Make sure to document every step of the in-silico and lab experiments. Make sketches, screenshots, notes, drawings - anything that helps you - and others understand the experiment.
Your Documentation should help you - and others - to understand the topic. Don’t be afraid to add things that don’t work. Show your failures - and how you overcame them. Your Documentation should be a description of the amazing journey you are on!
Overview
Ethics, safety, and security are essential considerations throughout (and beyond!) this class. We have therefore designed the Class Assignment this week to give you a strong foundation, and then will ask you to reflect each week and in the design of your final project.
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
Purpose: What is done now and what changes are you proposing?
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
• By helping respond
Foster Lab Safety
• By preventing incident
• By helping respond
Protect the environment
• By preventing incidents
• By helping respond
Other considerations
• Minimizing costs and burdens to stakeholders
• Feasibility?
• Not impede research
• Promote constructive applications
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
Assignment (Final Project) – Due as part of your Final Project presentation (not Feb 10)
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
As part of your final project, design one or more strategies to ensure that your project, and what it enables, contributes to growing an ethical biological future.
Assignment (Lab Preparation) — DUE BY START OF FEB 10 LECTURE
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
(Not Applicable)
Lab Training (failure to complete this will jeopardize your acceptance into the course)
Complete Lab Specific Training in Person.
Complete Safety Training in Atlas
Navigate to atlas.mit.edu and on the right-hand side, click “Learning Center”
Head to the Course Catalog and find the following two courses:
General Biosafety for Researchers (EHS00260w)
Managing Hazardous Waste (EHS00501w)
Assignment (Week 2 Lecture Prep) — DUE BY START OF FEB 10 LECTURE
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
In preparation for Week 2’s lecture on “DNA Read, Write, and Edit," please review these materials:
Lecture 2 slides as posted below.
The associated papers that are referenced in those slides.
In addition, answer these questions in each faculty member’s section:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the
length of the human genome. How does biology deal with that discrepancy?
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the
reasons that all of these different codes don’t work to code for the protein of interest?
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals
and how does this affect your view of the “Lysine Contingency”?
[Given slides #2 & 4 (AA:NA and NA:NA codes)] What code would you suggest for AA:AA interactions?
[(Advanced students)] Given the one paragraph abstracts for these real 2026 grant programs
sketch a response to one of them or devise one of your own:
Assignment (Your HTGAA Website) — DUE BY START OF FEB 10 LECTURE
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
Begin personalizing your HTGAA website in in https://edit.htgaa.org/, starting with your homepage —
fill in the template with information about yourself, or remove what’s there and make it your own. Be creative!
As with all assignments in HTGAA, be sure to write up every part of this Homework on your HTGAA website in order to receive credit.
Important
In order to continue in this course you need two things:
This homework completed and written up on your HTGAA website on pages.htgaa.org — make sure you’ve checked your
published website on pages.htgaa.org and are happy with how it shows up there; if your homework is not visible on
your pages.htgaa.org website course staff won’t see it and you will not be selected to continue in the course!
For this week only, AFTER your homework is complete and published on pages.htgaa.org, fill out the
Homework 1 Completion form which David emailed out just after
Lecture 1. This Google form expresses your interest in continuing with the course.
Without both of these you will not be accepted into HTGAA!
Synthetic Genomics: Options for Governance This is an older but useful report for thinking about a variety of options for the governance of biotechnology that inspired this week’s homework
National Security Commission on Emerging Biotechnology: This U.S. Congressional Commission will produce its first “comprehensive” report at the end of 2024 but has an “interim” 2023 report posted now, and they are currently soliciting input to guide national policy regulating biotech
iGEM 2020 Safety Hub: This page includes links to many useful resources including the WHO biosafety manual, the NIH guidelines and the CDC Biosafety in Microbial and Biomedical Laboratories Guide; additional information is available on the iGEM 2023 Responsibility page
Handbook for Community Biology Spaces: A handbook co-developed by community biolobabs, designed as a living document that can be updated and expanded by the community over time
DIYBio Ask a biosafety expert This page includes a portal where you can get your biosafety questions answered by professionals
Rooftop Solar and the Four Levers of Social Change: A blog post from Ethan Zuckerman considering different types of ways of regulating behavior, adopted in part from Lawrence Lessig’s book: Code 2.0, and explored in the context of energy consumption and production
Subsections of Week 1 (Feb 3)
Lab (Week 1) — Introduction to Pipetting and Dilutions
Overview
Objective
Welcome to HTGAA! This is our very first lab, and in this lab we will introduce students to the foundational techniques of pipetting and serial dilutions, critical for precise liquid handling and solution preparation in biological and chemical experiments.
This is a one-day lab with two protocols covered on mixing colors and dilution. By the end of the lab, students will confidently use pipettes, prepare solutions with desired concentrations, and troubleshoot common errors in pipetting.
Concepts Learned & Skills Gained
Students will:
Understand Units and Conversions: moles (mol), molarity (M), and conversions between µL, mL, and L.
Perform Serial Dilutions: Learn the stepwise dilution process to achieve specific solution concentrations.
Gain Pipetting Proficiency: Operate P20, P200, and P1000 pipettes accurately for volume transfers.
Visualize Mixing Outcomes: Use colors and absorbance measurements to observe concentration gradients.
Pre-Lab
Reading
Key Definitions
Here are some key definitions we’d like you to know before you get started.
Moles (mol): A unit representing $6.022 \times 10^{23}$ particles (atoms, molecules, etc.).
Molarity (M): Concentration defined as moles of solute per liter of solution (mol/L).
Conversions:
1 L = 1000 mL = 1,000,000 μL
1 M = 1000 mM = 1,000,000 μM
Planning Your Experiments
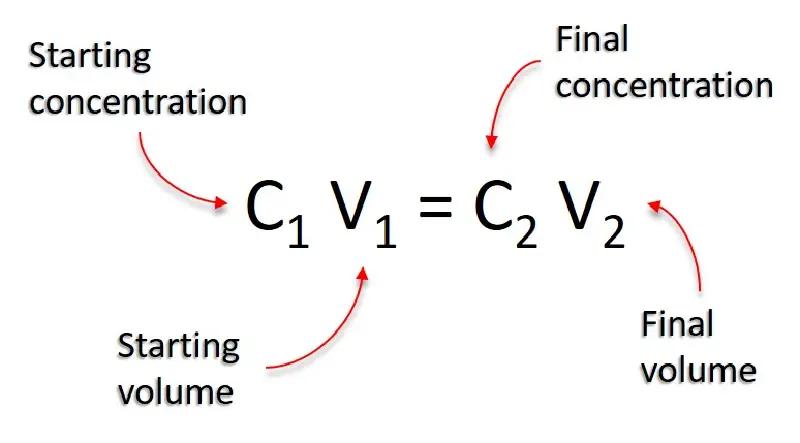
To calculate the volume of water needed for a dilution, use the formula: $$C_1 V_1 = C_2 V_2$$
$C_2$ : Final concentration (desired concentration).
$V_2$ : Final volume (total volume of the diluted solution).
Steps:
Rearrange the formula to calculate $V_1$: $$ V_1 = \frac{C_2 V_2}{C_1} $$
Calculate the volume of water (let’s call it $V_Water$) to add: $$ V_Water = V_2 - V_1 $$
Practice
Dilution Practice 1
Scenario: The stock concentration of a mystery substance (MS) is 5 M. Calculate how to dilute to 100 µM (0.1 mM):
Use sequential 1:499 and 1:99 dilution steps for accurate preparation.
Step 1: Dilute 5 M (5,000,000 µM) to 10,000 µM (500x dilution).
Step 2: Dilute 10,000 µM to 100 µM (100x dilution).
Dilution Practice 2
The stock concentration of a mystery substance (MS) is 5 M.
If the molar mass of MS is 532 g/mol, what’s the concentration of the stock concentration in g/mL? To make your life easier, you can use one of many online calculators.
You will perform a serial dilution to get 100 uM of MS. Devise a plan to dilute a 5 M MS solution to 100 uM. How many dilution steps will we need? Which tubes should we use? Which pipettes?
Fill out the following chart to prepare a final reaction with 60 uL reaction volume. Why did we make 100 uM MS if we actually need 40 uM MS? Why not prepare 40 uM in serial dilutions?
Reagent
Stock concentration
Desired concentration
Volume
Loading dye
6X
1X
MS
100 uM
40 uM
dH2O
n/a
n/a
Note
Please fill this out before coming to lab.
Additional resources
You must watch or be able to understand the following videos:
Mysterious substance (food coloring with water), henceforth: MS
Red, Blue and Yellow food coloring solutions
Gel loading dye (commonly used reagents for loading gels, strong purple color)
Part 1: Mixing Color
Prepare tubes with red, yellow, and blue food coloring solutions OR watercolor
Take ten tubes and mark them with numbers 1 to 6
Tube 1, 2 and 3: add 500 uL each red, yellow, and blue solution to the tube.
Tube 4: add 220 uL red solution to the tube, and add 220 uL yellow solution.
Try adding this in 2 steps: add 200 uL first, and then 20 uL. Discard your tips after you add one color!
Tube 5: add 525 uL yellow solution to the tube, and add 525 uL blue solution.
Tube 6: add 155 uL red solution to the tube, and add 155 uL blue solution.
Now you have a rainbow! You can try mixing other colors with the solutions.
Try plating different volumes (e.g. 1uL, 2uL, 5uL, 10uL) on a petri plate to make some designs and build your intuitive understanding of these volumes.
Part 2: Performing Serial Dilution
Perform serial dilutions to get 100 uM (0.1 mM) of MS.
Every time you mix in liquid, pipette up and down three or four times to ensure the two liquids are mixed thoroughly.
Mark each tube with its respective concentration using a pen.
Prepare a final reaction of 60 uL based on your table in the pre-lab.
Bonus: Take 20 uL from the final reaction and pipette it to a pre-prepared gel well. Wells are a bit trickier because they are thin and your pipette tip will puncture the gel if you’re not careful. Be gentle!
This week explores the read–write–edit toolkit: sequencing and synthesis workflows, restriction digests and gel electrophoresis, and early genome-editing frameworks.
Make sure to document every step of the in-silico and lab experiments. Make sketches, screenshots, notes, drawings… anything that helps you - and others - understand the experiment.
Your documentation should help you - and others - to understand the topic. Don’t be afraid to add things that don’t work. Show your failures - and how you overcame them. Your Documentation should be a description of the amazing journey you are on!
Part 0: Basics of Gel Electrophoresis
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
Attend or watch all lecture and recitation videos. Optionally watch bootcamp.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
[Example from our group homework, you may notice the particular format — The example below came from UniProt]
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
Lysis protein DNA sequence atggaaacccgattccctcagcaatcgcagcaaactccggcatctactaatagacgccggccattcaaacatgaggattacccatgtcgaagacaacaaagaagttcaactctttatgtattgatcttcctcgcgatctttctctcgaaatttaccaatcaattgcttctgtcgctactggaagcggtgatccgcacagtgacgactttacagcaattgcttacttaa
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Lysis protein DNA sequence with Codon-Optimization ATGGAAACCCGCTTTCCGCAGCAGAGCCAGCAGACCCCGGCGAGCACCAACCGCCGCCGCCCGTTCAAACATGAAGATTATCCGTGCCGTCGTCAGCAGCGCAGCAGCACCCTGTATGTGCTGATTTTTCTGGCGATTTTTCTGAGCAAATTCACCAACCAGCTGCTGCTGAGCCTGCTGGAAGCGGTGATTCGCACAGTGACGACCCTGCAGCAGCTGCTGACCTAA
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
3.5. [Optional] How does it work in nature/biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level.
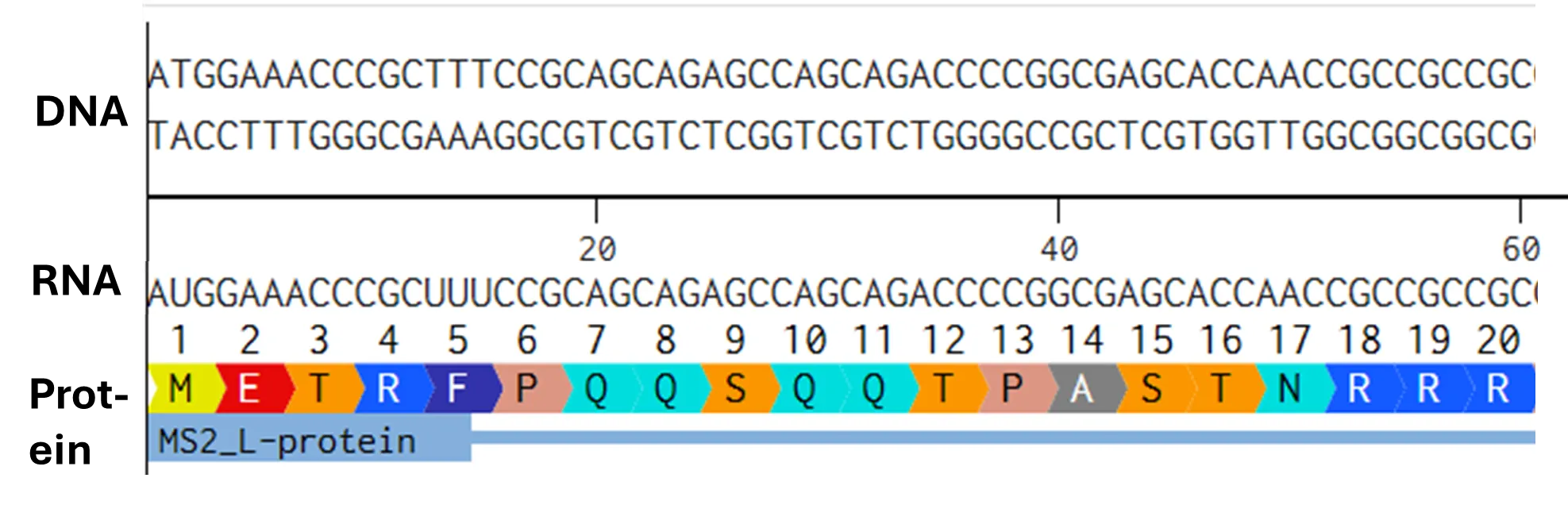
Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below.
[Example shows the biomolecular flow in central dogma from DNA to RNA to Protein] Special note that all “T” were transcribed into “U” and that the 3-nt codon represents 1-AA.
Rearranged snapshot of MS2 L-protein information flow from DNA to RNA to Protein. Captured from Ice’s Benchling and stitched together in a ppt
Part 4: Prepare a Twist DNA Synthesis Order
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
This is a practice exercise, not necessarily your real Twist order!
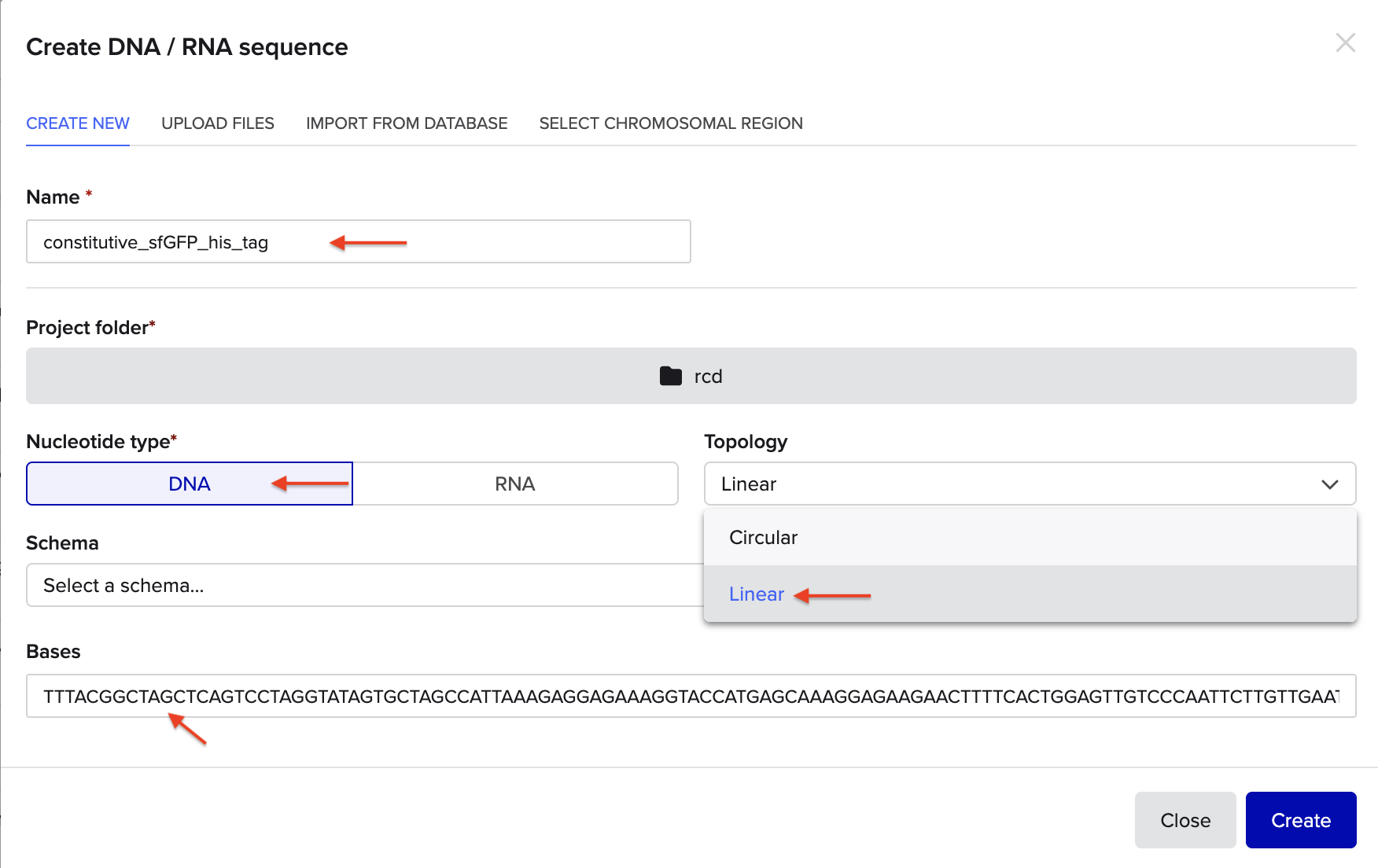
For example, let’s make a sequence that will make E. coli glow fluorescent green under UV light by constitutively (always) expressing sfGFP (a green fluorescent protein):
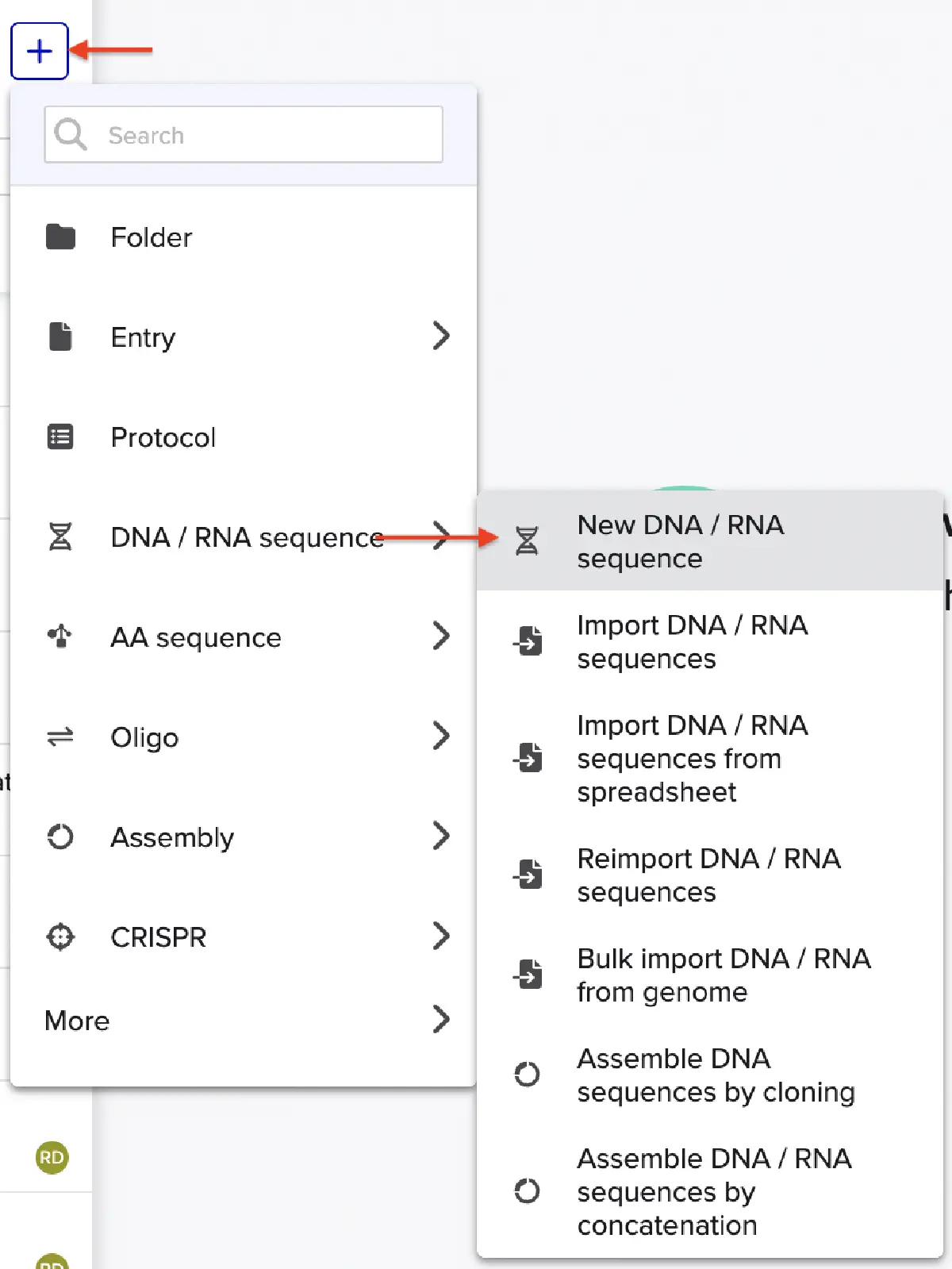
In Benchling, selectNew DNA/RNA sequence
Give your insert sequence a name and select DNA with a Linear topology (this is a linear sequence that will be inserted into a circular backbone vector of our choosing).
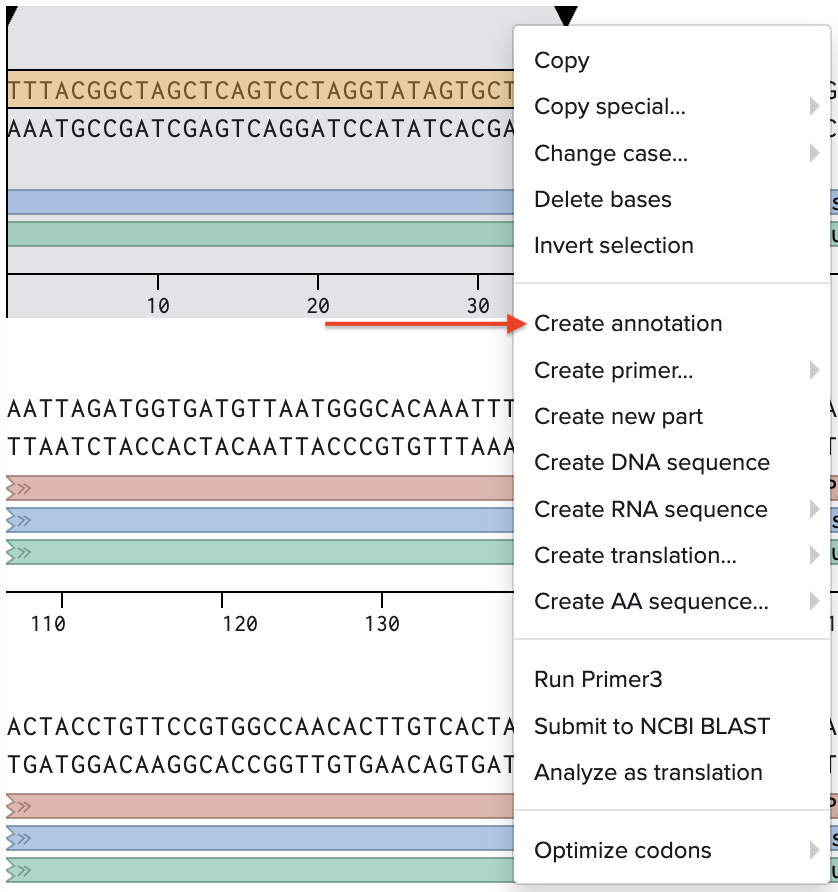
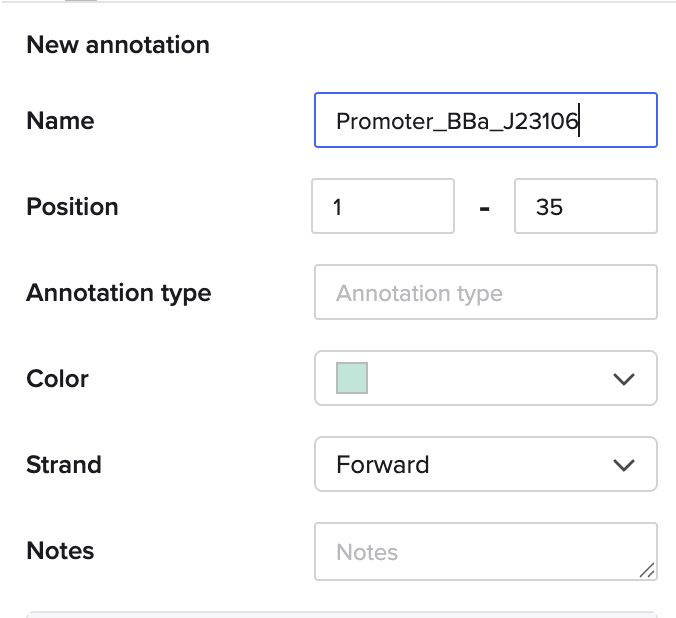
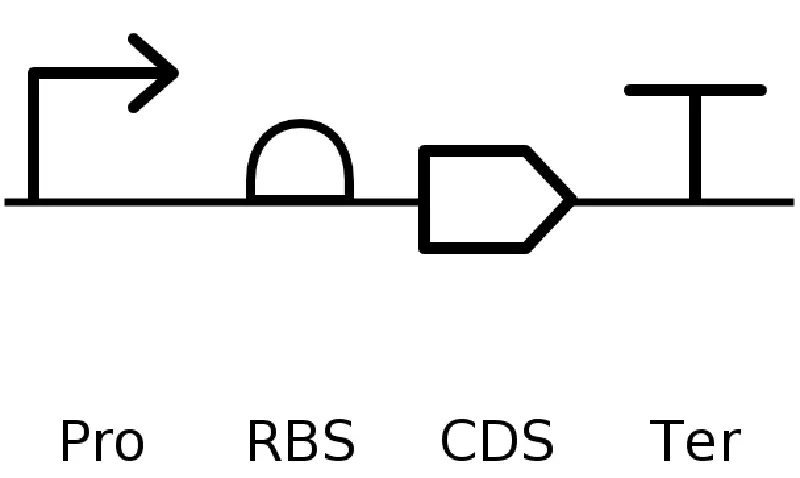
Go through each piece of the given DNA sequences highlighted below (Promoter, RBS, Start Codon, Coding Sequence, His Tag, Stop Codon, Terminator) and paste the sequences into the Benchling file one after the other (replacing the coding sequence with your codon optimized DNA sequence of interest!). Each time you add a new piece of the sequence, make sure to annotate by right clicking over the sequence and creating an annotation that describes what each piece (e.g., Promoter, RBS, etc.) is (see image below).
RBS (e.g. BBa_B0034 with spacers for optimal expression): CATTAAAGAGGAGAAAGGTACC
Start Codon: ATG
Coding Sequence (your codon optimized DNA for a protein of interest, sfGFP for example): AGCAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCAATTCTTGTTGAATTAGATGGTGATGTTAATGGGCACAAATTTTCTGTCCGTGGAGAGGGTGAAGGTGATGCTACAAACGGAAAACTCACCCTTAAATTTATTTGCACTACTGGAAAACTACCTGTTCCGTGGCCAACACTTGTCACTACTCTGACCTATGGTGTTCAATGCTTTTCCCGTTATCCGGATCACATGAAACGGCATGACTTTTTCAAGAGTGCCATGCCCGAAGGTTATGTACAGGAACGCACTATATCTTTCAAAGATGACGGGACCTACAAGACGCGTGCTGAAGTCAAGTTTGAAGGTGATACCCTTGTTAATCGTATCGAGTTAAAGGGTATTGATTTTAAAGAAGATGGAAACATTCTTGGACACAAACTCGAGTACAACTTTAACTCACACAATGTATACATCACGGCAGACAAACAAAAGAATGGAATCAAAGCTAACTTCAAAATTCGCCACAACGTTGAAGATGGTTCCGTTCAACTAGCAGACCATTATCAACAAAATACTCCAATTGGCGATGGCCCTGTCCTTTTACCAGACAACCATTACCTGTCGACACAATCTGTCCTTTCGAAAGATCCCAACGAAAAGCGTGACCACATGGTCCTTCTTGAGTTTGTAACTGCTGCTGGGATTACACATGGCATGGATGAGCTCTACAAA
7x His Tag (Let’s add a 7×His tag at the C-terminus of the protein to enable protein purification from E. coli): CATCACCATCACCATCATCAC
Once you’ve completed this, click on Linear Map to preview the entire sequence. If you intend to have a TA review a sequence in the future, this is a good way to verify that all sections are annotated!
This is not required for this exercise, but to share your design with others, please ensure that link sharing is turned on!
(Optional) Share your final sequence link with a TA for review!
This insert sequence you built is commonly referred to as an expression cassette in molecular biology (a sequence you can drop into any vector and it’ll perform its function). Go ahead and download the FASTA file for the sequence you made.
It’s helpful to visualize DNA designs using SBOL Canvas (Synthetic Biology Open Language) to convey your designs. Here’s an example of what you just annotated in Benchling:
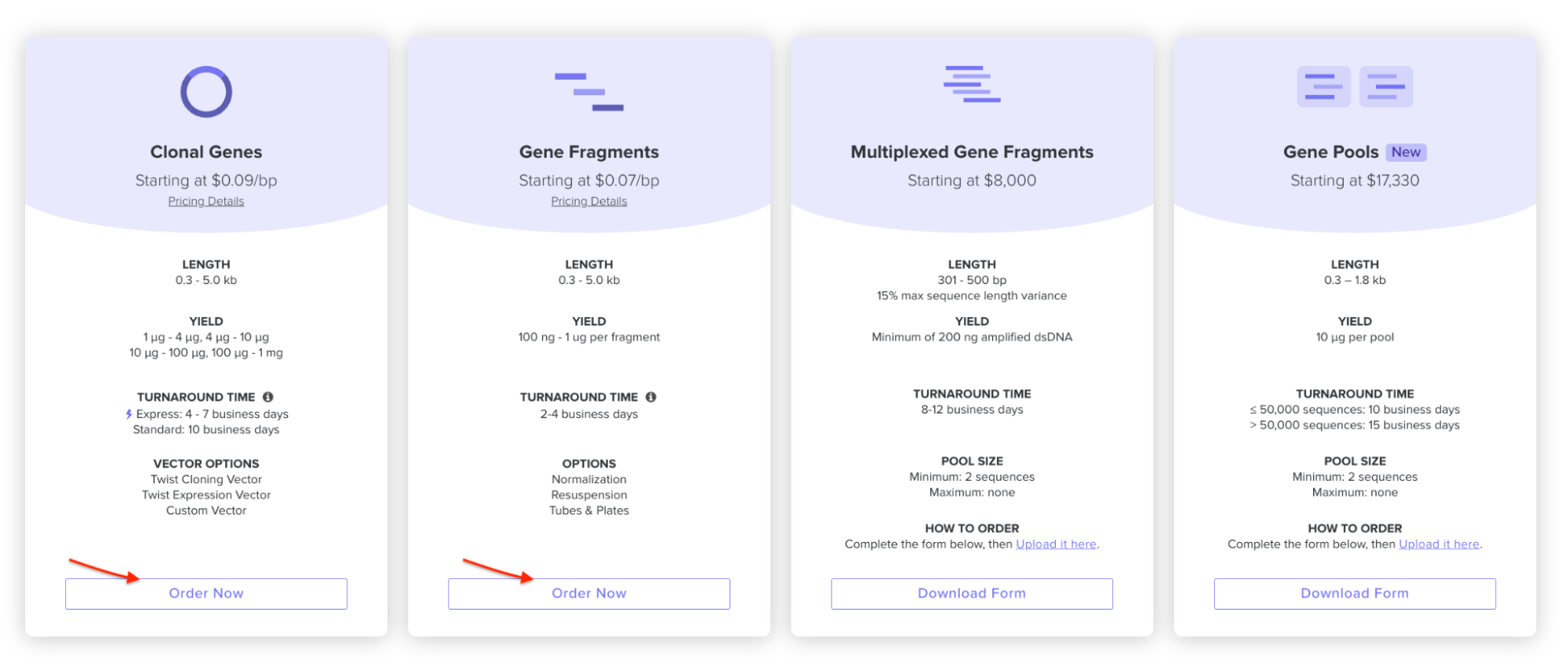
4.3. On Twist, Select The “Genes” Option
4.4. Select “Clonal Genes” option
For this demonstration, we’ll choose Clonal Genes. You’ll select clonal genes or gene fragments depending on your final project.
Historically, HTGAA projects using clonal genes (circular DNA) have reached experimental results 1-2 weeks quicker because they can be transformed directly into E. coli without additional assembly.
Gene fragments (linear DNA) offer greater design flexibility but typically require an assembly or cloning step prior to transformation. An advantage is If designed with the appropriate exonuclease protection, gene fragments can be used directly in cell-free expression.
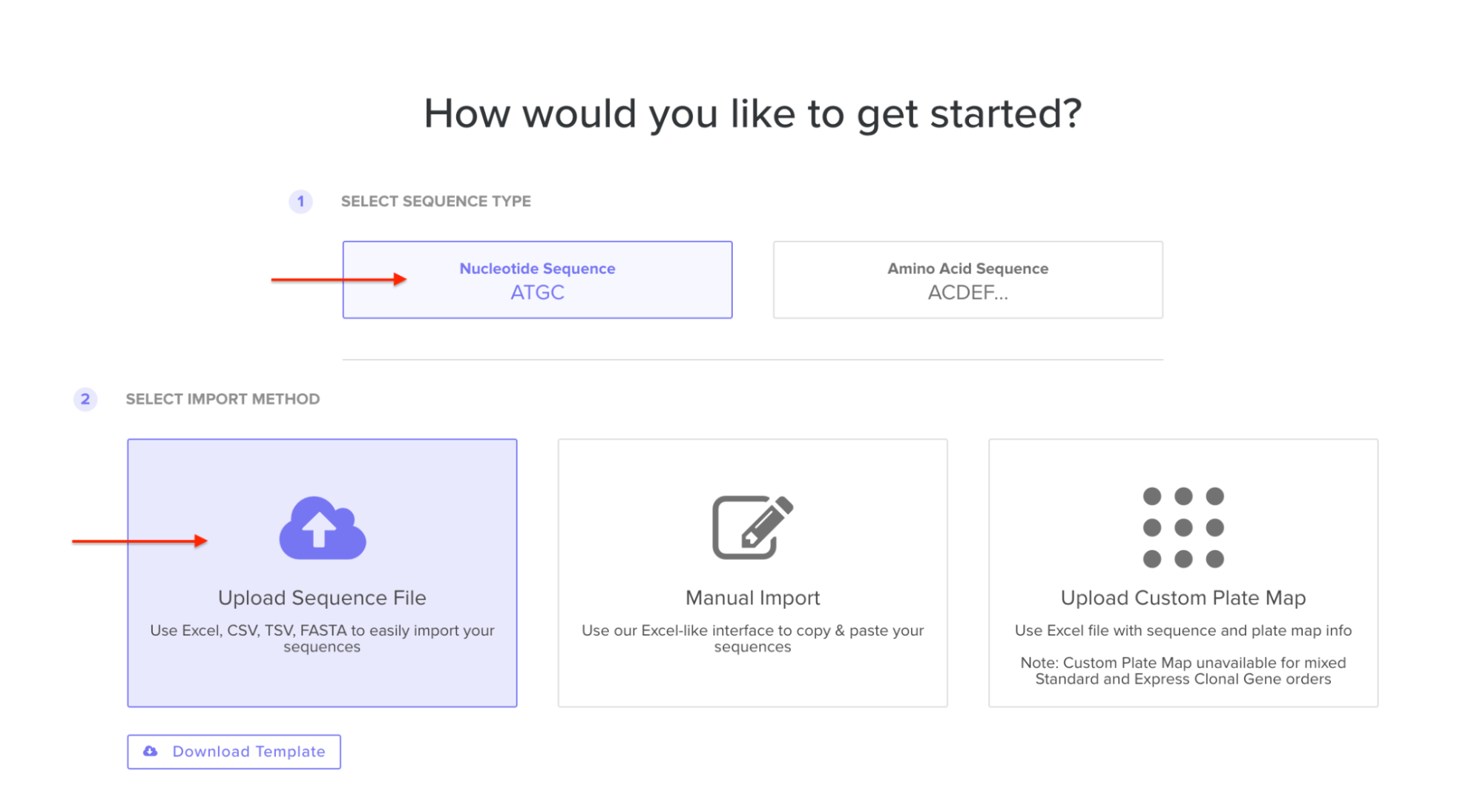
4.5. Import your sequence
You just took an amino acid sequence of interest and converted it into DNA, codon optimized it, and built an expression cassette around it! Choose the Nucleotide Sequence option and Upload Sequence File to upload your FASTA file.
4.6. Choose Your Vector
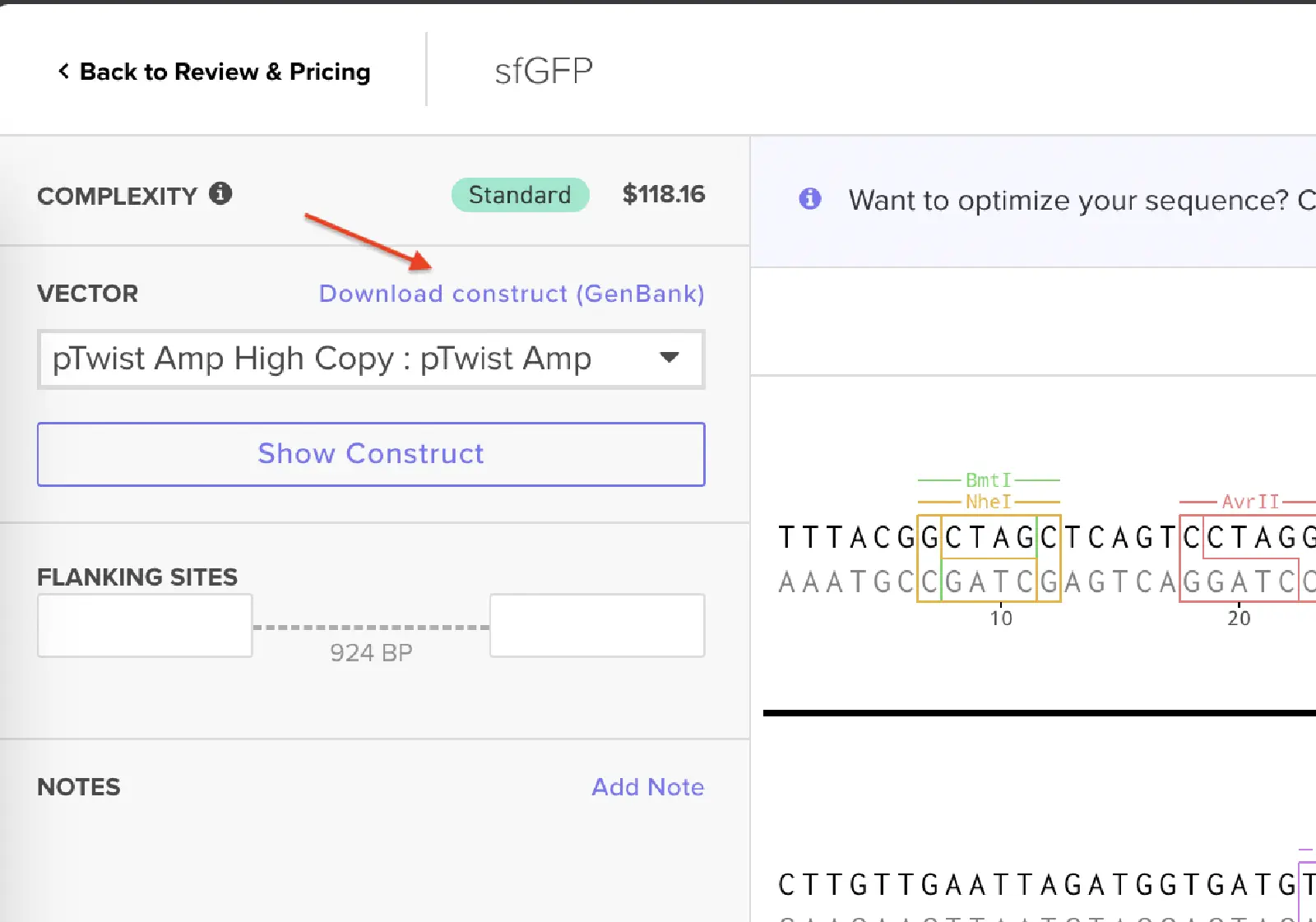
Since we’re ordering a clonal gene, you will need to refer to Twist’s Vector Catalog to choose your circular backbone. You can think of this as taking your linear expression cassette for your protein of interest, and completing the rest of the circle!
The backbone confers many special properties like antibiotic resistance, an origin of replication, and more. Discuss with your node to decide on appropriate antibiotic options. At MIT/Harvard, you can use Ampicillin, Chloramphenicol, or Kanamycin resistance.
Twist vectors do not contain restriction sites near the insert fragment, so make sure to flank your design with cut sites if you are intending to extract this DNA insert fragment later.
For this demonstration, choose a Twist cloning vectors like pTwist Amp High Copy.
Click into your sequence and select download construct (GenBank) to get the full plasmid sequence:
Go back to your Benchling account. Inside of a folder, click the import DNA/RNA sequence button and upload the GenBank file you just downloaded.
This is the plasmid you just built with your expression cassette included. Congratulations on building your first plasmid!
Important
For your final projects, remember to include:
Fully annotated Benchling insert fragment
Desired Twist cloning vector
Part 5: DNA Read/Write/Edit
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
What is the output of your chosen sequencing technology?
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
See some famous examples of DNA design
DNA origami by Paul W. K. Rothemund, California Institute of Technology, 2004. 100 nanometers in diameter.
(ii) What technology or technologies would you use to perform this DNA synthesis and why? Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
Colossal Biosciences Inc., a biotechnology company using genetic engineering to de-extinct various historic animals such as the woolly mammoth, dodo, and dire wolf.
(ii) What technology or technologies would you use to perform these DNA edits and why? Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Reading & Resources (click to expand)
Resources
DNA Sequencing at 40: Past, Present, and Future (2017) Shendure, J., Balasubramanian, S., Church, G. et al.https://doi.org/10.1038/nature24286
Base editors contain a nicking or dead Cas9 enzyme fused to a deaminase.
a.) PAM requirement: Base editors contain a nicking or dead Cas9 enzyme fused to a deaminase. For designing your guide RNA for base editing you will therefore have a PAM requirement like you would have for any Cas9 experiment.
b.) Deamination window: An additional design constraint is that the sequence window in which deamination occurs is only a few base pairs long. You can find information on the deamination windows in the review below (even though some new editors are not included).
BE4 and ABE7.10 are good starting points and both use SpCas9 with NGG Pam requirement. Base editors with other PAM sites have been constructed too.
TALEN
For TALENs, you can assume no sequence restrictions – One of the technology’s previous restrictions was a T starting base, but this has since been overcome. In contrast to the CRISPR/Cas technologies above, your DNA sequence is recognized through interactions between the DNA and the TALEN: each TAL in the array recognizes one base.
(Note: In order to introduce a double strand break, you will need to design to TALENs targeting the opposing strands.)
Gel Purification of DNA: after DNA gel electrophoresis, cutting a band of DNA out of the agarose gel allows isolation and purification of a specific DNA fragment:
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept.
If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead.
Ask for help early!
If you are having any trouble with scripting, contact your TAs as soon as possible for help. Do not wait until your scheduled robot time slot or you may not be able to complete this assignment!
If the Python component is proving too problematic even with AI and human assistance, download the full Python script from the GUI website and submit that:
Use the download icon pointed to by the red arrow in this diagram.
If you use AI to help complete this homework or lab, document how you used AI and which models made contributions.
Sign up for a robot time slot if you are at MIT/Harvard/Wellesley or at a Node offering Opentrons automation. The Python script you created will be run on the robot to produce your work of art!
At MIT/Harvard? Lab times are on Thursday Feb.19 between 10AM and 6PM.
Post-Lab Questions — DUE BY START OF FEB 24 LECTURE
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
One of the great parts about having an automated robot is being able to precisely mix, deposit, and run reactions without much intervention, and design and deploy experiments remotely.
For this week, we’d like for you to do the following:
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
While your description/project idea doesn’t need to be set in stone, we would like to see core details of what you would automate. This is due at the start of lecture and does not need to be tested on the Opentrons yet.
Example 1: You are creating a custom fabric, and want to deposit art onto specific parts that need to be intertwined in odd ways. You can design a 3D printed holder to attach this fabric to it, and be able to deposit bio art on top. Check out the Opentrons 3D Printing Directory.
Example 2: You are using the cloud laboratory to screen an array of biosensor constructs that you design, synthesize, and express using cell-free protein synthesis.
Echo transfer biosensor constructs and any required cofactors into specified wells.
Bravo stamp in CPFS reagent master mix into all wells of a 96-well / 384-well plate.
Multiflo dispense the CFPS lysate to all wells to start protein expression.
PlateLoc seal the plate.
Inheco incubate the plate at 37°C while the biosensor proteins are synthesized.
XPeel remove the seal.
PHERAstar measure fluorescence to compare biosensor responses.
Final Project Ideas — DUE BY START OF FEB 24 LECTURE
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
As explained in this week’s recitation, add 1-3 slides with 3 ideas you have for an Individual Final Project in the appropriate
slide deck
for MIT/Harvard/Wellesley students
or for Commited Listeners. Be sure to put your name on your slide(s); for CLs, also put your city and country on your slide(s) and be sure you’re putting your slide(s) in your Node’s section of the deck.
Lab work this week is contained within the homework assignment below.
Homework: Protein Design I — DUE BY START OF MAR 3 LECTURE
Objective:
Learn basic concepts:
amino acid structure
3D protein visualization
the variety of ML-based design tools
Brainstorm as a group how to apply these tools to engineer a better bacteriophage (setting the stage for the final project).
Part A. Conceptual Questions
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Why are there only 20 natural amino acids?
Can you make other non-natural amino acids? Design some new amino acids.
Where did amino acids come from before enzymes that make them, and before life started?
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Can you discover additional helices in proteins?
Why are most molecular helices right-handed?
Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation?
Why do many amyloid diseases form β-sheets?
Can you use amyloid β-sheets as materials?
Design a β-sheet motif that forms a well-ordered structure.
Part B: Protein Analysis and Visualization
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
Briefly describe the protein you selected and why you selected it.
Identify the amino acid sequence of your protein.
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
Does your protein belong to any protein family?
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
Are there any other molecules in the solved structure apart from protein?
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
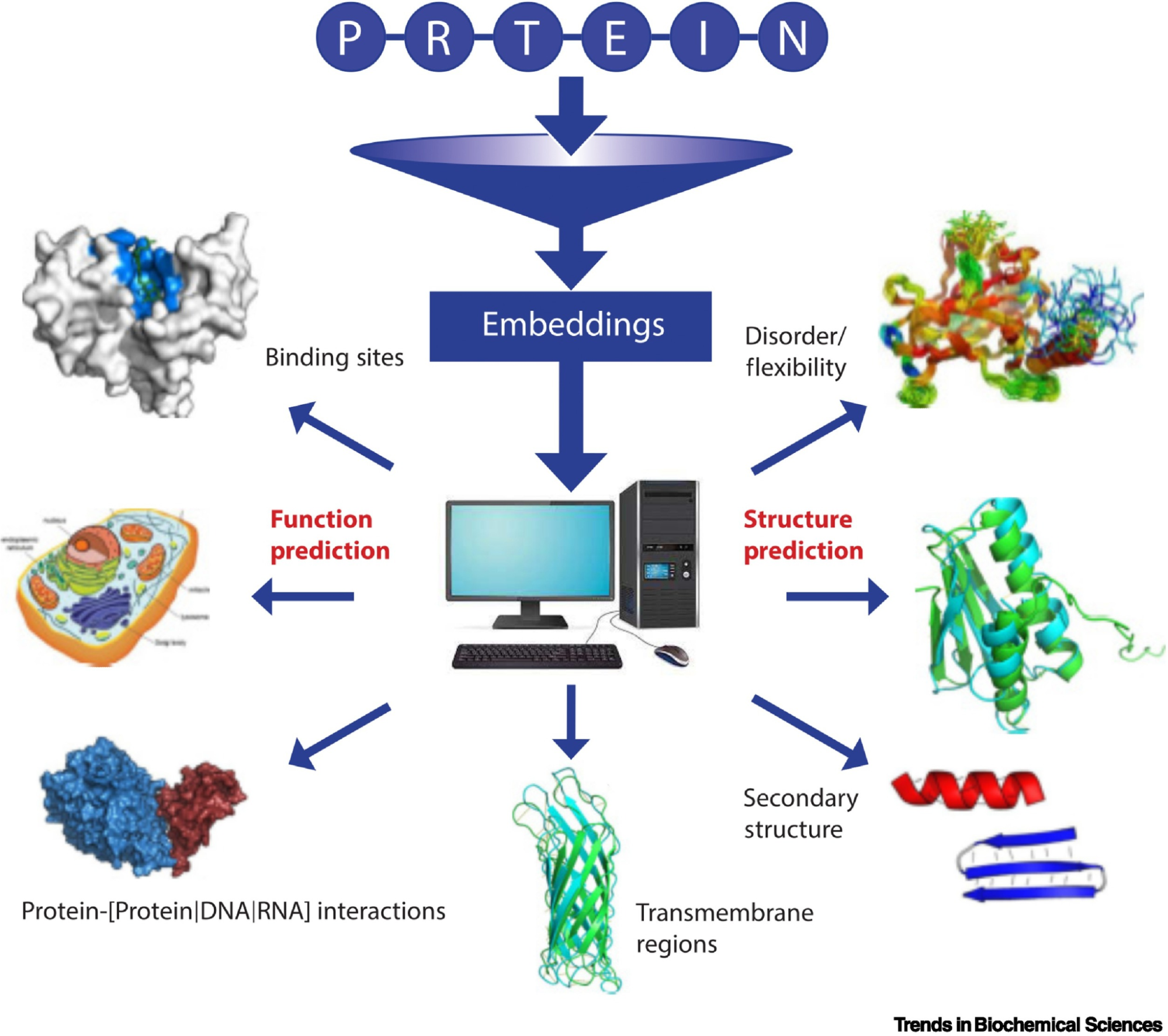
C1. Protein Language Modeling
Picture Source: Bordin, Nicola et al (2023). Novel machine learning approaches revolutionize protein knowledge. Trends in Biochemical Sciences, Volume 48, Issue 4, 345 - 359
Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Place your protein in the resulting map and explain its position and similarity to its neighbors.
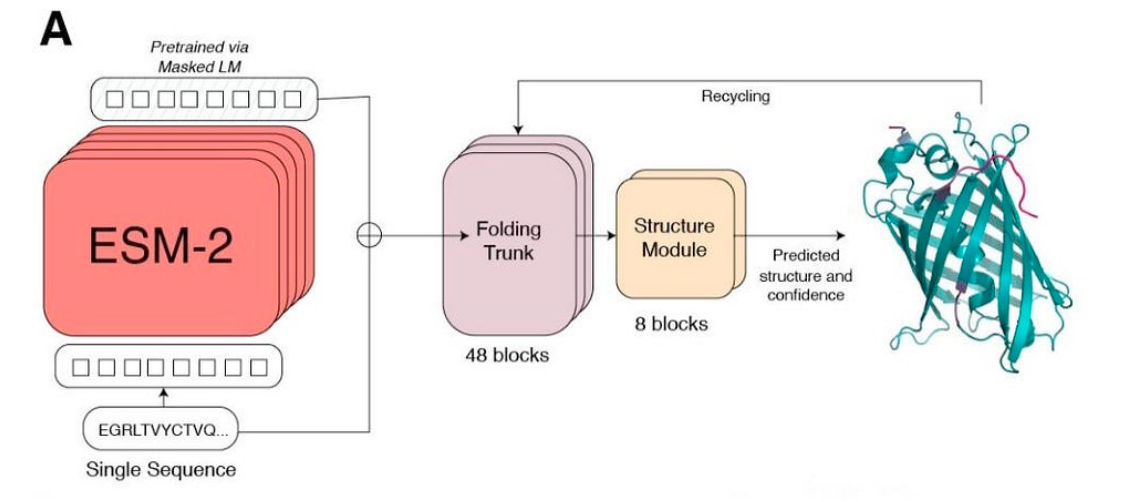
C2. Protein Folding
Picture Source: Lin et al (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model.
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
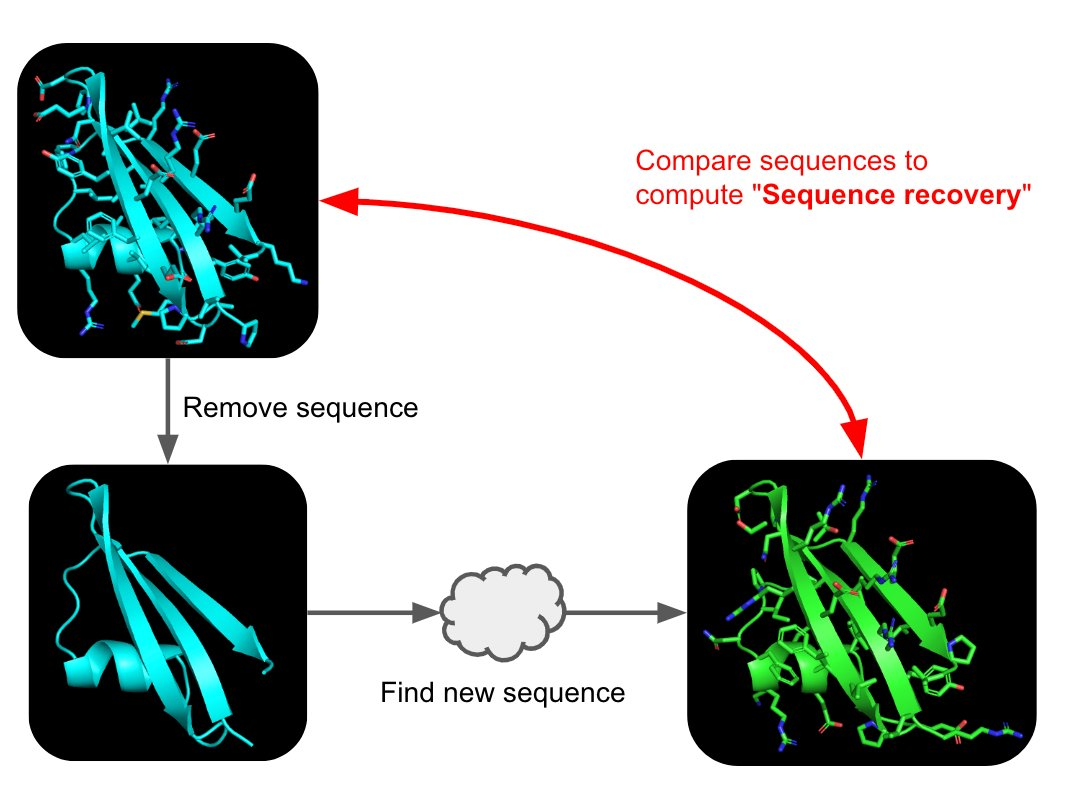
C3. Protein Generation
Picture Source: 1. Post from Sergey Ovchinnikov 2. Roney, Ovchinnikov et al (2022). State-of-the-art estimation of protein model accuracy using AlphaFold. Phys. Rev. Lett. 129, 238101
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Input this sequence into ESMFold and compare the predicted structure to your original.
Part D. Group Brainstorm on Bacteriophage Engineering
Assignees for this section
MIT/Harvard students
Optional
Committed Listeners
Required
Find a group of ~3–4 students
Read through the Phage Reading material listed under “Reading & Resources” below.
Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
Why do you think those tools might help solve your chosen sub-problem?
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
NGLViewer: NGL Viewer is a collection of tools for web-based molecular graphics. WebGL is employed to display molecules like proteins and DNA/RNA with a variety of representations.
Chimera: A highly extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles.
Lab work this week is contained within the homework assignment below.
Homework — DUE BY START OF MAR 10 LECTURE
Part A: SOD1 Binder Peptide Design (From Pranam)
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide!
For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a
MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
This homework requires computation that might take you a while to run, so please get started early.
Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
What are some factors that determine primer annealing temperature during PCR?
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
How does the plasmid DNA enter the E. coli cells during transformation?
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Model this assembly method with Benchling or Asimov Kernel!
Assignment: Asimov Kernel
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
Week 7 — Genetic Circuits Part II: Neuromorphic Circuits
This week covers neuromorphic genetic circuits, showing how engineered gene networks can implement neural-network
“perceptron”-like computation and learning.
Lecture (Tues, Mar 17)
Genetic Circuits Part II: Neuromorphic Circuits (▶️Recording) Ron Weiss
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
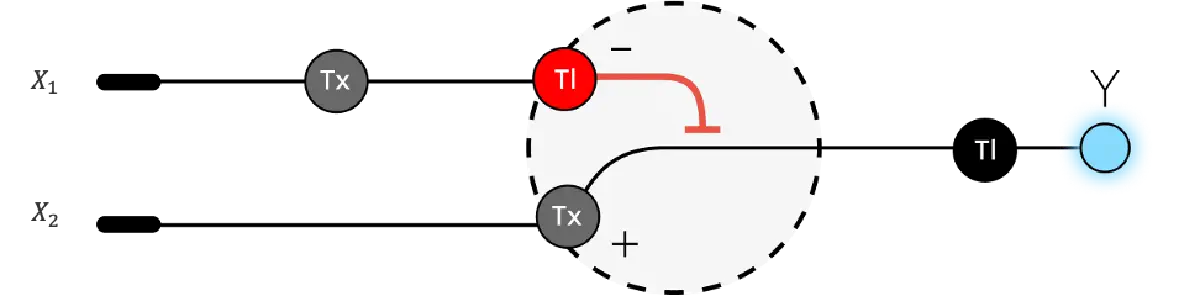
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Submit this Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs. DUE MARCH 20 FOR MIT/HARVARD/WELLESLEY STUDENTS
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
Homework Part A: General and Lecturer-Specific Questions
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Describe the main components of a cell-free expression system and explain the role of each component.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output?
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Could this function be realized by genetically modified natural cell?
Describe the desired outcome of your synthetic cell operation.
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
What would you encapsulate inside? Enzymes, small molecules.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
How will you measure the function of your system?
Example solution
Based on: Lentini, R. et al., 2014. Nat comm, 5, p.4012.
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output? Expand the sensing capacity of bacteria. Input: theophylline (inert to bacteria). Output of the SMC: IPTG. Output of the whole system: GFP produced in bacteria.
(Theophyline aptamer reference: *Martini, L. & Mansy, S.S., 2011. Cell-like systems with riboswitch controlled gene expression. Chemical Communications, 47(38), p.10734.*)
Could this function be realized by cell-free Tx/Tl alone, without encapsulation? No. If the IPTG were not encapsulated, it would go into the bacteria without the need of theophylline-induced membrane channel synthesis, thus the synthetic cell actuator would not exist.
Could this function be realized by genetically modified natural cell? Yes, in this particular case: the theophylline aptamer could be incorporated into a transformed gene. This lacks generality though – it is easier to make SMC than modify bacteria, so in this system a single bacteria reporter can be used to detect various small molecules.
Describe the desired outcome of your synthetic cell operation. In the presence of SMC, bacteria sense theophylline.
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of? Phospholipids + cholesterol.
What would you encapsulate inside? Enzymes, small molecules. cell-free Tx/Tl system, IPTG, gene for membrane transporter under the control of theophylline aptamer.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian) Bacterial, because of the theophylline riboswitch used as SMC input.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?) The membrane is permeable to the input molecule (theophylline), the output is IPTG that will cross the membrane via the membrane pore created after theophyline-initiated gene expression.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids: POPC, cholesterol
Enzymes: bacterial cell-free Tx/Tl
Genes: a-hemolysin (aHL) to encapsulate in SMC
Biological cells: *E.coli* transformed with GFP under T7 promoter and a lac operator
How will you measure the function of your system? Measure GFP output of the cells via flow cytometry. Alternatively, use enzymatic reporter, like luciferase, and measure bulk output of the enzyme.
Artificial cells translate chemical signals for E. coli. (a) In the absence of artificial cells (circles), E. coli (oblong) cannot sense theophylline. (b) Artificial cells can be engineered to detect theophylline and in response release IPTG, a chemical signal that induces a response in E. coli.
Homework question from Peter Nguyen
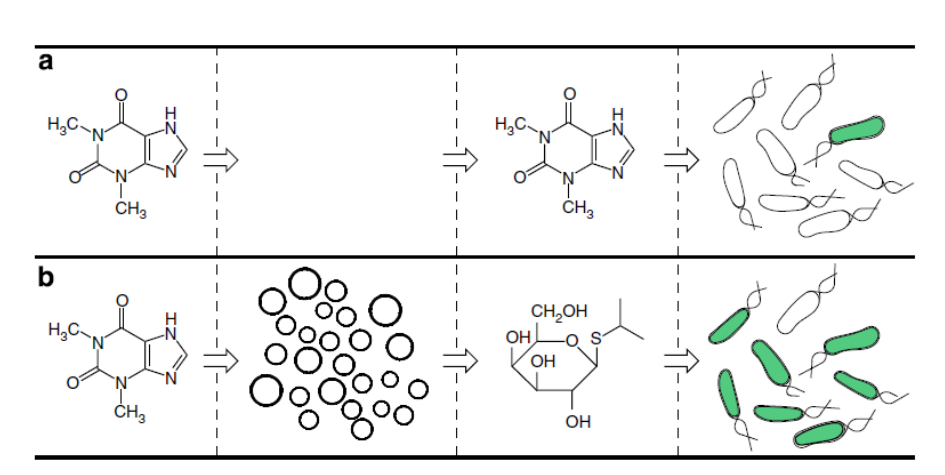
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
How will the idea work, in more detail? Write 3-4 sentences or more.
What societal challenge or market need will this address?
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Homework Part B: Individual Final Project
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
We’d like students to start exploring their final project in depth this week! Of your three Aims, for this week you should have at least Aim 1 decided and written down.
Put your chosen final project slide in the appropriate slide deck following the instructions on slide 1:
First Twist order deadline for MIT/Harvard/Wellesley students is Friday, April 3 at 11PM ET
First Twist order deadline for Committed Listeners is Friday, April 10 at 11PM ET. (Your Node Lead will place the Twist order, so please work with them to finalize your constructs and ordering decisions.)
This lecture presents a range of advanced technologies to do precision
measurement of proteins at atomic scales, characterizing chemical
composition, and detecting protein sequence and structure.
Homework is partly based on data that will be generated in the Waters Immerse Lab in Cambridge, MA. Students will characterize green fluorescent protein (eGFP, a recombinant protein standard) structure (primary, secondary/tertiary) in the lab using liquid chromatography and mass spectrometry, as well as Keyhole Limpet Hemocyanin (KLH) oligomeric states using charge detection mass spectrometry (CDMS). Data generated in the lab needed to do the homework is included both within this document and in the Appendix of the laboratory protocol.
Homework: Final Project
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Homework: Waters Part I — Molecular Weight
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence: MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Determine $z$ for each adjacent pair of peaks $(n, n+1)$ using:
$$ {\large z} = {\Large \frac{\frac{m}{z_{n+1}}}{\frac{m}{z_n} - \frac{m}{z_{n+1}}}} $$
Determine the MW of the protein using the relationship between $\frac{m}{z_n}$, $MW$, and $z$
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
$$ \text{Accuracy} = \frac{|MW_{\text{experiment}} - MW_{\text{theory}}|}{MW_{\text{theory}}} $$
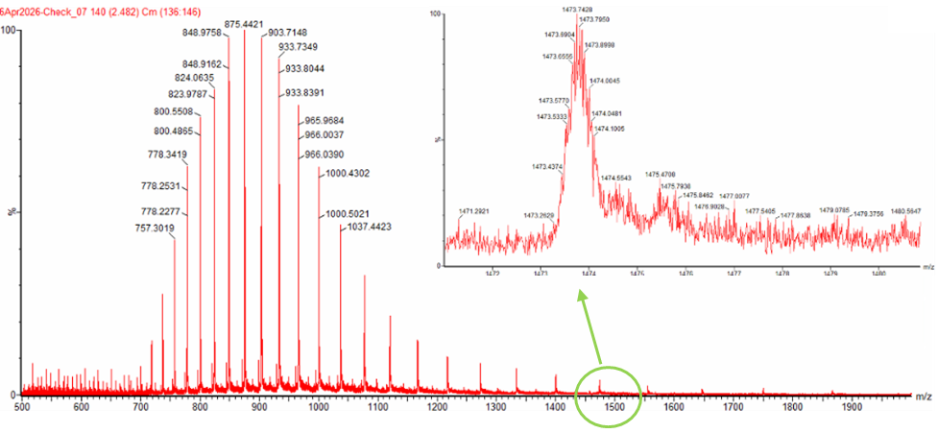
Figure 1. Mass Spectrum of intact eGFP protein from the Waters Xevo G3 LC-MS (a mass spectrometer with 30,000 resolution) with individual charge state peaks labeled with $\frac{m}{z}$ values.
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
Homework: Waters Part II — Secondary/Tertiary structure
Assignees for this section
MIT/Harvard students
Optional but highly recommended
Committed Listeners
Optional but highly recommended
We will analyze eGFP in its native, folded state and compare it to its denatured, unfolded state on a quadrupole time-of-flight MS. We will be doing MS-only analysis (no liquid chromatography, also known as “direct infusion” experiments) on the Waters Xevo G3-QToF MS.
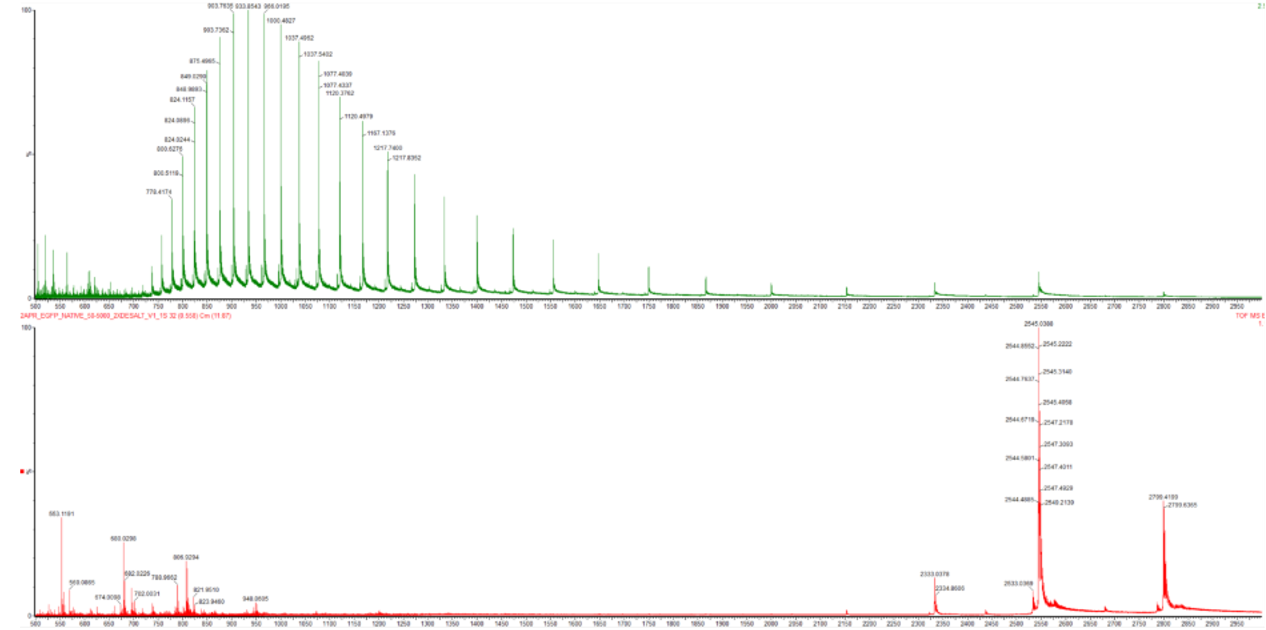
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
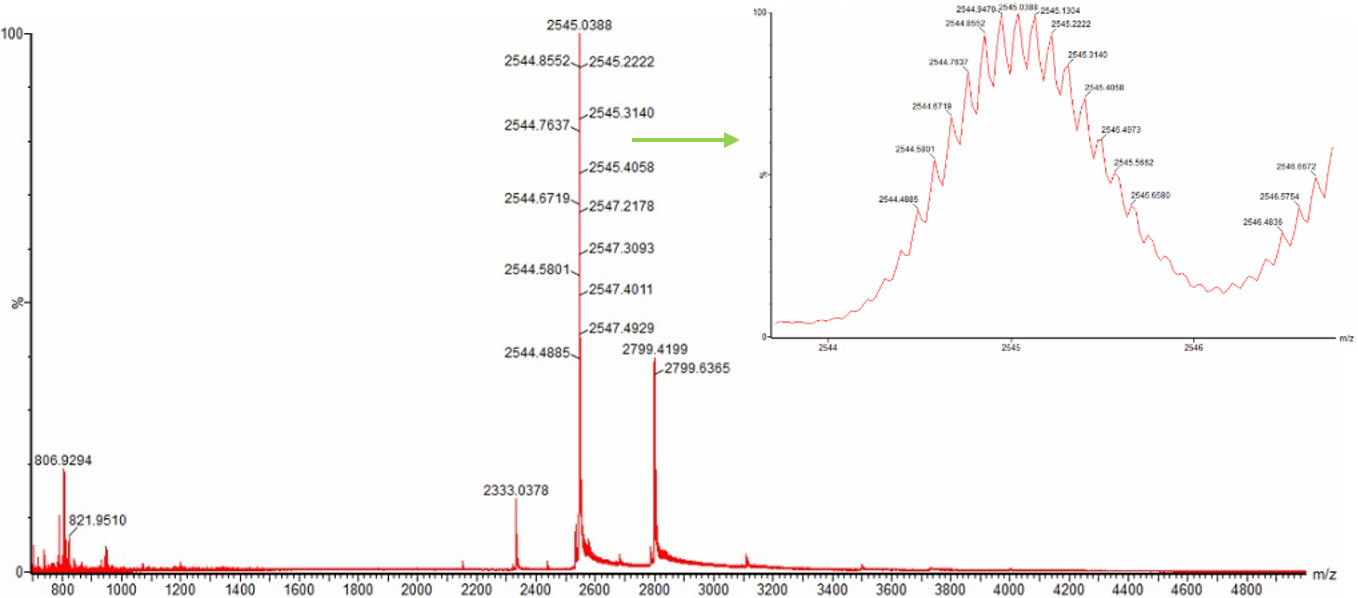
Figure 2. Comparison of the mass spectra between denatured (top) and native (bottom) eGFP standard on the Waters Xevo G3 QTof MS.
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800 $\frac{m}{z}$? What is the charge state? How can you tell?
Figure 3. Native eGFP mass spectrum from the Waters Xevo G3 Q-Tof MS. The inset is a zoomed-in view of the charge state at ~2800 $\frac{m}{z}$ on a mass spectrometer with 30,000 resolution.
Homework: Waters Part III — Peptide Mapping - primary structure
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
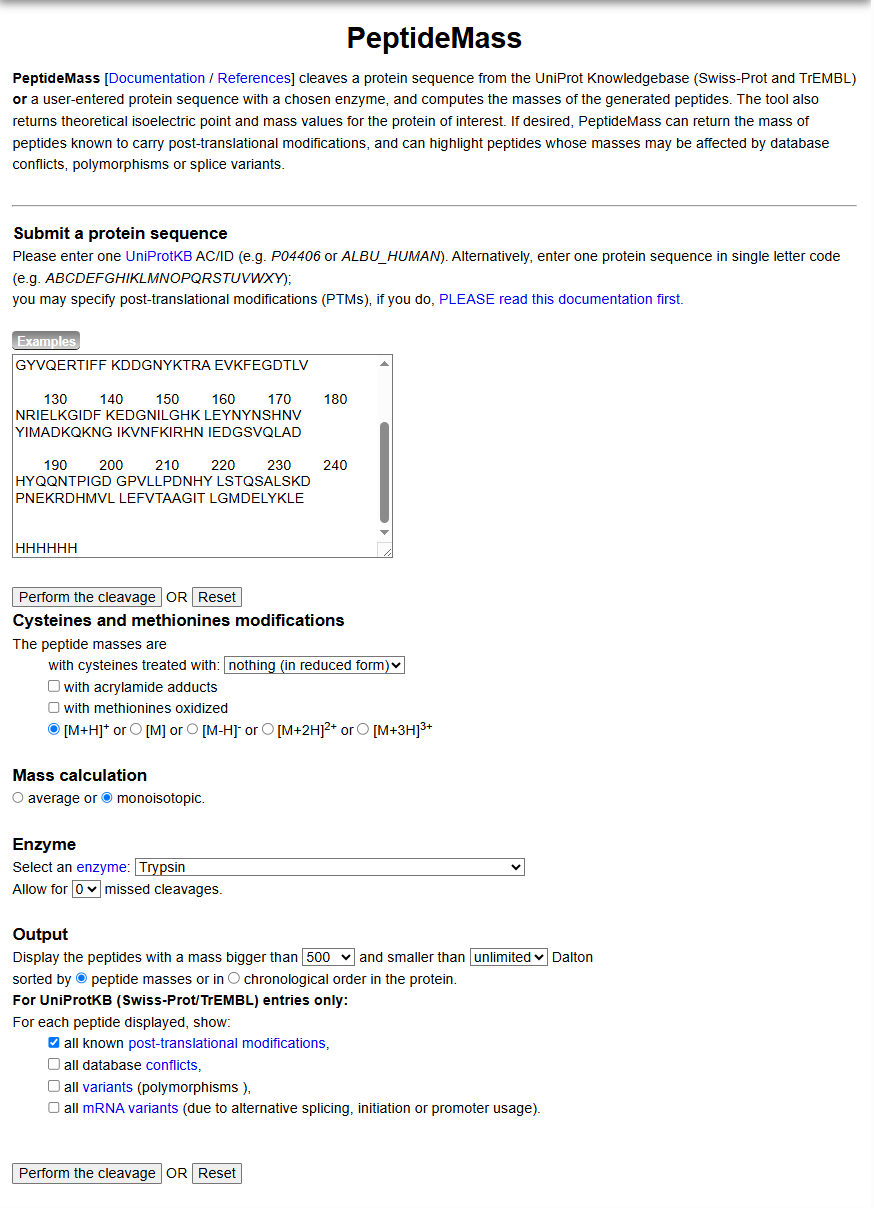
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
How many peptides will be generated from tryptic digestion of eGFP?
Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
Figure 4. Example conditions for predicting the number of tryptic peptides from the eGFP standard. Please replicate all parameters shown above.
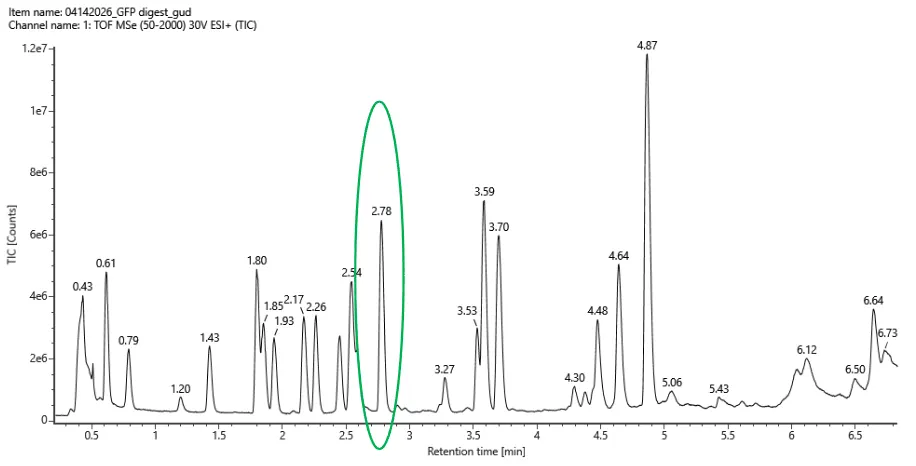
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
Figure 5a. Total ion chromatogram (TIC) of the eGFP peptide map. The peak at 2.78 minutes is circled, and its MS data is shown in the mass spectrum in Figure 5b, below.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
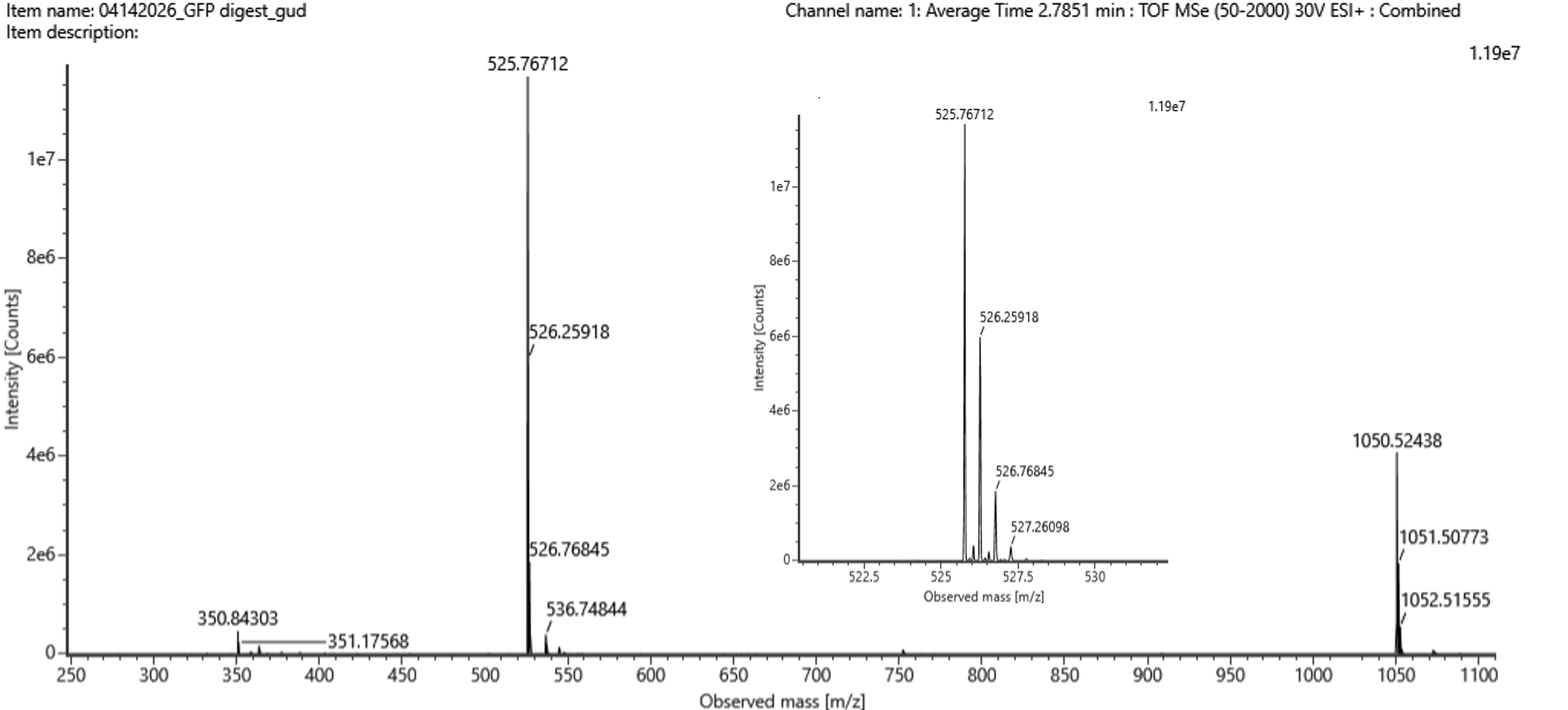
Identify the mass-to-charge ($\frac{m}{z}$) of the peptide shown in Figure 5b. What is the charge ($z$) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ($\small{[M\!\!+\!\!H]^+}$) based on its $\frac{m}{z}$ and $z$.
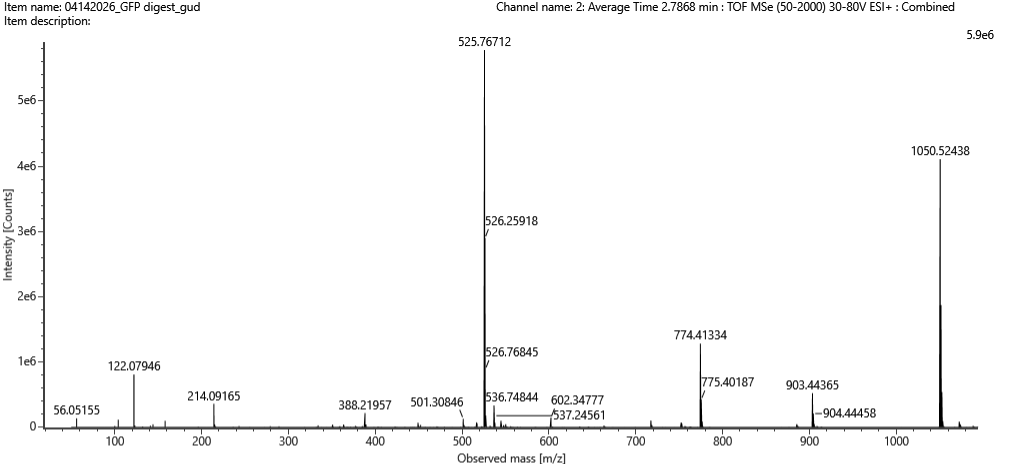
Figure 5b. Mass spectrum figure to show $\frac{m}{z}$ for the chromatographic peak at 2.78 min from Figure 5a above. The inset is a zoom-in of the peak at $\frac{m}{z}$ 525.76, to discern the isotope peaks.
Figure 5c. Fragmentation spectrum of the peptide eluting at retention time 2.78 minutes in Figure 5a (above).
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
(Recall that $ \text{Accuracy} = \frac{|MW_{\text{experiment}} - MW_{\text{theory}}|}{MW_{\text{theory}}} $ )
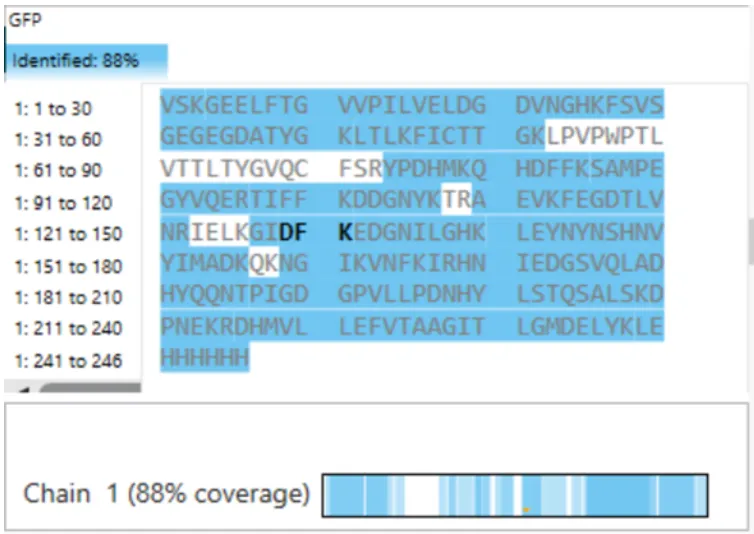
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
Figure 6. Amino Acid Coverage Map of eGFP based on BioAccord LC-MS peptide identification data.
Bonus Peptide Map Questions
Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c? (HINT: Use your results from Question 2 above to match the peptide molecular weight that is closest to that shown in Figure 5b. Copy and paste its sequence into this tool online to predict the fragmentation pattern based on its amino acid sequence: http://db.systemsbiology.net/proteomicsToolkit/FragIonServlet.html. What is the sequence of the eGFP peptide that best matches the fragmentation spectrum in Figure 5c?
Does the peptide map data make sense, i.e. do the results indicate the protein is the eGFP standard? Why or why not? Consult with Figure 6, which depicts the % amino acid coverage of peptides positively identified using their calculated mass and fragmentation pattern.
Homework: Waters Part IV — Oligomers
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
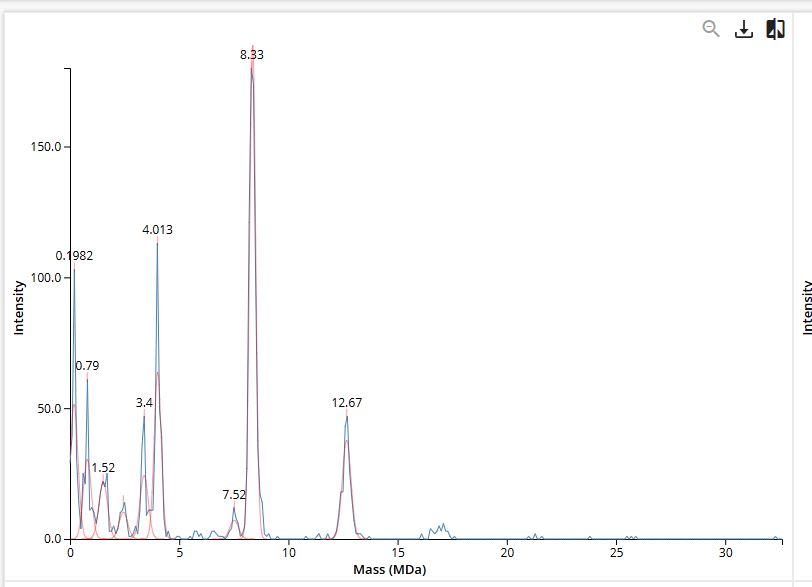
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS).
CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
7FU Decamer
8FU Didecamer
8FU 3-Decamer
8FU 4-Decamer
Polypeptide Subunit Name
Subunit Mass
7FU
340 kDa
8FU
400 kDa
Table 1: KLH Subunit Masses
Figure 7. Mass spectrum of Keyhole Limpet Hemocyanin (KLH) acquired on the CDMS.
Homework: Waters Part V — Did I make GFP?
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Cloud laboratories are making science accessible, affordable, and reproducible. Our aim this semester is to showcase how they can enable human creativity at scale, and how they provide a platform for collaboration and community.
How To Grow (Almost) Anything is about synthetic biology, bioengineering, robotics, automation, art, and AI. But it is also about friendship, shared purpose, and the freedom to build beyond what we know and to be inspired by what can be. To that end, the goal with this cloud lab unit and homework assignment is to inspire collaboration and creativity while designing a scientifically rigorous cell-free fluorescent protein optimization experiment together.
As you plan for final projects, you may want to refer to the provided non-exhaustive list of common Nebula protocols and their parameters in the “Reading & Resources” section below.
Homework — DUE BY START OF APR 28 LECTURE
Info
Note that this homework is due a week later than it ordinarily would due to its release a week later than normal.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse.
If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
Make a note on your HTGAA webpages including:
what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
what you liked about the project, and
what about this collaborative art experiment could be made better for next year.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
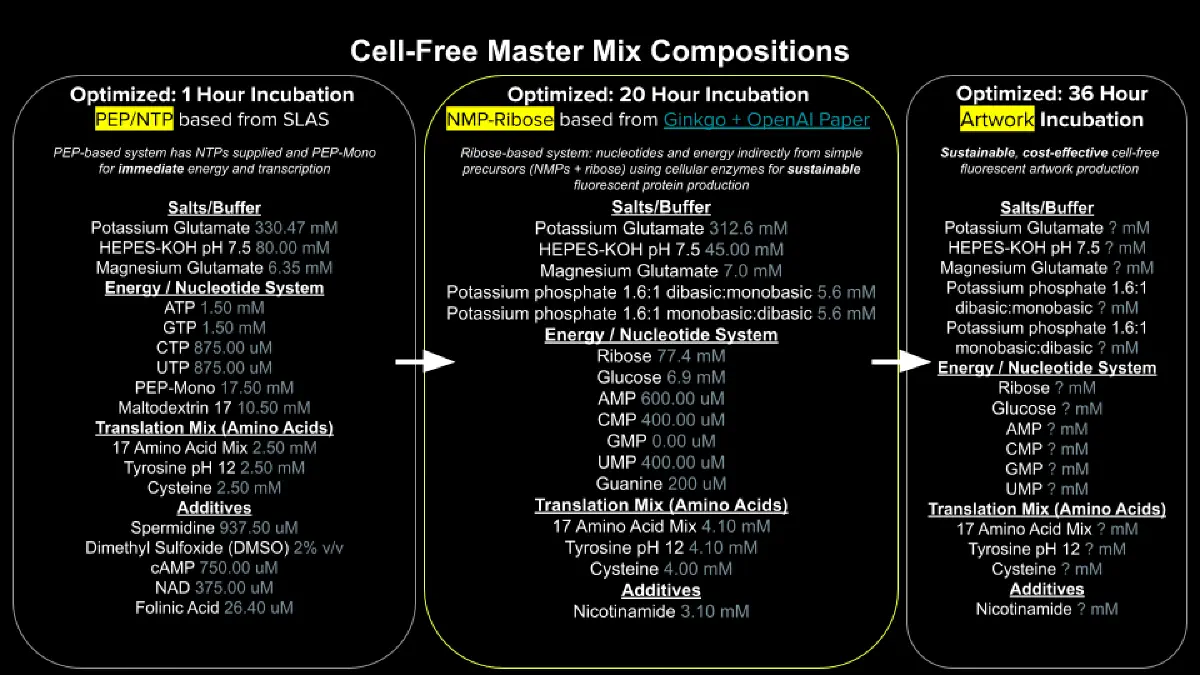
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
Salts/Buffer
Potassium Glutamate
HEPES-KOH pH 7.5
Magnesium Glutamate
Potassium phosphate monobasic
Potassium phosphate dibasic
Energy / Nucleotide System
Ribose
Glucose
AMP
CMP
GMP
UMP
Guanine
Translation Mix (Amino Acids)
17 Amino Acid Mix
Tyrosine
Cysteine
Additives
Nicotinamide
Backfill
Nuclease Free Water
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Assignees for this section
MIT/Harvard students
Required
Committed Listeners
Required
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!).
The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
Assignees for this section
MIT/Harvard students
Optional
Committed Listeners
Optional
Ginkgo Nebula Cloud Laboratory Rendering, 2025
Use this simulation tool to create an interesting looking cloud lab out of the Ginkgo Reconfigurable Automation Carts. This is just a minimal implementation so far, but I would love to see some fun designs!
Tip
Note from Ronan: If you are interested in helping me build out future HTGAA cloud lab software, please fill out this form!
Week 13 — AI, SynBio, and Scaling Health Innovation (ARPA-H)
This week covers designing, programming, and fabricating engineered living materials — such as self-healing concretes,
adaptive biofilms, and responsive biomaterials — by integrating genetic circuit design, materials science, and bioprocess
engineering.
Lecture (Tues, Apr 28)
AI, SynBio, and Scaling Health Innovation (ARPA-H) (▶️Recording) Renee Wegrzyn


















![DNA-based digital data storage technology. Source: Archives in DNA: Workshop Exploring Implications of an Emerging Bio-Digital Technology through Design Fiction - Scientific Figure on ResearchGate. Available from: https://www.researchgate.net/figure/DNA-based-digital-data-storage-technology_fig1_353128454 [accessed 11 Feb 2025]](/2026a/course-pages/weeks/week-02/image4.png)