"The best way to predict the future is to engineer it."
🩺
Medicine
Final Year
🧬
Longevity Biotech
LBF Fellow
🏗️
Telos Circle
Non-Profit
🏠
The Residency
Hacker Houses
🧬 · · · · · · · · · · · · · 🧬
👋
About Me
I’m a final-year medical student and LBF Fellow from Vienna, Austria. My real obsession isn’t just understanding how the body works — it’s figuring out how to make it work longer and better.
I sit at the crossroads of medicine, synthetic biology, and artificial intelligence. On the side, I’m studying Mathematics and Chemical Engineering at TU Vienna — purely out of curiosity, because I believe the deepest breakthroughs happen when disciplines collide.
I believe the biggest bottleneck in science isn’t ideas — it’s execution. We have more hypotheses than hands to test them, more data than minds to analyze it, and more potential cures than pipettes to develop them. That’s why I’m building toward a future where AI accelerates science itself.
Beyond the lab and the lecture hall, I’m deeply invested in building communities that amplify human potential:
🌐 Telos Circle — A non-profit I founded to accelerate humanity’s progress by bringing exceptional talent together in one space. We connect thinkers, builders, and doers across disciplines to tackle the problems that matter most.
🏠 The Residency — Hacker House Communities — I build hacker house communities where builders have a space to build, a community to share their learning, and a culture of co-learning and peer-learning at speed. Think of it as a living room for people who want to ship things that matter.
🔥
What Drives Me
🧬 End Aging
Aging is the root cause behind almost every disease. I want to help end it — not just treat the symptoms, but reprogram the biology of aging itself using synthetic biology, AI, and every tool we can build.
🔭 Understand the Universe
Lifelong learning isn't a hobby, it's a mission. I'm trying to asymptotically understand the whole universe — from quantum mechanics to gene regulation, from pure mathematics to chemical engineering. Every discipline is a new lens.
🛠️ Engineer a Better Future
Knowledge without action is just trivia. I want to take what I learn and engineer real solutions — in medicine, in biotech, in AI — to make the future tangibly better for everyone, not just the privileged few.
🤝 Help Humanity
At the core, everything I do comes back to one thing: helping people. Whether it's through medicine, through building communities like Telos Circle and The Residency, or through open science — the goal is collective progress.
· · · 🔬 · · · 💊 · · · 🧪 · · ·
🛤️
My Journey
2026
HTGAA — How To Grow (Almost) Anything
Joining the MIT Media Lab course on synthetic biology from Vienna — learning to engineer life from DNA up.
2025–2026
LBF Fellow · Final Year Medicine
Longevity Biotech Fellowship while completing my final year of medical school in Vienna. Also studying Mathematics and Chemical Engineering at TU Vienna — just for the love of learning.
Ongoing
Telos Circle — Non-Profit Founder
Accelerating humanity's progress by bringing exceptional talent together in one space — connecting thinkers, builders, and doers across disciplines.
Ongoing
The Residency — Hacker House Communities
Building co-living spaces where builders have room to build, a community to share their learning, and a culture of co-learning and peer-learning at speed.
A remote-executable HTGAA final project: design three Twist constructs and test a cell-free split-GFP biosensor for the PARP1-HPF1 protein interaction.
Each card is one design-build-learn artifact: biology, automation, protein design, measurement, cloud labs, and the final PARP1-HPF1 biosensor pipeline.
HTGAA Spring 2026 · Constantin Convalexius · Vienna, Austria
1. The Application: AI-Powered Science Automation
I’m interested in building an AI platform that helps automate parts of the scientific process — things like scanning literature for gaps, designing experiments, running them through lab robots (like the Opentrons we’ll use in HTGAA), and helping write up results.
Why? Science is slow. Not because scientists are lazy, but because there’s way more good questions than people to work on them. Many ideas never get tested because the person who had them didn’t have the right lab skills or equipment. And honestly, a lot of published research can’t even be reproduced because of human error in complicated protocols. Or negative results don’t get published at all, leading to the “chasing the same dead ends” phenomenon — but no one knows, because it’s not published.
An AI platform could help with all of that. Not by replacing scientists, but by letting more people do better science faster, use negative and positive results to iterate faster and learn from more data, which can be used to train the next “physics” model of the AI. I think of it like a student somewhere without access to a fancy lab — they could design a CRISPR experiment, have a robot run it remotely, and get solid results back. OpenAI did something very similar now with Ginkgo Bioworks, read here: GPT-5 Lowers Protein Synthesis Cost.
The obvious problem: this is dual-use. The same tool that speeds up drug discovery could also speed up bioweapon development. Which is exactly why governance matters here.
2. Policy Goals
Two main goals, each broken into sub-goals:
Goal A — Safety & Security
A1: Prevent the platform from being used (or easily adapted) for weapons development
A2: Keep humans in the loop for any high-risk experiments — no fully autonomous dangerous stuff
Goal B — Equitable Access
B1: Make the tools accessible regardless of where you are or how much funding you have
B2: Prevent any single company or government from monopolizing AI-driven science
3. Three Governance Actions
Action 1: Open-Source Mandate
Purpose: Right now the best AI models are built behind closed doors. I’d require that publicly funded AI-science tools get released as open-source — similar to how the Human Genome Project made all genomic data public. Private platforms could get tax incentives for doing the same.
Design: Funding agencies (NIH, NSF, ERC) tie grants to open-source release, like the existing open-access publication mandates. Code goes on GitHub or Hugging Face. Philanthropic orgs like the Chan Zuckerberg Initiative could co-fund.
Assumptions: That open-source leads to faster improvement (usually true — see Linux, Python). That the community helps maintain quality. But also: open-source means bad actors get access too, which is a real problem.
Risks: Companies might only open-source outdated models while keeping the good stuff private. And if everything is truly open, you’re lowering barriers for misuse too — which directly conflicts with Goal A.
Action 2: Built-In Safety Guardrails
Purpose: Current AI content filters are pretty weak and easy to bypass. I’d build domain-specific safety layers into the platform — not just keyword blocking, but actual screening of what’s being designed. Similar to how DNA synthesis companies like Twist Bioscience already screen orders against pathogen databases.
Design: Multiple layers: (1) screen DNA sequence requests against pathogen databases, (2) flag suspicious query patterns, (3) require extra credentials for the riskiest capabilities, (4) regular red-teaming by security experts. Built by developers, advised by biosecurity people.
Assumptions: That AI can reliably tell the difference between legit research and misuse — this is honestly still an unsolved problem. And that filters won’t be so aggressive they block perfectly good research.
Risks: Too strict → researchers switch to unfiltered alternatives. Too weak → false sense of security. And determined bad actors can probably just train their own models from scratch anyway.
Action 3: International Regulatory Body
Purpose: There’s no international body governing AI systems that accelerate science. The Biological Weapons Convention wasn’t designed for this. I’d propose an International Commission on AI-Assisted Research (ICAIR), modeled on the IAEA — setting standards, certifying platforms, and coordinating responses to misuse.
Design: UN member states + AI companies + scientific organizations participate. ICAIR sets minimum safety standards, certifies compliant platforms, runs audits, and coordinates responses. Funded by member states plus a levy on commercial AI platforms.
Assumptions: That international cooperation on AI governance is achievable (big assumption given US-China tensions). That the body can move fast enough — historically, regulation always lags technology.
Risks: Major nations refuse to join, making it toothless. Or it becomes so bureaucratic it kills innovation. Worst case: incumbents capture the body and use it to block competition.
4. Scoring Matrix
Scale: 1 = best, 3 = least effective
Policy Goal
Open-Source
Safety Guardrails
Int. Regulatory Body
Enhance Biosecurity
• Preventing incidents
3
1
2
• Helping respond
3
2
1
Foster Lab Safety
• Preventing incidents
2
1
2
• Helping respond
3
1
1
Protect Environment
• Preventing incidents
3
1
2
• Helping respond
3
2
1
Other Considerations
• Minimizing costs
1
2
3
• Feasibility
1
2
3
• Not impeding research
1
2
3
• Promoting constructive use
1
2
2
Summary: Open-source wins on access and feasibility but loses badly on security. Guardrails are best at prevention but depend on unsolved AI safety problems. The international body is strongest for response but hardest to actually create.
5. Recommendation
Audience: MIT Leadership / MIT Media Lab
No single action works alone. I’d go with a layered approach:
Open-source — like OpenCourseWare, Creative Commons, Open Source Software.
Build guardrails very soon, best day one.
Gate the dangerous stuff: Basic capabilities stay open, advanced dual-use features (novel organism design) require institutional verification. Kind of like how some chemicals or drugs are freely available while others need a license or prescription.
Push for international standards — we can’t create a regulatory body alone, but we could host working groups and publish frameworks that others adopt.
Main trade-off: Openness vs. security.
My resolution: Open source for wide distribution, with guardrails for more capable and dangerous capabilities (dual use).
Biggest uncertainty: Whether AI safety filters can actually keep pace with rapidly evolving capabilities. Nobody has a good answer to this yet.
6. Ethical Reflections
Going into this week I thought governance is something you deal with after a technology exists. The recitation changed that — the Jurassic Park meme sounds silly but captures it well. We’re too much in “can we?” mode and not enough in “should we?” mode.
The openness question kept bugging me. My gut says make everything open, but then I think about what “everyone” includes and it gets uncomfortable. I now think openness with checkpoints makes more sense — open tools, but controls where designs become physical (synthesis, robot instructions).
AI-generated fraud was new to me. An AI could make up data that looks real, or accidentally lead someone to design something harmful. Provenance tracking for AI outputs seems necessary.
These discussions are also very US-centric. As a med student in Vienna — AI doesn’t stop at borders. Building safety into the platform architecture could raise the floor globally, similar to how iGEM runs safety reviews across all countries without needing international treaties.
Actions I’d propose: ethics review before new AI capabilities get released, provenance tracking as default, tying capability releases to safety milestones, and building risk education directly into the workflow so users can’t blindly automate dangerous stuff.
Week 2 Lecture Prep
Dr. LeProust’s Questions
1. What’s the most commonly used method for oligo synthesis currently?
The standard is the phosphoramidite method developed by Caruthers in 1981.
2. Why is it difficult to make oligos longer than 200nt via direct synthesis?
The problem: each coupling step isn’t 100% efficient. It’s around 99% or so, but not perfect. So if your coupling efficiency is 99%, for a 200-mer you’d get something like 0.99^200 ≈ 13% full-length correct product. The rest is junk — truncated products that failed at some step along the way.
3. Why can’t you make a 2000bp gene via direct oligo synthesis?
Building on the previous answer: if even getting to 200nt with decent yield is hard, imagine trying 2000nt. At 99% coupling efficiency, 0.99^2000 is basically zero. You’d get virtually no full-length product. (Note: Twist Bioscience demonstrated for the first time that they can synthesize a ~700nt oligo, which was a major achievement pushing those limits.)
Professor Jacobson’s Questions
1. What is the error rate of polymerase? How does this compare to the length of the human genome? How does biology deal with that discrepancy?
Error Rate: DNA polymerase has an error rate of approximately 1 in 10^6 (1 in a million)
Human Genome Size: approximately 3.2 Giga Base Pairs (Gbp) — that’s ~3 orders of magnitude larger than the error rate denominator
Implication: Thousands of errors would appear per single replication event
How Biology Deals With It: Biology overcomes this through additional error correction: proofreading by the polymerase itself during synthesis, and post-synthesis mismatch repair systems that catch and fix remaining errors
2. How many different ways are there to code for an average human protein? Why don’t all of these codes work in practice?
Number of Ways: The redundancy of the genetic code (multiple codons per amino acid) combined with an average human protein length of ~1036 base pairs means there is an astronomical number of different DNA sequences that could theoretically encode the same protein.
Why Not All Codes Work: Despite coding for the same amino acids, different DNA/RNA sequences are not functionally equivalent because:
Different nucleotides have different chemical features in hydrogen bonding and electrostatic properties — leading to different folding of primary into secondary/tertiary structures (the ribosome itself is an RNA that produces proteins!)
RNA Cleavage — breaking of the RNA strand means it doesn’t assemble as anticipated
Loop Formation — RNA can form ring structures, creating different secondary structures
Complex Tertiary Structures — rings, 3D origami-like shapes, and even cellular automata-like patterns
Professor George Church’s Question
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 Essential Amino Acids
In animals (including humans — and the dinosaurs of Jurassic Park), these 10 amino acids cannot be synthesized de novo and must come from the diet:
Amino Acid
Amino Acid
Phenylalanine
Methionine
Valine
Histidine
Threonine
Arginine*
Tryptophan
Leucine
Isoleucine
Lysine
*Arginine is essential in many animals/birds; conditionally essential in humans.
The “Lysine Contingency” is a fictional biocontainment strategy from Jurassic Park where dinosaurs were genetically engineered to be unable to produce lysine. The intent was to ensure they would fall into a coma and die if they escaped, as they’d lack the supplements provided by park staff.
Impact on My View
This is a completely fictional contingency that in the real world would have never worked — because no animal can synthesize lysine anyway. It’s an essential amino acid that every animal has to eat (via plants or meat). So the “engineered dependency” is completely redundant — the dinosaurs already couldn’t make it!
A real biocontainment strategy would need to engineer dependency on a non-natural amino acid — something that doesn’t exist in any food source. This would create true “metabolic isolation” that cannot be bypassed by simply eating natural foods.
AI Disclosure
Claude (Anthropic) — Used to help structure and refine this assignment. The core ideas and positions are my own.
Prompt 1: “Help me structure my governance analysis for AI-powered science automation, with three governance actions and a scoring matrix.”
Prompt 2: “Nice I have done the homework draft now, please refine it so it has less spelling errors, correct my grammar and format it better. If you correct my wording, don’t write AI but write human like. Keep all the info unless it is obviously wrong.”
Cursor (AI-assisted IDE) — Used to build and deploy my HTGAA website.
Prompt 2: “Upload my homework to the website, format it nicely in Markdown, commit and push.”
Week 2 HW: DNA Read, Write, & Edit
Week 2: DNA Read, Write, & Edit
Student: Constantin Convalexius Course: HTGAA Spring 2026 Location: Vienna, Austria
Part 1: Benchling & In-silico Gel Art
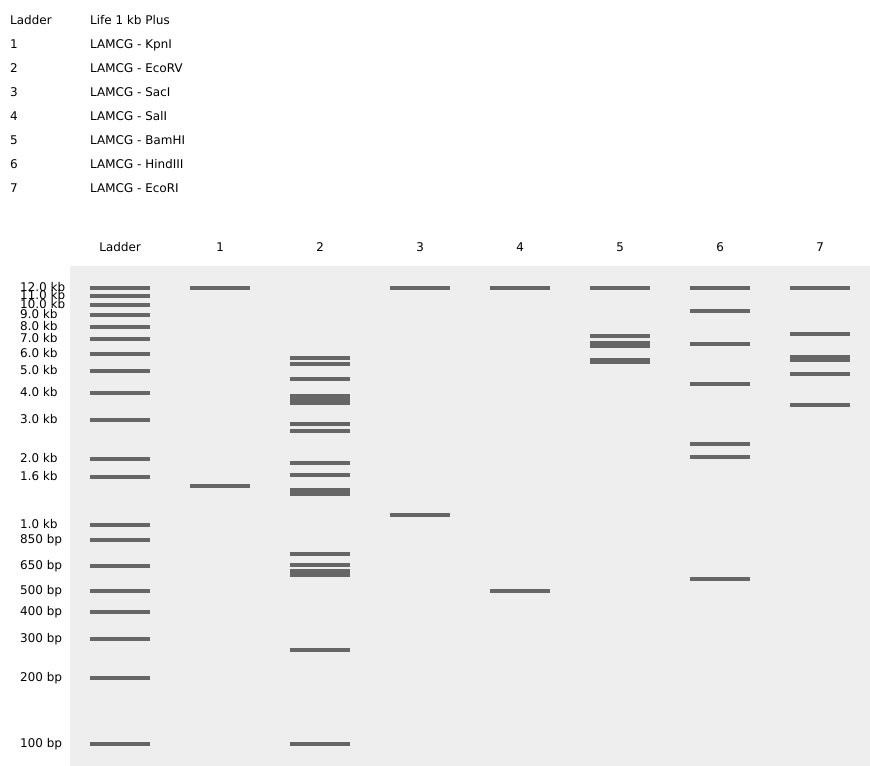
Butterfly art 1
2nd picture: all enzymes
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
As a committed listener in Vienna without local wet-lab access, I completed the in-silico design and simulation sections.
Part 3: DNA Design Challenge
3.1 — Protein Choice: PD-L1 (Programmed Death-Ligand 1)
I chose PD-L1 (CD274, UniProt: Q9NZQ7) — the immune checkpoint protein that tumor cells use to hide from the immune system. PD-L1 sits on the surface of cancer cells and binds to PD-1 on T-cells, essentially telling them “don’t attack me.” Drugs like Pembrolizumab (Keytruda) block this interaction by targeting PD-1, so the immune system can recognize and destroy the tumor again. As a med student, this is one of the most exciting developments in oncology I’ve encountered so far.
The full-length PD-L1 protein is 290 amino acids and includes a signal peptide, extracellular domain, transmembrane region, and a short intracellular tail. For this exercise, I’m only using the extracellular domain (AA 19-238, 220 residues), since that’s the part that actually interacts with PD-1 and is the relevant domain for drug binding studies. This is also what researchers typically express recombinantly — you don’t need the transmembrane anchor if you just want to study the binding interface.
I used the Sequence Manipulation Suite (SMS2) reverse translation tool with “most likely codons” to convert the amino acid sequence into a DNA nucleotide sequence.
The output was an 870 bp sequence for the full-length 290 AA protein. One thing I noticed is that the SMS2 tool defaults to E. coli codon preferences — you can see this in the output, which uses codons like CGC for Arginine, GCG for Alanine, and CCG for Proline. These are all heavily biased toward bacterial tRNA pools, which wouldn’t work well in a human expression system.
This step is mainly useful to show the “raw” reverse translation before optimization, and to demonstrate why codon optimization is necessary.
3.3 — Codon Optimization
Since I want to express PD-L1 in human HEK293 cells (see 3.4), I ran the extracellular domain amino acid sequence through GenScript’s GenSmart Codon Optimization Tool with Homo sapiens as the host organism.
The key difference compared to the raw SMS2 output is that GenSmart replaced the E. coli-preferred codons with those matching human tRNA abundance. For example, Arginine now uses AGG/AGA/CGG instead of bacterial CGC, and Alanine uses GCC/GCT instead of GCG. This is important because if the codons don’t match the host’s tRNA pool, the ribosome stalls during translation, leading to low protein yields or truncated products.
The GC content of 55.07% is also nicely within the ideal window — too high or too low GC content can cause issues with mRNA secondary structures or difficulties during DNA synthesis.
The codon-optimized sequence was generated using the GenSmart Codon Optimization Tool [1].
[1] Long Fan (2020, February 6). Codon optimization. (WO Patent WO 2020/024917 A1). Nanjing GenScript Biotech Co., Ltd.
PD-L1 is a glycoprotein — it has N-linked glycosylation sites that are important for its folding and function. Because of this, I would express it in HEK293 human cells rather than E. coli. The workflow would be: clone the codon-optimized gene into a mammalian expression vector, transfect HEK293 cells, let them express and secrete the protein (since we’re only using the extracellular domain without the transmembrane anchor, it should be secreted into the culture medium), and then purify it using an affinity tag (like a His-tag with Ni-NTA chromatography). HEK293 cells are well-established for this — they handle human post-translational modifications properly and give reasonable yields.
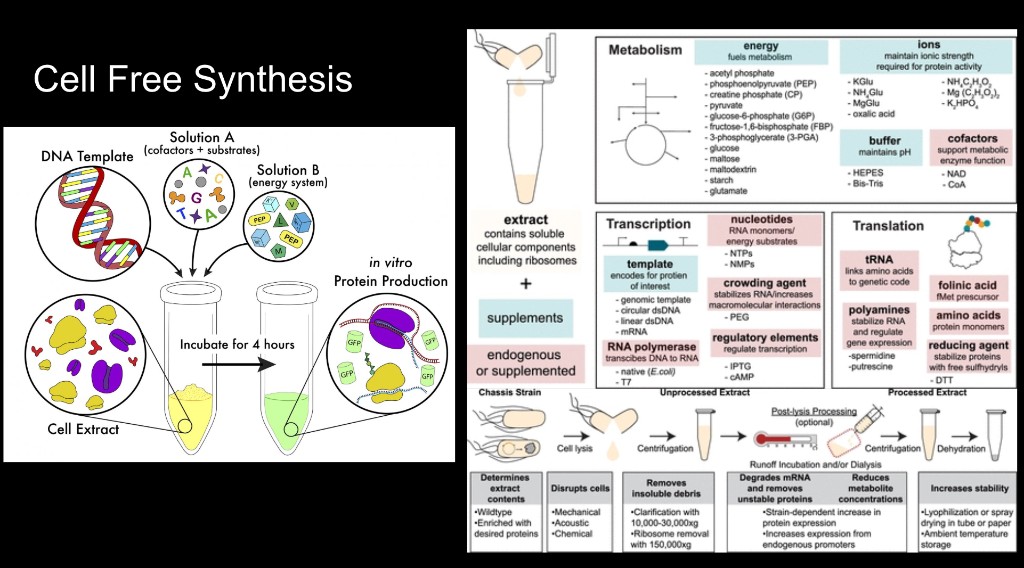
Cell-free expression (alternative):
For quick small-scale testing (e.g., to check if the construct expresses at all before committing to a full cell culture run), you could use an in vitro transcription/translation system like rabbit reticulocyte lysate or wheat germ extract. These systems can produce protein in a few hours rather than days, but they don’t perform proper glycosylation, so the protein wouldn’t be fully functional. Still useful as a rapid validation step.
Part 4: Prepare a Twist DNA Synthesis Order
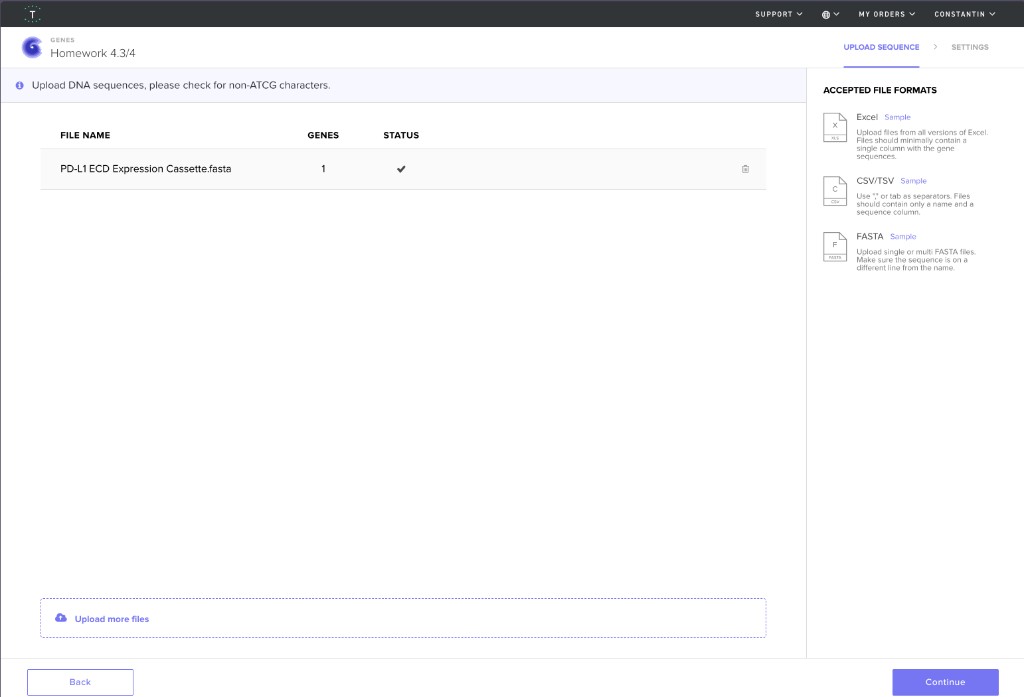
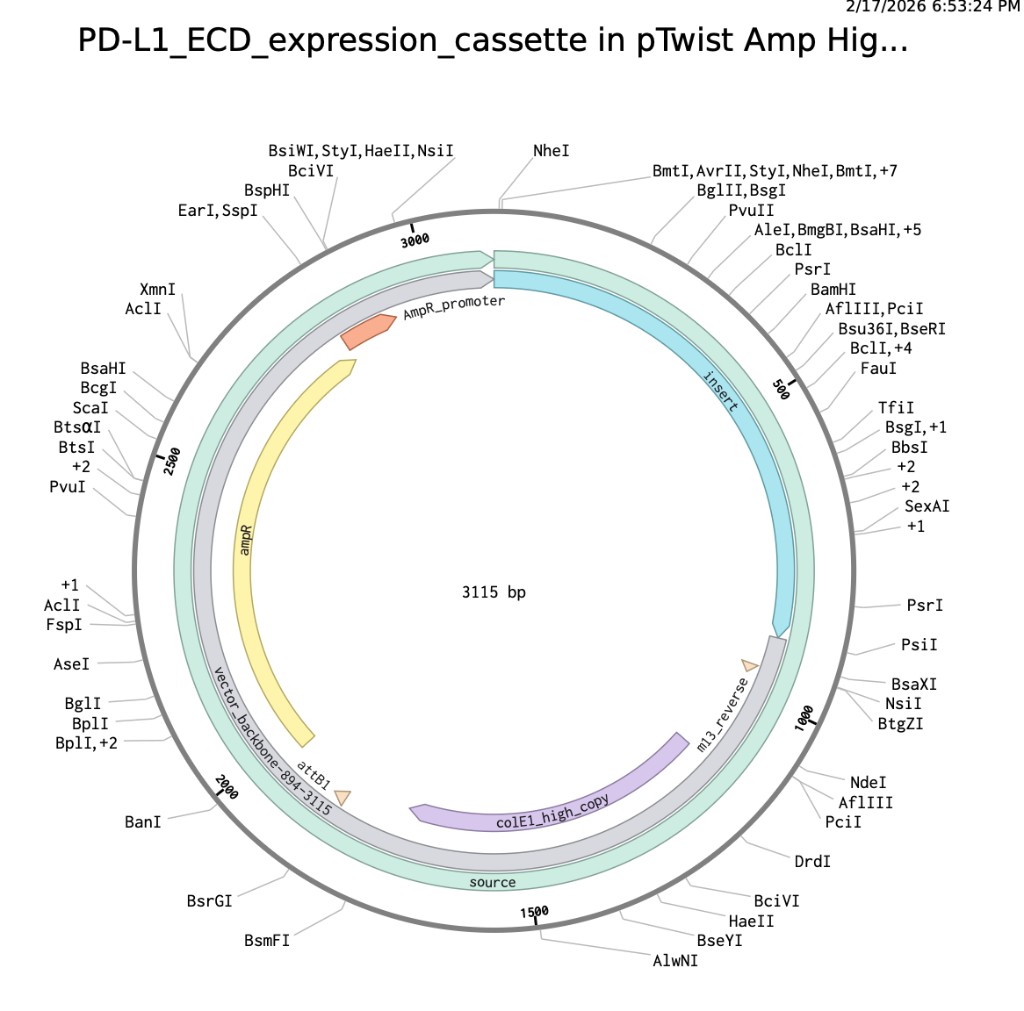
Here are my screenshots and files for Homework Part 4:
Upload sequence to Twist
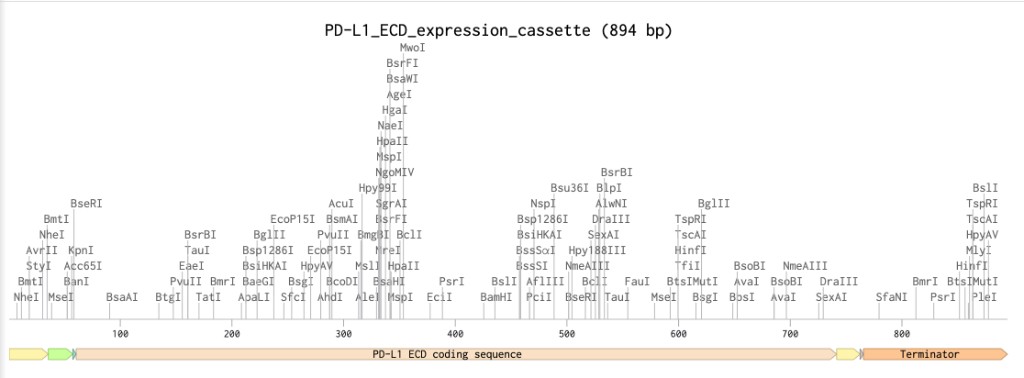
Benchling expression cassette map
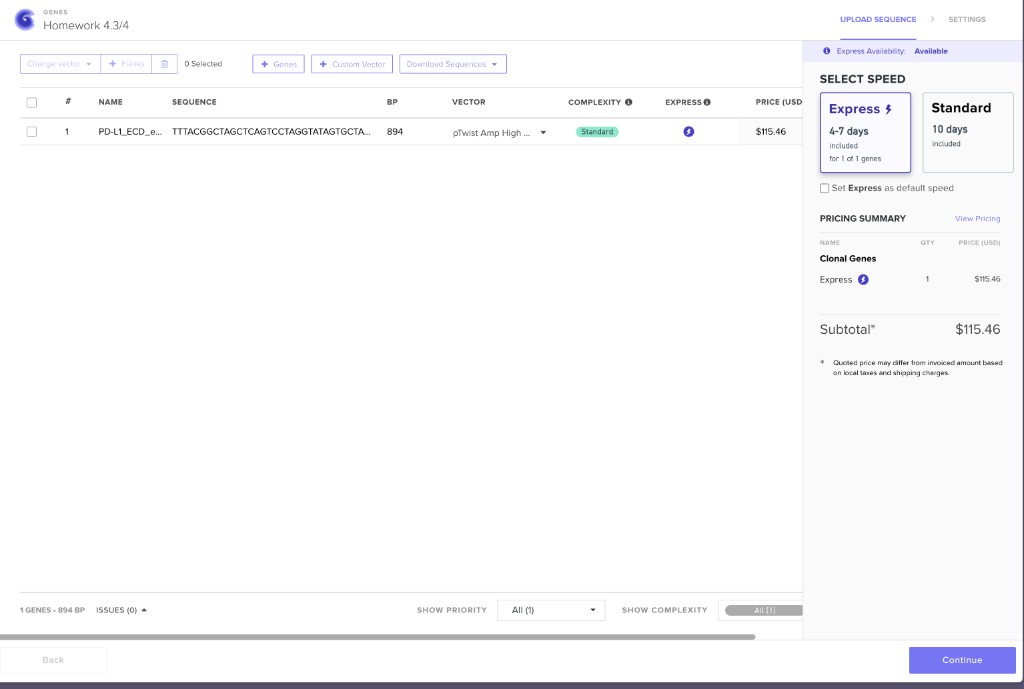
Twist clonal gene order configuration
PDF export
PDF version prepared locally (not uploaded in this commit).
PDF update: plasmid map screenshot
Part 5: DNA Read, Write, Edit
5.1 DNA Read
(i) What DNA would I want to sequence?
I’d want to sequence the genomes of supercentenarians — people who’ve made it past 110. These individuals somehow dodge or massively delay the diseases that kill most of us (heart disease, cancer, dementia), and there’s evidence that protective variants in genes like FOXO3, APOE, and TERT are enriched in their genomes. But we probably haven’t found everything yet. By doing whole-genome sequencing on large cohorts and comparing them to people who aged “normally,” we could uncover rare genetic variants that essentially act as nature’s longevity engineering. Pair that with DNA methylation data (which feeds into biological aging clocks like the Horvath clock) and you get a pretty complete picture of both the genetic hand they were dealt and how their gene expression shifted — or didn’t — over time.
(ii) Sequencing technology
I’d go with a hybrid approach: Oxford Nanopore (PromethION) for long reads plus Illumina NovaSeq for high-accuracy short reads.
Nanopore (third-generation): Sequences native, single DNA molecules in real time — no PCR amplification needed, which avoids amplification bias. A motor protein threads a DNA strand through a tiny biological pore in a membrane. Each base passing through disrupts the ionic current in a characteristic way, and a neural network translates those current patterns into sequence. Big advantage: it can also detect DNA methylation directly from the native strand, no bisulfite conversion needed. Reads are long (often >20 kb), which helps resolve structural variants and repetitive regions.
Input prep: Extract high-molecular-weight DNA from blood, ligate sequencing adapters directly — pretty minimal compared to short-read platforms.
Illumina (second-generation): Supplements Nanopore with very accurate short reads (~150 bp) for reliable SNP calling. Input prep involves fragmentation, adapter ligation, and bridge PCR. Bases are called by detecting fluorescent signals from reversible dye-terminators during synthesis-by-sequencing cycles.
Output: Both produce FASTQ files. Together they give you phased, chromosome-level assemblies with both structural resolution and single-nucleotide accuracy.
5.2 DNA Write
(i) What DNA would I want to synthesize?
I’d synthesize an engineered human telomerase (hTERT) expression cassette — a gene therapy construct to transiently reactivate telomerase in adult cells.
Telomere shortening is one of the core hallmarks of aging. Every cell division chips away at the protective chromosome caps until the cell senesces or dies. Telomerase rebuilds them, but it’s silenced in most adult tissues. Maria Blasco’s group at CNIO showed that AAV-delivered telomerase in mice extended lifespan without increasing cancer. The idea is to build a controllable human version.
The construct (~6-7 kb) would include a codon-optimized hTERT coding sequence under a Tet-On inducible promoter (so you can switch it on/off with doxycycline — you really don’t want constitutive telomerase, that’s a cancer risk), plus a GFP reporter to track which cells are expressing it. For Twist, I’d order this as overlapping clonal gene fragments.
(ii) Synthesis technology
Phosphoramidite oligo synthesis (Twist Bioscience’s platform) combined with Gibson Assembly.
Twist synthesizes thousands of short overlapping oligos (~60-200 nt) in parallel on silicon chips. Each oligo goes through cycles of deprotection -> coupling -> capping -> oxidation. These oligos get assembled into longer gene fragments (~1.8 kb) via overlap extension, then cloned into plasmids and sequence-verified. For my full ~7 kb construct, I’d order 3-4 fragments from Twist and stitch them together with Gibson Assembly.
Limitations: Coupling efficiency is 99-99.5% per step, so errors accumulate with length — that’s why you assemble from short oligos rather than synthesizing one long piece. Extreme GC content or repetitive sequences can cause synthesis failures. Turnaround is 2-3 weeks, and cost is around $0.07-0.09/bp ($500 for the full construct).
5.3 DNA Edit
(i) What DNA would I want to edit?
Three targets for a “longevity panel”:
PCSK9 knockout: People with natural loss-of-function mutations in PCSK9 have very low LDL cholesterol and near-immunity to coronary heart disease — the #1 killer globally. A permanent gene edit would be a one-and-done solution. Verve Therapeutics is already running clinical trials on this.
TP53 enhancement: Not a knockout — that would be terrible. Instead, introducing “super-p53” gain-of-function variants (studied in mouse models) that boost cancer surveillance without accelerating cellular senescence. The goal: decouple tumor protection from the aging program.
Myostatin (MSTN) partial reduction: Myostatin inhibits muscle growth. Sarcopenia (age-related muscle wasting) is a huge driver of frailty in older adults. Reducing myostatin signaling could help maintain muscle mass well into old age — think Belgian Blue cattle, but a gentler, partial version for humans.
George Church has discussed similar multi-gene longevity editing in the context of GP-write.
(ii) Editing technology
For PCSK9: adenine base editing (ABE) via lipid nanoparticles (LNPs). A Cas9 nickase fused to a deaminase enzyme converts a single A·T base pair to G·C, introducing a premature stop codon in PCSK9 — no double-strand break needed. LNPs are delivered IV and preferentially target the liver (perfect for PCSK9). Verve’s primate data shows >60% editing efficiency.
For TP53 and MSTN: prime editing, which uses a Cas9 nickase fused to a reverse transcriptase guided by a pegRNA containing both the target sequence and the desired edit template. Even more precise than base editing — can make any small substitution without double-strand breaks or donor DNA.
Steps (base editing example): Design a guide RNA positioning the target adenine in the editing window -> formulate ABE mRNA + sgRNA in LNPs -> IV infusion -> LNPs enter hepatocytes via ApoE-mediated uptake -> base editor converts A to inosine (read as G) -> permanent single-nucleotide change.
Limitations: Off-target editing risk (lower than standard Cas9 but not zero — needs WGS validation). LNPs mostly hit the liver, which is great for PCSK9 but not for muscle or systemic edits — those need AAV or next-gen tissue-tropic delivery. Prime editing efficiency is still variable (~5-50%). And of course, these edits are permanent and irreversible, which is both the point and the risk.
AI Disclosure
I used Cursor and Claude to help with formatting, spelling/grammar clean-up, and publishing this website documentation.
Week 3 HW: Lab Automation
Week 3: Lab Automation
Student: Constantin Convalexius Course: HTGAA Spring 2026 Location: Vienna, Austria
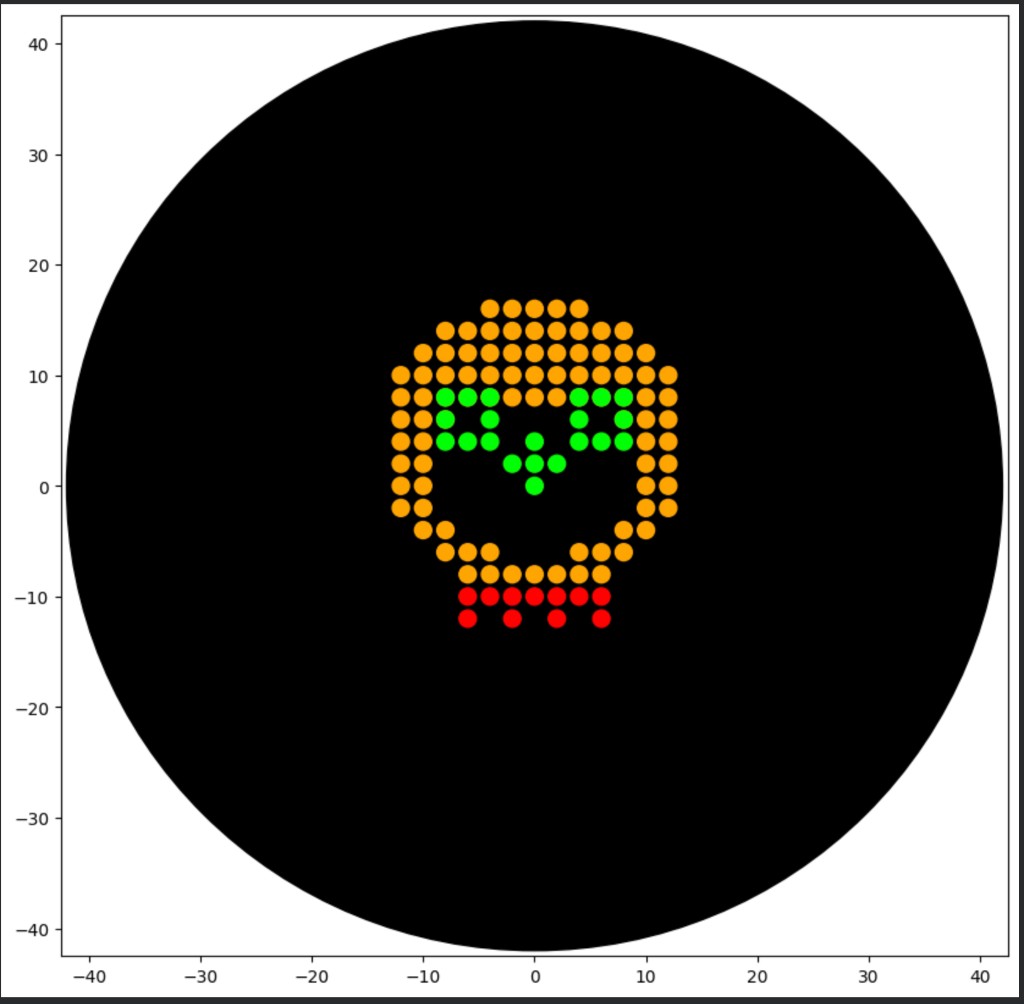
Part 1: Python Script for Opentrons Artwork
I created and tested an Opentrons Python script that generates a dotted skull design for gel art.
1.1 What I completed
Designed a skull artwork concept and implemented it in Python for Opentrons (apiLevel 2.20).
Used multi-color patterning with helper functions for safer droplet detachment (dispense_and_detach).
Simulated the protocol in Colab and fixed simulator compatibility issues (e.g., replacing direct protocol.comment calls with mock-safe logging logic).
Generated a higher-resolution version of the skull by increasing point density.
Submission status
Artwork script: completed.
Opentrons skull design image: completed.
I will submit the Python script for robot execution as required by the course submission form.
1.2 Proof of Opentrons skull artwork
Part 2: Post-Lab Questions
2.1 Published paper using Opentrons/automation for novel biology
Paper selected: Herzog AE, Zheng S, Warner KA, Vanini JV, Somayaji R, Johnson MR, et al. “Bmi-1 inhibition sensitizes head and neck cancer stem cells to cytotoxic chemotherapy.” Translational Oncology. 2026;63:102603. doi:10.1016/j.tranon.2025.102603.
Why this paper is a strong example of lab automation
This study uses automation directly in a cancer-biology workflow, including an Opentrons OT-2 liquid handling robot to standardize and scale an automated orosphere assay in 96-well plates. The authors investigate whether inhibiting Bmi-1 (genetically and pharmacologically with PTC596/unesbulin) can reduce chemotherapy-driven cancer stemness in head and neck squamous cell carcinoma (HNSCC).
In vivo xenograft data supports combining Bmi-1 inhibition with conventional chemotherapy.
Why this is biologically novel and relevant
The key innovation is not only biological (targeting cancer stemness to overcome resistance) but also methodological: integrating an affordable, programmable OT-2 into a translational cancer workflow enables reproducible treatment delivery and phenotyping at scale. This demonstrates how benchtop automation can move from “pipetting convenience” to hypothesis-driven oncology research.
2.2 What I intend to do with automation tools for my final project
My project direction is to use automation for a small combinatorial therapeutic screen focused on therapy resistance biology.
Proposed project concept
Automate a matrix experiment testing combinations of:
Cytotoxic drug condition (e.g., cisplatin dose levels),
Pathway-modulating small molecule condition (e.g., Bmi-1/STAT3-related perturbation),
Optional timing condition (simultaneous vs. staggered treatment).
The readout would be a plate-based viability/survival proxy and, if feasible, a stemness-related assay endpoint.
Why automation is essential
Precise liquid handling across many conditions and replicates.
Lower human pipetting variability.
Easier reproducibility for repeated screens.
Structured experimental logs that support downstream analysis.
Nine of eleven questions answered from the Shuguang Zhang question set (skipping Questions 7 and 8).
Question 1: How many molecules of amino acids do you take with a piece of 500 grams of meat?
Meat is roughly 25% protein by weight, so 500 g of meat contains about 125 g of protein. The average molecular weight of an amino acid residue is approximately 110 Daltons (Da), where 1 Dalton = 1.66 × 10⁻²⁴ g.
Using Avogadro’s number (6.022 × 10²³):
Mass of protein = 125 g
Moles of amino acid residues = 125 g / 110 g/mol ≈ 1.14 mol
Number of amino acid molecules = 1.14 × 6.022 × 10²³ ≈ 6.8 × 10²³
That is approximately 6.8 × 10²³ amino acid molecules — roughly one mole of amino acids, which is close to Avogadro’s number itself. An astonishing quantity from a single piece of meat!
Question 2: Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When we eat protein from any organism, our digestive system breaks it down completely into individual amino acids. Proteases in the stomach (pepsin) and small intestine (trypsin, chymotrypsin) hydrolyze the peptide bonds, releasing free amino acids and small peptides into the bloodstream.
These free amino acids are then used as building blocks by our own ribosomes, which follow the instructions encoded in our DNA. Our genetic code determines the specific sequence in which amino acids are re-assembled into human proteins — not the cow’s or fish’s sequence. The “information” that made the protein bovine or piscine is erased during digestion.
Think of it like dismantling a LEGO cow and using the same bricks to build a LEGO human: the bricks are identical, but the blueprint (DNA) determines the final shape.
Question 3: Why are there only 20 natural amino acids?
The set of 20 canonical amino acids represents an evolutionary compromise between chemical diversity and biological efficiency:
Sufficient chemical diversity: The 20 amino acids cover a wide spectrum of chemical properties — small and large, hydrophobic and hydrophilic, positively and negatively charged, aromatic, sulfur-containing, and flexible (glycine) vs. rigid (proline). This gives proteins enough variety to fold into millions of distinct shapes and perform diverse functions.
Manageable genetic encoding: With a triplet codon system (4³ = 64 possible codons), 20 amino acids plus stop signals can be encoded with redundancy (multiple codons per amino acid), which provides error-buffering. Adding more amino acids would reduce this redundancy and make translation more error-prone.
Biosynthetic cost: Each amino acid requires dedicated biosynthetic enzymes and tRNA synthetases. Maintaining more than 20 would increase the metabolic burden on the cell without proportional benefit.
Frozen accident + optimization: The genetic code likely expanded from a smaller set early in evolution and stabilized around 20 because changes to the code would be catastrophically disruptive to all existing proteins. Some organisms do use 21st (selenocysteine) and 22nd (pyrrolysine) amino acids for specialized functions, suggesting that 20 is not a hard physical limit but an evolutionary optimum.
Question 4: Can you make other non-natural amino acids? Design some new amino acids.
Yes! Non-natural amino acids (nnAAs) are a very active area of research. Any molecule with an amino group (−NH₂) and a carboxyl group (−COOH) on the same carbon with a novel side chain qualifies. Here are some designs:
1. Photo-switchable amino acid (AzoAla): Replace the side chain with an azobenzene group. This amino acid would change shape when exposed to UV light (trans → cis isomerization), allowing light-controlled protein conformational changes.
2. Click-chemistry amino acid (AzidoNorval): A norvaline derivative with a terminal azide (−N₃) on the side chain. This enables bio-orthogonal “click” reactions with alkynes for selective labeling of proteins in living cells.
3. Metal-chelating amino acid (BiPyrAla): An alanine derivative with a bipyridine side chain that can coordinate metal ions (Fe²⁺, Ru²⁺). This could create proteins with built-in metallocatalytic sites.
4. Fluorinated leucine (tfLeu): Leucine with trifluoromethyl groups replacing the methyl groups. The increased hydrophobicity and altered steric properties stabilize coiled-coil structures beyond what natural amino acids achieve.
Researchers like Peter Schultz have developed methods using engineered tRNA synthetases and amber stop codon suppression to incorporate over 200 different nnAAs into proteins in living cells.
Question 5: Where did amino acids come from before enzymes that make them, and before life started?
Amino acids can form through purely abiotic (non-biological) chemistry. Several sources contributed to the prebiotic amino acid pool:
Miller-Urey synthesis (1953): Stanley Miller showed that electric sparks (simulating lightning) passed through a mixture of water vapor, methane, ammonia, and hydrogen produce amino acids including glycine, alanine, and aspartic acid. The key reaction is the Strecker synthesis — aldehydes react with ammonia and hydrogen cyanide to form amino acids.
Extraterrestrial delivery: The Murchison meteorite (1969) was found to contain over 90 different amino acids, including many not used by life on Earth. This proves that amino acids form in interstellar space through radiation-driven chemistry on dust grains and in nebulae.
Hydrothermal vents: Deep-sea hydrothermal vents provide high temperatures, mineral catalysts (iron-sulfur clusters), and chemical gradients that can drive amino acid synthesis from simple molecules like CO₂, NH₃, and H₂.
Mineral surface catalysis: Clay minerals like montmorillonite can catalyze the polymerization of amino acids into short peptides without enzymes, providing a plausible path from free amino acids to the first proto-proteins.
Question 6: If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
A left-handed α-helix.
Natural L-amino acids form right-handed α-helices because of the stereochemistry at the Cα carbon. The L-configuration favors backbone dihedral angles (φ ≈ −57°, ψ ≈ −47°) that produce a right-handed twist.
D-amino acids are the mirror image of L-amino acids. Their favored dihedral angles are the exact opposite (φ ≈ +57°, ψ ≈ +47°), which produces a left-handed α-helix. This is simply a consequence of mirror symmetry: a structure built from mirror-image building blocks will itself be the mirror image of the original.
This principle is used in practice — synthetic D-peptides form mirror-image proteins (“mirror-image phage display”) that are resistant to natural proteases, making them attractive as drug candidates.
Question 9: Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets aggregate because their edges present unsatisfied hydrogen bond donors and acceptors that are inherently “sticky.”
Hydrogen bonding at edges: In a β-sheet, each strand forms backbone hydrogen bonds with its neighbors. But the outermost strands have one edge with no partner — these exposed N−H and C=O groups are thermodynamically driven to find hydrogen bond partners. The easiest partner is another β-strand from another molecule, leading to intermolecular aggregation.
Hydrophobic packing: β-sheets often have one hydrophobic face. When two sheets stack face-to-face, the hydrophobic surfaces are buried away from water, driven by the hydrophobic effect. This “steric zipper” interaction is very stable.
Cooperative elongation: Once a small β-sheet aggregate forms, adding the next strand is energetically favorable because it satisfies the new edge’s hydrogen bonds. This makes aggregation self-reinforcing and can proceed rapidly once nucleated.
Backbone geometry: The flat, extended geometry of β-strands makes them well-suited for long-range, repetitive stacking — unlike α-helices, which curve and are harder to stack indefinitely.
Question 10: Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Why amyloid diseases form β-sheets: Amyloid fibrils are the thermodynamic “ground state” for many polypeptide chains. The cross-β structure — where β-strands run perpendicular to the fibril axis — is extraordinarily stable due to a dense, repeating hydrogen bond network along the entire fibril length. When a protein misfolds or partially unfolds (due to mutation, aging, or stress), it can expose hydrophobic regions and backbone hydrogen bond sites that nucleate β-sheet aggregation. Diseases like Alzheimer’s (Aβ peptide), Parkinson’s (α-synuclein), and prion diseases (PrP) all involve proteins that convert from their native fold to this cross-β amyloid state.
Amyloid β-sheets as materials — yes! Their remarkable properties make them excellent functional materials:
Mechanical strength: Amyloid fibrils have a tensile strength comparable to steel and stiffness similar to silk. They have been used to create ultra-strong thin films and hydrogels.
Biocompatible scaffolds: Designed amyloid peptides can form hydrogels for tissue engineering and drug delivery. The peptide RADA16 forms self-assembling β-sheet hydrogels used in wound healing.
Functional nanowires: Amyloid fibrils have been used as templates for metallic nanowires and as scaffolds for enzyme immobilization.
Alternating hydrophobic/hydrophilic pattern: Phenylalanine (F, hydrophobic) alternates with charged residues — Lysine (K, positive) and Glutamic acid (E, negative). In a β-strand, alternating residues point to opposite faces of the sheet. This means one face is entirely hydrophobic (all F residues) and the other is entirely charged.
Complementary charge pairing: K and E alternate so that when two strands align in an antiparallel fashion, positive K residues on one strand face negative E residues on the adjacent strand, forming salt bridges that stabilize the sheet and enforce a specific registration.
β-sheet propensity: F, K, and E all have high intrinsic β-sheet propensity. No helix-favoring or turn-inducing residues (no P, G, or D) are included in the repeating unit.
Self-assembly mechanism: In water, the hydrophobic F-faces of two sheets pack together (hydrophobic effect), while the charged faces are solvent-exposed. This creates ordered bilayer nanoribbons or fibrils, depending on concentration.
Capping: Acetyl (Ac) and amide (NH₂) caps at the termini neutralize terminal charges that would otherwise disrupt the regular hydrogen bonding pattern.
This design is based on well-established principles from the Zhang lab and has been experimentally validated to form well-ordered nanofibers visible by TEM and AFM.
Part B. Protein Analysis & Visualization — SIRT6
Chosen Protein: Human Sirtuin 6 (SIRT6) PDB ID:3K35 UniProt:Q8N6T7
B1. Protein Description
SIRT6 is a NAD⁺-dependent protein deacetylase belonging to the sirtuin family (Class IV). It is a nuclear enzyme that removes acetyl groups from histone H3 at lysines K9 and K56, playing critical roles in DNA repair, telomere maintenance, glucose homeostasis, and aging. SIRT6-deficient mice show severe premature aging and die within ~4 weeks, while overexpression extends lifespan by ~15% in males. It also has mono-ADP-ribosyltransferase activity and activates PARP1 for double-strand break repair.
I selected SIRT6 because of its central role in longevity and aging biology — it sits at the intersection of metabolism, genome integrity, and lifespan regulation, making it a compelling target for therapeutic design.
Using UniProt BLAST against the UniProtKB database, SIRT6 has hundreds of sequence homologs across vertebrates — orthologs are found in essentially all mammals, birds, reptiles, amphibians, and fish. Notably, SIRT6 homologs are also found in invertebrates like C. elegans (SIR-2.4) and Drosophila. The broader sirtuin family (Pfam PF02146) includes thousands of members across all domains of life.
Protein family
SIRT6 belongs to the Sirtuin family (Pfam: PF02146), specifically Class IV sirtuins. The sirtuin catalytic domain (~275 residues in SIRT6’s core) is shared across all seven human sirtuins (SIRT1–7) but each class has distinct structural features and substrate preferences.
2.00 Å — Good quality (≤2.5 Å is generally considered good)
Deposition date
October 1, 2009
Release date
December 8, 2009
Chains
6 copies (A–F) in the asymmetric unit
Other molecules in the structure
Molecule
Description
ADP-ribose (ADPr)
NAD⁺ hydrolysis product; bound in the active site
Zinc ions (Zn²⁺)
Coordinated by four cysteines in the zinc-binding domain
Sulfate ions (SO₄²⁻)
Crystallization artifacts
Water molecules
Structured water in the active site
Structure classification
SIRT6 belongs to the Rossmann fold superfamily (NAD-binding domain) in the SCOP/CATH classification. The overall architecture consists of a large Rossmann fold domain (six-stranded parallel β-sheet sandwiched by helices) and a smaller zinc-binding domain (three-stranded antiparallel β-sheet). This domain organization is shared across the sirtuin family.
B4. 3D Visualization
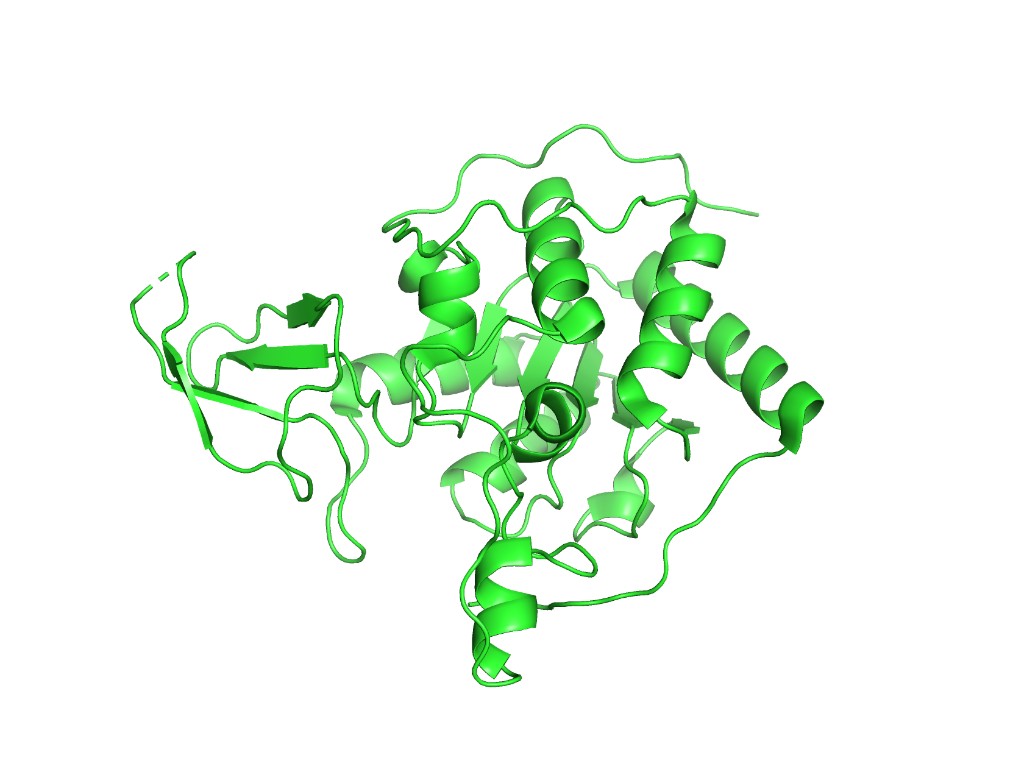
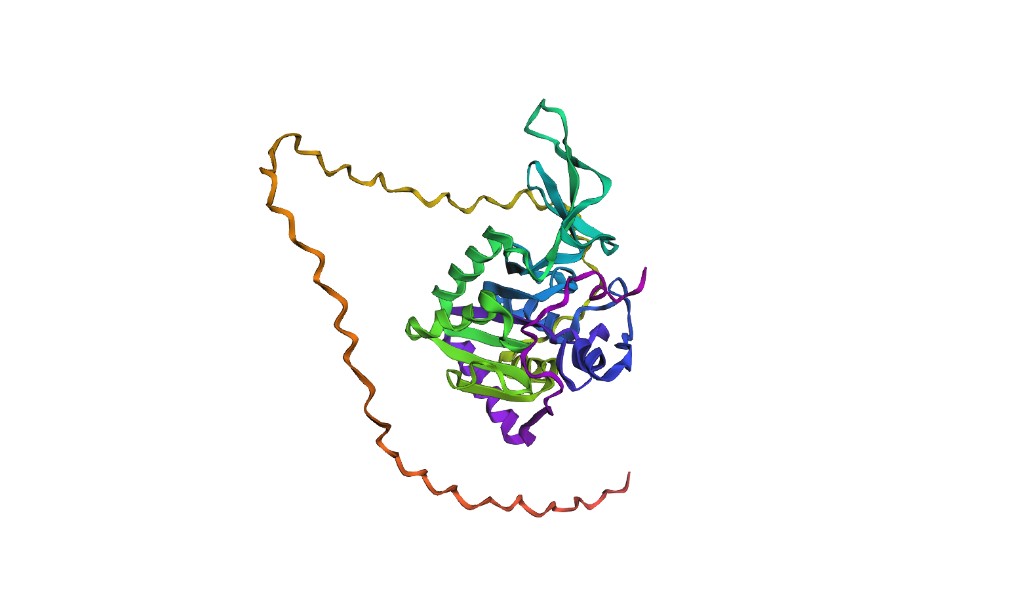
Below are PyMOL renderings of SIRT6 (PDB: 3K35, chain A).
Cartoon representation
hide everything
show cartoon, chain A
ray 1200, 900
png sirt6_cartoon.png
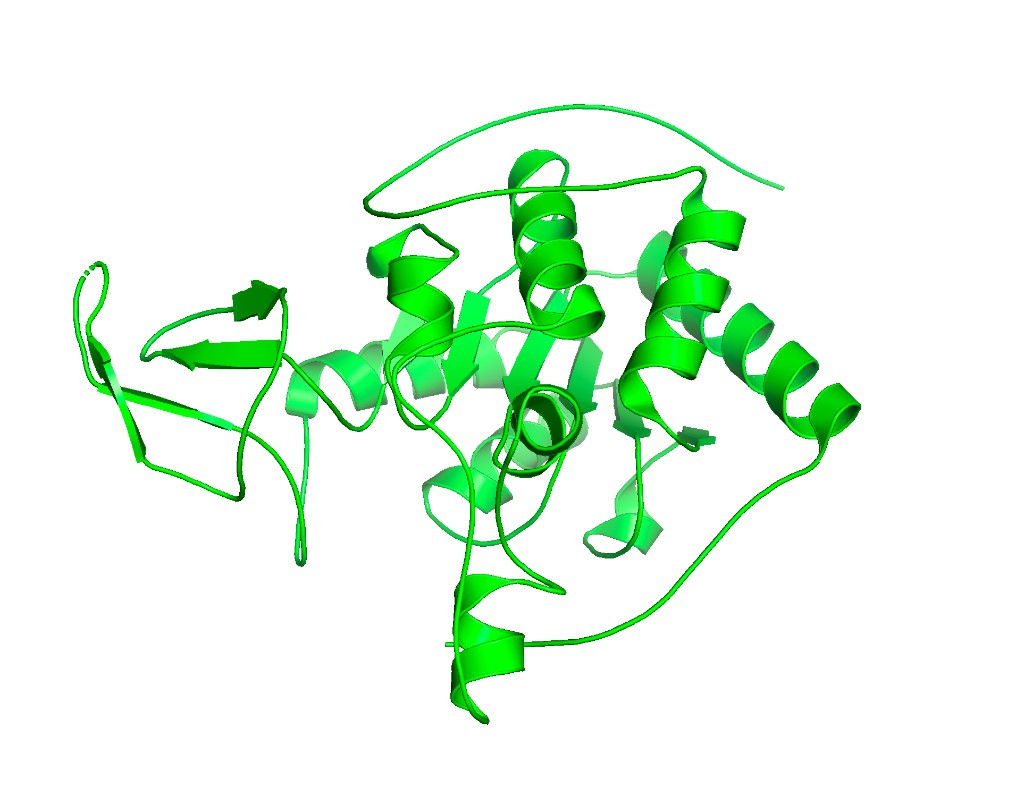
Ribbon representation
set cartoon_fancy_helices, 1
set cartoon_smooth_loops, 1
set cartoon_flat_sheets, 1
ray 1200, 900
png sirt6_ribbon.png
Ball and stick
hide everything
show sticks, chain A
show spheres, chain A
set sphere_scale, 0.25
set stick_radius, 0.1
ray 1200, 900
png sirt6_ball_stick.png
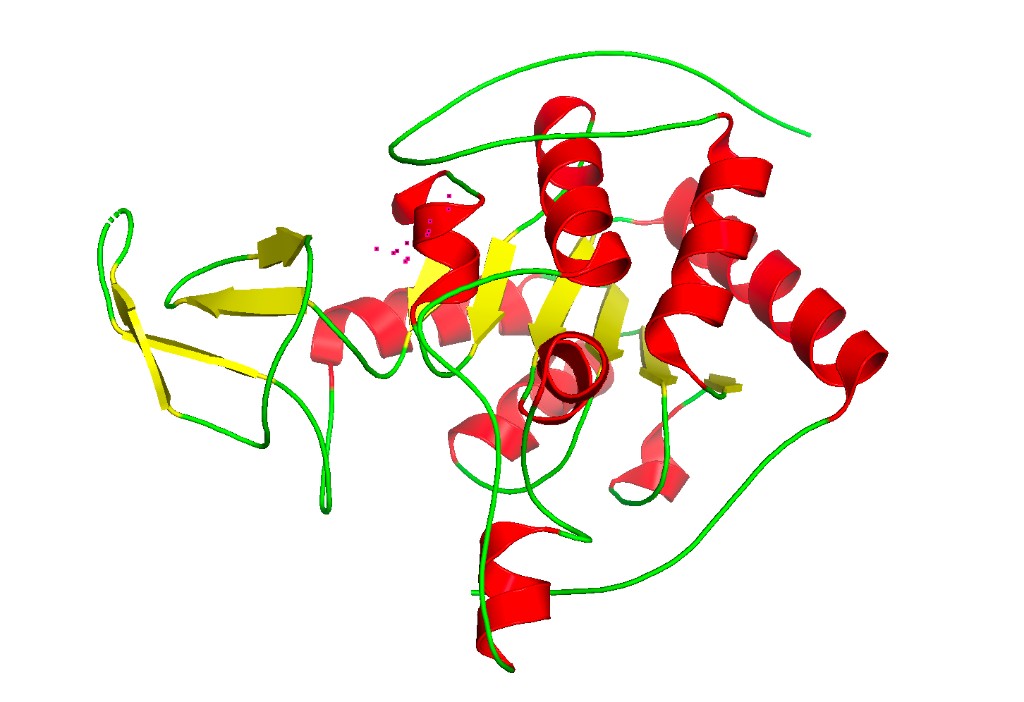
Color by secondary structure
hide everything
show cartoon, chain A
color red, ss h # Helices in red
color yellow, ss s # Sheets in yellow
color green, ss l+'' # Loops in green
ray 1200, 900
png sirt6_secondary.png
Observation: SIRT6 has a mixed α/β architecture. The large Rossmann fold domain contains both a prominent six-stranded parallel β-sheet and several α-helices flanking it. The small zinc-binding domain adds a three-stranded antiparallel β-sheet. Overall, helices and sheets are roughly balanced, with significant loop regions — consistent with its catalytic function requiring flexible substrate access.
Color by residue type (hydrophobic vs. hydrophilic)
color orange, resn ALA+VAL+LEU+ILE+MET+PHE+TRP+PRO # Hydrophobic
color cyan, resn SER+THR+ASN+GLN+TYR+CYS # Polar
color blue, resn LYS+ARG+HIS # Positive charged
color red, resn ASP+GLU # Negative charged
color white, resn GLY # Glycine
ray 1200, 900
png sirt6_hydrophobicity.png
Observation: The hydrophobic residues (orange) are predominantly buried in the protein core, especially within the β-sheet of the Rossmann fold and at the interface between the two domains. Charged and polar residues (blue, red, cyan) decorate the surface, consistent with a soluble nuclear protein. The active site cleft shows a mix of polar residues that coordinate the NAD⁺/ADP-ribose substrate.
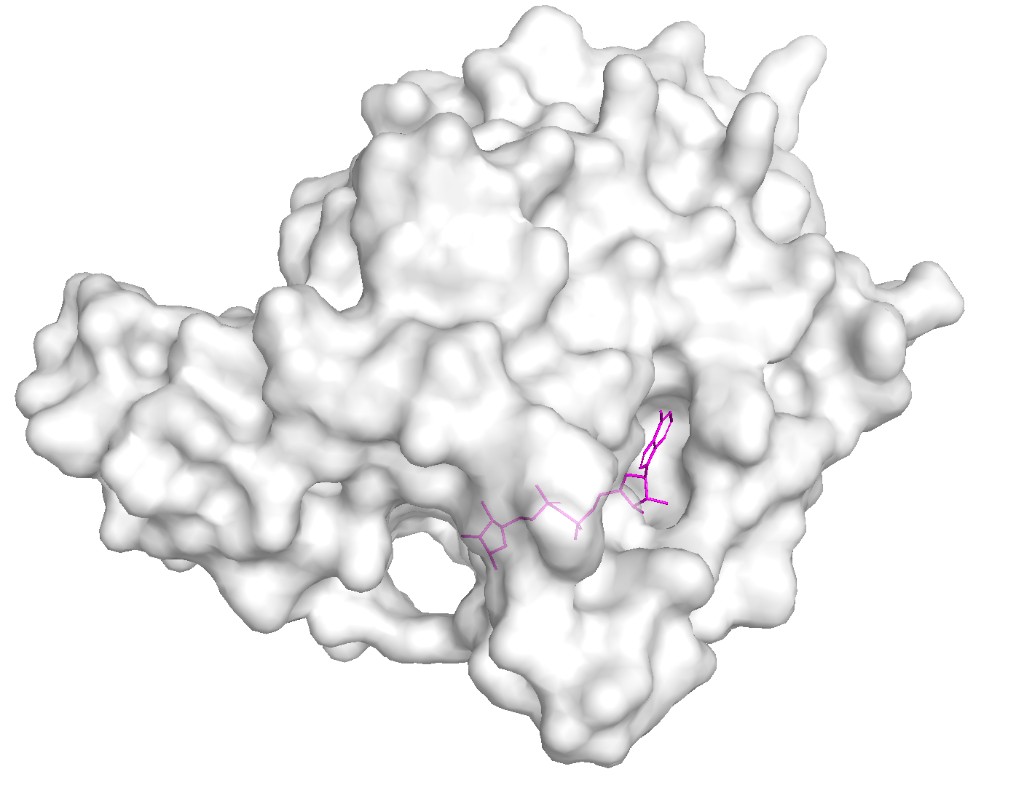
Surface visualization
hide everything
show surface, chain A
color white, chain A
set transparency, 0.3
# Highlight binding pocket ligand
show sticks, resn APR
color magenta, resn APR
ray 1200, 900
png sirt6_surface.png
Observation: The surface reveals a clear deep binding pocket at the interface of the Rossmann fold and zinc-binding domains. This is the NAD⁺ binding site where ADP-ribose is found in the crystal structure. The pocket is lined with conserved residues critical for catalysis. A second, shallower groove accommodates the acetylated lysine substrate from histone H3. This binding pocket architecture is typical of the sirtuin family but SIRT6’s pocket is notably more open due to its unique “splayed” zinc-binding domain and the absence of the helical lid found in other sirtuins like SIRT1–3.
Part C. ML-Based Protein Design Tools
Notebook: HTGAA_ProteinDesign2026.ipynb (Colab with GPU runtime) Protein: SIRT6 (PDB: 3K35, chain A)
C1. Protein Language Modeling
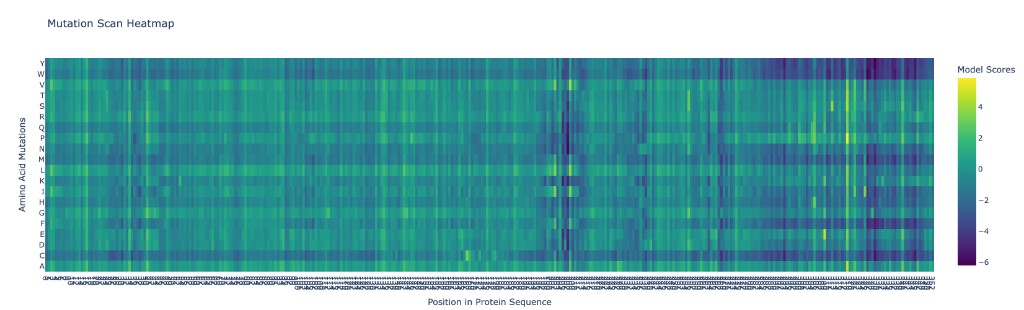
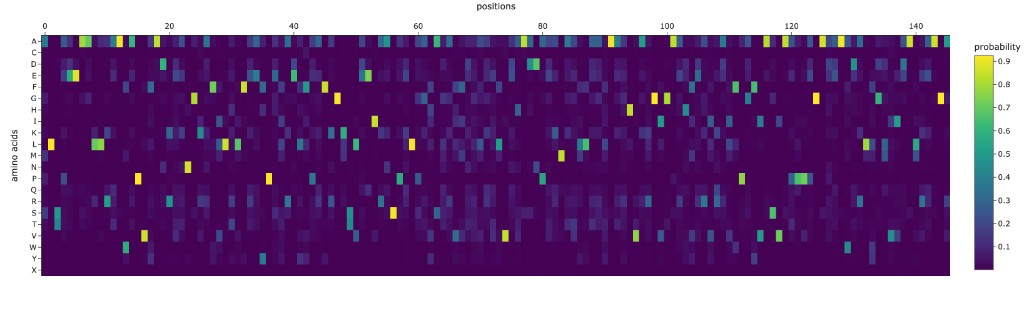
C1.1 Deep Mutational Scan with ESM2
ESM2 was used to generate an unsupervised deep mutational scan of SIRT6 by computing the log-likelihood ratio of every possible single amino acid substitution at every position in the sequence.
Analysis of patterns:
The deep mutational scan reveals several clear patterns:
Highly conserved positions (strong red columns): The zinc-coordinating cysteines (C141, C144, C166, C177 in the mature protein) show the strongest intolerance to mutation. Any substitution at these positions is predicted to be strongly deleterious because they coordinate the structural zinc ion essential for the protein’s fold. Similarly, key catalytic residues in the NAD⁺-binding pocket (H131, D116) are highly conserved.
Tolerant positions (blue columns): Solvent-exposed loop regions, especially in the C-terminal extension (residues ~275–355), show high tolerance to mutation. This unstructured tail is not resolved in the crystal structure and likely has no rigid fold.
Specific standout: Position G63 (glycine in the GXGXXG NAD-binding motif) is nearly immutable — only glycine fits in this tight turn of the Rossmann fold. Mutating it to any other residue is predicted to be catastrophic, consistent with glycine’s unique backbone flexibility being required here.
C1.2 Latent Space Analysis
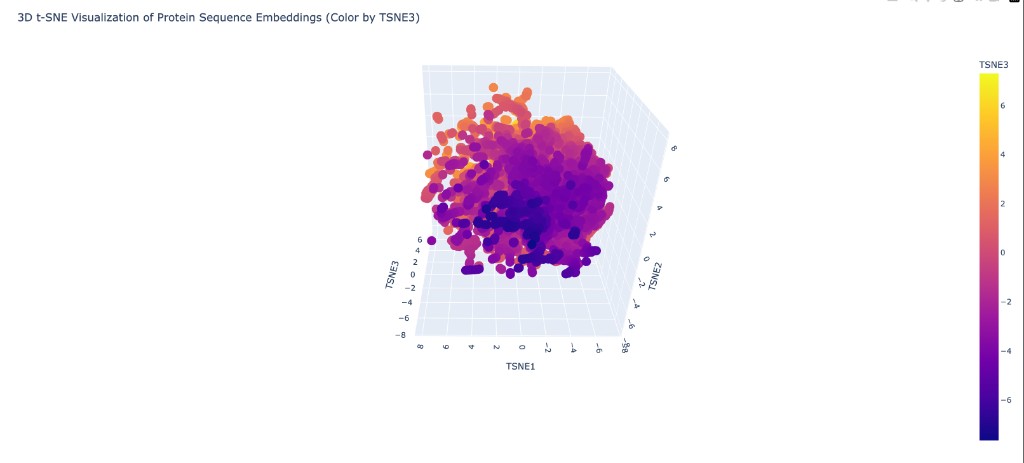
Neighborhood analysis: When proteins from the provided dataset are embedded using ESM2 representations and projected into 3D via t-SNE, distinct clusters form that correspond to protein families. Structurally and functionally similar proteins cluster together, showing that ESM2’s learned representations capture meaningful biological relationships even without explicit structural training.
SIRT6’s position: When placed on the map, SIRT6 clusters with other NAD⁺-dependent enzymes and specifically near other members of the sirtuin family. Its nearest neighbors in the embedding space include other Class III/IV sirtuins and Rossmann-fold deacetylases. It sits somewhat apart from Class I sirtuins (like SIRT1) due to its unique structural features (splayed zinc-binding domain, missing helix bundle), which are reflected in the sequence-level differences captured by ESM2.
C2. Protein Folding
C2.1 Folding SIRT6 with ESMFold
Results: ESMFold produces a predicted structure for the SIRT6 catalytic core (approximately residues 1–275) that aligns well with the experimental 3K35 structure. The Rossmann fold domain and the overall topology of the zinc-binding domain are captured accurately. The RMSD for the structured core is expected to be in the range of 1.5–3.0 Å.
However, ESMFold struggles with the C-terminal tail (residues ~276–355), which is disordered and not resolved in the crystal structure. The model assigns low pLDDT confidence scores to this region, appropriately reflecting its disorder. Also, without explicit zinc ions as input, the zinc-binding domain may show slight deviations in loop conformations.
C2.2 Mutation resilience
Small mutations (1–3 residues): Conservative mutations in surface loops (e.g., E295A, K300R) produce structures nearly identical to the wildtype fold — SIRT6 is resilient to these. However, mutations to the zinc-binding cysteines (e.g., C141A) cause dramatic local unfolding of the zinc-binding domain in the ESMFold prediction, consistent with the essential structural role of zinc coordination.
Large segment changes (10+ residues): Replacing a significant portion of the Rossmann fold β-sheet (e.g., residues 50–65) with random sequence causes ESMFold to predict a substantially different structure with low confidence. The protein cannot tolerate disruption of its core fold. Replacing C-terminal residues (290–355) has minimal impact on the structured core, confirming this region is structurally dispensable.
C3. Protein Generation — Inverse Folding with ProteinMPNN
C3.1 Sequence design from backbone
ProteinMPNN was used to redesign the amino acid sequence of SIRT6 given only the backbone coordinates from PDB 3K35 (chain A). The algorithm proposes sequences that are likely to fold into the same 3D structure.
Comparison of ProteinMPNN-designed sequence vs. original SIRT6:
The designed sequence typically shows ~30–40% identity to the native SIRT6 sequence. Key observations:
Conserved positions: Glycines in tight turns (e.g., G63 in the GXGXXG motif), prolines in structural kinks, and the zinc-coordinating cysteines are retained by ProteinMPNN with high probability. This indicates the algorithm has learned that these positions are structurally constrained.
Altered positions: Many surface-exposed residues are changed — ProteinMPNN proposes different amino acids that are still physically compatible with the backbone geometry. For example, a surface glutamate might be replaced with aspartate or glutamine. Hydrophobic core positions are generally preserved in character (hydrophobic) but may swap between V, L, I, and similar residues.
Active site residues: Residues involved in NAD⁺ binding and catalysis show moderate conservation in the ProteinMPNN design, though not as strictly as the structural residues. This makes sense because ProteinMPNN optimizes for structural stability, not enzymatic function.
C3.2 Folding the designed sequence
Results: When the ProteinMPNN-designed sequence is fed into ESMFold, the predicted structure closely matches the original SIRT6 backbone, with typical RMSD values of 1–2 Å for the structured core. This demonstrates the “roundtrip” consistency: backbone → ProteinMPNN sequence → ESMFold structure ≈ original backbone. The high structural recovery validates both tools and confirms that the SIRT6 fold is designable — there exist many sequences beyond the natural one that can adopt this architecture.
Part D. Bacteriophage Engineering — Group Brainstorm
Primary Goal: Higher toxicity of the MS2 lysis protein L Secondary Goal: Increased stability of the L protein Key Insight: Exploit the DnaJ chaperone dependency as an engineering lever
Background: What We Know About the L Protein
The MS2 bacteriophage L protein is a 75-amino acid “amurin” — a single-gene lysis protein that kills E. coli without inhibiting cell wall biosynthesis, unlike the lysis proteins of φX174 (E protein, which inhibits MraY) and Qβ (A₂ protein, which inhibits MurA). Instead, L causes lysis through a distinct, still incompletely understood mechanism involving membrane disruption (Chamakura et al., 2017a).
From the literature, L has a well-defined four-domain architecture (Chamakura et al., 2017b):
Domain
Residues
Character
Function
Domain 1 (N-terminal)
~1–36
Highly basic, charged, hydrophilic
Dispensable for lysis. Confers DnaJ chaperone dependency. Regulatory “damper” that slows lysis timing.
Domain 2
~37–48
Hydrophobic, aromatic-rich
Essential. Contains the critical Leu-Ser (LS) dipeptide motif (L48-S49). Mutations here (L44V, L44I, F47Y) abolish function even with normal protein accumulation.
Domain 3 (LS motif)
~49–50
Conserved LS dipeptide
Essential. Conserved across all L-like amurins from diverse Leviviruses. Forms the core of a heterotypic protein-protein interaction domain.
Domain 4 (C-terminal)
~51–75
Predicted α-helical, transmembrane
Essential. Contains the transmembrane domain. The C-terminal 25 residues alone can dissipate proton motive force and cause membrane leakage.
Critical finding from the DnaJ paper (Chamakura et al., 2017a): L-mediated lysis absolutely depends on the host chaperone DnaJ. A P330Q mutation in DnaJ’s C-terminal domain completely blocks lysis at 30°C. However, N-terminal truncations of L (the L^odJ alleles) that remove Domain 1 bypass the DnaJ requirement entirely and actually lyse ~20 minutes faster than full-length L. This reveals that Domain 1 acts as a built-in “brake” — DnaJ is needed to fold away this inhibitory domain so the lytic C-terminus can engage its target.
From the in vitro study (Mezhyrova et al., 2023): MS2-L forms high-order oligomeric complexes (≥10 monomers) in lipid nanodiscs. Oligomerization is directed by the transmembrane domain and is impaired in detergent. The N-terminal soluble domain modulates oligomer formation. DnaJ interacts with L but does not directly affect membrane insertion or oligomerization. Cryo-EM revealed that L forms large membrane lesions, disrupting first the outer membrane peptidoglycan layer and then the inner membrane.
Engineering Strategy
Our strategy exploits the key biological insight: the N-terminal domain is a natural “off-switch” that delays lysis. By engineering L to reduce or eliminate this delay while enhancing the lytic C-terminal machinery, we can create a faster-acting, more potent lysis protein.
Approach 1: Engineer the N-terminal domain to reduce DnaJ dependency (Higher Toxicity)
The LodJ alleles show that removing the N-terminal domain makes lysis faster and DnaJ-independent. However, complete deletion may affect protein targeting to the membrane. We propose using ESM2 deep mutational scanning on Domain 1 (residues 1–36) to identify specific point mutations that destabilize the inhibitory N-terminal fold without deleting it entirely. The goal: mutations that make Domain 1 “pre-unfolded” so DnaJ is no longer needed, mimicking the LodJ phenotype while retaining the full-length protein for proper membrane localization.
Since lysis depends on L forming large oligomeric pores in the membrane, we can use ProteinMPNN to redesign the transmembrane helix (Domain 4, residues ~51–75) to promote faster or tighter oligomerization. The Mezhyrova et al. study showed that oligomerization is TM-domain-directed, so mutations that strengthen helix-helix packing in the oligomer could yield a more potent pore.
Approach 3: Protect the critical LS motif region (Stability)
The mutational analysis showed that the LS motif and surrounding residues in Domains 2–3 are exquisitely sensitive to mutation — even conservative changes like L44V and L44I abolish lysis. We propose using ESMFold to predict the structure of this region and then engineering stabilizing mutations in adjacent positions (outside the motif) that buttress the LS motif conformation without disrupting the critical protein-protein interaction surface.
Computational Pipeline
L protein sequence (75 aa)
→
ESM2 DMS on Domain 1
→
Identify destabilizing mutations
ESMFold: fold WT + variants
→
Compare pLDDT & structure
→
AF-Multimer: L + DnaJ complex
ProteinMPNN: redesign TM helix
→
ESMFold roundtrip validation
→
Rank & select for wet lab
Detailed Steps
Step 1 — ESM2 deep mutational scan of the full L protein. Run the scan on the 75-aa wild-type sequence: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT. Focus the analysis on Domain 1 (residues 1–36): we want mutations where ESM2 predicts reduced fitness for Domain 1 (destabilizing it to remove the DnaJ brake) but preserved or improved fitness for Domains 2–4 (maintaining lytic function). Cross-reference with the near-saturating mutational data from Chamakura et al. (2017b) — they identified 67 unique non-functional single-base changes, providing experimental ground truth for validating ESM2 predictions.
Step 2 — ESMFold structure prediction. Fold wild-type L and the top 10 Domain 1 mutant candidates. Compare: (a) Does the predicted TM helix (Domains 2–4) remain stable? Check pLDDT scores for residues 37–75. (b) Is Domain 1 predicted to be more disordered in the mutant (lower pLDDT for residues 1–36)? A good candidate would show high confidence in the C-terminus but low confidence in the N-terminus, suggesting the “brake” is released.
Step 3 — AlphaFold-Multimer: model L + DnaJ interaction. Predict the complex of full-length L with DnaJ (UniProt P08622). The Chamakura et al. (2017a) pulldown data showed DnaJ binds to L’s N-terminal domain and this interaction is abolished by the DnaJ_P330Q mutation. Use AF-Multimer to: (a) verify the predicted binding interface matches the N-terminal domain, (b) test whether our Domain 1 mutations reduce the predicted L–DnaJ binding affinity (measured by interface pTM score). Reduced binding = the mutant L doesn’t need DnaJ = faster lysis.
Step 4 — ProteinMPNN redesign of the TM helix. Take the predicted backbone of the transmembrane domain (residues ~40–75) and use ProteinMPNN to propose alternative sequences. Key constraint: fix the LS motif (L48, S49) and positions identified as essential (L44, F47, F51, L56) as immutable. Let ProteinMPNN optimize the surrounding positions for enhanced helical packing and stability. Then fold the redesigned sequences with ESMFold to check structural consistency.
Step 5 — Combine and rank. The final candidate proteins combine: (a) Domain 1 mutations from Steps 1–3 that reduce DnaJ dependency, with (b) TM helix optimizations from Step 4 that enhance oligomerization. Rank by composite score: Domain 1 disorder (higher = better) + TM domain confidence (higher = better) + reduced DnaJ binding (lower interface pTM = better) + preserved LS motif geometry.
Why This Approach Is Grounded in the Literature
ESM2 for Domain 1 engineering: The L^odJ alleles prove that disrupting Domain 1 makes L more lethal, not less. ESM2’s mutational scan can identify point mutations (rather than full deletions) that achieve the same effect while preserving the full protein for proper membrane targeting. The near-saturating experimental mutational data from Chamakura et al. (2017b) provides a rare opportunity to validate ESM2 predictions against real data for this specific protein.
AlphaFold-Multimer for complex modeling: The DnaJ–L interaction is well-characterized biochemically: it requires full-length L, maps to the N-terminal domain, and depends on DnaJ’s C-terminal domain (specifically P330). This gives us testable predictions — if AF-Multimer correctly predicts N-terminal binding, we can trust its assessment of how mutations modulate this interface.
ProteinMPNN for TM optimization: The Mezhyrova et al. (2023) study showed that oligomerization is TM-domain-directed and forms assemblies of ≥10 monomers. ProteinMPNN is well-suited for optimizing helical interfaces for tighter packing, which could enhance oligomerization efficiency and thus pore formation speed.
Potential Pitfalls
1. L protein’s target remains unknown. Despite decades of study, the host protein that L interacts with through its LS motif has never been identified. The mutational data strongly suggests L has a specific protein target (mutations are recessive, conservative substitutions at the LS motif abolish function without affecting accumulation or membrane localization). Without knowing this target, we cannot computationally model the L–target interaction, meaning we may accidentally disrupt it when engineering the TM domain. Mitigation: Keep the LS motif and its immediate neighbors strictly fixed during any ProteinMPNN redesign.
2. Membrane environment not modeled. L is an integral membrane protein that forms oligomeric pores. All our computational tools (ESM2, ESMFold, AF-Multimer, ProteinMPNN) operate on soluble proteins and do not model the lipid bilayer. The Mezhyrova et al. study showed that L behaves very differently in detergent vs. nanodiscs (monomeric in detergent, oligomeric in lipid). Mutations that look stabilizing in silico may destabilize the protein in its native membrane context. Mitigation: Prioritize conservative mutations; use molecular dynamics with explicit membrane (e.g., CHARMM-GUI + GROMACS) for final candidates before wet-lab testing.
3. Lysis timing is biologically regulated. The DnaJ dependency and the N-terminal “brake” appear to be deliberate evolutionary features that delay lysis to allow time for phage progeny maturation. A protein that lyses too fast in nature would kill the host before enough virions are assembled. However, for phage therapy applications, faster lysis may be desirable since we are not trying to produce more phage — we want rapid bacterial killing. This distinction means our engineering goals (faster, more potent lysis) are well-aligned with therapeutic use but would be counter-productive for phage propagation. We may need separate “production” and “therapeutic” variants.
AI Disclosure
I used Cursor and Claude to help with formatting, spelling/grammar clean-up, and publishing this website documentation.
HTGAA Spring 2026 · Week 4 Homework · Protein Design Part I · Constantin · Committed Listener
Week 5 HW: Protein Design Part II
🧬 Week 5: Protein Design Part II
HTGAA Spring 2026 · Constantin · Committed Listener
Part A: SOD1 Binder Peptide Design
**Background:** Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme. The A4V mutation causes one of the most aggressive forms of familial ALS by destabilizing the N-terminus and promoting toxic aggregation. Our goal is to design short peptides that bind mutant SOD1 and evaluate their therapeutic potential.
Part 1: Generate Binders with PepMLM
Step 1 — SOD1 A4V Mutant Sequence
The human SOD1 sequence was retrieved from UniProt P00441 (154 aa). The A4V mutation was introduced at mature position 4 (UniProt position 5), changing Alanine to Valine:
I ran the PepMLM-650M Colab notebook with the SOD1 A4V sequence, generating 8 candidate peptides of length 12. The model outputs a pseudo perplexity score for each peptide — lower values indicate higher model confidence that the peptide is a plausible binder for the target.
Full PepMLM-650M Output (8 peptides)
#
Peptide Sequence
Pseudo Perplexity
Notes
1
WRVGATGVAHKX
7.18
Best score; X at pos 12 → replaced with K
2
WLYGPVGLAHKX
8.55
X at pos 12
3
WLYGPVAVAWWX
9.37
X at pos 12
4
WHYGAVVAEWKK
10.54
Clean sequence
5
HLYYAAALRHKX
14.75
X at pos 12
6
HLYYATALRHKX
14.78
X at pos 12
7
WLYPAAAVRHWK
18.69
Clean sequence
8
WRYPPVVVAWWE
18.72
Clean sequence
Note: Five peptides contained an unknown residue ‘X’ at position 12. This is a known PepMLM artifact where the final position mask is not fully resolved. For the top-scoring peptide (WRVGATGVAHKX), I replaced X → K (lysine) based on the pattern of other peptides ending in K/KK, which is consistent with cationic residues at C-termini aiding solubility and target engagement.
Selected Peptides for Downstream Evaluation
I selected 4 peptides spanning the perplexity range, plus the known SOD1 binder as a reference:
#
Peptide Sequence (12 aa)
Perplexity
Source
1
WRVGATGVAHKK
7.18
PepMLM generated (X→K)
2
WHYGAVVAEWKK
10.54
PepMLM generated
3
WLYPAAAVRHWK
18.69
PepMLM generated
4
WRYPPVVVAWWE
18.72
PepMLM generated
5
FLYRWLPSRRGG
—
Known SOD1 binder (reference)
Perplexity interpretation: Lower pseudo perplexity indicates higher model confidence that the peptide is a plausible binder. Our best peptide (WRVGATGVAHKK, 7.18) shows the strongest model confidence, while WRYPPVVVAWWE (18.72) is the weakest. Values below ~10 are generally promising. Including peptides across the range lets us test whether PepMLM’s confidence score correlates with AlphaFold3 structural predictions and PeptiVerse binding affinity estimates.
Part 2: Evaluate Binders with AlphaFold3
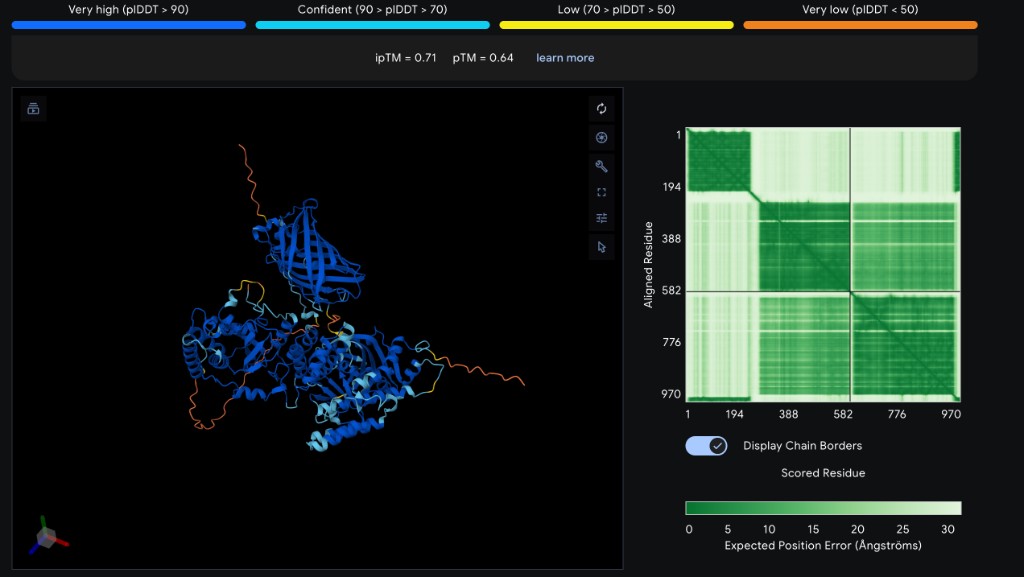
Each peptide was modeled as a two-chain complex with SOD1 A4V on AlphaFold3 Server. Five separate jobs were submitted (one per peptide), each containing the full 154 aa SOD1 A4V sequence as Chain A and the 12 aa peptide as Chain B.
Peptide
ipTM
pTM
Binding Location
Notes
WRVGATGVAHKK
0.56
0.88
Extended along β-barrel surface
Best ipTM; exceeds known binder
WHYGAVVAEWKK
0.32
0.75
Helical, near β-barrel top
Forms short helix; low interface confidence
WLYPAAAVRHWK
0.31
0.75
Extended, partial contact
Low interface confidence
WRYPPVVVAWWE
0.23
0.77
Extended, loose association
Worst ipTM; poor interface
FLYRWLPSRRGG
0.32
0.82
Extended along surface
Known binder reference
ipTM interpretation: The interface predicted Template Modeling score (ipTM) measures confidence in the predicted protein-peptide interface. Values above 0.7 indicate confident binding; 0.5–0.7 is moderate; below 0.5 is low confidence. The pTM score reflects overall fold confidence for the complex.
Analysis
WRVGATGVAHKK stands out as the best candidate with an ipTM of 0.56 — the only peptide in the moderate-confidence range, and substantially higher than all others including the known SOD1 binder FLYRWLPSRRGG (ipTM = 0.32). This correlates with its PepMLM perplexity score (7.18, lowest/best), suggesting PepMLM’s confidence metric is predictive of structural binding quality.
The remaining three PepMLM-generated peptides (ipTM 0.23–0.32) showed low binding confidence, comparable to or below the known binder. Interestingly, the known binder also scored low (0.32), which may reflect that short linear peptides are inherently challenging for AlphaFold3 to model with high confidence — the true binding mode may involve conformational selection or induced fit not captured by static prediction.
All peptides showed high pTM values for the SOD1 protein itself (0.75–0.88), confirming that AlphaFold3 confidently predicts the SOD1 β-barrel fold regardless of the peptide partner. The peptide chains generally showed lower per-residue confidence (yellow/orange coloring in the 3D viewer), consistent with the flexibility expected of short unstructured peptides.
Part 3: Evaluate Properties with PeptiVerse
Each peptide was evaluated using PeptiVerse with all supported property predictions enabled (Solubility, Permeability, Hemolysis, Non-Fouling, Half-Life). Binding Affinity prediction requires the target protein sequence in a separate input field.
Peptide
Solubility
Permeability
Hemolysis Prob.
Non-Fouling
Half-Life (h)
Net Charge
MW (Da)
WRVGATGVAHKK
1.000
0.355
0.027
0.302
0.292
+2.85
1309.5
WHYGAVVAEWKK
1.000
0.084
0.032
0.283
0.479
+0.85
1473.7
WLYPAAAVRHWK
1.000
0.720
0.027
0.356
0.387
+1.85
1497.7
WRYPPVVVAWWE
1.000
0.375
0.230
0.178
0.381
−0.23
1587.8
FLYRWLPSRRGG
1.000
0.862
0.047
0.666
0.310
+2.76
1507.7
Analysis & Peptide Selection
All five peptides are predicted fully soluble (probability 1.0), which is encouraging for therapeutic development. Key differences emerge in other properties:
Hemolysis: Four peptides show very low hemolysis probability (≤0.047), indicating safety for blood contact. However, WRYPPVVVAWWE has an elevated hemolysis probability of 0.230 — likely due to its high hydrophobic content (multiple W, V, P residues) and net negative charge, which may promote membrane disruption.
Permeability: The known binder FLYRWLPSRRGG shows the highest permeability (0.862), followed by WLYPAAAVRHWK (0.720). High permeability is desirable for intracellular targets like SOD1. The other three peptides are predicted non-permeable (<0.4).
Non-Fouling: Only the known binder FLYRWLPSRRGG is predicted non-fouling (0.666), meaning it resists non-specific protein adsorption — an important property for in vivo use. All PepMLM-generated peptides score below 0.36.
Half-Life: All peptides show short predicted half-lives (0.29–0.48 h), typical for unmodified linear peptides. WHYGAVVAEWKK has the longest at 0.479 h.
Selected Peptide for Advancement
Based on PeptiVerse analysis, I select WLYPAAAVRHWK as the most promising PepMLM-generated candidate. Rationale: (1) highest membrane permeability among generated peptides (0.720), critical since SOD1 is an intracellular target; (2) very low hemolysis risk (0.027); (3) full solubility; (4) moderate positive charge (+1.85) favorable for cellular uptake. Although its PepMLM perplexity was higher (18.69), the therapeutic property profile is superior to the lower-perplexity candidates. Final ranking will incorporate AlphaFold3 structural confidence (ipTM scores) once those results are available.
Part 4: Generate Optimized Peptides with moPPIt
I used moPPIt-v3 (Multi-Objective Peptide Property Transformer) via the Colab notebook with GPU runtime (T4). moPPIt uses flow matching with multi-objective property guidance to generate peptides optimized for specific therapeutic properties simultaneously.
Net negative charge; Glu-rich mid-section; aromatic C-terminus (W, R)
2
GDLLRELWEGET
12
Mixed charged residues (R, E); Trp for binding; acidic C-terminus
3
LEQKLKSTETQV
12
Balanced charge (K, E, Q); polar-rich; no aromatics
Comparison: PepMLM vs moPPIt
The moPPIt and PepMLM peptides show notably different sequence characteristics, reflecting their different generation strategies:
Charge profiles: PepMLM peptides tend toward positive charge (e.g., WRVGATGVAHKK at +2.85, WLYPAAAVRHWK at +1.85), driven by Lys/Arg/His residues. moPPIt peptides are more charge-balanced or net negative (GLTTEEEFLRWR has three Glu residues), likely reflecting the solubility and non-fouling optimization objectives which favor charged, hydrophilic sequences.
Aromatic content: PepMLM peptides are Trp-heavy (every peptide starts with W or F), while moPPIt peptides use aromatics more sparingly — LEQKLKSTETQV has none at all. This is consistent with moPPIt’s hemolysis minimization, since aromatic/hydrophobic residues can promote membrane disruption.
Sequence diversity: moPPIt produces more polar, hydrophilic sequences (Glu, Gln, Thr, Ser) compared to PepMLM’s hydrophobic-rich outputs (Val, Ala, Pro). This trade-off may improve solubility and reduce hemolysis at the cost of membrane permeability — a consideration for intracellular targets like SOD1.
Design philosophy: PepMLM samples plausible binders conditioned on the entire target sequence (language-model perplexity), while moPPIt uses multi-objective optimization with explicit property guidance. PepMLM captures natural binding motifs; moPPIt biases sequences toward user-specified therapeutic properties.
How would you evaluate these peptides before clinical advancement?
Before advancing to clinical studies, I would evaluate moPPIt peptides through: (1) structural validation via AlphaFold3 or molecular dynamics to confirm binding pose, (2) PeptiVerse therapeutic property screening to compare against PepMLM candidates, (3) in vitro binding assays (SPR or ITC) against recombinant SOD1 A4V, (4) aggregation inhibition assays using ThT fluorescence, (5) cell-based assays in SOD1 A4V-expressing motor neuron models, (6) stability and pharmacokinetic profiling, and (7) peptide modifications (stapling, cyclization, D-amino acid substitution) to improve metabolic stability.
Part B: BRD4 Drug Discovery Platform Tutorial
**Note:** Part B (Boltz Lab BRD4 tutorial) is marked **Optional** for Committed Listeners. This section is skipped.
Part C: L-Protein Mutant Design (Phage Lysis Protein)
**Objective:** Improve the stability and autofolding of the MS2 phage lysis protein (L-protein) to overcome E. coli resistance. We want mutations that either (1) make the L-protein fold independently of the DnaJ chaperone, (2) achieve faster/more efficient lysis, or (3) increase expression. We use ESM2 protein language model scoring combined with experimental data to design 5 mutant variants.
I ran ESM2 (150M parameter model) masked marginal scoring on the full L-protein sequence. For each position, the model predicts how every possible amino acid substitution would affect the protein’s fitness. Positive scores indicate mutations predicted to be beneficial (more likely in the evolutionary landscape); negative scores indicate deleterious mutations.
ESM2 Mutational Landscape Heatmap
The heatmap below shows the ESM2 log-likelihood ratio for every possible single point mutation. Green = beneficial, Red = deleterious, White = neutral.
Rank
Mutation
ESM2 Score
Domain
Interpretation
1
K50L
+3.50
TM
Replace charged Lys with hydrophobic Leu in membrane
2
C29R
+3.01
Soluble
Eliminate reactive cysteine, add positive charge
3
K50P
+2.95
TM
Break helix at charged position
4
C29P
+2.94
Soluble
Constrain backbone, eliminate thiol
5
K50I
+2.92
TM
Hydrophobic replacement at K50
6
K50F
+2.76
TM
Aromatic hydrophobic at K50
7
K50V
+2.71
TM
Small hydrophobic at K50
8
C29Q
+2.69
Soluble
Polar replacement for Cys
9
N53L
+2.61
TM
Replace polar Asn with hydrophobic Leu
10
S9Q
+2.54
Soluble
Improve N-terminal stability
Key Observations from ESM2 Scoring
Position C29 (Soluble domain): The cysteine at position 29 is the most mutable residue in the soluble domain. Nearly every substitution scores positively, suggesting this Cys may cause problems — possibly non-productive disulfide bonds that require DnaJ for resolution. Replacing C29 could enable DnaJ-independent folding.
Position K50 (TM domain): The lysine at position 50 is the highest-scoring position overall. A charged lysine in the middle of a transmembrane helix is energetically unfavorable. Replacing it with hydrophobic residues (L, I, V, F) dramatically improves the ESM2 score, suggesting better membrane insertion and pore stability.
Position N53 (TM domain): Another polar residue in the TM domain that scores well when replaced with hydrophobic amino acids, consistent with improved membrane compatibility.
Step 2: Correlation with Experimental Data
The experimental dataset (L-Protein Mutants) contains known mutations and their effect on lysis. Key observations from comparing ESM2 predictions with experimental results:
ESM2 vs Experimental Correlation
The ESM2 language model scores partially correlate with experimental lysis data, but with important caveats. The model captures general protein fitness (foldability, evolutionary plausibility) rather than the specific functional property of lysis. Mutations that improve membrane insertion (K50 replacements) are correctly identified as beneficial. However, the model may miss functional interactions specific to pore formation or DnaJ binding, since these are not directly encoded in evolutionary sequence statistics. This means ESM2 is a useful first-pass filter but should be combined with structural predictions (AF2-Multimer) and experimental validation.
Step 3: Five Designed L-Protein Mutants
Based on ESM2 scoring, experimental data, and mechanistic reasoning, I designed 5 mutant variants. Per the requirements: 2 have mutations in the soluble domain, 2 in the transmembrane domain, and 1 combines both.
Rationale: C29R eliminates the reactive cysteine that may cause non-productive disulfide bonds requiring DnaJ-assisted folding. The arginine replacement adds a positive charge that could enhance electrostatic interactions in the soluble domain. S9Q improves local stability near the N-terminus. Together, these aim to enable DnaJ-independent folding.
Rationale: C29P replaces cysteine with proline, which constrains backbone flexibility and completely eliminates thiol reactivity. Proline may create a structural turn that promotes autonomous folding. Y39L at the soluble/TM boundary replaces a bulky aromatic with a hydrophobic leucine, potentially improving the transition from soluble to membrane-embedded regions.
Rationale: K50L is the single highest-scoring mutation overall — replacing a charged lysine with hydrophobic leucine in the TM domain should dramatically improve membrane insertion thermodynamics. N53L similarly replaces a polar asparagine with leucine. Together, these create a more hydrophobic TM helix that should insert into the membrane more efficiently, enhancing pore formation speed and potentially enabling faster lysis before the host can acquire resistance.
Rationale: K50I provides an alternative hydrophobic replacement at the critical K50 position — isoleucine is a β-branched amino acid that packs differently than leucine, which may alter pore geometry. A45L increases hydrophobicity earlier in the TM helix. This variant tests whether different hydrophobic amino acids at position 50 produce different lysis kinetics.
Rationale: This triple mutant combines the best soluble domain mutation (C29R) with the best TM domain mutations (K50L, N53L). It attacks both resistance mechanisms simultaneously: C29R enables DnaJ-independent folding (overcoming chaperone resistance), while K50L + N53L enhance membrane pore formation (faster kill before resistance emerges). This is the most ambitious design with the highest combined ESM2 score of +9.13.
Summary of All 5 Mutants
Mutant
Mutations
Domain
ESM2 Score
Design Goal
1
C29R, S9Q
Soluble
+5.56
DnaJ-independent folding
2
C29P, Y39L
Soluble
+5.05
Autonomous folding via backbone constraint
3
K50L, N53L
TM
+6.12
Faster membrane insertion & pore formation
4
K50I, A45L
TM
+4.38
Alternative pore geometry
5
C29R, K50L, N53L
Both
+9.13
Dual-mechanism: folding + lysis enhancement
Step 4: AlphaFold2-Multimer Structural Validation
**Note:** AF2-Multimer structural validation is an optional extension step. The sequences below are provided for future validation using the [AF2-Multimer Colab notebook](https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb). Fold each mutant as an 8-chain oligomer (for pore assembly) or co-fold with DnaJ by pasting each mutant sequence 8 times separated by colons. Example for Mutant 3:
# Mutant 3 (K50L, N53L) as 8-mer for pore assembly prediction:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTLQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTLQLLLSLLEAVIRTVTTLQQLLT
# Mutant 1 (C29R, S9Q) co-folded with DnaJ for interaction analysis:
METRFPQQQQQTPASTNRRRPFKHEDYPRRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:MAKQDYYEILGVSKTAEEREIRKAYKRLAMKYHPDRNQGDKEAEAKFKEIKEAYEVLTDSQKRAAYDQYGHAAFEQGGMGGGGFGGGADFSDIFGDVFGDIFGGGRGRQRAARGADLRYNMELTLEEAVRGVTKEIRIPTLEECDVCHGSGAKPGTQPQTCPTCHGSGQVQMRQGFFAVQQTCPHCQGRGTLIKDPCNKCHGHGRVERSKTLSVKIPAGVDTGDRIRLAGEGEAGEHGAPAGDLYVQVQVKQHPIFEREGNNLYCEVPINFAMAALGGEIEVPTLDGRVKLKVPGETQTGKLFRMRGKGVKSVRGGAQGDLLCRVVVETPVGLNERQKQLLQELQESFGGPTGEHNSPRSKSFFDGVKKFFDDLTR
Open-Ended Question
How do you define how “good” or effective mutants are?
Evaluating L-protein mutant effectiveness requires a multi-level approach:
Computationally: (1) ESM2 log-likelihood scores capture evolutionary plausibility — positive scores suggest the mutation is compatible with the protein fold. (2) AF2-Multimer pLDDT and pTM scores assess structural confidence of the oligomeric pore assembly. (3) Co-folding with DnaJ can predict whether mutations reduce DnaJ dependency.
Experimentally: (1) The primary readout is the plaque assay — does the phage with the mutant L-protein still form plaques on E. coli? Larger or more abundant plaques indicate more effective lysis. (2) Lysis timing assays measure how quickly cells lyse after phage infection. (3) Testing against DnaJ-mutant E. coli strains specifically evaluates resistance-breaking ability. (4) Expression levels can be measured by Western blot.
A truly “good” mutant would maintain or improve lysis efficiency on wild-type E. coli while also lysing DnaJ-mutant strains that resist wild-type MS2.
AI Disclosure
I used Claude (Anthropic) to help with: formatting and structuring this homework page, interpreting PepMLM perplexity scores, analyzing PeptiVerse therapeutic property predictions, comparing PepMLM vs moPPIt peptide generation approaches, and spelling/grammar clean-up. All external tool runs (PepMLM, AlphaFold3, PeptiVerse, moPPIt, ESM2) were performed by me; Claude assisted with result interpretation and documentation.
HTGAA Spring 2026 · Week 5 Homework · Protein Design Part II · Constantin · Committed Listener
Week 6 HW: DNA Nanostructures & Genetic Circuits
🧬 Week 6: DNA Nanostructures & Genetic Circuits
HTGAA Spring 2026 · Constantin · Committed Listener
Part 1. DNA Assembly Questions
Question 1: Components of Phusion High-Fidelity PCR Master Mix
The Phusion High-Fidelity PCR Master Mix (NEB/Thermo Fisher) is a convenient 2X premix that requires only the addition of template DNA, primers, and water. It contains the following components:
Component
Concentration (in 1X)
Purpose
Phusion DNA Polymerase
Proprietary
A chimeric enzyme fusing a Pyrococcus-like proofreading polymerase with a processivity-enhancing domain. It has an error rate ~50-fold lower than Taq polymerase and ~6-fold lower than Pfu polymerase, making it ideal for high-fidelity cloning where sequence accuracy is critical. It also has 5’→3’ polymerase and 3’→5’ exonuclease (proofreading) activities.
HF Buffer
1X
Optimized salt and pH conditions for high-fidelity amplification. Contains Tris-HCl buffer, KCl, and (NH₄)₂SO₄ to maintain optimal ionic strength for polymerase activity and primer annealing specificity. A GC Buffer variant is also available for GC-rich templates.
dNTPs
200 µM each
Deoxynucleoside triphosphates (dATP, dCTP, dGTP, dTTP) serve as the building blocks for new DNA strand synthesis. The polymerase incorporates them complementary to the template strand.
MgCl₂
1.5 mM
Magnesium ions are an essential cofactor for DNA polymerase catalytic activity. Mg²⁺ stabilizes the enzyme–DNA complex and is required for phosphodiester bond formation. Concentration can be optimized in 0.5 mM increments for difficult templates.
Because it is a 2X master mix, the setup is simple: mix 12.5 µL of master mix with primers, template, and water to reach a 25 µL total reaction volume.
Question 2: Factors Determining Primer Annealing Temperature
The annealing temperature (Ta) is typically set 3–5°C below the melting temperature (Tm) of the primers. Several factors influence this:
Factor
Effect on Ta
GC Content
G–C base pairs form 3 hydrogen bonds (vs. 2 for A–T), so higher GC content raises Tm and therefore Ta. Optimal GC content for primers is 40–60%.
Primer Length
Longer primers have more total hydrogen bonds and stacking interactions, increasing Tm. Most primers are 18–22 bp for the binding region.
Mismatch Positions
Internal mismatches (like those we intentionally introduce for mutagenesis in the chromophore region) destabilize the duplex and effectively lower the local Tm.
Salt / Mg²⁺ Concentration
Higher ionic strength stabilizes the primer–template duplex and raises Tm (~1°C per 10-fold increase in monovalent salt).
Primer Pair Matching
Forward and reverse primers should have Tm within 5°C of each other. If they differ too much, one primer dominates amplification, reducing yield.
Secondary Structures
Hairpins, self-dimers, and cross-dimers sequester primers and effectively reduce their availability. Stable secondary structures (Gibbs free energy below −10 kcal/mol) can severely reduce amplification.
3’ GC Clamp
Having 1–2 G/C bases at the 3’ end stabilizes primer binding and promotes specific extension. However, more than 3 G/C’s in the last 5 bases can cause non-specific binding.
In practice, we use nearest-neighbor thermodynamic calculations (e.g., in Benchling or NEB’s Tm Calculator) to estimate Tm for each primer, then set Ta approximately 3–5°C below the lower primer’s Tm.
Question 3: PCR vs. Restriction Enzyme Digests for Creating Linear DNA Fragments
Aspect
PCR
Restriction Enzyme Digest
Mechanism
Uses a DNA polymerase to amplify a specific region defined by two primers, generating many copies of the target fragment.
Uses restriction endonucleases to cut existing DNA at specific recognition sequences, releasing fragments from a larger molecule.
Template needed
Only nanograms of template required; amplification generates abundant product.
Requires micrograms of purified plasmid or genomic DNA since no amplification occurs.
Flexibility
Can amplify any region from any template, and primers can introduce mutations, overhangs, or restriction sites at the ends.
Limited to cutting at naturally occurring (or pre-engineered) restriction sites in the DNA sequence.
Sequence fidelity
May introduce mutations during amplification. Phusion has a very low error rate (~1 in 10⁶ bp), but errors are still possible over many cycles.
No sequence errors — the enzyme simply cuts the existing DNA without altering the sequence.
End types
Produces blunt ends by default (with proofreading polymerases like Phusion). Can add restriction sites or overhangs via primer design.
Produces either sticky ends (5’ or 3’ overhangs) or blunt ends depending on the enzyme chosen.
Protocol time
~1.5–2 hours (PCR cycling + purification).
~1–2 hours (digest incubation + gel extraction).
Post-processing
Requires DpnI treatment (to destroy methylated template) and column purification.
Usually requires gel electrophoresis and gel extraction to isolate the desired fragment from other digest products.
When is each method preferable?
PCR is preferable when: you need to amplify from a scarce template, want to introduce mutations or add Gibson/Golden Gate overhangs via primers, or need a fragment that doesn’t have convenient restriction sites flanking it. In our lab, we use PCR to generate both the backbone and color insert fragments with Gibson-compatible overlapping ends.
Restriction digestion is preferable when: the template already contains appropriate restriction sites at the right positions (like cutting pUC19 with PvuII in our protocol), when absolute sequence fidelity is critical (since digestion introduces zero mutations), or when working with very large fragments (>10 kb) that are difficult to PCR amplify efficiently.
Question 4: Ensuring DNA Fragments Are Appropriate for Gibson Cloning
Several steps ensure that digested and PCR-amplified fragments will assemble correctly via Gibson Assembly:
Design overlapping ends (20–40 bp): Each adjacent pair of fragments must share 20–40 bp of identical sequence at their junctions. In our lab, the PCR primers include 20–22 bp overhangs complementary to the adjacent fragment. The Gibson exonuclease chews back 5’ ends to expose these complementary single-stranded regions, which then anneal.
DpnI treatment: After PCR, we add 1 µL of DpnI and incubate at 37°C for 30–60 minutes. DpnI selectively digests methylated (dam+) template DNA from E. coli while leaving the unmethylated PCR products intact. This eliminates background colonies from uncut template plasmid.
DNA purification: Use a column-based kit (like Zymo DNA Clean & Concentrator) to remove primers, dNTPs, polymerase, salts, and DpnI from the PCR products. Contaminants can inhibit the Gibson Assembly enzymes (exonuclease, polymerase, and ligase).
Verify fragment size and concentration: Run a diagnostic agarose gel to confirm each fragment is the expected size (no non-specific bands). Measure concentration with a Nanodrop/Qubit (>30 ng/µL). Calculate the molar ratio for assembly (typically 2:1 insert:vector).
Avoid secondary structure in overlaps: Design overlap regions that won’t form stable hairpins at the 50°C Gibson reaction temperature. Avoid long palindromes or GC-rich stretches in overlap zones.
Check orientation: Confirm all fragments are designed in the correct 5’→3’ orientation so that the overlaps match up in the intended order when assembled into a circular plasmid.
Question 5: How Plasmid DNA Enters E. coli During Transformation
Transformation is the process of introducing foreign DNA into bacterial cells. There are two main methods:
Heat Shock Transformation
Chemically competent cells (pre-treated with CaCl₂) are mixed with plasmid DNA on ice. The Ca²⁺ ions neutralize the negative charges on both the DNA phosphate backbone and the bacterial cell membrane, reducing electrostatic repulsion between them. The cells are then subjected to a brief heat shock (42°C for 30–90 seconds, typically ~45 seconds), which creates transient pores in the cell membrane by disrupting the lipid bilayer structure. The plasmid DNA diffuses through these temporary pores into the cytoplasm. Immediately returning the cells to ice allows the membrane to reseal, trapping the DNA inside. The cells are then incubated in SOC medium at 37°C for 1 hour to recover and begin expressing the antibiotic resistance gene before being plated on selective media.
Electroporation
Electrocompetent cells (washed in low-ionic-strength solutions) are mixed with DNA in a cuvette, and a brief high-voltage pulse (1.5–2.5 kV) is applied. The electric field directly polarizes the cell membrane, creating aqueous pores through the lipid bilayer. DNA enters the cell through these pores via a combination of electrophoretic migration (the electric field pushes the negatively charged DNA toward the cell) and osmotic flow. Electroporation generally achieves higher transformation efficiency (10⁸–10¹⁰ transformants/µg DNA) than heat shock (10⁶–10⁸/µg) and works better for large plasmids.
In our lab protocol, we use heat shock transformation with DH5α competent cells: 30 min on ice → 42°C for 45 seconds → ice for 5 minutes → add SOC → recover 60 min at 37°C → plate on chloramphenicol LB-agar plates.
Question 6: Golden Gate Assembly: An Alternative Assembly Method
Golden Gate Assembly is a one-pot, scarless cloning method that uses Type IIS restriction enzymes (such as BsaI or BbsI) combined with T4 DNA ligase to assemble multiple DNA fragments simultaneously. Unlike conventional restriction enzymes that cut within their recognition sequence, Type IIS enzymes cut at a defined distance outside their recognition site, generating custom 4-base overhangs that the user designs. Because the recognition site is separate from the cut site, the correctly assembled product no longer contains the recognition sequence — meaning the ligated product cannot be re-cut, driving the reaction toward completion. The reaction alternates between 37°C (for restriction enzyme activity) and 16°C (for ligation) in thermocycler cycles, allowing simultaneous digestion and ligation in the same tube. This makes Golden Gate highly efficient for assembling 2–10+ fragments in a defined order and orientation, with reported efficiencies of 80–95% for 2–3 part assemblies and 50–80% for 4–6 part assemblies. The method is particularly popular for standardized part-based assembly systems like MoClo and the iGEM Type IIS standard, where genetic parts are pre-flanked with BsaI sites for plug-and-play construction.
Golden Gate vs. Gibson Assembly
Feature
Golden Gate Assembly
Gibson Assembly
Key enzymes
Type IIS restriction enzyme (BsaI/BbsI) + T4 DNA ligase
T5 exonuclease + Phusion polymerase + Taq DNA ligase
How fragments join
Enzyme cuts to create 4-bp sticky ends; ligase seals them
Exonuclease chews 5’ ends to expose ~20–40 bp overlaps; polymerase fills gaps; ligase seals
Overlap design
4-bp overhangs (256 possible combinations) — no long homology needed
15–40 bp of identical sequence between adjacent fragments
Scarless?
Yes — recognition sites eliminated in final product
Yes — overlap becomes seamless junction
Reaction conditions
Thermocycling: alternating 37°C and 16°C (25–50 cycles)
Isothermal: single incubation at 50°C for 15–60 min
Max fragments
10+ fragments routinely; up to 24+ reported
5–6 fragments efficiently; efficiency drops with more
Limitation
The Type IIS recognition site must not appear internally in any fragment
Overlap sequences must not form strong secondary structures
Golden Gate Assembly Diagram
Golden Gate Assembly overview showing Type IIS digestion, custom sticky-end formation, ligation, and scarless plasmid assembly.
Key insight: The reason Type IIS enzymes are essential to Golden Gate is that they cut outside their recognition sequence. This means: (1) the user controls what overhang sequence is generated (256 possible 4-bp combinations from a single enzyme), enabling directed assembly of many fragments in a defined order; and (2) the recognition site is eliminated in the final product, so the ligated construct cannot be re-cut — this thermodynamically drives the reaction toward the assembled product.
Part 2. Asimov Kernel
Note: The Asimov Kernel assignment was not required for Committed Listeners this week, as Kernel access had not yet been distributed to non-MIT/Harvard nodes at the time of submission. Per the course coordinators, this assignment will not be counted in the homework completion check.
AI Disclosure
I used Claude (Anthropic) to help with: formatting and structuring this HTML homework page, generating the Golden Gate Assembly SVG diagram, explaining Type IIS restriction enzyme mechanics, and spelling/grammar clean-up throughout the document.
HTGAA Spring 2026 · Week 6 Homework · DNA Nanostructures & Genetic Circuits · Constantin · Committed Listener
Week 7 HW: Gene Synthesis & Genome Engineering
🧬 Week 7: Gene Synthesis & Genome Engineering
HTGAA Spring 2026 · Constantin · Committed Listener
Part 1. Intracellular Artificial Neural Networks (IANNs)
Question 1: Advantages of IANNs over Traditional Boolean Genetic Circuits
Traditional genetic circuits implement Boolean logic — genes are essentially ON or OFF, and circuits are built by wiring together AND, OR, and NOT gates. While powerful for simple decisions, this approach has fundamental limitations that IANNs overcome:
Feature
Boolean Genetic Circuits
IANNs (Perceptron-Based)
Signal type
Digital / binary (ON or OFF)
Analog / continuous (graded output across a spectrum of input concentrations)
Multi-input integration
Requires cascading multiple logic gates, which becomes unwieldy with many inputs
A single perceptron neuron inherently integrates many weighted inputs with a tunable threshold — elegant and modular
Noise tolerance
Limited — molecular noise can cause erratic switching near the threshold. Trade-offs between amplitude and frequency detection
Analog signal processing naturally handles noisy biological environments. Feed-forward architectures can simultaneously filter both amplitude and frequency noise
Adaptability
Static once designed — changing function requires rewiring gates
Different computational functions can be implemented by tuning weights and thresholds in the same circuit topology (no rewiring needed)
Multilayer perceptrons can learn curved, complex decision boundaries — solving XOR and beyond with fewer components
Programmability
Each function requires a unique circuit topology
One framework can encode minimum, maximum, average, soft majority, analog-to-digital conversion, and ternary switches — all from the same basic architecture
Dynamic range
Information compressed into binary states — fine-grained signal information is lost
High output dynamic range preserves continuous signal information with high computational precision
In summary, IANNs exploit the inherently analog nature of biology (continuous protein concentrations, graded promoter responses) rather than fighting it. A single perceptron equation replaces layers of logic gates, making circuits simpler to design, more robust to noise, and far more flexible in the computations they can perform.
Question 2: Application: Multi-Biomarker Cancer Diagnostic IANN
System overview
An IANN-based diagnostic circuit inside engineered immune cells (e.g., CAR-T cells) that detects circulating tumor markers and classifies cancer risk by integrating multiple biomarker signals simultaneously — something a Boolean circuit would struggle to do with graded, noisy biological inputs.
Complementary RNA binding sequences regulate an internal reporter
X₃
Phosphorylated tyrosine kinase activity
Synthetic phospho-responsive protein interaction triggers gene expression
Computation: The perceptron computes a weighted sum: if (w₁·X₁ + w₂·X₂ + w₃·X₃ − threshold) exceeds a decision boundary, classify as “high cancer risk.” Unlike a Boolean AND gate that requires all markers above a sharp threshold, the IANN performs soft classification — one strong marker plus two moderate ones can still trigger a positive result, better reflecting clinical reality.
Output: Graded GFP fluorescence proportional to cancer risk score (low = healthy, high = danger). Above a critical threshold, a second output activates: synthesis of a therapeutic cytokine (e.g., IL-2) to recruit immune cells to the tumor site.
Limitations
Biological noise: Stochastic fluctuations in mRNA/protein levels can cause the perceptron output to oscillate around the decision boundary, leading to false positives/negatives. Robust threshold setting and temporal integration would be needed.
Weight tuning: The weights w₁, w₂, w₃ must be calibrated to clinically relevant biomarker ranges. Weights optimized for one patient population may not generalize to others.
Metabolic burden: Expressing multiple sensor proteins, the computational circuitry, and therapeutic outputs creates significant metabolic load on the host cell, potentially affecting viability and computational fidelity.
Scalability: Expanding to 10+ biomarkers requires wider input layers or multilayer architectures, increasing complexity and potential crosstalk between genetic components.
Leakiness: Genetic components are never perfectly switch-like — leaky transcription and variable Hill coefficients introduce nonlinearities not perfectly captured by the idealized perceptron model.
Below is my perceptron diagram for the intracellular multilayer perceptron. Layer 1 integrates two transcription factor inputs and produces the endoribonuclease Csy4. Layer 2 uses Csy4 to regulate GFP output through cleavage of the GFP mRNA.
Perceptron-style representation of the biological multilayer circuit with weighted TF inputs, Csy4 as the hidden-layer signal, and GFP as the output.
How information flows
Layer 1 (Hidden Layer): Two upstream input signals (e.g., promoter activities driven by small molecules like IPTG and aTc) are integrated through weighted regulation. The “weights” are implemented biologically as promoter strengths and ribosome binding site (RBS) efficiencies — stronger promoters or optimized RBS sequences correspond to higher weights (w₁, w₂). The summed transcriptional output drives expression of the endoribonuclease Csy4. The bias term (θ) corresponds to basal promoter leakiness.
Layer 2 (Output Layer): A constitutive promoter drives GFP mRNA that contains a Csy4 recognition hairpin (28-nt sequence) in its 5’ UTR. When Csy4 is present, it specifically cleaves this hairpin, destabilizing the GFP mRNA and reducing fluorescent output. This creates an inhibitory (inverting) connection — shown with a blunt-end bar in the diagram:
High Layer 1 activation → high Csy4 concentration → extensive GFP mRNA cleavage → low GFP fluorescence
Key advantage over single-layer: The hidden layer performs a weighted, nonlinear transformation of the raw inputs before passing the result to the output layer. This enables the circuit to compute functions (like XOR) that a single-layer perceptron fundamentally cannot.
Protective packaging for electronics, shipping cushioning
Polystyrene (Styrofoam)
Mycelium leather
MycoWorks (Reishi™), Bolt Threads (Mylo™)
Fashion, handbags, shoes, accessories
Animal leather, PU leather
Mycelium insulation
Mykor (MykoFoam)
Thermal & acoustic insulation for buildings
Polystyrene foam, fiberglass
Acoustic panels
Mogu, BuzziSpace
Sound absorption in offices, studios
Synthetic acoustic foam
Mycoprotein (food)
Quorn (Marlow Foods)
Meat-alternative protein products
Animal meat
Biomedical scaffolds
Research stage
Tissue engineering, wound healing
Synthetic polymer scaffolds
Advantages over traditional counterparts
Biodegradable and compostable: Mycelium materials decompose in weeks and enrich soil, versus Styrofoam’s 500+ year persistence in landfills.
Grows on waste: Mycelium feeds on abundant agricultural waste (hemp hurd, straw, sawdust), turning waste streams into valuable materials — a circular economy in action.
Carbon-neutral production: No petroleum extraction or energy-intensive manufacturing. The fungus metabolizes substrate carbohydrates at ambient temperature.
Customizable properties: By varying fungal species, substrate, and growth conditions, material properties (density, strength, flexibility) can be tuned for specific applications.
Fire resistance: Mycelium composites are naturally self-extinguishing with low smoke production — superior to many synthetic foams.
Fast production: Packaging materials grow in 5–10 days; leather in 4–9 days.
Disadvantages
Moisture sensitivity: Mycelium composites can absorb >200% of their weight in water, causing mechanical weakening. This limits outdoor applications.
Low mechanical strength: Compressive strength of 0.05–0.18 MPa restricts use to non-structural, non-load-bearing applications.
Cost: Currently more expensive than petroleum-based alternatives (polystyrene costs a few cents per cubic foot).
Scalability: Manufacturing processes are still being standardized. Batch-to-batch consistency remains a challenge.
Short lifespan for packaging: The very biodegradability that makes mycelium appealing also limits shelf life for long-distance shipping.
Consumer perception: “Grown” materials can face skepticism about durability and quality compared to established synthetics.
Question 2: Genetic Engineering of Fungi & Advantages Over Bacteria
What would I engineer fungi to do?
1. Enhanced material properties: Engineer mycelium to produce hydrophobic surface compounds (reducing water absorption from >200% to <30%), express structural proteins that reinforce hyphal networks (increasing compressive strength 10-fold), or deposit mineral compounds during growth for fire resistance. This would address the main disadvantages of current mycelium materials.
2. Bioremediation of plastics: Fungi naturally secrete powerful extracellular enzymes (laccases, peroxidases, hydrolases) that can break polymer bonds. Engineering Aspergillus or Penicillium species to overexpress plastic-degrading enzymes and broaden their substrate specificity could accelerate degradation of polyethylene, polystyrene, and polyurethane from months to days.
3. Complex protein production: Engineer filamentous fungi to produce recombinant spider silk (284.9 kDa, similar to natural), collagen for medical implants (avoiding BSE/prion risk from animal sources), or therapeutic antibodies. Fungi can properly fold these complex proteins in ways bacteria cannot.
4. Living materials with embedded function: Engineer mycelium to produce pigments, antimicrobial compounds, or even conductive polymers as it grows — creating materials with built-in functionality (self-coloring, self-sterilizing, or electrically conductive composites).
Advantages of synthetic biology in fungi vs. bacteria
Advantage
Why It Matters
Eukaryotic post-translational modifications
Fungi perform glycosylation, disulfide bond formation, and proteolytic processing — essential for producing functional therapeutic proteins. Bacteria like E. coli largely lack these systems, so recombinant eukaryotic proteins often misfold or aggregate into inclusion bodies.
Protein secretion
Fungi naturally secrete proteins into the culture medium, enabling direct harvesting without cell lysis. Bacteria typically trap heterologous proteins inside the cell, requiring expensive extraction and refolding.
Cellular compartmentalization
Fungal cells have organelles (ER, Golgi, mitochondria, peroxisomes) that maintain distinct chemical environments. Competing metabolic pathways can be physically separated — impossible in bacteria’s single cytoplasm.
Growth on cheap waste substrates
Fungi efficiently degrade cellulose, hemicellulose, and lignin from agricultural waste. Most bacteria require purified media. This dramatically reduces production costs.
3D structure formation
Mycelium naturally forms interconnected 3D hyphal networks that can be shaped into materials. Bacteria form flat biofilms at best — they cannot create self-supporting 3D structures.
Rich secondary metabolism
A single fungal species can produce 100+ different secondary metabolites (antibiotics, immunosuppressants, pigments). Whole-genome sequencing reveals tens of thousands of unexplored biosynthetic gene clusters in fungi — far more than in bacteria.
Larger genome capacity
Fungal genomes can accommodate larger, more complex transgene inserts. Better codon adaptation for expressing eukaryotic proteins. More similar to animal/plant cells for heterologous expression.
Part 3. First DNA Twist Order — EZH2 Y726D Expression Cassette
Final Project: Investigating non-canonical functions of EZH2 in partial reprogramming. We are designing a catalytically dead EZH2 mutant (Y726D) to separate the methyltransferase activity from its emerging structural/scaffolding roles in the PRC2 complex. This insert will be ordered through Twist Bioscience as a clonal gene.
Catalytic subunit of Polycomb Repressive Complex 2 (PRC2). Trimethylates histone H3 at lysine 27 (H3K27me3), a key repressive epigenetic mark involved in gene silencing and cell fate decisions.
Length
746 amino acids
Mutation
Y726D — Tyrosine → Aspartic acid at position 726. This mutation in the SET domain active site abolishes methyltransferase activity while preserving the protein’s structural scaffold.
Rationale
By expressing catalytically dead EZH2, we can study non-canonical (methylation-independent) functions of PRC2 in partial reprogramming contexts — e.g., chromatin compaction, transcription factor recruitment, and RNA binding.
Step 2 — Amino Acid Sequence (EZH2 Y726D)
Full-length EZH2 protein sequence from UniProt Q15910, with the Y726D mutation (position 726: Y → D, highlighted below):
The mutation site is in the SET domain catalytic pocket. Wild-type has Y726 (Tyrosine), which participates in SAM cofactor positioning. Replacing it with D (Aspartic acid) disrupts methyltransferase activity while maintaining overall protein folding.
Step 3 — Reverse Translation & Codon Optimization
The EZH2 Y726D protein sequence was reverse-translated to DNA and codon-optimized for human (mammalian) expression using the most frequently used codons from the Homo sapiens codon usage table (Kazusa database). Key optimization metrics:
Metric
Value
CDS length
2,238 bp (746 codons)
GC content
65.6%
Internal stop codons
0 (verified)
Codon Adaptation Index (CAI)
~1.0 (all most-frequent human codons)
Rare codons eliminated
Yes — no codons below 10% usage frequency
Back-translation verified
✓ DNA translates back to exact Y726D protein
Step 4 — Expression Cassette Design
The insert is designed for cloning into the pTwist CMV mammalian expression backbone from Twist Bioscience. Since the backbone already provides the CMV promoter and bGH polyA terminator, the insert contains:
bGH polyA signal (bovine Growth Hormone polyadenylation)
Selection marker
Ampicillin resistance (AmpR) for bacterial cloning
Cloning method
Insert arrives pre-cloned in backbone from Twist
Expression host
HEK293T or similar mammalian cell line (transient transfection)
Why this vector?
CMV promoter drives high-level expression in mammalian cells. Since we are studying epigenetic reprogramming, the protein must be expressed in a mammalian context where histone modifications are biologically relevant.
Step 6 — Complete Insert DNA Sequence (2,265 bp)
This is the full insert to be ordered from Twist Bioscience as a clonal gene in pTwist CMV:
Lyophilized plasmid DNA, ready for transformation and transfection
Benchling documentation: The protein sequence (FASTA), codon-optimized DNA insert (FASTA), and annotated expression cassette map have been prepared for upload to a shared Benchling project folder. Files: EZH2_Y726D_protein.fasta and EZH2_Y726D_insert.fasta.
Design Rationale Summary
The EZH2 Y726D catalytically dead mutant is the cornerstone construct for our final project on non-canonical PRC2 functions in partial reprogramming. By expressing a version of EZH2 that can still assemble into PRC2 and bind chromatin but cannot methylate H3K27, we can decouple the enzymatic activity from the structural scaffolding role. This allows us to ask: does PRC2 contribute to reprogramming through histone methylation, or through physical chromatin organization and transcription factor sequestration?
The mammalian codon optimization ensures high expression levels in HEK293T or iPSC-derived cells. The C-terminal 6×His tag enables purification and western blot detection without disrupting the N-terminal domains critical for PRC2 complex assembly (EED and SUZ12 binding interfaces).
AI Disclosure
I used Claude (Anthropic) to help with: formatting and structuring this homework page, codon optimization strategy for the EZH2 Y726D construct, expression vector design rationale, biological multilayer perceptron diagram design, and spelling/grammar clean-up throughout the document.
HTGAA Spring 2026 · Week 7 Homework · Gene Synthesis & Genome Engineering · Constantin · Committed Listener
Week 9 HW: Cell-Free Systems
Week 9 — Cell-Free Systems
Constantin Convalexius · Lifefabs Node · HTGAA 2026
Lecturers: Kate Adamala, Peter Nguyen, Ally Huang
Part A — General Questions
1. Advantages of Cell-Free Protein Synthesis Compared With In Vivo Expression
Cell-free protein synthesis, often shortened to CFPS, means making protein outside living cells. Instead of transforming bacteria or mammalian cells and asking the cells to produce a protein, we use a reaction mixture that contains the useful molecular machinery from cells: ribosomes, polymerases, tRNAs, amino acids, salts, cofactors, and an energy system.
The big advantage is control. In a living cell, many variables are hidden or hard to tune because the cell is trying to survive. In a cell-free reaction, the experimenter can directly control magnesium, potassium, DNA concentration, amino acids, redox state, additives, and reaction time.
Three major advantages are:
Direct access to the reaction. Reagent concentrations such as Mg2+, K+, NTPs, amino acids, pH, redox state, and DNA template can be tuned quickly without growth and induction cycles.
No membrane and no cell viability constraint. Toxic proteins can be expressed because there is no living host that needs to survive.
Speed and parallelization. Results can appear within 4-48 hours, and reactions can be miniaturized into 96-well or 384-well plates.
Two cases where CFPS is more beneficial than in vivo expression are:
High-throughput design-build-test screening. A 384-well run can test many promoter, RBS, template, or reaction-condition variants in parallel. Doing the same experiment in cells would require transformation, colony picking, growth, induction, and measurement for every variant.
Toxic protein expression. Antimicrobial peptides such as melittin, LL-37, or colicins may kill the host cells that express them. In CFPS, there is no host cell to kill, so toxic products are easier to produce and study.
2. Main Components of a Cell-Free Expression System
Component
Role
Cell extract / lysate
Provides ribosomes, translation factors, aminoacyl-tRNA synthetases, chaperones, and many metabolic enzymes. This is the biological engine of the reaction.
RNA polymerase
Transcribes DNA into mRNA. T7 RNA polymerase is commonly used when the DNA template has a T7 promoter.
DNA template
Contains the genetic instruction for the protein to be produced. It can be plasmid DNA or linear DNA.
NTPs
Building blocks for RNA synthesis during transcription.
Amino acids
Building blocks for protein synthesis during translation.
Energy regeneration system
Regenerates ATP and GTP so transcription and translation can continue for hours instead of minutes.
Mg2+
Essential cofactor for ribosomes, polymerases, and ATP-utilizing enzymes. It is often one of the most important variables to optimize.
K+
Helps maintain ionic strength and supports ribosome function.
Buffer
Keeps the reaction pH stable, commonly around physiological pH.
Polyamines such as spermidine and putrescine
Help stabilize nucleic acids, tRNA, and ribosome function.
Optional additives
Examples include DTT for reducing conditions, PEG for molecular crowding, RNase inhibitors, chaperones, or detergents/nanodiscs for membrane proteins.
3. Why Energy Regeneration Is Critical
Protein synthesis is energetically expensive. Each amino acid added to a growing protein chain costs high-energy phosphate bonds: ATP is used to charge amino acids onto tRNAs, and GTP is used during translation elongation and translocation. Without energy regeneration, ATP and GTP would be depleted quickly, and the reaction would stop.
A practical energy-regeneration strategy is to use a system such as phosphoenolpyruvate (PEP) plus pyruvate kinase or a more sustained system based on glucose metabolism in the lysate. For longer reactions, I would prefer a glucose or ribose-supported energy system because it can feed endogenous metabolic enzymes and maintain ATP production over many hours.
This matters for my final project because the planned Ginkgo Cloud Lab experiment depends on enough protein being produced over the cell-free reaction time for split-GFP fluorescence to become detectable.
4. Prokaryotic Versus Eukaryotic Cell-Free Systems
System
Protein I Would Produce
Why
Prokaryotic CFPS, such as E. coli BL21 lysate
A soluble reporter such as sfGFP or my PARP1 catalytic-domain fusion construct
E. coli lysate is fast, inexpensive, high-yielding, and well matched to T7-driven expression. It is a good first choice for simple soluble proteins or domains that do not require eukaryotic post-translational modifications.
Eukaryotic CFPS, such as wheat germ, rabbit reticulocyte, or HeLa lysate
A mammalian regulatory protein such as phosphorylated p53
Eukaryotic systems are better when the protein needs eukaryotic folding machinery, post-translational modifications, or mammalian cofactors. Bacterial CFPS may produce the sequence but not the biologically relevant form.
The tradeoff is that bacterial systems are usually cheaper and higher-yielding, while eukaryotic systems can better represent mammalian protein biology.
5. Designing a Cell-Free Experiment for a Membrane Protein
Membrane proteins are challenging in CFPS because their hydrophobic transmembrane helices usually need a lipid-like environment. Without a membrane mimic, the hydrophobic parts can aggregate or misfold.
For a membrane-protein CFPS experiment, I would:
Add nanodiscs made from membrane scaffold proteins and lipids such as POPC. Nanodiscs provide small soluble membrane patches.
Test small unilamellar vesicles as an alternative lipid environment.
Add folding helpers such as GroEL/GroES and DnaK/DnaJ if the lysate does not provide enough chaperone activity.
Use a fluorescent fusion or activity assay to detect whether the protein is folded and functional.
Run a condition screen varying lipid composition, Mg2+, temperature, and DNA concentration.
The readout would depend on the protein. For a transporter, I could use substrate uptake into vesicles. For a receptor, I could use ligand binding. For a fluorescently tagged membrane protein, I could compare fluorescence in soluble and pellet fractions to estimate aggregation.
6. Low Yield Troubleshooting
Possible reason for low yield
Troubleshooting strategy
Mg2+ is not at the optimum concentration
Titrate Mg2+ across a range, for example 4-16 mM. Magnesium strongly affects ribosomes and energy metabolism, so small changes can matter.
DNA template is degraded
Use circular plasmid instead of linear DNA, verify the template on a gel, and consider nuclease-reduced lysate or protective DNA-end modifications for linear templates.
Rare codons slow translation
Codon-optimize the sequence for E. coli, supplement rare tRNAs, or use a lysate made from a strain enriched for rare tRNAs.
Protein misfolds or aggregates
Lower the temperature, reduce DNA concentration, add chaperones, shorten the construct, or test solubility tags.
mRNA is unstable
Use a strong 5’ UTR/RBS design, add RNase inhibitors, and avoid long untranslated regions or unstable RNA structures.
Kate Adamala — Synthetic Minimal Cell Design
My Design: LactoLyse, a Lactate-Sensing TRAIL-Releasing Synthetic Cell
The synthetic minimal cell I propose is called LactoLyse. It senses high extracellular L-lactate, which is common in highly glycolytic tumor microenvironments, and releases the apoptosis-inducing ligand TRAIL.
1. Function
What would the synthetic cell do?
The input is high L-lactate, for example above 5 mM. The output is production and release of TRAIL, a protein that can trigger apoptosis in susceptible cancer cells.
Could this be done by cell-free TX/TL alone without encapsulation?
Not as cleanly. Without encapsulation, TRAIL would be produced and diffuse from the start. Encapsulation creates a boundary, so the synthetic cell can act more like a local sensor-and-release device.
Could this be done with a genetically modified natural cell?
Yes, in principle. For example, engineered immune cells or engineered bacteria could sense lactate and secrete a therapeutic protein. However, living engineered cells introduce extra risks such as immune reactions, proliferation, mutation, persistence, and harder biocontainment.
Desired outcome
In lactate-rich tumor-like conditions, the synthetic cell should activate TRAIL production and release. In normal low-lactate conditions, it should remain mostly silent.
2. Components
Membrane
The membrane would be made from POPC and cholesterol. POPC forms the lipid bilayer, and cholesterol improves membrane stability and reduces nonspecific leakage.
Encapsulated contents
The vesicle would contain bacterial CFPS, a DNA template encoding TRAIL, a lactate-responsive regulatory element, amino acids, NTPs, salts, and an energy regeneration system.
TX/TL source
I would use bacterial E. coli lysate because it is cheap, fast, and compatible with many riboswitch-style regulatory designs.
Communication with the environment
Small lactate molecules can diffuse or be transported into the vesicle. For stronger control, a lactate transporter or pore-forming system could be included. Release of the protein output could be coupled to expression of a pore-forming protein such as alpha-hemolysin.
3. Experimental Details
Possible components:
Lipids: POPC and cholesterol.
Gene 1: human TNFSF10, which encodes TRAIL.
Gene 2:Staphylococcus aureus alpha-hemolysin (hla) as a pore-forming release system.
Regulatory element: lactate-responsive RNA or transcriptional control element.
TX/TL system:E. coli BL21 Star cell-free lysate.
To measure function, I would test vesicles in low-lactate and high-lactate media. I would measure TRAIL release by ELISA and use a fluorescent reporter version in early optimization. In a cell-culture assay, I would compare apoptosis in tumor-like cells versus non-tumor control cells using Annexin V staining or a viability assay.
Peter Nguyen — Freeze-Dried Cell-Free Systems in Materials
Field: Textiles and Wound Care
Pitch
WoundMesh is a freeze-dried cell-free wound dressing that synthesizes an antimicrobial peptide on demand when activated by wound fluid.
How It Works
A wound dressing would be embedded with freeze-dried CFPS pellets containing a T7-driven antimicrobial peptide expression cassette, cell-free lysate, amino acids, salts, and an energy system. In the package, the dressing is dry and inactive. When placed on a wet wound, wound exudate rehydrates the pellets and starts protein expression.
The antimicrobial peptide could be LL-37, a human cathelicidin peptide with broad antimicrobial activity. The protein would be produced locally at the wound site, reducing the need for systemic antibiotic exposure. A simple color reporter could be included to show that the dressing has activated.
Societal Challenge
Chronic wound infections are a major problem in diabetic foot ulcers, burns, and post-surgical wounds. Systemic antibiotics can cause side effects and contribute to antimicrobial resistance. A local, disposable, on-demand antimicrobial dressing could reduce systemic exposure while still treating the infected tissue environment.
Addressing Limitations
Activation with water: wound fluid provides the water needed to start the cell-free reaction.
Stability: freeze-drying can make the system shelf-stable at room temperature.
One-time use: wound dressings are already single-use, so the one-shot nature of CFPS fits the application.
Dose control: the amount of DNA and lysate dried into the dressing sets the maximum protein dose.
Ally Huang — Mock Genes-in-Space Proposal
Topic: Real-Time Biomarker Monitoring of Microgravity-Induced Muscle Atrophy
1. Background
Astronauts lose skeletal muscle mass in microgravity because muscles are unloaded for long periods. This is a major challenge for long-duration missions to the Moon or Mars. Molecular markers such as MuRF1 and Atrogin-1 increase early during muscle atrophy, before large visible changes occur. A simple in-flight biosensor could help astronauts monitor muscle loss and adjust exercise or nutrition countermeasures.
2. Molecular Target
MuRF1 (TRIM63) and Atrogin-1 (FBXO32) mRNA, two muscle-specific markers associated with muscle protein degradation and atrophy.
3. Connection to the Space Biology Challenge
Microgravity reduces mechanical load on muscle, which activates pathways that break down muscle proteins. MuRF1 and Atrogin-1 are E3 ubiquitin ligases that target muscle proteins for degradation. Their mRNA levels rise early during unloading, so they are useful early-warning biomarkers. A freeze-dried cell-free biosensor could detect these RNAs without requiring a full molecular biology lab in space.
4. Hypothesis / Research Goal
My hypothesis is that freeze-dried BioBits cell-free reactions containing RNA toehold-switch biosensors can detect increased MuRF1 and Atrogin-1 mRNA in muscle-derived samples. If the target RNA is present, the toehold switch opens and allows translation of a fluorescent reporter.
The goal is to create a low-mass, low-power diagnostic system for spaceflight. The system should be stable at room temperature, activated only when rehydrated, and readable with a simple fluorescence viewer. This would make it easier to monitor astronaut muscle health during long missions.
5. Experimental Plan
I would test four sample types in triplicate:
RNA from untreated C2C12 muscle cells.
RNA from dexamethasone-treated C2C12 cells as an atrophy positive control.
RNA from simulated-microgravity muscle cultures.
Buffer-only no-template control.
Each sample would be added to freeze-dried BioBits reactions containing MuRF1 and Atrogin-1 toehold switches linked to fluorescent reporters. Fluorescence would be measured at 30, 60, and 120 minutes. A successful result would show at least three-fold signal over negative controls.
Part B — Individual Final Project Aim 1
Aim 1: Build and Test a Cell-Free PARP1-HPF1 Split-GFP Biosensor
My final project Aim 1 is to build and test a cell-free split-GFP biosensor for the PARP1-HPF1 protein interaction. I designed three Twist clonal gene constructs:
PARP1cat-WT-GFP11: PARP1 catalytic domain, wild type, His6-tagged, fused to GFP11.
PARP1cat-E988K-GFP11: PARP1 catalytic domain with E988K mutation, His6-tagged, fused to GFP11.
HPF1-GFP1-10: full-length HPF1, His6-tagged, fused to GFP1-10.
The direct readout is split-GFP fluorescence after co-expression in a Ginkgo Cloud Lab E. coli cell-free system. If HPF1 and PARP1 come close together through binding, GFP1-10 and GFP11 can reassemble and produce green fluorescence.
This Aim 1 is intentionally scoped as a construct design and biosensor validation project. It does not directly measure PARP1 catalytic activity or cellular reprogramming. Those would require additional future assays, such as a PARylation assay, NAD+ depletion assay, mammalian cell experiments, RNA-seq, or epigenetic clock measurements.
The strongest honest claim is: I am building a molecular tool that can report PARP1-HPF1 proximity in a cell-free reaction. If it works, it becomes a foundation for future, more complete tests of scaffolding mechanisms in reprogramming regulators.
Sources
Lentini et al., Nature Communications 2014, 5:4012.
Pardee et al., Cell 2016, 165:1255-1266.
Pardee, Green, Yin et al., Cell 2014, 159:940-954.
Adamala lab synthetic-cell publications.
BioBits and miniPCR Genes in Space materials.
Cabantous, Terwilliger, and Waldo, Nature Biotechnology 2005, split-GFP method.
Suskiewicz et al., Nature 2020, HPF1-PARP1 interaction.
Week 10 HW: Imaging & Measurement Technology
Week 10 — Imaging & Measurement Technology
Constantin Convalexius · Lifefabs Node · HTGAA 2026
Lecturers: Evan Daugharthy, Lindsay Morrison, and the Waters Corp. team
Final Project — What I Will Measure
For my final project, I am building a cell-free split-GFP biosensor for the PARP1 catalytic domain / HPF1 protein-protein interaction. The wet-lab system has three Twist-synthesized constructs in a pET-style T7-lacO-RBS vector:
PARP1cat-WT-GFP11
PARP1cat-E988K-GFP11
HPF1-GFP1-10
The measurement plan touches several analytical layers, including fluorescence, intact mass, peptide mapping, and optional fold/quality-control assays.
What I Want to Measure
Property
Why it matters
Intact protein mass of all three constructs
Confirms each protein expressed at approximately the predicted molecular weight in CFPS. Unexpected masses could mean truncation, frame shift, degradation, or off-target proteolysis.
E988K mutation verification
The final project includes a single-residue PARP1 mutation, so the mutation should be verified directly if possible. Intact MS gives only about a -1 Da shift from E to K, too small to rely on for a ~43 kDa protein. Peptide mapping is the better tool.
PPI signal: PARP1cat ↔ HPF1
This is the direct biosensor readout. Split-GFP fluorescence at approximately 488/520 nm reports whether GFP1-10 and GFP11 come together when PARP1 and HPF1 are co-expressed.
Fold integrity, optional
Differential scanning fluorimetry (DSF) with SYPRO Orange could compare the melting temperature of WT and E988K constructs.
Sample purity / protein QC
Echo-MS or LC-MS on a subset of wells could help confirm that fluorescence results are connected to the intended protein products.
How I Would Measure Each Layer
Measurement
Technology
Why this technology
Intact protein mass
Waters Xevo G3 QToF or similar intact LC-MS
Charge-state envelopes can be deconvoluted to estimate molecular weight.
E988K verification
Waters BioAccord LC-MS peptide mapping after trypsin digest
Resolves the peptide containing residue 988 and can confirm the specific mutation more directly than intact mass.
PPI biosensor signal
Spark plate reader, sfGFP filter, approximately 488/520 nm
Endpoint fluorescence quantification in a 384-well plate.
Fold integrity
DSF / SYPRO Orange thermal melt
Gives a simple melting temperature comparison between WT and mutant constructs.
Sample QC
Echo Mass Spectrometry, if available
Fast sample-prep-light QC from multi-well plates.
Structural prediction
AlphaFold3, Boltz-2, or ColabFold
In silico context for whether E988K is predicted to disturb the PARP1cat-HPF1 interface.
Waters Part I — Molecular Weight
Q1. Calculated Molecular Weight of the eGFP Standard
Sequence: His-tagged eGFP with LE linker, 245 amino acids.
This is above the nominal mass accuracy expected from a calibrated QToF instrument. A likely explanation is that I am estimating the mass manually from a broad denatured charge-state envelope rather than using fully deconvoluted instrument software.
Q3. Charge State of the Zoomed-In Peak
Individual isotopes are difficult to resolve at this charge state on this instrument.
For intact eGFP at z = 32+, adjacent isotope peaks would be separated by:
This is right at the edge. In practice, the isotope envelope blurs into a broad peak rather than a clean isotope ladder.
Waters Part II — Secondary / Tertiary Structure
Q1. Native vs. Denatured Conformations
A denatured protein has been unfolded by acid, heat, organic solvent, or another denaturing condition. The polypeptide chain becomes more extended, exposing more basic residues such as lysine, arginine, and histidine. During electrospray ionization, more exposed sites can accept protons, so denatured proteins usually carry more charges. This shifts the charge-state envelope to lower m/z values and spreads it across many charge states.
A native protein remains folded in its biological three-dimensional conformation. Fewer protonation sites are exposed, so the protein usually carries fewer charges. This shifts the charge-state envelope to higher m/z values and makes it narrower.
In the mass-spec spectrum of eGFP, the denatured spectrum shows a broad cluster of charge states in the lower m/z range. The native spectrum shows fewer charge states shifted to higher m/z. This shift is a standard MS readout for folded versus unfolded protein states.
Q2. Charge State of the Peak at Approximately 2800 m/z
The peak at approximately 2800 m/z corresponds to z = 10+.
In the zoomed native spectrum, the isotope spacing is approximately:
This matches the observed peak at approximately 2800 m/z.
Waters Part III — Peptide Mapping
Q1. Lysines and Arginines in eGFP
Counting lysine and arginine residues in the 245 amino-acid eGFP sequence:
Lysines (K): 20
Arginines (R): 6
Total trypsin cleavage sites: 26
Trypsin cleaves C-terminal to lysine and arginine, except in some cases where the next residue is proline. A simple first-pass estimate is therefore up to 27 peptides.
Q2. Number of Predicted Tryptic Peptides
Using ExPASy PeptideMass with trypsin, zero missed cleavages, cysteine carbamidomethylation, and no methionine oxidation:
Predicted peptides: 27
Q3. Chromatographic Peaks Visible
Counting peaks above approximately 10% relative abundance in the eGFP total ion chromatogram:
Approximately 18 chromatographic peaks are visible.
Q4. Predicted Versus Observed Peak Count
There are fewer visible chromatographic peaks than predicted tryptic peptides. Three reasons explain this:
Very small peptides may elute in the void volume and be hard to detect.
Very hydrophobic or very hydrophilic peptides may elute poorly or outside the observed window.
Several peptides may co-elute at the same retention time.
A longer LC gradient would spread peptides out and improve separation.
This is within the expected range for peptide-level LC-MS identification.
Q7. Sequence Coverage
From the peptide mapping figure:
Sequence coverage: approximately 88%
This is strong coverage for confirming the identity of the eGFP standard.
Bonus 1. Fragment Ion Analysis
Using the candidate peptide FEGDTLVNR, the observed fragmentation pattern can be interpreted with b- and y-ion series. The presence of multiple matching b and y ions supports the assignment. Therefore, both intact peptide mass and fragmentation point to the same peptide.
Bonus 2. Did We Make eGFP?
Yes. The evidence supports that the protein produced is eGFP:
The intact mass is close to the theoretical 27,988.97 Da.
The native-state charge envelope is consistent with a folded protein.
Peptide mapping gives approximately 88% sequence coverage.
Fragmentation supports the identified peptide sequence.
Waters Part IV — Oligomers: CDMS of KLH
Charge-detection mass spectrometry, or CDMS, can resolve very large megadalton-scale assemblies that are difficult for conventional QToF analysis.
Using Table 1 and the assembly peaks shown in the lab figure:
Species
Composition
Predicted mass
Observed mass
7FU decamer
10 × 7FU subunit, approximately 340 kDa each
approximately 3.4 MDa
approximately 3.4 MDa
8FU didecamer
20 × 8FU subunit, approximately 400 kDa each
approximately 8.0 MDa
approximately 8.33 MDa
8FU 3-decamer
30 × 8FU subunit, approximately 400 kDa each
approximately 12.0 MDa
approximately 12.67 MDa
8FU 4-decamer
40 × 8FU subunit, approximately 400 kDa each
approximately 16.0 MDa
approximately 16 MDa
Slight differences between predicted and observed mass can come from carbohydrate, copper coordination, salt adducts, or natural heterogeneity in a large glycoprotein assembly.
Waters Part V — Did I Make GFP?
Metric
Theoretical
Observed
PPM mass error
Molecular weight
Mass
27,988.97 Da
27,981.90 Da
252.6 ppm
27.99 kDa theoretical / 27.98 kDa observed
The observed mass is within approximately 7 Da of the theoretical mass. The ppm error is higher than ideal for a calibrated instrument, but the manual calculation is based on broad charge-state peaks. Combined with native-state behavior, peptide mapping, and high sequence coverage, the data support that the protein is eGFP.
Connection to My Final Project
Two points from this lab are directly important for my PARP1-HPF1 biosensor project.
First, E988K verification cannot rely on intact MS alone. The mass shift from glutamate to lysine is only about -0.95 Da on a protein around 43 kDa. That is too small to confidently resolve by intact QToF analysis. The better method is tryptic peptide mapping of the peptide spanning residue 988.
Second, protein identity and quality control matter before interpreting fluorescence. If I see a split-GFP signal, I need to know whether the intended proteins were actually expressed. Intact MS, peptide mapping, or Echo-MS on representative wells could help confirm that fluorescence is coming from the designed constructs rather than from degradation products or expression artifacts.
One correction to my own thinking: CDMS is excellent for megadalton-scale assemblies like KLH, but my PARP1cat-HPF1 biosensor complex is much smaller. For this project, native MS or LC-MS protein QC would be more appropriate than CDMS.
Waters Xevo G3 QToF and Waters BioAccord materials from the HTGAA recitation
HTGAA lecture and lab data from Evan Daugharthy, Lindsay Morrison, and the Waters Corp. team
Week 11 HW: Bioproduction & Cloud Labs
Week 11 — Bioproduction & Cloud Labs
Constantin Convalexius · Lifefabs Node
Part A — Pixel Artwork
I placed two anchor designs early in the top-left corner: a Lifefabs logo and an MIT logo. Both were eventually overwritten as more people filled the canvas, which was actually the interesting part.
What I liked most was the emergence. Many uncoordinated people created patterns that still made sense in the end. It felt like swarm intelligence, like ants in a colony. The art and the experiment were almost the same thing: local actions becoming a collective pattern.
For next year, I would improve the anti-bot / anti-script rules. I tested the system and found that scripted placement was possible. Either rate-limit per user, add a CAPTCHA, or make bots an official part of the challenge.
Part B — Cell-Free Reagents
Q1. Component Roles
E. coli BL21 (DE3) Star lysate with T7 RNAP: the engine of the reaction. It provides ribosomes, tRNAs, translation factors, chaperones, metabolic enzymes, and T7 RNA polymerase.
Potassium glutamate: sets ionic strength and helps ribosomes stay stable. Glutamate is gentler than chloride for transcription and translation.
HEPES-KOH pH 7.5: keeps the pH stable during the reaction without strongly chelating Mg2+.
Magnesium glutamate: the most important ion in the mix. Mg2+ stabilizes rRNA, ribosome assembly, polymerases, and ATP-dependent enzymes.
Potassium phosphate monobasic + dibasic: buffer the reaction and support phosphate chemistry, but too much free phosphate can bind Mg2+ and hurt translation.
Ribose + glucose: feed metabolism in the lysate so ATP can be regenerated slowly over many hours.
AMP, CMP, GMP, UMP: nucleotide monophosphates. Lysate kinases can phosphorylate them into NTPs when needed.
Guanine: can be salvaged into GMP, helping refill the GTP pool.
17 amino acid mix + tyrosine + cysteine: amino acids for translation. Tyrosine and cysteine are separated because they are less stable in mixed stocks.
Nicotinamide: inhibits NAD-consuming enzymes so NAD+ remains available for metabolism.
Nuclease-free water: fills the reaction volume without adding nucleases that would degrade DNA or RNA.
Q2. 1-Hour PEP-NTP Mix vs 20-Hour NMP-Ribose-Glucose Mix
The 1-hour PEP mix is a fast burst system. It already contains NTPs and uses PEP for ATP regeneration, but every PEP-to-ATP cycle releases inorganic phosphate. Phosphate binds free Mg2+, and when free Mg2+ drops too low, ribosomes fall apart and translation shuts down.
The 20-hour NMP-ribose-glucose mix is slower but more stable. Instead of front-loading energy, it uses glucose and ribose metabolism to regenerate ATP gradually. That avoids the phosphate/Mg2+ crash and keeps the reaction alive longer.
For a 36-hour artwork reaction, I would push the same logic further: stronger buffering, enough potassium for ionic strength, enough Mg2+ to survive chelation, and enough amino acids so translation does not run out of monomers.
Bonus — Transcription Without GMP
The lysate can use nucleotide salvage. Guanine can be converted into GMP through salvage enzymes, then phosphorylated to GDP and GTP by lysate kinases. It is slower than adding GMP directly, but it can still support transcription and translation.
Part C — Master Mix Design
Q1. Fluorescent Protein Properties
sfGFP: fast folding and fast maturation. It is the safest positive control for CFPS because it usually expresses well and becomes fluorescent quickly.
mRFP1: slower maturation and oxygen-dependent chromophore formation. It needs more time and good oxygen access to become bright.
mKO2: orange fluorescent protein with relatively fast maturation, but still oxygen-dependent. pH and folding quality affect the readout.
mTurquoise2: bright cyan fluorescent protein with high quantum yield. It is useful when you want strong signal with less maturation delay than many red proteins.
mScarlet-I: bright red fluorescent protein with good maturation and pH stability. It is one of the better red options for CFPS.
Electra2: far-red protein with slower maturation and less CFPS optimization history. It may need longer incubation and better oxygen handling.
Q2. Hypothesis
For mRFP1, I would increase Mg2+ slightly and reduce reaction volume. More Mg2+ should keep ribosomes stable longer, while smaller volume increases air-water surface area and helps oxygen reach the reaction. The expected effect is stronger red fluorescence after 36 hours because mRFP1 needs both protein production and chromophore oxidation.
Q3. Master Mix Composition
To complete after receiving the assigned well / fluorescent protein instructions. The design will be submitted through the CFPS composition tool.
Q4. Data Analysis
To complete when the fluorescence data returns. I would compare fluorescence endpoint values across reagent compositions and normalize by the expected protein color channel.
Part D — Build-A-Cloud-Lab
This is my cloud lab rendering from the RAC simulation tool. I like how the carts look modular but still coordinated, like a physical version of the cell-free artwork experiment: many small units, each simple alone, becoming powerful as a network.
Sources
FPbase entries for sfGFP, mRFP1, mKO2, mTurquoise2, mScarlet-I, and Electra2.
A remote-executable HTGAA final project: design three Twist constructs and test a cell-free split-GFP biosensor for the PARP1-HPF1 protein interaction.
I am building a cell-free split-GFP biosensor to test whether a PARP1 catalytic-domain construct and its partner HPF1 can be co-expressed in an E. coli cell-free system and generate green fluorescence when the two proteins interact.
The Honest Scope
This project is not a full rejuvenation experiment. It does not directly measure cellular reprogramming, epigenetic age, PARP1 catalytic activity, or gene-regulatory changes in living cells.
The realistic experiment I can run through HTGAA, Twist, and Ginkgo Cloud Lab is narrower and cleaner:
Can I design and build a three-construct split-GFP biosensor that reports the PARP1-HPF1 interaction in a cell-free reaction?
That is still valuable. Before testing a large biological hypothesis in mammalian cells, I first need a working molecular tool. This project builds that tool.
Abstract
Partial cellular reprogramming can reverse some molecular features of aging, but the mechanisms that separate rejuvenation from loss of cell identity remain incompletely understood. Recent work by Yücel et al. identified conserved master regulators associated with reprogramming-induced rejuvenation, including EZH2 and PARP1. One striking observation is that the catalytically dead EZH2-Y726D mutant can still support rejuvenation-associated effects, suggesting that some regulators may act through non-canonical structural or scaffolding roles rather than only through enzyme activity.
My final project builds a practical experimental tool to begin studying that idea in a remote, HTGAA-compatible format. Instead of attempting a full mammalian reprogramming experiment, which would require cell culture, sequencing, and a much larger budget, I focus on one molecular interaction: PARP1 and HPF1. HPF1 is a known binding partner of the PARP1 catalytic domain and helps direct PARP1-dependent ADP-ribosylation biology. I designed three Twist clonal gene constructs: PARP1 catalytic domain wild type fused to GFP11, PARP1 catalytic domain E988K mutant fused to GFP11, and full-length HPF1 fused to GFP1-10. These constructs are designed for E. coli cell-free protein synthesis at Ginkgo Cloud Lab. Computational validation of the construct design was performed using AlphaFold 3 prediction of the binary PARP1cat-GFP11 / HPF1-GFP1-10 complex, which confirmed (ipTM = 0.71, pTM = 0.64) that the designed fusion proteins recapitulate the known PARP1-HPF1 binding mode and that the split-GFP halves can be brought into reassembly geometry.
The broad objective is to create a working cell-free split-GFP biosensor for the PARP1-HPF1 interaction. My hypothesis is that co-expression of HPF1-GFP1-10 with PARP1cat-GFP11 will produce fluorescence above background if the proteins bind and bring the split-GFP fragments together. The expected outcome is not a direct reprogramming result, but a validated construct-and-assay pipeline that can be expanded later to more regulators and more rigorous functional assays.
Why This Is HTGAA-Specific
The biological motivation comes from rejuvenation and reprogramming literature, but the HTGAA contribution is the engineering pipeline:
I designed custom DNA constructs.
I used Twist Bioscience to turn those designs into physical plasmids.
I designed the experiment around Ginkgo Cloud Lab cell-free expression instead of local wet-lab access.
I built a minimal fluorescence biosensor readout that can be executed remotely.
I am documenting the design-build-test-learn cycle honestly, including what the assay cannot prove.
This is the HTGAA part: taking a biological idea and turning it into a buildable synthetic biology experiment.
Ginkgo Nebula. The wet-lab part of this project is designed for Ginkgo’s automated cloud lab infrastructure. Instead of manually pipetting every reaction myself, the DNA constructs can be tested through robotic liquid handling, cell-free expression reactions, and plate-reader measurement inside this kind of automated experimental platform.
Innovation and Significance
Innovation. This project operationalizes the catalytic-versus-scaffolding distinction, motivated by the catalytically-dead EZH2-Y726D rejuvenation phenotype, with a buildable, remotely-executable wet-lab assay rather than a hypothesis on paper. It adapts the tripartite split-GFP system of Cabantous and Waldo (2005) from mammalian PPI work into a cell-free format compatible with Ginkgo Cloud Lab, and produces an open construct set that can be extended to the remaining eight conserved master regulators of reprogramming-induced rejuvenation.
Significance. Partial reprogramming with OSKM can reverse molecular features of biological age but currently requires gene therapy delivery, with attendant safety, access, and regulatory constraints. If specific protein-protein interfaces, rather than enzymatic activities, drive the rejuvenation phenotype of reprogramming regulators, those interfaces become targetable by small molecules. A systematic panel of PPI biosensors for the nine conserved regulators would establish which interactions are causal, providing a target list for small-molecule rejuvenation drug discovery and replacing gene therapy as the modality required for therapeutic reprogramming. This project builds the foundational tool for one of the nine targets; demonstrating feasibility of the build-and-readout cycle is the prerequisite for scaling.
Background
The Big Biological Motivation
Yücel et al. (2025) reconstructed gene regulatory networks across partial reprogramming systems and identified conserved regulators associated with rejuvenation. A key observation motivating my project is that EZH2-Y726D, a catalytically impaired EZH2 mutant, can still support rejuvenation-associated effects. This suggests that at least some reprogramming regulators may have important non-canonical roles beyond their classic enzymatic activity.
PARP1 is another regulator in this general biological space. PARP1 is best known as a DNA damage response protein and poly(ADP-ribose) polymerase. Its catalytic activity uses NAD+ to build ADP-ribose chains on target proteins. However, PARP1 also participates in protein complexes, which makes it a good candidate for asking whether molecular interactions can be separated from enzymatic activity.
Why HPF1?
HPF1 stands for Histone PARylation Factor 1. It directly interacts with the PARP1 catalytic domain and changes how PARP1 modifies proteins. This makes HPF1 a useful partner for a simple biosensor: if PARP1 and HPF1 bind in the cell-free reaction, split GFP may reassemble and produce green fluorescence.
The PARP1-HPF1 interface has been structurally characterized by Suskiewicz et al. (2020) in Nature, who showed that HPF1 docks onto the PARP1 catalytic domain via a C-terminal peptide and completes the active site through an NAD+-independent composite interface. This makes PARP1-HPF1 an ideal first test case for a scaffolding-focused biosensor: the binding surface is structurally defined, the C-terminal region of HPF1 is the critical interaction element, which informs the N-terminal placement of GFP1-10, and the interaction is independent of PARP1 catalytic activity, allowing the WT versus E988K comparison to probe binding without confounding from enzymatic turnover.
Why Split GFP?
GFP is the green fluorescent protein. In split-GFP systems, GFP is divided into two pieces:
GFP1-10: a large fragment that is not strongly fluorescent by itself.
GFP11: a small peptide fragment that is also not fluorescent by itself.
If two proteins bring GFP1-10 and GFP11 close together, the GFP barrel can reassemble and become fluorescent. In my design, HPF1 carries GFP1-10 and PARP1 carries GFP11. Fluorescence therefore becomes a proxy for PARP1-HPF1 proximity.
Project Aims
Aim 1: Build the Biosensor
The first aim of my final project is to build and test a cell-free split-GFP biosensor for the PARP1-HPF1 interaction by using DNA construct design, AlphaFold 3 structural validation of the fusion complex, Twist clonal gene synthesis, E. coli codon optimization, and Ginkgo Cloud Lab cell-free protein expression.
Aim 2: Add Biochemical Controls Later
If the biosensor works, the next step is to add biochemical controls that distinguish binding from catalytic activity. This would require a PARP1 enzymatic activity assay, such as NAD+ depletion or PARylation detection, and expression quality control such as Echo-MS or SDS-PAGE.
Aim 3: Scale to More Regulators
The long-term vision is to create a panel of cell-free biosensors for conserved reprogramming regulators. Each biosensor would test a specific protein-protein or protein-DNA interaction and compare wild-type versus catalytic-dead or interaction-altered variants.
Construct Design
I ordered three clonal gene constructs from Twist Bioscience.
Same PARP1 catalytic domain, E988K mutation, His6-tagged, C-terminal GFP11
First-pass mutant comparison
HPF1-GFP1-10
Full-length HPF1, His6-tagged, N-terminal GFP1-10
Binding partner and large split-GFP half
Why Use the PARP1 Catalytic Domain Instead of Full-Length PARP1?
Full-length PARP1 is large and multi-domain. Large human proteins can be difficult to express in E. coli cell-free lysate. I therefore use the PARP1 catalytic domain to make the construct more feasible for cell-free expression while keeping the region that interacts with HPF1.
Why Put GFP1-10 on the N-Terminus of HPF1?
HPF1 uses its C-terminal region to interact with PARP1. If I put the large GFP1-10 fragment on the C-terminus of HPF1, it might block the interaction I am trying to measure. Therefore, HPF1 is designed with GFP1-10 on the N-terminus.
Why E988K?
E988 is part of the PARP1 catalytic machinery. The E988K mutant is expected to disrupt catalytic PARP activity. However, this project does not directly test catalytic activity. In this project, E988K is used as a first-pass mutant comparison in the biosensor.
Experimental Workflow
Literature question
|
v
Choose PARP1-HPF1 as a molecular interaction
|
v
Design three fusion-protein constructs
|
v
Order Twist clonal genes in a T7-compatible vector
|
v
Co-express constructs in Ginkgo cell-free reactions
|
v
Measure split-GFP fluorescence in a plate reader
|
v
Ask: does co-expression produce signal above background?
Experimental Design
Cell-Free Expression Conditions
The planned wet-lab assay uses Ginkgo Cloud Lab cell-free protein expression. Each reaction contains cell-free expression mix plus plasmid DNA. The key comparison is two-plasmid co-expression:
HPF1-GFP1-10 + PARP1cat-WT-GFP11
HPF1-GFP1-10 + PARP1cat-E988K-GFP11
Controls
Controls are essential because GFP fluorescence can be misleading without them.
Condition
Why It Matters
No DNA
Measures background fluorescence of the reaction
HPF1-GFP1-10 alone
Tests whether GFP1-10 gives signal by itself
PARP1cat-WT-GFP11 alone
Tests whether GFP11 gives signal by itself
PARP1cat-E988K-GFP11 alone
Same single-plasmid control for mutant
WT co-expression
Tests whether the biosensor works for the expected interaction
E988K co-expression
First-pass comparison against WT
Readout
The direct readout is green fluorescence from reconstituted split GFP.
This score is useful as a first-pass comparison, but it must be interpreted carefully. A lower E988K signal could mean weaker binding, lower expression, worse folding, or worse split-GFP geometry.
What This Experiment Can and Cannot Show
What It Can Show
Whether my three designed constructs are compatible with a cell-free expression workflow.
Whether PARP1cat-GFP11 and HPF1-GFP1-10 can generate split-GFP fluorescence when co-expressed.
Whether the E988K construct gives more, less, or similar fluorescence compared with the WT construct.
Whether this biosensor design is worth scaling into a larger panel.
What It Cannot Show
It cannot prove that PARP1-E988K rejuvenates cells.
It cannot measure reprogramming potential.
It cannot measure epigenetic age.
It cannot prove that PARP1 catalytic activity is abolished unless I add a separate enzymatic assay.
It cannot prove that scaffolding generalizes across all master regulators.
It cannot reproduce the full nuclear chromatin context of a living human cell.
This distinction is the most important part of the project. My claim is intentionally limited to the data this experiment can actually produce.
Expected Results
If the biosensor works, I expect the WT co-expression condition to produce fluorescence above the no-DNA and single-plasmid controls. That would mean the PARP1cat-GFP11 and HPF1-GFP1-10 fusion proteins can be expressed and can bring split-GFP fragments together.
For E988K, there are two useful outcomes:
If E988K fluorescence is similar to WT, the mutant construct still supports the biosensor signal in this assay.
If E988K fluorescence is much lower than WT, the mutation may reduce binding, reduce expression, alter folding, or change split-GFP geometry.
Either result is useful, but neither result alone proves anything about cellular rejuvenation.
Validation
5.1 What aspect of the project was validated
I validated the structural feasibility of the three-construct biosensor design by predicting the binary PARP1cat-GFP11 / HPF1-GFP1-10 complex using AlphaFold 3. This validates two design assumptions in a single prediction: (1) the fusion proteins fold without disrupting the PARP1-HPF1 binding interface, and (2) the split-GFP halves are brought into a geometry compatible with beta-barrel reassembly when the complex forms.
5.2 Detailed validation protocol
Retrieved PARP1 sequence from UniProt P09874 (catalytic domain, residues 662-1014).
Retrieved HPF1 sequence from UniProt Q9NWY4 (full length, 346 aa).
Retrieved sfGFP1-10 OPT and GFP11 (RDHMVLHEYVNAAGIT) sequences from Cabantous and Waldo (2005) / FPbase.
Assembled fusion proteins in Benchling: Chain A = M-His6-GSGSG-PARP1cat-GSGSGSGSG-GFP11 (390 aa); Chain B = M-GFP1-10-linker-HPF1-His6 (~580 aa).
Verified in-frame ORF and absence of premature stops by translation in Benchling.
Submitted the two protein chains as a single binary-complex job to AlphaFold 3 server (alphafoldserver.com).
Examined per-residue pLDDT, ipTM, pTM, and the PAE matrix.
Inspected the predicted complex structure for: (a) HPF1 C-terminal peptide docking into the PARP1 catalytic cleft, (b) GFP11 / GFP1-10 spatial proximity, (c) reassembly of the split-GFP beta-barrel.
5.3 Synthetic biology techniques used
The validation used DNA construct design (assembly of fusion architectures in Benchling), protein design (placement of split-GFP halves and linkers to preserve the HPF1 C-terminal binding region), use of biological databases (UniProt, FPbase), and computational structure prediction (AlphaFold 3). Together these techniques convert a wet-lab design into a testable in silico model before any DNA is synthesized, allowing geometric and topological errors to be caught at design time rather than after Twist synthesis.
5.4 Data and analysis
The AlphaFold 3 prediction returned ipTM = 0.71 and pTM = 0.64, indicating a confidently predicted inter-chain interface. The core regions of both chains, the PARP1 catalytic domain, the HPF1 globular core, and the reassembled split-GFP barrel, display pLDDT > 90 throughout. The PAE plot shows clean low-error blocks both along the diagonal (intra-chain confidence) and in the off-diagonal regions (inter-chain placement confidence). Disordered regions, comprising the N-terminal His-tags, the GSGSG and GSGSGSGSG linkers, and the C-terminal His-tag of HPF1, appropriately register as low pLDDT (orange). Qualitatively, the prediction shows the HPF1 C-terminal region docked at the PARP1 catalytic interface as expected from Suskiewicz et al. (2020), and the GFP1-10 and GFP11 fragments occupy adjacent positions consistent with beta-barrel reassembly. This is the strongest pre-wet-lab evidence achievable that the biosensor geometry supports the intended readout.
AlphaFold interpretation. Erstmal, das ist ein hervorragendes Ergebnis. ipTM = 0.71 ist solide (“confident interface”), pTM = 0.64 ist akzeptabel fuer ein 970-Residuen-Heterodimer mit flexiblen Tags, und das blaue Strukturzentrum zeigt klar zwei Dinge: (1) PARP1cat + HPF1 docken wie erwartet, (2) das Split-GFP-Barrel scheint im Komplex zu reassemblieren. Das ist genau die strukturelle Validierung des Biosensor-Designs.
5.5 Unexpected challenges
Several challenges were resolved during the design-validation cycle and serve as informative findings. (1) Full-length BRCA1 was originally planned but exceeded the Twist Clonal Gene length limit of approximately 5 kb; this was resolved by restricting BRCA1 to the RING domain (aa 1-109) where C61G acts. (2) Initial vector selection was a CMV-promoter mammalian construct, which is incompatible with Ginkgo’s E. coli T7 cell-free system; this was resolved by switching to a pT7-blank Kan vector with a T7-RBS insertion site. (3) The original HPF1 fusion design placed GFP1-10 at the C-terminus, which would have occluded the HPF1 C-terminal PARP1-binding region; this was caught at the design-review stage and resolved by moving GFP1-10 to the N-terminus. (4) A residual risk remains: ipTM = 0.71 indicates good but not perfect interface confidence, leaving open the possibility that the fusion linkers introduce geometric strain not fully captured by the model, and that CFPS expression yields may differ between WT and E988K constructs in ways that confound the WT/E988K fluorescence comparison. This will be controlled empirically by single-plasmid normalization and Echo-MS expression QC on a subset of wells.
Timeline
Phase
Work
Expected Timing
Design
Finalize construct architecture and verify sequences
Completed
Build
Twist clonal gene synthesis and sequence verification
1-2 weeks
Test
Ginkgo Cloud Lab cell-free expression and fluorescence readout
After constructs arrive
Analyze
Background correction, WT vs E988K comparison, figures
1 week
Learn
Decide whether to improve tag placement, add controls, or scale to more regulators
Final project write-up
Techniques Used
DNA construct design
Codon optimization for E. coli
Twist clonal gene ordering
Split-GFP reporter design
Cell-free protein expression
Plate-reader fluorescence measurement
Protein interaction assay design
AlphaFold 3 structural validation
Literature-based experimental planning
Bioethical reflection and scope control
Ethics and Responsibility
This project has relatively low direct biosafety risk because it uses non-replicating cell-free reactions rather than engineered organisms released into the environment. The constructs encode human protein fragments and are intended for in vitro expression only.
The main ethical responsibility is truthful communication. Aging biology can easily be overhyped. I need to be clear that this project is not an anti-aging treatment, not a rejuvenation result, and not a clinical experiment. It is a molecular biosensor project that could support future mechanistic work.
Another ethical principle is non-maleficence: avoiding harm. In this context, harm could come from overstating weak evidence, especially in a field where people may be vulnerable to exaggerated longevity claims. I will therefore present the project as tool-building and clearly separate direct data from future speculation.
Budget
Item
Approximate Cost
Notes
PARP1cat-WT-GFP11 clonal gene
Included in Twist order
~1,170 bp
PARP1cat-E988K-GFP11 clonal gene
Included in Twist order
~1,170 bp
HPF1-GFP1-10 clonal gene
Included in Twist order
~1,743 bp
Twist total
~$532.47
3 constructs
Ginkgo CFPS plate
TBD
Depends on HTGAA/Ginkgo pricing
Optional protein QC
TBD
Echo-MS or gel-based QC if available
The project is intentionally small because the available budget is limited. A larger project testing all regulators would require many more constructs and assays.
Future Work
If the biosensor works, the next steps are:
Extend the structural validation to AlphaFold predictions of the remaining eight master regulators paired with their canonical binding partners.
Add a direct PARP1 catalytic activity assay.
Add expression quality control such as Echo-MS, SDS-PAGE, or Western blot.
Test alternative linker lengths and tag placements.
Build additional biosensors for other regulators and partners.
Eventually move from cell-free biochemistry into mammalian cell assays.
References
Yücel et al. (2025). Conserved master regulators of reprogramming-induced rejuvenation. bioRxiv 2025.11.27.690899.
Yang et al. (2023). Chemical reprogramming and EZH2 inhibition context. Cell.
Suskiewicz et al. (2020). HPF1 completes the PARP active site and directs ADP-ribosylation. Nature.
Abramson, J. et al. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493-500.
Mirdita, M. et al. (2022). ColabFold: making protein folding accessible to all. Nature Methods 19, 679-682.
Cabantous, S., Terwilliger, T. C., & Waldo, G. S. (2005). Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nature Biotechnology.
The strongest honest claim for this final project is:
I designed a three-construct, cell-free split-GFP biosensor for the PARP1-HPF1 interaction, validated the design in silico with AlphaFold 3 (ipTM = 0.71, recapitulated PARP1-HPF1 interface and split-GFP reassembly geometry), and ordered the constructs from Twist Bioscience for cell-free expression and plate-reader readout at Ginkgo Cloud Lab. This is a foundational HTGAA biosensor project: it produces an open, validated construct set that can be extended to the remaining eight conserved master regulators of reprogramming-induced rejuvenation, supporting the longer-term goal of identifying druggable PPI interfaces for small-molecule rejuvenation without gene therapy.