Week 6 HW: DNA Nanostructures & Genetic Circuits
🧬 Week 6: DNA Nanostructures & Genetic Circuits
Part 1. DNA Assembly Questions
Question 1: Components of Phusion High-Fidelity PCR Master Mix
The Phusion High-Fidelity PCR Master Mix (NEB/Thermo Fisher) is a convenient 2X premix that requires only the addition of template DNA, primers, and water. It contains the following components:
| Component | Concentration (in 1X) | Purpose |
|---|---|---|
| Phusion DNA Polymerase | Proprietary | A chimeric enzyme fusing a Pyrococcus-like proofreading polymerase with a processivity-enhancing domain. It has an error rate ~50-fold lower than Taq polymerase and ~6-fold lower than Pfu polymerase, making it ideal for high-fidelity cloning where sequence accuracy is critical. It also has 5’→3’ polymerase and 3’→5’ exonuclease (proofreading) activities. |
| HF Buffer | 1X | Optimized salt and pH conditions for high-fidelity amplification. Contains Tris-HCl buffer, KCl, and (NH₄)₂SO₄ to maintain optimal ionic strength for polymerase activity and primer annealing specificity. A GC Buffer variant is also available for GC-rich templates. |
| dNTPs | 200 µM each | Deoxynucleoside triphosphates (dATP, dCTP, dGTP, dTTP) serve as the building blocks for new DNA strand synthesis. The polymerase incorporates them complementary to the template strand. |
| MgCl₂ | 1.5 mM | Magnesium ions are an essential cofactor for DNA polymerase catalytic activity. Mg²⁺ stabilizes the enzyme–DNA complex and is required for phosphodiester bond formation. Concentration can be optimized in 0.5 mM increments for difficult templates. |
Because it is a 2X master mix, the setup is simple: mix 12.5 µL of master mix with primers, template, and water to reach a 25 µL total reaction volume.
Question 2: Factors Determining Primer Annealing Temperature
The annealing temperature (Ta) is typically set 3–5°C below the melting temperature (Tm) of the primers. Several factors influence this:
| Factor | Effect on Ta |
|---|---|
| GC Content | G–C base pairs form 3 hydrogen bonds (vs. 2 for A–T), so higher GC content raises Tm and therefore Ta. Optimal GC content for primers is 40–60%. |
| Primer Length | Longer primers have more total hydrogen bonds and stacking interactions, increasing Tm. Most primers are 18–22 bp for the binding region. |
| Mismatch Positions | Internal mismatches (like those we intentionally introduce for mutagenesis in the chromophore region) destabilize the duplex and effectively lower the local Tm. |
| Salt / Mg²⁺ Concentration | Higher ionic strength stabilizes the primer–template duplex and raises Tm (~1°C per 10-fold increase in monovalent salt). |
| Primer Pair Matching | Forward and reverse primers should have Tm within 5°C of each other. If they differ too much, one primer dominates amplification, reducing yield. |
| Secondary Structures | Hairpins, self-dimers, and cross-dimers sequester primers and effectively reduce their availability. Stable secondary structures (Gibbs free energy below −10 kcal/mol) can severely reduce amplification. |
| 3’ GC Clamp | Having 1–2 G/C bases at the 3’ end stabilizes primer binding and promotes specific extension. However, more than 3 G/C’s in the last 5 bases can cause non-specific binding. |
In practice, we use nearest-neighbor thermodynamic calculations (e.g., in Benchling or NEB’s Tm Calculator) to estimate Tm for each primer, then set Ta approximately 3–5°C below the lower primer’s Tm.
Question 3: PCR vs. Restriction Enzyme Digests for Creating Linear DNA Fragments
| Aspect | PCR | Restriction Enzyme Digest |
|---|---|---|
| Mechanism | Uses a DNA polymerase to amplify a specific region defined by two primers, generating many copies of the target fragment. | Uses restriction endonucleases to cut existing DNA at specific recognition sequences, releasing fragments from a larger molecule. |
| Template needed | Only nanograms of template required; amplification generates abundant product. | Requires micrograms of purified plasmid or genomic DNA since no amplification occurs. |
| Flexibility | Can amplify any region from any template, and primers can introduce mutations, overhangs, or restriction sites at the ends. | Limited to cutting at naturally occurring (or pre-engineered) restriction sites in the DNA sequence. |
| Sequence fidelity | May introduce mutations during amplification. Phusion has a very low error rate (~1 in 10⁶ bp), but errors are still possible over many cycles. | No sequence errors — the enzyme simply cuts the existing DNA without altering the sequence. |
| End types | Produces blunt ends by default (with proofreading polymerases like Phusion). Can add restriction sites or overhangs via primer design. | Produces either sticky ends (5’ or 3’ overhangs) or blunt ends depending on the enzyme chosen. |
| Protocol time | ~1.5–2 hours (PCR cycling + purification). | ~1–2 hours (digest incubation + gel extraction). |
| Post-processing | Requires DpnI treatment (to destroy methylated template) and column purification. | Usually requires gel electrophoresis and gel extraction to isolate the desired fragment from other digest products. |
When is each method preferable?
PCR is preferable when: you need to amplify from a scarce template, want to introduce mutations or add Gibson/Golden Gate overhangs via primers, or need a fragment that doesn’t have convenient restriction sites flanking it. In our lab, we use PCR to generate both the backbone and color insert fragments with Gibson-compatible overlapping ends.
Restriction digestion is preferable when: the template already contains appropriate restriction sites at the right positions (like cutting pUC19 with PvuII in our protocol), when absolute sequence fidelity is critical (since digestion introduces zero mutations), or when working with very large fragments (>10 kb) that are difficult to PCR amplify efficiently.
Question 4: Ensuring DNA Fragments Are Appropriate for Gibson Cloning
Several steps ensure that digested and PCR-amplified fragments will assemble correctly via Gibson Assembly:
Design overlapping ends (20–40 bp): Each adjacent pair of fragments must share 20–40 bp of identical sequence at their junctions. In our lab, the PCR primers include 20–22 bp overhangs complementary to the adjacent fragment. The Gibson exonuclease chews back 5’ ends to expose these complementary single-stranded regions, which then anneal.
DpnI treatment: After PCR, we add 1 µL of DpnI and incubate at 37°C for 30–60 minutes. DpnI selectively digests methylated (dam+) template DNA from E. coli while leaving the unmethylated PCR products intact. This eliminates background colonies from uncut template plasmid.
DNA purification: Use a column-based kit (like Zymo DNA Clean & Concentrator) to remove primers, dNTPs, polymerase, salts, and DpnI from the PCR products. Contaminants can inhibit the Gibson Assembly enzymes (exonuclease, polymerase, and ligase).
Verify fragment size and concentration: Run a diagnostic agarose gel to confirm each fragment is the expected size (no non-specific bands). Measure concentration with a Nanodrop/Qubit (>30 ng/µL). Calculate the molar ratio for assembly (typically 2:1 insert:vector).
Avoid secondary structure in overlaps: Design overlap regions that won’t form stable hairpins at the 50°C Gibson reaction temperature. Avoid long palindromes or GC-rich stretches in overlap zones.
Check orientation: Confirm all fragments are designed in the correct 5’→3’ orientation so that the overlaps match up in the intended order when assembled into a circular plasmid.
Question 5: How Plasmid DNA Enters E. coli During Transformation
Transformation is the process of introducing foreign DNA into bacterial cells. There are two main methods:
Heat Shock Transformation
Chemically competent cells (pre-treated with CaCl₂) are mixed with plasmid DNA on ice. The Ca²⁺ ions neutralize the negative charges on both the DNA phosphate backbone and the bacterial cell membrane, reducing electrostatic repulsion between them. The cells are then subjected to a brief heat shock (42°C for 30–90 seconds, typically ~45 seconds), which creates transient pores in the cell membrane by disrupting the lipid bilayer structure. The plasmid DNA diffuses through these temporary pores into the cytoplasm. Immediately returning the cells to ice allows the membrane to reseal, trapping the DNA inside. The cells are then incubated in SOC medium at 37°C for 1 hour to recover and begin expressing the antibiotic resistance gene before being plated on selective media.
Electroporation
Electrocompetent cells (washed in low-ionic-strength solutions) are mixed with DNA in a cuvette, and a brief high-voltage pulse (1.5–2.5 kV) is applied. The electric field directly polarizes the cell membrane, creating aqueous pores through the lipid bilayer. DNA enters the cell through these pores via a combination of electrophoretic migration (the electric field pushes the negatively charged DNA toward the cell) and osmotic flow. Electroporation generally achieves higher transformation efficiency (10⁸–10¹⁰ transformants/µg DNA) than heat shock (10⁶–10⁸/µg) and works better for large plasmids.
In our lab protocol, we use heat shock transformation with DH5α competent cells: 30 min on ice → 42°C for 45 seconds → ice for 5 minutes → add SOC → recover 60 min at 37°C → plate on chloramphenicol LB-agar plates.
Question 6: Golden Gate Assembly: An Alternative Assembly Method
Golden Gate Assembly is a one-pot, scarless cloning method that uses Type IIS restriction enzymes (such as BsaI or BbsI) combined with T4 DNA ligase to assemble multiple DNA fragments simultaneously. Unlike conventional restriction enzymes that cut within their recognition sequence, Type IIS enzymes cut at a defined distance outside their recognition site, generating custom 4-base overhangs that the user designs. Because the recognition site is separate from the cut site, the correctly assembled product no longer contains the recognition sequence — meaning the ligated product cannot be re-cut, driving the reaction toward completion. The reaction alternates between 37°C (for restriction enzyme activity) and 16°C (for ligation) in thermocycler cycles, allowing simultaneous digestion and ligation in the same tube. This makes Golden Gate highly efficient for assembling 2–10+ fragments in a defined order and orientation, with reported efficiencies of 80–95% for 2–3 part assemblies and 50–80% for 4–6 part assemblies. The method is particularly popular for standardized part-based assembly systems like MoClo and the iGEM Type IIS standard, where genetic parts are pre-flanked with BsaI sites for plug-and-play construction.
Golden Gate vs. Gibson Assembly
| Feature | Golden Gate Assembly | Gibson Assembly |
|---|---|---|
| Key enzymes | Type IIS restriction enzyme (BsaI/BbsI) + T4 DNA ligase | T5 exonuclease + Phusion polymerase + Taq DNA ligase |
| How fragments join | Enzyme cuts to create 4-bp sticky ends; ligase seals them | Exonuclease chews 5’ ends to expose ~20–40 bp overlaps; polymerase fills gaps; ligase seals |
| Overlap design | 4-bp overhangs (256 possible combinations) — no long homology needed | 15–40 bp of identical sequence between adjacent fragments |
| Scarless? | Yes — recognition sites eliminated in final product | Yes — overlap becomes seamless junction |
| Reaction conditions | Thermocycling: alternating 37°C and 16°C (25–50 cycles) | Isothermal: single incubation at 50°C for 15–60 min |
| Max fragments | 10+ fragments routinely; up to 24+ reported | 5–6 fragments efficiently; efficiency drops with more |
| Limitation | The Type IIS recognition site must not appear internally in any fragment | Overlap sequences must not form strong secondary structures |
Golden Gate Assembly Diagram
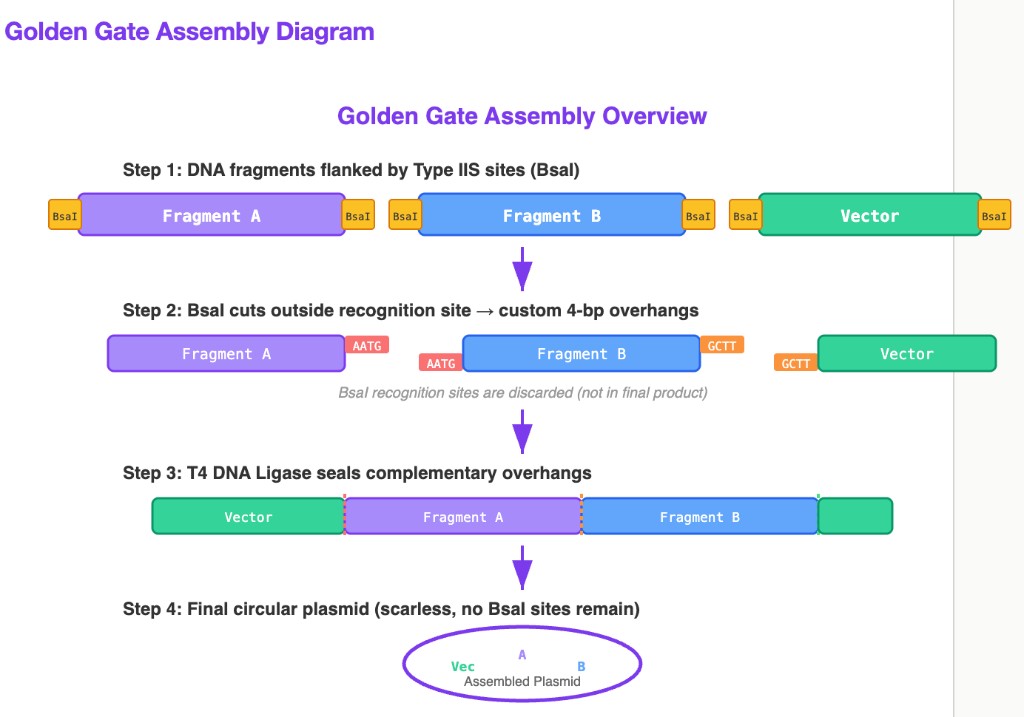
Key insight: The reason Type IIS enzymes are essential to Golden Gate is that they cut outside their recognition sequence. This means: (1) the user controls what overhang sequence is generated (256 possible 4-bp combinations from a single enzyme), enabling directed assembly of many fragments in a defined order; and (2) the recognition site is eliminated in the final product, so the ligated construct cannot be re-cut — this thermodynamically drives the reaction toward the assembled product.
Part 2. Asimov Kernel
AI Disclosure
I used Claude (Anthropic) to help with: formatting and structuring this HTML homework page, generating the Golden Gate Assembly SVG diagram, explaining Type IIS restriction enzyme mechanics, and spelling/grammar clean-up throughout the document.
HTGAA Spring 2026 · Week 6 Homework · DNA Nanostructures & Genetic Circuits · Constantin · Committed Listener