Week 04 HW: Protein Design part I

Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average, an amino acid is ~100 Daltons)
500 grams of protein, approximately, has 20% of protein.
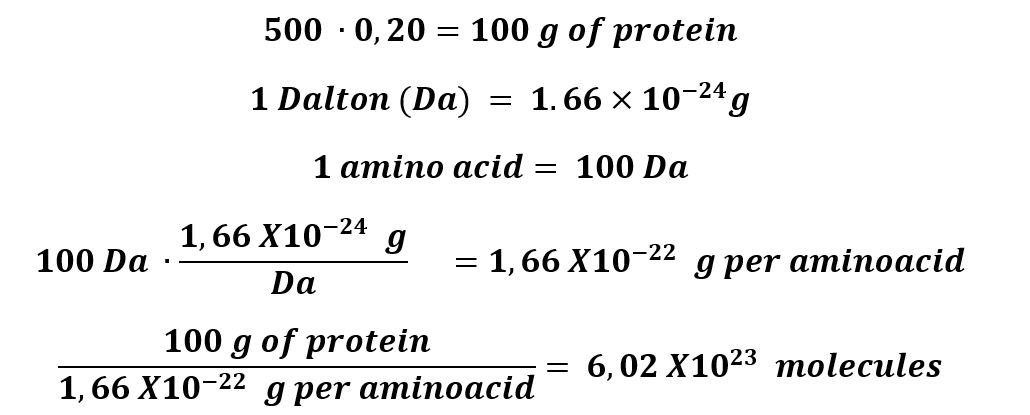
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
This phenomenon occurs because humans are living beings with a special anatomy; indeed, we have a relatively smaller colon and larger small intestine, which shows that our system is prepared to process high-protein diets. These characteristics, along with others like gastric acidity, allow humans to ingest beef and fish, and thought-out gastric system becomes a big part of food, especially meat, in amino acids that our body can use to synthesize proteins that we need. This is why it is important to have a balanced diet with an adequate amount of protein.
3. Why are there only 20 natural amino acids?
It is not like just existing 20 amino acids; in fact, there might be different combinations of amino acids. However, nature is wise and decided the combinations for the 20 natural amino acids that we know, due to several reasons.
Criteria for selecting amino acids:
Choice of atoms: Amino acids need to be made of atoms that are abundant on Earth, such as C, H, N, O, and S.
Functional groups: Due to the selection of atoms is important that the functional groups form hydrogen bonds and electrostatic interactions. Like Amides, amines, hydroxyls, carboxyls, and carbon–nitrogen bonds.
Biosynthetic cost: Protein synthesis is the process that uses the largest amount of energy in a cell. Scientists have measured the cost of biosynthesis of each amino acid, measured in terms of glucose and ATP molecules. For example, Leu costs only 1 ATP, but its isomer Ile costs 11. Nature chooses the most effective cost option.
Solubility: Amino acids need to be soluble in high concentrated aqueous environment.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, scientists have been doing that for years. And for this educational exercise, I would like to design a fluorescent amino acid. A fluorescent molecule typically has a conjugated system with one or more aromatic rings.
The base structure of amino acids is:
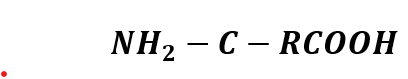
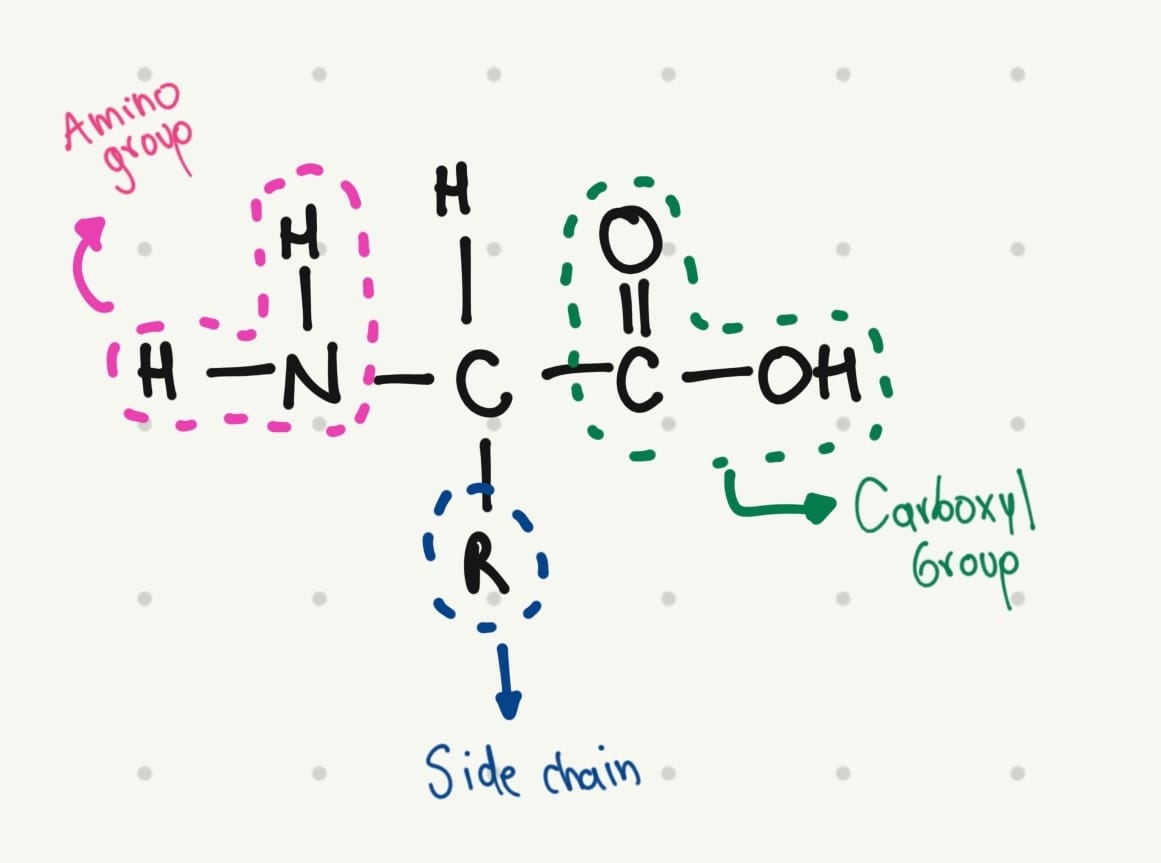
That’s why I thought in a simple structure:
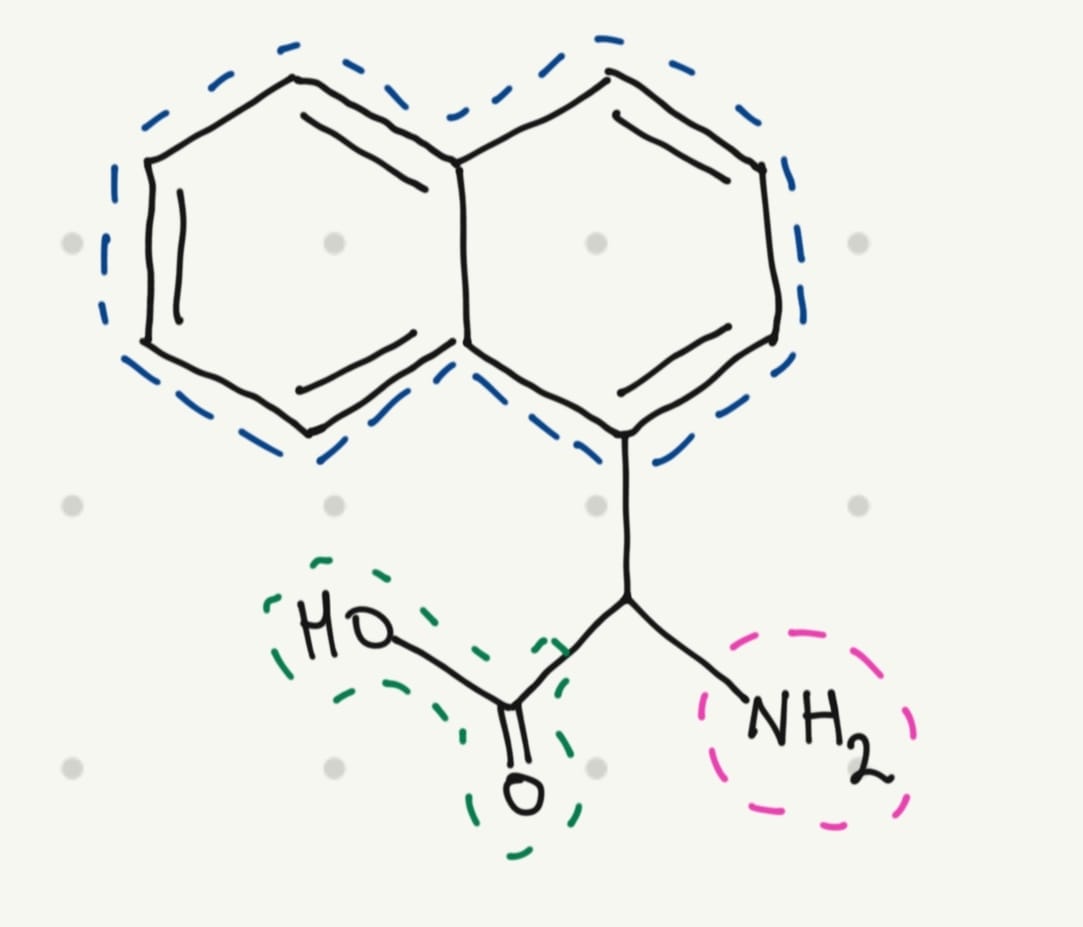
5. Where did amino acids come from before enzymes that make them, and before life started?
Today many amino acids are synthesized by metabolic and biosynthesis pathways. However, in the earliest years of life (between 4000 and 3500 million years), they were synthesized by chemical synthesis.
This hypothesis was proven by Miller and Urey in 1953, when they performed an experiment to recreate the conditions of primordial Earth in a flask. They create an atmosphere with ammonia, hydrogen, methane, and water vapor, plus electrical sparks. They found that new molecules were formed. Specifically, these molecules result in eleven standard amino acids.
In conclusion, in the beginning, amino acids were synthesized due to the environmental conditions; today, they are synthesized by biosynthesis.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
There exists a concept known as chirality, which is the property of an object that is not superimposed on its mirror image. This means that molecules with chirality have an asymmetric carbon, making them mirror images of each other. One good example of this phenomenon is your hands; they are the mirror image of each other, but they cannot be superimposed.
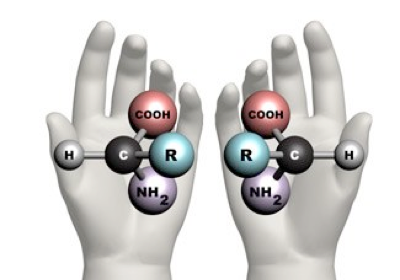 Taken from: https://www.maths.ox.ac.uk/node/14490
Taken from: https://www.maths.ox.ac.uk/node/14490
Natural proteins are made of L-amino acids. When these amino acids form an α-helix, it is right-handed, but following the idea of chirality, if D-amino acids form the α-helix, it will be left-handed.
7. Can you discover additional helices in proteins?
Yes, indeed, scientists have been developing new forms of helices for years. They have identified only 1,000 distinct protein folds in nature; however, they are developing different modifications of these natural folds. For example, researchers have identified alternative helical conformations such as 3₁₀-helices and π-helices.
They have also been trying to fold random amino acid sequences. All these methods are great, but the results might be inaccurate and do not represent a standardized process.
For this reason, they are presenting a new computational method for generating packings of secondary structures, which will facilitate the search for novel protein folds.
8. Why are most molecular helices right-handed?
Besides the natural chirality of amino acids that form proteins, several influencing factors determine why most molecular helices are right-handed. The alpha helix structure is more stable because it uses the hydrogen bond between the C=O and N-H groups of the main chain to stabilize it. Although these bonds can form in both right-handed and left-handed alpha helices, they are more favorable in a right-handed alpha helix, because it requires less energy due to reduced steric clashes between the side chains and the main chain.
9. Why do β-sheets tend to aggregate? And what is the driving force for β-sheet aggregation?
β-Sheets are polypeptide strands connected by hydrogen bonds of adjacent backbone amides; these bonds are stronger and perpendicular, especially when the strands are aligned in opposite directions.
These characteristics provide the strands with the capacity to extend in a planar and stable structure due to the hydrogen bonds, which means that β-Sheets can interact with other β-Sheets, leading to aggregation.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
1. Briefly describe the protein you selected and why you selected it.
I chose the Dopamine Transporter (DAT) because one of my interests is the addiction area. As a pharmacist, I acknowledge that people with chronic pain are more vulnerable to developing addiction. But this problem can be presented in other individuals whose use abused drugs.
This transporter has a special role in dopamine homeostasis because it is the one responsible for the reuptake of dopamine from the synaptic space. The DAT is the major target of the most common drug of abuse, especially psychostimulants. When we do pleasurable activities, there are signaling pathways that create action potential, which indicate the release of neurotransmitters, among them dopamine, in the synaptic space.
After the action potential disappears, the DAT has the responsibility of maintaining homeostasis and the reuptake of dopamine to maintain the balance. However, abused drugs affect this process in different ways.
Alcohol, nicotine, and heroin increase the action potential, leading to a major release of dopamine. Cocaine and methamphetamine bind to the DAT and block the reuptake of dopamine.
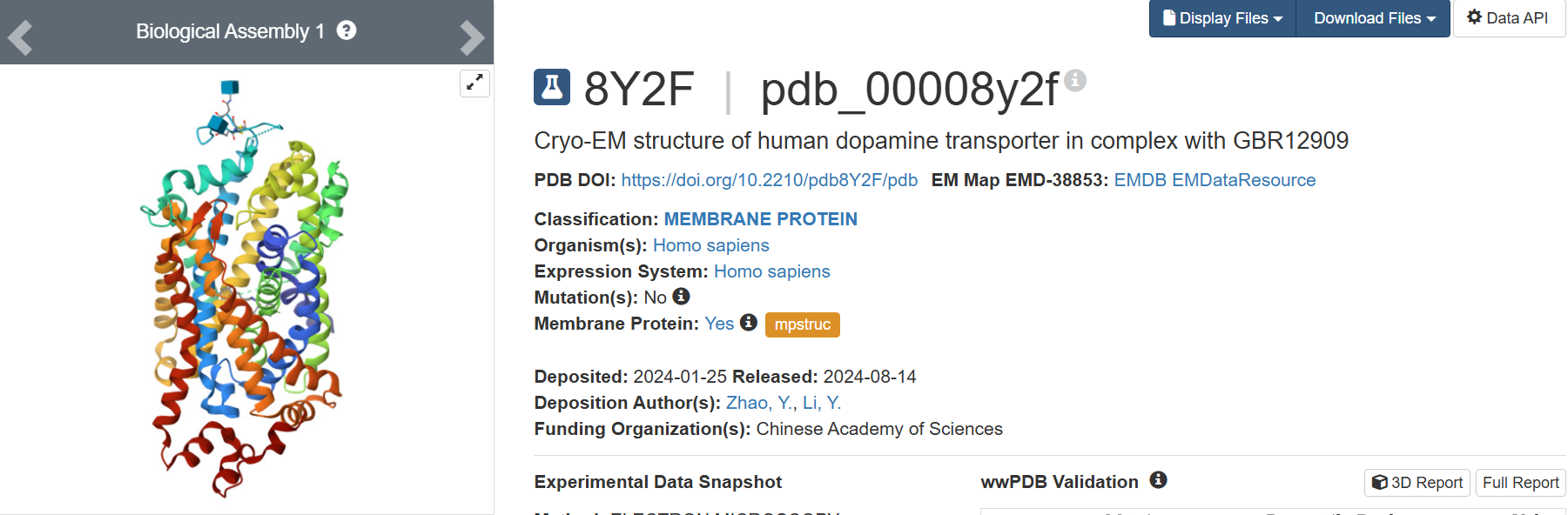
I found the structure in PDB title: 8Y2F | pdb_00008y2f Cryo-EM structure of human dopamine transporter in complex with GBR12909
2. Identify the amino acid sequence of your protein.
sp|Q01959|SC6A3_HUMAN Sodium-dependent dopamine transporter OS=Homo sapiens OX=9606 GN=SLC6A3 PE=1 SV=1
MSKSKCSVGLMSSVVAPAKEPNAVGPKEVELILVKEQNGVQLTSSTLTNPRQSPVEAQDRETWGKKIDFLLSVIGFAVDLANVWRFPYLCYKNGGGAFLVPYLLFMVIAGMPLFYMELALGQFNREGAAGVWKICPILKGVGFTVILISLYVGFFYNVIIAWALHYLFSSFTTELPWIHCNNSWNSPNCSDAHPGDSSGDSSGLNDTFGTTPAAEYFERGVLHLHQSHGIDDLGPPRWQLTACLVLVIVLLYFSLWKGVKTSGKVVWITATMPYVVLTALLLRGVTLPGAIDGIRAYLSVDFYRLCEASVWIDAATQVCFSLGVGFGVLIAFSSYNKFTNNCYRDAIVTTSINSLTSFSSGFVVFSFLGYMAQKHSVPIGDVAKDGPGLIFIIYPEAIATLPLSSAWAVVFFIMLLTLGIDSAMGGMESVITGLIDEFQLLHRHRELFTLFIVLATFLLSLFCVTNGGIYVFTLLDHFAAGTSILFGVLIEAIGVAWFYGVGQFSDDIQQMTGQRPSLYWRLCWKLVSPCFLLFVVVVSIVTFRPPHYGAYIFPDWANALGWVIATSSMAMVPIYAAYKFCSLPGSFREKLAYAIAPEKDRELVDRGEVRQFTLRHWLKV
• How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
620 aminoacids
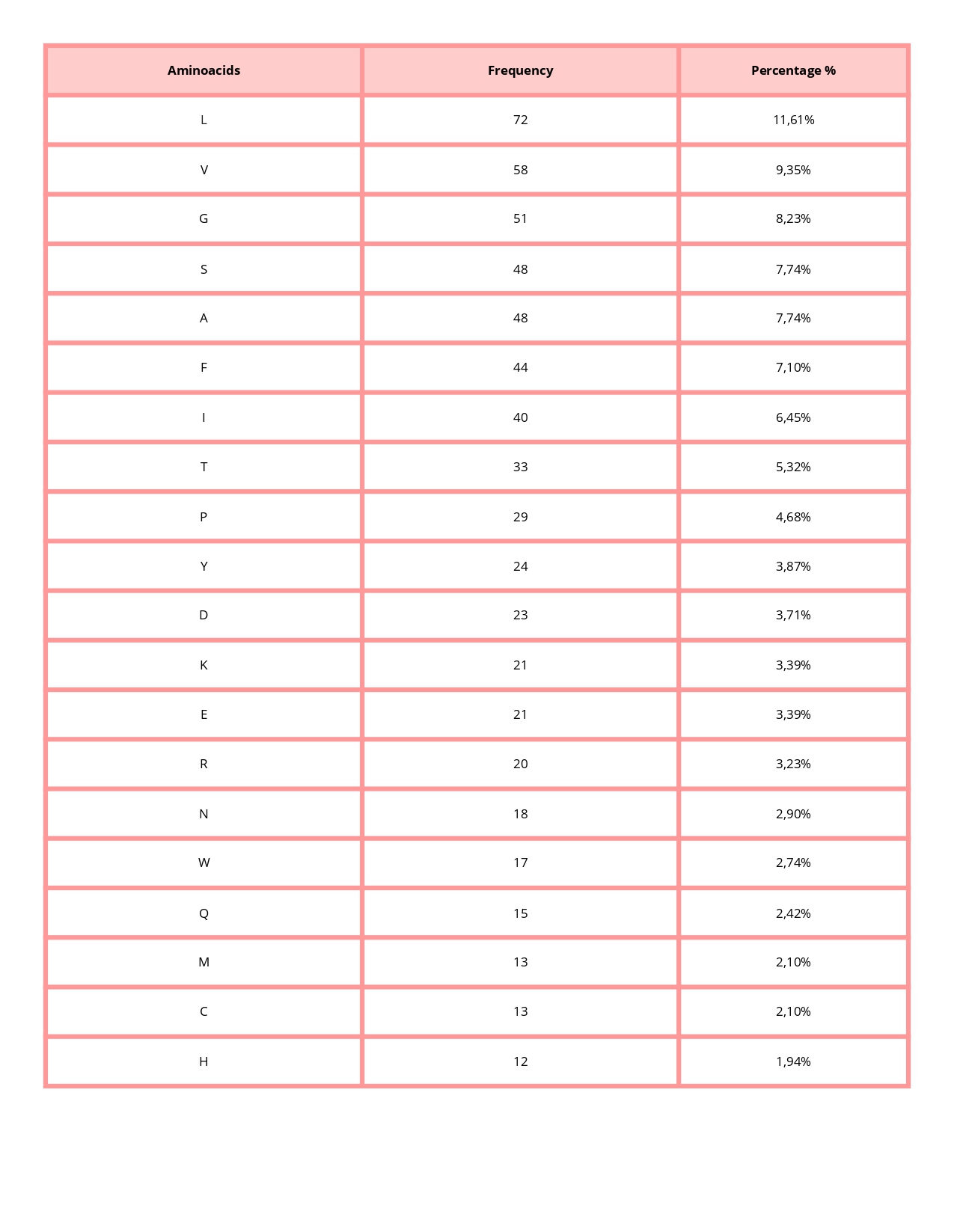
The most common amino acid is: L (Leucine), which appears 72 times.
• How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
Uniprot’s BLAST tool found 250 homologs

• Does your protein belong to any protein family?
Yes, it is a member of the monoamine transporter family (MAT), which is the family of proteins responsible for regulating neurotransmitter concentrations.
3. Identify the structure page of your protein in RCSB
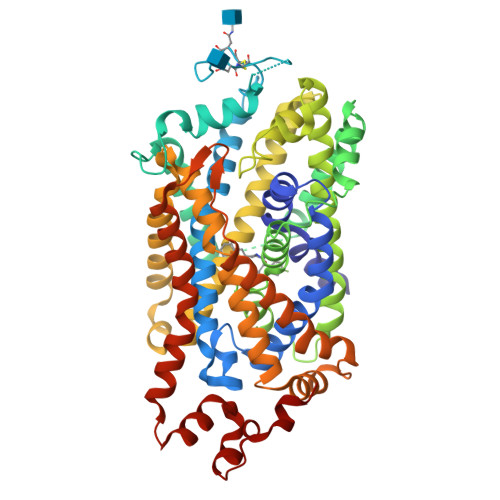
• When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The 8Y2F structure of the human Dopamine Transporter was deposited in the PDB on January 25, 2024 and published on August 14, 2024.
Resolution: 2.97 Å
The best resolution in electron microscopy for protein structure determination is between 1.25 Å - 2.00, however, one value of 2.97 Å is accurate but might be losing some details.
• Are there any other molecules in the solved structure apart from protein?
Yes, 2 small ligands:
1. Vanoxerine (ID: A1D5S): C28 H32 F2 N2 O – Chains: B
2. 2-acetamido-2-deoxy-beta-D-glucopyranose (ID:NAG): C8 H15 N O6 – Chains: C and D
• Does your protein belong to any structure classification family?
Membrane protein
4. Open the structure of your protein in any 3D molecule visualization software:
• PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
• Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
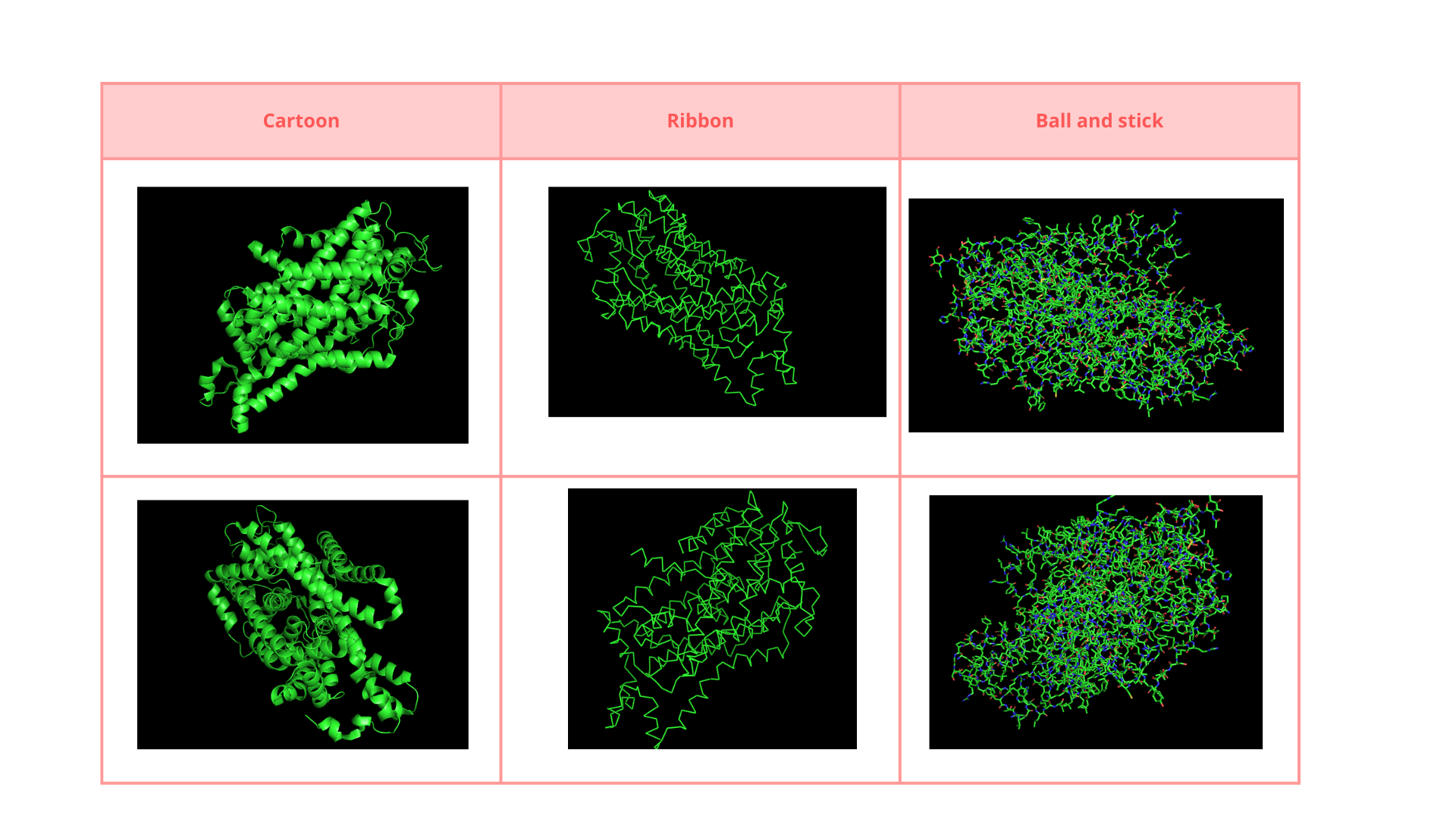
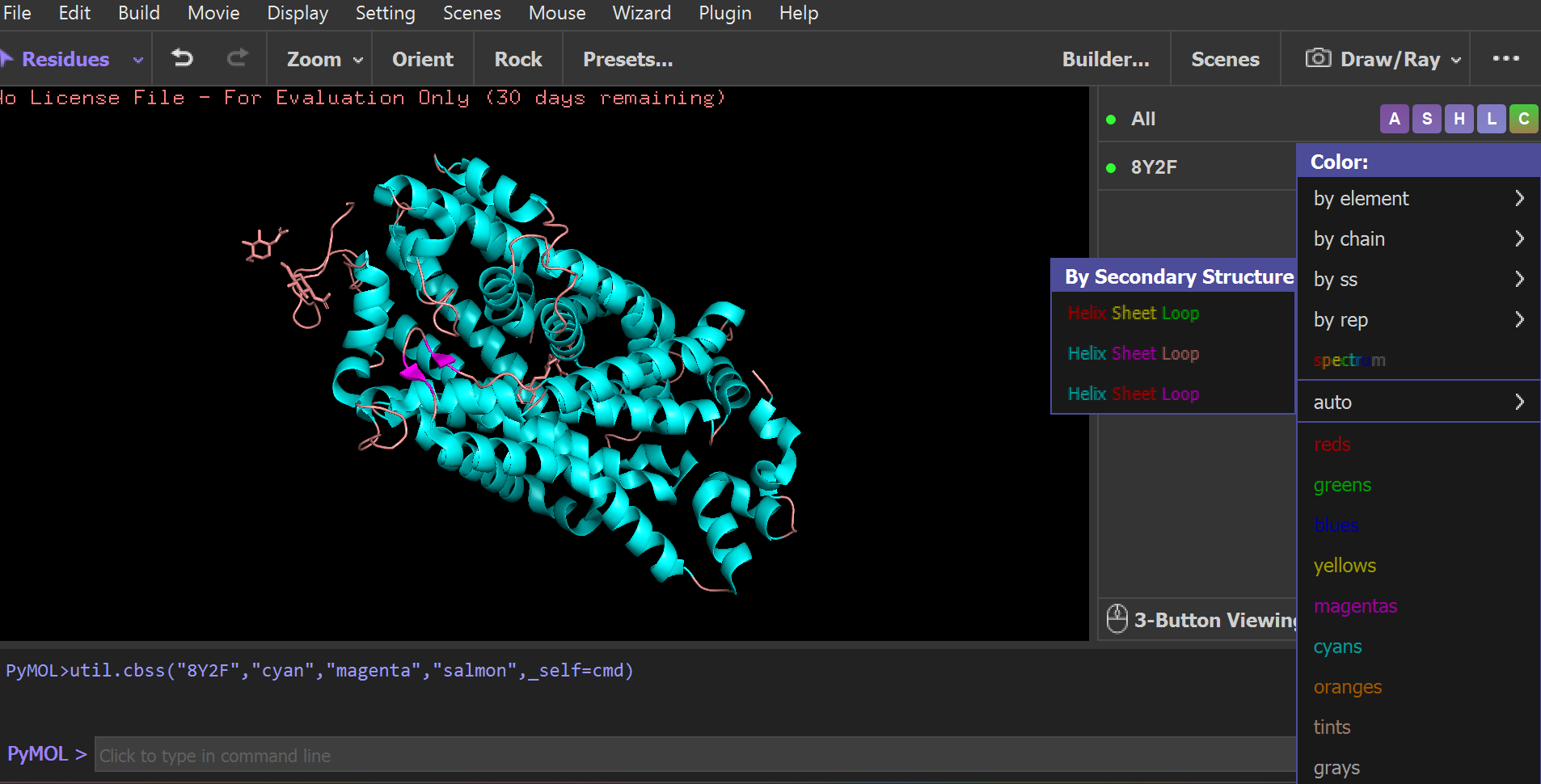
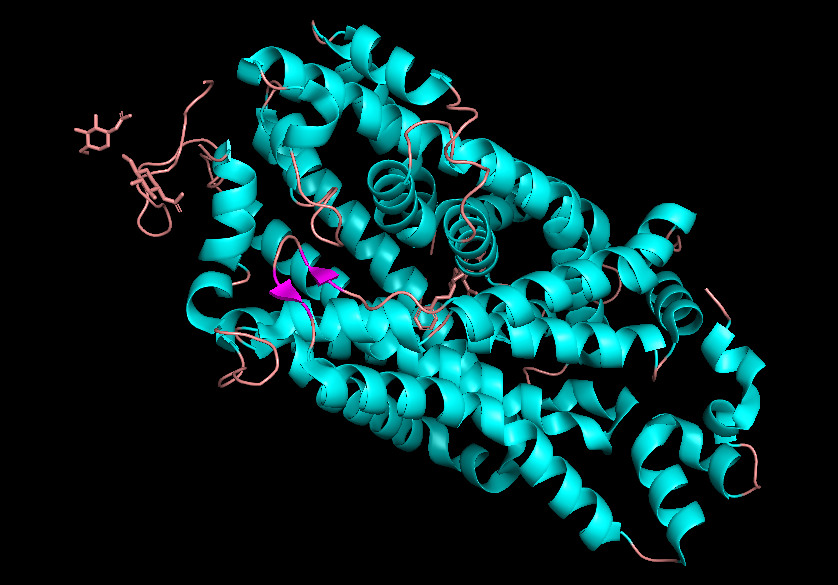
• Color the protein by secondary structure. Does it have more helices or sheets?
• Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
The surface of the protein was colored by residue type using util.cbag().
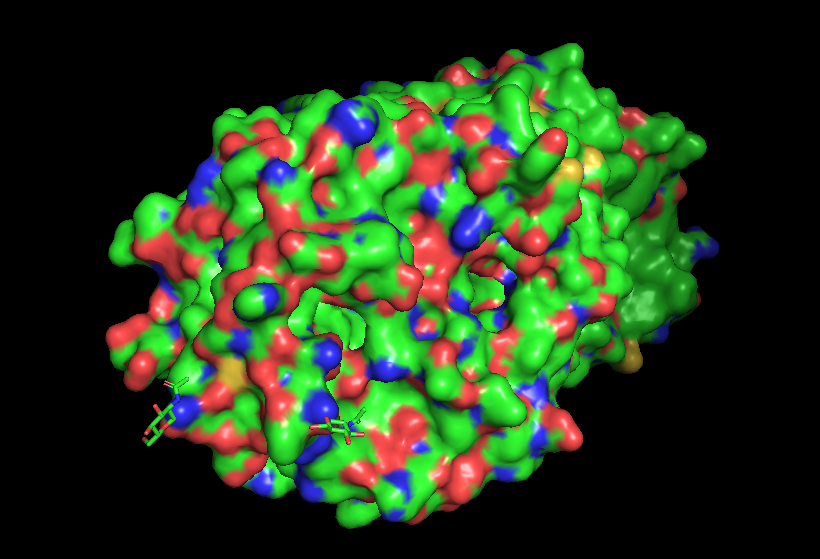
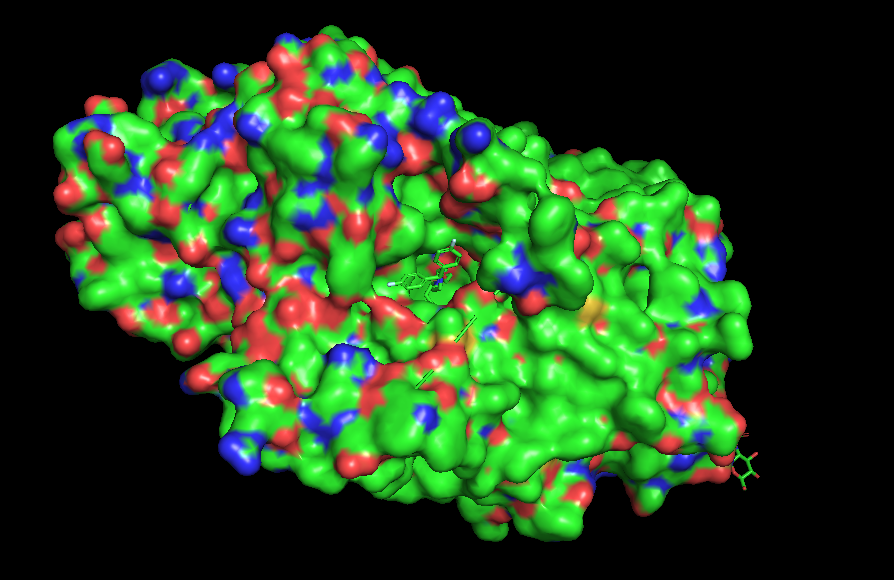
Green 🟢 → hydrophobic residues
Red 🔴 → negatively charged residues (Asp, Glu)
Blue 🔵 → positively charged residues (Lys, Arg, His)
The protein surface shows a mixture of hydrophobic (green) and charged residues (red and blue). Hydrophobic residues are abundant, while charged residues are distributed across the surface.
The combination of opposite charges can stabilize electrostatic interactions. The green patches on the surface could indicate interaction with another protein or membrane.
• Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Yes, it has a binding pocket, which is correct, as this is a transport protein.
C1. Protein Language Modeling
Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
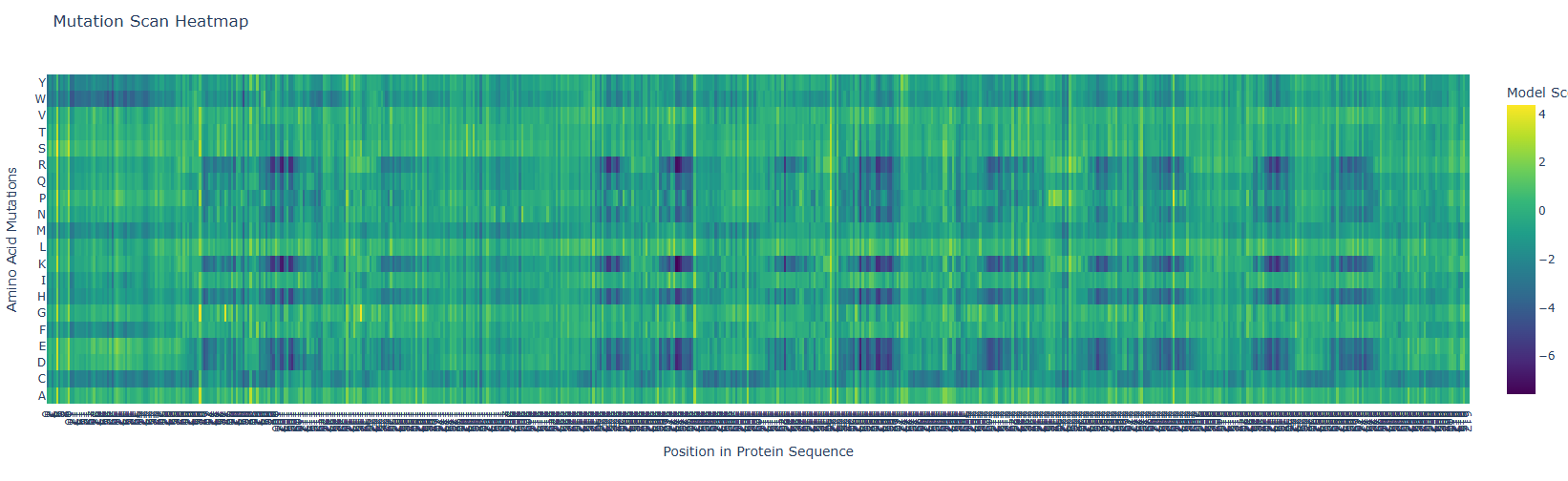
First, it is important to consider the model score:
Yellow 🟡(~4): Favorable mutation
Green 🟢 (~0): neutral mutation or tolerable mutation, which means that there is no affectation of the protein activity.
Dark blue 🔵 (~-6 a -7): Unfavorable mutation, makes the protein unstable and affects its function.
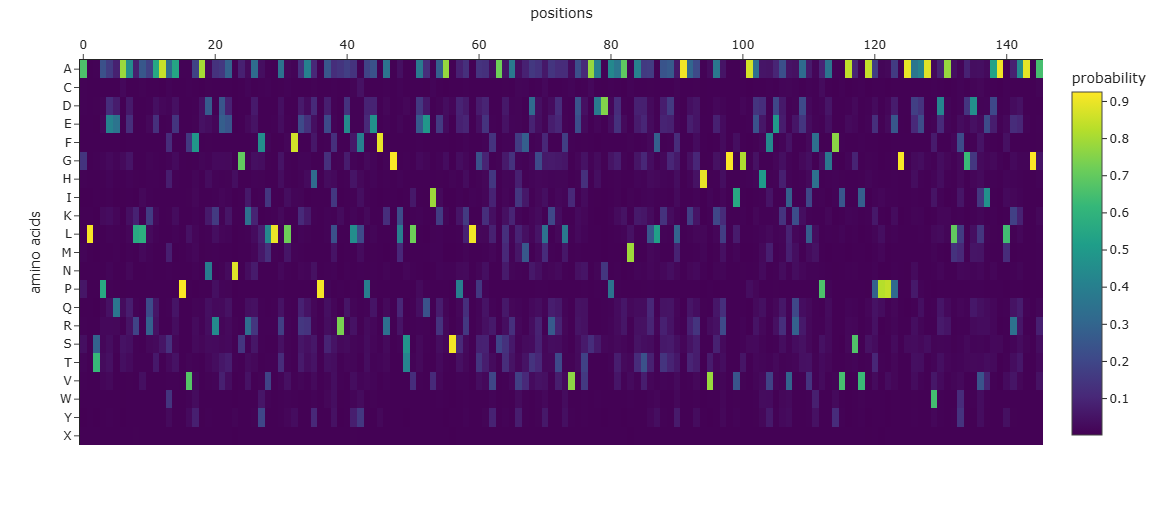
a. Can you explain any particular pattern? (Choose a residue and a mutation that stands out)
In the next picture, I highlight the patrons that I found interesting:
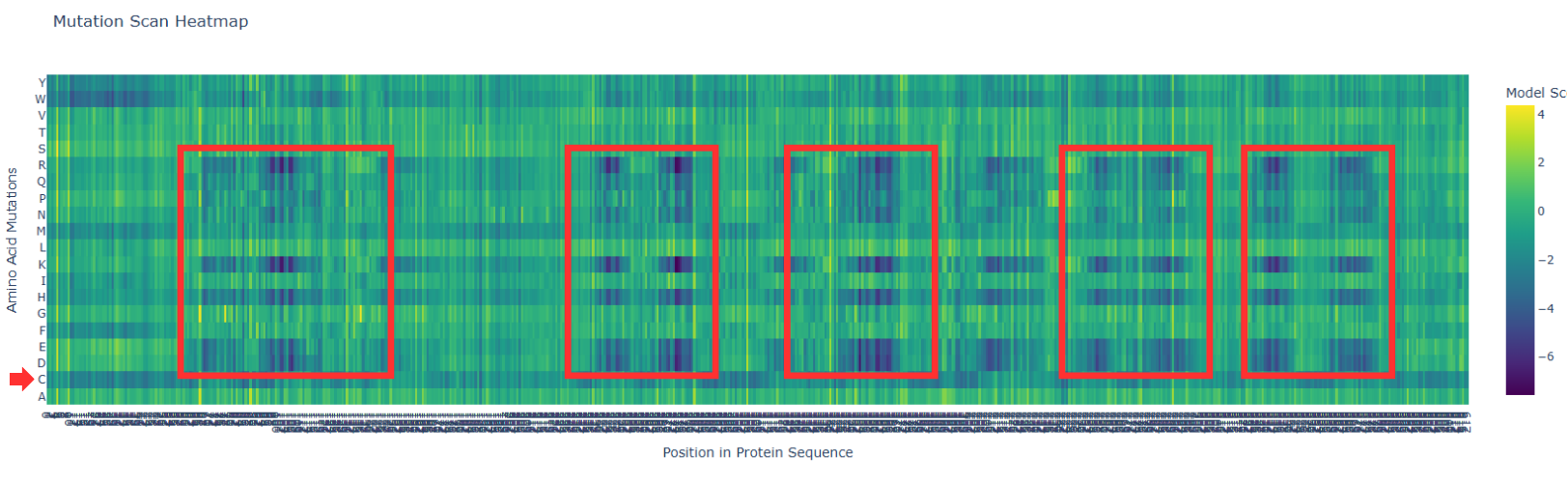
I). Some specific columns with a purple color, that appear symmetrical and in specific zones of the proteins. Especially some amino acids like R (Arginine), K (Lysine), H (Histidine), E (Glutamic acid), D (aspartic acid), in different positions in the entire chain. Regarding this information, I establish the hypothesis that these positions are fundamental for protein function, and mutations in these zones might affect protein function, or, in general, they will be unfavorable.
II). In the row of amino acid Cysteine, many of the different positions are blue, which means that the model of ESM2 considers that this amino acid is unfavorable for most of the positions in the chain. This might affect the function of the protein, since this mutation is found in most of the protein; it is reasonable to believe that Cys is not the best amino acid for this type of protein.
2. Latent Space Analysis
a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
Protein sequences from the provided dataset were embedded using Colab and executing the cells corresponding to Latent Space Analysis. The result is a figure where we can visualize and compare protein similarity in latent space.
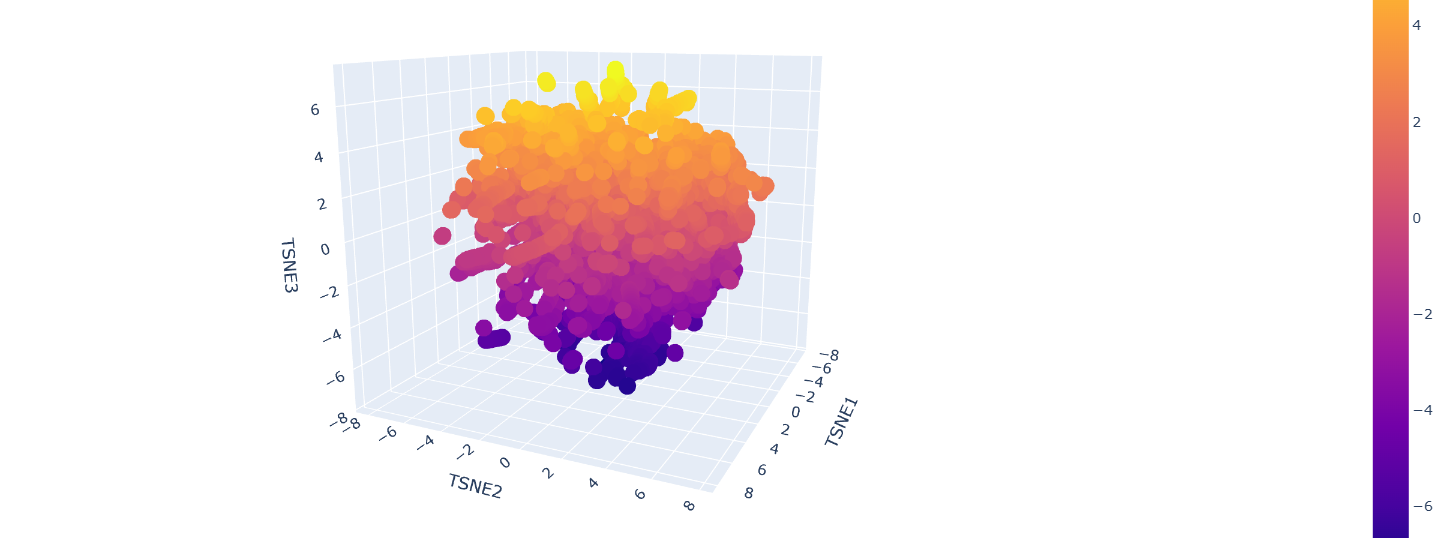
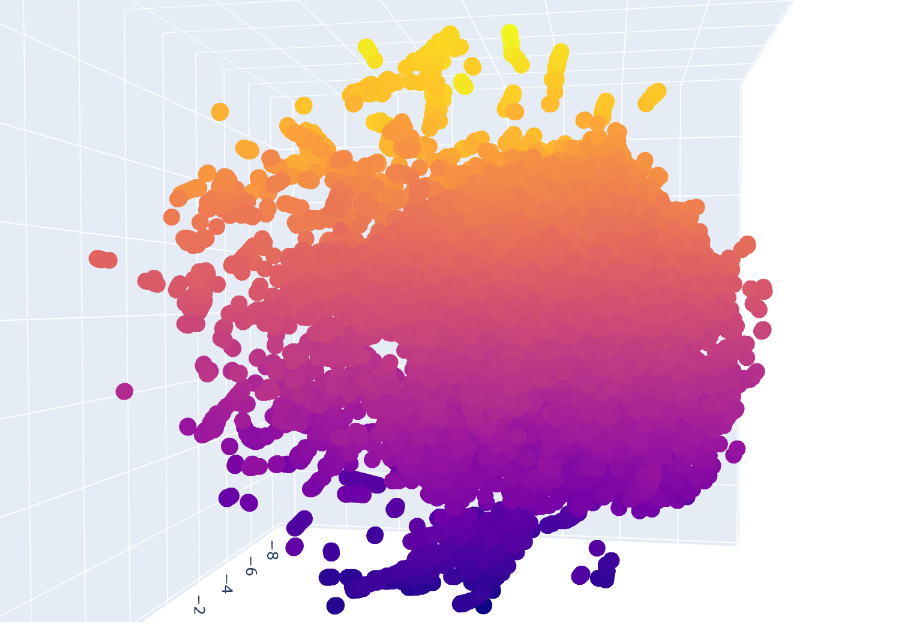
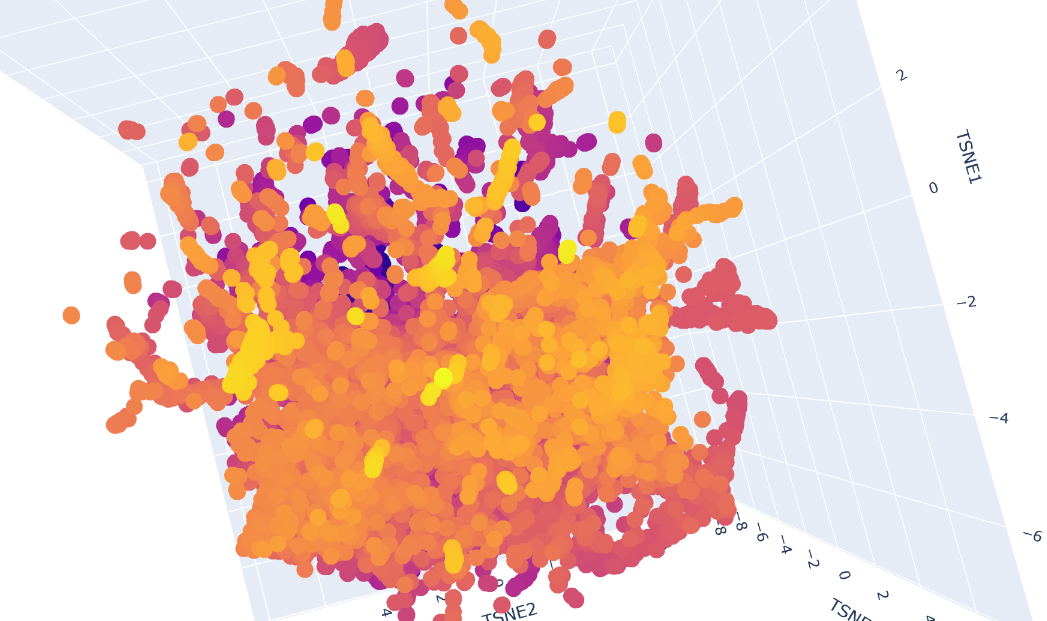
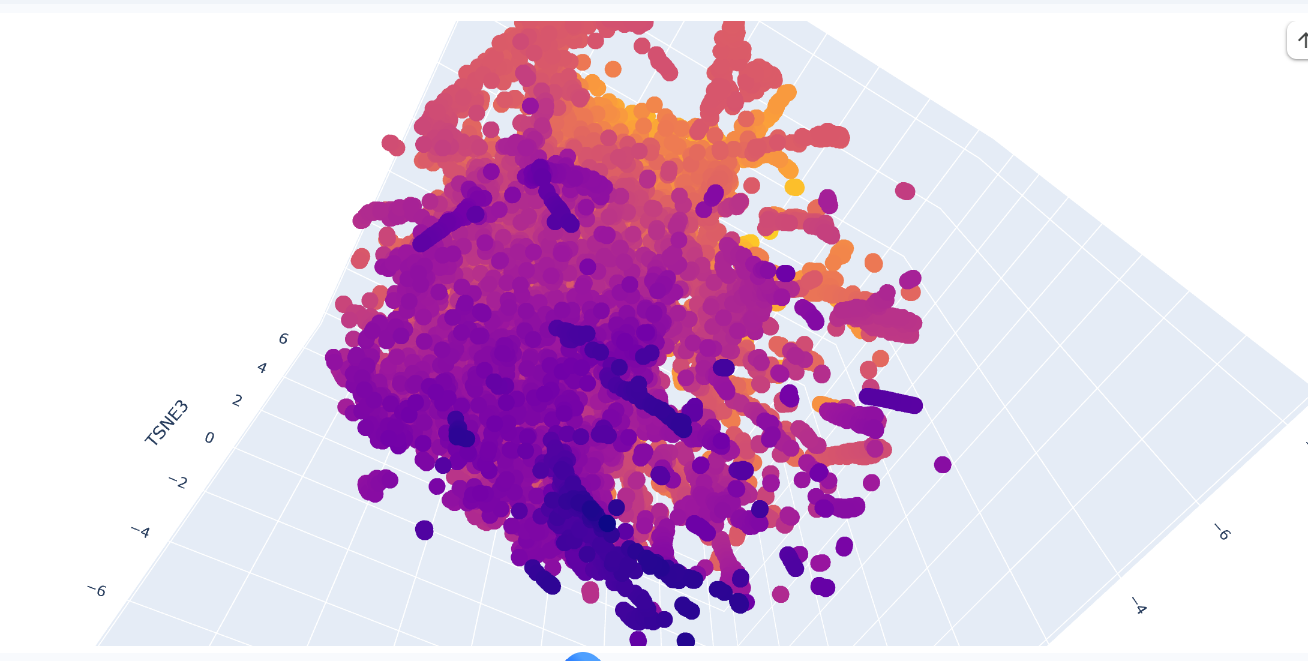
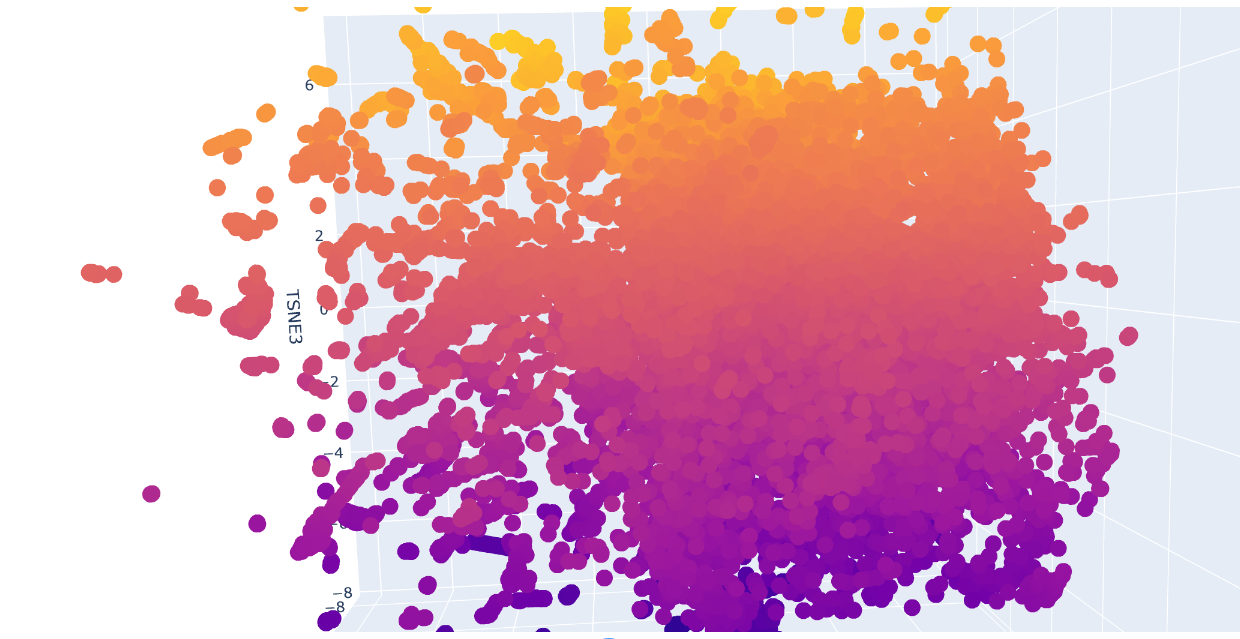
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
Inside the figure, we have three characteristics used to embed and compare the proteins. TSNE1, TSNE2, and TSNE3, the colors are provided by the last one.
Yes, there are some clusters of proteins, especially at the top, where the overall set is larger.
At the bottom of the figure, there are a few clusters, but these clusters are more separate between them. This performance suggests that at the top, there are proteins sharing features. In contrast, the smaller clusters at the bottom probably represent unique proteins or very different proteins. For example, Beta-defensin, BD, and Phrixotoxin are similar proteins because they share some parts of the structure, even though their function is different.
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.

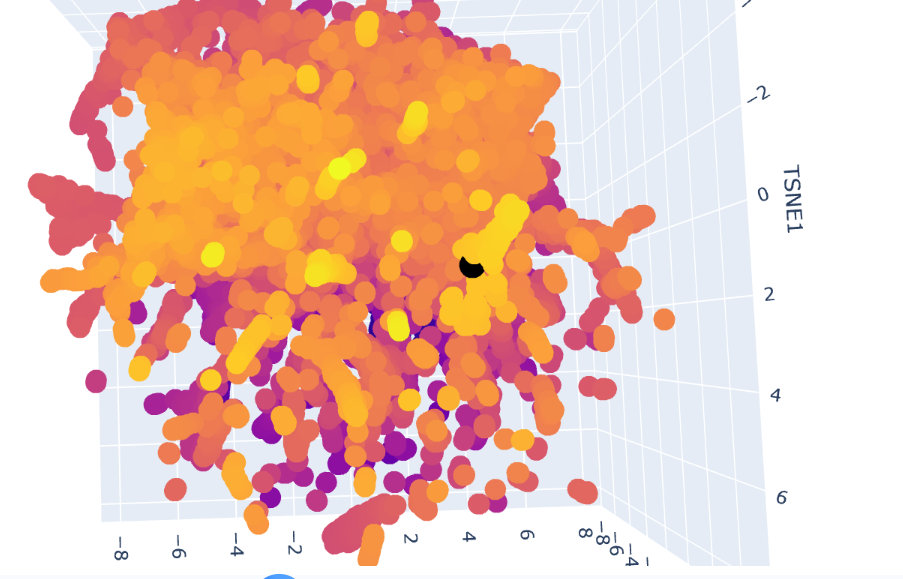
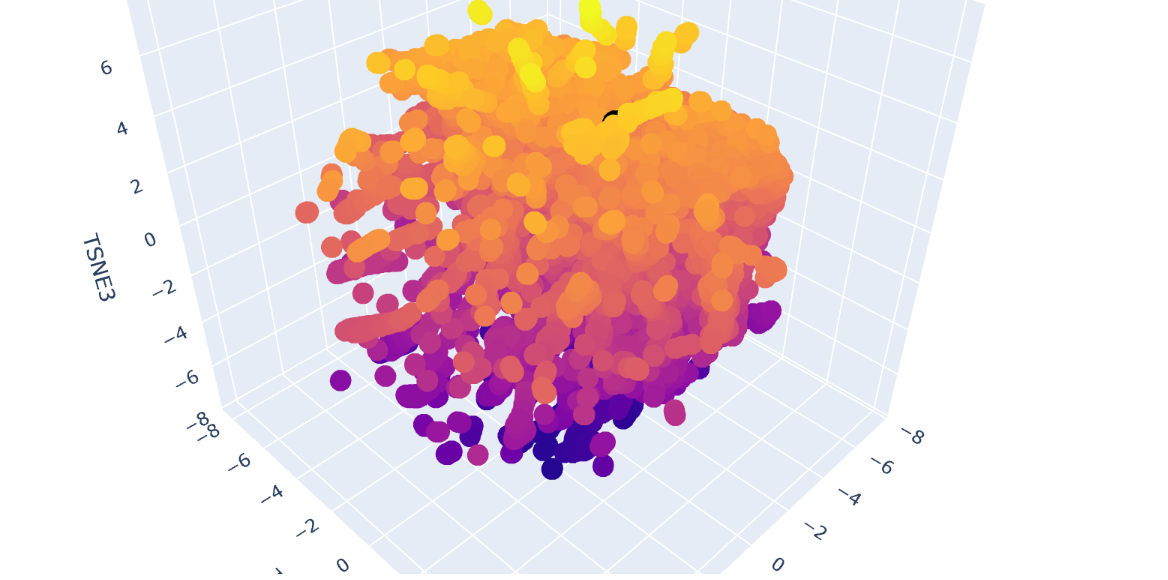

The Dopamine Transporter (DAT) is at the top of the 3D latent space representation, clearly identifiable as a black dot. We can see that it is not isolated and it is close to the central cluster. This suggests that it is not an atypical protein. This expectation is based on the fact that DAT is a membrane protein, and these proteins are common in nature.
A closer inspection of its near proteins: Ionotropic glutamate receptor 2 (GluR2), Vacuolar ATP synthase subunit a (Saccharomyces cerevisiae), MurE (UDP-N-acetylmuramyl tripeptide synthetase), and Threonine deaminase (Escherichia coli). These proteins belong to different functional classes and organisms
This variety of proteins supports the hypothesis that, in latent space analysis, the position of DAT might indicate that it shares structural characteristics with other proteins, especially hydrophobic domains, and that their positions do not necessarily indicate functional similarity.
C2. Protein Folding
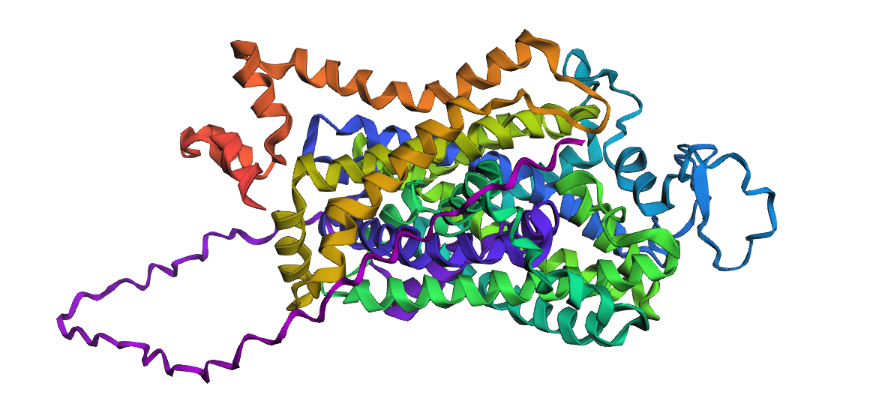
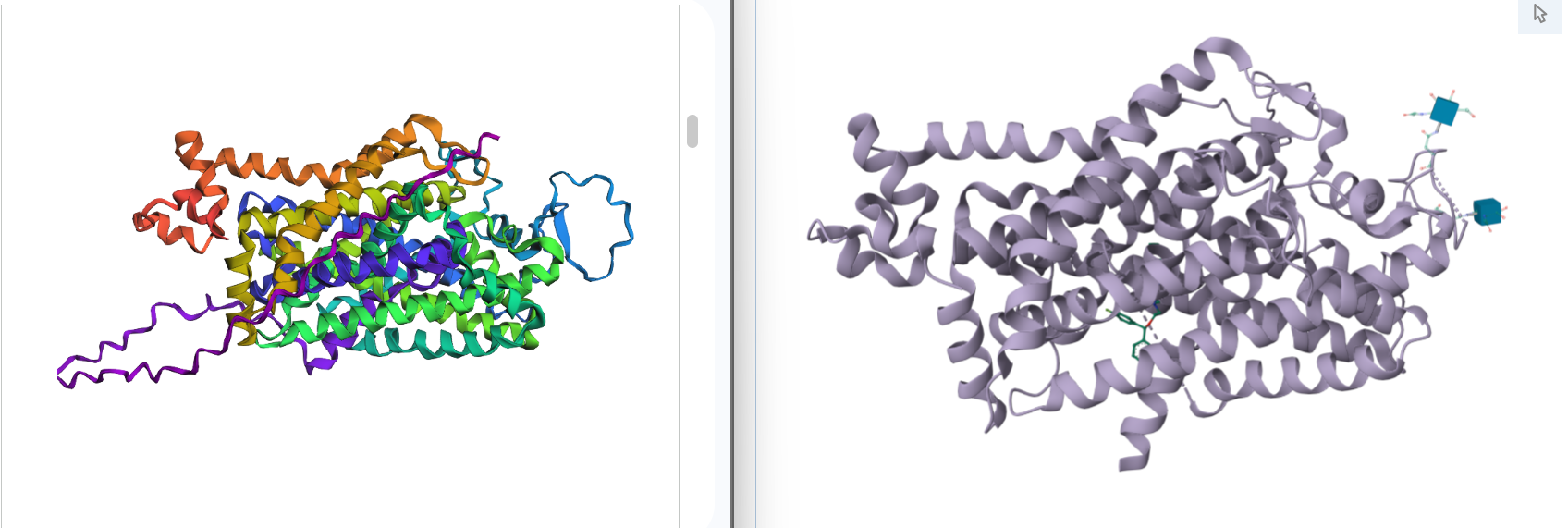
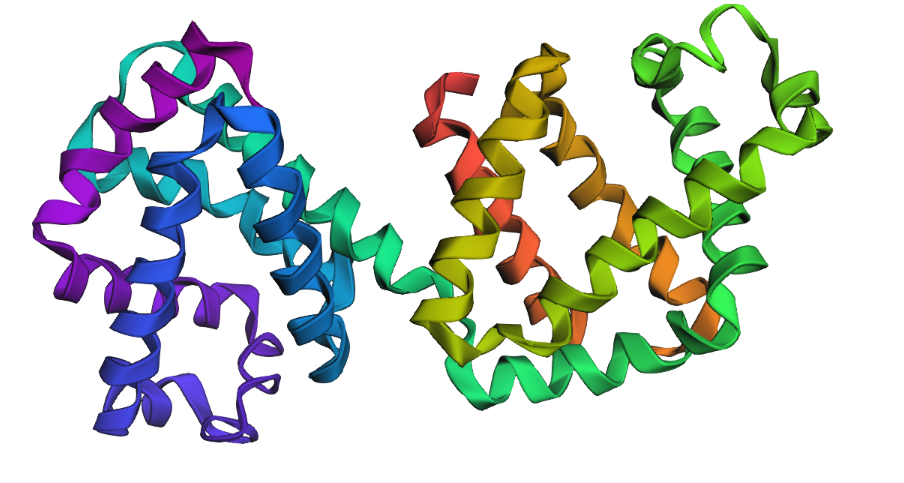
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Yes, you can see it in the image below that the helices match, and the general disposition coincides. Moreover, Protein Folding with ESMFold provides us with data that allows us to conclude that the structure obtained is accurate.
Structure obtained
Comparation
1. Total sequence length: 620 amino acids
2. Predicted Template Modeling (pTM): 0,905
Score estimating global fold accuracy, high confidence structures pTM > 0.7
3. Predicted Local Distance Difference Test (pLDDT): 91.395
Confidence score over all residues, high confidence structures pLDDT > 90
2. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
As it was determined using the mutation scan, there are some positions in the chain where modifications might result in unfavorable effects for the protein. I try some mutations:
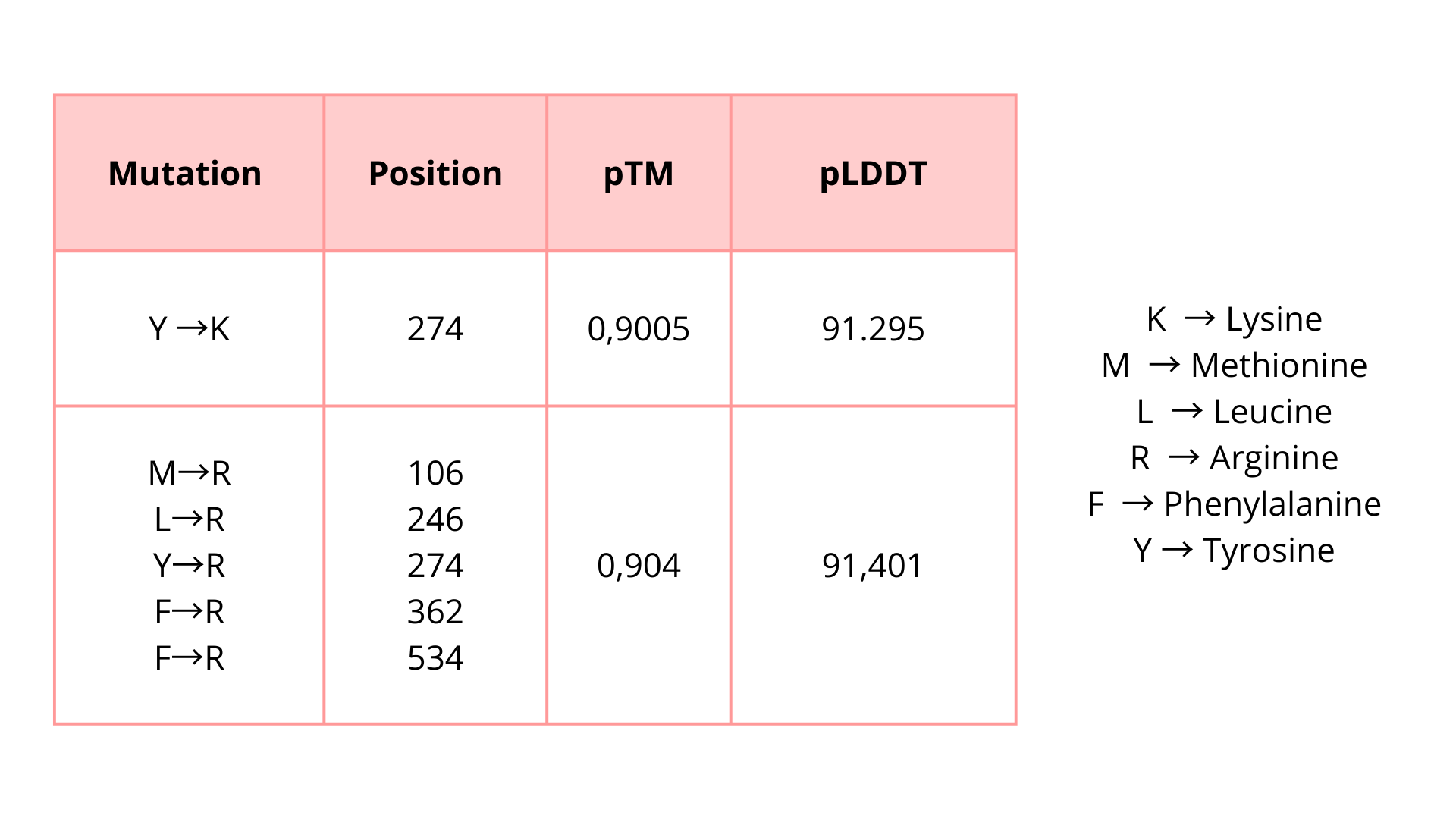
I introduced these mutations in critical zones to evaluate if these modifications will affect the protein function unfavorably. Based on the predicted pTM and pLDDT scores, the modified protein appears to maintain a high-confidence structural model. These results suggest that the protein may tolerate these substitutions without major structural disruption.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
SLSAAEADLAGKSWAPVFANKNANGLDFLVALFEKFPDSANFFADFKGKSVADIKASPKLRDVSSRIFTRLNEFVNNAANAGKMSAMLSQFAKEHVGFGVGSAQFENVRSMFPGFVASVAAPPAGADAAWTKLFGLIIDALKAAGAALTPEQAALLRAAAAPVFANREANGKAFLLALFAAHPALRELFPEFAGLSLAEIAASPKLGEVATAVFDGLRTLVATADDPAAMATLLAALAAAHVARGIGAAHFEAVRALHPAFVASVAPPPPGAAAAWDALFGDVIAALRAAGA
2. Input this sequence into ESMFold and compare the predicted structure to your original.
Part D. Group Brainstorm on Bacteriophage Engineering
1. Find a group of ~3–4 students
2. Read through the Phage Reading material listed under “Reading & Resources” below.
3. Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest) Higher titers (medium) Higher toxicity of lysis protein (hard)
4. Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
Why do you think those tools might help solve your chosen sub-problem?
5. Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
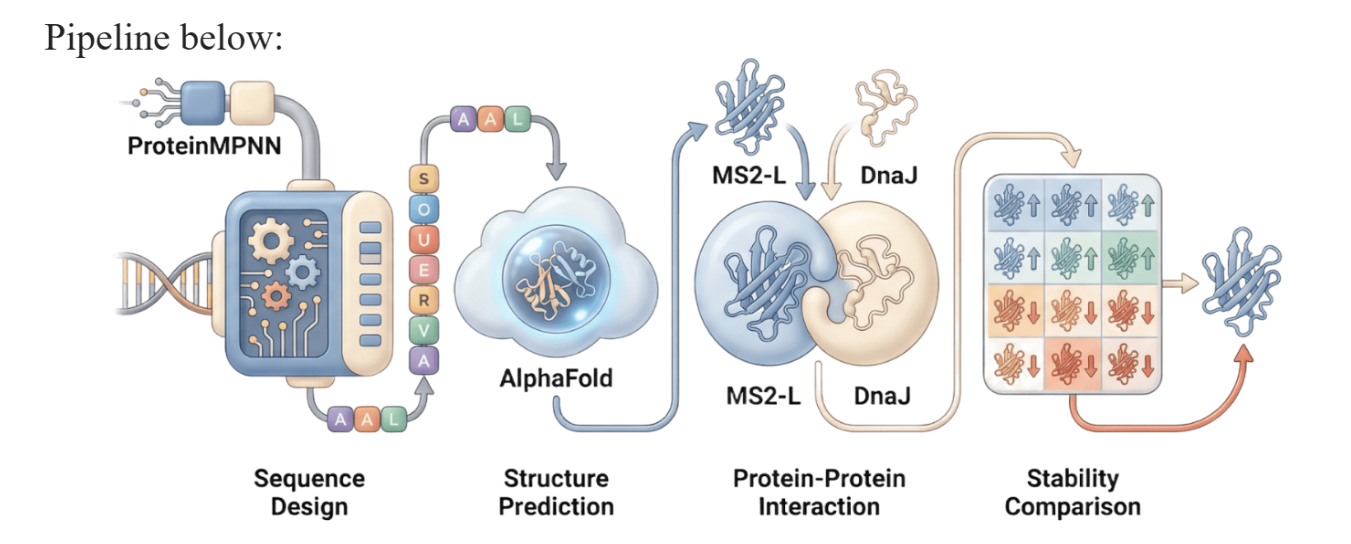
6. Include a schematic of your pipeline.
7. This resource may be useful: HTGAA Protein Engineering Tools
Each individually put your plan on your HTGAA website
Include your group’s short plan for engineering a bacteriophage
Names: Danna Betancourt, Rodrigo Arredondo, Valeria Q. Ortega, Jessica Wu
As discussed in “Phage Therapy: Past, Present and Future”, phage therapy represents an interesting alternative to antibiotic treatments, especially as recent developments allow researchers to engineer bacteriophages and their proteins. Our final group project for HTGAA Spring 2026 focuses on improving the bacteriophage MS2’s ability to kill its host bacteria E. coli by engineering its lysis protein MS2-L.
As an interdisciplinary team with different levels of experience in biotechnology, we propose increasing the stability of MS2-L. The lysis protein relies on the chaperone DnaJ for proper protein folding, a process E. coli can disrupt. However, it has been previously demonstrated that mutations deleting the N-terminal half of the MS2-L remove its dependence on DnaJ while also accelerating bacterial lysis. We believe this direction is promising for discovering variants that have structural stability within its host.
Our proposed approach begins with ProteinMPNN to look for alternative amino acid sequences that will improve the stability of MS2-L, then the sequences can be evaluated using AlphaFold and AlphaFold-Multimer to verify compatibility with their biological function and their interaction with DnaJ, with Alphafold specialized to model oligomeric complexes like MS2 and AlphaFold-Multimer tailored to predict protein-protein interactions like the one between MS2 and DnaJ.
Lastly, we must identify promising sequences for experimentation. We can do this by comparing variants quantitatively, e.g. using a deep mutational scan to see how each variant holds up when introduced to point mutations. This will narrow our candidate list to the most promising candidates for synthesis and experimental validation, reducing costs and promoting data-informed decision-making.
Any pitfalls are tied to the reliability of our tools; computational predictions of stability may not fully reflect protein behavior. For example, AlphaFold-Multimer has a systematic bias toward interactions between ordered protein regions, with a reduced accuracy for disordered regions and transient interactions such as those of a chaperone and its complex.
We are also held back by a narrow scope. Phage therapy depends on several biological variables beyond a single protein, and there is currently a lack of pharmacokinetic and pharmacodynamic studies on phage therapy. This means that we can make MS2-L more stable, but other factors could limit the effectiveness of the bacteriophage.
References
- Ajomiwe, Nneka, et al. “Protein Nutrition: Understanding Structure, Digestibility, and Bioavailability for Optimal Health.” Foods, vol. 13, no. 11, 1 Jan. 2024, p. 1771, www.mdpi.com/2304-8158/13/11/1771, https://doi.org/10.3390/foods13111771.
- Alila Medical Media. “Mechanism of Drug Addiction in the Brain, Animation.” YouTube, 11 Sept. 2014, www.youtube.com/watch?v=NxHNxmJv2bQ.
- “Amino Acids, Evolution| Learn Science at Scitable.” Nature.com, 2026, www.nature.com/scitable/topicpage/an-evolutionary-perspective-on-amino-acids-14568445/?error=server_error. Accessed 4 Mar. 2026.
- “Antiparallel and Parallel Beta Sheets.” Pearson.com, 2022, www.pearson.com/channels/biochemistry/learn/jason/protein-structure/antiparallel-and-parallel-beta-sheets.
- “Beta Sheet - an Overview | ScienceDirect Topics.” Www.sciencedirect.com, www.sciencedirect.com/topics/neuroscience/beta-sheet.
- Bu, Mengfei, et al. “Dynamic Control of the Dopamine Transporter in Neurotransmission and Homeostasis.” Npj Parkinson’s Disease, vol. 7, no. 1, 5 Mar. 2021, pp. 1–11, www.nature.com/articles/s41531-021-00161-2, https://doi.org/10.1038/s41531-021-00161-2.
- Cheng, Zhiming, et al. “Fluorescent Amino Acids as Versatile Building Blocks for Chemical Biology.” Nature Reviews Chemistry, vol. 4, no. 6, 13 May 2020, pp. 275–290, https://doi.org/10.1038/s41570-020-0186-z.
- Clemente-Suárez, Vicente Javier, et al. “Human Digestive Physiology and Evolutionary Diet: A Metabolomic Perspective on Carnivorous and Scavenger Adaptations.” Metabolites, vol. 15, no. 7, 4 July 2025, pp. 453–453, mdpi.com/2218-1989/15/7/453, https://doi.org/10.3390/metabo15070453.
- Data, Protein. “RCSB PDB - 8Y2F: Cryo-EM Structure of Human Dopamine Transporter in Complex with GBR12909.” Rcsb.org, 2024, www.rcsb.org/structure/8Y2F. Accessed 4 Mar. 2026.
- Emberly, Eldon G, et al. “Designability of α-Helical Proteins.” Proceedings of the National Academy of Sciences, vol. 99, no. 17, 12 Aug. 2002, pp. 11163–11168, https://doi.org/10.1073/pnas.162105999.
- “ESM Metagenomic Atlas | Meta AI.” Esmatlas.com, 2025, esmatlas.com/about.
- “ESMFold.” BioLM, 2023, biolm.ai/models/esmfold/. Accessed 4 Mar. 2026.
- Niesel, David. “Biomolecules Are Left or Right Handed.” Medical Discovery News (Mdnews), 8 Apr. 2025, www.utmb.edu/mdnews/podcast/episode/biomolecules-are-left-or-right-handed.
- Nowick, James S. “Exploring β-Sheet Structure and Interactions with Chemical Model Systems.” Accounts of Chemical Research, vol. 41, no. 10, 1 Oct. 2008, pp. 1319–1330, www.ncbi.nlm.nih.gov/pmc/articles/PMC2728010/, https://doi.org/10.1021/ar800064f.
- Parnas, M. Laura, and Roxanne Vaughan. “DAT, Dopamine Transporter.” XPharm: The Comprehensive Pharmacology Reference, 2007, pp. 1–10, www.sciencedirect.com/topics/medicine-and-dentistry/dopamine-transporter, https://doi.org/10.1016/b978-008055232-3.60441-6.
- Robinson, Scott W., et al. “Bioinformatics: Concepts, Methods, and Data.” Handbook of Pharmacogenomics and Stratified Medicine, 2014, pp. 259–287, https://doi.org/10.1016/b978-0-12-386882-4.00013-x.
- Uniprot.“UniProt.” UniProt, 2026, www.uniprot.org/blast/uniprotkb/ncbiblast-R20260301-002658-0868-42734055-p1m/overview. Accessed 4 Mar. 2026.
- Yip, Ka Man, et al. “Atomic-Resolution Protein Structure Determination by Cryo-EM.” Nature, vol. 587, 21 Oct. 2020, pp. 1–5, www.nature.com/articles/s41586-020-2833-4, https://doi.org/10.1038/s41586-020-2833-4.
- Zeppelin, Talia, et al. “Effect of Palmitoylation on the Dimer Formation of the Human Dopamine Transporter.” Scientific Reports, vol. 11, no. 1, 18 Feb. 2021, https://doi.org/10.1038/s41598-021-83374-y. Accessed 4 Mar. 2023.
- Zhu, J., and M. Reith. “Role of the Dopamine Transporter in the Action of Psychostimulants, Nicotine, and Other Drugs of Abuse.” CNS & Neurological Disorders - Drug Targets, vol. 7, no. 5, 1 Nov. 2008, pp. 393–409, https://doi.org/10.2174/187152708786927877.