Week 10 HW: Measurement Technology

Homework: Final Project
For your final project:
1. Please identify at least on6e (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
The main measurable aspect of this project is the presence or absence of the A118G polymorphism in the OPRM1 gene.
2. Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
The detection system is based on a CRISPR-Cas13 biosensor coupled to a Broccoli RNA aptamer. The elements measured include the presence of the target mutation and the resulting fluorescence signal.
When the mutant sequence (G118) is present, the crRNA guides Cas13 to specifically recognize the target RNA. This activates Cas13’s collateral cleavage activity, which degrades a blocking RNA sequence. Once the blocker is degraded, the Broccoli aptamer can fold into its active structure and bind to its fluorophore (e.g., DFHBI), producing a fluorescent signal (ON state).
In contrast, if the wild-type sequence (A118) is present, Cas13 is not activated, the blocker remains intact, and the aptamer does not fluoresce (OFF state).
Fluorescence will be measured using a fluorimeter or plate reader, allowing detection of signal presence or absence, and potentially signal intensity. Additionally, gel electrophoresis may be used to confirm RNA integrity and Cas13-mediated cleavage.
3. What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
- CRISPR-Cas13 system: Used for sequence-specific recognition of RNA and discrimination between the A118 and G118 variants. Upon recognition of the mutant sequence, Cas13 becomes activated and induces collateral RNA cleavage.
- Cell-free expression system: The biosensor will be implemented in a cell-free transcription-translation system, which allows controlled expression and interaction of the RNA components without the use of living cells. This system provides a rapid and tunable platform for biosensing.
- Fluorescent RNA aptamer (Broccoli): Functions as a reporter that emits fluorescence upon proper folding and binding to a fluorophore such as DFHBI, indicating activation of the system.
- Fluorescence detection (fluorimeter or plate reader): Used to measure the biosensor output, enabling qualitative (ON/OFF).
Homework: Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
1. Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence:
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
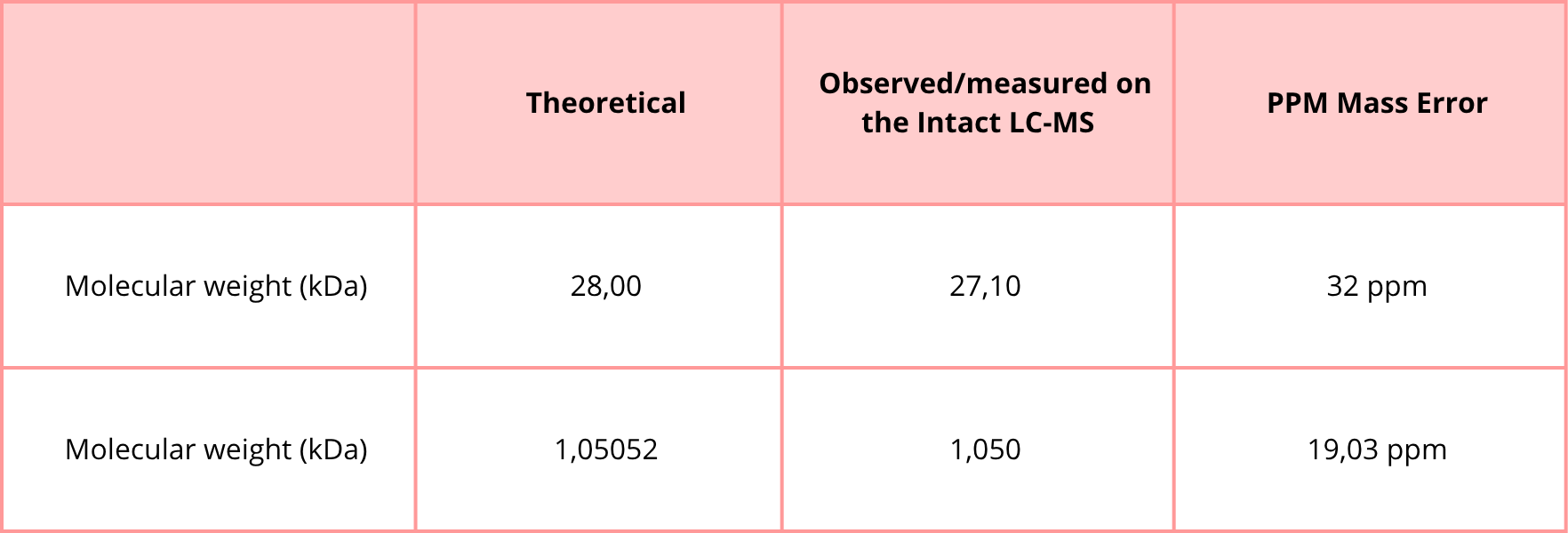
The calculated molecular weight of eGFP based on its amino acid sequence is approximately 28,006.60 Da (~28.0 kDa), as determined using the ExPASy Compute pI/Mw tool. This value includes the C-terminal His-tag present in the sequence.
2. Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Figure 1 Mass Spectrum of intact eGFP protein from the Waters Xevo G3 LC-MS (a mass spectrometer with 30,000 resolution) with individual charge state peaks labeled with m/z values.
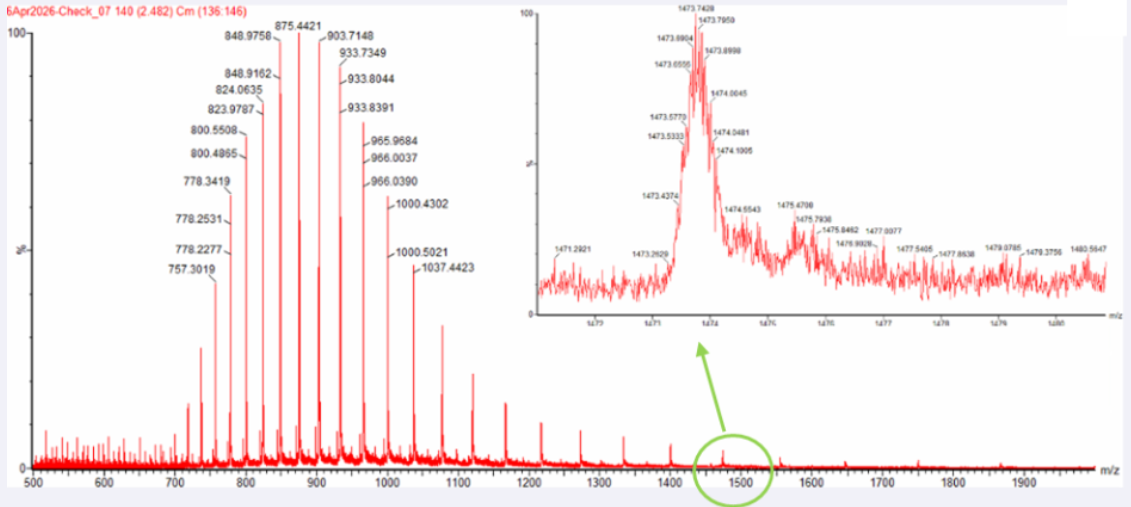
2.1 Determine z for each adjacent pair of peaks (n, n + 1) using:
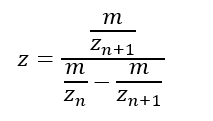
Peak 1: 875,4
Peak 2: 903,7
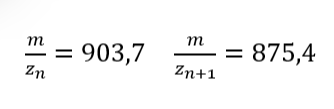
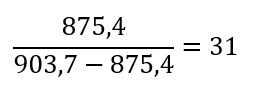
2.2 Determine the MW of the protein using the relationship between m/zn, MW, and Z

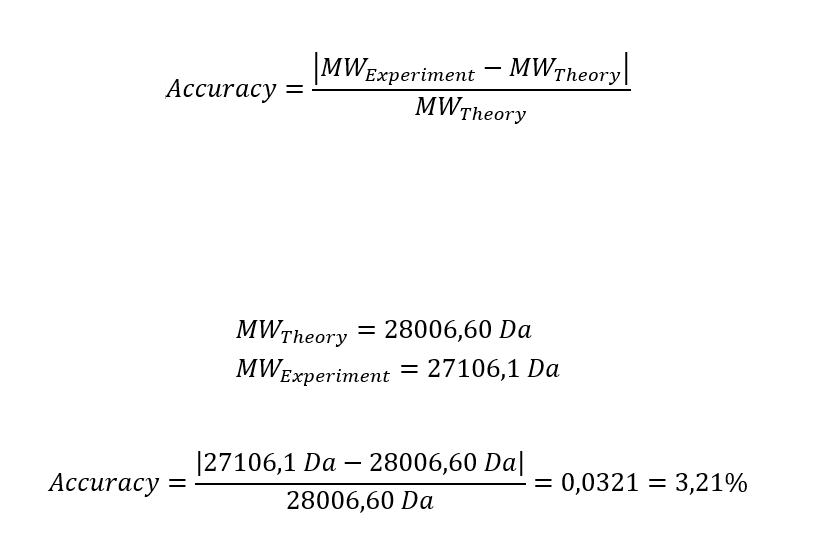
2.3 Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
Homework: Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
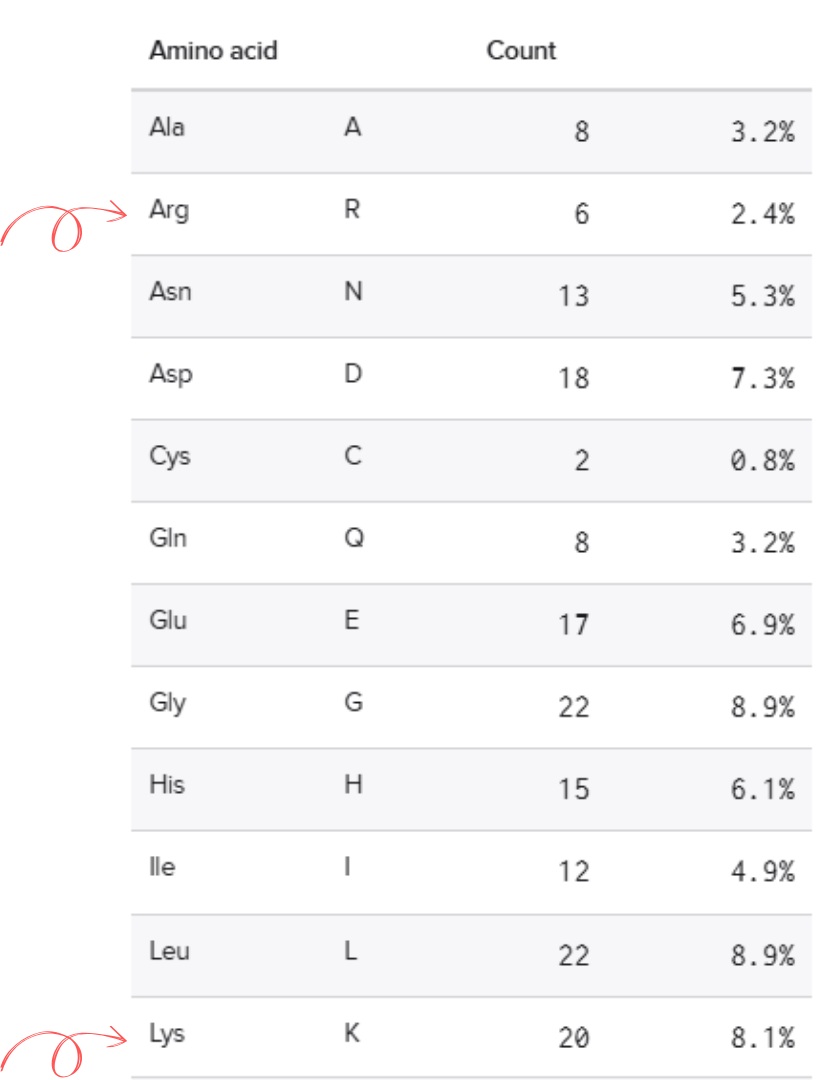
1. How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).


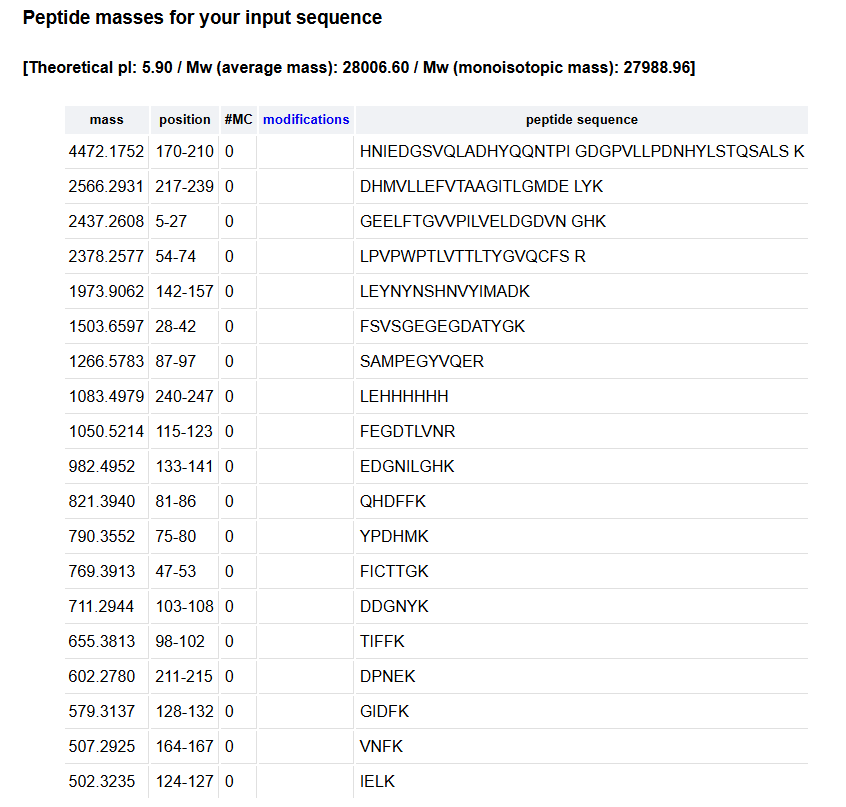
2. How many peptides will be generated from tryptic digestion of eGFP?
2.1 Navigate to https://web.expasy.org/peptide_mass/
2.2 Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
2.3 Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
Using Expasy, We obtained 19 peptides.
3. Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
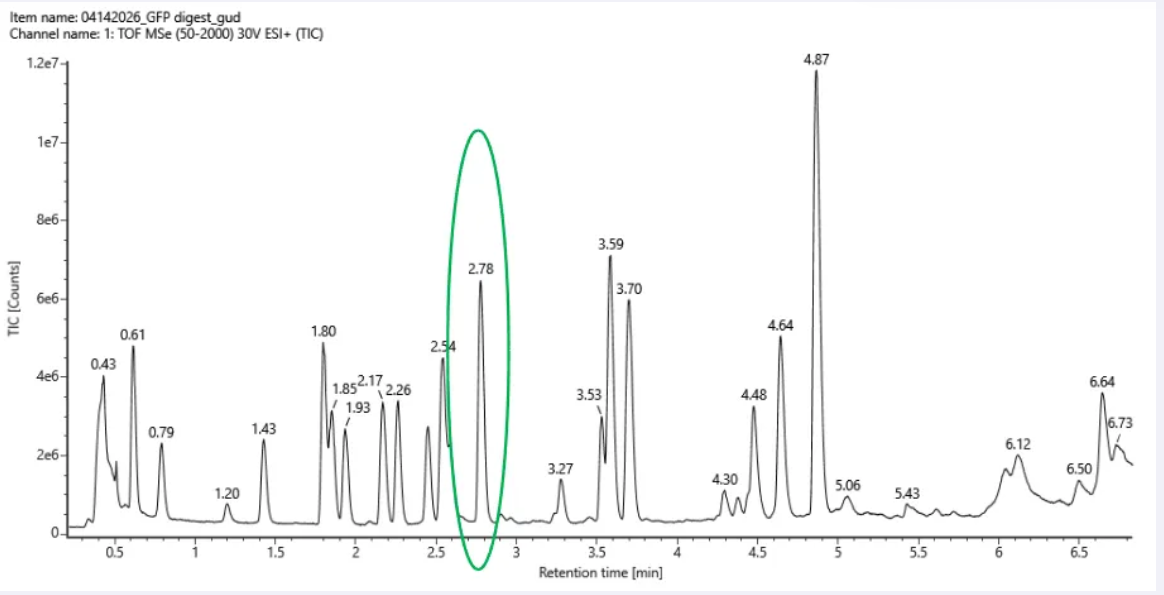 Figure 5a. Total ion chromatogram (TIC) of the eGFP peptide map. The peak at 2.78 minutes is circled, and its MS data is shown in the mass spectrum in Figure 5b, below.
Figure 5a. Total ion chromatogram (TIC) of the eGFP peptide map. The peak at 2.78 minutes is circled, and its MS data is shown in the mass spectrum in Figure 5b, below.
Approximately 17 chromatographic peaks above 10% relative abundance between 0.5 and 6 minutes.
4. Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
Assuming all the peaks are peptides, the number of peaks does not match the number of peptides predicted from question 2. This is because, in question 2, we obtain 19 peptides, but only 17 significant peaks are observed in the chromatogram. This discrepancy may be due to several factors, including co-elution of peptides, low-abundance peptides falling below the detection limit, incomplete digestion, or ionization efficiency differences between peptides, in addition to experimental noise.
5. Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ( [M+H]+) based on its m/z and z.
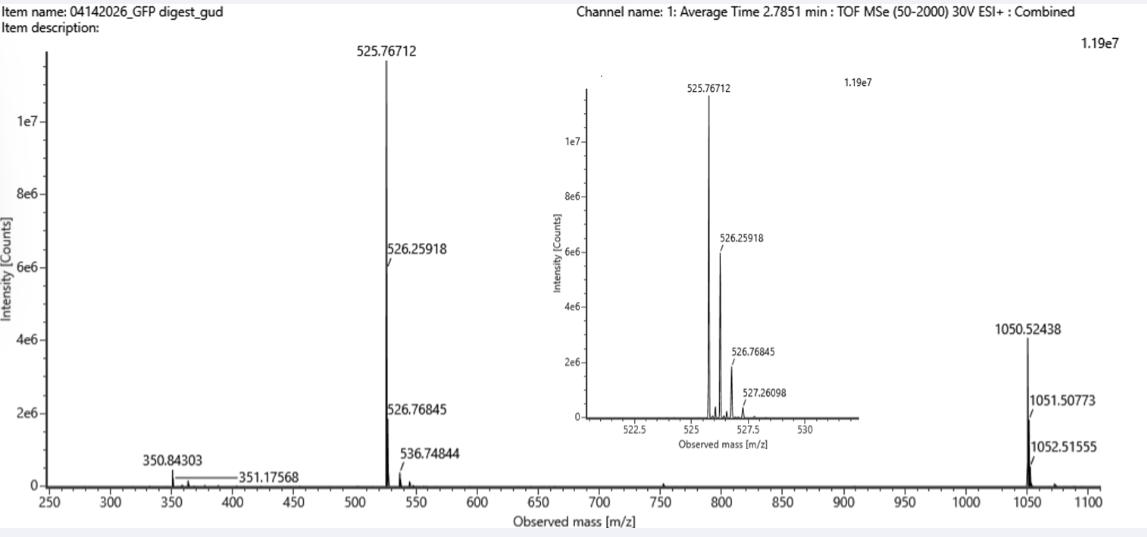 Figure 5b. Mass spectrum figure to show m/z for the chromatographic peak at 2.78 min from Figure 5a above. The inset is a zoom-in of the peak at m/z 525.76, to discern the isotope peaks.
Figure 5b. Mass spectrum figure to show m/z for the chromatographic peak at 2.78 min from Figure 5a above. The inset is a zoom-in of the peak at m/z 525.76, to discern the isotope peaks.
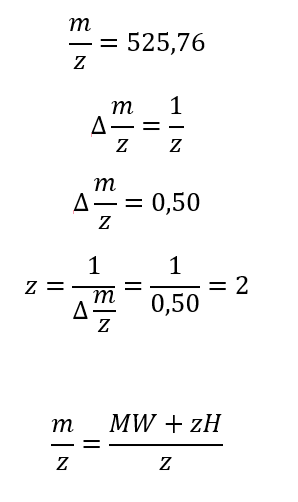

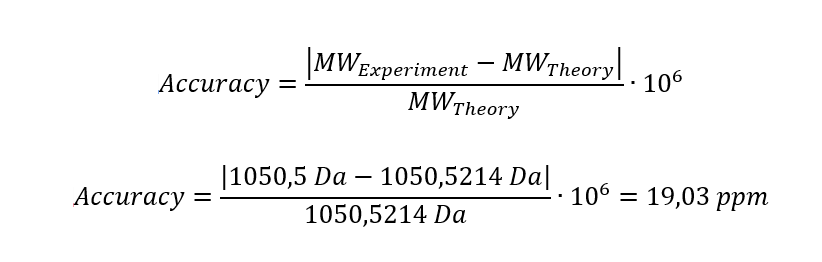
6. Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
In the table of point 2, I obtained the peptide FEGDTLVNR, with a mass of 1050.5214 Da. This peptide may correspond to the one shown in Figure 5b, whose experimental mass is 1050.5 Da.
7. What is the percentage of the sequence that is confirmed by peptide mapping?
The percentage of the sequence confirmed by peptide mapping is 88%.
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution.
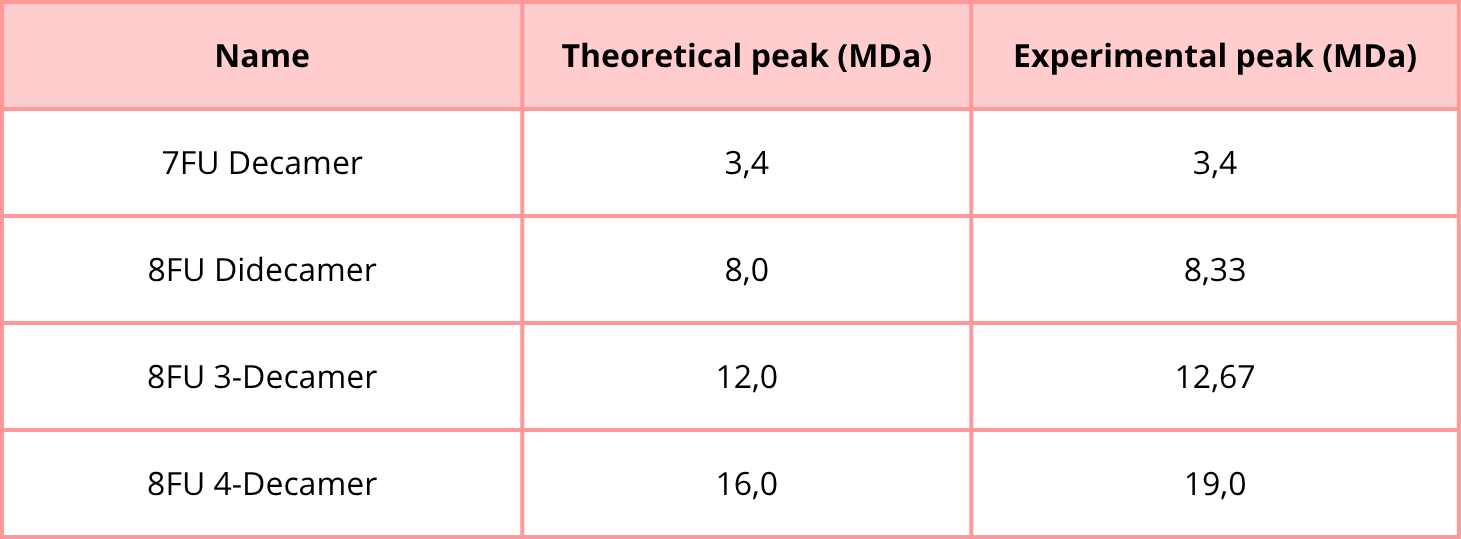
1. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
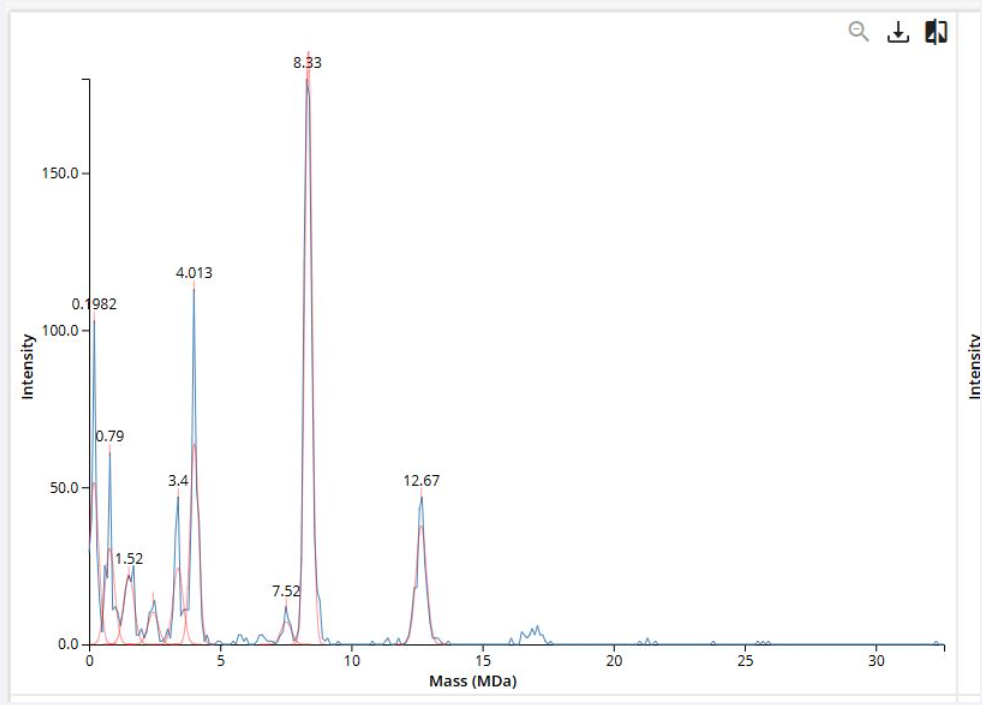 Figure 7. Mass spectrum of Keyhole Limpet Hemocyanin (KLH) acquired on the CDMS.
Figure 7. Mass spectrum of Keyhole Limpet Hemocyanin (KLH) acquired on the CDMS.
Polypeptide Subunit Name ⭢ 7FU ⭢ Subunit Mass ⭢ 340 kDa
Polypeptide Subunit Name ⭢8FU ⭢ Subunit Mass ⭢ 400 kDa
- 7FU Decamer
- 8FU Didecamer
- 8FU 3-Decamer
- 8FU 4-Decamer
Homework: Waters Part V — Did I make GFP?
1. Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.