Week 4 HW: Protein Design Part 1
Part A. Conceptual Questions
Question from Shuguang Zhang:
Question 1
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
According to the European Commission (2021), if we are talking about beef, meat has a protein percentage of 16.9%.
Therefore, in 500 grams of beef, we can find 84,5 grams of protein.
Considering the transformation 1 Dalton = 1 g/mol and that proteins are just chains of amino acids, we can get that in these 84,5 grams of protein there are 0,845 moles of amino acids (84.5 grams of amino acids * (1 mol / 100 grams of amino acids)).
Finally using the conversion 1 mol = 6.022 × 1023 molecules, we obtain that in this piece there are 5.09 × 1023 molecules of aminoacids.
Question 2
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans do not become cows when eating beef or fish when eating fish because food is broken down into its basic molecules during digestion.
Additionally, food does not have any transformative effect on our DNA. Our cells synthesize proteins based on our own genetic information, stored in our DNA and expressed through RNA. Therefore, although we use nutrients derived from other organisms, the instructions that determine who we are, come exclusively from our own genome.
Question 3
Why are there only 20 natural amino acids?
There is still no clear answer to this; however, many scientists hypothesize that it is due to natural selection. The 20 natural amino acids likely emerged because of the conditions of early Earth: its chemical composition, biochemical processes, and the functional utility these amino acids provided for primitive metabolisms. There are additional ideas in this area, but there is still much to investigate.
Question 4
Can you make other non-natural amino acids? Design some new amino acids.
Yes. To design these we can first expand the genetic code using 4-base codons (example CCCG). Then we have to perform chemical loading of theses aa in tRNAs.
Question 5
Where did amino acids come from before enzymes that make them, and before life started?
Before enzymes and life existed, amino acids most likely originated from prebiotic synthesis on early Earth. Experiments such as the Miller–Urey experiment, which replicated primitive environmental conditions, demonstrated that amino acids can be synthesized from simple molecules like methane, ammonia, and water under those conditions.
Additionally, it is believed that amino acids could also have formed through other prebiotic pathways, such as the Strecker synthesis (the reaction of aldehydes with ammonia and hydrogen cyanide) and salt-induced peptide formation.
Finally, extraterrestrial delivery is also considered a possible source, as amino acids have been found in certain meteorites.
Question 6
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
As α-helix using L-amino acids are right-handed, I will expect that if we use D-amino acids these helices would be left-handed.
In the literature, I found that homochiral decapeptides with D-aminoacids do form left-handed an α-helices.
Question 7
Why are most molecular helices right-handed?
Because overall they are formed from L-amino acids that inherently produce right-handed.
Additionally, these helices have greater thermodynamics and kinetic stability and can play critical functional or specific roles.
Question 8
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
There is a combination of factors that promotes this aggregation, which encompasses thermodynamic factors, such as entropy changes, as well as amino acid sequence and solvent conditions.
The main driving force overall is hydrophobic interactions. The hydrophobic faces of β-sheets tend to associate with each other in order to reduce their exposure to the surrounding solvent. This reduction in solvent exposure stabilizes the aggregated structure.
Question 9
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Many amyloid diseases involve the formation of β-sheets because β-sheet structures are highly stable. This stability promotes aggregation, leading to fibril formation and protein misfolding.
Yes, β-sheets can be used as materials. They are employed in biomaterials such as hydrogels due to their favorable properties, including good biocompatibility, strong mechanical strength, and more.
Part B. Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
I select the protein: Human Ferrochelatase
Recently I have been reading about therapeutics that target mitochondria. Specifically in the paper of Corson Lab they mention about this protein that when is inhibited causes mitochondrial dysfunction and this is benefitial for anti-angiogenesis purposes in retinal pathologies. So I choose it because im interested in its therapeutic applications.
2. Identify the amino acid sequence of your protein
>sp|P22830|HEMH_HUMAN Ferrochelatase, mitochondrial OS=Homo sapiens OX=9606 GN=FECH PE=1 SV=2 MRSLGANMAAALRAAGVLLRDPLASSSWRVCQPWRWKSGAAAAAVTTETAQHAQGAKPQV QPQKRKPKTGILMLNMGGPETLGDVHDFLLRLFLDRDLMTLPIQNKLAPFIAKRRTPKIQ EQYRRIGGGSPIKIWTSKQGEGMVKLLDELSPNTAPHKYYIGFRYVHPLTEEAIEEMERD GLERAIAFTQYPQYSCSTTGSSLNAIYRYYNQVGRKPTMKWSTIDRWPTHHLLIQCFADH ILKELDHFPLEKRSEVVILFSAHSLPMSVVNRGDPYPQEVSATVQKVMERLEYCNPYRLV WQSKVGPMPWLGPQTDESIKGLCERGRKNILLVPIAFTSDHIETLYELDIEYSQVLAKEC GVENIRRAESLNGNPLFSKALADLVHSHIQSNELCSKQLTLSCPLCVNPVCRETKSFFTS QQL
How long is it? What is the most frequent amino acid?
The sequence contains 422 amino acids.
The most frequent amino acid in this sequence is Leucine (L).
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs. Using the Blast tool: https://www.uniprot.org/blast/uniprotkb/ncbiblast-R20260304-130448-0471-13468649-p1m/overview
I found 250 coincidences, these are basically are the analogues of ferrochelatase in other species. The ones with a higher coincidence, are from closely related species such as gorillas, chimpanzees and more.
Does your protein belong to any protein family? Yes, it belong to the ferrochelatase family.
3. Identify the structure page of your protein in RCSB
https://www.rcsb.org/3d-view/1HRK
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The crystal structure of human ferrochelatase was solved in 2001 and released on June 22, 2001. The structure was published by Wu et al. in Nature Structural Biology.
It was determined using X-ray diffraction, with a resolution of 2.0 Å (high quality).
Are there any other molecules in the solved structure apart from protein?
Yes, besides the protein chains, there are additional molecules listed as CHD, FES, and O.
- FES → Iron–sulfur cluster (2Fe–2S cluster). Important for structural stability and possibly regulation.
- CHD → A ligand molecule bound in the structure. Porphyrin-related compound (a heme precursor or analog bound in the active site).
- O (often shown as O or HOH) → Oxygen atoms from water molecules. These are crystallographic water molecules resolved in the X-ray structure.
Does your protein belong to any structure classification family? Using the CATCH classification system (class, architecture, topology, homology). I found that this protein does belong to some classification families. Here you can see my findings: https://www.cathdb.info/search/by_text?q=P22830
Specifically I found that it is assigned to the CATH superfamilies 3.40.50.140 and 3.40.50.1400. Which means: Class 3 (α/β proteins), with a 3-layer (αβα) sandwich architecture and Rossman fold topology.
There are some other families as well.
4. Open the structure of your protein in any 3D molecule visualization software:
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
I use Pymol for the visualization purposes.
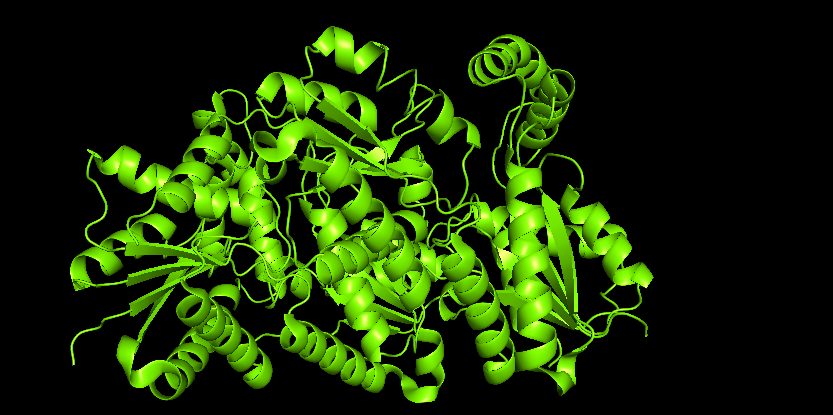 Figure 1. Cartoon
Figure 1. Cartoon
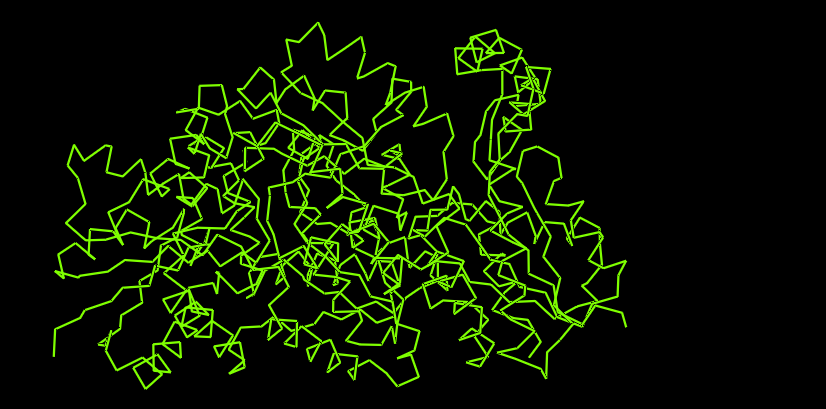 Figure 2. Ribbon
Figure 2. Ribbon
 Figure 3. Ball and stick
Figure 3. Ball and stick
Color the protein by secondary structure. Does it have more helices or sheets?
It is color by the legend: red helices, yellow sheets and green loops and others. Visually it appears that it has more red (helices).
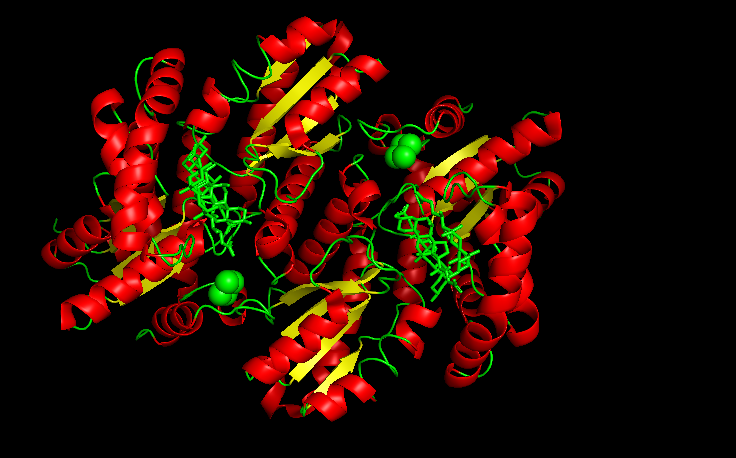
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
First I color following this legend:
- Hydrophobic residues
- ALA, VAL, LEU, ILE, MET, PHE, TRP, PRO (colored orange)
- Polar uncharged residues
- SER, THR, ASN, GLN, TYR, CYS (colored cyan)
- Negatively charged residues
- ASP, GLU (colored red)
- Positively charged residues
- LYS, ARG, HIS (colored blue)
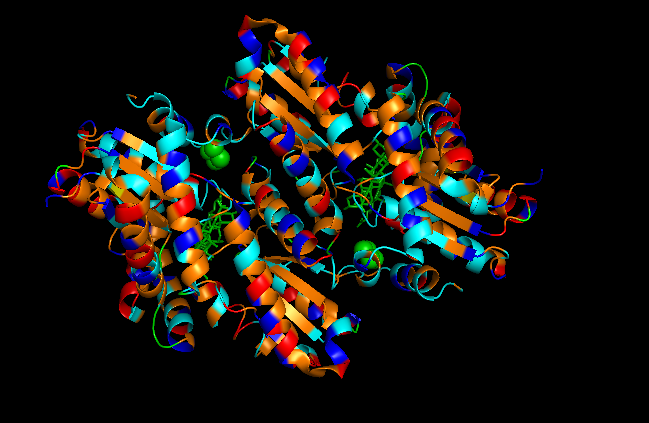
To understand the distribution of hydrophobic vs hydrophilic residues, then I color following this legend:
- Hydrophobic residues
- ALA, VAL, LEU, ILE, MET, PHE, TRP, PRO (colored cyan)
- Hydrophilic residues
- SER, THR, ASN, GLN, TYR, CYS, ASP, GLU, LYS, ARG, HIS (colored magenta)
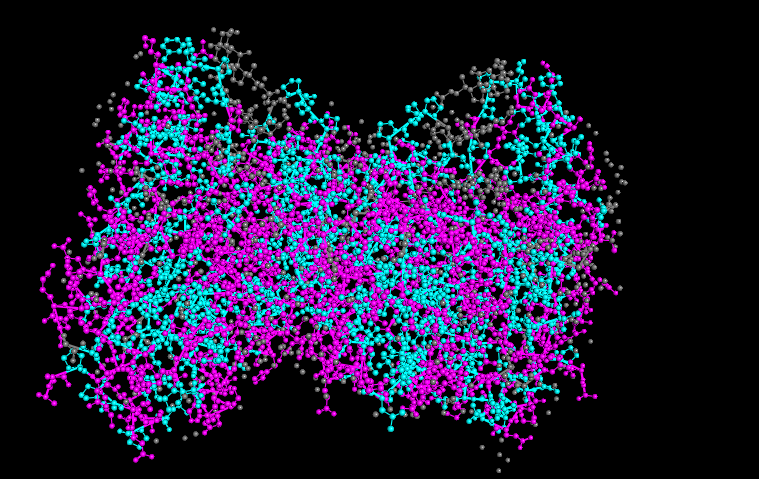
It seems like the hydrophobic residues are not fully just in the core of the protein, like it is common in some proteins. This is because it is associated with the inner mitochondrial membrane, allowing partial interaction with the lipid environment.
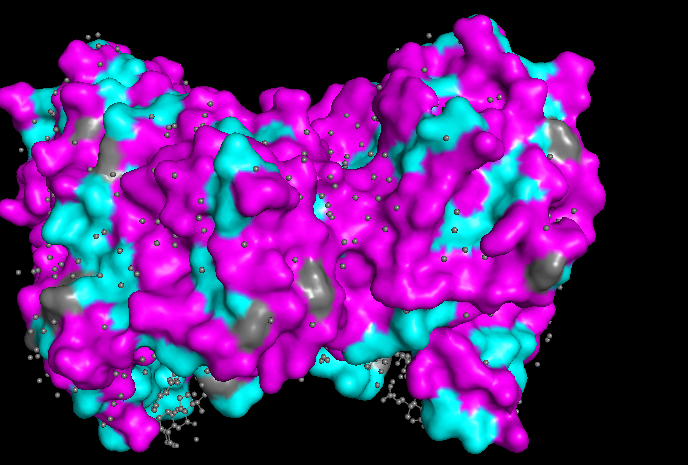
Watching the surface of the protein, we can see more magenta (hydrophilic) which is common but also some cyan (hydrophobic)
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
With this 2 disctint views of the protein we can see clearly 2 huge pockets.
The ferrochelatase is a homodimer, so in each chain it has a binding pocket.
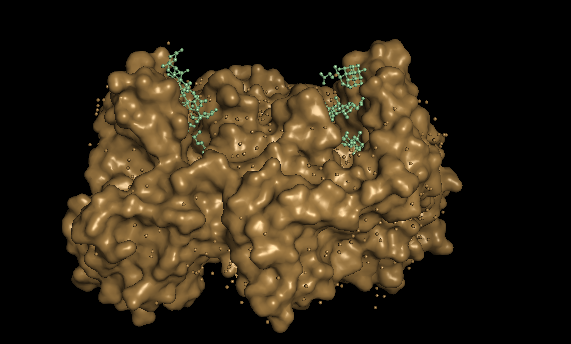
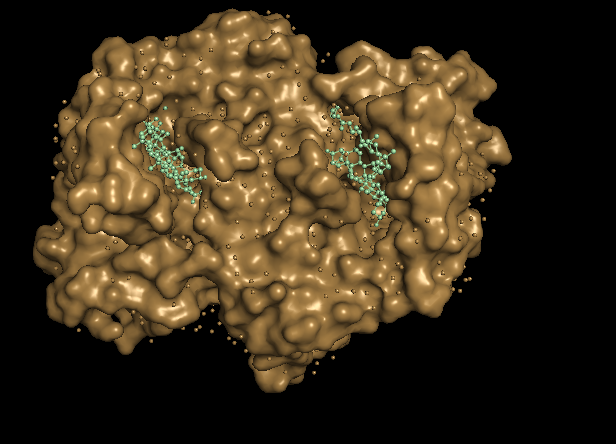
Part C. Using ML-based Protein Design Tools
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
Choose your favorite protein from the PDB.
We will now try multiple things in the three sections below:
C1. Protein Language Modeling
Deep Mutational Scans
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
c. (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
Latent Space Analysis
a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein folding
Folding a protein
a. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
b. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
C3. Protein Generation
Inverse-Folding a protein
Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
a. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
b. Input this sequence into ESMFold and compare the predicted structure to your original.
Part D. Group Brainstorm on Bacteriophage Engineering
Main goal chosen to address computationally
We’ll try to slightly delay the timing for lysis, so the virion assembly improved and we get higher titers.
One page proposal

Link for the detailed brainstorm, plan and references:
https://docs.google.com/document/d/1ey8aSVH0GR7EnGLjnGY2CeJfcK8cSaQCqVYApxdl8WY/edit?usp=sharing
References
European Commission. (2021, January 22). Average protein content in g/100 g and % of food energy from protein in animal-derived raw foods. Knowledge4Policy Health Promotion Knowledge Gateway. https://knowledge4policy.ec.europa.eu/health-promotion-knowledge-gateway/dietary-protein-animal-1a_en
Fernández, A. (2005). What factor drives the fibrillogenic association of β-sheets?. FEBS letters, 579(29), 6635-6640.
Jurkowski, M., Kogut, M., Sappati, S., & Czub, J. (2024). Why are left-handed G-quadruplexes scarce?. The Journal of Physical Chemistry Letters, 15(11), 3142-3148.
Li, C., Qin, R., Liu, R., Miao, S., & Yang, P. (2018). Functional amyloid materials at surfaces/interfaces. Biomaterials science, 6(3), 462-472.
Pascal, R., & Boiteau, L. (2007). Origins of Life: Emergence of Amino Acids. Wiley Encyclopedia of Chemical Biology, 1-7.
Philip, G. K., & Freeland, S. J. (2011). Did evolution select a nonrandom “alphabet” of amino acids?. Astrobiology, 11(3), 235-240.
Shepherd, N. E., Hoang, H. N., Abbenante, G., & Fairlie, D. P. (2009). Left-and right-handed alpha-helical turns in homo-and hetero-chiral helical scaffolds. Journal of the American Chemical Society, 131(43), 15877-15886.
Sisido, M., Ninomiya, K., Ohtsuki, T., & Hohsaka, T. (2005). Four-base codon/anticodon strategy and non-enzymatic aminoacylation for protein engineering with non-natural amino acids. Methods, 36(3), 270-278.