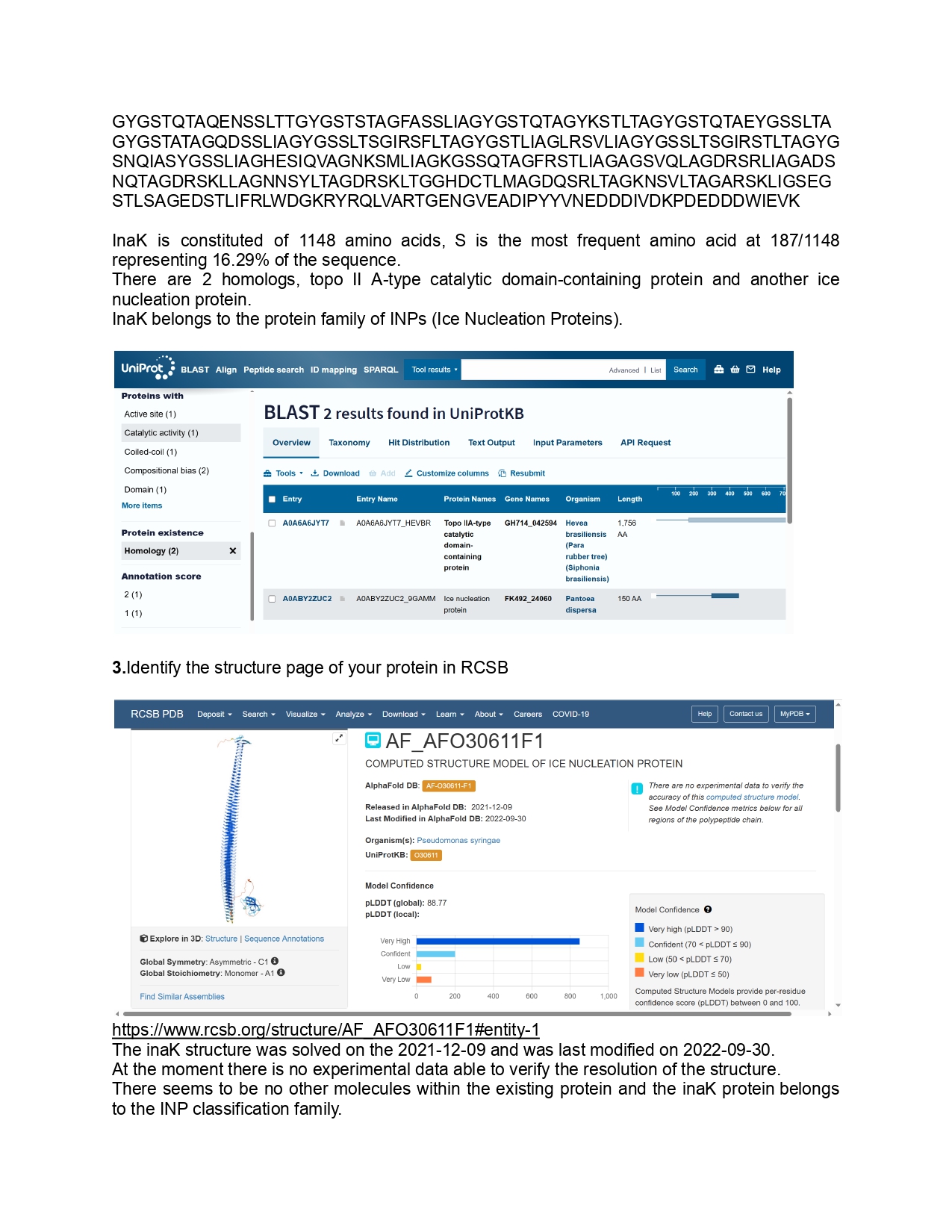
As for this project I aim to use inaK for ice production I would like to measure the ice nucleation ratio and efficiency of the inaK protein. Additionally, I would like to measure the temperatures inaK can resist to, on its own and as a supplement to an ice sample. If my initial experiments are successful I would like to measure the inaK ratio innoculated into ice to find the most optimal inaK quantity needed.
Week 11 Bioproduction & Cloud Lab Part A - The 1.536 pixel art work canvas, collective artwork
1.Contribute at least one pixel to the global artwork
I added early on a pixel towards the top left corner. I do not have much to say about this section of the work except maybe understanding the full purpose of this exercise.
Subsections of Homework
Week 1 HW: Principles and Practices
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
• By helping respond
Foster Lab Safety
• By preventing incident
• By helping respond
Protect the environment
• By preventing incidents
• By helping respond
Other considerations
• Minimizing costs and burdens to stakeholders
• Feasibility?
• Not impede research
• Promote constructive applications
Week 2 HW:DNA Read, Write & Edit
Week 3 HW:Lab Automation
Week 4 HW: Protein Design Part 1
Week 5 HW: Protein Design Part 2
Week 6 HW: Genetic Circuits Part 1
HTGAA Week 6 Genetic circuit part 1
DNA Assembly
1.What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix is a concentrated DNA polymerase solution containing a high fidelity reaction buffer, MgCI (Magnesium chloride) and dNTPs ( deoxynucleoside triphosphates). It is used in PCR as polymerase solution can help fill the gaps in the sequence cloned during Gibson or HiFi assembly. The reaction buffer serves as a stabilizer and MgCI and dNTPs serve as building blocks for PCR.
Reference list
BioChain Institute Inc. (2024). Biochain Institute Inc. [online] Biochain Institute Inc. Available at: https://www.biochain.com/blog/using-dntp-in-polymerase-chain-reaction-pcr/.
New England Biolabs (2026). [online] Neb.com. Available at: https://www.neb.com/en-gb/products/m0531-phusion-high-fidelity-pcr-master-mix-with-hf-buffer [Accessed 23 Mar. 2026].
2.What are some factors that determine primer annealing temperature during PCR?
The primer annealing temperature during PCR will be determined by the melting temperature ( Tm) of primers and according to the primer design guidelines in the protocol the binding region 18-22 bp at a 52-58°C Tm allows for primers pairs to have 5°C between each other.
3.There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
The aim of PCR is to amplify a specific DNA sequence in order to duplicate it a large amount of times and restriction enzyme digests uses enzymes to cut DNA of a chosen sequence sites into smaller sequence fragments. The PCR allows to copy the DNA while the restriction enzyme digest allows to cut DNA (insert and vector, so the plasmid) to create compatible fragment sides enabling better ligation, they are often used as a combination in cloning.
PCR uses heat-stable DNA polymerase and primers in order to stimulate a DNA replication and the restriction enzyme digests uses endonuclease enzymes to break phosphodiester bonds.
Restriction enzyme digests tend to be the preferred method as it is an easier protocol, it is a single step incubation protocol and less machinery. PCR requires precise primers, binders, restriction digests and temperatures, there are a lot more steps to the protocol and a slight lack of precision in any part of the protocol or the media and the PCR might be incorrect. However, the PCR does produce very large amounts of DNA sequencing.
4.How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To make sure a DNA sequence is suitable for the Gibson cloning one can verify that the DNA sequence has homologous primers and high-fidelity amplification in order to check that there are overlapping homologous sequences which will allow for a seamless fragment assembly.
Primer overlaps should have 20-40 homologous bp, preferably with a higher ratio of GC amino acids which favor stable annealing, between fragments an overlap of 15-30 bp is sufficient but the more there are fragments the longer the overlap sections should be.
5.How does the plasmid DNA enter the E. coli cells during transformation?
There are two main ways for E. coli to enter the cells during transformation:
On one hand, a heat shock, a chemical transformation where heating the cell and DNA mixture abruptly forms a thermal current allowing to shape temporary pores in the bacterial cell letting the plasmid pass through.
On the other hand, electroporation, creating those same pores in the bacterial cell for the plasmid to pass through but this time using high electrical voltage.
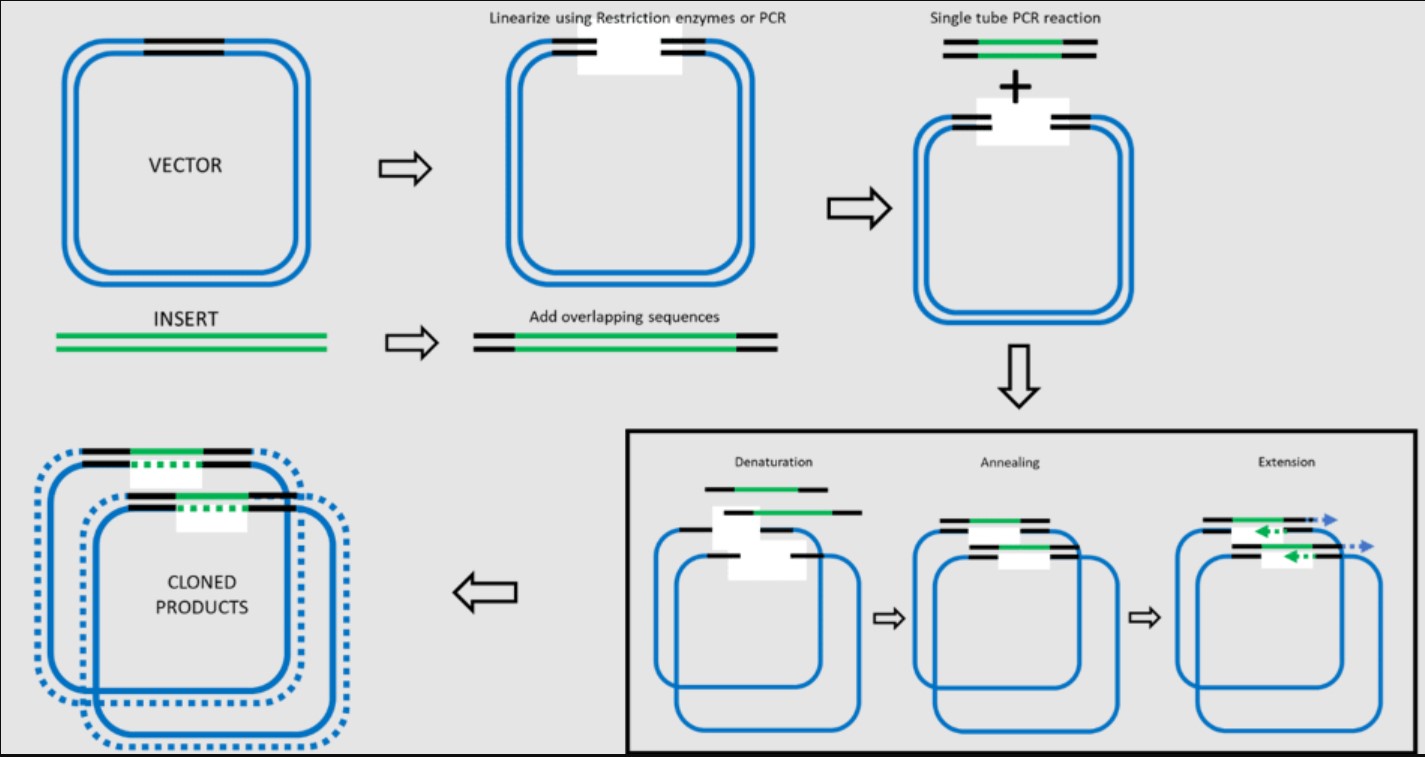
6.Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Another DNA assembly method is the CPEC, Circular Polymerase Extension Cloning, an in vivo and in vitro technology that uses PCR to amplify and extend and overlap sequence fragments. Due to its overlapping quality it allows for inserts or assembly without using restriction enzymes. This technology relies on homologs, the fragments are first amplified in PCR with homologous ends between them, the homologous region give the possibility to anneal and extend each section by the DNA polymerase through a second PCR round, then the DNA can be recombined creating larger DNA sequences which can then be introduced to the plasmid.
Reference list
Bitesize Bio. (2019). CPEC– a Quick and Inexpensive Cloning Strategy. [online] Available at: https://bitesizebio.com/44113/cpec-a-quick-and-inexpensive-cloning-strategy/.
Chao, R., Yuan, Y. and Zhao, H. (2014). Recent advances in DNA assembly technologies. FEMS Yeast Research, p.n/a-n/a. doi:https://doi.org/10.1111/1567-1364.12171.
Asimov Kernel N/A as we were not given access
Week 7 HW: Genetic Circuits Part 2
HTGAA Week 7 Genetic circuits part 2
Part 1: Intracellular Artificial Neural Networks
1.What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
The advantage of IANNs over Boolean genetic circuits is their analog sensing and nonlinear processing capabilities, while traditional genetic circuits function as an On and Off system the IANNs can work in a continuous way due to its broader and stronger range of nonlinear inputs. Additionally, the IANNs have better pattern recognition and generalization technology, they can detect more complex patterns in data and can organise them better than the Boolean can.The IANNs are also less likely to fail and have a better error tolerance than the Boolean which is very susceptible to fail if there is a single issue with a gate. Moreover, the IANNs have a better scalable capacity due to the protein splicing mechanism allowing to create a multiple input-output circuit. The IANN technology can also better adapt and evolve to its environment compared to the Boolean which has a fixed function in its environment. Finally, IANNs have a strong memory quality that can be passed through to subsequent generations within the cell due stoichiometric cleavage and splicing which is irreversible.
Reference list
Ana Halužan Vasle and Miha Moškon (2024). Synthetic biological neural networks: From current implementations to future perspectives. Biosystems, 237, pp.105164–105164. doi:https://doi.org/10.1016/j.biosystems.2024.105164.
Anastassov, S., Filo, M. and Khammash, M. (2024a). Inteins: A Swiss army knife for synthetic biology. Biotechnology Advances, 73, p.108349. doi:https://doi.org/10.1016/j.biotechadv.2024.108349.
Anastassov, S., Filo, M. and Khammash, M. (2024b). Inteins: A Swiss army knife for synthetic biology. Biotechnology Advances, 73, p.108349. doi:https://doi.org/10.1016/j.biotechadv.2024.108349.
Claus Kadelka, Taras-Michael Butrie, Hilton, E., Kinseth, J. and Haris Serdarevic (2024). A meta-analysis of Boolean network models reveals design principles of gene regulatory networks. Science Advances, 10(2). doi:https://doi.org/10.1126/sciadv.adj0822.
Gao, Y., Wang, L. and Wang, B. (2023). Customizing cellular signal processing by synthetic multi-level regulatory circuits. Nature communications, [online] 14(1). doi:https://doi.org/10.1038/s41467-023-44256-1.
Ilia, K. and Del Vecchio, D. (2022). Squaring a Circle: To What Extent Are Traditional Circuit Analogies Impeding Synthetic Biology? GEN Biotechnology, 1(2), pp.150–155. doi:https://doi.org/10.1089/genbio.2021.0014.
Karkalos, N.E. and Markopoulos, A.P. (2017). Modeling of hard machining. [online] Available at: https://www.sciencedirect.com/topics/mathematics/artificial-neural-network.
Wang, H., Wang, L., Zhong, B. and Dai, Z. (2022). Protein Splicing of Inteins: A Powerful Tool in Synthetic Biology. Frontiers in Bioengineering and Biotechnology, 10. doi:https://doi.org/10.3389/fbioe.2022.810180.
2.Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
The IANN genetic circuit can be use in cancer theranostics as they have the ability to process multiple biomarkers simultaneously and within a tumour can clearly identify and distinguish a cancerous cell from a healthy one. Additionally, the IANN is able to recognise the cancer patterns and can trigger the synthesis of an anti-cancer drug.
The IANN circuit used for cancer theranostics requires many inputs in order to increase precision:
MicroRNAs which allow to classify cancerous cells
TSAs which are Tumor Specific Antigens
TAAs which are Tumor Associated Antigens, they work specifically on the cancerous tissue
TME which is a Tumor Microenvironment
Enzymes
NIR which is an external stimuli such as light, magnetic field or ultrasounds which server a switch to trigger the therapy is a specific area of the body
The outputs for an IANN circuit used for cancer theranostics presents itself as a localized response:
Apoptosis triggering which leads to the programmed cell death induced by the drug delivery
Drug delivery of anticancer agents in a controlled and chosen way and area
Bioimaging through fluorescence, NIR signals or MRI
PTT or PDT heat generated chemical reaction which kills tumor cells
Creation of immune stimulatory proteins and antibodies
The IANN faces many limits however in cancer theranostics use, firstly, it is a very costly technology and is difficult to scale as it requires using and reproducing many inorganic nanoparticles in a consistent way.
One of the main issues the IANN presents is the biosafety issue as it uses inorganic nanoparticles which lack biodegradability which cause systemic toxicity and long-term retention in the liver, spleen and kidney which can lead to further toxic side effects. Furthermore, because some nanoparticles do not degrade it can cause premature drug release or loss of diagnostic functionality. The IANN also does not provide very clear imagery. Moreover, the IANN faces difficulties penetrating deep cancerous tissue, this physiological barrier issue is made worse by the binding of nanoparticles to the cancerous cells on the outer edge of a tumour which restrict even more the penetration into inner areas of the tumor tissue. Lastly, IANN can lead to skin discoloration because of its high concentration of metallic nanoparticles.
Reference List
Aminolroayaei, F., Shahbazi‐Gahrouei, D., Shahbazi‐Gahrouei, S. and Rasouli, N. (2021). Recent nanotheranostics applications for cancer therapy and diagnosis: A review. IET Nanobiotechnology, 15(3), pp.247–256. doi:https://doi.org/10.1049/nbt2.12021.
Ausländer, S. and Fussenegger, M. (2016). Engineering Gene Circuits for Mammalian Cell–Based Applications. Cold Spring Harbor Perspectives in Biology, 8(7), p.a023895. doi:https://doi.org/10.1101/cshperspect.a023895.
Brijendra Kumar Kashyap, Singh, V., Manoj Kumar Solanki, Kumar, A., Janne Ruokolainen and Kavindra Kumar Kesari (2023). Smart Nanomaterials in Cancer Theranostics: Challenges and Opportunities. ACS omega, 8(16), pp.14290–14320. doi:https://doi.org/10.1021/acsomega.2c07840.
Chen, H. and Zhao, Y. (2018). Applications of Light-Responsive Systems for Cancer Theranostics. ACS Applied Materials & Interfaces, 10(25), pp.21021–21034. doi:https://doi.org/10.1021/acsami.8b01114.
Chen, J., Fu, S., Zhang, C., Liu, H. and Su, X. (2022). DNA Logic Circuits for Cancer Theranostics. Small, 18(20). doi:https://doi.org/10.1002/smll.202108008.
Jiang, L., Fu, Z., Ye, B., Feng, X., Chen, Z., Chen, Q., Long, Y., Wang, S. and Deng, G. (2025). Metal nanoparticles in cancer theranostics: from synthesis to tumor microenvironment-responsive applications. Drug Delivery, 32(1). doi:https://doi.org/10.1080/10717544.2025.2565480.
Kang, S., Gil, Y.-G., Min, D.-H. and Jang, H. (2020). Nonrecurring Circuit Nanozymatic Enhancement of Hypoxic Pancreatic Cancer Phototherapy Using Speckled Ru–Te Hollow Nanorods. ACS Nano, 14(4), pp.4383–4394. doi:https://doi.org/10.1021/acsnano.9b09974.
Li, Y., Zhang, X., Wang, J., Wang, K., Li, B., Qiao, X., He, W., Cai, J., Liu, D. and Yang, L.-L. (2025). Leveraging adenosine triphosphate for cancer theranostics. Theranostics, [online] 15(10), pp.4708–4733. doi:https://doi.org/10.7150/thno.106291.
Moon Sung Kang, Kwon, M., Jang, H.-S., Jeong, S., Han, D.-W. and Ki Su Kim (2022). Biosafety of inorganic nanomaterials for theranostic applications. Emergent materials, 5(6), pp.1995–2029. doi:https://doi.org/10.1007/s42247-022-00426-3.
Sergeeva, O.V., Luo, L. and Guiseppi-Elie, A. (2025). Cancer theragnostics: closing the loop for advanced personalized cancer treatment through the platform integration of therapeutics and diagnostics. Frontiers in Bioengineering and Biotechnology, 12. doi:https://doi.org/10.3389/fbioe.2024.1499474.
Tânia F.G.G. Cova, Freitas, D. and Odebrecht, S. (2019). Computational Approaches in Theranostics: Mining and Predicting Cancer Data. 11(3), pp.119–119. doi:https://doi.org/10.3390/pharmaceutics11030119.
Weranga Rajapaksha, Riya Khetan, Ian, Blencowe, A., Garg, S., Albrecht, H. and Gillam, T.A. (2024). Future theranostic strategies: emerging ovarian cancer biomarkers to bridge the gap between diagnosis and treatment. Frontiers in Drug Delivery, 4. doi:https://doi.org/10.3389/fddev.2024.1339936.
Xie, Z., Wroblewska, L., Prochazka, L., Weiss, R. and Benenson, Y. (2011). Multi-input RNAi-based logic circuit for identification of specific cancer cells. Science (New York, N.Y.), [online] 333(6047), pp.1307–11. doi:https://doi.org/10.1126/science.1205527.
Zuo, Y., Li, P., Wang, W., Xu, C., Xu, S., Herman, Sun, J., Jin, G., Wang, W., Ryan, Jacky and Tang, B.Z. (2024). Tumor Site‐Specific In Vivo Theranostics Enabled by Microenvironment‐Dependent Chemical Transformation and Self‐Amplifying Effect. Advanced Science, 12(4), pp.e2409506–e2409506. doi:https://doi.org/10.1002/advs.202409506.
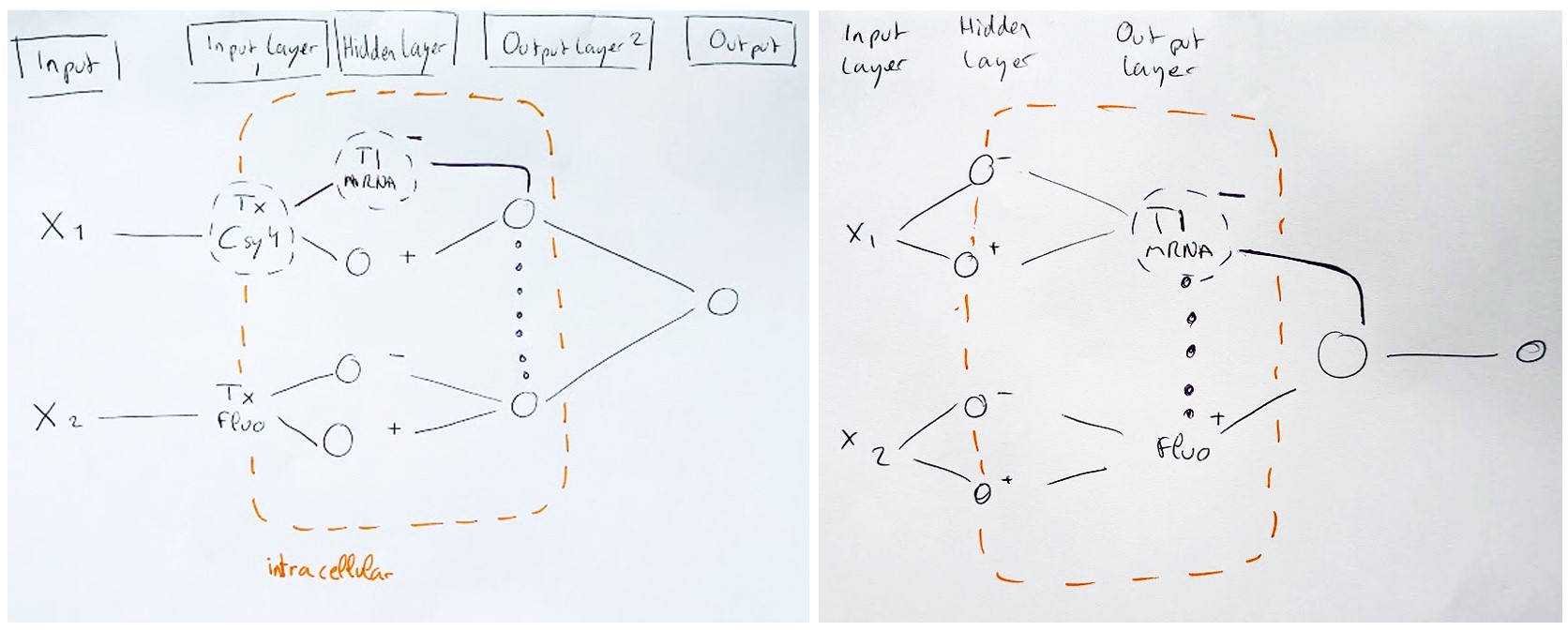
3.Draw a diagram for an intercellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Part 2 Fungal Materials
1.What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
One of the most common examples of fungal materials today is mycelium packaging, mycelium is the main fungal strand used in biodesign. It is able to grow within a mold and once cooked it dies but the shape given is preserved.
Mycelium is non harmful to the environment and biodegradable which has inspired many companies to launch mycelium packaging with the aim to replace and reduce plastic use in packaging. Mycelium grows rapidly but still requires the time to grow compared to plastic packaging which can be produced instantly. Mycelium is a living organism and holds a higher risk in production. Mycelium materials however require no chemical input which is better for the environment, the producer and the consumer, eliminating the risk of chemicals and microplastics. Furthermore, mycelium material packaging is currently more costly than producing plastic but the cost gap is reducing as mycelium materials are being scaled up according to a mycelium packaging market report of 2025.
Companies such as Grown Bio already offer viable alternatives to packaging of all sorts. They offer a range of packaging of all shapes and qualities (some with reinforced protective design) which one could buy directly or they offer the possibility to grow one’s own packaging.
2.What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
In the continuation of mycelium packaging I would love to explore whether one could genetically modify the mycelium to have active properties as well. Maybe mycelium packaging could also have cold properties allowing to store products which require to be kept in the cold. Many mycelium species, such as the most commonly used oyster mushroom (Pleurotus ostreatus), can withstand freezing temperatures and simply go into a dormant state. Typically in packaging mycelium would be killed with heat to stop its growth and preserve its shape, however, could the mycelium be kept alive, genetically modified with ice nucleation proteins, put in a dormant state because of the freezing temperature which would still stun its growth and therefore preserve its shape and then be used a cold packaging system, which in its end of life could still be biodegradable or contribute actively to nature.
It appears there would be two ways to genetically modify mycelium to produce cold. Mycelium is already used successfully as a host in synthetic biology. Additionally, it seems that one could introduce ice nucleotides to the genetic code of mycelium DNA and the mycelium would accept it. An experiment of adding ice nucleotide proteins to water which was then fed to mycelium has already been done, with the aim to study freezing in mycelium (Schwidetzky et al., 2023). Secondly, certain mycelium strands (including the commonly used oyster mushroom) already appear to contain ice nucleotide allowing them to resist freezing temperatures, one could explore genetically modifying the mycelium to express this protein in a more active way allowing it to produce a freezing quality.
Fungi can produce complex molecules and proteins better than bacteria, they are also able to produce much more enzymes than bacteria making purification processes easier. Fungi are also eukaryotic organisms which allows them to perform complex post-translational modifications like protein folding offer a bigger potential for synthetic biology. Additionally, fungi naturally produce more secondary metabolites than bacteria such as terpenoids, polyketides and alkaloids which are commonly used in pharmaceutical research and development. Moreover, fungi genomes naturally contain biosynthetic gene clusters (BGCs) which are used in synthetic biology to engineer new-to-nature chemicals.
Fungi can be easier to work with as they are able to live off a wider range of feedstock and grow rapidly as well as being robust cultures able to adapt to harsh environments and withstand a range of PH levels or a range of temperatures.
Reference list
Awasthi, S., Alam, M.I. and Pal, D.B. (2025). Importance of Utilizing Fungus Rather Than Bacteria for Biomass Valorization. Fungal Biology, pp.107–140. doi:https://doi.org/10.1007/978-3-031-82599-6_5.
CATALEX BIO. (2025). Fungal vs Bacterial Enzymes: Industrial Applications & Selection Guide | Catalex Bio Enzyme Manufacturer & Supplier. [online] Available at: https://catalexbio.com/fungal-vs-bacterial-enzymes-comparison-guide/.
Cordero, B., Ellie Rose Mattoon, Ramos, Z. and Casadevall, A. (2023). The hypothermic nature of fungi. PNAS, 120(19). doi:https://doi.org/10.1073/pnas.2221996120.
Eufemio, R.J., Rojas, M., Shaw, K., de Almeida Ribeiro, I., Guo, H.-B., Renzer, G., Belay, K., Liu, H., Suseendran, P., Wang, X., Fröhlich-Nowoisky, J., Pöschl, U., Bonn, M., Berry, R.J., Molinero, V., Vinatzer, B.A. and Meister, K. (2026). A previously unrecognized class of fungal ice-nucleating proteins with bacterial ancestry. Science Advances, 12(11). doi:https://doi.org/10.1126/sciadv.aed9652.
Garg, S. (2025a). The importance of fungal biotechnology for sustainable applications. Trends in Biotechnology, [online] 0(0). doi:https://doi.org/10.1016/j.tibtech.2025.06.010.
Garg, S. (2025b). The importance of fungal biotechnology for sustainable applications. Trends in Biotechnology, [online] 0(0). doi:https://doi.org/10.1016/j.tibtech.2025.06.010.
Hinneburg, H., Gu, S. and Naseri, G. (2025). Fungal Innovations—Advancing Sustainable Materials, Genetics, and Applications for Industry. Journal of Fungi, 11(10), p.721. doi:https://doi.org/10.3390/jof11100721.
Jo, C., Zhang, J., Tam, J.M., Church, G.M., Khalil, A.S., Segrè, D. and Tang, T.-C. (2023a). Unlocking the magic in mycelium: Using synthetic biology to optimize filamentous fungi for biomanufacturing and sustainability. Materials Today Bio, 19, p.100560. doi:https://doi.org/10.1016/j.mtbio.2023.100560.
Jo, C., Zhang, J., Tam, J.M., Church, G.M., Khalil, A.S., Segrè, D. and Tang, T.-C. (2023b). Unlocking the magic in mycelium: Using synthetic biology to optimize filamentous fungi for biomanufacturing and sustainability. Materials Today Bio, 19, p.100560. doi:https://doi.org/10.1016/j.mtbio.2023.100560.
Ma, D., Yang, G., Mu, L. and Li, C. (2011). Tolerance of ectomycorrhizal fungus mycelium to low temperature and freezing–thawing. Canadian Journal of Microbiology, 57(4), pp.328–332. doi:https://doi.org/10.1139/w11-001.
Moreno-Giménez, E., Mónica Gandía, Zara Sáez, Manzanares, P., Yenush, L., Orzáez, D., Marcos, J.F. and Garrigues, S. (2023). FungalBraid 2.0: expanding the synthetic biology toolbox for the biotechnological exploitation of filamentous fungi. Frontiers in Bioengineering and Biotechnology, 11. doi:https://doi.org/10.3389/fbioe.2023.1222812.
Nielsen, J. (2013). Production of biopharmaceutical proteins by yeast. Bioengineered, 4(4), pp.207–211. doi:https://doi.org/10.4161/bioe.22856.
Raymond, J.A. and Janech, M.G. (2009). Ice-binding proteins from enoki and shiitake mushrooms. Cryobiology, 58(2), pp.151–156. doi:https://doi.org/10.1016/j.cryobiol.2008.11.009.
Sanitá Lima, M. and Coutinho de Lucas, R. (2022). Co-cultivation, Co-culture, Mixed Culture, and Microbial Consortium of Fungi: An Understudied Strategy for Biomass Conversion. Frontiers in Microbiology, 12. doi:https://doi.org/10.3389/fmicb.2021.837685.
Schwidetzky, R., Ingrid, Bothen, N., Backes, A.T., DeVries, A.L., Bonn, M., Fröhlich-Nowoisky, J., Molinero, V. and Meister, K. (2023a). Functional aggregation of cell-free proteins enables fungal ice nucleation. Proceedings of the National Academy of Sciences of the United States of America, 120(46). doi:https://doi.org/10.1073/pnas.2303243120.
Schwidetzky, R., Ingrid, Bothen, N., Backes, A.T., DeVries, A.L., Bonn, M., Fröhlich-Nowoisky, J., Molinero, V. and Meister, K. (2023b). Functional aggregation of cell-free proteins enables fungal ice nucleation. Proceedings of the National Academy of Sciences of the United States of America, 120(46). doi:https://doi.org/10.1073/pnas.2303243120.
Shankar, M.P., Hamza, A., Khalad, A., Shanthi, G., Kuppireddy, S. and Kumar, D.S. (2024). Engineering mushroom mycelium for a greener built environment: Advancements in mycelium-based biocomposites and bioleather. Food Bioscience, [online] 62, p.105577. doi:https://doi.org/10.1016/j.fbio.2024.105577.
Week 9 HW: Cell Free Systems
HTGAA Week 9 Cell Free Systems
Part A General and Lecture specific questions
General Homework Questions
1.Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Time : CFPS can be executed in very little time, a couple of hours however in vivo methods will take a few days to a few weeks. Time is a key part of efficient research as less time is wasted waiting to synthesize proteins to experiment on.
System : CFPS is an open access system as it has no membrane and gives direct access to the molecule which one wants to work on, it offers more flexibility. It also allows for better control over the synthesis as one can specifically choose which components to input in a protein and better predict protein folding, this simplifies monitoring as well. Versus an in vivo method which has a membrane and therefore a closed system, this means the host and its other components will also react to any modification making it harder to control and direct, the cell’s functioning can often get in the way of the synthesis or create unpredicted issues.
Tolerance: CFPS can tolerate high rates of toxic and difficult proteins as it is non living, however, in vivo technologies are more sensitive to toxins as it is likely to harm the host. CFPS also works using non-natural components offering a wider spectrum of possibilities.
Complexity : CFPS is a simple PCR based procedure while in vivo requires more complex cloning and transformation steps.
Reference List
Silverman, A. D., et al. (2020). Quarto: A User’s Guide to Cell-Free Protein Synthesis. Nature Reviews Genetics.
Gregorio, N. E., et al. (2019). Cell-free microbial synthesis of proteins. Frontiers in Bioengineering and Biotechnology.
2.Describe the main components of a cell-free expression system and explain the role of each component.
There are four core components to a CFPS system which are the catalytic engine, the instruction template, the energy source and the biochemical blocks.
At first a “soup” is created by breaking a cell pen and removing debris, this gives us the heart of the system which provides us with the molecules required for transcription and translation like ribosomes or tRNAs.
The DNA instruction template is the blueprint for the CFPS and it is very flexible as it can function with a plasmid but it can also work using PCR linear PCR products which allows to reduce cloning time in the preparatory stages.
Compared to most synthesis technologies requiring a lot of energy to keep the cell alive and the ribosomes active, CFPS can be fueled by ATPs and GTPs which are high energy sources.
Biochemical building blocks and buffers stabilize the CFPS reaction by adding to the solution amino acids to build the chain, the RNA polymerase and often magnesium or potassium salts which can help stabilize the protein folding.
Reference List
Silverman, A. D., Karim, A. S., & Jewett, M. C. (2020). Quarto: A User’s Guide to Cell-Free Protein Synthesis. Nature Reviews Genetics.
Hodgman, C. E., & Jewett, M. C. (2012). Cell-free protein synthesis: The state of the art. Biotechnology and Bioengineering.
Caschera, F., & Noireaux, V. (2014). Synthesis of 2.3 mg/ml of GFP with an all-E. coli cell-free transcription-translation system. Biological Engineering.
Shimizu, Y., et al. (2001). Cell-free translation reconstituted with purified components. Nature Biotechnology.
Tinafar, A., et al. (2019). A Manual of Cell-Free Protein Synthesis Systems. Frontiers in Bioengineering and Biotechnology.
3.Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Protein synthesis is a very energy taxing process. An issue in CFPS is the ATP “leaks” caused by enzymes breaking down enzymes to ADP (without the ribosome using it) and inorganic phosphate, the regeneration of energy allows to preserve fuel for the synthesis rather than it being wasted by enzymes.
A method of not just adding an excess of ATP is adding a substrate fuel which could be Phosphoenolpyruvate (PEP) and Pyruvate Kinase (PK). The substrate PEP acts as a battery which contains the phosphate group. Once the ribosome processes the ATP into an ADP the PK is able to directly collect the phosphate from the PEP and link it back onto the ADP changing it back to an ATP creating a steady loop preserving ATP levels consistent till all the PEP is processed.
Reference List
Silverman, A. D., et al. (2020). Nature Reviews Genetics.
Caschera, F., & Noireaux, V. (2014). Biological Engineering.
4.Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Both prokaryotic and eukaryotic cell free systems aim to produce protein from a DNA template.
Prokaryotic systems have high speed and yield rates while eukaryotic systems are slower and produce less proteins.
In prokaryotic systems the transcription and translation happen in the same place, they are coupled, eukaryotic systems are decoupled and can require extra steps.
While the eukaryotic cell free expression is quite costly the prokaryotic based system is inexpensive and simpler to set up and run.
However, the eukaryotic system can offer advanced protein folding and more complex modifications as it is able to mimic human-like post-translational modification. The prokaryotic cannot offer natural modifications and creates basic protein folding.
Unless an experiment requires human-like testing I would choose the prokaryotic cell free expression system as it is simpler, less expensive and more prolific. Additionally, if an experiment requires more stability and better control, the fact that prokaryotic systems cannot offer modifications might also make it more predictable and easier to work with.
The INP inaK protein I have been working with would be suitable for prokaryotic cell-free expression as it is a highly repetitive protein which is commonly found in Pseudomonas Syringae, a prokaryote, but if I use an E. coli extract then the codon bias is the same as the native bacteria so the ribosome will read with ease the repetitive portions of the sequence. Moreover, inaK only requires a membrane to anchor to, it does need additional complex compounds. Furthermore, I would aim for a high concentration of inaK for my project in order to produce more ice, which then is more suitable to the high yield of prokaryotic systems.
For a eukaryotic cell-free expression system I would choose a complex human protein such as the tPA, tissue plasminogen activator (a clot-buster), which have more complex modifications and toxins, it would benefit from a more advanced and control protein folding method.
Globally, if a protein has a bacterial origin or has a simple structure and needs to be produced in a large quantity then the prokaryotic systems are more suitable. Eukaryotic systems are useful for complex and human or mammal proteins.
Reference List
Zemella, A., et al. (2015). Cell-Free Protein Synthesis: Pros and Cons of Prokaryotic and Eukaryotic Systems. ChemBioChem.
Silverman, A. D., et al. (2020). Quarto: A User’s Guide to Cell-Free Protein Synthesis. Nature Reviews Genetics.
Endo, Y., & Sawasaki, T. (2006). A cell-free protein synthesis system for high-throughput proteomics. Journal of Structural and Functional Genomics. (Focusing on Wheat Germ advantages).
5.How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
I will be using inaK as it is a membrane protein and use a prokaryotic system.
The first issue I will be faced with is that the inaK N-terminal is highly hydrophobic and might create aggregates if I don’t offer it a lipid surface to link to.
I would start with a circular plasmid sourced using Twist and Benchling.
For the expression system I might use an E. coli based S30 extract because the inaK is a bacterial protein and the codon bias and translation speed using the E. coli ribosomes will be naturally optimized for its repetitive sequence. In this case, the transcription and translation will be coupled to ensure the protein folds as it is produced.
I would include PEP and PK for energy regeneration.
Next, as I need to provide the inaK with a new membrane to latch on to I could select a liposome host membrane which might mimic their initial membrane closely, it is structured as small bubbles of phospholipids which should help the first hydrophobic issue.
To determine where exactly to produce the inaK I might have to run a few optimization matrix experiments whether tuning temperature, optimizing lipid to protein ratio to avoid aggregations or a diluted solution, and magnesium and potassium concentrations as membrane proteins are sensitive to charge and a wrong ratio could make the ribosome collapse.
In the end I would have to evaluate the success ratio of my system. The purpose of the inaK is to freeze water, I could place a drop of the cell free solution on a cold plate and observe which solution (supposing I would run a few) would freeze at the highest temperature.
Reference List
Hartmann, et al. (2022). Overcoming bottlenecks for in vitro synthesis of ice nucleating protein InaZ.
Silverman, A. D., et al. (2020). Quarto: A User’s Guide to Cell-Free Protein Synthesis. Nature Reviews Genetics.
Henrich, et al. (2015). Analyzing the specialized lipid environment of membrane proteins.
6.Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
A low yield result can be common in CFPS.
This can be caused by energy exhaustion, this system is very energy consuming and the PEP might have been consumed too rapidly or the ATP consuming enzymes too active in the sample leading to the ribosome to not have enough energy to produce enough protein. To resolve this a higher concentration of the energy substrate could be used.
The DNA template might also be damaged or degraded, causing a blueprint issue. If a linear PCR template is used rather than a plasmid it can happen that enzymes might interfere or damage the ends of the template causing no proteins to be created. Using a controlled plasmid should help avoid this issue, it is possible to make a linear PCR into a circular plasmid through ligations.
There might also be an ionic imbalance within the solution which can be due to an unstable ribosome or DNA sequence, this imbalance might work for certain proteins but maybe not the one studied. For instance, mRNA and DNA are very dependent on magnesium and potassium which requires finding the perfect ion balance for the protein in the solution. Adjusting the magnesium ratios can help, this can be done by running multiple tubes with different magnesium percentages.
Reference List
Silverman, A. D., et al. (2020). Quarto: A User’s Guide to Cell-Free Protein Synthesis. Nature Reviews Genetics.
Sun, Z. Z., et al. (2013). Linear DNA for rapid prototyping. ACS Synthetic Biology.
Caschera, F., & Noireaux, V. (2014). Synthesis of 2.3 mg/ml of GFP. Biological Engineering.
Questions from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
1.Pick a function and describe it.
a.What would your synthetic cell do? What is the input and what is the output?
My synthetic cell design will aim to be used to synthesize inaK (using a prokaryotic cell design) and allow it to more efficiently catalyze ice. Using CFPS for inaK synthesis enables me to control and organise the proteins to sit on the membrane surface in high concentration. Ice nucleation is a surface dependent process and through CFPS I could target the ice nucleation in active sites or clusters which will be more effective than loose individual proteins.
The input is what I would give the cell to stay active. I would input a codon optimized plasmid containing the inaK sequence serving as genetic instructions. I would also input the standard set of 20 amino acids in order to build the inaK protein chains. I would input an ATP for energy (with PEP and PK to have energy regeneration). Finally, I would input magnesium and potassium salts to stabilize the internal soup of the cell.
If I am designing a prokaryotic cell design then the transcription and translation are coupled and happen simultaneously.
The output of this design creates a functional change, primarily the folded inaK proteins anchors to this new membrane and forms an ice active surface. The proteins are physically aligned in a new structure allowing for the crystal lattice of surrounding water molecules. Finally, there would be inorganic phosphate and heat outputs which will be a result of the ATP function.
Reference List
Noireaux, V., & Libchaber, A. (2004). A vesicle bioreactor as a step toward an artificial cell. PNAS.
Silverman, A. D., et al. (2020). Quarto: A User’s Guide to Cell-Free Protein Synthesis. Nature Reviews Genetics.
b.Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
This design could be realized by cell-free Tx/Tl without encapsulation but it will change the function of the synthesis. If I remove the membrane then the inaK depending on it to efficiently catalyze ice would significantly change. The ice nucleation would decrease as I would lose the opportunity to design and arrange clusters but the protein can still technically be synthesized using the 20 amino acids and the DNA template and it would still catalyze ice to some extent. The main restriction will be that the hydrophobic N-terminal will have no membrane to latch onto and it will create disorder aggregates disturbing the ice formation.
As much as this synthesis can technically be realized without encapsulation it would not be beneficial or very useful to the experiment.
Reference List
Schmid, S., et al. (2016). Probing the ice-nucleation activity of the Pseudomonas syringae InaK protein. (Discussing the importance of membrane context).
Noireaux, V., & Libchaber, A. (2004). A vesicle bioreactor as a step toward an artificial cell. PNAS.
c.Could this function be realized by genetically modified natural cell?
Yes this function could be realized in a genetically modified natural cell, E. coli was used as a living cell before CFPS became a common technique. Here the inaK could be inserted as plasmid into a lab strain of E. coli to serve as a host. The advantage would be that it is energy self-sustainning as it is living,however, it has a higher complexity rate as there are many other interfering proteins and it has a high bio-safety risk as living GMOs can escape and multiply.
A cell free system would need a controlled energy input but has a simpler and safer function. The synthesis efficiency is also much higher and more robust in CFPS than in natural cell.
Reference List
In a Natural Cell: The surface of a living E. coli is crowded with lipopolysaccharides (LPS), flagella, and other proteins. These can “get in the way” of the InaK anchors, potentially lowering the density of the ice-active patches.
In a Synthetic Cell: You can create a “naked” lipid bilayer. This allows the InaK proteins to pack together tightly without any biological “clutter,” often leading to a more efficient ice-nucleation point.
d.Describe the desired outcome of your synthetic cell operation.
Ideally the inaK protein would catalyze ice in a more efficient and organized manner which could be used in glaciers to help preserve and rebuild their natural ice formation and be more resistant to warmer temperatures.
2.Design all components that would need to be part of your synthetic cell.
a.What would be the membrane made of?
To create a suitable cell and membrane for the inaK protein and its ice nucleation function I would start with a primary lipid mix of 70% POPE (palmitoyl oleoyl sn glycero phosphoethanolamine), helping creating necessary curvature and lateral pressure in membrane thanks to its cone shape, and 30% POPG (palmitoyl oleoyl sn glycero phosphoglycerol), provides negatively charged lipids to help inaK proteins be inserted in the membrane. The common mixture for mimicking an E.coli or Pseudomonas (inaK is commonly found in Pseudomonas Syringae) membrane is composed of PE (phosphatidylethanolamine) and PG (phosphatidylglycerol). This method would prevent aggregation of inaK as the hydrophobic N-terminal cannot insert itself into too stiff of a membrane. Furthermore, the lateral pressure of the membrane will be imitated and this tension helps push the protein together to cluster the inaK and enable ice nucleation at a higher temperature.
Reference List
Schmid et al. (2016) found that the ice-nucleation activity of InaK is highly dependent on being embedded in a lipid bilayer rather than just being free-floating, specifically noting that PE/PG mixes provided the most “natural” environment for bacterial anchors.
b.What would you encapsulate inside? Enzymes, small molecules.
In order for the cell not to just be a lipid membrane bubble I will add a variety of synthetic cell internal organs which ideally would focus on improving the speed of the transcription and translation system as the inaK is a long and repetitive protein.
The cell will encapsulate the genetic blueprint, the DNA plasmid containing the inaK codon optimized gene sequence. Additionally, there should be a strong promoter and double terminator to process large amounts of mRNA better and to ensure the RNA polymerase end in a specific chosen section preventing useless loss of energy.
I could add a PURE system to the cell replacing a cell lysate, this is a purified protein synthesis mix including 10S ribosomes (builds the protein), T7 RNA polymerase (turns DNA to mRNA), 36 essential enzymes to charge tRNA and translate information ( aminoacyl, IF,EF,RF) and tRNAs (transfer RNAs moving amino acids to the ribosome).
Moreover, I will add the energy source for the PURE system to work. This will include the set of 20 amino acids to build the inaK protein chain, NTPs (ATP, GTP, CTP and UTP) as energy for building the protein sequence and the secondary energy substrate, here creatine phosphate combined with creatine kinase commonly used in PURE systems to keep the ATP charged.
To ensure the protein gets to the membrane wall correctly and efficiently I might add SRPs (signal recognition particles) which will connect to the inaK and guide to the membrane and chaperones (DnaK, DnaJ and Grpe) which help with protein folding and prevent the inaK anchor to create clumps before reaching the membrane.
Finally, to stabilize the cell I will add magnesium salts.
I used Gemini to help me organize my information.
Reference List
Noireaux, V., & Libchaber, A. (2004). A vesicle bioreactor as a step toward an artificial cell. PNAS. This paper provides the foundational protocol for encapsulating T7-based expression systems inside POPE/POPG vesicles.
Cui, Y., Chen, X., Wang, Z. and Lu, Y. (2022) ‘Cell-free PURE system: evolution and achievements’, Biodesign Research, 2022, art. no. 9847014. doi: 10.34133/2022/9847014.
c.Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
For the inaK CFPS I have chosen a prokaryotic system using an E. coli based PURE method, because it is bacterial the codon language between the Pseudomonas prokaryotic bacteria origin of the inaK and the synthetic prokaryotic cell will match and the ribosome function will be more efficient. The Tx/TI system will then be coupled which works better for a membrane protein and here my hydrophobic anchor will find the membrane immediately avoiding complications. Additionally, in this case a mammalian system might struggle with the repetitiveness of my sequence.
Using a prokaryotic E. coli PURE design will give me a higher yield.
Reference List
Cui, Y., et al. (2022). ‘Cell-free PURE system: evolution and achievements’, Biodesign Research. This paper confirms that the E. coli-based PURE system is the most effective defined platform for expressing and inserting bacterial membrane proteins.
d.How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The cell will communicate mainly with its external environment through the protein action itself, the inaK has an ice nucleating function which will translate as a physical frozen output, the surrounding water molecules will also align and will themselves freeze.
Further membrane modifications could also allow for more communication with its environment whether through adding pores to the membrane to create an ATP reactive cell if it detects surrounding ATPs, if not it would stay dormant.
3.Experimental details
a.List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
inaK: protein derived from Pseudomonas syringae, target gene
T7 RNA polymerase : transcription (if the PURE system doesn’t already contain a protein, the gene is used to produce polymerase to transcribe inaK)
secY, secE, secG : translocation genes
POPE: lipid building blocks
POPG : lipid building block
70S ribosomes: translation
20 amino acids: translation
tRNA: translation
SRP: connects inaK to membrane
hlyA : communication gene (used if wanted to form pores in membrane)
Used Gemini to help me organize my information.
Reference List
Cui, Y. et al. (2022) ‘Cell-free PURE system: evolution and achievements’, Biodesign Research. (For the PURE components).
Noireaux, V. and Libchaber, A. (2004) ‘A vesicle bioreactor as a step toward an artificial cell’, PNAS. (For the POPE/POPG and $\alpha$HL pore strategy).
Li, Q. et al. (2012) ‘Characterization of the ice nucleation protein InaK’, Journal of Biological Chemistry. (For the inaK gene details).
b.How will you measure the function of your system?
I could try protein localization through fluorescence protease protection assay using fluorescent tags at the inaK terminals.
I could measure the freezing efficiency once again through a droplet trial, testing droplets of multiple versions of the solution when they freeze at what temperature and how resistant they are to temperature.
Homework question from Peter Nguyen
1.Write a one-sentence summary pitch sentence describing your concept.
I could use cell free systems adapted to producing inaK in order to directly inoculate glaciers with the aim to preserve and boost ice formation, it would help glaciers rebuild and be more resistant to increasing temperatures caused by climate change. However, I would be interested in pushing the idea of geotextiles already helping preserve glaciers and design a living material, with inaK with a boosted ice nucleation function to create proactive glacier covering actively working to rebuild and preserve glacier ice.
2.How will the idea work, in more detail? Write 3-4 sentences or more.
The inaK would be synthesized through a cell free model, using an alternative to E. coli which could resist and be active in sub-zero conditions (to stay active in glaciers), such as Oleispira antarctica a psychrophiles bacteria which has evolved to have specialized ribosomes and enzymes able to remain flexible and functional in a frozen environment. Oleispira antarctica contains unique chaperone (Cpn60 and Cpn10) preventing protein misfolds in frozen temperatures. This cell design would include pores in the membrane so it can stay alive in the textile by having an ATP source of input.
These then freeze dried cells would be put into a textile (inoculated during the making of the textile), the textile can be brought to location and installed on the glacier and then be rehydrated to allow ice nucleation of the glacier to begin. This would permit me to create a live material that would be dormant in production and transportation and control its freezing function (preventing the textile from accidentally freezing its surrounding).
Note that because I am working with ice nucleation there might be challenges in freeze drying these cells.
Reference List
Ferrer, M. et al. (2003) ‘Low temperature-induced systems failure in Escherichia coli: Insights from rescue by cold-adapted chaperones’, Journal of Biological Chemistry.
Cui, Y. et al. (2022) ‘Cell-free PURE system: evolution and achievements’, Biodesign Research.
D’Amico, S. et al. (2006) ‘Psychrophilic microorganisms: challenges for life’, EMBO reports.
3.What societal challenge or market need will this address?
This addresses the environmental, social and political issue of melting glaciers caused by climate change, which only increases the power of climate change as glaciers are key factors in slowing climate change. We are actively losing biodiversities and ecosystems and doing very little about it. It is not seen as a profitable income so little motivation is inputted. However, in the longterm, this irreversible damage done to our nature will actively make climate change worse, and there will be many destructive environmental, social and economic consequences driven by this overlooked issue.
4.How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Working at very large scale, scale of the glacier, it can be a challenge to efficiently rehydrate the living material as it would be very energy consuming and costly to do it manually, but, if the living geotextile is strategically implemented at the right time of the year (early spring, already when they glacier coverings are usually installed) then nature itself through rain could activate the material naturally. The aim is to limit the human labor impact and simply give nature a tool to reinforce what it already knows how to do.
Considering the one time use issue, geotextile coverings which are already used to protect glaciers are removed and installed yearly according to their natural ice melting and forming cycles. The next step of my research would be to find a way to keep the textile created and reabsorb it with new inaK cell free protein systems when it is needed next. The goal is to create a regenerative textile and closed loop system to avoid waste through one time solutions.
Homework question from Ally Huang
1.Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
I am interested in exploring the purpose of ice nucleation in cell free design, freeze dried or not, taking the shape of a multipurpose textile which can be used as an alternative to current voluminous refrigeration tools or a freezing textile to activate. In space stations like the ISS a lot of research relies on lab samples being preserved in sub zero temperatures, from human research samples to organisms or protein crystals. Within research some experiments need cold induced phase changes to be activated or triggered. This technology could also be used for food or medical supplies.
2.Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
The genetic target of this project would be the inaK ice nucleating protein, commonly found in Pseudomonas syringae, with a wide potential of freezing functions.
3.Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
The challenge is optimizing cold packaging and storage systems, a material which would require less space or a material able to be activated once in space again having less constraints in terms of space while travelling. The inaK offers a variety of possibilities in a cell free system whether freeze dried or not as it has a focused and controlled function to freeze. According to the development of the product it can be chosen at what temperature it freezes or activates and how resistant it can be to external temperatures. Creating a highly controlled and bespoke design for certain use in space allows for better control on the research done in space, every aspect of the research can be tailored in hopes to improve success rates of experiments. InaK is a relatively easy INP to work with.
4.Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
I am interested in creating polyvalent designs with multiple usages and applications, this project aims to find an optimal alternative refrigerating system which can have bespoke qualities specific to in space research. As small of a detail it might seem every aspect and tool of experiments impacts the result of research and can lead to better efficiency, results or unexpected breakthroughs. During a space mission all equipment has to be optimized due to lack of space and need for many items and a polyvalent tool that can respond to a wide range of uses can help with the space optimization.
5.Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
I would design a cell free system for the inaK ice nucleating protein, freeze dry some and then create living textiles, some active and some dormant. The practicality of a textile is that it can be molded, cut, sewn, layered to adapt to any existing object which would then need a freezing function.
I can control the amount of inaK for the freezing rate needed, experiment with the different temperatures it can freeze at and the different temperatures it can stay frozen at, I can explore the threshold of the inaK.
I would then test the reactivation rates, how much water is needed and how long it would take.
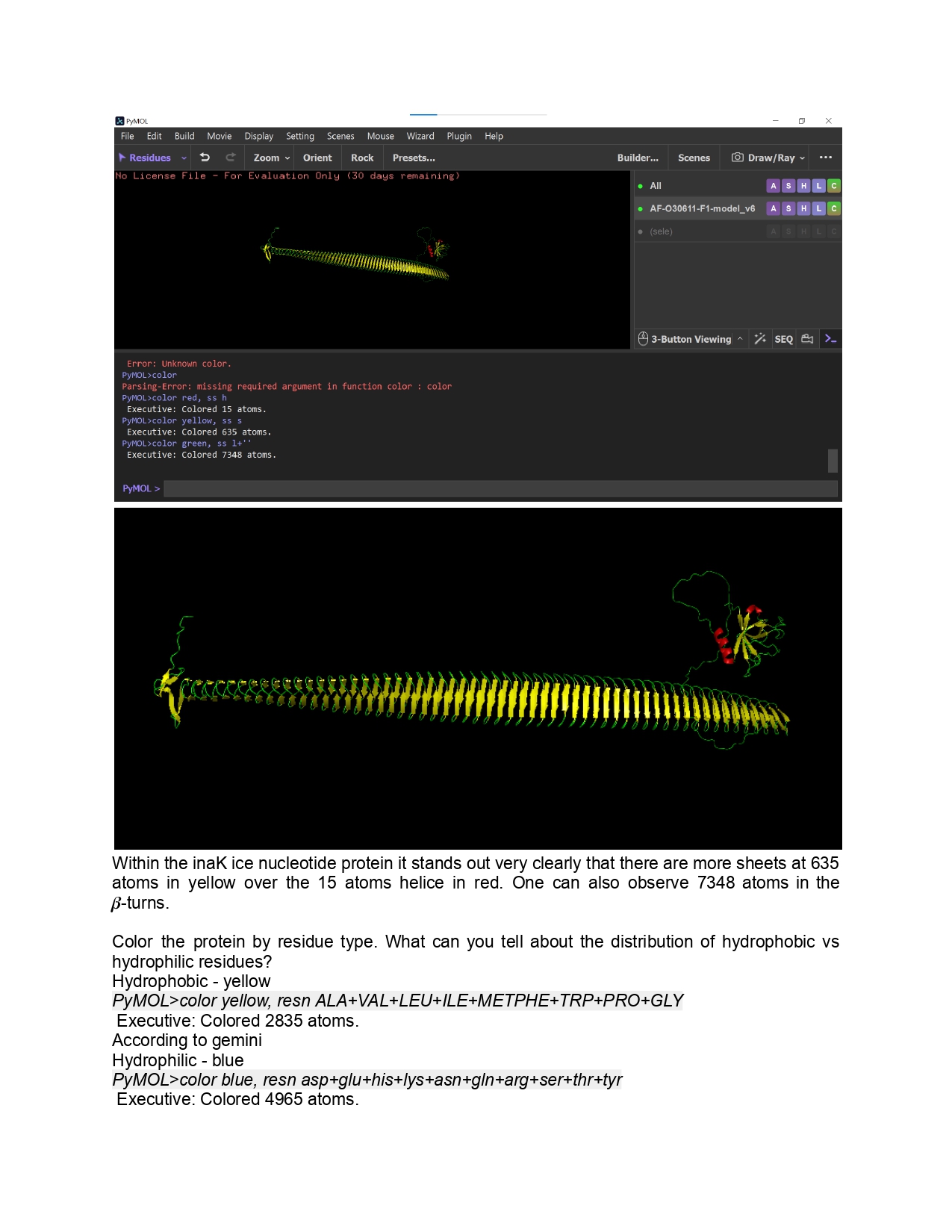
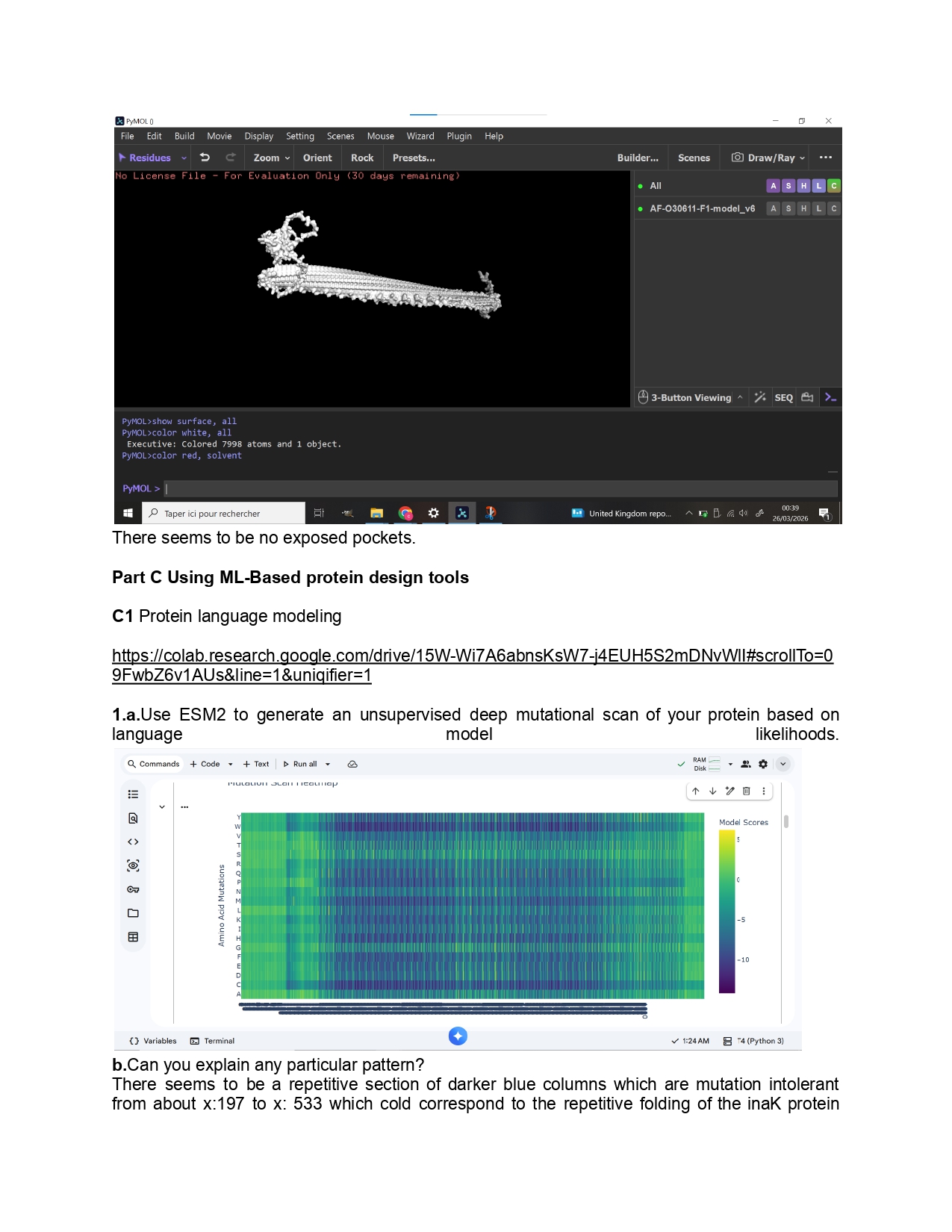
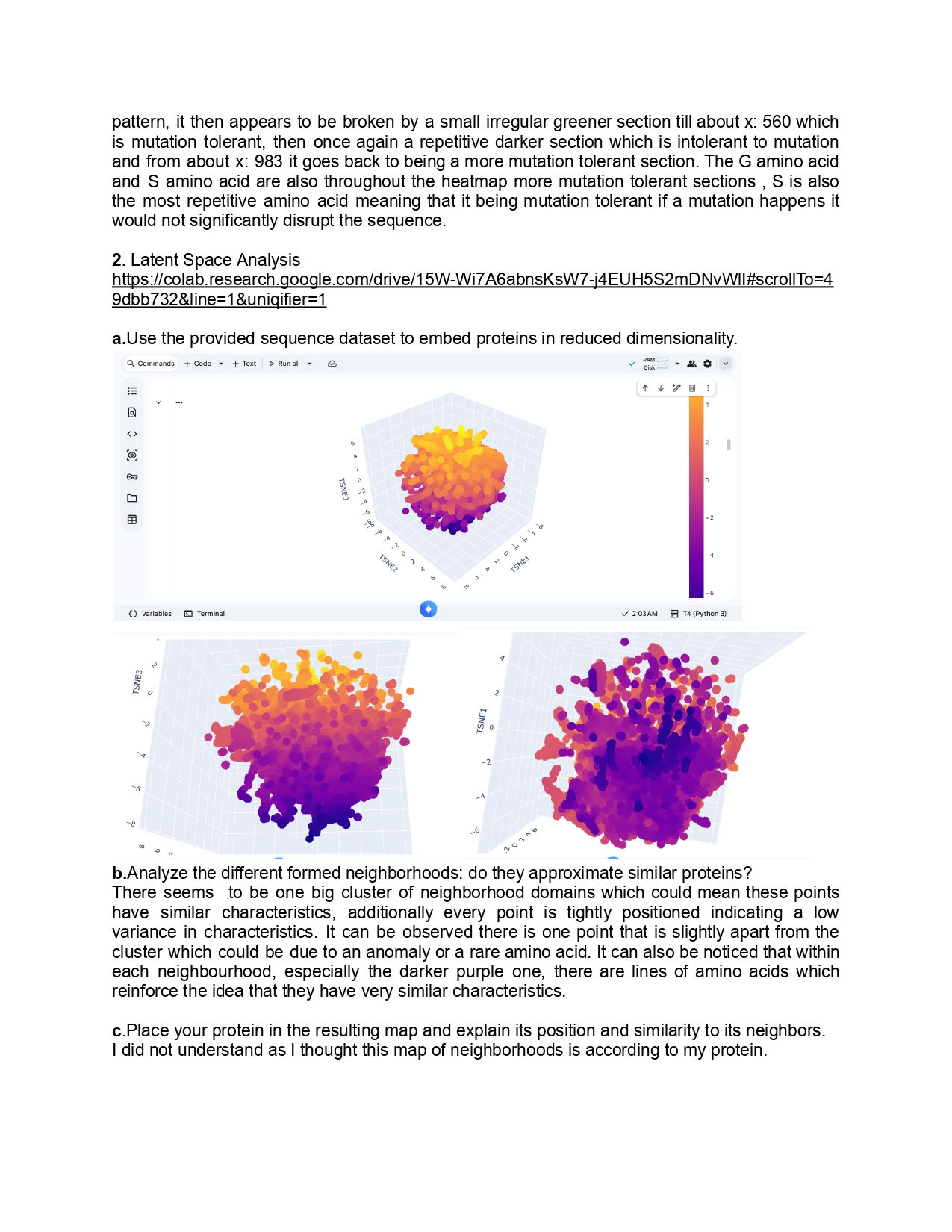
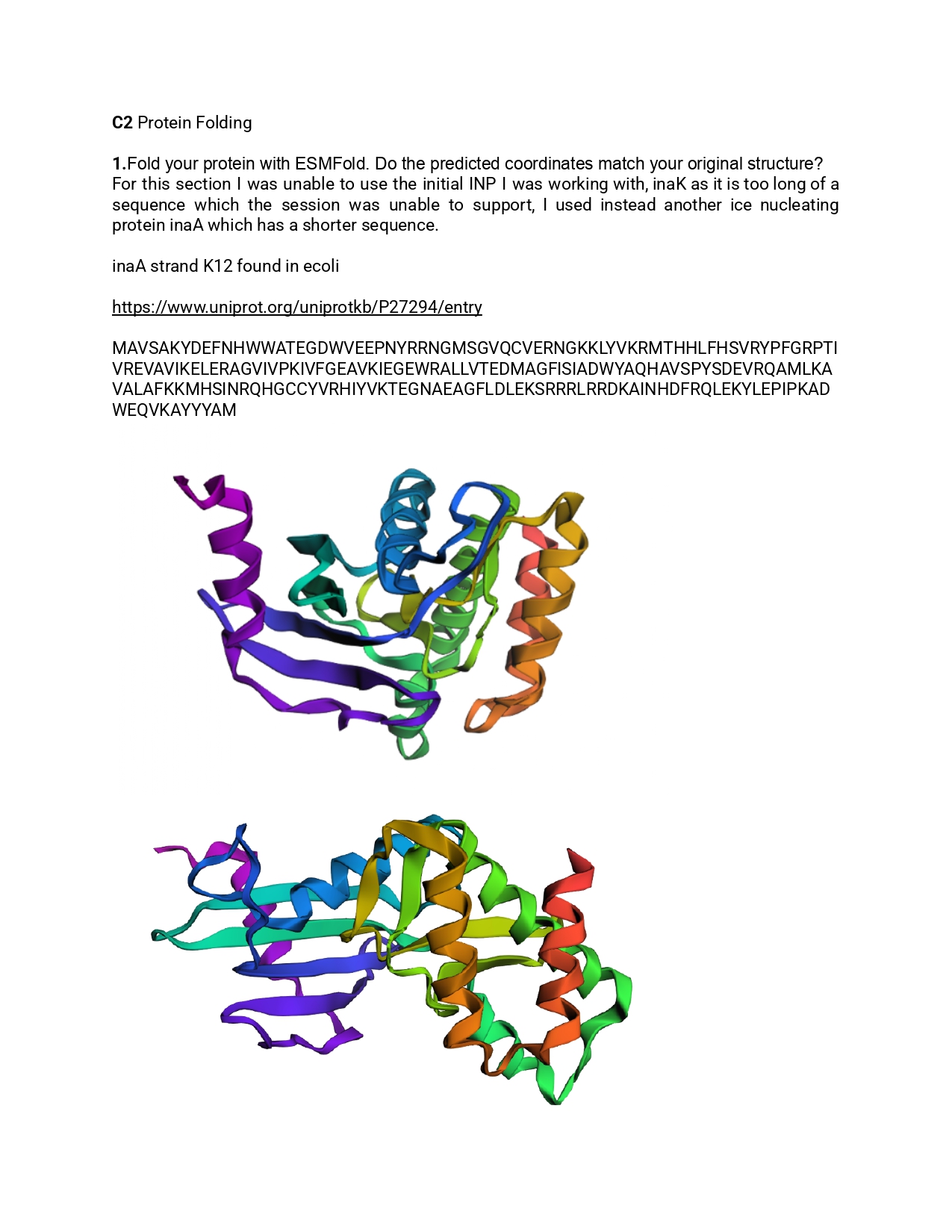
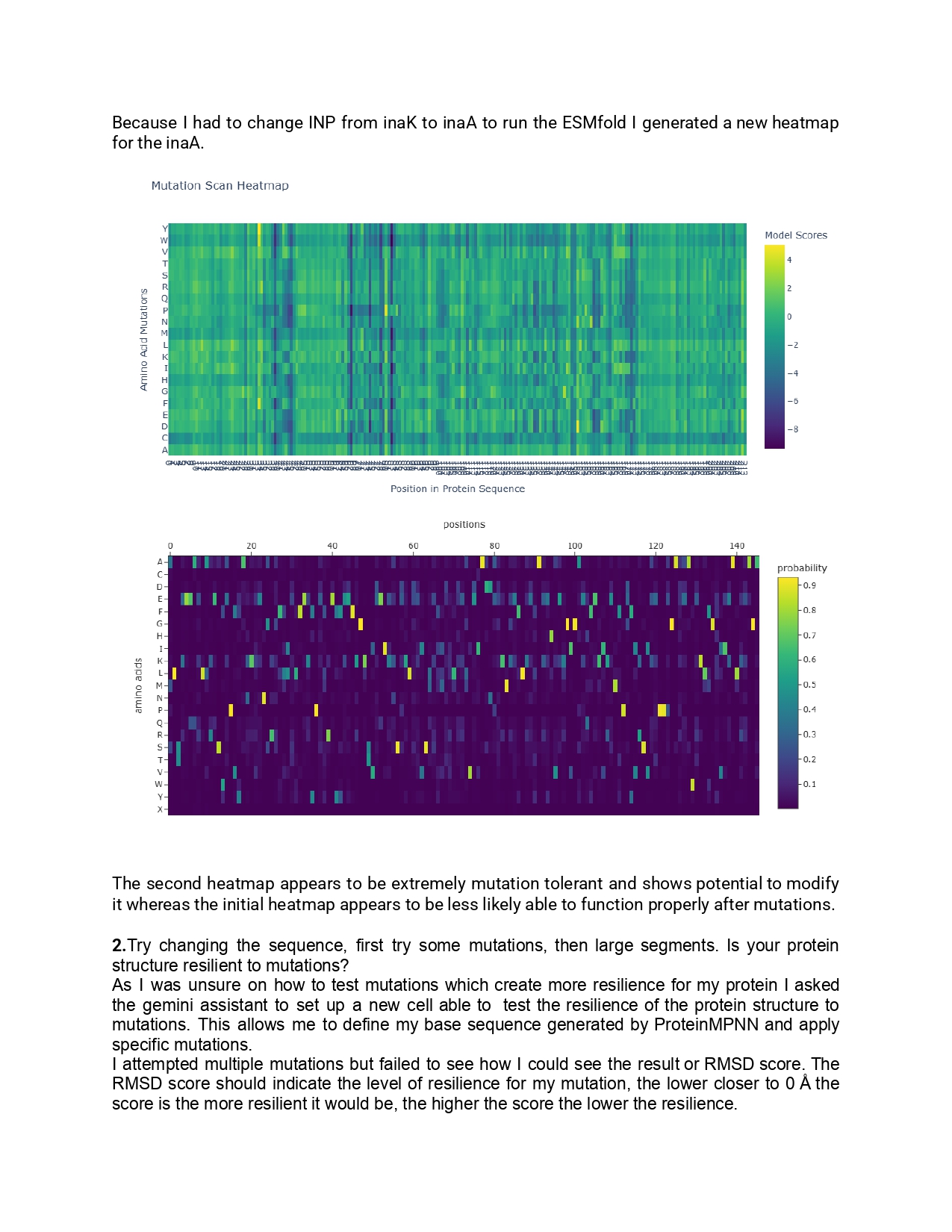
1.Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
As for this project I aim to use inaK for ice production I would like to measure the ice nucleation ratio and efficiency of the inaK protein. Additionally, I would like to measure the temperatures inaK can resist to, on its own and as a supplement to an ice sample.
If my initial experiments are successful I would like to measure the inaK ratio innoculated into ice to find the most optimal inaK quantity needed.
2.Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
To measure the ice production ratio and efficiency of inaK I could use differential scanning calorimetry (DSC) which measures the difference in the amount of heat required to increase the temperature in the sample compared to a reference. It measures the ice nucleation ratio by calculating the enthalpy which is the area below the peak, allowing me to understand precisely how much of the water in the cell is being converted to ice. It measures the efficiency of ice nucleation by creating an exothermic peak ( release of a burst of energy) and analysing how high of a temperature the inaK can still function. This should give me information on thermodynamic efficiency.
I can also measure nucleation temperature through a droplet freezing assay for smaller samples allowing me to test a multitude of potential solution mixes. Here a high speed camera paired with a cooling plate ( a Linkam for example) can allow me to assess how fast a droplet of a solution containing inaK can freeze. Testing this on multiple samples containing different amounts of inaK will give me a spectrum of freezing capacity to find the most optimal ratio of inaK. This experiment could be coupled with an infrared thermography technology which will capture the heat spike and nucleation rate of an inaK and understand how fast the ice nucleation spreads through the cell membrane.
Reference List
Schmid, D. et al. (2016) ‘A high-throughput assay for the characterization of ice-nucleating proteins’, Biophysical Journal. This study outlines the specific use of droplet assays to quantify InaK efficiency.
3.What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
For these experiments I will use a Differential Scanning Calorimeter for the DSC, a high precision camera, a cooling plate and IR thermography.
Waters Part 1 - Molecular Weight
For this section I used a combination of the tools provided, my knowledge and AI assistance as I have trouble understanding math related work.
Based on the predicted amino acid sequence of eGFP and any known modifications, what is the calculated molecular weight ?
According to Expasy I found that this sequence has a theoretical pI/Mw of 5.90 / 28006.60.
To calculate the molecular weight of this sequence I referred to the standard isotopic mass of amino acids and subtracting H²O for each peptide bond. For this amino acid sequence I found that :
-eGFP of 238 AA = 26.735.6 Da
-LE Linker of 2 AA = 242.3 Da
-x-His Tag of 6 AA = 822.8 Da
Resulting in a total molecular weight of 27800.7 Da
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (figure 1) and:
MW : molecular weight in Daltons
m/z : value of the peak on the x-axis of the spectrum
z : integer charge state of said peak
H+ : mass of a proton
a.Determine z for each adjacent pair of peaks $(n, n+1)$ using: $$ {\large z} = {\Large \frac{\frac{m}{z_{n+1}}}{\frac{m}{z_n} - \frac{m}{z_{n+1}}}} $$
Here I am using the adjacent peaks at 875.4421 and 903.7148.
m:zn = 875.4421
m:zn+1 = 903.7148
The lower the m/z the higher the charge versus the higher the m/z the lower the charge.
Using the provided formula
z = 903.7148 : (903.7148 – 875.4421) = 903.7148 : 28.2727 = 31.96
Charge states must be integers and the charge state for the peak at 903.7148 is z= 31
So, the peak at 875.4421 has a charge state of z+1=32
b.Determine the MW of the protein using the relationship between $\frac{m}{z_n}$, $MW$, and $z$
The base equation for the peaks in this figure is
m:z = (MW + (z x H+)) : z
Here I rearrange the formula to find MW
MW = z x (m:z) - (z x H+)
If z=31, then,
MW = 31 x(903.7148) - (31x1.008)
MW = 28 015.1588 – 31.248
MW = 27 983.91 Da
c.Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using: $$ \text{Accuracy} = \frac{|MW_{\text{experiment}} - MW_{\text{theory}}|}{MW_{\text{theory}}} $$
Using the provided formula and substituting the values accordingly,
If,
Accuracy = (MW experimental - MWtheory) : MW theory
Then,
Accuracy = (27 982.90 - 27 782.70) : 27 782.70
Accuracy = 200.20 : 27 782.70
Accuracy = 0.0072
=0.72%
3.Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
One can observe the charge state in the zoomed in peak but not using the previous calculation method using the adjacent peak method used for the entire spectrum.
The zoomed in area presents an isotopic cluster of one charge state, therefore, the observation is focused on isotopic resolution. The other peaks represent different amounts of protons.
So, here, yes I can see the charge state using isotopic resolution because the peaks are distinct and separate and the instrument has high enough of a resolution to expose the isotopes separately. It is because the instrument used here is of high precision that we are able to have high resolution, if a lesser precise instrument were used then it would be very difficult to see the charge state.
The formula to calculate isotopic spacing here would be
z = 1 : Δm/z
Waters Part 2 - secondary & tertiary structure
1.Please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (figure 2)?
There is a structural difference between the native and the denatured protein conformations, the native protein conformation is a tightly folded protein, a unique three dimensional structure unique to its biological environment. The structure is compact but is held by weak non-covalent bonds. In contrast, a denatured protein conformation is the unfolded structure once the weak bonds are broken, the structure is flexible and unpredictable resulting in a random coil shape. The denatured protein no longer has a function compared to the native protein conformation.
A mass spectrometer scans the protein shape by measuring its mass and charge rather than measuring the shape directly.
The top green graph corresponds to the denatured confirmation and the bottom red chart corresponds to the native conformation.
The denatured conformation graph shows crowded peaks in the 500 to 1200 m/z range, these are low m/z values. A lower m/z equals a higher z charge and when a protein is unfolded into a random coil then the basic residues are exposed to a proton rich environment and can easily be protonated creating a protein with a very high net positive charge.
The native conformation has larger m/z values with peaks at 2545 and 2799, the higher the m/z value the lower the z charge. When the protein is still tightly folded the basic residues are buried in the hydrophobic core protecting it from its environment meaning protons cannot impact them so the protein is less protonated.
2.Zooming into the native mass spectrum of the eGFP from the Waters Xevo G3 QTof or MS (figure 3), can you discern the charge state of the peak at ~2800? What is the charge state? How can you tell?
It is possible to discern the charge state of the peak at ~2800 m/z because instrument provides a high resolution even if the image focuses on the 2525 m/z peak, we are still able to see the isotopic distribution by measuring the distance between the individual isotopes and we would be able to calculate the charge state.
Measuring the charge state of the peak at ~2800 m/z using isotopic spacing,
In a mass spectrum the individual isotopes of the same molecule differs by approximately 1 Dalton,
The distance between the isotopes here on the x-axis Δm/z can be calculated using the following formula,
Δm/z = 1: z
Zooming in at the 2799.4199 peak same as for the 2545 peak (with a peak separation of precisely 0.1 m/z) then,
z = 1: 0.1 = 10
So the charge state of the peak at ~2800 m/z is +10
Waters Part 3 - Peptide mapping, primary structure
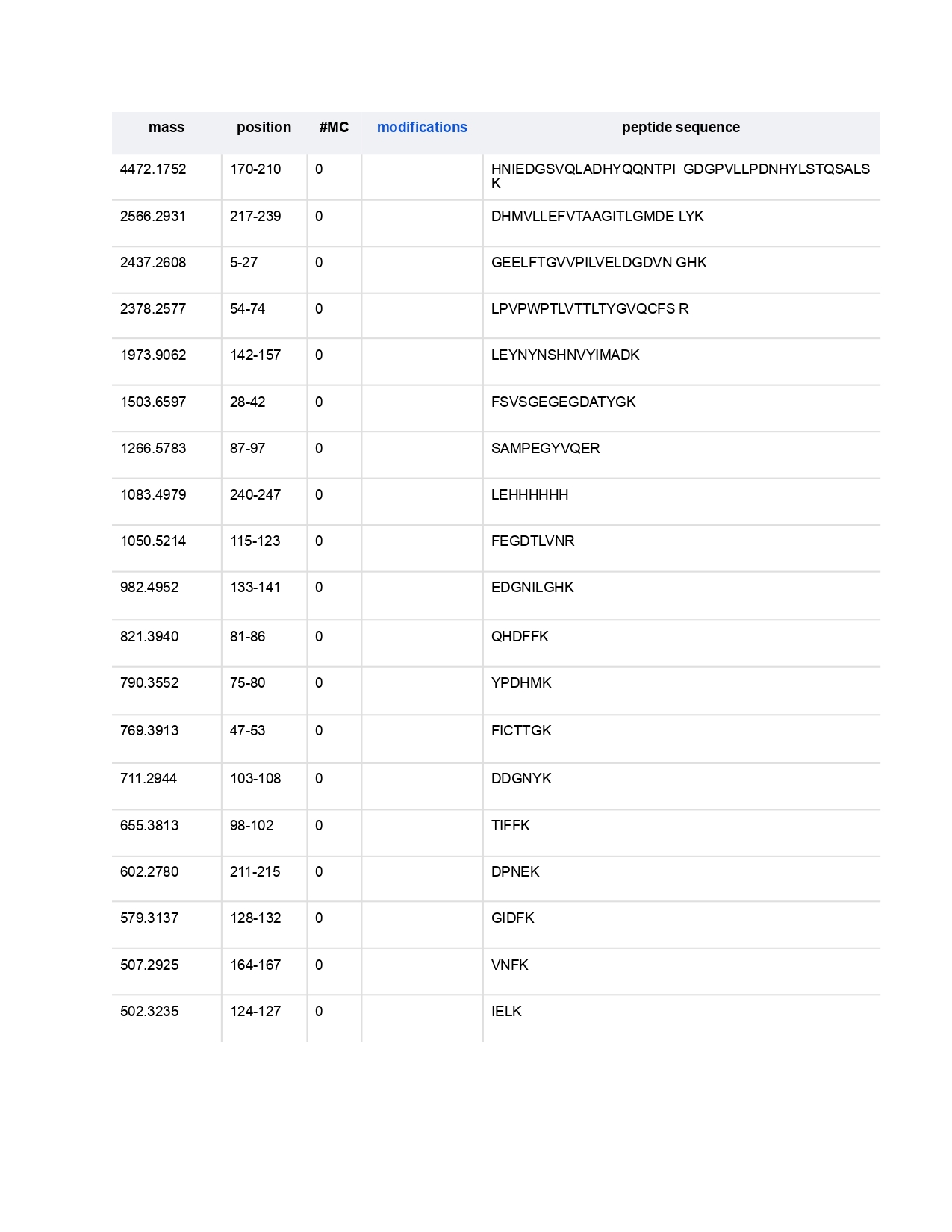
1.How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking the biochemical properties tab will show you a count for each amino acid).
2.How many peptides will be generated from tryptic digestion of eGFP?
I found 19 peptides were generated from this sequence using the trypsin.
3.Based on the LC-MS data for the Peptide Map data generated in the lab ( please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are>10% relative abundance.
Between 0.5 and 6 minutes there are 14 distinct peaks above 10% relative abundance in this figure.
4.Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
In comparison to the amount of predicted peptides, 19, there are fewer peptides in the chromatogram.
5.Identify the mass-to-charge (m:z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H]+) based on its m:z and z.
The z charge of the most abundant peak in this peptide is m/z = 525.76712
To calculate the charge state,
First isotope peak 525.76712
Second isotope peak 526.25918
Calculating the spacing,
(Δm/z):526.25918 - 525.76712 = 0.49206
Using the formula,
z = 1: 0.49206≈2.03
So the charge state z of the most abundant peak of the peptide is of +2
Calculating the mass of the singly charged form of the peptide ([M+H]+),
First I need to calculate the neutral mass M using M=zx(m/z)-(zx1.00727),
1.00727 Da is the mass of a proton,
So,
M = 2 x (525.76712) - (2x1.00727)
M = 1051.53424 - 2.01454 = 1049.5197 Da
([M+H]+) can be calculated by adding one proton mass back to the neutral mass,
[M+H]+ = 1049.5197 + 1.00727 = 1050.5270 Da
6.Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm. (Recall that Accuracy = (MW experimental - MWtheory) : MW theory)
To identify the peptide I consider that the experimental neutral molecular sight (MWexperimental) was calculated for the peak 525.76712 m/z and equaled MW: 1049.5197 Da, I will use the following peptide sequence as a theoretical tryptic digest as a comparison, LPDNHYLSTQSALSK, and considering theoretical MW (MWtheory) : 1049.5393 Da. This peptide corresponds to the residues 139–153 of the eGFP protein.
To calculate the mass accuracy, error in ppm (parts per million) I will use the following formula,
Accuracy (ppm) = ((MWexperimental - MW theory) : MWtheory) x 106
Now adding the values,
MW experimental = 1049.5197
MW theory = 1049.5393
Accuracy (ppm) = ((1049.5197 - 1049.5393) : 1049.5393) x 106
Accuracy (ppm) = (-0.0196) : 1049.5393) x 106
Accuracy (ppm) = –18.67ppm
7.What is the percentage of the sequence that is confirmed by peptide mapping ? (see figure 6)
The percentage of the sequence that is confirmed by peptide mapping seems to be indicated at 88%, the blue highlighted areas are confirmed amino acids in the sequence.
Waters Part 4 - Oligomers
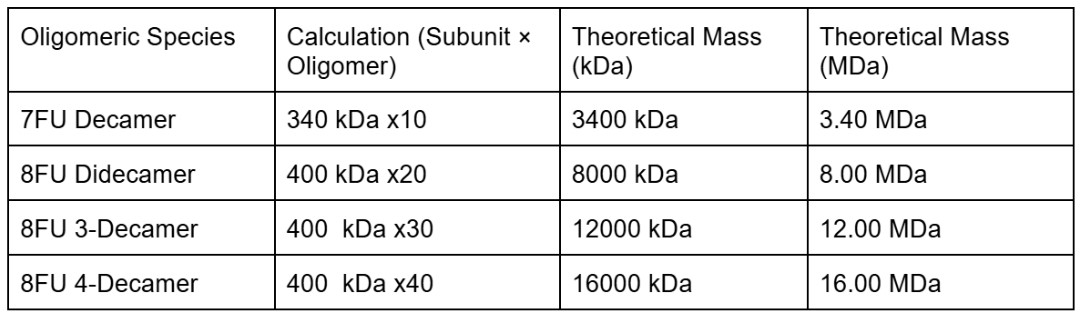
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
-7FU Decamer
-8FU Didecamer
-8FU 3-Decamer
-8FU 4-Decamer
I will first calculate the theoretical mass for the species using the given measures 7FU = 340 kDa and 8FU = 400 kDa. The axis of the spectrum is in MDa (megadaltons) where 1MDa = 1000 kDa.
I can then identify the peaks on the spectrum
-7FU Decamer ≈ 3.40 MDa is the peak labeled 3.4 on the spectrum
-8FU Didecamer ≈ 8.00 MDa correspond to the peak labeled 8.33
-8FU 3-Decamer ≈ 12.00 MDa matches the peak labeled 12.67
-8FU 4-Decamer ≈ 16.00 MDa would appear in the cluster of small unlabeled peaks around 16 to 17 MDa on the far right side of the chart
Waters part 5 - Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Week 11 HW: Building genomes
Week 11 Bioproduction & Cloud Lab
Part A - The 1.536 pixel art work canvas, collective artwork
1.Contribute at least one pixel to the global artwork
I added early on a pixel towards the top left corner. I do not have much to say about this section of the work except maybe understanding the full purpose of this exercise.
Part B - Cell Free protein synthesis, cell free reagents
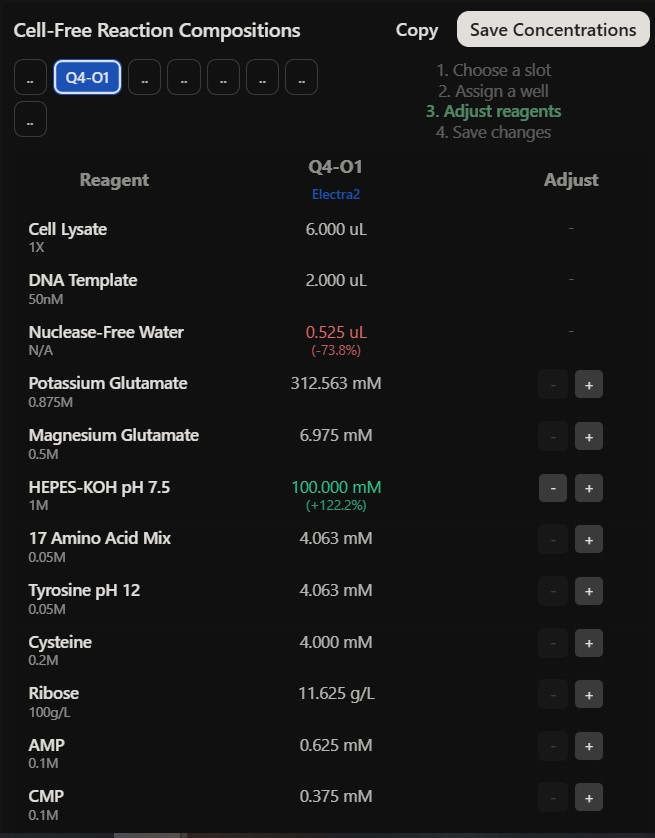
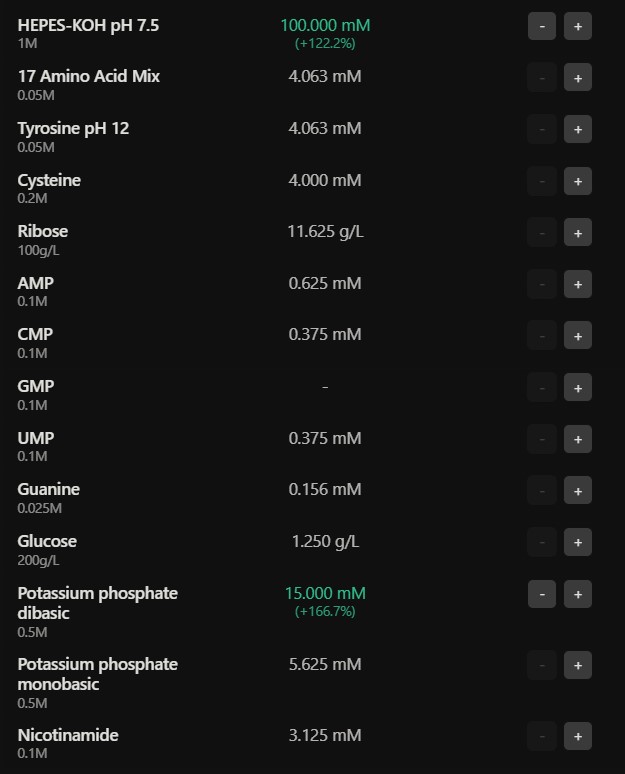
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase) : offers the base molecular machinery such as ribosomes, tRNAs and enzymes for translation, the Star Lysate strain reduces mRNA degradation, and, the T7 Polymerase drives high level transcription from T7 promoters
Salts / Buffer
Potassium Glutamate : primary salt that maintains ionic strength and provides potassium ions essential to ribosomal function and protein to nucleic acid exchange
HEPES-KOH pH 7.5 : chemical buffer which helps maintain a stable physiological pH which affects enzymatic function of the transcription and translation machinery
Magnesium Glutamate : magnesium ions are vital contributors to stabilizing the ribosome structure and enabling catalytic activity of the polymerases kinases
Potassium phosphate, monobasic and dibasic : functions as a secondary pH buffer and a source of inorganic phosphate essential for the regeneration of high energy molecules such as ATP
Energy / Nucleotide system
Ribose : serve as a carbon backbone precursor for the synthesis of nucleotides, allowing for regeneration of NTPs essential for transcription and energy transfer
Glucose : primary metabolic energy source fueled through glycolysis allowing to regenerate the ATP and GTP essential to the good functioning of protein synthesis
AMP / CMP / UMP : offers nucleotide building blocks for RNA synthesis and can be converted into triphosphate such as ATP, CTP, UTP needed in transcription
GMP : from the lack of GMP might demonstrate a dependency on salvage pathways to generate GTP essential to translation
Guanine : precursor for GMP/GTP synthesis through salvage pathways helpful to RNA synthesis and ribosomal function
Translation Mix (Amino acids)
17 Amino Acid Mix : provide the base building blocks to synthesize the polypeptide chain
Tyrosine : supplied separately because of its solubility limitations, becomes an essential building block for protein synthesis once it is adapted into a usable form
Cysteine : added separately due to its oxidation limitations, it is an essential compound in forming disulfide bonds in proteins
Additives
Nicotinamide : serves as a precursor for NAD+ / NADH synthesis reinforcing redox balance and metabolic reactions occurring in energy regeneration
Backfill
Nuclease Free Water : is used to adjust all the components to the desired the final reaction volume while it avoids degradation of DNA / RNA by nuclease and ensures stable transcription and translation processes
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the slide.
The main difference between the two master mix results in found in the energy and nucleotide sourcing a the 1-hour mix makes use of the PEP and pre-synthesized NTPs for instant and high burst protein synthesis compared to the 20-hour mix uses the ribose, glucose and NMPs as precursors to regenerate energy and nucleotides throughout time. Therefore, the 1-hour mix is designed for speed and rapid prototyping in contrast to the 20-hour mix allows to better optimize the cost for effectiveness by using the Lysate’s metabolic pathway to support the reaction for an extended period of time.
Part C - Planning the global experiment, cell-free master mix design
1.Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP : provides robust and rapid protein folding therefore the protein is less likely to aggregate enabling it to offer a strong fluorescent readout even if fused to complex proteins
mRFP1 : is a protein with a longer maturation time signifying the fluorescence develops slower after translation and might have a delayed signaling time in shorter experiments, it has a low acidity tolerance
mKO2 : is fast maturing and has a relative acidity tolerance, meaning the fluorescence will be less visible in a lower pH context but the fluorescence could increase in longer cell free reactions
mTurqoise2 : is a cyan protein with for a high quantum yield and high photostability making the fluorescence outread a great signal no matter the length of the reaction, it is very sensitive to pH
mScarlet_I : engineered for fast maturing and high brightness allowing for stronger fluorescence signals compared to older red proteins
Electra2 : protein engineered for very fast maturation making it very useful for fast consuming energy systems in an experiment where a rapid output is needed before the mix’s energy is used up
2.Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Could the mTurquoise2 yield be increased or accelerated through pH stabilisation.
The reagents HEPES-KOH would be increased to 100mM and Potassium Phosphate would be increased to 15mM.
This adjustment should increase the capacity of the buffer within the mater mix and should neutralize organic acid by products such as lactate and acetate generated during the 36-hour metabolism of glucose and ribosome. Because mTurquoise2 is very reactive to pH, preserving the pH environment at 7.5 would prevent the typically occurring rapid cooling of the cyan signal which usually occurs as the mix acidifies over time. Therefore, the high quantum of yield of mTurquoise2 is complete and optimized leading to a bright and stable cyan readout which won’t dim as the energy levels decrease.
3.The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
)
)
)
)
)
Copy of reagent doses
Reagent Preset:
Variant 1 Variant 2 (O1) Variant 3 Variant 4 Variant 5 Variant 6 Variant 7 Variant 8
Cell Lysate 6.000 uL 6.000 uL (-) 6.000 uL (-) 6.000 uL (-) 6.000 uL (-) 6.000 uL (-) 6.000 uL (-) 6.000 uL (-) 6.000 uL (-)
DNA Template 0.000 mM 2.000 uL (-) 2.000 uL (-) 2.000 uL (-) 2.000 uL (-) 2.000 uL (-) 2.000 uL (-) 2.000 uL (-) 2.000 uL (-)
Nuclease-Free Water 2.000 uL 2.000 uL (-) 0.525 uL (-73.8%) 2.000 uL (-) 2.000 uL (-) 2.000 uL (-) 2.000 uL (-) 2.000 uL (-) 2.000 uL (-)
Potassium Glutamate 312.563 mM 312.563 mM (-) 312.563 mM (-) 312.563 mM (-) 312.563 mM (-) 312.563 mM (-) 312.563 mM (-) 312.563 mM (-) 312.563 mM (-)
Magnesium Glutamate 6.975 mM 6.975 mM (-) 6.975 mM (-) 6.975 mM (-) 6.975 mM (-) 6.975 mM (-) 6.975 mM (-) 6.975 mM (-) 6.975 mM (-)
HEPES-KOH pH 7.5 45.000 mM 45.000 mM (-) 100.000 mM (+122.2%) 45.000 mM (-) 45.000 mM (-) 45.000 mM (-) 45.000 mM (-) 45.000 mM (-) 45.000 mM (-)
17 Amino Acid Mix 4.063 mM 4.063 mM (-) 4.063 mM (-) 4.063 mM (-) 4.063 mM (-) 4.063 mM (-) 4.063 mM (-) 4.063 mM (-) 4.063 mM (-)
Tyrosine pH 12 4.063 mM 4.063 mM (-) 4.063 mM (-) 4.063 mM (-) 4.063 mM (-) 4.063 mM (-) 4.063 mM (-) 4.063 mM (-) 4.063 mM (-)
Cysteine 4.000 mM 4.000 mM (-) 4.000 mM (-) 4.000 mM (-) 4.000 mM (-) 4.000 mM (-) 4.000 mM (-) 4.000 mM (-) 4.000 mM (-)
Ribose 11.625 g/L 11.625 g/L (-) 11.625 g/L (-) 11.625 g/L (-) 11.625 g/L (-) 11.625 g/L (-) 11.625 g/L (-) 11.625 g/L (-) 11.625 g/L (-)
AMP 0.625 mM 0.625 mM (-) 0.625 mM (-) 0.625 mM (-) 0.625 mM (-) 0.625 mM (-) 0.625 mM (-) 0.625 mM (-) 0.625 mM (-)
CMP 0.375 mM 0.375 mM (-) 0.375 mM (-) 0.375 mM (-) 0.375 mM (-) 0.375 mM (-) 0.375 mM (-) 0.375 mM (-) 0.375 mM (-)
GMP - - (-) - (-) - (-) - (-) - (-) - (-) - (-) - (-)
UMP 0.375 mM 0.375 mM (-) 0.375 mM (-) 0.375 mM (-) 0.375 mM (-) 0.375 mM (-) 0.375 mM (-) 0.375 mM (-) 0.375 mM (-)
Guanine 0.156 mM 0.156 mM (-) 0.156 mM (-) 0.156 mM (-) 0.156 mM (-) 0.156 mM (-) 0.156 mM (-) 0.156 mM (-) 0.156 mM (-)
Glucose 1.250 g/L 1.250 g/L (-) 1.250 g/L (-) 1.250 g/L (-) 1.250 g/L (-) 1.250 g/L (-) 1.250 g/L (-) 1.250 g/L (-) 1.250 g/L (-)
Potassium phosphate dibasic 5.625 mM 5.625 mM (-) 15.000 mM (+166.7%) 5.625 mM (-) 5.625 mM (-) 5.625 mM (-) 5.625 mM (-) 5.625 mM (-) 5.625 mM (-)
Potassium phosphate monobasic 5.625 mM 5.625 mM (-) 5.625 mM (-) 5.625 mM (-) 5.625 mM (-) 5.625 mM (-) 5.625 mM (-) 5.625 mM (-) 5.625 mM (-)
Nicotinamide 3.125 mM 3.125 mM (-) 3.125 mM (-) 3.125 mM (-) 3.125 mM (-) 3.125 mM (-) 3.125 mM (-) 3.125 mM (-) 3.125 mM (-)
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total : 20 μL reaction