Week 2 HW: DNA Read, Write, & Edit
Week 2: DNA Read, Write, & Edit
Student: Constantin Convalexius
Course: HTGAA Spring 2026
Location: Vienna, Austria
Part 1: Benchling & In-silico Gel Art
Butterfly art 1

2nd picture: all enzymes
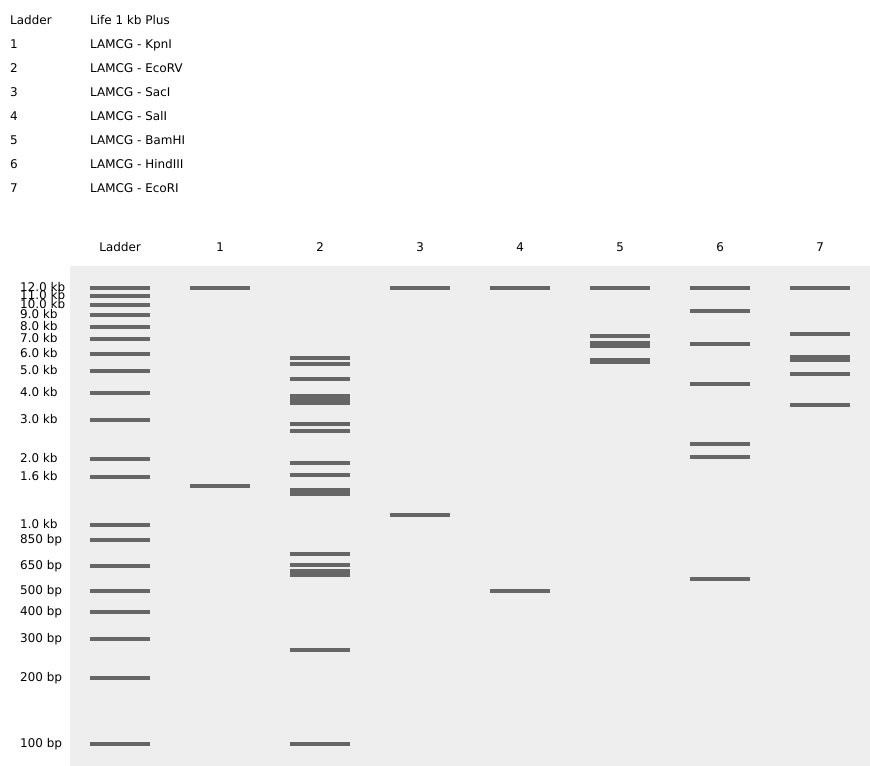
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
As a committed listener in Vienna without local wet-lab access, I completed the in-silico design and simulation sections.
Part 3: DNA Design Challenge
3.1 — Protein Choice: PD-L1 (Programmed Death-Ligand 1)
I chose PD-L1 (CD274, UniProt: Q9NZQ7) — the immune checkpoint protein that tumor cells use to hide from the immune system. PD-L1 sits on the surface of cancer cells and binds to PD-1 on T-cells, essentially telling them “don’t attack me.” Drugs like Pembrolizumab (Keytruda) block this interaction by targeting PD-1, so the immune system can recognize and destroy the tumor again. As a med student, this is one of the most exciting developments in oncology I’ve encountered so far.
The full-length PD-L1 protein is 290 amino acids and includes a signal peptide, extracellular domain, transmembrane region, and a short intracellular tail. For this exercise, I’m only using the extracellular domain (AA 19-238, 220 residues), since that’s the part that actually interacts with PD-1 and is the relevant domain for drug binding studies. This is also what researchers typically express recombinantly — you don’t need the transmembrane anchor if you just want to study the binding interface.
Protein sequence (extracellular domain):
3.2 — Reverse Translation
I used the Sequence Manipulation Suite (SMS2) reverse translation tool with “most likely codons” to convert the amino acid sequence into a DNA nucleotide sequence.
The output was an 870 bp sequence for the full-length 290 AA protein. One thing I noticed is that the SMS2 tool defaults to E. coli codon preferences — you can see this in the output, which uses codons like CGC for Arginine, GCG for Alanine, and CCG for Proline. These are all heavily biased toward bacterial tRNA pools, which wouldn’t work well in a human expression system.
This step is mainly useful to show the “raw” reverse translation before optimization, and to demonstrate why codon optimization is necessary.
3.3 — Codon Optimization
Since I want to express PD-L1 in human HEK293 cells (see 3.4), I ran the extracellular domain amino acid sequence through GenScript’s GenSmart Codon Optimization Tool with Homo sapiens as the host organism.
Results:
| Parameter | Value |
|---|---|
| Input | 227 AA (extracellular domain) |
| Output | 681 bp optimized DNA |
| GC content | 55.07% (ideal range: 30-70%) |
| Host organism | Homo sapiens (Human) |
Optimized DNA sequence:
The key difference compared to the raw SMS2 output is that GenSmart replaced the E. coli-preferred codons with those matching human tRNA abundance. For example, Arginine now uses AGG/AGA/CGG instead of bacterial CGC, and Alanine uses GCC/GCT instead of GCG. This is important because if the codons don’t match the host’s tRNA pool, the ribosome stalls during translation, leading to low protein yields or truncated products.
The GC content of 55.07% is also nicely within the ideal window — too high or too low GC content can cause issues with mRNA secondary structures or difficulties during DNA synthesis.
The codon-optimized sequence was generated using the GenSmart Codon Optimization Tool [1].
[1] Long Fan (2020, February 6). Codon optimization. (WO Patent WO 2020/024917 A1). Nanjing GenScript Biotech Co., Ltd.
3.4 — Production Technologies
Cell-dependent expression (primary approach): HEK293 cells
PD-L1 is a glycoprotein — it has N-linked glycosylation sites that are important for its folding and function. Because of this, I would express it in HEK293 human cells rather than E. coli. The workflow would be: clone the codon-optimized gene into a mammalian expression vector, transfect HEK293 cells, let them express and secrete the protein (since we’re only using the extracellular domain without the transmembrane anchor, it should be secreted into the culture medium), and then purify it using an affinity tag (like a His-tag with Ni-NTA chromatography). HEK293 cells are well-established for this — they handle human post-translational modifications properly and give reasonable yields.
Cell-free expression (alternative):
For quick small-scale testing (e.g., to check if the construct expresses at all before committing to a full cell culture run), you could use an in vitro transcription/translation system like rabbit reticulocyte lysate or wheat germ extract. These systems can produce protein in a few hours rather than days, but they don’t perform proper glycosylation, so the protein wouldn’t be fully functional. Still useful as a rapid validation step.
Part 4: Prepare a Twist DNA Synthesis Order
Here are my screenshots and files for Homework Part 4:
Upload sequence to Twist
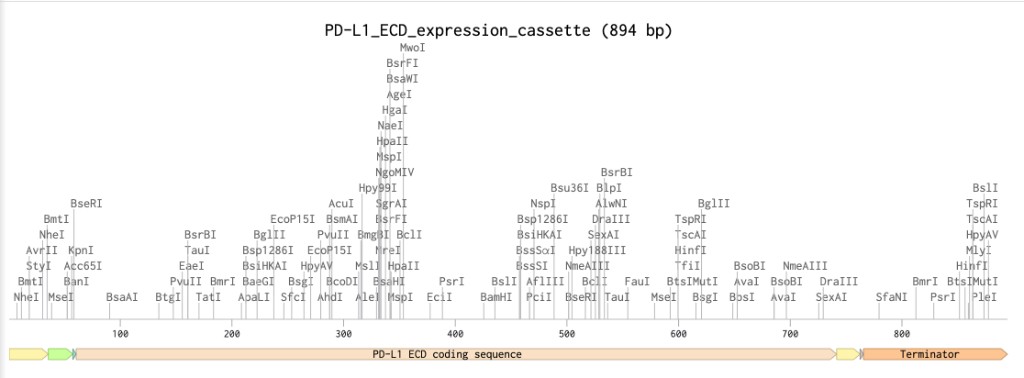
Benchling expression cassette map
Twist clonal gene order configuration
PDF export
PDF version prepared locally (not uploaded in this commit).
PDF update: plasmid map screenshot
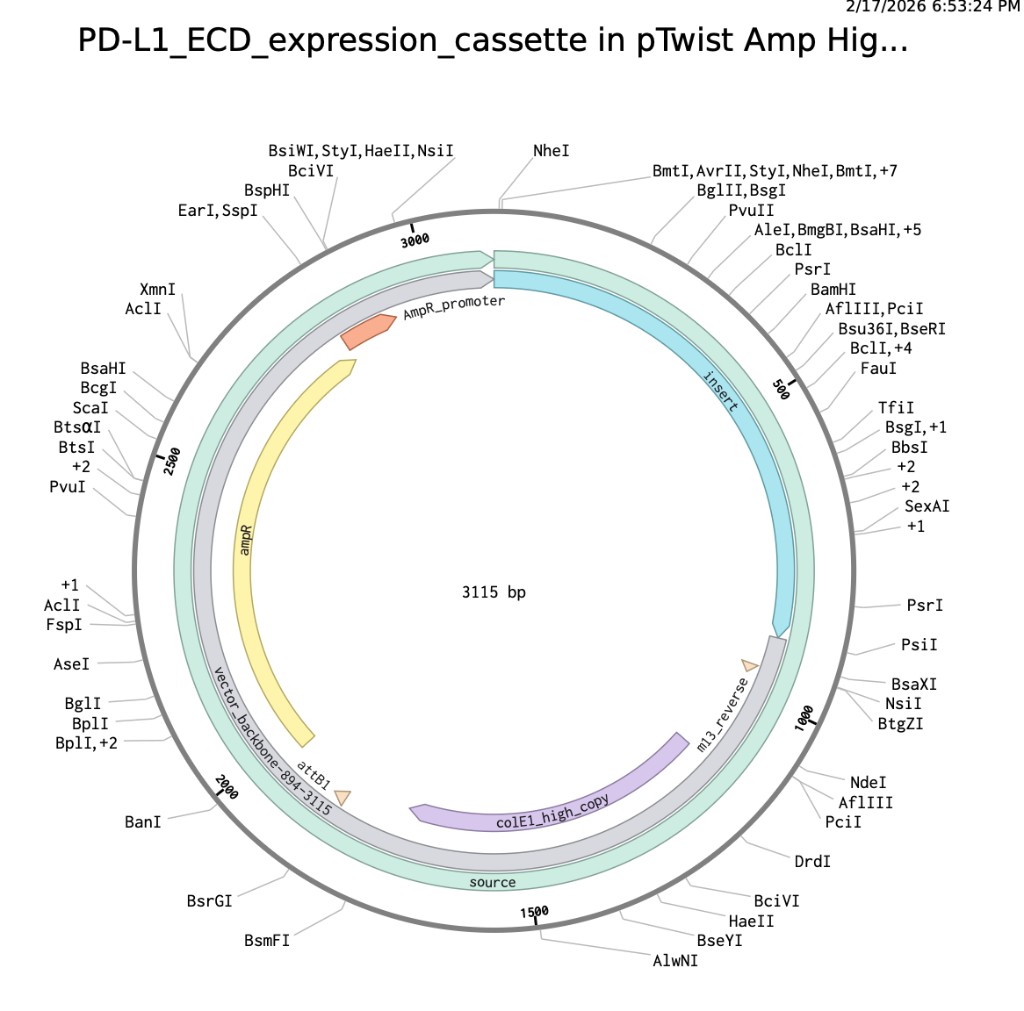
Part 5: DNA Read, Write, Edit
5.1 DNA Read
(i) What DNA would I want to sequence?
I’d want to sequence the genomes of supercentenarians — people who’ve made it past 110. These individuals somehow dodge or massively delay the diseases that kill most of us (heart disease, cancer, dementia), and there’s evidence that protective variants in genes like FOXO3, APOE, and TERT are enriched in their genomes. But we probably haven’t found everything yet. By doing whole-genome sequencing on large cohorts and comparing them to people who aged “normally,” we could uncover rare genetic variants that essentially act as nature’s longevity engineering. Pair that with DNA methylation data (which feeds into biological aging clocks like the Horvath clock) and you get a pretty complete picture of both the genetic hand they were dealt and how their gene expression shifted — or didn’t — over time.
(ii) Sequencing technology
I’d go with a hybrid approach: Oxford Nanopore (PromethION) for long reads plus Illumina NovaSeq for high-accuracy short reads.
Nanopore (third-generation): Sequences native, single DNA molecules in real time — no PCR amplification needed, which avoids amplification bias. A motor protein threads a DNA strand through a tiny biological pore in a membrane. Each base passing through disrupts the ionic current in a characteristic way, and a neural network translates those current patterns into sequence. Big advantage: it can also detect DNA methylation directly from the native strand, no bisulfite conversion needed. Reads are long (often >20 kb), which helps resolve structural variants and repetitive regions.
Input prep: Extract high-molecular-weight DNA from blood, ligate sequencing adapters directly — pretty minimal compared to short-read platforms.
Illumina (second-generation): Supplements Nanopore with very accurate short reads (~150 bp) for reliable SNP calling. Input prep involves fragmentation, adapter ligation, and bridge PCR. Bases are called by detecting fluorescent signals from reversible dye-terminators during synthesis-by-sequencing cycles.
Output: Both produce FASTQ files. Together they give you phased, chromosome-level assemblies with both structural resolution and single-nucleotide accuracy.
5.2 DNA Write
(i) What DNA would I want to synthesize?
I’d synthesize an engineered human telomerase (hTERT) expression cassette — a gene therapy construct to transiently reactivate telomerase in adult cells.
Telomere shortening is one of the core hallmarks of aging. Every cell division chips away at the protective chromosome caps until the cell senesces or dies. Telomerase rebuilds them, but it’s silenced in most adult tissues. Maria Blasco’s group at CNIO showed that AAV-delivered telomerase in mice extended lifespan without increasing cancer. The idea is to build a controllable human version.
The construct (~6-7 kb) would include a codon-optimized hTERT coding sequence under a Tet-On inducible promoter (so you can switch it on/off with doxycycline — you really don’t want constitutive telomerase, that’s a cancer risk), plus a GFP reporter to track which cells are expressing it. For Twist, I’d order this as overlapping clonal gene fragments.
(ii) Synthesis technology
Phosphoramidite oligo synthesis (Twist Bioscience’s platform) combined with Gibson Assembly.
Twist synthesizes thousands of short overlapping oligos (~60-200 nt) in parallel on silicon chips. Each oligo goes through cycles of deprotection -> coupling -> capping -> oxidation. These oligos get assembled into longer gene fragments (~1.8 kb) via overlap extension, then cloned into plasmids and sequence-verified. For my full ~7 kb construct, I’d order 3-4 fragments from Twist and stitch them together with Gibson Assembly.
Limitations: Coupling efficiency is 99-99.5% per step, so errors accumulate with length — that’s why you assemble from short oligos rather than synthesizing one long piece. Extreme GC content or repetitive sequences can cause synthesis failures. Turnaround is 2-3 weeks, and cost is around $0.07-0.09/bp ($500 for the full construct).
5.3 DNA Edit
(i) What DNA would I want to edit?
Three targets for a “longevity panel”:
- PCSK9 knockout: People with natural loss-of-function mutations in PCSK9 have very low LDL cholesterol and near-immunity to coronary heart disease — the #1 killer globally. A permanent gene edit would be a one-and-done solution. Verve Therapeutics is already running clinical trials on this.
- TP53 enhancement: Not a knockout — that would be terrible. Instead, introducing “super-p53” gain-of-function variants (studied in mouse models) that boost cancer surveillance without accelerating cellular senescence. The goal: decouple tumor protection from the aging program.
- Myostatin (MSTN) partial reduction: Myostatin inhibits muscle growth. Sarcopenia (age-related muscle wasting) is a huge driver of frailty in older adults. Reducing myostatin signaling could help maintain muscle mass well into old age — think Belgian Blue cattle, but a gentler, partial version for humans.
George Church has discussed similar multi-gene longevity editing in the context of GP-write.
(ii) Editing technology
For PCSK9: adenine base editing (ABE) via lipid nanoparticles (LNPs). A Cas9 nickase fused to a deaminase enzyme converts a single A·T base pair to G·C, introducing a premature stop codon in PCSK9 — no double-strand break needed. LNPs are delivered IV and preferentially target the liver (perfect for PCSK9). Verve’s primate data shows >60% editing efficiency.
For TP53 and MSTN: prime editing, which uses a Cas9 nickase fused to a reverse transcriptase guided by a pegRNA containing both the target sequence and the desired edit template. Even more precise than base editing — can make any small substitution without double-strand breaks or donor DNA.
Steps (base editing example): Design a guide RNA positioning the target adenine in the editing window -> formulate ABE mRNA + sgRNA in LNPs -> IV infusion -> LNPs enter hepatocytes via ApoE-mediated uptake -> base editor converts A to inosine (read as G) -> permanent single-nucleotide change.
Limitations: Off-target editing risk (lower than standard Cas9 but not zero — needs WGS validation). LNPs mostly hit the liver, which is great for PCSK9 but not for muscle or systemic edits — those need AAV or next-gen tissue-tropic delivery. Prime editing efficiency is still variable (~5-50%). And of course, these edits are permanent and irreversible, which is both the point and the risk.
AI Disclosure
I used Cursor and Claude to help with formatting, spelling/grammar clean-up, and publishing this website documentation.