Subsections of Homework
Week 01 HW: Principles and Practices

This week we evaluate the principles and governance policies for our new ideas. It is important to avoid the misuse of these new developments.
1. First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
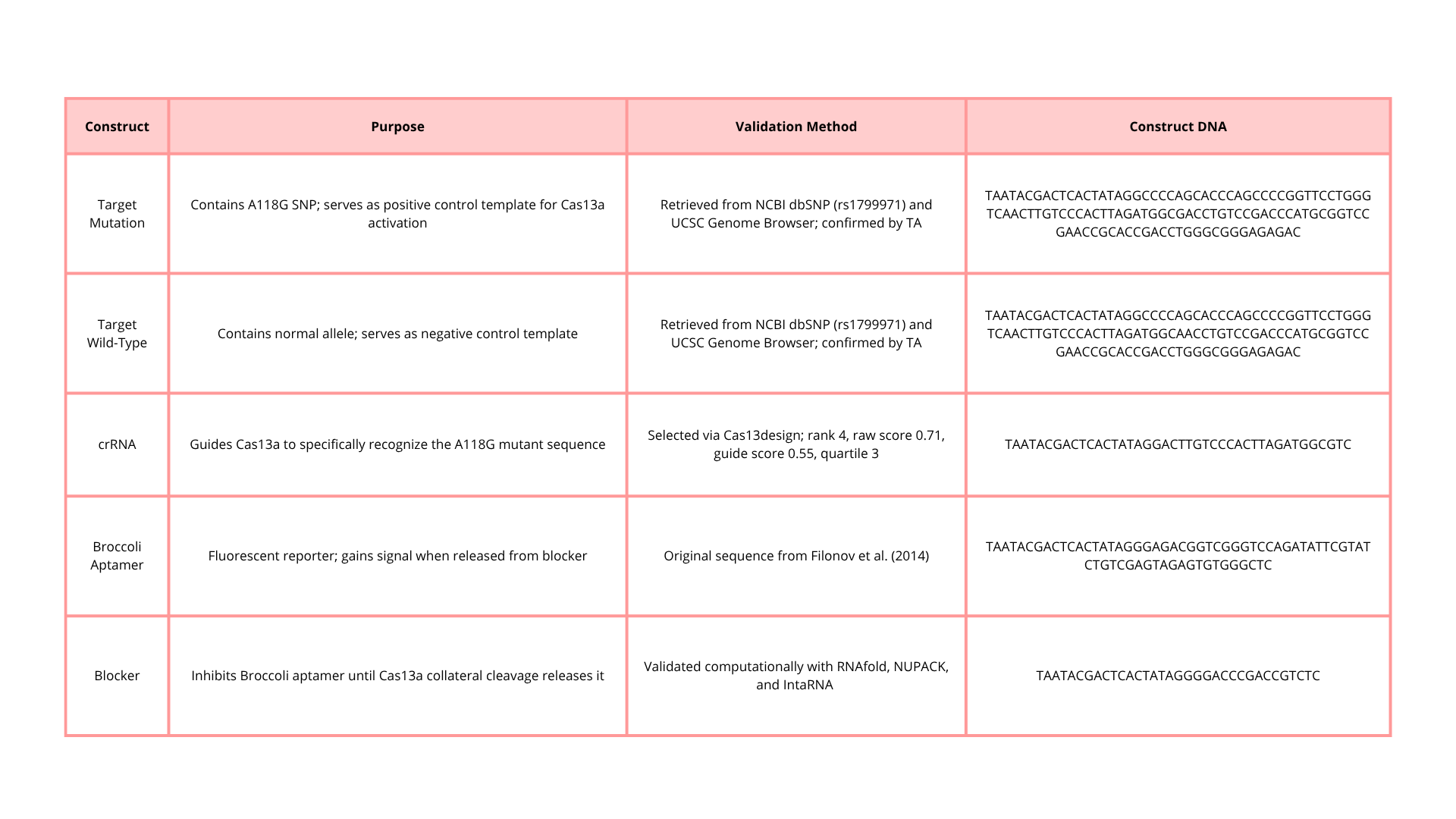
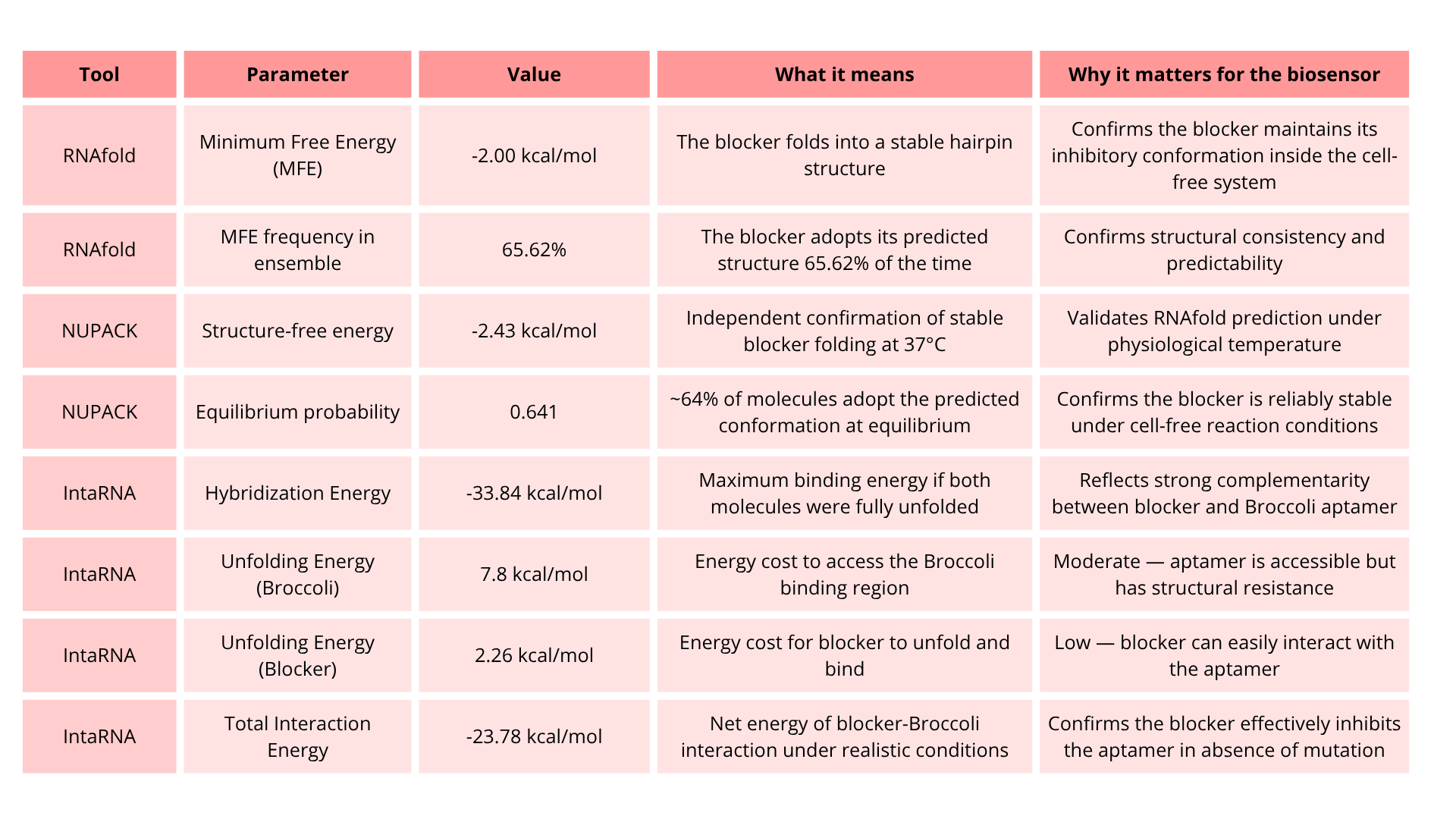
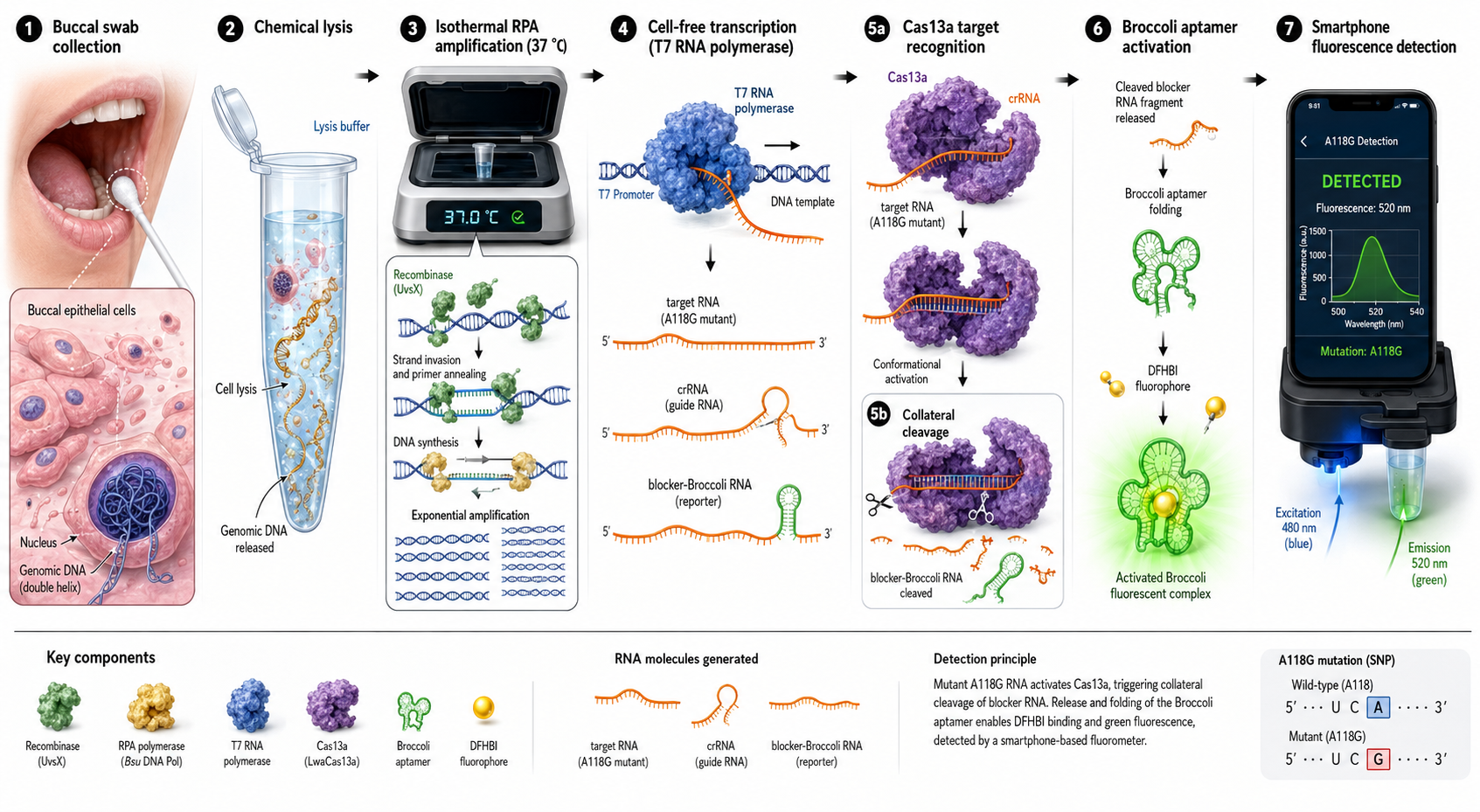
Portable Biosensor for Rapid Identification of Animal Venom Toxicity Profiles in Emergency Settings
This idea came about because I am from Colombia, where in some rural areas access to health care is a challenge, especially in emergencies. In low- and middle-income countries, it is common for people to encounter dangerous and venomous animals such as snakes, scorpions, spiders, jellyfish, and venomous fish. In many cases, treating a person who has been bitten or stung is difficult because doctors and emergency teams need to know the species and characteristics of the animal to provide appropriate care.
Identifying the animal that bit or stung a person is challenging because sometimes patients are unconscious, or, in general, people do not have the knowledge to identify the animal or describe its characteristics.
In these kinds of situations, the time window is crucial because if the time between the bite and receiving proper medical attention is too long, the chances of successfully treating the person decrease.
In this context, it is essential to develop a tool that allows identification of the type of toxin affecting a person, for example, neurotoxic, hemotoxic, cytotoxic, or myotoxic.
The use of a portable biosensor for rapid identification of animal venom toxicity profiles will allow identification of the type of venom or toxin affecting a person among the following: neurotoxic, hemotoxic, cytotoxic, or myotoxic. This will improve positive outcomes because it will be easier to treat the symptoms and administer the appropriate antivenom.
The general idea is that the portable biosensor identifies the type of venom using lateral flow assays (LFA) with antibodies directed against families of toxins. For example, snake venom is composed of a variety of proteins and enzymes; however, there are four main groups of proteins classified according to their activity.
Three-finger toxin (3FTx) family:
- Neurotoxic effects (causing paralysis)
- Cytotoxic effects
Phospholipases (PLA2s):
- Neurotoxic effects
- Cytotoxic effects (through direct/indirect plasma membrane disruption)
Metalloproteases (SVMPs):
- Hemotoxic effects (cause severe hemorrhage, inflammation, and coagulopathy)
Serine proteases (SVSPs):
These types of proteins are present in most snake species, which provides an advantage by reducing the impact of geographical variation, because the antibodies used by this sensor will be specific to the protein family rather than to a single type of snake venom.
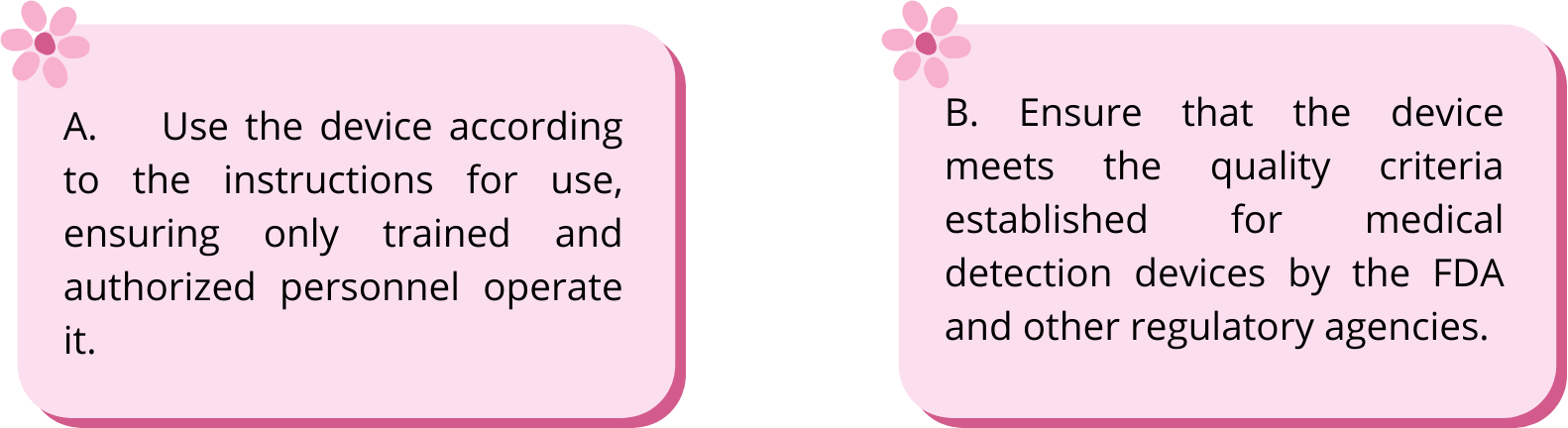
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
A. Use the device according to the instructions for use, ensuring only trained and authorized personnel operate it.
Specific objectives:
- Identify the secondary effects or risks associated with the use of this device
- Recommend that users comply with safety protocols and instructions.
B. Ensure that the device meets the quality criteria established for medical detection devices by the FDA and other regulatory agencies.
Specific objectives
- Identify the countries where this technology is urgently needed and therefore comply with the parameters established by their respective regulatory agency.
- Verify device accuracy and reliability
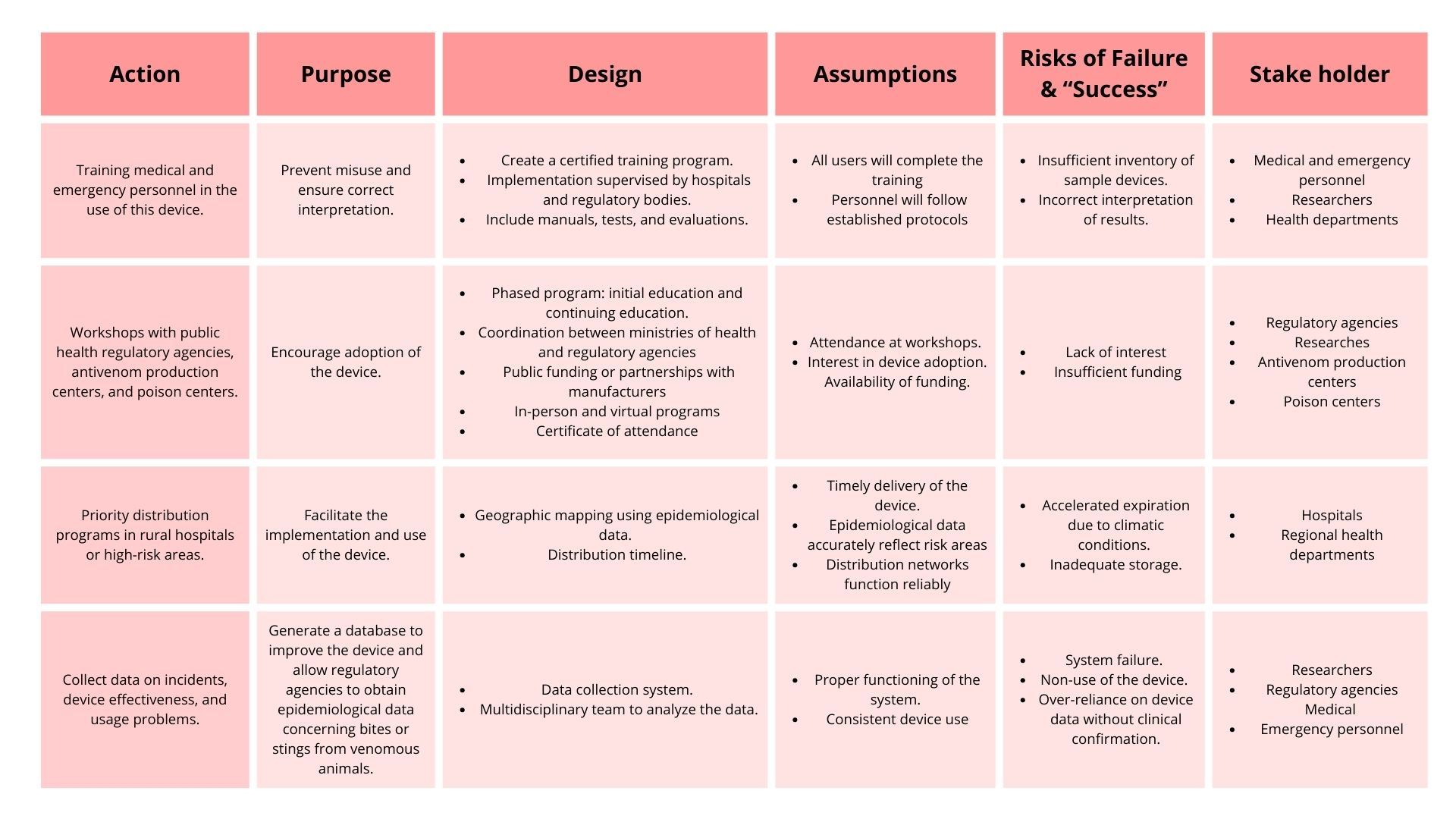
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
Purpose: What is done now and what changes are you proposing?
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals.
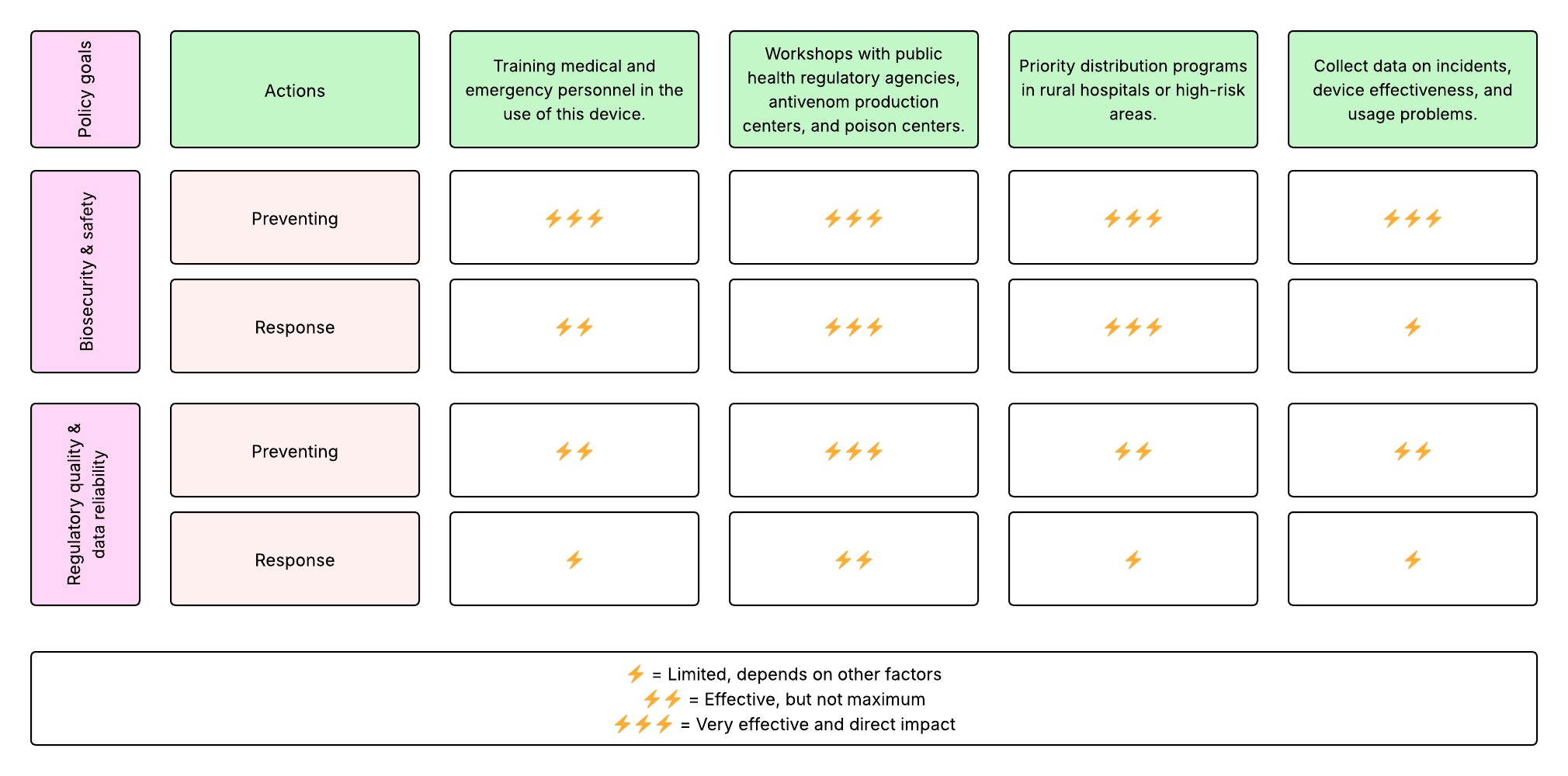
5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
Based on the scoring of each governance action, these options ranked highest. Consequently, they would be prioritized:
- Workshops with public health regulatory agencies, antivenom production centers, and poison centers.
- Priority distribution programs in rural hospitals or high-risk areas.
These actions have a direct influence on the two governance policies:
The priority distribution programs ensure that populations in high-risk areas have access to this device first, as they are the people who need it most. However, it is important to recognize that these priority programs require funding from regulatory agencies, hospitals, and health departments in order to function properly and avoid, for example, delays because of weather conditions or areas of difficult access, especially communities in middle- and low-income countries where there are some paramilitary groups.
As well, this is relevant for the medical care team in order to learn how to access, use, and interpret the results of the device.
Regarding this idea, the workshops will require interest from researchers, the population, health care teams, emergency teams, and paramilitary groups. Additionally, they will require the availability of time to develop the workshops and meetings to monitor the continuing evaluation of the device and its use in the community.
The prioritization of these two actions assumes that all resources will be available consistently, which is uncertain, especially in remote areas. This is why it is important to consider these actions as a whole and not as separate actions. If there is no interest in promoting the device from regulatory agencies, antivenom production centers, and poison centers, it will be difficult for populations in remote areas to begin using the device.
These two combined ideas will promote the safe use of the device and save lives in emergencies because it allows the identification of the type of toxin affecting a person after a bite or sting. Moreover, the medical team will provide adequate medical attention.
The target population for this recommendation is national health ministries, regulatory agencies, hospitals, and emergency teams responsible for device distribution and training.

Warm-up Questions for Week 2
In preparation for Week 2’s lecture on “DNA Read, Write, and Edit," please review these materials:
Lecture 2 slides as posted below.
The associated papers that are referenced in those slides.
In addition, answer these questions in each faculty member’s section:
Homework Questions from Professor Jacobson:
1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome? How does biology deal with that discrepancy?
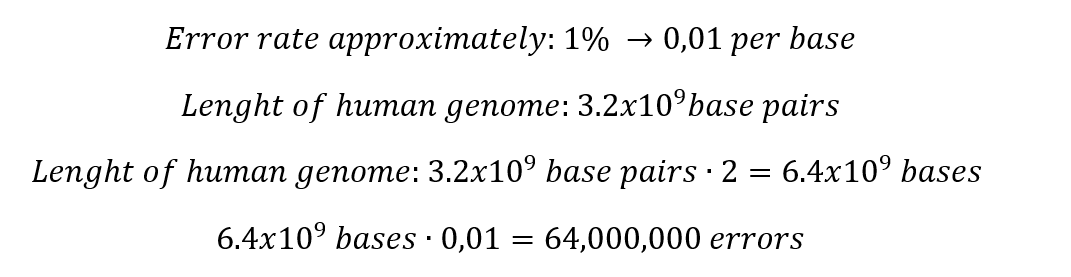
Which means that in the total length of the human genome, the polymerase fails in only 64 million bases. Indeed, this situation generates some discrepancy, but it is important to consider that 99% of the human genome is copied successfully.
When some mismatches occur, the polymerase has several ways to correct these mistakes.
- 3'-exonuclease activity
- Mismatch repair:Fixes mistakes in DNA after replication or transcription, a repair mechanism for endogenous damage
- Base excision: This method works by excising a single damaged base or nucleotide
- Nucleotide excision:Use the mechanism Cut and patch
2. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
In the present day, we recognize approximately 500 amino acids; however, only 20 are considered in the Protein synthesis. Some scientists believe that this occurs due to a matter of evolution; they explain that evolution prioritized modifications that provided advantages to survive.
In the beginning of the process, there are 4 nucleotides in DNA: A, T, C, and G, A, U, C, and G in RNA, these for nucleotides form codons of three nucleotides each, which means that:
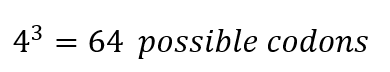
From these 64 codons, 61 correspond to amino acids for protein synthesis, and 3 are used as stop signals in the process. Which means that initially we have 20 elements to correspond to 61 options, consequently there are several codons which codify for 1 amino acid.
Indeed, to identify how many ways there might be to code 1 protein, it is necessary to know the number of amino acids in the protein.
For example, the Hemoglobin has 146 amino acids in one chain; basically, the calculation would be done like this.
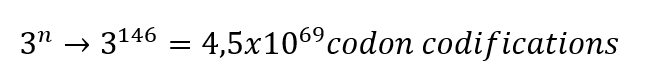
This is the theory of degeneracy in the genetic code. This theory suggests that the unequal distribution of 64 codons would be caused by nature to maintain stable C : H ratios.
Although there are several ways to code a protein, not all of them work properly; this is because, during the protein synthesis process, there are a variety of codons more commonly used than others.
This might cause a problem when the process tries to use codons used rarely, as it will be difficult for the ribosomes decode these types of codons. This theory is known as Codon usage bias.
Homework Questions from Dr. LeProust:
1. What’s the most commonly used method for oligo synthesis currently?
- Solid-phase phosphoramidite synthesis
It occurs on a solid support held between filters, in columns that allow all reagents and solvents to pass through freely.
Principal component: Solid supports
These are insoluble particles of 50-200 μm in diameter; the oligonucleotide makes bonds with these supports during the synthesis process.
The two most common materials used in solid supports are Controlled-pore glass (CPG) and Polystyrene (PS). Both methods work perfectly when synthesizing oligonucleotides up to 150 bases in length. After that limit, the efficiency rate decreases.
One nucleotide is added per synthesis cycle; the whole process includes approximately 14 steps.
This is because the growing oligonucleotide blocks the pores, which means that the diffusion of reagents will decrease. Additionally, in the case of PS with oligonucleotides longer than 40 bases, the efficiency rate will decrease because long chains generate steric hindrance.
Advantages:
- Occurs in columns that allow reagents and solvents to pass through freely
- Quick completion
- No purification is required
- The whole process might be automated
2. Why is it difficult to make oligos longer than 200nt via direct synthesis?
After reaching the limit, we can identify the principal disadvantage of the process, which is that the growing oligonucleotide blocks the pores, and long chains generate steric hindrance, which means that the diffusion of reagents will decrease.
The efficiency of the process is approximately 95.6% per 10 base chain length, which means that if you have a chain of 100nt, and you try to add, for example, 10 more bases, approximately 4 bases of the chain might fail, leaving the chain with 96nt. Imagine that you want to continue the process: you try to add 10 more bases, but again, probably 4 bases will be stuck, which leaves you with a chain of approximately 92 bases.
This is what scientists call coupling efficiency. At the end of the process, a chain of 200nt will have an efficiency rate of 36.9 %, which is less than half.
3. Why can’t you make a 2000bp gene via direct oligo synthesis?
A gene of 2000 bp has more than 200nt, which means that if the efficiency rate is less than half with just 200nt, then the efficiency rate of a gene of that length will be close to 0%.
Homework Question from George Church:
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
1. [Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
10 Essential amino acids
- Histidine (His)
- Isoleucine (Ile)
- Leucine (Leu)
- Lysine (Lys)
- Methionine (Met)
- Phenylalanine (Phe)
- Threonine (Thr)
- Tryptophan (Trp)
- Valine (Arg)
Lysine Contingency:
Lysine is the first limiting amino acid, which means that it is the amino acid present in food in the lowest quantity, compared to the amount needed by the body. In short, the quantity of lysine present in foods is less than the quantity required.
However, we talk about lysine contingency even though there are nine other essential amino acids, because lysine has a fundamental role in the proper functioning of the body.
- Acts as a substrate for post-translational modifications and the synthesis of a variety of substances used and produced by the body to work properly
- Fundamental to adequate muscle performance
- Possible Herpes Simplex Infections Therapy
- Modulation of Immune and Cardiovascular Functions
- Cancer Therapy
- Elimination of Harmful Substances
- Osteoporosis Therapy
- Wound Healing
When we detail all these uses of lysine, it is possible to notice that since this amino acid is extremely relevant, its availability in the diet is low. Therefore, it makes sense to establish it as a principal amino acid and consider its importance as an advantage in science, for example, developing an organism that depends mostly on lysine; this might be a way of controlling the activity of this organism.
Other applications could be developing new therapies to treat diseases or even indicators of diseases.
References
- Tasoulis, T., & Isbister, G. (2017). A review and database of snake venom proteomes. Toxins, 9(9), 290. https://doi.org/10.3390/toxins9090290
Alonso, L. L., Slagboom, J., Casewell, N. R., Samanipour, S., & Kool, J. (2025). Categorization and Characterization of Snake Venom Variability through Intact Toxin Analysis by Mass Spectrometry. Journal of Proteome Research, 24(3), 1329–1341. https://doi.org/10.1021/acs.jproteome.4c00923
- Miller, E. (n.d.). GeNotes. GeNotes. https://www.genomicseducation.hee.nhs.uk/genotes/knowledge-hub/genome/
- Marteijn, J. A., Lans, H., Vermeulen, W., & Hoeijmakers, J. H. J. (2014). Understanding nucleotide excision repair and its roles in cancer and ageing. Nature Reviews Molecular Cell Biology, 15(7), 465–481. https://doi.org/10.1038/nrm3822
- Behura, S. K., & Severson, D. W. (2012). Codon usage bias: causative factors, quantification methods and genome‐wide patterns: with emphasis on insect genomes. Biological Reviews/Biological Reviews of the Cambridge Philosophical Society, 88(1), 49–61. https://doi.org/10.1111/j.1469-185x.2012.00242.x
- On the origin of degeneracy in the genetic code. (2019, October). The Royal Society Publishing. https://royalsocietypublishing.org/rsfs/article/9/6/20190038/35070/On-the-origin-of-degeneracy-in-the-genetic
- Ye, S., & Lehmann, J. (2022). Genetic code degeneracy is established by the decoding center of the ribosome. Nucleic Acids Research, 50(7), 4113–4126. https://doi.org/10.1093/nar/gkac171
- Qian, Y., Zhang, R., Jiang, X., & Wu, G. (2021). The constraints between amino acids influence the unequal distribution of codons and protein sequence evolution. Royal Society Open Science, 8(6), 201852. https://doi.org/10.1098/rsos.201852
- ATDBio - Nucleic Acids Book - Chapter 5: Solid-phase oligonucleotide synthesis. (n.d.). ATDBio - Nucleic Acids Book. https://atdbio.com/nucleic-acids-book/Solid-phase-oligonucleotide-synthesis
- Holeček, M. (2025). Lysine: sources, metabolism, physiological importance, and use as a supplement. International Journal of Molecular Sciences, 26(18), 8791. https://doi.org/10.3390/ijms26188791
- Lopez, M. J., & Mohiuddin, S. S. (2024, April 30). Biochemistry, essential amino acids. StatPearls - NCBI Bookshelf. https://www.ncbi.nlm.nih.gov/books/NBK557845/
- MMDB. (n.d.). 2H35: Solution structure of Human normal adult hemoglobin. https://www.ncbi.nlm.nih.gov/Structure/pdb/2H35
Week 02 HW: DNA Read, write, and edit

This week, we evaluated the different methods to read, write, and edit DNA.
Part 1: Benchling & In-silico Gel Art
Make a free account at benchling.com
Import the Lambda DNA.
Simulate Restriction Enzyme Digestion with the following Enzymes:
- EcoRI
- HindIII
- BamHI
- KpnI
- EcoRV
- SacI
- Sall
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
I decided to elaborate on the design of a square wine glass. This design is new, and it provides a fresh look for wine.
To make this design, I used 3 types of restriction enzymes:
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
Assignees for the following sections
MIT/Harvard students = RequiredCommitted Listeners = Optional (for those with Lab access)
Part 3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
[Example from our group homework, you may notice the particular format — The example below came from UniProt]
/>sp|P03609|LYS_BPMS2 Lysis protein OS=Escherichia phage MS2 OX=12022 PE=2 SV=1 METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLL EAVIRTVTTLQQLLT
I chose kinesin (Kinesin-1 heavy chain, KIF5B). I was impressed by its movement because, during my career, I have seen different animations representing it. This protein walks along microtubules to transport cellular cargo.
 Retrieved from: https://www.chemistryworld.com/research/walking-proteins-tiny-steps-measured-with-germanium-nanospheres/4013257.article
Retrieved from: https://www.chemistryworld.com/research/walking-proteins-tiny-steps-measured-with-germanium-nanospheres/4013257.article
This is important to me because, although we know that life exists beneath our skin, this kind of animation is a great way to improve our understanding of life. Consequently, I want to increase my knowledge of this protein, especially because this protein is in several cellular components.
>sp|P33176|KINH_HUMAN Kinesin-1 heavy chain OS=Homo sapiens OX=9606 GN=KIF5B PE=1 SV=1 MADLAECNIKVMCRFRPLNESEVNRGDKYIAKFQGEDTVVIASKPYAFDRVFQSSTSQEQVYNDCAKKIVKDVLEGYNGTIFAYGQTSSGKTHTMEGKLHDPEGMGIIPRIVQDIFNYIYSMDENLEFHIKVSYFEIYLDKIRDLLDVSKTNLSVHEDKNRVPYVKGCTERFVCSPDEVMDTIDEGKSNRHVAVTNMNEHSSRSHSIFLINVKQENTQTEQKLSGKLYLVDLAGSEKVSKTGAEGAVLDEAKNINKSLSALGNVISALAEGSTYVPYRDSKMTRILQDSLGGNCRTTIVICCSPSSYNESETKSTLLFGQRAKTIKNTVCVNVELTAEQWKKKYEKEKEKNKILRNTIQWLENELNRWRNGETVPIDEQFDKEKANLEAFTVDKDITLTNDKPATAIGVIGNFTDAERRKCEEEIAKLYKQLDDKDEEINQQSQLVEKLKTQMLDQEELLASTRRDQDNMQAELNRLQAENDASKEEVKEVLQALEELAVNYDQKSQEVEDKTKEYELLSDELNQKSATLASIDAELQKLKEMTNHQKKRAAEMMASLLKDLAEIGIAVGNNDVKQPEGTGMIDEEFTVARLYISKMKSEVKTMVKRCKQLESTQTESNKKMEENEKELAACQLRISQHEAKIKSLTEYLQNVEQKKRQLEESVDALSEELVQLRAQEKVHEMEKEHLNKVQTANEVKQAVEQQIQSHRETHQKQISSLRDEVEAKAKLITDLQDQNQKMMLEQERLRVEHEKLKATDQEKSRKLHELTVMQDRREQARQDLKGLEETVAKELQTLHNLRKLFVQDLATRVKKSAEIDSDDTGGSAAQKQKISFLENNLEQLTKVHKQLVRDNADLRCELPKLEKRLRATAERVKALESALKEAKENASRDRKRYQQEVDRIKEAVRSKNMARRGHSAQIAKPIRPGQHPAASPTHPSAIRGGGAFVQNSQPVAVRGGGGKQV
Tool: Uniprot
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
[Example: Get to the original sequence of phage MS2 L-protein from its genome phage MS2 genome - Nucleotide - NCBI]
Lysis protein DNA sequence/
atggaaacccgattccctcagcaatcgcagcaaactccggcatctactaatagacgccggccattcaaacatgaggattacccatgtcgaagacaacaaagaagttcaactctttatgtattgatcttcctcgcgatctttctctcgaaatttaccaatcaattgcttctgtcgctactggaagcggtgatccgcacagtgacgactttacagcaattgcttacttaa
P33176|KINH_HUMAN Kinesin-1 heavy chain protein DNA sequence 2889 bases
atggcggatctggcggaatgcaacattaaagtgatgtgccgctttcgcccgctgaacgaaagcgaagtgaaccgcggcgataaatatattgcgaaatttcagggcgaagataccgtggtgattgcgagcaaaccgtatgcgtttgatcgcgtgtttcagagcagcaccagccaggaacaggtgtataacgattgcgcgaaaaaaattgtgaaagatgtgctggaaggctataacggcaccatttttgcgtatggccagaccagcagcggcaaaacccataccatggaaggcaaactgcatgatccggaaggcatgggcattattccgcgcattgtgcaggatatttttaactatatttatagcatggatgaaaacctggaatttcatattaaagtgagctattttgaaatttatctggataaaattcgcgatctgctggatgtgagcaaaaccaacctgagcgtgcatgaagataaaaaccgcgtgccgtatgtgaaaggctgcaccgaacgctttgtgtgcagcccggatgaagtgatggataccattgatgaaggcaaaagcaaccgccatgtggcggtgaccaacatgaacgaacatagcagccgcagccatagcatttttctgattaacgtgaaacaggaaaacacccagaccgaacagaaactgagcggcaaactgtatctggtggatctggcgggcagcgaaaaagtgagcaaaaccggcgcggaaggcgcggtgctggatgaagcgaaaaacattaacaaaagcctgagcgcgctgggcaacgtgattagcgcgctggcggaaggcagcacctatgtgccgtatcgcgatagcaaaatgacccgcattctgcaggatagcctgggcggcaactgccgcaccaccattgtgatttgctgcagcccgagcagctataacgaaagcgaaaccaaaagcaccctgctgtttggccagcgcgcgaaaaccattaaaaacaccgtgtgcgtgaacgtggaactgaccgcggaacagtggaaaaaaaaatatgaaaaagaaaaagaaaaaaacaaaattctgcgcaacaccattcagtggctggaaaacgaactgaaccgctggcgcaacggcgaaaccgtgccgattgatgaacagtttgataaagaaaaagcgaacctggaagcgtttaccgtggataaagatattaccctgaccaacgataaaccggcgaccgcgattggcgtgattggcaactttaccgatgcggaacgccgcaaatgcgaagaagaaattgcgaaactgtataaacagctggatgataaagatgaagaaattaaccagcagagccagctggtggaaaaactgaaaacccagatgctggatcaggaagaactgctggcgagcacccgccgcgatcaggataacatgcaggcggaactgaaccgcctgcaggcggaaaacgatgcgagcaaagaagaagtgaaagaagtgctgcaggcgctggaagaactggcggtgaactatgatcagaaaagccaggaagtggaagataaaaccaaagaatatgaactgctgagcgatgaactgaaccagaaaagcgcgaccctggcgagcattgatgcggaactgcagaaactgaaagaaatgaccaaccatcagaaaaaacgcgcggcggaaatgatggcgagcctgctgaaagatctggcggaaattggcattgcggtgggcaacaacgatgtgaaacagccggaaggcaccggcatgattgatgaagaatttaccgtggcgcgcctgtatattagcaaaatgaaaagcgaagtgaaaaccatggtgaaacgctgcaaacagctggaaagcacccagaccgaaagcaacaaaaaaatggaagaaaacgaaaaagaactggcggcgtgccagctgcgcattagccagcatgaagcgaaaattaaaagcctgaccgaatatctgcagaacgtggaacagaaaaaacgccagctggaagaaagcgtggatgcgctgagcgaagaactggtgcagctgcgcgcgcaggaaaaagtgcatgaaatggaaaaagaacatctgaacaaagtgcagaccgcgaacgaagtgaaacaggcggtggaacagcagattcagagccatcgcgaaacccatcagaaacagattagcagcctgcgcgatgaagtggaagcgaaagcgaaactgattaccgatctgcaggatcagaaccagaaaatgatgctggaacaggaacgcctgcgcgtggaacatgaaaaactgaaagcgaccgatcaggaaaaaagccgcaaactgcatgaactgaccgtgatgcaggatcgccgcgaacaggcgcgccaggatctgaaaggcctggaagaaaccgtggcgaaagaactgcagaccctgcataacctgcgcaaactgtttgtgcaggatctggcgacccgcgtgaaaaaaagcgcggaaattgatagcgatgataccggcggcagcgcggcgcagaaacagaaaattagctttctggaaaacaacctggaacagctgaccaaagtgcataaacagctggtgcgcgataacgcggatctgcgctgcgaactgccgaaactggaaaaacgcctgcgcgcgaccgcggaacgcgtgaaagcgctggaaagcgcgctgaaagaagcgaaagaaaacgcgagccgcgatcgcaaacgctatcagcaggaagtggatcgcattaaagaagcggtgcgcagcaaaaacatggcgcgccgcggccatagcgcgcagattgcgaaaccgattcgcccgggccagcatccggcggcgagcccgacccatccgagcgcgattcgcggcggcggcgcgtttgtgcagaacagccagccggtggcggtgcgcggcggcggcggcaaacaggtg
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
[Example from Codon Optimization Tool | Twist Bioscience while avoiding Type IIs enzyme recognition sites BsaI, BsmBI, and BbsI]
Lysis protein DNA sequence with Codon-Optimization
ATGGAAACCCGCTTTCCGCAGCAGAGCCAGCAGACCCCGGCGAGCACCAACCGCCGCCGCCCGTTCAAACATGAAGATTATCCGTGCCGTCGTCAGCAGCGCAGCAGCACCCTGTATGTGCTGATTTTTCTGGCGATTTTTCTGAGCAAATTCACCAACCAGCTGCTGCTGAGCCTGCTGGAAGCGGTGATTCGCACAGTGACGACCCTGCAGCAGCTGCTGACCTAA
Tool: https://www.idtdna.com/CodonOpt
Codon Optimization chain
ATG GCT GAT CTC GCT GAA TGT AAC ATC AAA GTG ATG TGC CGC TTT CGC CCC TTG AAC GAA TCA GAG GTG AAC CGC GGG GAC AAA TAC ATC GCC AAG TTT CAG GGG GAA GAT ACC GTG GTG ATT GCT TCT AAA CCT TAT GCG TTT GAT CGG GTG TTC CAG TCC TCA ACC TCC CAA GAA CAG GTG TAT AAC GAT TGT GCA AAG AAG ATC GTT AAA GAT GTT CTT GAG GGT TAC AAT GGC ACT ATC TTT GCC TAT GGC CAG ACT TCA TCC GGA AAG ACA CAC ACT ATG GAG GGC AAA CTT CAT GAT CCA GAG GGA ATG GGC ATC ATT CCA CGG ATT GTT CAG GAC ATA TTC AAC TAT ATA TAC AGC ATG GAC GAG AAC CTC GAG TTT CAT ATC AAG GTG AGC TAC TTC GAG ATC TAT CTC GAT AAA ATC CGG GAT CTT TTG GAT GTG TCT AAA ACT AAT CTG TCC GTT CAC GAG GAC AAG AAC AGA GTG CCC TAT GTG AAA GGG TGC ACC GAA CGG TTC GTG TGT TCA CCC GAC GAG GTC ATG GAT ACC ATT GAC GAG GGC AAA TCT AAC AGG CAT GTG GCT GTG ACC AAC ATG AAC GAG CAT AGC AGT AGG TCT CAT TCT ATA TTT CTG ATT AAT GTC AAG CAG GAG AAC ACC CAG ACT GAA CAG AAA TTG TCA GGC AAA CTC TAT CTG GTC GAC CTC GCA GGG AGC GAA AAG GTT TCC AAG ACA GGC GCA GAA GGC GCT GTG CTT GAC GAA GCC AAG AAT ATC AAC AAG TCC CTG AGC GCT CTT GGA AAC GTG ATA TCA GCC CTC GCC GAG GGC TCT ACG TAC GTT CCA TAT CGG GAT TCT AAA ATG ACC CGG ATC CTC CAA GAT TCC CTT GGA GGC AAC TGC AGG ACA ACA ATC GTC ATC TGT TGC AGT CCC TCT TCT TAC AAT GAG TCT GAA ACT AAG TCT ACT CTC CTG TTT GGG CAG AGA GCC AAG ACT ATA AAG AAT ACT GTG TGC GTC AAT GTG GAG CTG ACA GCG GAG CAG TGG AAG AAA AAA TAT GAA AAA GAA AAG GAA AAG AAT AAG ATC CTC AGA AAT ACC ATT CAG TGG CTT GAA AAC GAG CTG AAT AGG TGG AGG AAT GGC GAG ACT GTG CCC ATC GAC GAG CAG TTC GAT AAG GAG AAG GCT AAT TTG GAG GCG TTT ACA GTG GAT AAG GAT ATT ACA TTG ACA AAT GAC AAA CCA GCC ACC GCC ATT GGA GTA ATC GGC AAT TTT ACC GAT GCT GAG AGA AGG AAA TGC GAG GAG GAA ATC GCA AAG CTC TAT AAG CAA CTC GAT GAT AAG GAC GAG GAA ATC AAC CAA CAG TCC CAA CTC GTT GAA AAA CTG AAA ACA CAG ATG CTC GAC CAG GAA GAG CTG CTG GCC TCC ACT AGG CGG GAT CAG GAT AAT ATG CAG GCC GAA CTG AAC AGA CTT CAG GCC GAG AAC GAC GCC TCA AAG GAG GAG GTA AAG GAG GTG CTG CAG GCC CTG GAG GAG CTG GCG GTT AAC TAT GAT CAA AAG AGT CAG GAG GTG GAG GAC AAG ACT AAG GAG TAC GAA CTG CTG TCC GAC GAG CTT AAC CAG AAG TCA GCC ACA CTT GCG AGC ATC GAT GCC GAG CTC CAG AAA CTG AAA GAG ATG ACG AAT CAT CAG AAA AAG AGG GCT GCT GAA ATG ATG GCA AGC CTG TTG AAA GAC CTG GCG GAG ATC GGA ATC GCC GTG GGG AAT AAT GAT GTG AAA CAG CCC GAA GGG ACC GGA ATG ATA GAC GAG GAG TTC ACA GTA GCC AGA CTG TAC ATA AGC AAG ATG AAA TCT GAG GTA AAA ACG ATG GTT AAG CGA TGT AAA CAG CTC GAG TCT ACA CAG ACC GAG AGT AAC AAA AAG ATG GAG GAA AAT GAG AAA GAA CTG GCC GCT TGC CAG CTG CGG ATA TCA CAG CAT GAG GCC AAG ATT AAA AGT CTT ACT GAA TAC TTG CAG AAT GTA GAG CAA AAG AAA CGG CAA CTG GAG GAA AGC GTG GAT GCC CTC TCA GAG GAA CTC GTG CAG CTC AGA GCC CAA GAA AAG GTT CAT GAG ATG GAG AAA GAG CAC CTT AAT AAA GTA CAG ACG GCC AAT GAA GTC AAA CAG GCT GTG GAA CAG CAG ATC CAG TCT CAC AGG GAG ACA CAC CAG AAG CAG ATA AGC TCA CTG AGG GAC GAA GTG GAA GCA AAA GCC AAG CTC ATC ACT GAT CTC CAA GAC CAG AAT CAG AAG ATG ATG CTT GAG CAG GAG CGA CTC CGA GTG GAG CAT GAA AAA TTG AAG GCA ACT GAC CAA GAG AAG TCT AGA AAA CTT CAC GAA CTC ACT GTG ATG CAG GAC CGC AGG GAG CAG GCG CGC CAA GAC CTG AAA GGA CTT GAA GAG ACT GTG GCT AAG GAG CTC CAG ACC CTC CAT AAT CTG CGG AAG CTG TTC GTT CAG GAT TTG GCC ACC AGA GTC AAA AAA AGT GCG GAA ATT GAT AGC GAT GAC ACT GGC GGC AGT GCC GCC CAG AAG CAA AAA ATT TCT TTC TTG GAG AAC AAC TTG GAA CAG CTG ACA AAG GTA CAC AAG CAG CTG GTG AGA GAT AAC GCT GAC CTC CGA TGC GAA CTC CCA AAG TTG GAG AAA AGA CTG CGG GCC ACA GCA GAG AGG GTT AAA GCC CTG GAG TCA GCT CTG AAA GAA GCT AAG GAG AAC GCC TCC AGG GAC AGA AAA CGG TAC CAG CAA GAG GTA GAC CGG ATT AAA GAG GCC GTC AGG TCC AAA AAC ATG GCA AGA AGG GGG CAT AGT GCC CAG ATC GCC AAA CCC ATT AGA CCC GGA CAA CAC CCC GCC GCA TCC CCT ACC CAC CCT TCT GCA ATT CGG GGT GGG GGA GCC TTC GTT CAG AAT AGT CAG CCT GTG GCC GTA CGC GGC GGC GGA GGT AAG CAG GTG
Why you need to optimize codon usage?
Last week, we discussed how one amino acid might codify for several codons. These preferences vary from one organism to another, which means that if you try to put the gene from one organism into another, the choice of codon used by the gene might be different from the one preferred by the organism. Consequently, the expression of the protein will be affected. This explains why the optimal codon sequence is fundamental to ensure the highest level of expression of one specific protein.
Which organism have you chosen to optimize the codon sequence for and why?
Human (Homo sapiens)
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
Cell-dependent method:
In this case, scientists use live cells and their biological capacity to produce one protein. This means that this method employs the central dogma of biology.
DNA encodes RNA → RNA encodes Protein → Amino Acids Encode Proteins
1. Transcription of DNA to RNA: During this step, the RNA polymerase uses the DNA strand of nucleic acids to produce an antiparallel RNA chain ( mRNA).
2. Translation of RNA to protein: Protein synthesis occurs in the cytoplasm with the help of ribosomes. These structures read the mRNA and incorporate each amino acid according to the codon sequence.
For example, scientists could use a bacterium to produce and purify a human protein. However, there are several disadvantages to this method:
Living cells are complex and require specific conditions to ensure their growth.
It is difficult to control the many variables that a cell possesses.
The whole process is expensive
Cell- free methods:
In this method, scientists follow the central dogma of biology, but in this case, protein synthesis occurs in a controlled environment outside the cell.
There are three components:
- Cell-free extract: Contains all the machinery from the cell to build proteins.
- DNA sequence: Provides genetic information to build the protein.
- Energy and Cofactors: Energy sources and supplies to facilitate the process
This method has several advantages:
- The process is fast; scientists might obtain one protein in a couple of hours.
- It is more flexible because scientists can improve the reaction to produce the protein, and they do not need to maintain a living cell.
- Less expensive, because it requires less maintenance in contrast to maintaining living cells
- Minimal contamination of protein.
These 2 methods have advantages and disadvantages, but without doubt, we can say that they improve our knowledge of life.
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account and a Benchling account
4.2. Build Your DNA Insert Sequence
4.3. On Twist, Select The “Genes” Option
4.4. Select “Clonal Genes” option
4.5. Import your sequence
4.6. Choose Your Vector
This is the plasmid you just built with your expression cassette included. Congratulations on building your first plasmid!
Part 5: DNA Read/Write/Edit
5.1 DNA Read
1. What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
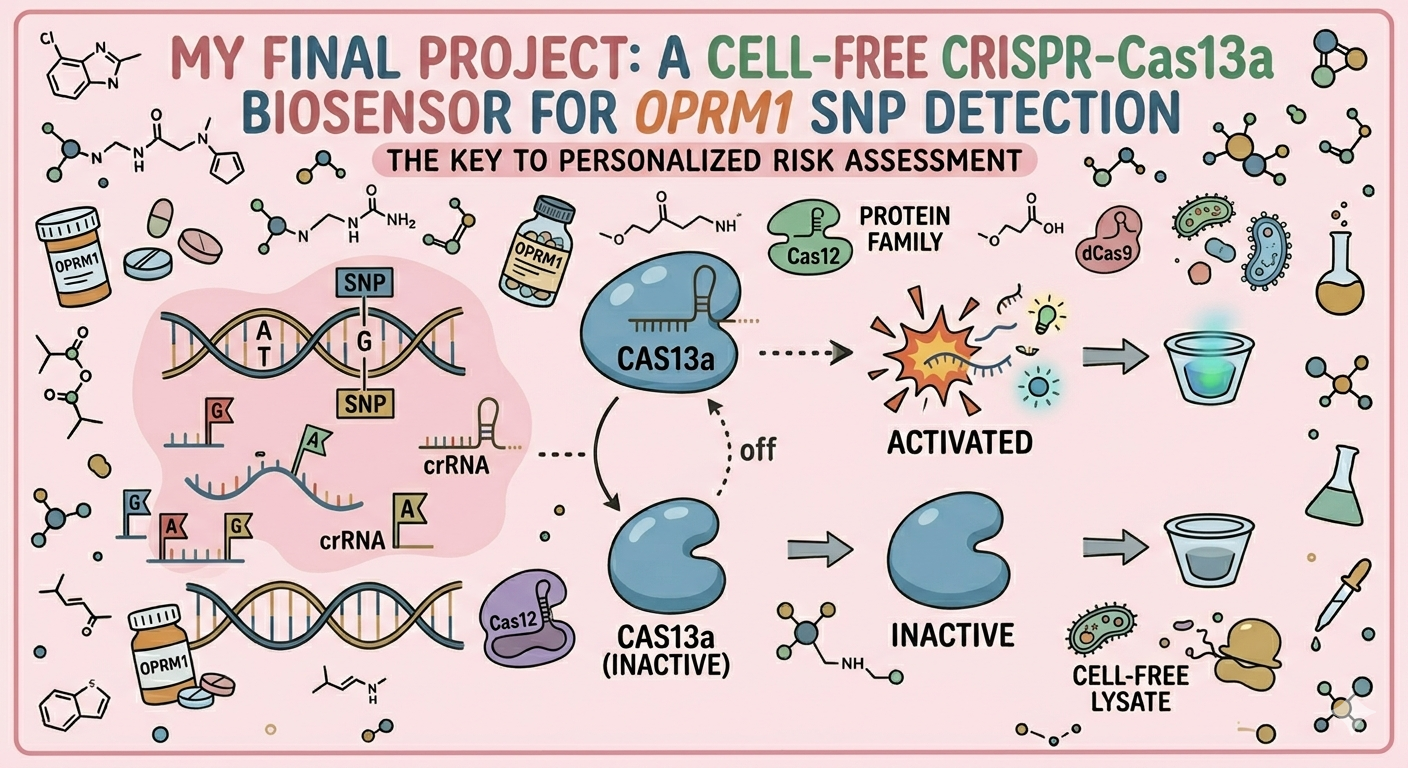
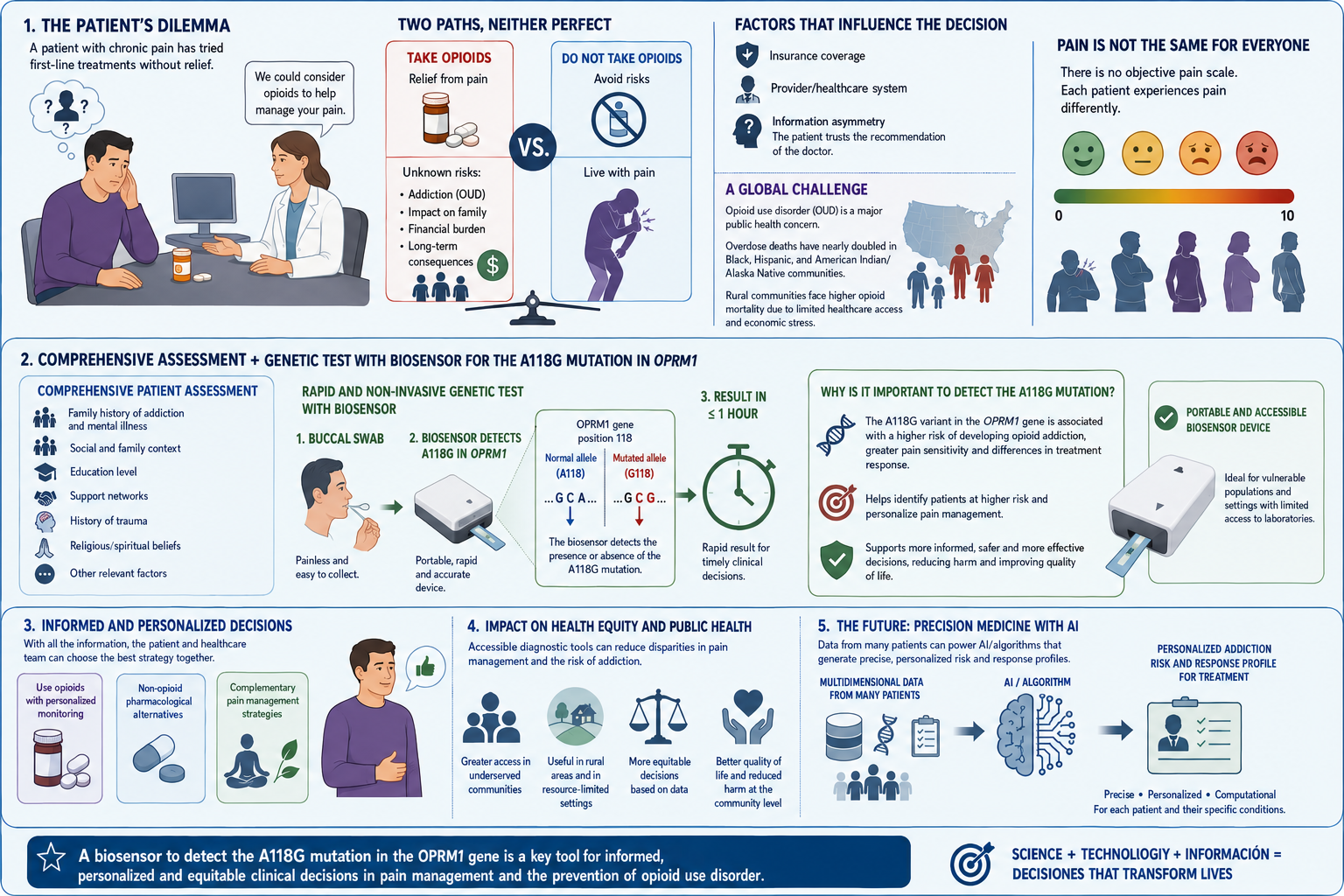
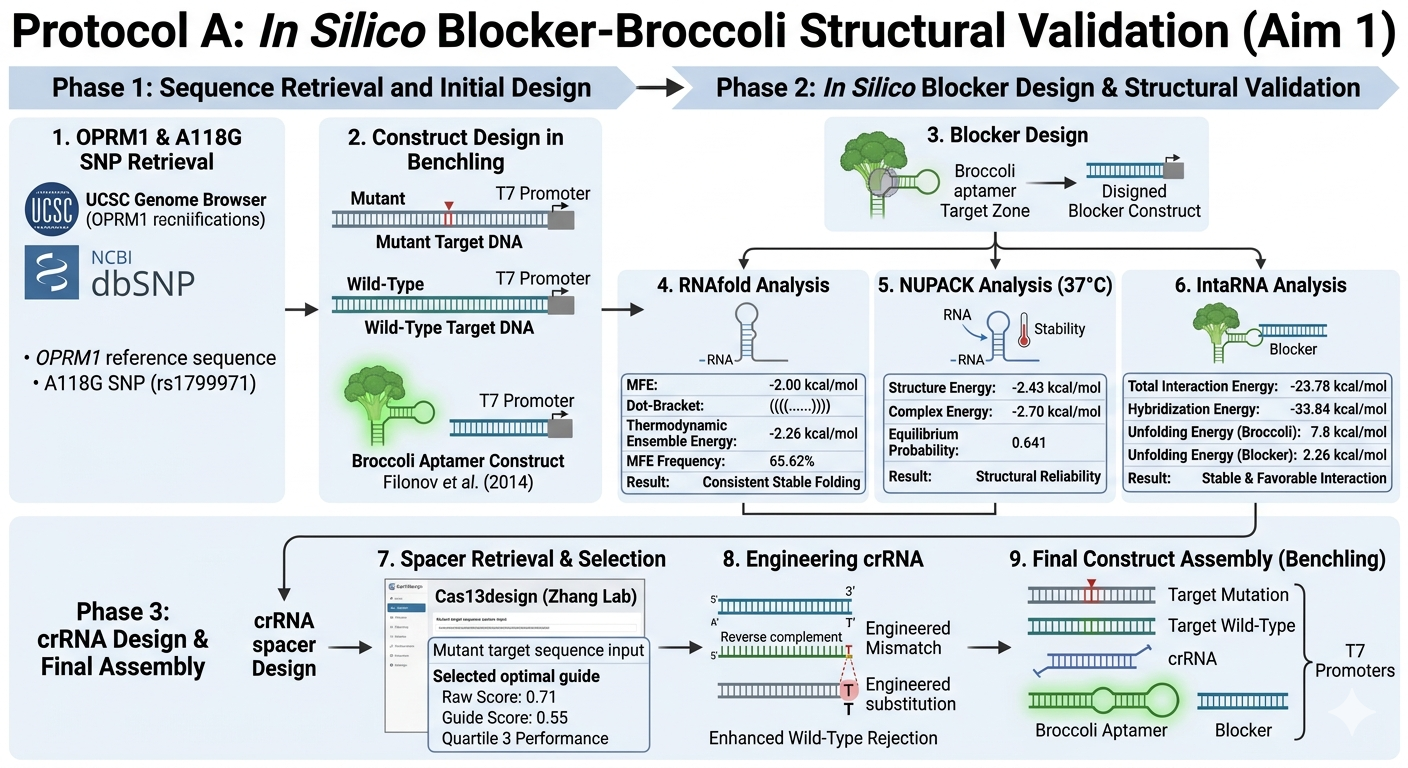
I want to read OPRM1 (Opioid Receptor Mu 1), which encodes the activity of opioid receptors in humans (MOR). MOR is the target of most opioid analgesics and other medicines related to pain management. Also, it has a fundamental role in dependence on other substances such as nicotine, cocaine, and alcohol.
Scientists have found several variations in this gene related to a major risk of addiction. In my opinion, as a pharmacist, with the opioid crisis and the emergence of new substances every day, it is important to be aware of possible addictions. Especially, because the result of addictions might be death. And as a healthcare team, we don’t desire that people die because of medicines whose primary purpose was to treat pain.
The objective of reading this gene is that if in one moment a patient goes to the doctor’s office suffering from chronic pain, the doctor will have the chance to sequence this specific gene in their patient and then know if their patient has a higher risk to develop addictions caused by variations or polymorphisms in this gene, especially A118G.
Consequently, the doctor will use this information and their knowledge to prescribe a lower dose of medicine with risk of addiction or may try to manage the pain of the patient by other methods, such as physical therapy, massages, or even other medicines from different therapeutic groups.
2. In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would like to perform sequencing on the OPRM1 gene using sequencing by synthesis (SBS). This is because this method allows the doctor to take a sample of DNA of the patient, and by using the fluorescent image obtained of each different color from nucleotides, we can compare the sequence obtained from the patient vs a normal sequence of the gene.
Also, answer the following questions:
2.1 Is your method first-, second-, or third-generation or other? How so?
Second-generation sequencing, because it allows sequencing multiple fragments at the same time, which brings several advantages, including fast and economic results.
2.2 What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
Input: Patient blood sample
- Purify the DNA sample
- Ensure that the sample is pure and undegraded
- Start library preparation:
→Cut the DNA sample into DNA fragments using high-frequency sound waves or enzymes
→Add adapters to each DNA fragment
→Assure that the library contains enough concentration to sequence
3. What are the essential steps of your chosen sequencing technology? How does it decode the bases of your DNA sample (base calling)?
In sequencing by synthesis (SBS), the DNA fragments are copied one base at a time. And each nucleotide is marked with a fluorescent dye. This produces an image with the flow cell.
3.1 What is the output of your chosen sequencing technology?
After obtaining the flow cell, they pass through a process of demultiplexing, obtaining different reads that will be organized based on a reference genome.
Consequently, scientists will compare the reference genome with the patient’s sample, and evaluate if there are any polymorphisms in OPRM1.
5.2 DNA Write
1.What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I would like to create a cellular sensor for opioids. The sensor will use a mu-opioid receptor (MOR), whose activity is regulated by the OPRM1 gene. The principal idea is that when the receptor is activated by high concentrations of opioids, it will trigger a genetic circuit that produces a fluorescent protein, causing the cell to have a visible glow that might be easy to detect and measure.
Higher glows indicate higher concentrations of opioids; this sensor is useful because it allows scientists to evaluate how different doses of opioids affect the activity of the receptors. Also, this sensor might be used in educational programs regarding the use of opioids to show people the activity in cells in a different way.
2. What technology or technologies would you use to perform this DNA synthesis and why?
I would like to use Twist Bioscience’s chip-based gene synthesis, because it allows precise and efficient synthesis. It is the easiest way, since I need to digitally design the sensor and submit the design to Twist for synthesis.
Additionally, my sensor is composed of several DNA fragments, including the gene OPRM1, promoters, fluorescent circuit, etc. This method synthesizes multiple DNA fragments on one chip. It is fast, economic, and accurate.
Also answer the following questions:
2.1 What are the essential steps of your chosen sequencing methods?
The essential types of Twist Bioscience’s chip-based gene synthesis are:
- Upload your gene sequence and configure your project
- DNA is synthesized at Twist
- DNA is assembled
- High-quality genes
One method to verify if the synthesis is correct might be SBS, previously described.
2.2 What are the limitations of your sequencing method (if any) in terms of speed, accuracy, and scalability?
Disadvantages of Sequencing by Synthesis (SBS):
- Sample preparation requires purification, PCR, and fragmentation
- Acquiring fluorescent dyes might be difficult and expensive, depending on the market.
- High initial instruments cost
- It is a second-generation method of synthesis, with short read lengths in contrast to third-generation methods
- This method has great potential for scalability
5.3 DNA Edit
1. What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
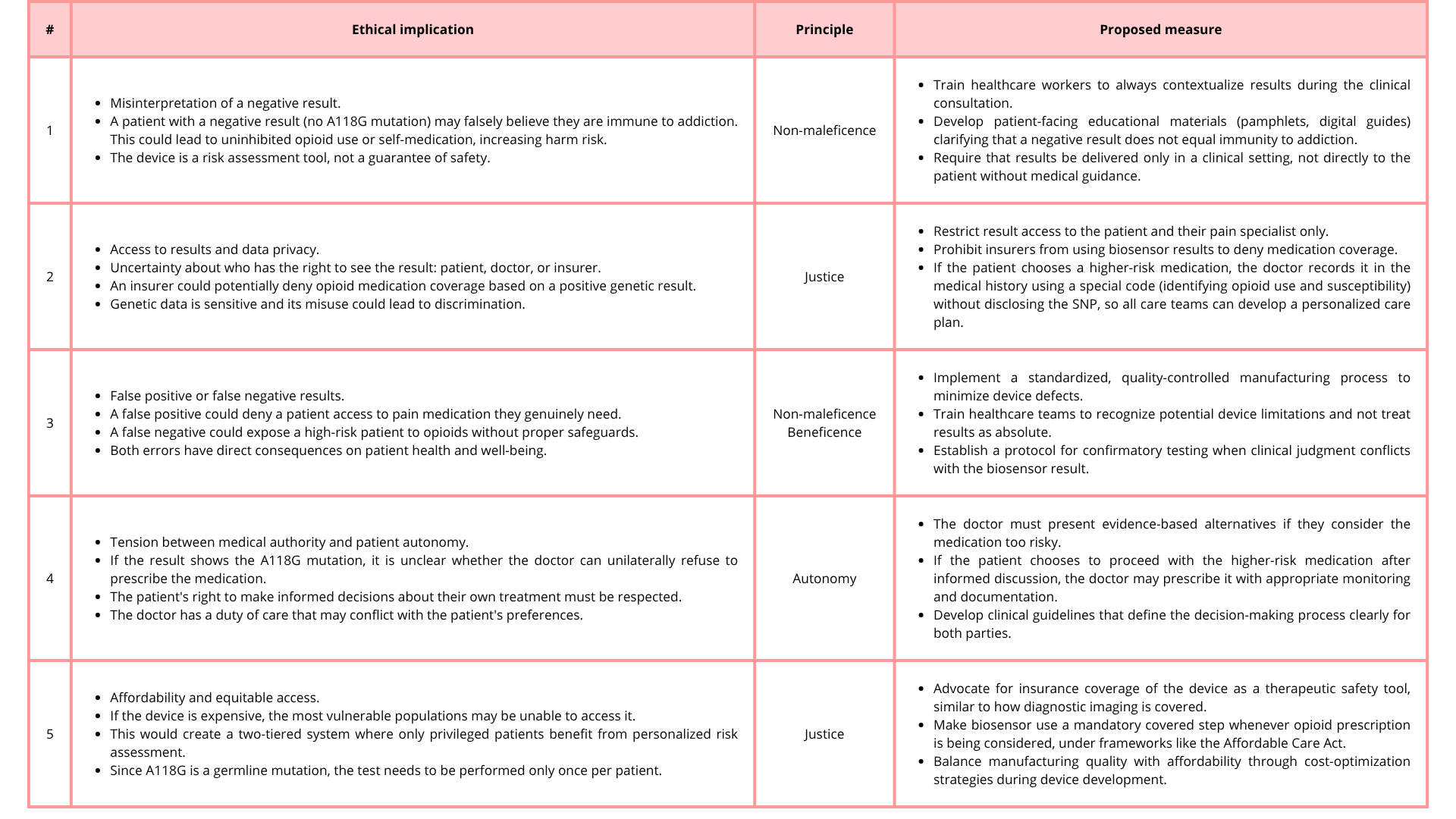
I would like to edit and correct the OPRM1 gene in patients with the A118G polymorphism to reduce the risk of addiction. However, it is important to remember that addictions are multifactorial conditions, which means that reducing the risk does not eliminate it.
I believe that this is a good framework, especially for patients with chronic pain whose better option to manage the pain is opioids. This approach, with adequate monitoring and supervision by doctors and family members, might reduce the incidence of addictions.
2. What technology or technologies would you use to perform these DNA edits and why?
I would like to use CRISPR-Cas9, because it is an editing technology frequently used today, and there are some protocols defined, even though this technology is not widely used for editing humans. It is a well-known technology.
Also, answer the following questions:
2.1 How does your technology of choice edit DNA? What are the essential steps?
It has 2 parts:
Cas9 protein: Cut DNA
Guide RNA: Recognize the site of DNA to be edited
C= Clustered
R= Regularly
I= Interspaced
S= Short
P= Palindromic
R= Repeats
- CRISPR/Cas9 complex formation
- CRISPR/Cas9 complex attaches to the target DNA sequence and induces a double-strand break (DSB) at the specific site
- Insertion of donor DNA and results in the transformed DNA sequence
2.2 What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
- Identify the sequence of the human genome that is causing the disease or problem.
- Create a specific RNA
- Introducing the complex CRISPR-Cas9 to the cells
- The CRISPR-Cas9 complex can edit the sequence by eliminating, modifying, or inserting a new sequence.
- Use cells as the biological system.
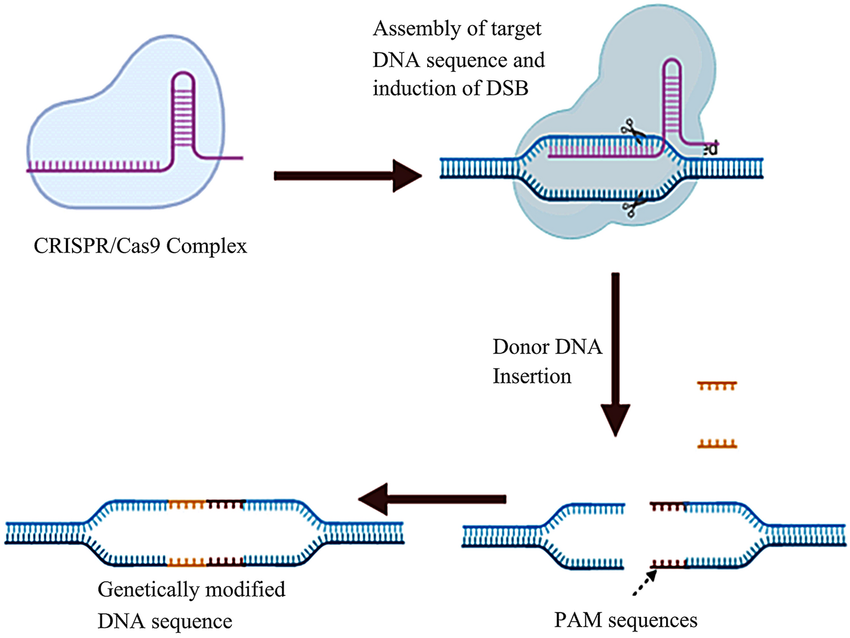 Retrieved from: https://www.researchgate.net/figure/Mechanism-of-the-CRISPR-cas9-system-The-first-step-in-this-process-is-the-CRISPR-Cas9_fig2_362382684
Retrieved from: https://www.researchgate.net/figure/Mechanism-of-the-CRISPR-cas9-system-The-first-step-in-this-process-is-the-CRISPR-Cas9_fig2_362382684
2.3 What are the limitations of your editing methods (if any) in terms of efficiency or precision?
- In terms of precision, CRISPR-Cas9 has a high frequency of off-target effects (OTEs), specifically ≥50%
- There is the possibility that CRISPR-Cas9 triggers apoptosis rather than objective gene editing.
- Immunotoxicity
References
- KIF5B kinesin family member 5B [Homo sapiens (human)] - Gene - NCBI. (2025). Nih.gov. https://www.ncbi.nlm.nih.gov/gene/3799
- P33176 KINH_HUMAN (Homo sapiens(human))- Gene- Retrieved February (2026) Uniprot. https://www.uniprot.org/uniprotkb/P33176/entry
- supreme_admin. (2025, March 31). Codon Optimization: Understanding the Basics | IDT. IDT. https://www.idtdna.com/page/support-and-education/decoded-plus/codon-optimization-the-basics-explained/
- Social Science, L. (2020, June 30). 3.4: DNA and Protein Synthesis. Social Sci LibreTexts. https://socialsci.libretexts.org/Courses/College_of_the_Canyons/Anthro_101%3A_Physical_Anthropology/03%3A_Cell_biology/3.04%3A_DNA_and_Protein_Synthesis
- Brookwell, A., Oza, J. P., & Caschera, F. (2021). Biotechnology Applications of Cell-Free Expression Systems. Life, 11(12), 1367. https://doi.org/10.3390/life11121367
- Technologies , I. D. (2015). Cell-Free Protein Synthesis Explained | IDT. Integrated DNA Technologies. https://www.idtdna.com/pages/applications/cell-free-protein-synthesis
- Medicine, N. L. of. (2025, November 25). OPRM1 opioid receptor mu 1 [Homo sapiens (human)] - Gene - NCBI. Www.ncbi.nlm.nih.gov. https://www.ncbi.nlm.nih.gov/gene/4988
- Taqi, M. M., Faisal, M., & Zaman, H. (2019). OPRM1 A118G polymorphisms and its role in opioid addiction: Implication on severity and treatment approaches. Pharmacogenomics and Personalized Medicine, Volume 12, 361–368. https://doi.org/10.2147/pgpm.s198654
- ClevaLab. (2022, December 4). Next Generation Sequencing - A Step-By-Step Guide to DNA Sequencing. Www.youtube.com. https://www.youtube.com/watch?v=WKAUtJQ69n8
- Zhang, X., Jiang, X., Wang, Y., Chen, Q., Jiang, H., Zhang, H., Beltran, A., Yang, W., Chen, T., Liang, C., Cheng, N., Huang, Y., Ding, G., Xie, C., Gao, N., Liu, J., Xu, W., Huang, J., Cai, D., & Zhu, L. (2025). Scaling DNA synthesis with a microchip-based massively parallel synthesis system. Nature Biotechnology. https://doi.org/10.1038/s41587-025-02844-0
- Fuller, C. W., Middendorf, L. R., Benner, S. A., Church, G. M., Harris, T., Huang, X., Jovanovich, S. B., Nelson, J. R., Schloss, J. A., Schwartz, D. C., & Vezenov, D. V. (2009). The challenges of sequencing by synthesis. Nature Biotechnology, 27(11), 1013–1023. https://doi.org/10.1038/nbt.1585
- The Power of Silicon-Based DNA Synthesis- Retrieved February (2026)
Twist BioScience. https://www.twistbioscience.com/products/genes/gene-synthesis?tab=overview&utm_source=google&utm_medium=cpc&utm_campaign=PSR-GLBL-FY21-1791-GENES-Twist-Genes-Product&adgroup=114820677303&utm_term=gene%20fragment%20synthesis&utm_content=aud-1246333009810:kwd-366151829721&creative=747198843491&device=c&matchtype=b&location=9004247&gad_source=1&gad_campaignid=12061463038&gbraid=0AAAAADdPWR--SRjJbKP9Btyj804YD913x&gclid=CjwKCAiA-sXMBhAOEiwAGGw6LHgdq1r8sKVubeax3HNyhDZuKiraOMwMm2M6z5Vk7xDgdWaBj3uD5hoCwBUQAvD_BwE
- Jayachandran, M., Fei, Z., & Qu, S. (2022). Genetic advancements in obesity management and CRISPR-Cas9-based gene editing system. Molecular and Cellular Biochemistry, 478. https://doi.org/10.1007/s11010-022-04518-w
- Mayo Clinic. (2018). CRISPR Explained [YouTube Video]. In YouTube. https://www.youtube.com/watch?v=UKbrwPL3wXE
- Uddin, F., Rudin, C. M., & Sen, T. (2020). CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Frontiers in Oncology, 10(1387). https://doi.org/10.3389/fonc.2020.01387
Week 03 HW: Lab automation

Assignment: Python Script for Opentrons Artwork — DUE BY YOUR LAB TIME!
0. Your task this week is to Create a Python file to run on an Opentrons liquid handling robot.
1. Review this week’s recitation and this week’s lab for details on the Opentrons and programming it.
2. Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
- Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
- You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept.
- If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead.
4. If you use AI to help complete this homework or lab, document how you used AI and which models made contributions.
5. Sign up for a robot time slot if you are at MIT/Harvard/Wellesley or at a Node offering Opentrons automation. The Python script you created will be run on the robot to produce your work of art!
6. Submit your Python file via this form.
✨✨My code✨✨
https://colab.research.google.com/drive/1rOfQVambbO3m8ZcjPQd7lQDcfa-qjr_j?usp=sharing

My inspiration was this image of the little fox from the story of the little prince.
Post-Lab Questions — DUE BY START OF FEB 24 LECTURE
One of the great parts about having an automated robot is being able to precisely mix, deposit, and run reactions without much intervention, and design and deploy experiments remotely.
For this week, we’d like for you to do the following:
1. Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Title: Automation of protein crystallization scaleup via Opentrons 2 liquid handling
Publication: SLAS Technology, 2025
This article is impressive because it explains that protein crystallization is an important process; however, there are several variables that are difficult to control to execute this process, especially on a small scale. When humans develop this process, it might be difficult, exhausting, and the outcomes may be inaccurate.
Consequently, as a new way to resolve this problem, scientists have developed a new approach optimizing protein crystallization trials at the multi-microliter scale with the Opentrons-2 liquid handling robot.
Scientists explain that although there are different robots on the market whose objective is to improve this process, these robots are expensive and their programming is exclusive. On the other hand, we have Opentrons, which is a robot with automation for several purposes, and it can be programmed using Python.
With Python scripts, scientists compare the efficacy and accuracy of the process developed by Opentrons OT-2 vs the manual method.
The materials they used included:
- Opentrons OT-2
- Crystallization plates (sitting drop 24-well): for forming the protein crystals.
- Protein solutions: the proteins they wanted to crystallize.
- Precipitating and buffer solutions: substances that help the crystals form.
Steps
- Plate Preparation
- The wells of the crystallization plate were placed in specific positions on the robot deck
- Robot Programming
- Python was used to instruct the robot on how to move liquids
- Pick up protein from one container.
- Pick up buffer/precipitate from another container.
- Mix small drops into the wells of the plate.
- Running Assays
- The robot performed all pipetting automatically, drop by drop.
Some plates contained different combinations of protein and precipitate to test various conditions simultaneously.
They illustrate the process with the following images:
2. Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
While your description/project idea doesn’t need to be set in stone, we would like to see core details of what you would automate. This is due at the start of lecture and does not need to be tested on the Opentrons yet.
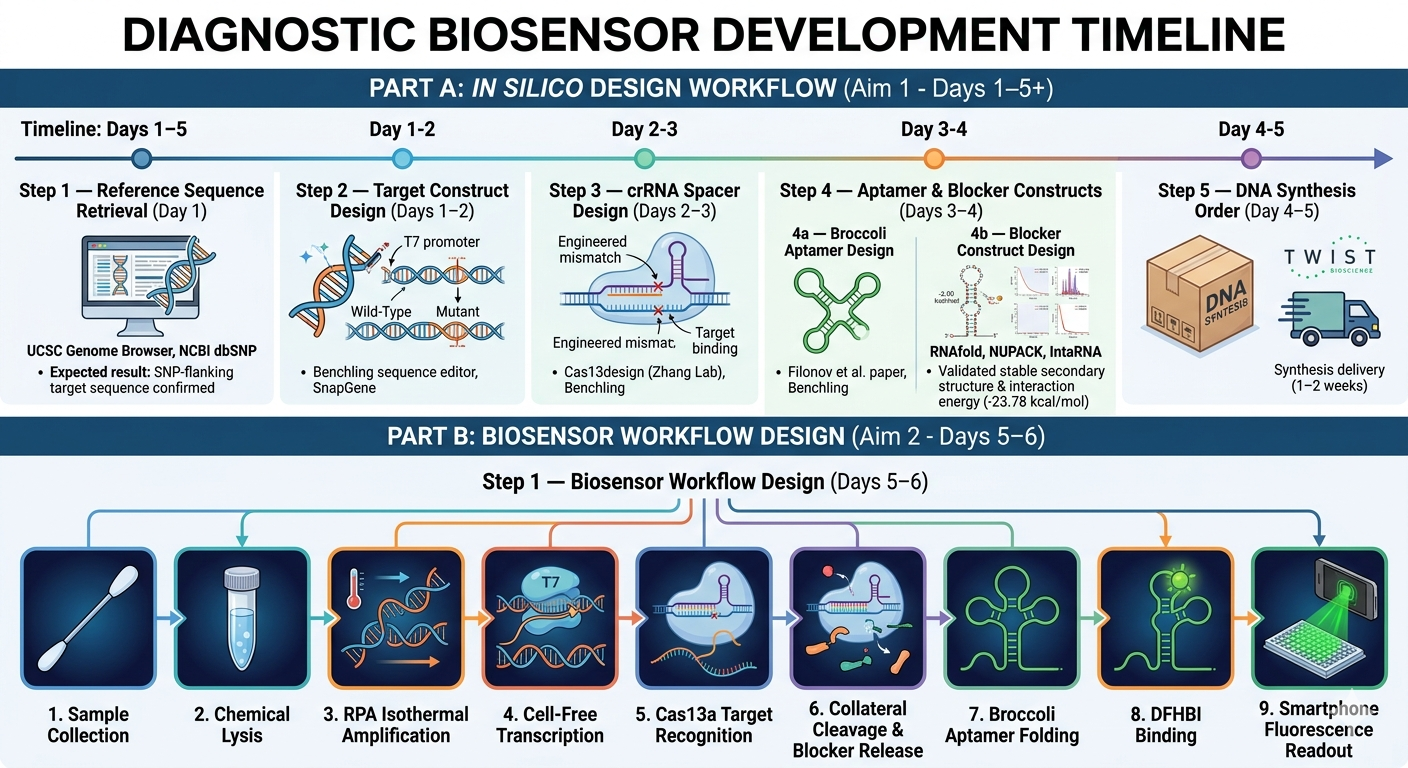
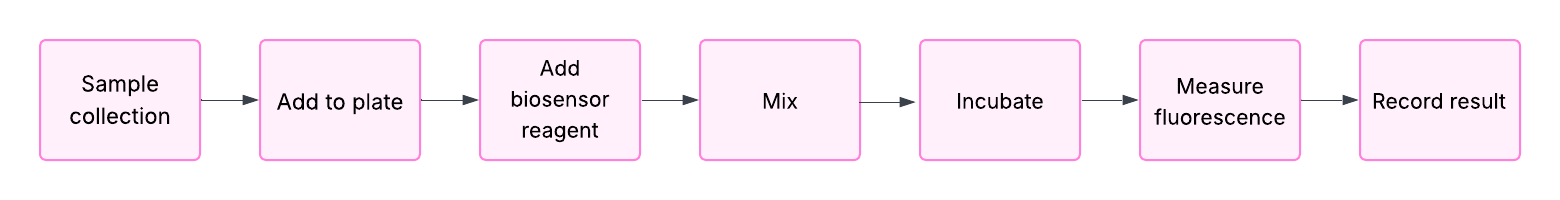
For my project, I want to develop a biosensor. I plan to automate sample preparation and measurement using a liquid-handling robot (like Opentrons) and a plate reader (like PHERAstar).
This automation will be used only during laboratory development to ensure reproducible and accurate results. However, this automation will not be included in the final product, because the main goal of my biosensor is that it will be used in ambulatory settings.
Final Project Ideas — DUE BY START OF FEB 24 LECTURE
As explained in this week’s recitation, add 1-3 slides in your Node’s section of this slide deck with 3 ideas you have for an Individual Final Project. Be sure to put your name, city, and country on your slide!



References
- DeRoo, Jacob B., et al. “Automation of Protein Crystallization Scaleup via Opentrons-2 Liquid Handling.” SLAS Technology, vol. 32, June 2025, p. 100268, pmc.ncbi.nlm.nih.gov/articles/PMC12229254/, https://doi.org/10.1016/j.slast.2025.100268. Accessed 12 Dec. 2025.
- Taqi, M. M., Faisal, M., & Zaman, H. (2019). OPRM1 A118G polymorphisms and its role in opioid addiction: Implication on severity and treatment approaches. Pharmacogenomics and Personalized Medicine, Volume 12, 361–368. https://doi.org/10.2147/pgpm.s198654
- Tasoulis, T., & Isbister, G. (2017). A review and database of snake venom proteomes. Toxins, 9(9), 290. https://doi.org/10.3390/toxins9090290
- Alonso, L. L., Slagboom, J., Casewell, N. R., Samanipour, S., & Kool, J. (2025). Categorization and Characterization of Snake Venom Variability through Intact Toxin Analysis by Mass Spectrometry. Journal of Proteome Research, 24(3), 1329–1341. https://doi.org/10.1021/acs.jproteome.4c00923
Week 04 HW: Protein Design part I

Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average, an amino acid is ~100 Daltons)
500 grams of protein, approximately, has 20% of protein.
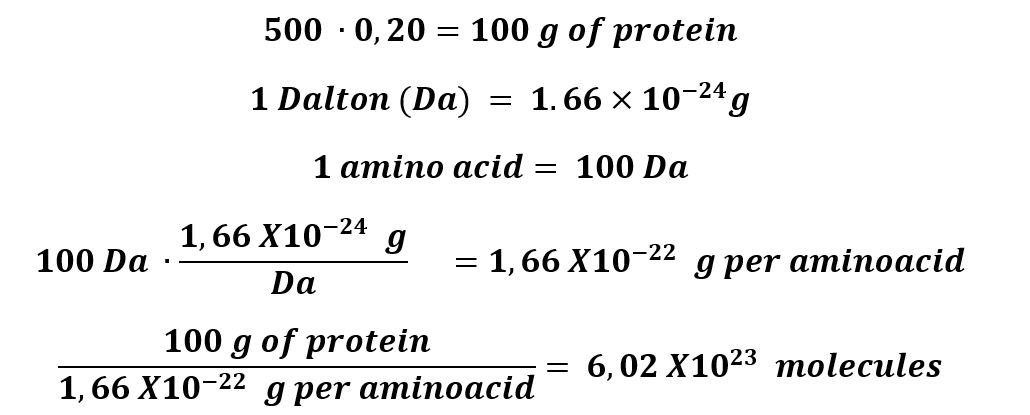
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
This phenomenon occurs because humans are living beings with a special anatomy; indeed, we have a relatively smaller colon and larger small intestine, which shows that our system is prepared to process high-protein diets.
These characteristics, along with others like gastric acidity, allow humans to ingest beef and fish, and thought-out gastric system becomes a big part of food, especially meat, in amino acids that our body can use to synthesize proteins that we need.
This is why it is important to have a balanced diet with an adequate amount of protein.
3. Why are there only 20 natural amino acids?
It is not like just existing 20 amino acids; in fact, there might be different combinations of amino acids. However, nature is wise and decided the combinations for the 20 natural amino acids that we know, due to several reasons.
Criteria for selecting amino acids:
Choice of atoms: Amino acids need to be made of atoms that are abundant on Earth, such as C, H, N, O, and S.
Functional groups: Due to the selection of atoms is important that the functional groups form hydrogen bonds and electrostatic interactions. Like Amides, amines, hydroxyls, carboxyls, and carbon–nitrogen bonds.
Biosynthetic cost: Protein synthesis is the process that uses the largest amount of energy in a cell. Scientists have measured the cost of biosynthesis of each amino acid, measured in terms of glucose and ATP molecules. For example, Leu costs only 1 ATP, but its isomer Ile costs 11.
Nature chooses the most effective cost option.
Solubility: Amino acids need to be soluble in high concentrated aqueous environment.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, scientists have been doing that for years. And for this educational exercise, I would like to design a fluorescent amino acid. A fluorescent molecule typically has a conjugated system with one or more aromatic rings.
The base structure of amino acids is:
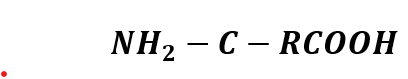
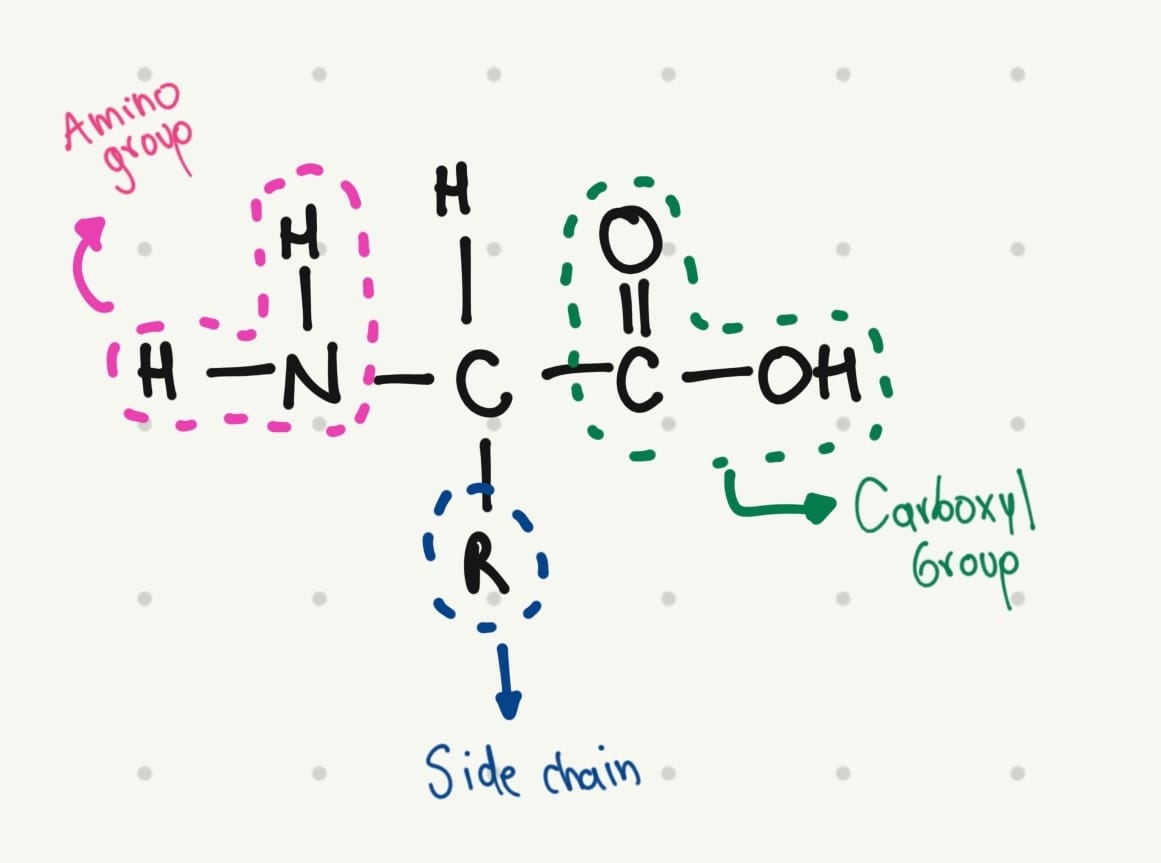
That’s why I thought in a simple structure:
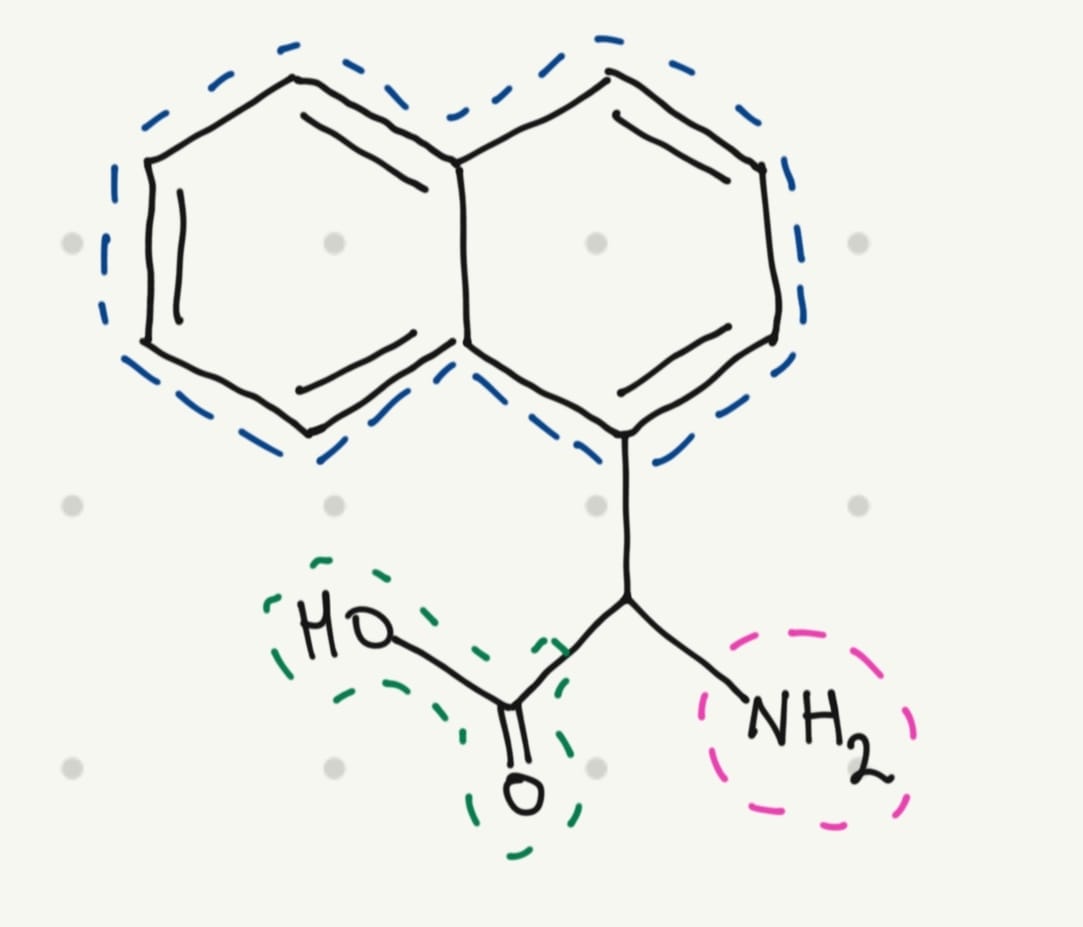
5. Where did amino acids come from before enzymes that make them, and before life started?
Today many amino acids are synthesized by metabolic and biosynthesis pathways. However, in the earliest years of life (between 4000 and 3500 million years), they were synthesized by chemical synthesis.
This hypothesis was proven by Miller and Urey in 1953, when they performed an experiment to recreate the conditions of primordial Earth in a flask.
They create an atmosphere with ammonia, hydrogen, methane, and water vapor, plus electrical sparks. They found that new molecules were formed.
Specifically, these molecules result in eleven standard amino acids.
In conclusion, in the beginning, amino acids were synthesized due to the environmental conditions; today, they are synthesized by biosynthesis.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
There exists a concept known as chirality, which is the property of an object that is not superimposed on its mirror image. This means that molecules with chirality have an asymmetric carbon, making them mirror images of each other.
One good example of this phenomenon is your hands; they are the mirror image of each other, but they cannot be superimposed.
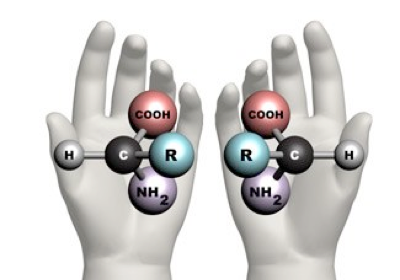 Taken from: https://www.maths.ox.ac.uk/node/14490
Taken from: https://www.maths.ox.ac.uk/node/14490
Natural proteins are made of L-amino acids. When these amino acids form an α-helix, it is right-handed, but following the idea of chirality, if D-amino acids form the α-helix, it will be left-handed.
7. Can you discover additional helices in proteins?
Yes, indeed, scientists have been developing new forms of helices for years. They have identified only 1,000 distinct protein folds in nature; however, they are developing different modifications of these natural folds. For example, researchers have identified alternative helical conformations such as 3₁₀-helices and π-helices.
They have also been trying to fold random amino acid sequences. All these methods are great, but the results might be inaccurate and do not represent a standardized process.
For this reason, they are presenting a new computational method for generating packings of secondary structures, which will facilitate the search for novel protein folds.
8. Why are most molecular helices right-handed?
Besides the natural chirality of amino acids that form proteins, several influencing factors determine why most molecular helices are right-handed.
The alpha helix structure is more stable because it uses the hydrogen bond between the C=O and N-H groups of the main chain to stabilize it. Although these bonds can form in both right-handed and left-handed alpha helices, they are more favorable in a right-handed alpha helix, because it requires less energy due to reduced steric clashes between the side chains and the main chain.
9. Why do β-sheets tend to aggregate? And what is the driving force for β-sheet aggregation?
β-Sheets are polypeptide strands connected by hydrogen bonds of adjacent backbone amides; these bonds are stronger and perpendicular, especially when the strands are aligned in opposite directions.
These characteristics provide the strands with the capacity to extend in a planar and stable structure due to the hydrogen bonds, which means that β-Sheets can interact with other β-Sheets, leading to aggregation.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
1. Briefly describe the protein you selected and why you selected it.
I chose the Dopamine Transporter (DAT) because one of my interests is the addiction area. As a pharmacist, I acknowledge that people with chronic pain are more vulnerable to developing addiction. But this problem can be presented in other individuals whose use abused drugs.
This transporter has a special role in dopamine homeostasis because it is the one responsible for the reuptake of dopamine from the synaptic space. The DAT is the major target of the most common drug of abuse, especially psychostimulants.
When we do pleasurable activities, there are signaling pathways that create action potential, which indicate the release of neurotransmitters, among them dopamine, in the synaptic space.
After the action potential disappears, the DAT has the responsibility of maintaining homeostasis and the reuptake of dopamine to maintain the balance. However, abused drugs affect this process in different ways.
Alcohol, nicotine, and heroin increase the action potential, leading to a major release of dopamine. Cocaine and methamphetamine bind to the DAT and block the reuptake of dopamine.
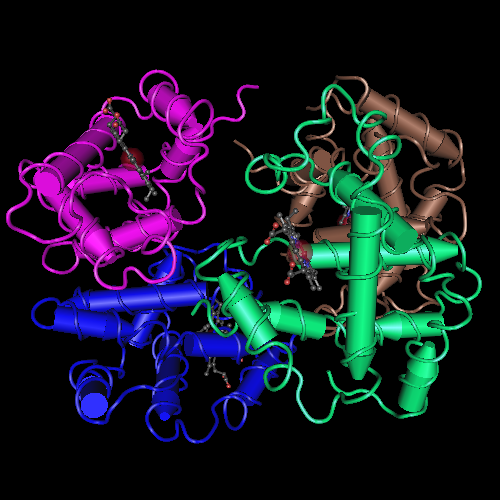
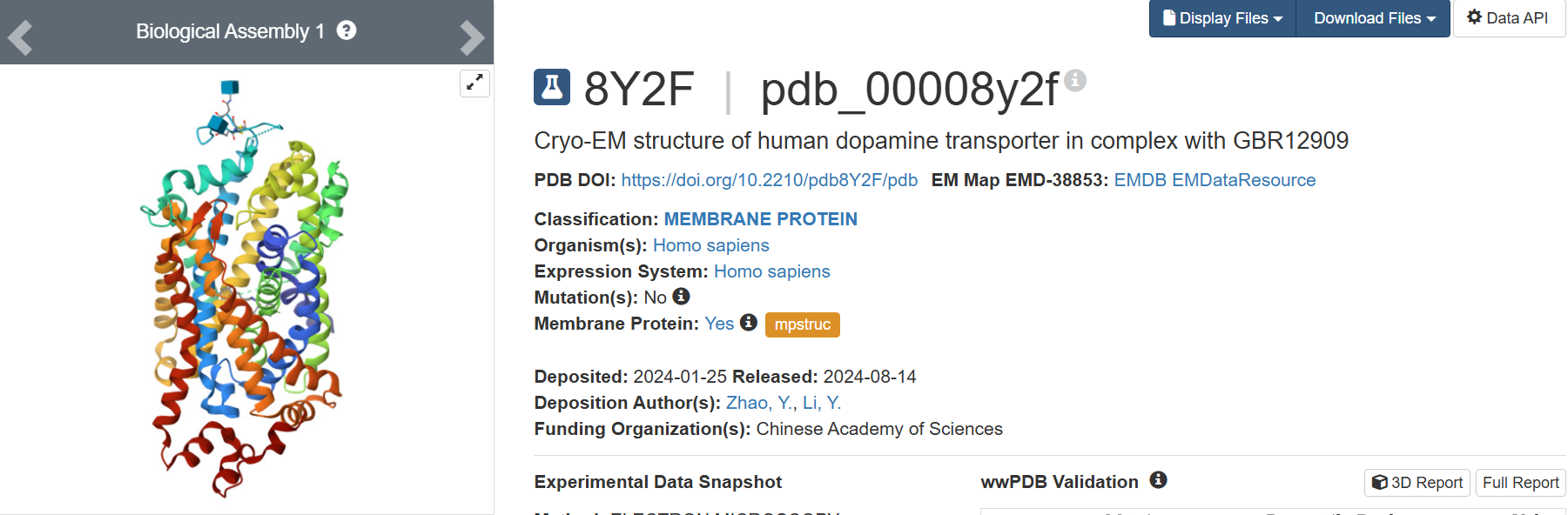
I found the structure in PDB title:
8Y2F | pdb_00008y2f
Cryo-EM structure of human dopamine transporter in complex with GBR12909
2. Identify the amino acid sequence of your protein.
sp|Q01959|SC6A3_HUMAN Sodium-dependent dopamine transporter OS=Homo sapiens OX=9606 GN=SLC6A3 PE=1 SV=1
MSKSKCSVGLMSSVVAPAKEPNAVGPKEVELILVKEQNGVQLTSSTLTNPRQSPVEAQDRETWGKKIDFLLSVIGFAVDLANVWRFPYLCYKNGGGAFLVPYLLFMVIAGMPLFYMELALGQFNREGAAGVWKICPILKGVGFTVILISLYVGFFYNVIIAWALHYLFSSFTTELPWIHCNNSWNSPNCSDAHPGDSSGDSSGLNDTFGTTPAAEYFERGVLHLHQSHGIDDLGPPRWQLTACLVLVIVLLYFSLWKGVKTSGKVVWITATMPYVVLTALLLRGVTLPGAIDGIRAYLSVDFYRLCEASVWIDAATQVCFSLGVGFGVLIAFSSYNKFTNNCYRDAIVTTSINSLTSFSSGFVVFSFLGYMAQKHSVPIGDVAKDGPGLIFIIYPEAIATLPLSSAWAVVFFIMLLTLGIDSAMGGMESVITGLIDEFQLLHRHRELFTLFIVLATFLLSLFCVTNGGIYVFTLLDHFAAGTSILFGVLIEAIGVAWFYGVGQFSDDIQQMTGQRPSLYWRLCWKLVSPCFLLFVVVVSIVTFRPPHYGAYIFPDWANALGWVIATSSMAMVPIYAAYKFCSLPGSFREKLAYAIAPEKDRELVDRGEVRQFTLRHWLKV
• How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
620 aminoacids
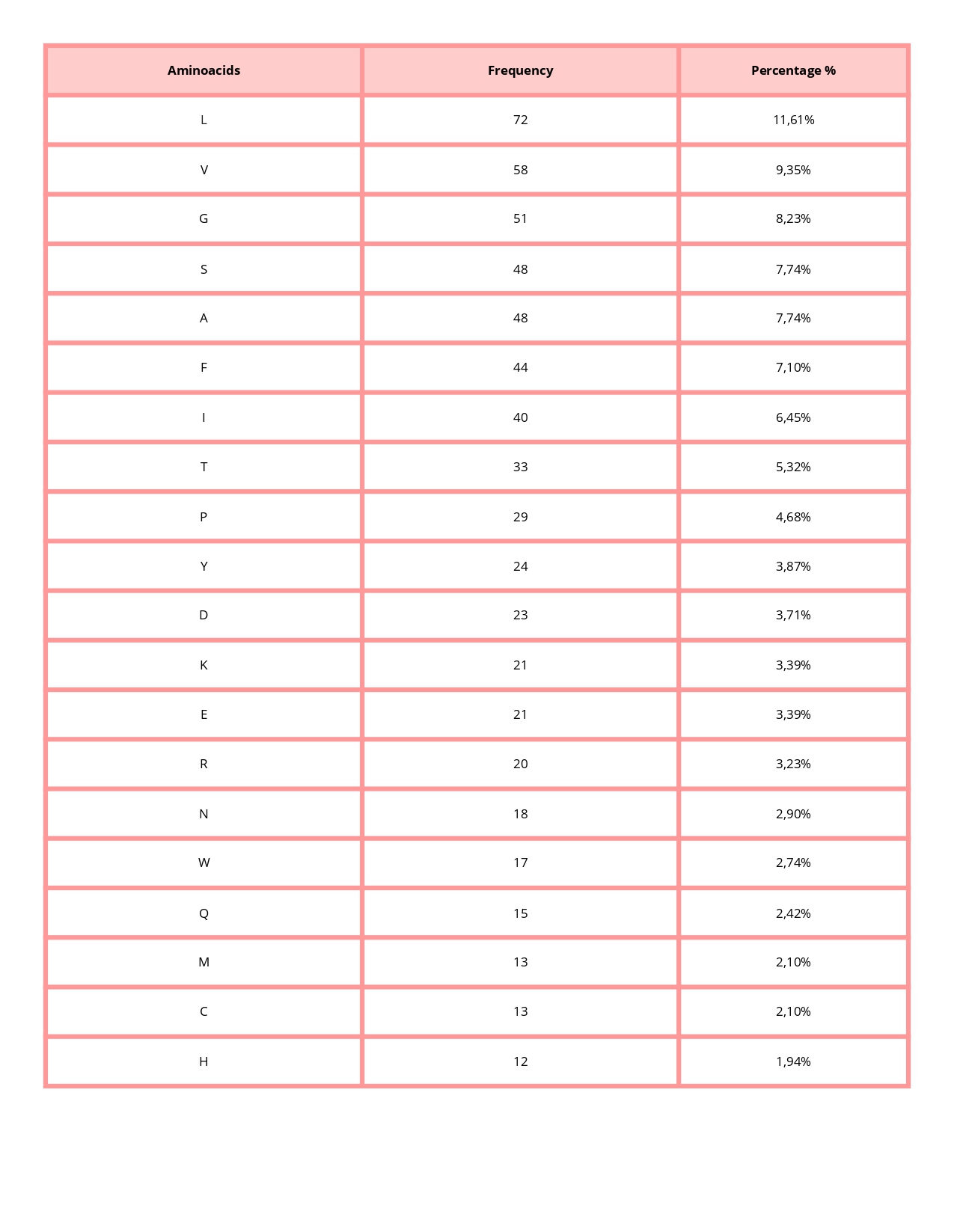
The most common amino acid is: L (Leucine), which appears 72 times.
• How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
Uniprot’s BLAST tool found 250 homologs

• Does your protein belong to any protein family?
Yes, it is a member of the monoamine transporter family (MAT), which is the family of proteins responsible for regulating neurotransmitter concentrations.
3. Identify the structure page of your protein in RCSB
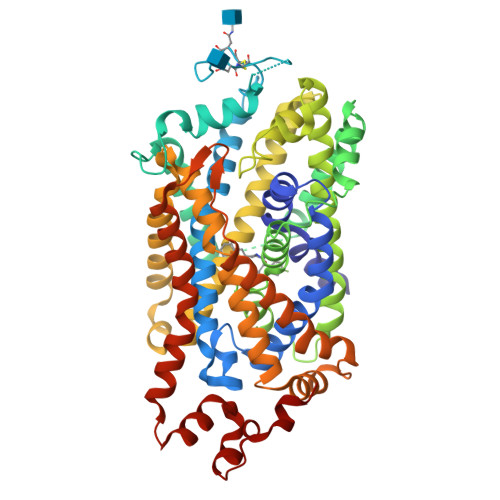
• When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The 8Y2F structure of the human Dopamine Transporter was deposited in the PDB on January 25, 2024 and published on August 14, 2024.
Resolution: 2.97 Å
The best resolution in electron microscopy for protein structure determination is between 1.25 Å - 2.00, however, one value of 2.97 Å is accurate but might be losing some details.
• Are there any other molecules in the solved structure apart from protein?
Yes, 2 small ligands:
1. Vanoxerine (ID: A1D5S): C28 H32 F2 N2 O – Chains: B
2. 2-acetamido-2-deoxy-beta-D-glucopyranose (ID:NAG): C8 H15 N O6 – Chains: C and D
• Does your protein belong to any structure classification family?
Membrane protein
4. Open the structure of your protein in any 3D molecule visualization software:
• PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
• Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
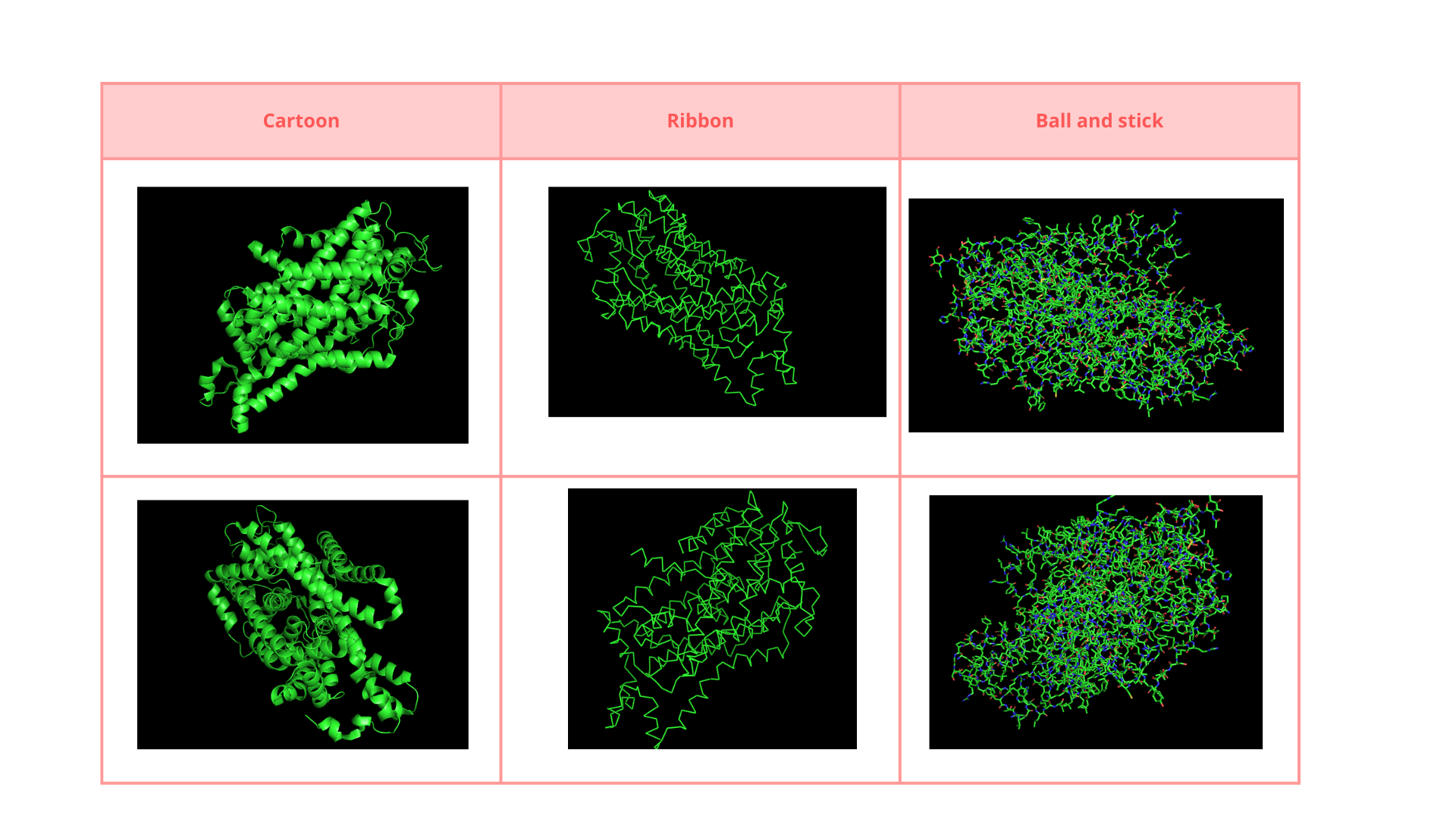
• Color the protein by secondary structure. Does it have more helices or sheets?
The structure is predominantly alpha-helical• Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
The surface of the protein was colored by residue type using util.cbag().
Green 🟢 → hydrophobic residues
Red 🔴 → negatively charged residues (Asp, Glu)
Blue 🔵 → positively charged residues (Lys, Arg, His)
The protein surface shows a mixture of hydrophobic (green) and charged residues (red and blue). Hydrophobic residues are abundant, while charged residues are distributed across the surface.
The combination of opposite charges can stabilize electrostatic interactions.
The green patches on the surface could indicate interaction with another protein or membrane.
• Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Yes, it has a binding pocket, which is correct, as this is a transport protein.
C1. Protein Language Modeling
Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
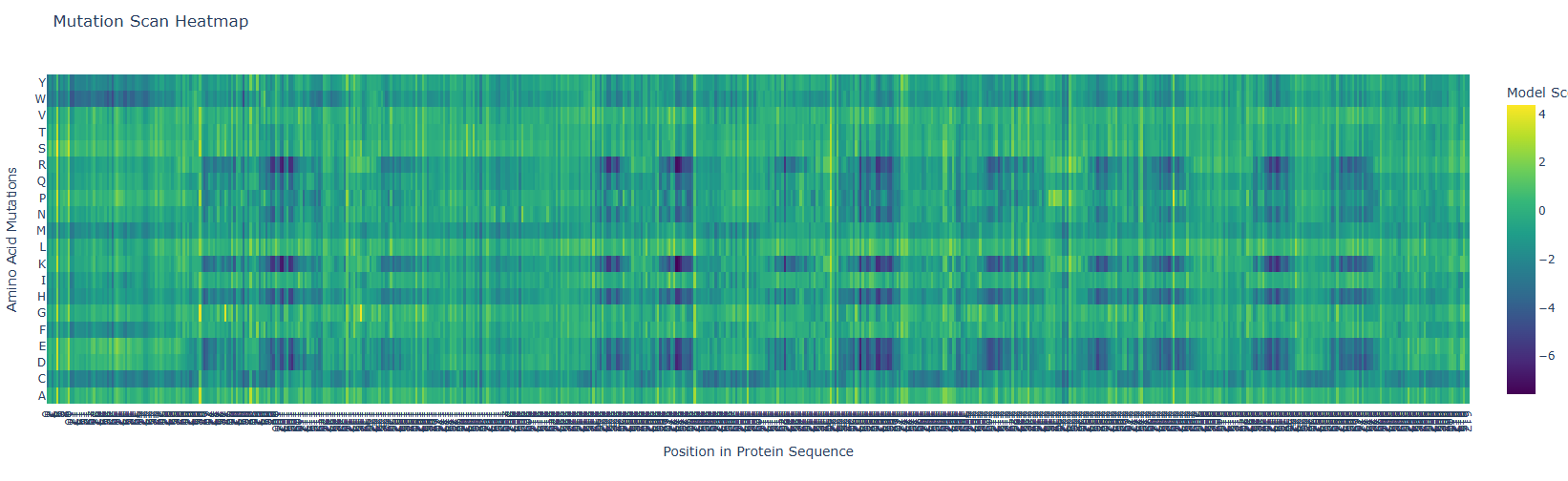
First, it is important to consider the model score:
Yellow 🟡(~4): Favorable mutation
Green 🟢 (~0): neutral mutation or tolerable mutation, which means that there is no affectation of the protein activity.
Dark blue 🔵 (~-6 a -7): Unfavorable mutation, makes the protein unstable and affects its function.
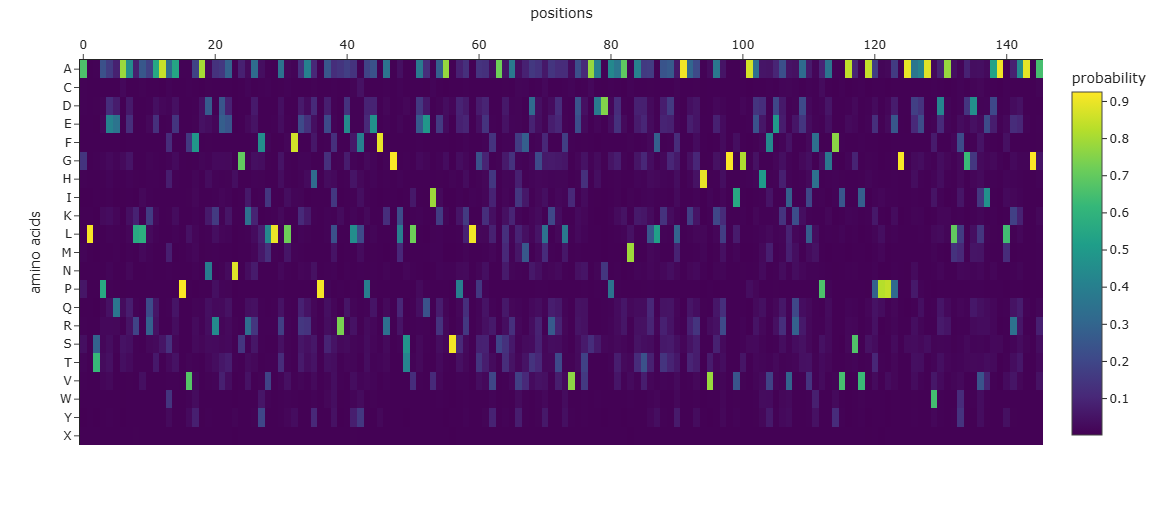
a. Can you explain any particular pattern? (Choose a residue and a mutation that stands out)
In the next picture, I highlight the patrons that I found interesting:
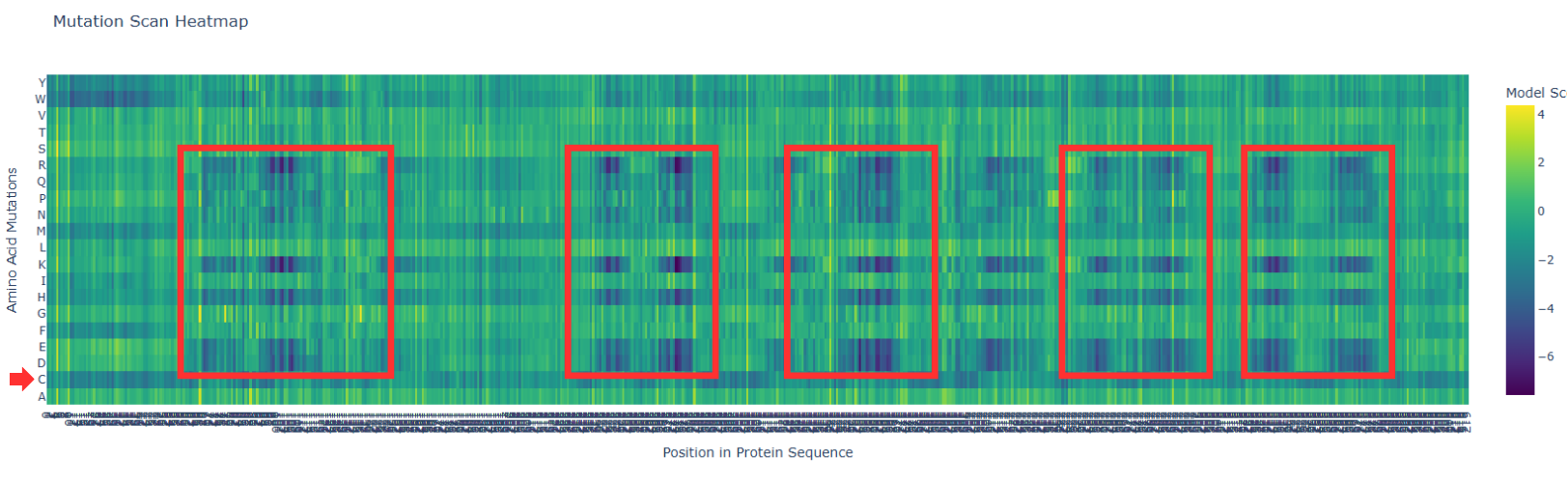
I). Some specific columns with a purple color, that appear symmetrical and in specific zones of the proteins. Especially some amino acids like R (Arginine), K (Lysine), H (Histidine), E (Glutamic acid), D (aspartic acid), in different positions in the entire chain.
Regarding this information, I establish the hypothesis that these positions are fundamental for protein function, and mutations in these zones might affect protein function, or, in general, they will be unfavorable.
II). In the row of amino acid Cysteine, many of the different positions are blue, which means that the model of ESM2 considers that this amino acid is unfavorable for most of the positions in the chain.
This might affect the function of the protein, since this mutation is found in most of the protein; it is reasonable to believe that Cys is not the best amino acid for this type of protein.
2. Latent Space Analysis
a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
Protein sequences from the provided dataset were embedded using Colab and executing the cells corresponding to Latent Space Analysis.
The result is a figure where we can visualize and compare protein similarity in latent space.
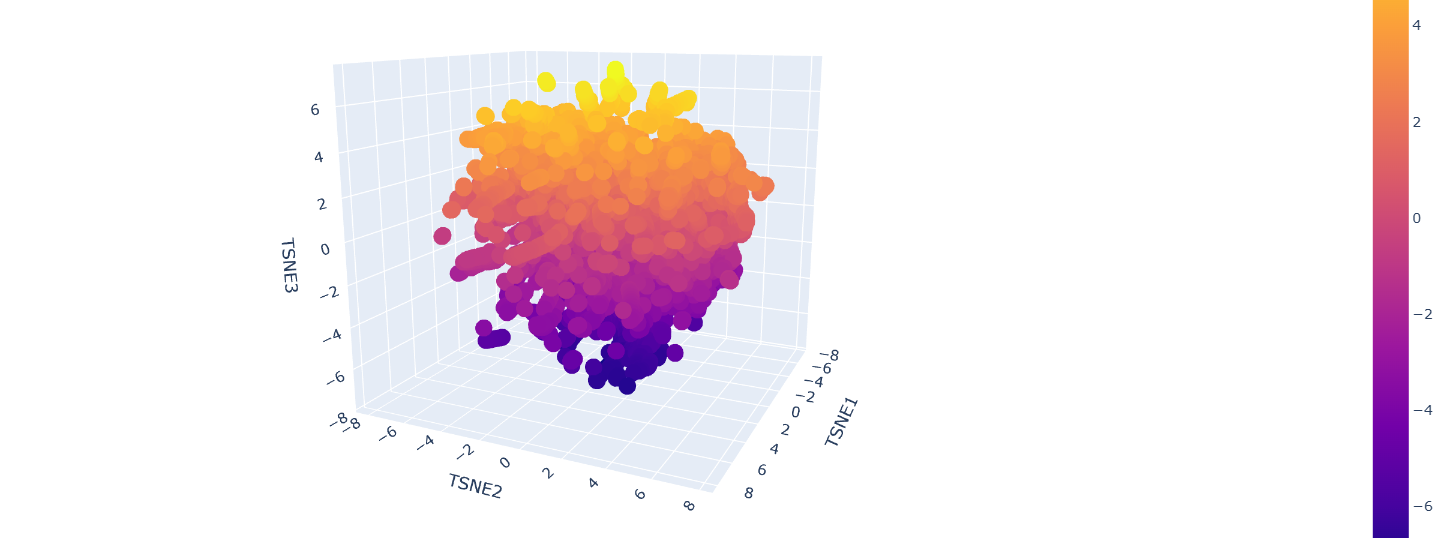
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
Inside the figure, we have three characteristics used to embed and compare the proteins.
TSNE1, TSNE2, and TSNE3, the colors are provided by the last one.
Yes, there are some clusters of proteins, especially at the top, where the overall set is larger.
At the bottom of the figure, there are a few clusters, but these clusters are more separate between them.
This performance suggests that at the top, there are proteins sharing features. In contrast, the smaller clusters at the bottom probably represent unique proteins or very different proteins.
For example, Beta-defensin, BD, and Phrixotoxin are similar proteins because they share some parts of the structure, even though their function is different.
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
The Dopamine Transporter (DAT) is at the top of the 3D latent space representation, clearly identifiable as a black dot.
We can see that it is not isolated and it is close to the central cluster. This suggests that it is not an atypical protein.
This expectation is based on the fact that DAT is a membrane protein, and these proteins are common in nature.
A closer inspection of its near proteins: Ionotropic glutamate receptor 2 (GluR2), Vacuolar ATP synthase subunit a (Saccharomyces cerevisiae), MurE (UDP-N-acetylmuramyl tripeptide synthetase), and Threonine deaminase (Escherichia coli). These proteins belong to different functional classes and organisms
This variety of proteins supports the hypothesis that, in latent space analysis, the position of DAT might indicate that it shares structural characteristics with other proteins, especially hydrophobic domains, and that their positions do not necessarily indicate functional similarity.
C2. Protein Folding
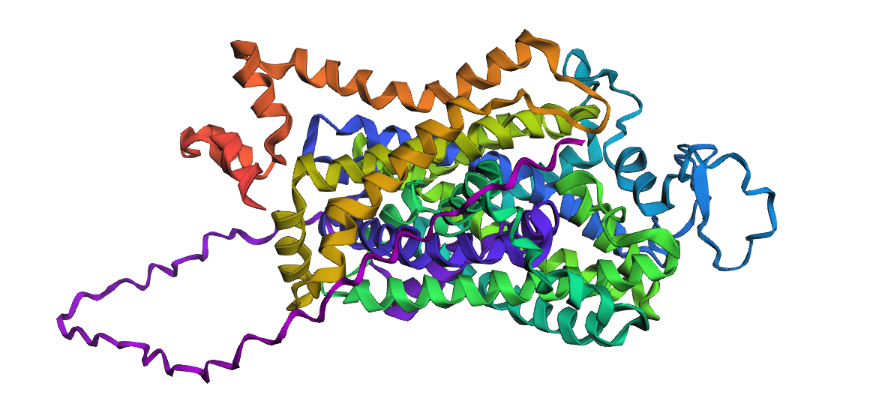
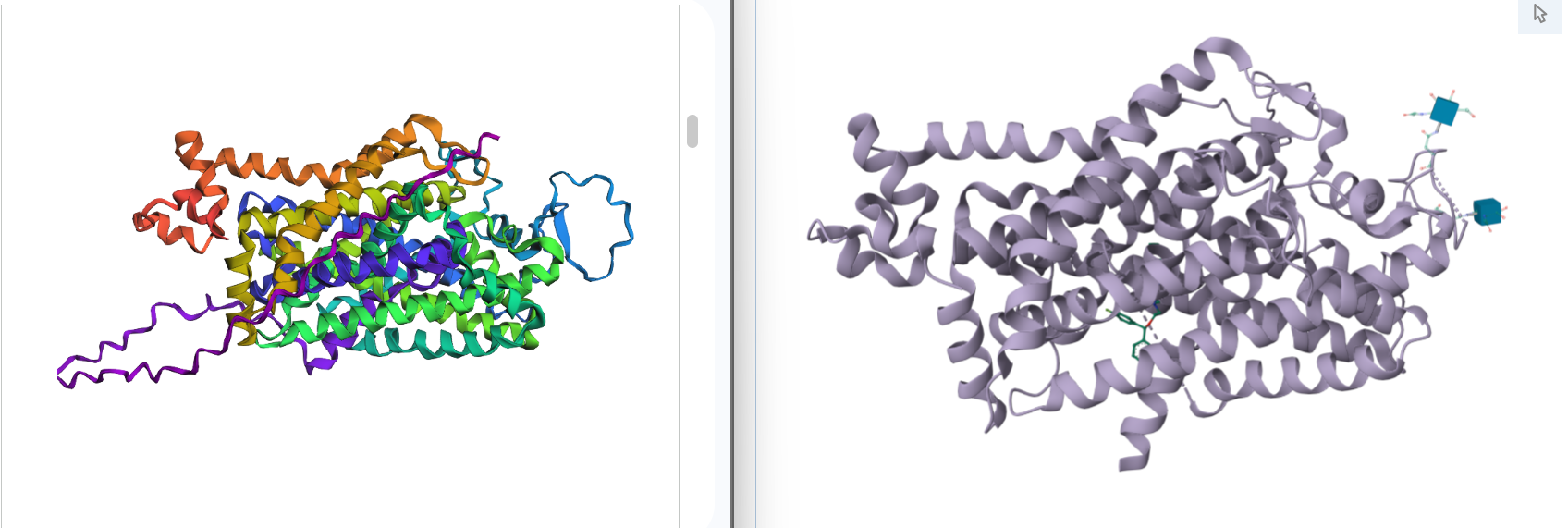
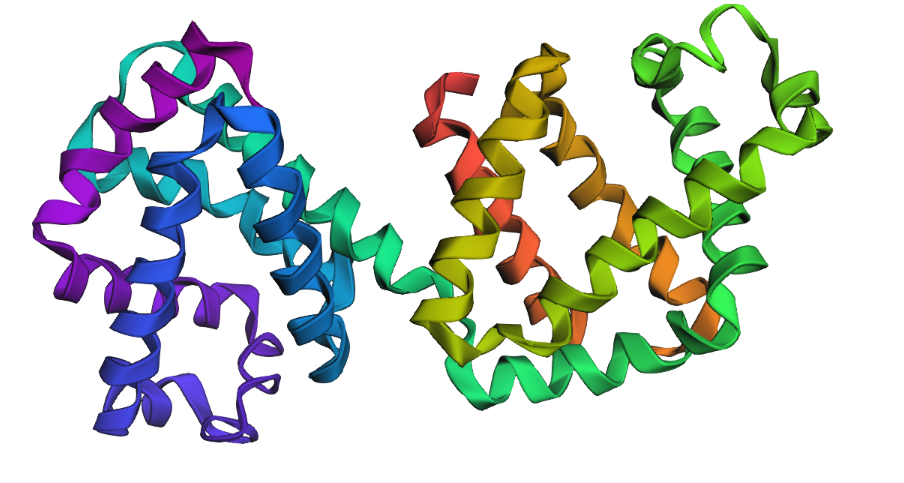
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Yes, you can see it in the image below that the helices match, and the general disposition coincides. Moreover, Protein Folding with ESMFold provides us with data that allows us to conclude that the structure obtained is accurate.
Structure obtained
Comparation
1. Total sequence length: 620 amino acids
2. Predicted Template Modeling (pTM): 0,905
Score estimating global fold accuracy, high confidence structures pTM > 0.7
3. Predicted Local Distance Difference Test (pLDDT): 91.395
Confidence score over all residues, high confidence structures pLDDT > 90
2. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
As it was determined using the mutation scan, there are some positions in the chain where modifications might result in unfavorable effects for the protein.
I try some mutations:
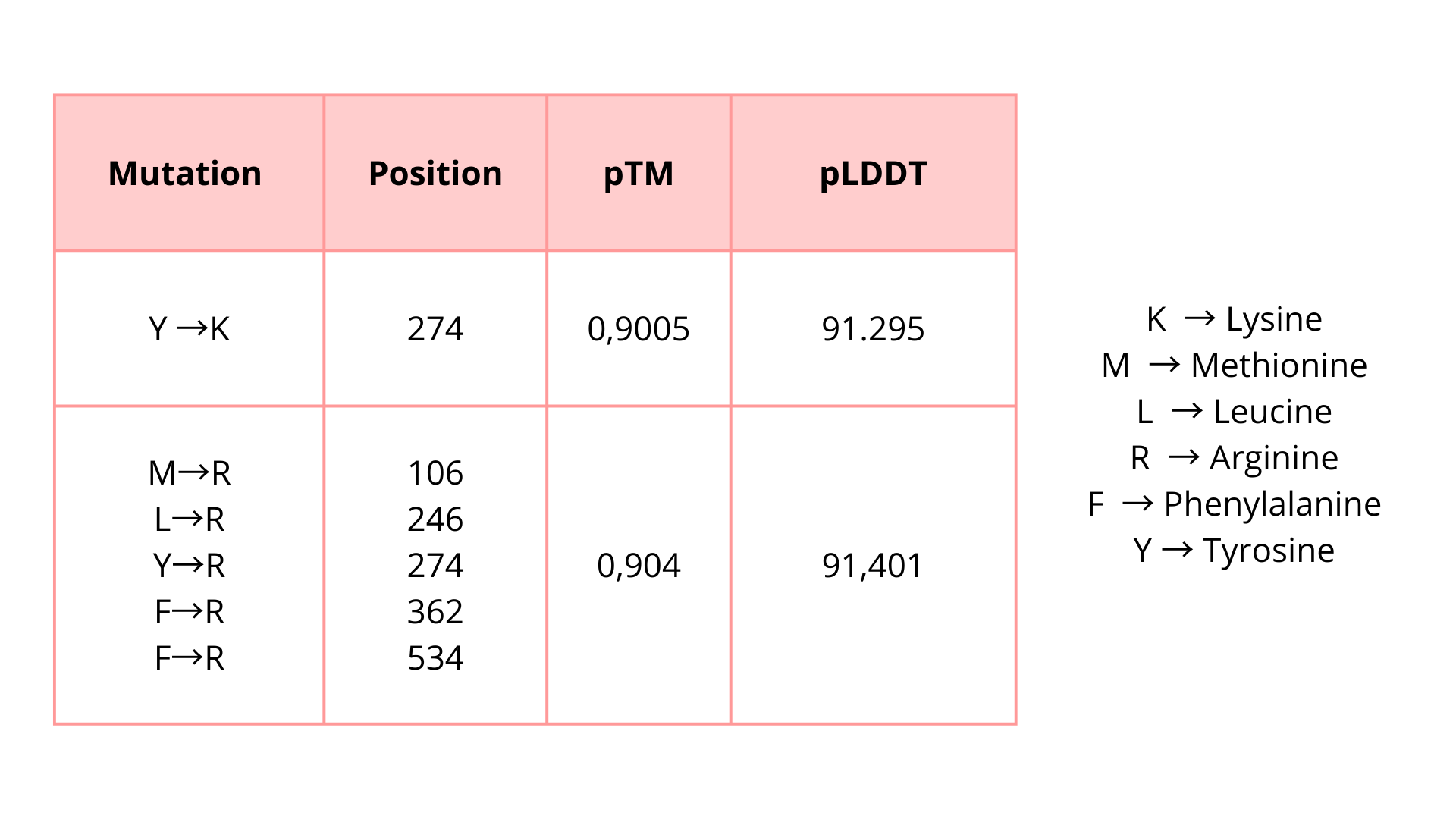
I introduced these mutations in critical zones to evaluate if these modifications will affect the protein function unfavorably. Based on the predicted pTM and pLDDT scores, the modified protein appears to maintain a high-confidence structural model. These results suggest that the protein may tolerate these substitutions without major structural disruption.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
SLSAAEADLAGKSWAPVFANKNANGLDFLVALFEKFPDSANFFADFKGKSVADIKASPKLRDVSSRIFTRLNEFVNNAANAGKMSAMLSQFAKEHVGFGVGSAQFENVRSMFPGFVASVAAPPAGADAAWTKLFGLIIDALKAAGAALTPEQAALLRAAAAPVFANREANGKAFLLALFAAHPALRELFPEFAGLSLAEIAASPKLGEVATAVFDGLRTLVATADDPAAMATLLAALAAAHVARGIGAAHFEAVRALHPAFVASVAPPPPGAAAAWDALFGDVIAALRAAGA
2. Input this sequence into ESMFold and compare the predicted structure to your original.
Part D. Group Brainstorm on Bacteriophage Engineering
1. Find a group of ~3–4 students
2. Read through the Phage Reading material listed under “Reading & Resources” below.
3. Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
4. Brainstorm Session
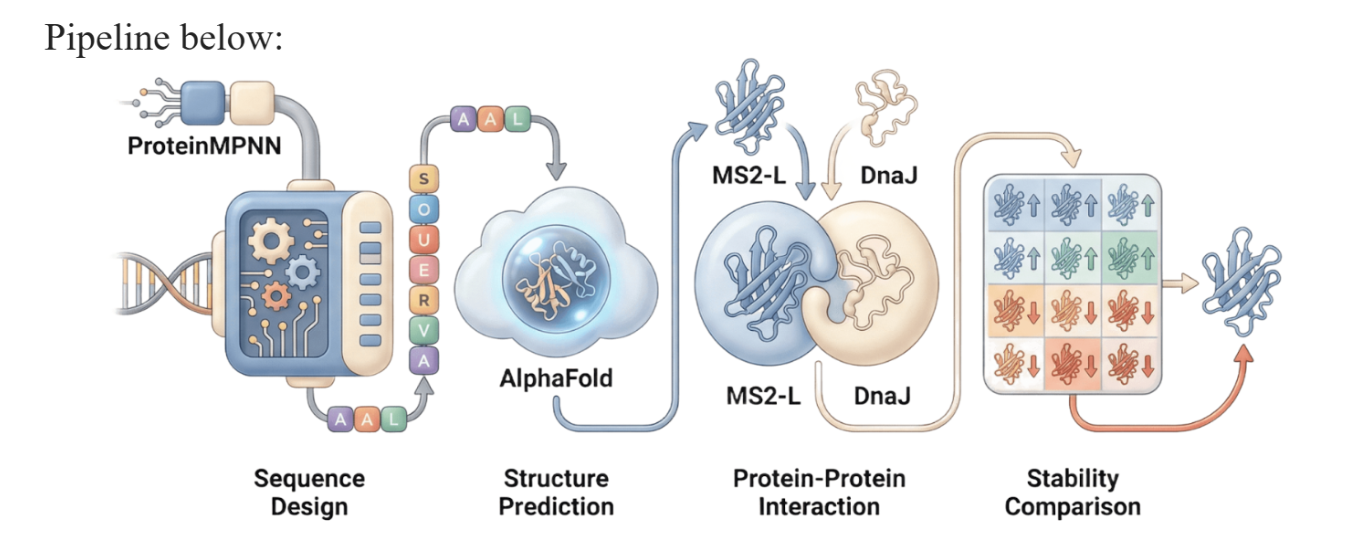
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
Why do you think those tools might help solve your chosen sub-problem?
5. Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
6. Include a schematic of your pipeline.
7. This resource may be useful: HTGAA Protein Engineering Tools
Each individually put your plan on your HTGAA website
Include your group’s short plan for engineering a bacteriophage
Names: Danna Betancourt, Rodrigo Arredondo, Valeria Q. Ortega, Jessica Wu
https://docs.google.com/document/d/1JUZVTdriMrHQLlgWFNaTYffs7yu_GVOmP1FvbnNvVl8/edit?tab=t.6qzjf868mf7r
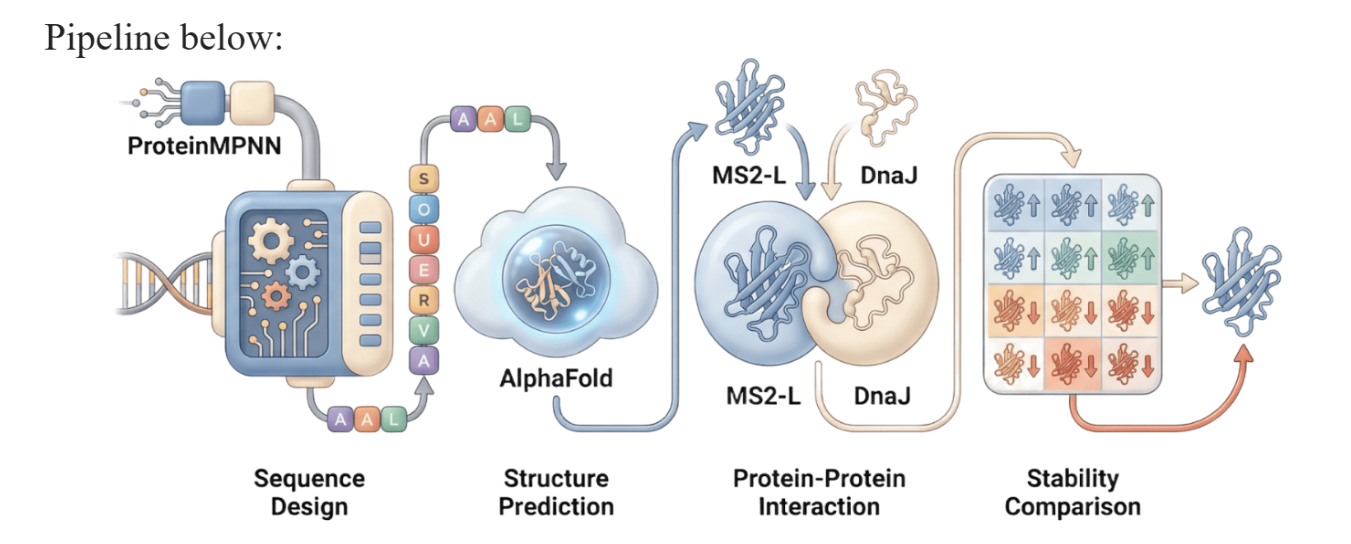
As discussed in “Phage Therapy: Past, Present and Future”, phage therapy represents an interesting alternative to antibiotic treatments, especially as recent developments allow researchers to engineer bacteriophages and their proteins. Our final group project for HTGAA Spring 2026 focuses on improving the bacteriophage MS2’s ability to kill its host bacteria E. coli by engineering its lysis protein MS2-L.
As an interdisciplinary team with different levels of experience in biotechnology, we propose increasing the stability of MS2-L. The lysis protein relies on the chaperone DnaJ for proper protein folding, a process E. coli can disrupt. However, it has been previously demonstrated that mutations deleting the N-terminal half of the MS2-L remove its dependence on DnaJ while also accelerating bacterial lysis. We believe this direction is promising for discovering variants that have structural stability within its host.
Our proposed approach begins with ProteinMPNN to look for alternative amino acid sequences that will improve the stability of MS2-L, then the sequences can be evaluated using AlphaFold and AlphaFold-Multimer to verify compatibility with their biological function and their interaction with DnaJ, with Alphafold specialized to model oligomeric complexes like MS2 and AlphaFold-Multimer tailored to predict protein-protein interactions like the one between MS2 and DnaJ.
Lastly, we must identify promising sequences for experimentation. We can do this by comparing variants quantitatively, e.g. using a deep mutational scan to see how each variant holds up when introduced to point mutations. This will narrow our candidate list to the most promising candidates for synthesis and experimental validation, reducing costs and promoting data-informed decision-making.
Any pitfalls are tied to the reliability of our tools; computational predictions of stability may not fully reflect protein behavior. For example, AlphaFold-Multimer has a systematic bias toward interactions between ordered protein regions, with a reduced accuracy for disordered regions and transient interactions such as those of a chaperone and its complex.
We are also held back by a narrow scope. Phage therapy depends on several biological variables beyond a single protein, and there is currently a lack of pharmacokinetic and pharmacodynamic studies on phage therapy. This means that we can make MS2-L more stable, but other factors could limit the effectiveness of the bacteriophage.
References
- Ajomiwe, Nneka, et al. “Protein Nutrition: Understanding Structure, Digestibility, and Bioavailability for Optimal Health.” Foods, vol. 13, no. 11, 1 Jan. 2024, p. 1771, www.mdpi.com/2304-8158/13/11/1771, https://doi.org/10.3390/foods13111771.
- Alila Medical Media. “Mechanism of Drug Addiction in the Brain, Animation.” YouTube, 11 Sept. 2014, www.youtube.com/watch?v=NxHNxmJv2bQ.
- “Amino Acids, Evolution| Learn Science at Scitable.” Nature.com, 2026, www.nature.com/scitable/topicpage/an-evolutionary-perspective-on-amino-acids-14568445/?error=server_error. Accessed 4 Mar. 2026.
- “Antiparallel and Parallel Beta Sheets.” Pearson.com, 2022, www.pearson.com/channels/biochemistry/learn/jason/protein-structure/antiparallel-and-parallel-beta-sheets.
- “Beta Sheet - an Overview | ScienceDirect Topics.” Www.sciencedirect.com, www.sciencedirect.com/topics/neuroscience/beta-sheet.
- Bu, Mengfei, et al. “Dynamic Control of the Dopamine Transporter in Neurotransmission and Homeostasis.” Npj Parkinson’s Disease, vol. 7, no. 1, 5 Mar. 2021, pp. 1–11, www.nature.com/articles/s41531-021-00161-2, https://doi.org/10.1038/s41531-021-00161-2.
- Cheng, Zhiming, et al. “Fluorescent Amino Acids as Versatile Building Blocks for Chemical Biology.” Nature Reviews Chemistry, vol. 4, no. 6, 13 May 2020, pp. 275–290, https://doi.org/10.1038/s41570-020-0186-z.
- Clemente-Suárez, Vicente Javier, et al. “Human Digestive Physiology and Evolutionary Diet: A Metabolomic Perspective on Carnivorous and Scavenger Adaptations.” Metabolites, vol. 15, no. 7, 4 July 2025, pp. 453–453, mdpi.com/2218-1989/15/7/453, https://doi.org/10.3390/metabo15070453.
- Data, Protein. “RCSB PDB - 8Y2F: Cryo-EM Structure of Human Dopamine Transporter in Complex with GBR12909.” Rcsb.org, 2024, www.rcsb.org/structure/8Y2F. Accessed 4 Mar. 2026.
- Emberly, Eldon G, et al. “Designability of α-Helical Proteins.” Proceedings of the National Academy of Sciences, vol. 99, no. 17, 12 Aug. 2002, pp. 11163–11168, https://doi.org/10.1073/pnas.162105999.
- “ESM Metagenomic Atlas | Meta AI.” Esmatlas.com, 2025, esmatlas.com/about.
- “ESMFold.” BioLM, 2023, biolm.ai/models/esmfold/. Accessed 4 Mar. 2026.
- Niesel, David. “Biomolecules Are Left or Right Handed.” Medical Discovery News (Mdnews), 8 Apr. 2025, www.utmb.edu/mdnews/podcast/episode/biomolecules-are-left-or-right-handed.
- Nowick, James S. “Exploring β-Sheet Structure and Interactions with Chemical Model Systems.” Accounts of Chemical Research, vol. 41, no. 10, 1 Oct. 2008, pp. 1319–1330, www.ncbi.nlm.nih.gov/pmc/articles/PMC2728010/, https://doi.org/10.1021/ar800064f.
- Parnas, M. Laura, and Roxanne Vaughan. “DAT, Dopamine Transporter.” XPharm: The Comprehensive Pharmacology Reference, 2007, pp. 1–10, www.sciencedirect.com/topics/medicine-and-dentistry/dopamine-transporter, https://doi.org/10.1016/b978-008055232-3.60441-6.
- Robinson, Scott W., et al. “Bioinformatics: Concepts, Methods, and Data.” Handbook of Pharmacogenomics and Stratified Medicine, 2014, pp. 259–287, https://doi.org/10.1016/b978-0-12-386882-4.00013-x.
- Uniprot.“UniProt.” UniProt, 2026, www.uniprot.org/blast/uniprotkb/ncbiblast-R20260301-002658-0868-42734055-p1m/overview. Accessed 4 Mar. 2026.
- Yip, Ka Man, et al. “Atomic-Resolution Protein Structure Determination by Cryo-EM.” Nature, vol. 587, 21 Oct. 2020, pp. 1–5, www.nature.com/articles/s41586-020-2833-4, https://doi.org/10.1038/s41586-020-2833-4.
- Zeppelin, Talia, et al. “Effect of Palmitoylation on the Dimer Formation of the Human Dopamine Transporter.” Scientific Reports, vol. 11, no. 1, 18 Feb. 2021, https://doi.org/10.1038/s41598-021-83374-y. Accessed 4 Mar. 2023.
- Zhu, J., and M. Reith. “Role of the Dopamine Transporter in the Action of Psychostimulants, Nicotine, and Other Drugs of Abuse.” CNS & Neurological Disorders - Drug Targets, vol. 7, no. 5, 1 Nov. 2008, pp. 393–409, https://doi.org/10.2174/187152708786927877.
Week 05 HW: Protein design part II

Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
1. Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
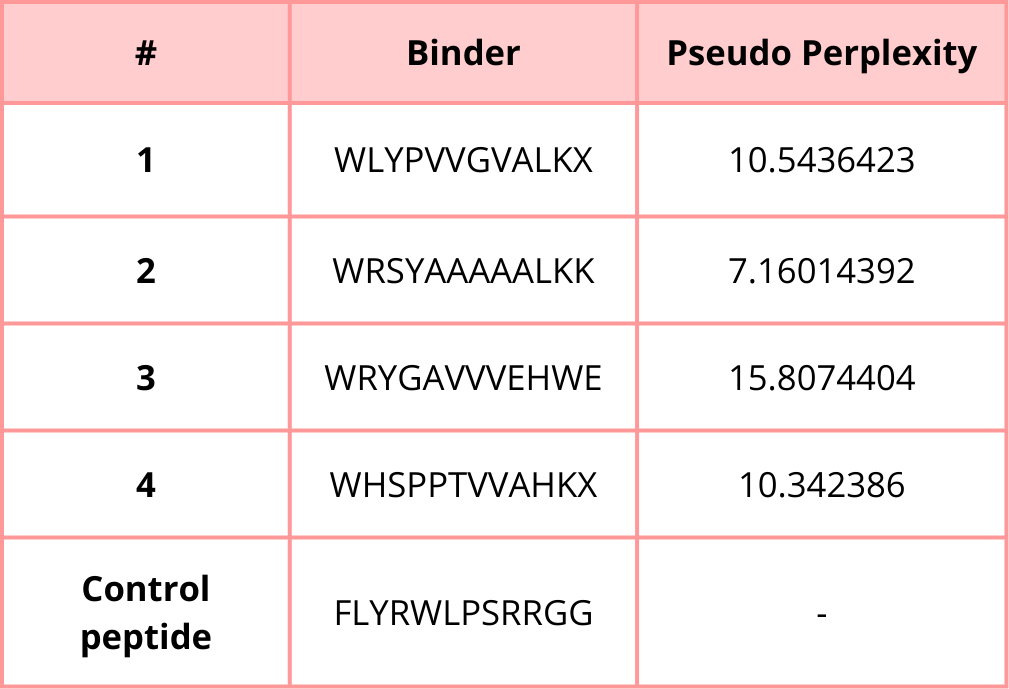
2. Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
3. Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
4. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
5. Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Part 2: Evaluate Binders with AlphaFold3
1. Navigate to the AlphaFold Server: alphafoldserver.com
2. For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
3. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
4. In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
Peptide 1
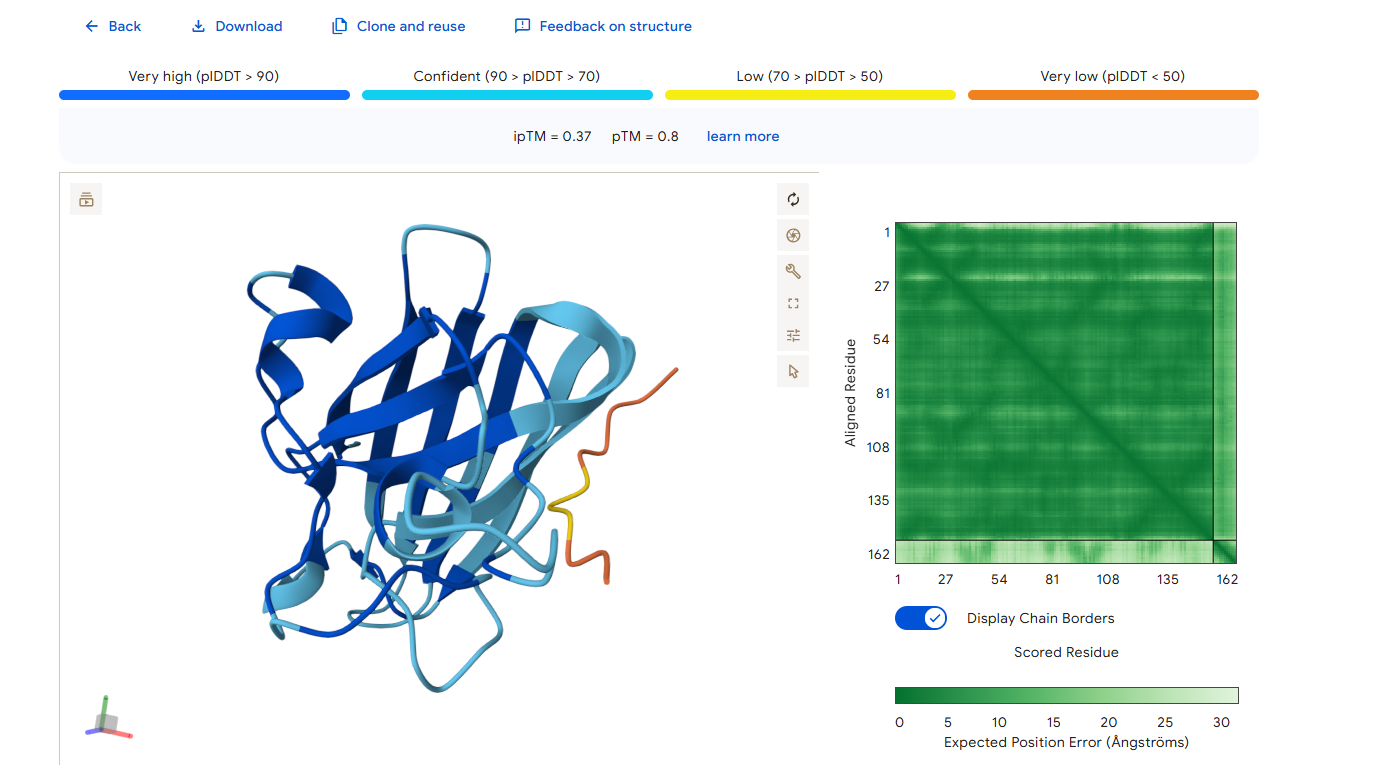
Peptide 2
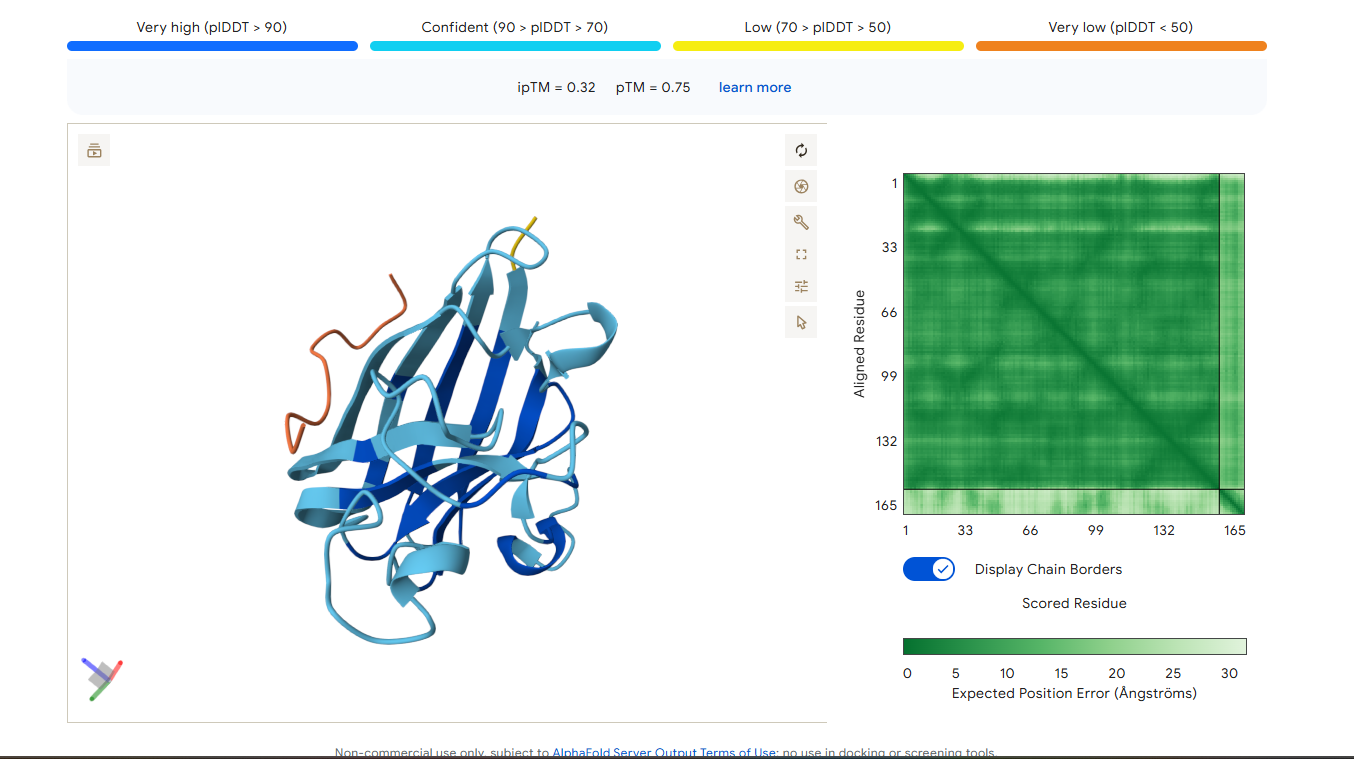
Peptide 3
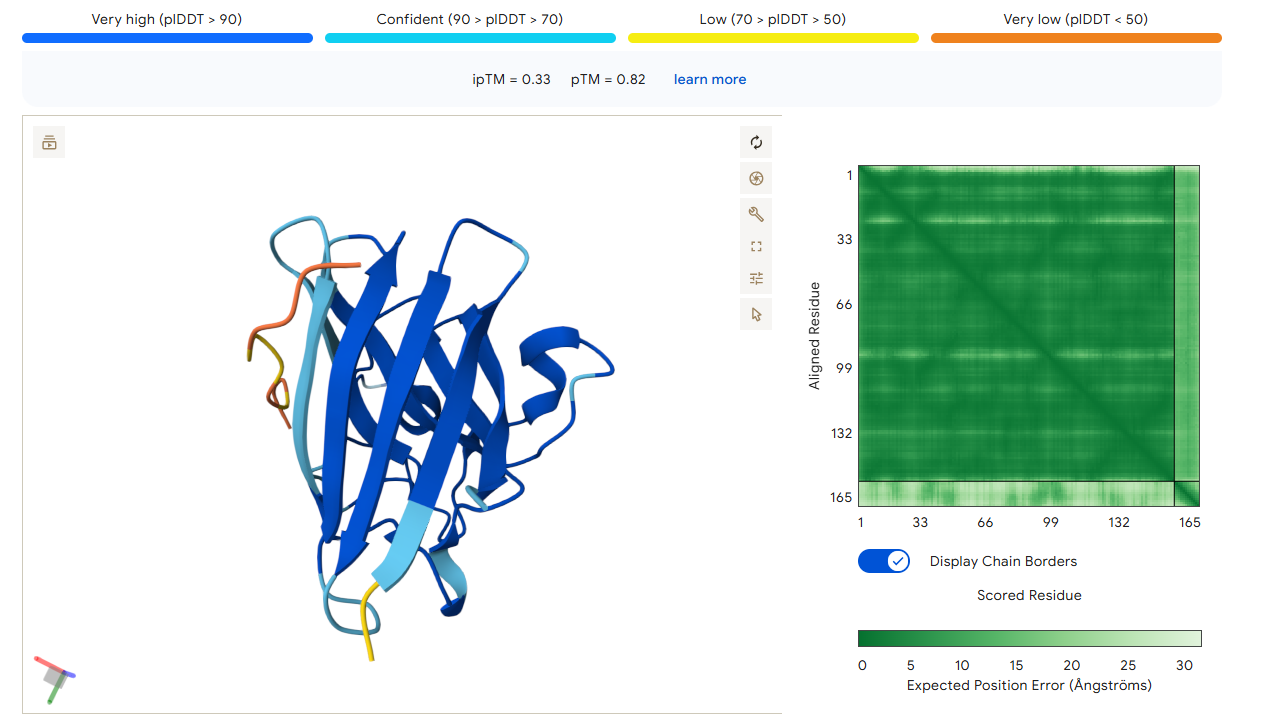
Peptide 4
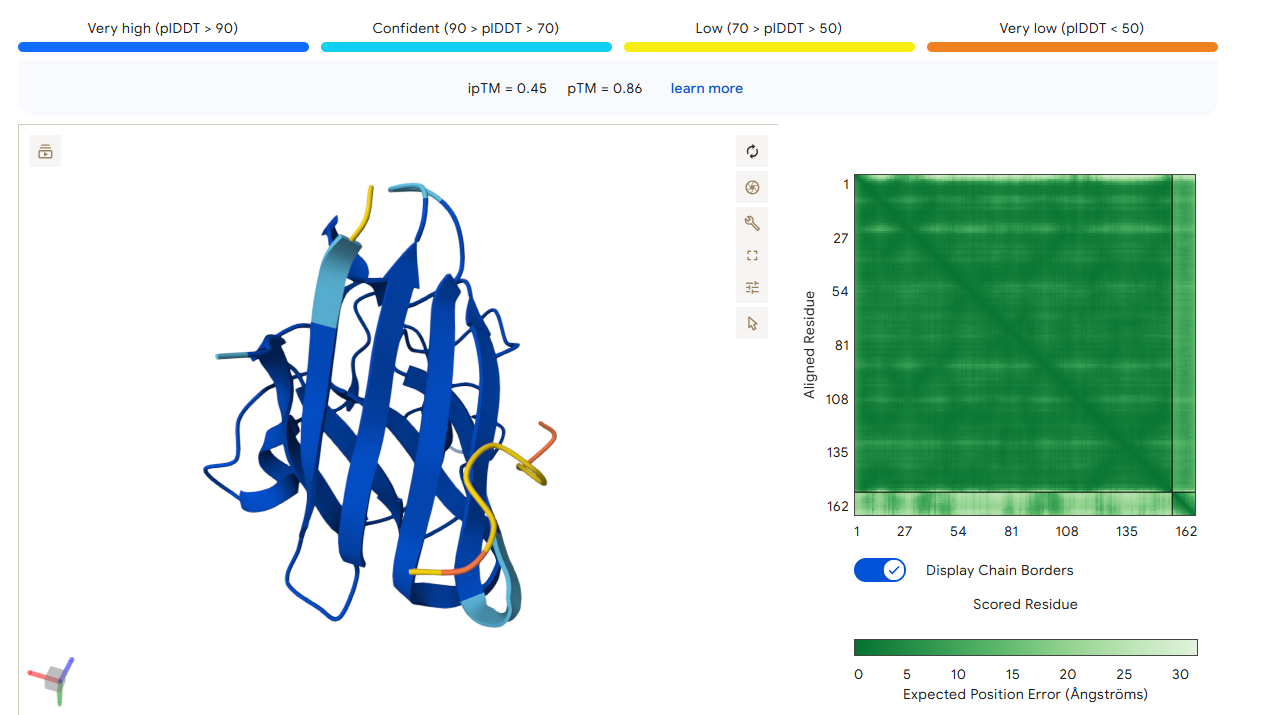
Control Peptide
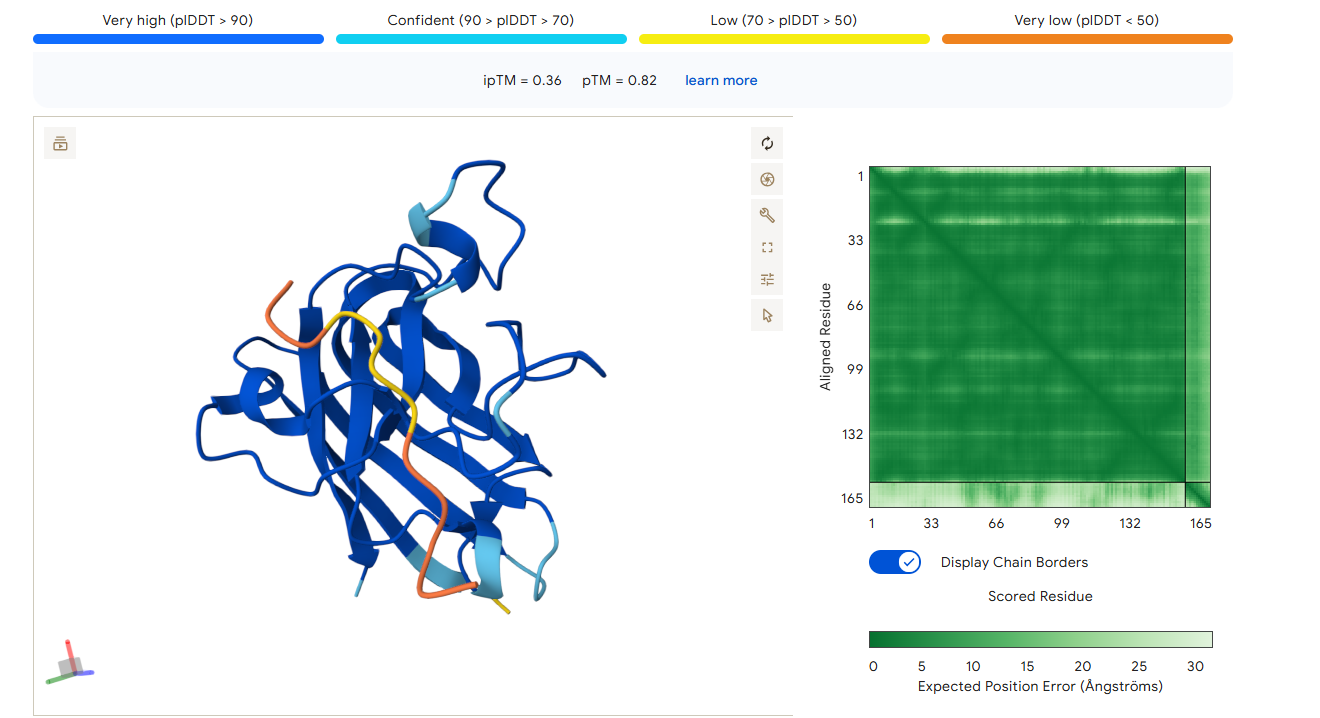
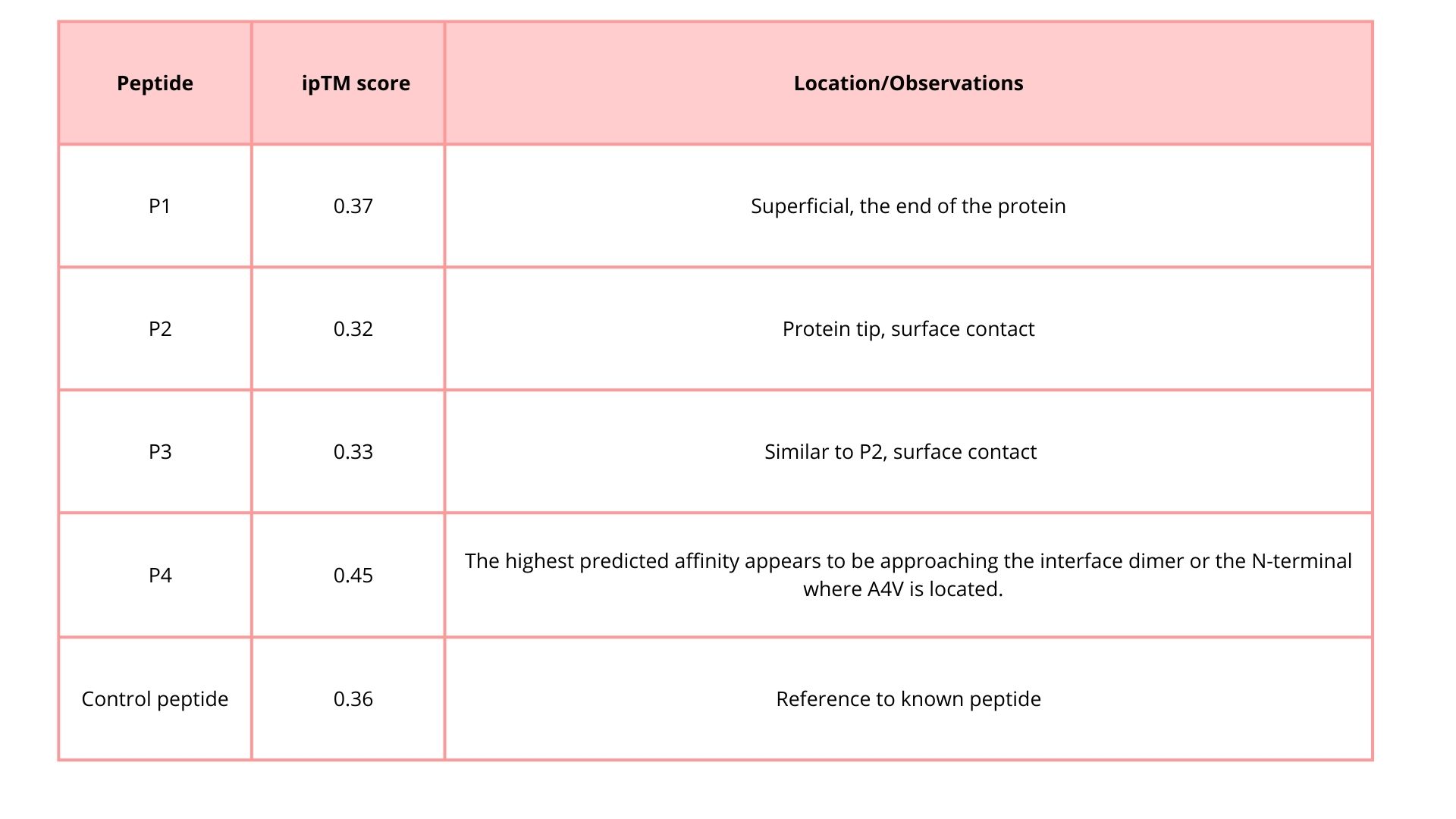
The ipTM score gives you an idea of the confidence in the interaction:
Values close to 1 → peptide binds in the predicted region.
Values close to 0 → low confidence, weak or doubtful interaction.
The ipTM score for peptide 2 is 0.32, indicating a relatively low affinity for the protein-peptide complex. Visually, the peptide is localized on the surface. This suggests that, although the peptide can bind to the protein, it is unlikely to directly influence protein stabilization.
The ipTM score for peptide 3 is 0.33, indicating a relatively low affinity for the protein-peptide complex. Visually, the peptide is localized on the surface. This suggests that, although the peptide can bind to the protein, it is unlikely to directly influence protein stabilization. The ipTM score of peptide 4 is 0.45, indicating a relatively low affinity, but higher than the other peptides, for the protein-peptide complex. It binds to the N-terminus, where the A4V mutation is located, or to the interface dimer, suggesting that it could interfere with SOD1 aggregation or stability.
The ipTM score of the control peptide is 0.36, indicating a relatively low affinity for the protein-peptide complex. Visually, the peptide is localized to the surface. This suggests that, although the peptide may bind to the protein, it is unlikely to directly influence protein stabilization.
In summary, among the four peptides, peptide 4 has the highest ipTM, which may be the best option.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
1. Paste the peptide sequence.
2. Paste the A4V mutant SOD1 sequence in the target field.
3. Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
Peptide 1

Peptide 2
Peptide 3
Peptide 4

Control Peptide
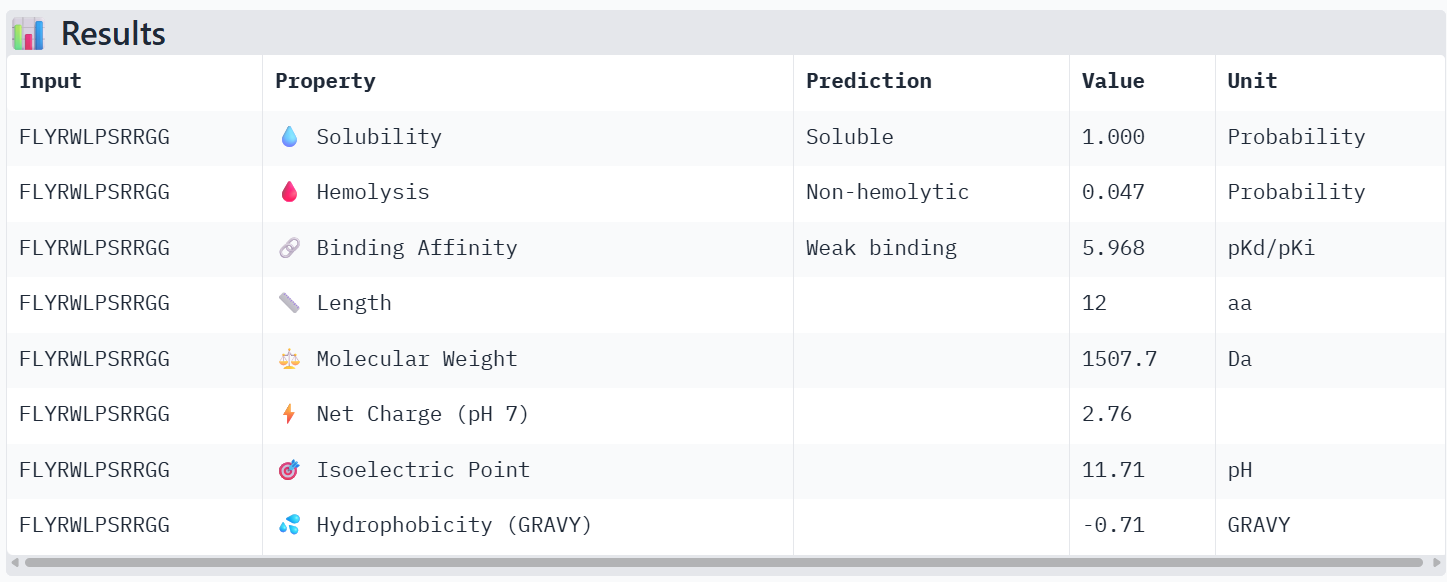
All the peptides show weak predicted binding affinity, non-hemolytic activity, and good solubility. The predicted pKd/pKi values are around ~6, which corresponds to weak to moderate binding on the logarithmic scale, where strong affinity interactions are typically associated with values ≥ 9. Peptide 4 shows slightly lower predicted binding affinity than the control peptide according to PeptiVerse, despite having the highest ipTM score in the AlphaFold structural prediction.
However, I still chose peptide 4 because, in AlphaFold, structural prediction has the best ipTM value, even though it is not the greatest. Moreover, in PeptiVerse the value is not high but either low, or it is the peptide with the lowest value of probability of hemolysis.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
1. Open the moPPit Colab linked from the HuggingFace moPPIt model card
2. Make a copy and switch to a GPU runtime.
3. In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
4. After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
In moPPIt, motif guidance was applied by specifying position 4 within the peptide sequence to encourage residues that may promote interaction with the target region. Additional objectives including affinity, solubility, and hemolysis were enabled to balance binding with therapeutic properties.
The peptides generated with PepMLM and moPPIt differ mainly in their design strategy and optimization objectives. PepMLM peptides are generated through sequence sampling conditioned on the target protein sequence. The model predicts plausible peptide binders but does not explicitly optimize specific therapeutic properties.
In contrast, moPPIt peptides design peptides that bind to specific residues on the target protein while simultaneously optimizing therapeutic properties. This approach produces peptides that are optimized for desirable biochemical and therapeutic characteristics.
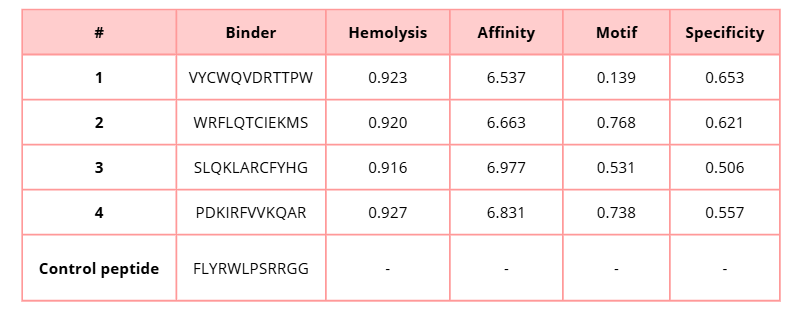
In this table, lower hemolysis values are preferable because they indicate a lower predicted risk of lysis. On the other hand, higher values for affinity, motif, and specificity are desirable, since they suggest stronger binding to the target protein, better motif compatibility, and greater binding specificity.
SLQKLARCFYHG shows the highest predicted affinity (6.977), suggesting stronger potential binding to the mutant SOD1 protein. However, PDKIRFVVKQAR presents a balanced profile with high affinity (6.831), strong motif score (0.738), and relatively good specificity (0.557), which may indicate a favorable interaction with the targeted binding site.
Similarly, WRFLQTCIEKMS also demonstrates good performance with high affinity (6.663) and the highest motif score (0.768) among the generated peptides, suggesting strong compatibility with the selected binding motif.
I would evaluate these peptides using:
- Computational validation
- In vitro binding assays
- Toxicity and hemolysis testing
Part C: Final Project: L-Protein Mutants
High-level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
L-Protein Engineering | Option 1: Mutagenesis
1. Designing these mutants with good computational confidence is hard. It will show you limitations of some of the structure based models. Ultimately, you can pick various combinations of mutations and get lab results and then decide to pick the next round of mutations, but this assay will not be easy to run at scale in this class.
2. Run this notebook to generate for each position in the amino acid sequence, a “score” for what would happen to the protein if you mutated into another amino acid. It can be positive or negative for the protein. We want to identify possible mutations that are “positive” If you run this notebook - you will see a .csv file in the sidebar. You can download it and look at it in the google sheets if that’s easier
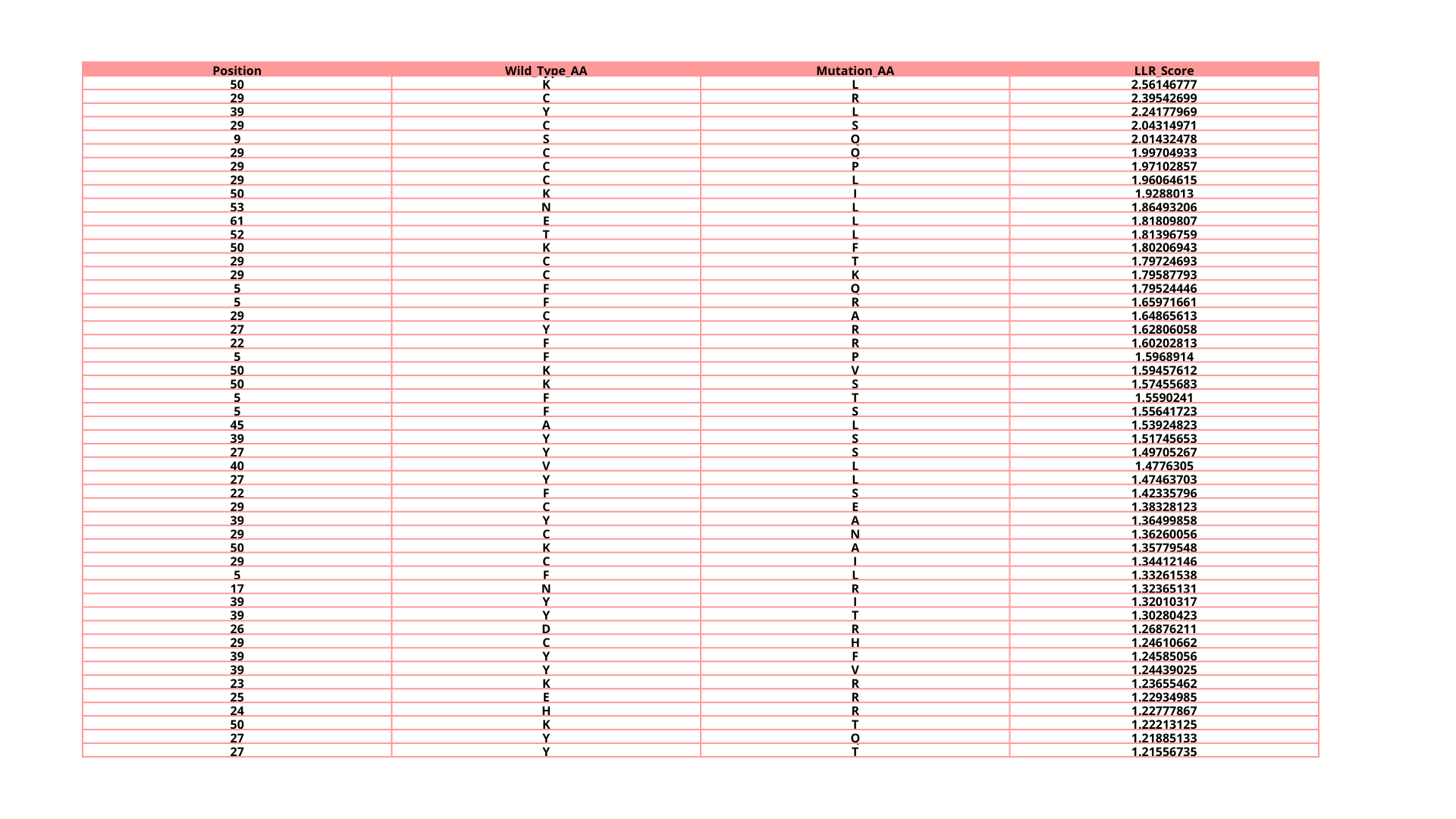
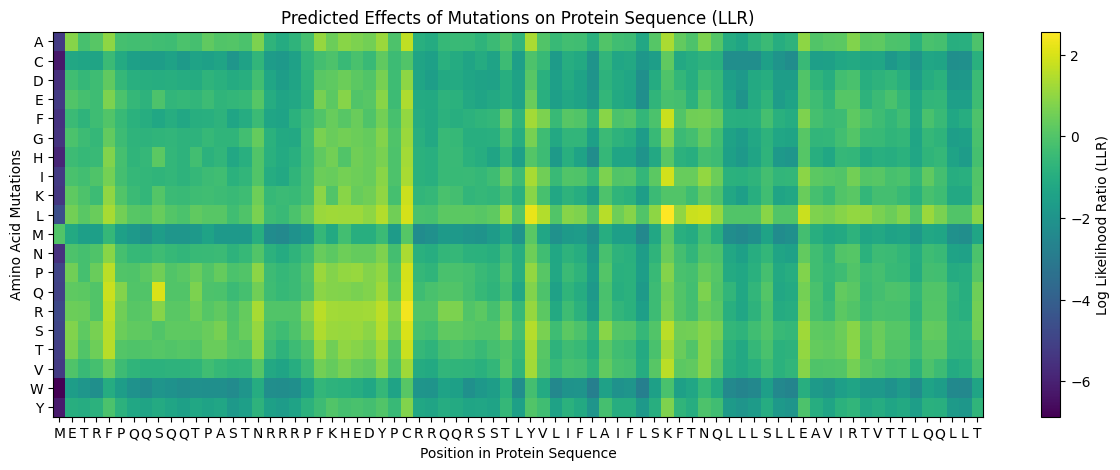
3. Use the experimental data here. This dataset contains information about mutants of the L-Protein and their effect on lysis in the lab - L-Protein Mutants
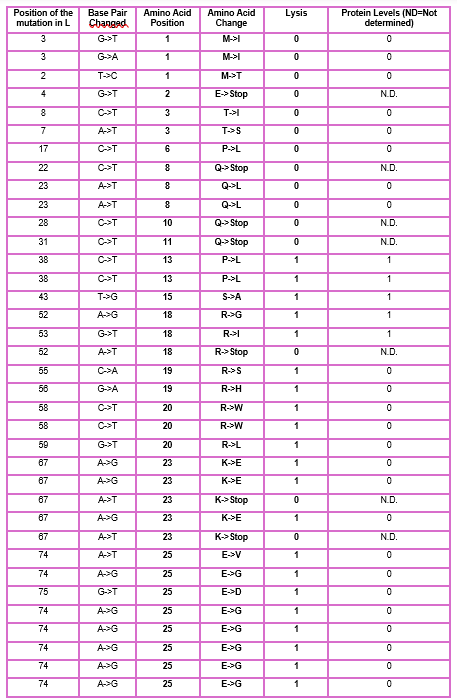
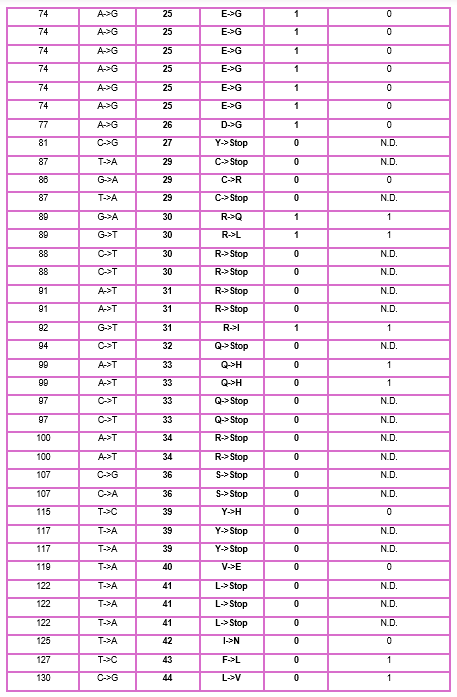
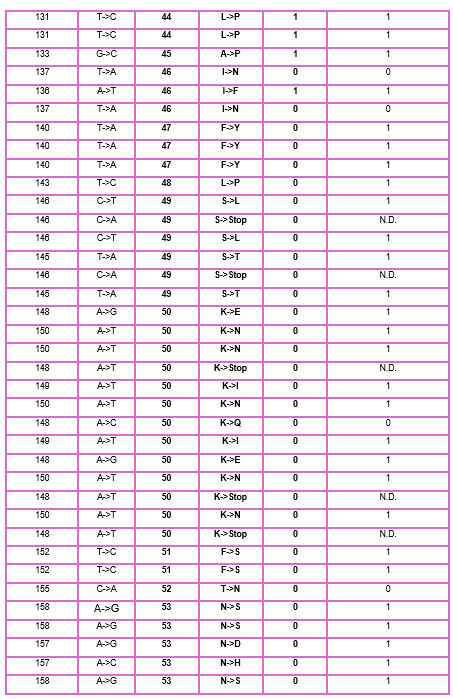
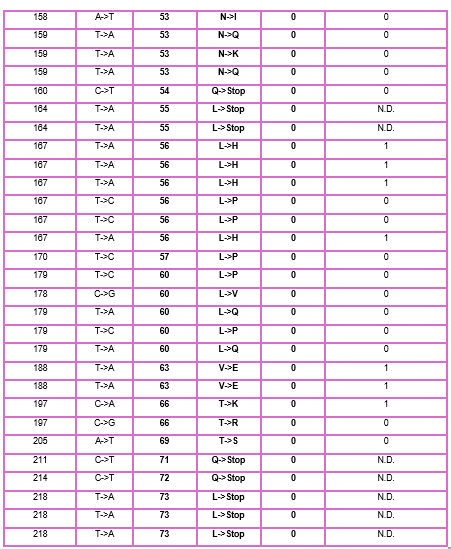
4. First check, does the experimental data correlate with the scores from the notebook in (b)? This should give you a clue on how well these language embeddings capture information about this protein sequence
When I compared the experimental data with the theoretical scores obtained from the Colab notebook, I observed that there is not a perfect correlation but there is a partial one, which means that I cannot find the same specific mutation in the experimental data; however, I found that in some positions, the change for other amino acids might be favorable to increase the Lysis protein activity indicating that the language embeddings capture some relevant structural and functional information about this protein.
5. Using information about the effect of protein mutations at these sites - both the scores and the experimental data in the drive, come up with 5 mutations for each student along with how you came up with them and why you believe they would work. 2 of the variants you submit must have mutations in the transmembrane region (refer to notes above on what amino acid positions these are) and 2 of them must be in the soluble region . Remember that you can also use the pBLAST to see which residues are conserved and not mutate them if you want to.
One easy way to generate sequence mutations could be to look for residue positions and mutations that have a positive mutational effect either in the experimental or have a positive score from step 1. And pick a combination of those mutations.
I decided to choose these mutations because they have lysis protein activity of 1 in experimental data and in the Colab notebook; their score is positive, which means that the mutations are favorable.
Transmembrane mutations (A45L and A45V): could influence the protein’s ability to insert into and form pores in the membrane.
Soluble DnaJ domain mutations (D26R, K23R, and E25R): enhance the protein’s stability or interactions without disrupting its overall structure.
These mutations were selected based on a combination of experimental evidence, computational predictions, and consideration of their location within functional regions of the protein.
6. You can utilize Af2_Multimer to generate a Multimeric Assembly; you can do this by making your query sequence as. We want to do this because - A running hypothesis for how this protein functions is that it assembles to make a perforation in the bacterial membrane.
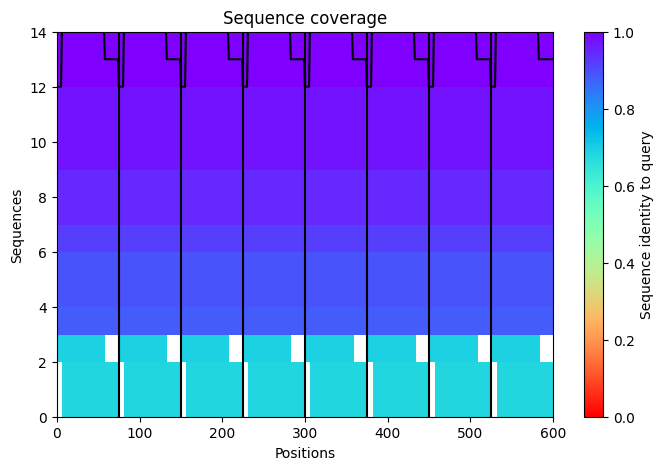
Week 06 HW: Genetic circuits part I: Assembly Technologies

DNA Assembly
Answer these questions about the protocol in this week’s lab:
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix is a mix that has several components that allow the user to add only the DNA template, primers, and water to perform PCR.
Advantages:
- Robust Reactions - Maximal success with minimal optimization.
- Extreme Fidelity - > 50X greater than Taq
- High Speed - Dramatically reduced extension times (10X faster than Pfu)
- High Yield - Increased product yield using minimal amount of enzyme.
- Versatile - Can be used for routine per as well as long or difficult templates.

2. What are some factors that determine primer annealing temperature during PCR?
PCR has three important steps:
1. Denaturation (95 °C) → the DNA separates into two strands.
2. Annealing (50–65 °C) → the primers attach to the DNA.
3. Extension (~72 °C) → the polymerase copies the DNA.
The annealing temperature is the temperature adequate for the primer to bind to the DNA.
If the temperature is not adequate, the primer might bind in the wrong place or not bind to the DNA.
Factors that determine the annealing temperature
1. Melting temperature (Tm)
The Tm is the temperature at which half of the primers are attached to the DNA and the other half are not.
If the Tm is high, you need a higher annealing temperature.
2. Content of GC
The DNA bases are bound by hydrogen bonds
A–T → 2 bonds (weak)
G–C → 3 bonds (strong)
Primers with more GC will have a Tm higher
Primers with fewer GC will have a Tm lower
3. Primer length
Long primer → Stronger bond → Higher Tm
Short primer → weakest bond → lowest Tm
4. Salt Concentration (Na⁺ or Mg²⁺)
Salts stabilize the interaction between DNA and primer.
More salt → the negative charges of the DNA are neutralized → the primer binds more easily.
More salt → increases Tm
Less salt → decreases Tm
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
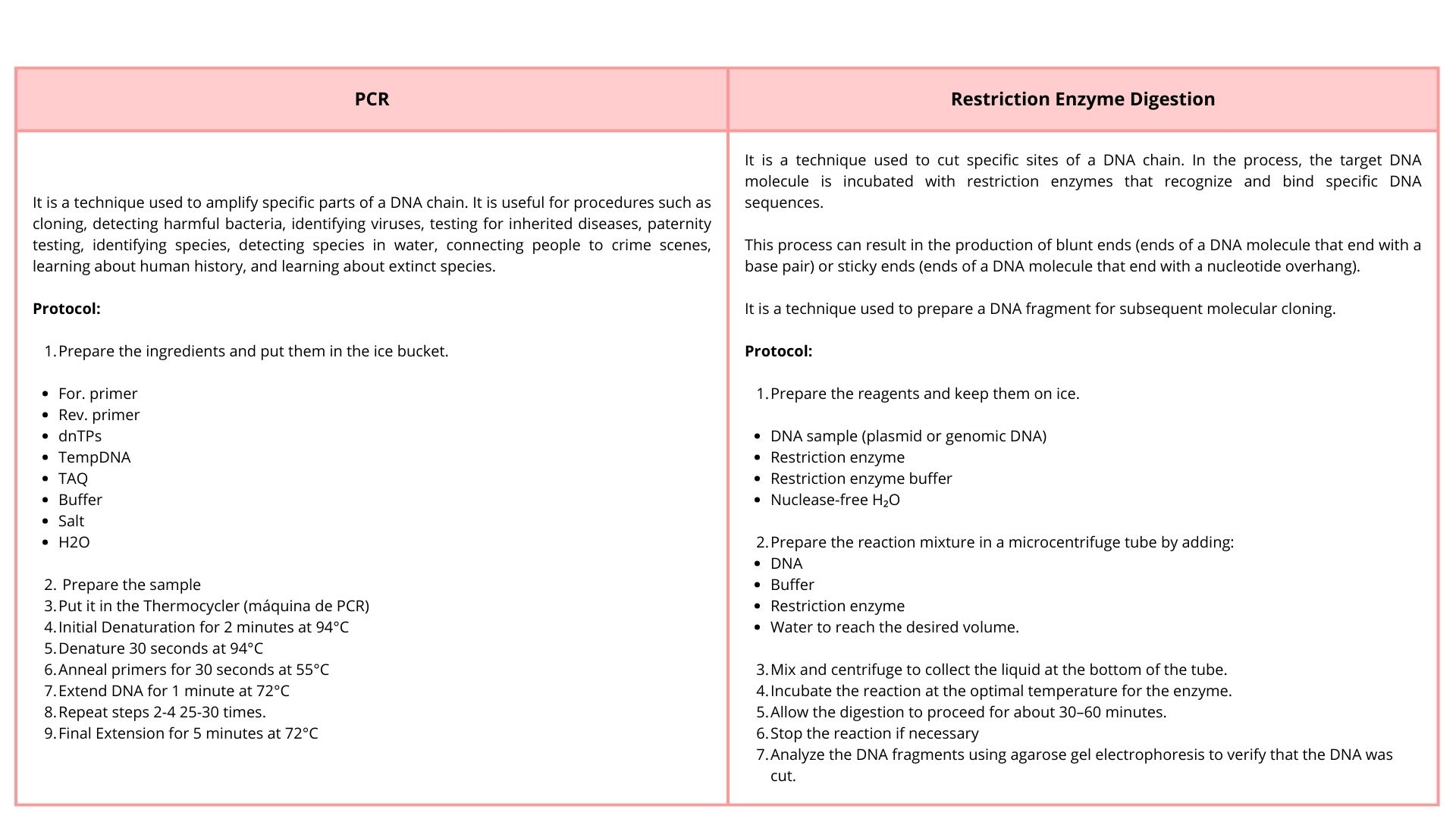
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Gibson cloning is a technique used to assemble multiple linear DNA fragments, since this technique does not require specific restriction sites and leaves no scar between joined fragments.
Researchers must ensure that the DNA fragments that they want to join are compatible, because adjacent segments should have identical sequences at the ends, approximately 30 bp that match the ends of adjacent fragments
5. How does the plasmid DNA enter the E. coli cells during transformation?
The process is known as bacterial transformation. In this technique, scientists use cells that have been treated with calcium chloride to allow plasmid DNA to attach to the cell membrane. These cells are called competent cells. A brief heat shock then creates temporary pores in the membrane, allowing the plasmid DNA to enter the cell. Another method is electroporation, which uses an electrical field to increase the permeability of the cell membrane, allowing plasmids to enter the cell. After transformation, the bacteria can be cultivated on agar plates.
6. Describe another assembly method in detail (such as Golden Gate Assembly)
- Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
- Model this assembly method with Benchling or Asimov Kernel!
Golden Gate Assembly is a molecular cloning method that allows the assembly of multiple DNA fragments in a single reaction. This method uses Type IIS restriction enzyme digestion, which cuts DNA outside of its recognition sequence, generating specific overhangs. These overhangs are designed with the objective that adjacent DNA fragments have complementary ends that can join in the correct order. DNA ligase joins the fragments to generate a single DNA molecule.
The principal advantage of this method is that the correct order of DNA fragments is ensured by designing specific complementary overhangs that only allow adjacent fragments to ligate together.
My assembly:
Assignment: Asimov Kernel
1. Create a Repository for your work
2. Create a blank Notebook entry to document the homework and save it to that Repository
3. Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
4. Create a blank Construct and save it to your Repository
5. Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
6. Search the parts using the Search function in the right menu
7. Drag and drop the parts into the Construct
8. Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
10. Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
11. Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
12. Explain in the Notebook Entry how you think each of the Constructs should function
13. Run the simulator and share your results in the Notebook Entry. If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
The repressilator consists of three transcription units arranged in a cyclic inhibitory network. Each gene produces a repressor that inhibits the next gene in the circuit. Simulation results show oscillatory behavior in protein concentrations over time, confirming the expected dynamic behavior of the repressilator.
Original construct
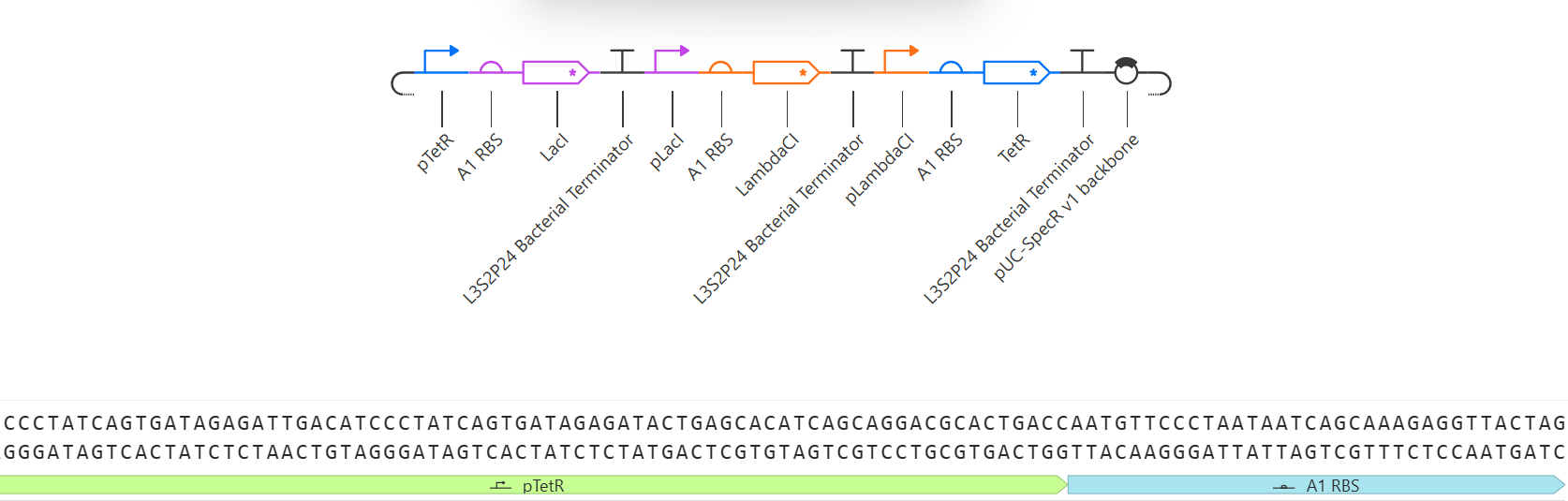
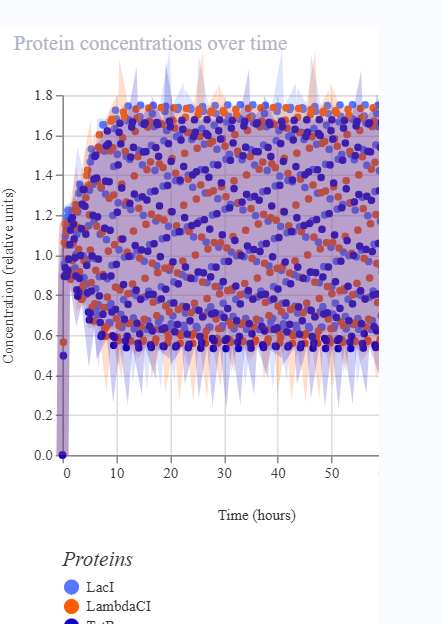
My copy
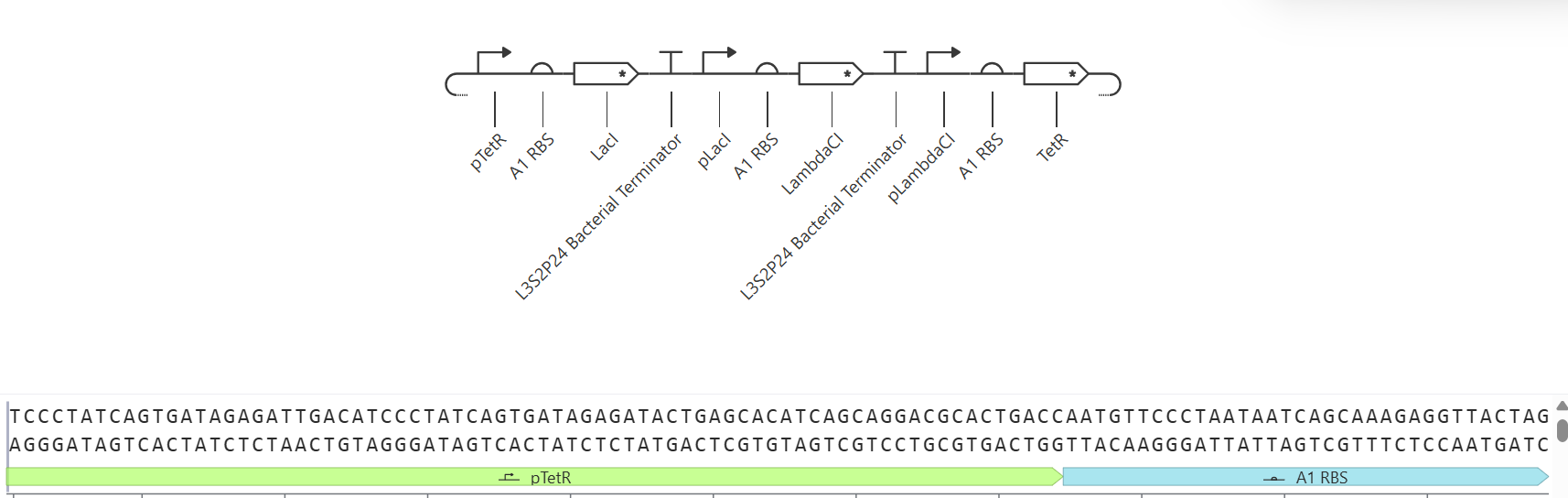
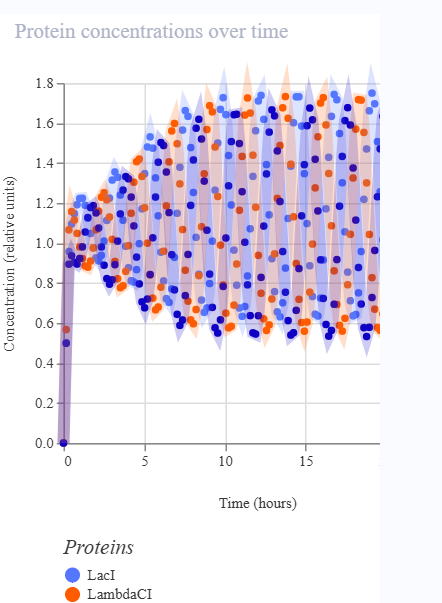
1. Construct A118G Prototype
- Activatable Promoter: Represents the region that detects the A118G polymorphism of the MPR1 gene.
- CDS Placeholder (LambdaCl): Simulates the reporter protein (equivalent to GFP) that indicates the presence of the SNP.
- Terminator: Terminates the transcription of the construct.
- When the A118G sequence is present, the activatable promoter is activated.
- This activation triggers the expression of the CDS placeholder (LambdaCl).
- The terminator ensures that transcription is completed correctly.
- The simulation illustrates the dynamics of the reporter protein as a function of promoter activation.
- The line representing LambdaCl rises when the promoter is activated, indicating the presence of the A118G SNP.
- If the promoter is not activated (SNP absent), the line remains low or at zero.
- There are no oscillations like those seen in the repressilator; the signal functions as an ON/OFF switch, reflecting the detection of the SNP.
- The construct effectively simulates a genetic biosensor: the presence of the A118G SNP translates into an observable signal (a rise in LambdaCl).
- It serves to visualize and document the biosensor's operation within the simulator, even though it does not utilize the actual GFP protein.
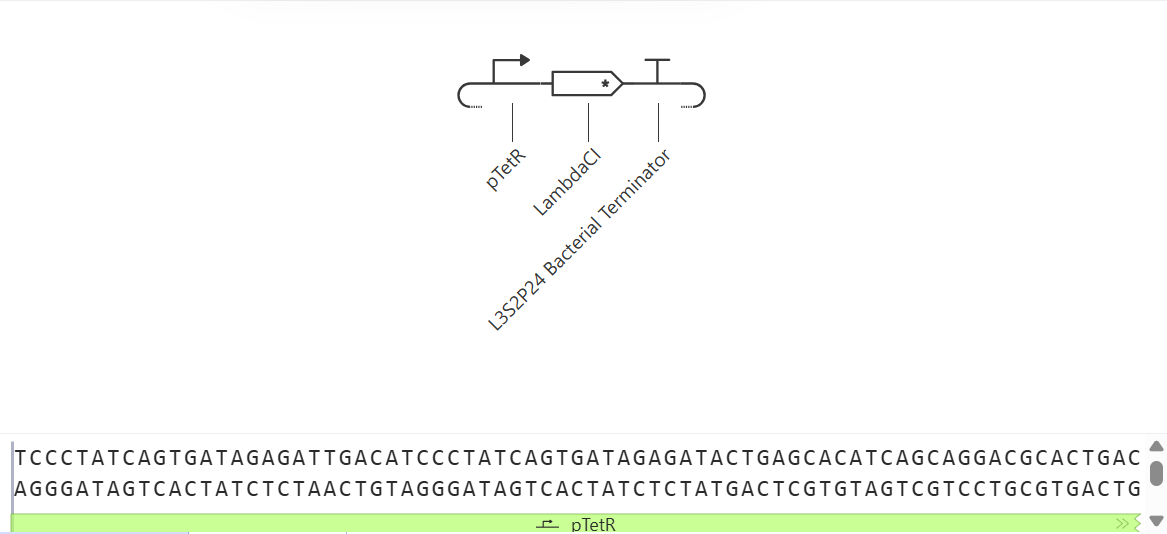
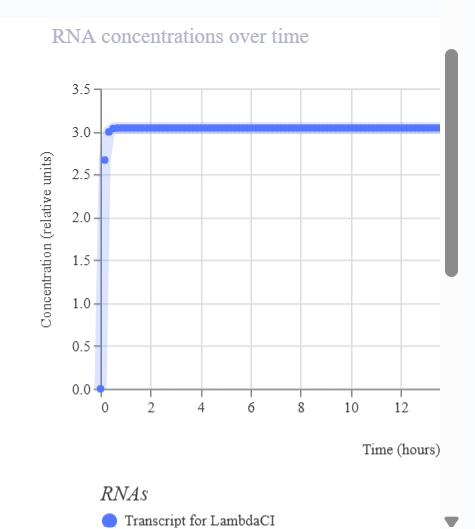
2. Biosensor A118G Amplification
- Activatable promoter: Represents the region that detects the A118G polymorphism of the MPR1 gene.
- TetR CDS placeholder: Functions as an intermediate repressor to simulate signal amplification within the biosensor.
- Reporter promoter: Controlled by TetR, it regulates the expression of the reporter.
- LambdaCl CDS placeholder: Simulates the reporter protein (equivalent to GFP).
- Terminator: Terminates the transcription of the construct.
- When the A118G sequence is present, the activatable promoter becomes active.
- This produces TetR, which regulates the reporter promoter.
- The regulation of the reporter (LambdaCl) simulates signal amplification, increasing the reporter's expression when the SNP is present.
- The terminator ensures that transcription is completed correctly.
- LambdaCl levels rise when the activatable promoter detects the SNP.
- The signal follows an ON/OFF pattern, representing the activation of the biosensor.
- The construct simulates signal amplification logic using an intermediate repressor.
- Although the graph does not differ significantly from that of the simple construct, the circuit illustrates how the signal could be amplified in a real-world design.
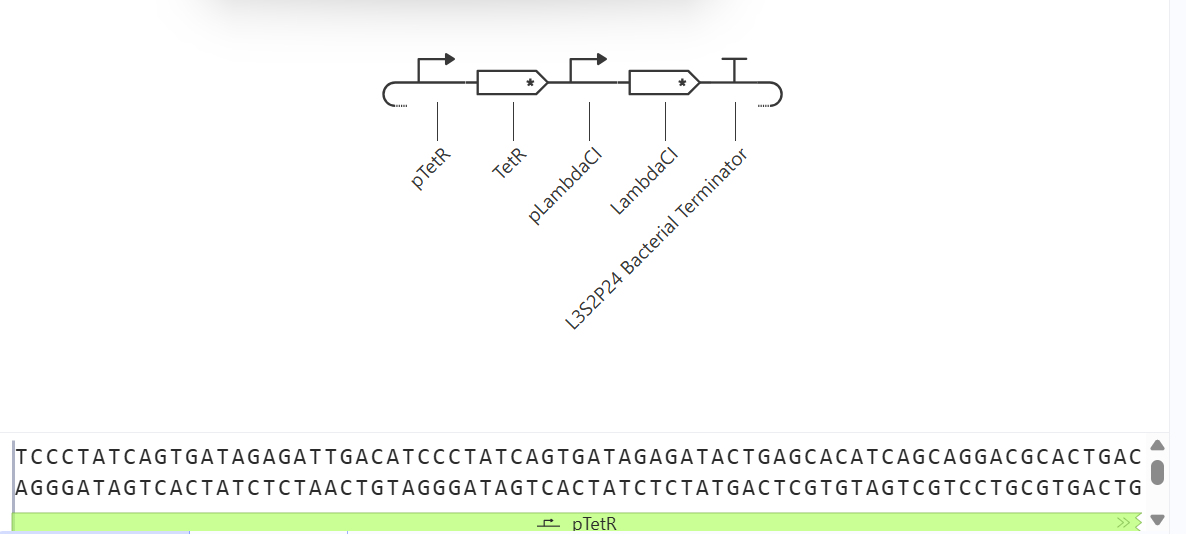
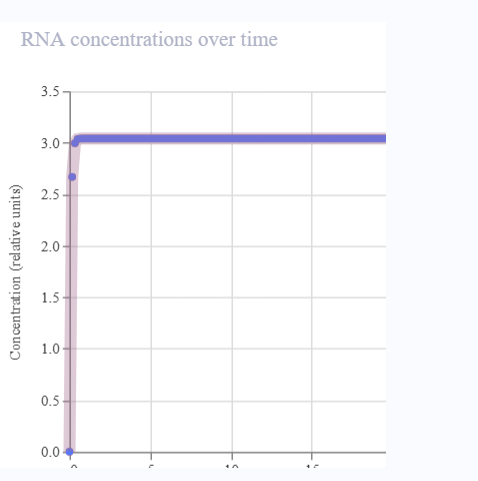
3. A118G AND-type Biosensor
- Activatable Promoter 1: Represents the detection of the A118G polymorphism of the MPR1 gene.
- Activatable Promoter 2: Represents an additional input signal (e.g., another marker or condition).
- LambdaCl CDS Placeholder: Simulates the reporter protein (equivalent to GFP).
- Terminator: Terminates the transcription of the construct.
- Expression of the reporter (LambdaCl) occurs only if both promoters are active.
- This simulates AND logic; both conditions must be met for a signal to be generated.
- The terminator ensures that transcription is completed correctly.
- LambdaCl levels rise only when both promoters are activated simultaneously.
- If only one is activated → the line remains low → no signal is generated.
- It functions as a highly specific switch: ON only when both conditions are met.
- This construct simulates a more specific genetic biosensor, capable of filtering out false signals.
- It allows for the demonstration of AND logic within Asimov, even though the promoters and CDS are placeholders.
- It is useful for illustrating how a more robust biosensor could be designed to detect the A118G SNP in conjunction with another input.
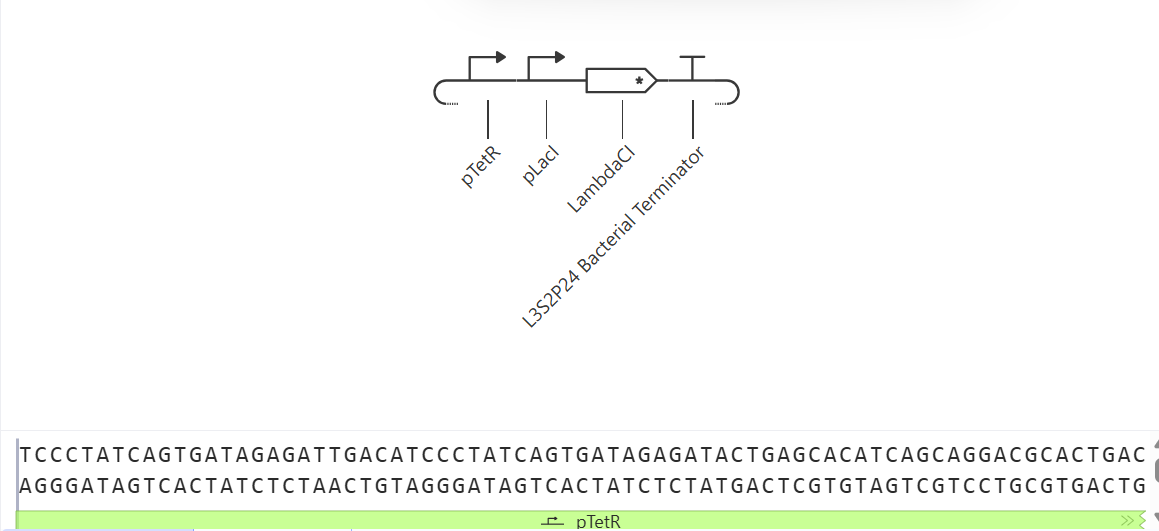
References
- Addgene. “Addgene: What Is Polymerase Chain Reaction (PCR).” Addgene.org, 2019, www.addgene.org/protocols/pcr/.
- ---. “Bacterial Transformation.” Addgene, 13 Nov. 2017, www.addgene.org/protocols/bacterial-transformation/.
- ---. “Molecular Biology Protocol - Restriction Digest of Plasmid DNA.” Addgene.org, 11 Oct. 2016, www.addgene.org/protocols/restriction-digest/.
- addgene. “Addgene: Gibson Assembly Protocol.” Www.addgene.org, www.addgene.org/protocols/gibson-assembly/.
- “Buffers for Biochemical Reactions.” Www.promega.com, www.promega.com/resources/guides/lab-equipment-and-supplies/buffers-for-biochemical-reactions/.
- “Deoxynucleotide Triphosphates (DNTP): Definition & Overview.” Www.excedr.com, www.excedr.com/resources/deoxynucleotide-triphosphates-dntp.
- “DNTPs (Deoxynucleotide Triphosphates).” Promega.com, 2026, www.promega.com/products/pcr/taq-polymerase/deoxynucleotide-triphosphates-dntps/?catNum=U1205. Accessed 22 Mar. 2026.
- “DNTPs: Structure, Role & Applications.” Baseclick GmbH, 15 Jan. 2025, www.baseclick.eu/science/glossar/dntps/.
- Excedr. “What Is the Role of MgCl2 in PCR Amplification Reactions?” Www.excedr.com, 19 Apr. 2022, www.excedr.com/resources/what-is-the-role-of-mgcl2-in-pcr.
- National Human Genome Research institute. “Reacción En Cadena de La Polimerasa (PCR) | NHGRI.” Genome.gov, 2024, www.genome.gov/es/genetics-glossary/Reacci%C3%B3n-en-cadena-de-la-polimerasa-PCR.
- New England Biolabs. “PhusionTM High-Fidelity PCR Master Mix with HF Buffer.” Neb.com, 2026, www.neb.com/en-us/products/m0531-phusion-high-fidelity-pcr-master-mix-with-hf-buffer?srsltid=AfmBOordry1wI142MJKbWwHlBq8WS18SoRym6puZTlpVRlkvbUOZgHqf. Accessed 22 Mar. 2026.
- ---. Neb.com, 2016, www.neb.com/en-us/applications/cloning-and-synthetic-biology/dna-preparation/restriction-enzyme-digestion?srsltid=AfmBOopkvxDAKNCNwCGI1NxjmySwglebU_iEos8Cy0py_cihV-FqNn1N. Accessed 22 Mar. 2026.
- “PCR Cycling Parameters—Six Key Considerations for Success - US.” Www.thermofisher.com, www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-cycling-considerations.html.
- “Phusion High-Fidelity PCR Master Mix with HF Buffer.” Www.thermofisher.com, www.thermofisher.com/order/catalog/product/F531L.
- “Restriction Enzymes Digestion | GenScript.” Genscript.com, 2026, www.genscript.com/what-is-restriction-digestion.html?__cf_chl_tk=crrewo623z2zjnCx9kOFdTqz0j9BoDdXu3XELXiUgz0-1773342635-1.0.1.1-gQT1okoakeY4JnNUIaPNcP6YhydQbvodS2WN._vGsHI. Accessed 22 Mar. 2026.
- SnapGene. “Golden Gate Assembly.” Www.snapgene.com, www.snapgene.com/guides/golden-gate-assembly.
- “The Plasmid Cloning Cycle - Snapgene.” Www.snapgene.com, www.snapgene.com/guides/the-plasmid-cloning-cycle.
- University of Utah. “PCR.” Utah.edu, 2000, learn.genetics.utah.edu/content/labs/pcr/.
Week 07 HW: Genetic circuits part II: Neuromorphic Circuits

Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Boolean network models contain a set of variables; each variable might have one of two possible values: false or true. In contrast, IANNs can approximate a wide range of nonlinear functions, obtaining multi-level outputs, which allows taking more precise decisions.
IANNs are designed to work with continuous signals, so they can be more robust to that variability than a circuit, making it easy to build big and complex systems. They can incorporate time and dynamics.
In conclusion, they are more flexible, compact, and accurate.
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
IANN has been used to detect Gynecological lesions. Scientists used a PAPANET system that successfully identified the atypical cells in the cervical smear.
The input to the IANN consists of digital images of cervical smears obtained from Pap tests. These images contain cells with varying morphological characteristics (normal and abnormal cells).
The IANN processes the images by reducing their dimensionality and extracting relevant features such as cell shape, size, texture, and nucleus-to-cytoplasm ratio. This is done automatically by the hidden layers without the need for manual feature selection by a pathologist.
The output of the system is a classification of the cells or the entire image, typically normal or abnormal (e.g., presence of atypical or precancerous cells). In some cases, the system may also assign a probability score indicating the likelihood of abnormality.
Using IANN, the entire image can be flattened to fewer than 100 pixels, and data can be extracted without bias; feature extraction does not require expert pathologists.
Limitations:
- This approach requires a team to implement an ANN in the laboratory. The team will consist of a data scientist, an engineer, and a cytologist (pathologist).
- The hidden layer acts as a “black box”, making it difficult to interpret, control, and optimize how the system processes inputs and generates outputs.
3. Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Assignment Part 2: Fungal Materials
1. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
1.Mycelium packaging foams
Companies grow mycelium packaging by inoculating agricultural waste (e.g., husks, stalks) with fungal mycelium, letting it bind into a solid, then drying it. It is molded into custom protective inserts that function similarly to expanded polystyrene (EPS) for shipping electronics, cosmetics, or food.
Advantages
- 100% biodegradable/compostable in natural conditions, no microplastics.
- Uses low-value agricultural byproducts as feedstock, reducing waste.
- Good impact protection and thermal insulation comparable to EPS for many uses.
Disadvantages
- Lower mechanical strength and durability than many petrochemical foams.
- More sensitive to moisture and prolonged damp conditions; can degrade faster.
- Scaling, consistency, and cost can be challenging compared with mature plastic supply chains.
2.Mycelium building and insulation materials
Mycelium is used as a building material in blocks/panels for non-load-bearing walls, cladding, and insulation. It is grown as a composite: mycelium binds plant fibers into rigid, lightweight bricks or panels.
Advantages
- Very good thermal insulation and sound absorption; traps more heat than fiberglass in some tests.
- Fire resistant and non-toxic when burned, unlike some synthetic foams.
- Lightweight, 100% biodegradable, and can be composted or returned to soil at end of life.
Disadvantages
- Pure mycelium has relatively low compressive strength; often unsuitable as a primary load-bearing structural material.
- Water resistance often decreases over time, especially if not well protected, leading to mold/humidity issues and shorter lifespan in damp environments.
- Mechanical performance is less predictable than concrete, steel, or engineered wood; more research and standards are needed.
3. Mycelium “leather” alternatives
Mycelium grown in controlled sheets or as fine composites is used as a leather alternative for fashion, accessories, and upholstery. Brands partner with biotech companies to supply sheet materials that can be cut and sewn like leather.
Advantages
- Animal-free and typically plastic-free or low-plastic, addressing ethical and some environmental concerns.
- Can be grown to targeted thickness and texture, and tuned via growth conditions and coatings.
- Potentially lower land and water use and lower greenhouse-gas emissions than livestock-based leather.
Disadvantages
- Raw mycelium sheets often lack the strength, flex resistance, and abrasion resistance of premium animal leather; they usually need reinforcement or coatings.
- Many commercial products rely on polyurethane (PU) or similar synthetic coatings for durability, which reduces overall biodegradability.
- Long-term durability, aging behavior, and large-scale cost competitiveness are still being proven.
2. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Objective: Engineering fungi to produce new antibiotics or variants of existing antibiotics that resistant bacteria cannot inactivate.
The concept involves modifying the genes or metabolic pathways of fungi that naturally produce antibiotics to create novel or more potent molecules.
Advantages of Using Fungi
- They can produce complex metabolites that bacteria or yeasts cannot.
- They allow for the modification of biosynthetic pathways more easily than plants do.
Limitations
- Potential toxicity or side effects associated with the new compound.
- Scaling up production to an industrial level can be complex.
Assignment Part 3: First DNA Twist Order
1. Review the Individual Final Project documentation guidelines.
2. Submit this Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs.
3. Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector that will be synthesized in on your website
Biosensor_A118G_GFP
This insert contains the A118G mutation recognition region, an inducible promoter activated by the binding of the protein of interest, the GFP reporter gene for visualization, and an intrinsic (rho-independent) terminator to stop transcription. Designed as a prototype biosensor for detecting SNPs in clinical samples
Backbone vector:
pUC19 (common example for expression in E. coli) — contains the ColE1 origin of replication and ampicillin resistance, compatible with the insertion of this construct and GFP expression testing.
GGTCAACTTGTCCCACTTAGATGGCGACCTGTCCGACCCATGCGGTCCGAAAACTGTTAATTAGTAGCCGAGCATATTACTACTCATTTCCTCTTCTTGAAAAGTGACCTCAACAGGGTTAAGAACAACTTAATCTACCCTACAATTACCCGTGTTTAAAAGACAGTCACCTCTCCCACTTCCACTACGTTGTATGCCTTTTGAATGGGAATTTAAATAAACGTGATGACCTTTTGATGGACAAGGTACCGGTTGTGAACAGTGATGAAAGAGAATACCACAAGTTACGAAAAGTTCTATGGGTCTAGTATACTTTGCCGTACTGAAAAAGTTCTCACGGTACGGGCTTCCAATACATGTCCTTTCTTGATATAAAAAGTTCTACTGCCCTTGATGTTCTGTGCACGACTTCAGTTCAAACTTCCACTATGGGAACAATTATCTTAGCTCAATTTTCCATAACTAAAATTTCTTCTACCTTTGTAAGAACCTGTGTTTAACCTTATGTTGATATTGAGTGTGTTACATATGTAGTACCGTCTGTTTGTTTTCTTACCTTAGTTTCAATTGAAGTTTTAATCTGTGTTGTAACTTCTACCTTCGCAAGTTGATCGTCTGGTAATAGTTGTTTTATGAGGTTAACCGCTACCGGGACAGGAAAATGGTCTGTTGGTAATGGACAGGTGTGTTAGACGGGAAAGCTTTCTAGGGTTGCTTTTCTCTCTGGTGTACCAGGAAGAACTCAAACATTGTCGACGACCCTAATGTGTACCGTACCTACTTGATATGTTTATCTCAATCTAACTACGCGGCGATACGCGTTGCGTAAATAAA


References
- Bangalore University. “E.coli Promoters.” Slideshare, 2018, es.slideshare.net/slideshow/ecoli-promoters/127916141#10. Accessed 22 Mar. 2026.
- Brophy, Jennifer A N, and Christopher A Voigt. “Principles of Genetic Circuit Design.” Nature Methods, vol. 11, no. 5, 29 Apr. 2014, pp. 508–520, https://doi.org/10.1038/nmeth.2926.
- Dey, Pranab. “Artificial Neural Network in Diagnostic Cytology.” Cytojournal, vol. 19, 2 Apr. 2022, p. 27, https://doi.org/10.25259/cytojournal_33_2021. Accessed 28 Nov. 2025.
- Elsacker, Elise, et al. “Recent Technological Innovations in Mycelium Materials as Leather Substitutes: A Patent Review.” Frontiers in Bioengineering and Biotechnology, vol. 11, 7 Aug. 2023, www.ncbi.nlm.nih.gov/pmc/articles/PMC10441217/, https://doi.org/10.3389/fbioe.2023.1204861.
- GSL Biotech LLC. “Tac Promoter Sequence and Map.” SnapGene, 2024, www.snapgene.com/plasmids/basic_cloning_vectors/tac_promoter.
- Hatkar, Aishwarya, and Aditi Lanke. “Mycelium: An Eco-Friendly Construction Material.” International Journal of Engineering Research & Technology, vol. 10, no. 3, 11 Feb. 2022, www.ijert.org/mycelium-an-eco-friendly-construction-material, https://doi.org/10.17577/IJERTCONV10IS03042.
- hugohek. “Grown-Design | Beautiful Products with Fungus and Biomass.” Grown.bio, 2022, www.grown.bio/.
- Katz, Leslie. “This Furniture Is Made of Fungus.” Forbes, 10 Oct. 2023, www.forbes.com/sites/lesliekatz/2023/10/10/this-furniture-is-made-of-fungi/.
- LLC, GSL Biotech. “GFP Sequence and Map.” Www.snapgene.com, www.snapgene.com/plasmids/fluorescent_protein_genes_and_plasmids/GFP.
- “Mycelium Building Material | Local Green US Hubs.” Local Green US Hubs, 2020, www.gogreenlocally.org/sahproject/mycelium-building-material. Accessed 22 Mar. 2026.
- Schwab, Julian D., et al. “Concepts in Boolean Network Modeling: What Do They All Mean?” Computational and Structural Biotechnology Journal, vol. 18, no. 18, 2020, pp. 571–582, https://doi.org/10.1016/j.csbj.2020.03.001.
- Sustainability Directory. “Mycelium Leather Commercialization Secures Durable, Plastic-Free, Animal-Free Luxury Materials → Fashion.” News → Sustainability Directory, 18 Oct. 2025, news.sustainability-directory.com/fashion/mycelium-leather-commercialization-secures-durable-plastic-free-animal-free-luxury-materials/. Accessed 22 Mar. 2026.
- ---. “What Are the Drawbacks of Mycelium? → Question.” Product → Sustainability Directory, 10 Apr. 2025, product.sustainability-directory.com/question/what-are-the-drawbacks-of-mycelium/.
- “Tema 10.” Web.uah.es, 2026, biomolq.web.uah.es/BM/Esquemas/Tema10.htm?utm_source=chatgpt.com. Accessed 22 Mar. 2026.
- You, Linlin, et al. “Structural Basis for Intrinsic Transcription Termination.” Nature, vol. 613, no. 7945, 1 Jan. 2023, pp. 783–789, www.nature.com/articles/s41586-022-05604-1, https://doi.org/10.1038/s41586-022-05604-1. Accessed 11 May 2023.
Week 09 HW: Cell-free systems
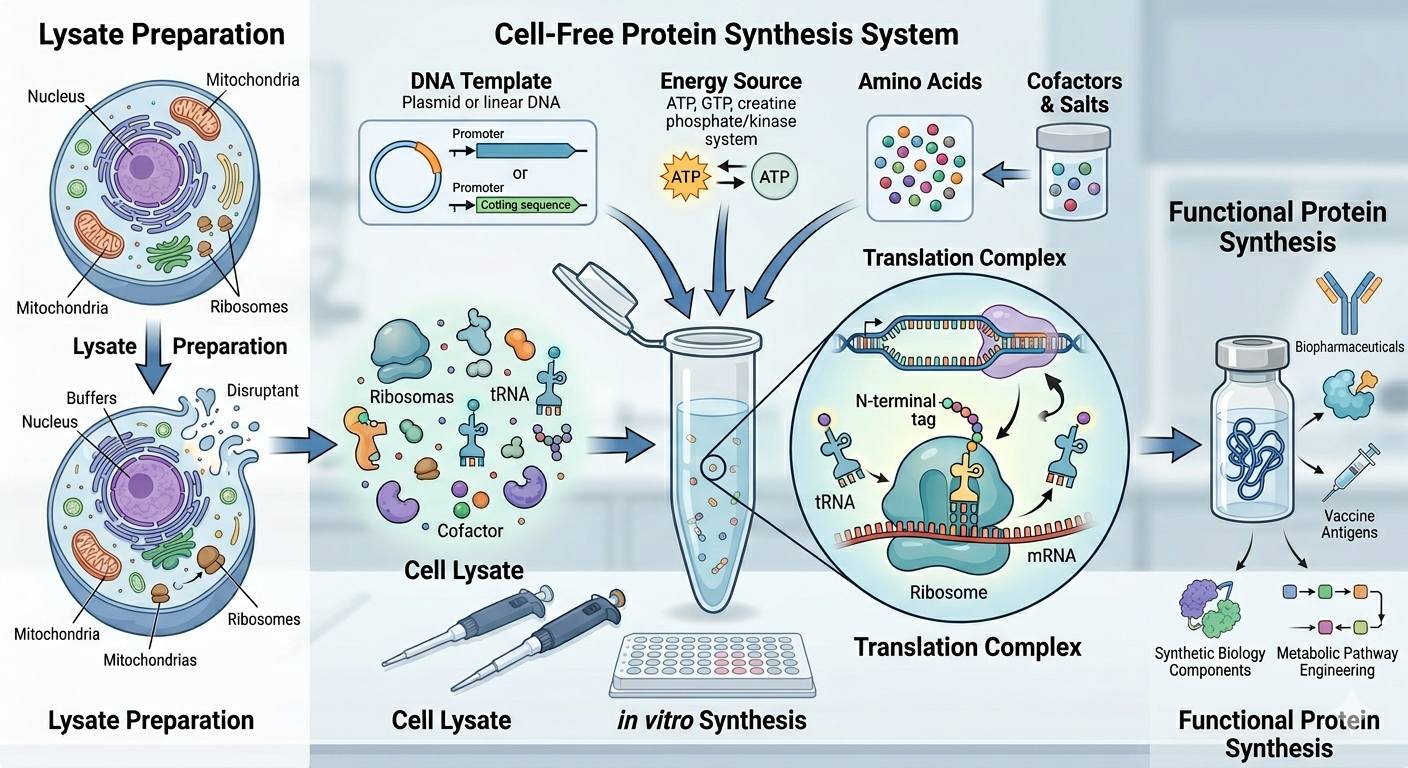
General homework questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis is usually better than traditional in vivo expression systems because it is an open system. It allows direct tuning of the reaction conditions, monitoring the process, and adding components like labels, chaperones, cofactors, or non-natural amino acids much more easily than in living cells.
Control and flexibility: This system avoids the limits of cell growth and cellular metabolism; consequently, its resources can be devoted to making the target protein. Meaning that, it is not constrained by cell viability, membrane transport, or homeostasis, so you can rapidly change variables such as buffer composition, template type, ion concentrations, folding aids, and amino acid content. That makes optimization faster and experimental control much tighter than in vivo expression, where many internal cellular processes are hard to isolate or adjust.
When is it especially beneficial?
Cell-free expressions are especially useful for:
- Toxic proteins, because there are no living host cells to damage
- Difficult-to-express proteins, specifically including some proteins that are unstable or rapidly degraded in cells
2. Describe the main components of a cell-free expression system and explain the role of each component.
Cell extract: This is the biological “machinery” of the system. It supplies ribosomes, tRNAs, initiation/elongation/termination factors, and other enzymes needed for transcription and translation.
Template DNA or mRNA: This provides the instructions for the protein you want to make.
Energy system: The reaction includes ATP/GTP and often an energy-regeneration module, because protein synthesis consumes a lot of energy.
Amino acids: These are the raw building blocks that get assembled into the target protein.
Salts and cofactors: Magnesium, potassium, and other cofactors help maintain ribosome structure and support enzymatic activity throughout the reaction.
Buffer system: The buffer keeps the pH and ionic conditions in a range during the process.
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy provision regeneration is required in cell-free systems because the whole process of protein synthesis requires a large amount of energy; the reaction will stop quickly if ATP is not continuously replenished.
Efficient regeneration also helps avoid the buildup of inhibitory byproducts, which can further suppress protein production.
A method used to maintain ATP is an acetyl phosphate/acetate kinase system, which can regenerate ATP continuously while also helping reduce phosphate accumulation in some formats.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Consequently, A prokaryotic cell-free system is a strong choice for sfGFP or another easy-to-express bacterial reporter protein because these proteins are compact, do not require elaborate post-translational processing, and can be produced efficiently in an E. coli extract.
A eukaryotic cell-free system might be more complex. Still, it is a better choice for human insulin, specifically proinsulin, because insulin production benefits from a more eukaryotic-like folding environment and processing workflow.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
I believe it is a good approach to develop an experiment where I can run a small factorial screen that varies three things:
- The DNA template amount
- The membrane-mimetic environment
- The reaction conditions, such as temperature and incubation time.
The main goal is to find the condition that gives the best balance between total yield and correctly folded, membrane-inserted protein, because membrane proteins often stall translation and aggregate before they fold properly.
Set up
Use a cell-free transcription/translation system, then compare a modest matrix of conditions in parallel. I suggest that a practical starting design is:
- 2 to 3 DNA concentrations
- 3 membrane environments: no membrane mimic, liposomes or microsomes, and nanodiscs
- 2 to 3 temperatures
- 2 harvest times
That provides a general screen while still testing the major variables known to affect membrane protein synthesis in cell-free systems.
It is necessary to track both expression and function, not just total protein.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
1. Pick a function and describe it.
a. What would your synthetic cell do? What is the input and what is the output?
Synthetic minimal cell design: OPRM1 A118G biosensor
My synthetic minimal cell would be a liposome-encapsulated cell-free biosensor designed to detect the A118G polymorphism in the OPRM1 gene. The function of the synthetic cell is to identify a specific single-nucleotide polymorphism in a DNA sample.
The input would be a DNA sample containing the target sequence, and the output would be a measurable fluorescent signal, such as GFP expression, only when the G allele is present.
Input: DNA containing the OPRM1 A118G sequence.
Output: Fluorescence or another visible signal indicating the presence of the G allele.
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
This function could be performed by cell-free Tx/Tl. However, a cell-free system inside a vesicle gives compartmentalization, protection of the reaction, and better control over the sensing environment.
c. Could this function be realized by a genetically modified natural cell?
Yes, this function could also be engineered in a genetically modified natural cell, such as E. coli or yeast. However, a synthetic minimal cell is better for a biosensor because it avoids the complexity of living metabolism and cell growth
d. Describe the desired outcome of your synthetic cell operation.
The desired outcome is a vesicle that can be used as a tiny diagnostic device: when it encounters the correct DNA sequence, it produces a fluorescent signal that can be measured externally.
2. Design all components that would need to be part of your synthetic cell.
a. What would be the membrane made of?
The membrane would be a lipid bilayer liposome made from phospholipids
b. What would you encapsulate inside? Enzymes, small molecules.
Inside the synthetic cell I would encapsulate:
- A cell-free Tx/Tl system from E. coli
- A DNA recognition module specific for the A118G polymorphism
- A reporter gene such as gfp
c. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
I would use a bacterial cell-free system, especially from E. coli, because it is well characterized, easy to use, and suitable for a minimal synthetic cell.
d. How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
- Allowing the DNA sample or small molecules to enter during assay setup
- Possibly using a membrane pore to permit small-molecule exchange
- Releasing a fluorescent signal that can be detected outside the vesicle
3. Experimental details
a. List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
- gfp for fluorescence reportin
- hla if a membrane pore is needed
- A synthetic DNA-sensing module designed to distinguish the A and G alleles of OPRM1
b. How will you measure the function of your system?
Fluorescence intensity
Comparison of samples with A allele, G allele, and no-template control
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
1. Write a one-sentence summary pitch sentence describing your concept.
A cell-free biosensor band for astronauts that detects toxic environmental exposure and produces an immediate visible signal
2. How will the idea work, in more detail? Write 3-4 sentences or more.
It will be a flexible wearable band or patch designed for astronauts or people working in extreme environments. The material would contain cell-free biosensors that become active when a small amount of water or a sample is added, and they would detect dangerous environmental toxins such as heavy metals or other harmful chemicals. When the target toxin is present, the cell-free system would express a reporter such as GFP, creating an easy-to-read signal.
3. What societal challenge or market need will this address?
This idea addresses the need for portable, low-maintenance safety monitoring in places where conventional sensors may be expensive or difficult to use. It could be valuable for astronauts, space missions, emergency responders, industrial workers, or anyone exposed to toxic environments.
4. How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
The main limitation of cell-free systems is that they typically require water to initiate and can lose activity over time. I would solve this by freeze-drying the biosensor inside the textile or material, so it stays stable during storage and activates only when needed. To make it reusable or longer-lasting, the band could use replaceable sensing patches.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
1. Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
In space exploration, microgravity affects cellular function, reducing proliferation and altering metabolism. Understanding how microgravity impacts cell growth is essential for astronaut health and for maintaining effective medications and microbial cultures.
BioBits® lets us test protein activity without using live cells. By measuring fluorescence under microgravity and UV radiation, we can see how these conditions affect cell growth. This experiment will show how space affects metabolism and growth in a simple, visual way.
2. Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
GFP protein expressed in the BioBits® system, used as a visual indicator of metabolic activity to measure how simulated cell growth changes under microgravity and UV radiation.
3. Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
GFP fluorescence reflects the metabolic activity of the BioBits® system, serving as an indicator of simulated cell growth: higher fluorescence indicates faster growth, while lower fluorescence indicates slower growth. By comparing fluorescence under microgravity and UV radiation, we can assess how these space conditions affect growth rates. This information is relevant for predicting effects on human cells, microbial cultures, and protein stability in space. Using GFP as an indicator allows quick, safe, and portable measurements, providing practical insights for protecting astronaut health and supporting biotechnological applications during long space missions.
4. Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Hypothesis:
Microgravity reduces the simulated cellular growth rate compared to UV radiation, with GFP fluorescence serving as a reliable indicator of metabolic activity.
Research goal:
To evaluate how microgravity and UV radiation affect simulated cell growth using BioBits®. This will help determine whether microgravity slows critical metabolic processes, which has direct implications for astronaut health, drug stability, and microbial culture productivity. We expect lower fluorescence under microgravity, indicating reduced activity, while UV exposure may produce a different pattern. This experiment will generate preliminary data to inform strategies that mitigate microgravity effects, such as optimizing antioxidants, nutrients, or cellular support systems for long-term missions.
5. Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Two BioBits® systems with GFP will be set up: one simulating microgravity and one exposed to UV radiation. Controls include a system without microgravity or UV, and one with standard GFP for calibration. Fluorescence will be measured every 10 minutes for 1 hour using the P51 Molecular Fluorescence Viewer. Fluorescence intensity will be compared between conditions to infer relative growth rates. Additional experiments may include antioxidants or nutrients to test mitigation strategies. Data will be recorded quantitatively and graphed to analyze patterns in metabolic activity and simulated growth under space conditions
Homework Part B: Individual Final Project
DNA Order
Project slide:

References
- Baldo, Thaisa A., et al. “Wearable and Biodegradable Sensors for Clinical and Environmental Applications.” ACS Applied Electronic Materials, 10 Dec. 2020, https://doi.org/10.1021/acsaelm.0c00735.
- ---. “Wearable and Biodegradable Sensors for Clinical and Environmental Applications.” ACS Applied Electronic Materials, 10 Dec. 2020, https://doi.org/10.1021/acsaelm.0c00735.
- Banks, Alice M., et al. “Key Reaction Components Affect the Kinetics and Performance Robustness of Cell-Free Protein Synthesis Reactions.” Computational and Structural Biotechnology Journal, vol. 20, 1 Jan. 2022, pp. 218–229, www.sciencedirect.com/science/article/pii/S2001037021005213, https://doi.org/10.1016/j.csbj.2021.12.013.
- ---. “Key Reaction Components Affect the Kinetics and Performance Robustness of Cell-Free Protein Synthesis Reactions.” Computational and Structural Biotechnology Journal, vol. 20, 1 Jan. 2022, pp. 218–229, www.sciencedirect.com/science/article/pii/S2001037021005213, https://doi.org/10.1016/j.csbj.2021.12.013.
- Borhani, Shayan G, et al. “On-Demand Insulin Manufacturing Using Cell-Free Systems with an “On-Column” Conversion Approach.” New Biotechnology, 1 June 2025, https://doi.org/10.1016/j.nbt.2025.06.002. Accessed 12 Sept. 2025.
- ---. “On-Demand Insulin Manufacturing Using Cell-Free Systems with an “On-Column” Conversion Approach.” New Biotechnology, 1 June 2025, https://doi.org/10.1016/j.nbt.2025.06.002. Accessed 12 Sept. 2025.
- Calhoun, Kara A., and James R. Swartz. “Energy Systems for ATP Regeneration in Cell-Free Protein Synthesis Reactions.” In Vitro Transcription and Translation Protocols, 2007, pp. 3–17, https://doi.org/10.1007/978-1-59745-388-2_1.
- “Cell-Free Expression Support—Troubleshooting | Thermo Fisher Scientific - US.” Thermofisher.com, 2025, www.thermofisher.com/ca/en/home/technical-resources/technical-reference-library/protein-expression-support-center/cell-free-expression-support/cell-free-expression-support-troubleshooting.html. Accessed 7 Apr. 2026.
- “Cell-Free Protein Expression System- CUSABIO.” Cusabio.com, 2026, www.cusabio.com/cell-free-expression-system.html. Accessed 7 Apr. 2026.
- “Cell-Free Protein Synthesis Explained | IDT.” Integrated DNA Technologies, 2015, www.idtdna.com/pages/applications/cell-free-protein-synthesis.
- Chauhan, Nidhi, et al. “A Review on Biosensor Approaches for the Detection of Hazardous Elements in Water.” Talanta Open, 1 Aug. 2025, pp. 100536–100536, https://doi.org/10.1016/j.talo.2025.100536.
- Grooms, Kelly. “Moving out of the Cell: Advantages of Cell-Free Protein Expression - Promega Connections.” Promega Connections, 22 Aug. 2016, www.promegaconnections.com/moving-out-of-the-cell-advantages-of-cell-free-protein-expression/.
- ---. “Moving out of the Cell: Advantages of Cell-Free Protein Expression - Promega Connections.” Promega Connections, 22 Aug. 2016, www.promegaconnections.com/moving-out-of-the-cell-advantages-of-cell-free-protein-expression/.
- Kim, D M, and J R Swartz. “Prolonging Cell-Free Protein Synthesis with a Novel ATP Regeneration System.” Biotechnology and Bioengineering, vol. 66, no. 3, 1999, pp. 180–8, pubmed.ncbi.nlm.nih.gov/10577472/.
- ---. “Prolonging Cell-Free Protein Synthesis with a Novel ATP Regeneration System.” Biotechnology and Bioengineering, vol. 66, no. 3, 1999, pp. 180–8, pubmed.ncbi.nlm.nih.gov/10577472/.
- Kim, Ho-Cheol, and Dong-Myung Kim. “Methods for Energizing Cell-Free Protein Synthesis.” Journal of Bioscience and Bioengineering, vol. 108, no. 1, July 2009, pp. 1–4, pubmed.ncbi.nlm.nih.gov/19577183/, https://doi.org/10.1016/j.jbiosc.2009.02.007.
- ---. “Methods for Energizing Cell-Free Protein Synthesis.” Journal of Bioscience and Bioengineering, vol. 108, no. 1, July 2009, pp. 1–4, pubmed.ncbi.nlm.nih.gov/19577183/, https://doi.org/10.1016/j.jbiosc.2009.02.007.
- Meyer, Conary, et al. “Designer Artificial Environments for Membrane Protein Synthesis.” Nature Communications, vol. 16, no. 1, 10 May 2025, www.nature.com/articles/s41467-025-59471-1, https://doi.org/10.1038/s41467-025-59471-1. Accessed 7 Apr. 2026.
- ---. “Designer Artificial Environments for Membrane Protein Synthesis.” Nature Communications, vol. 16, no. 1, 10 May 2025, www.nature.com/articles/s41467-025-59471-1, https://doi.org/10.1038/s41467-025-59471-1. Accessed 7 Apr. 2026.
- Nguyen, Peter Q., et al. “Wearable Materials with Embedded Synthetic Biology Sensors for Biomolecule Detection.” Nature Biotechnology, vol. 39, 28 June 2021, pp. 1–9, www.nature.com/articles/s41587-021-00950-3, https://doi.org/10.1038/s41587-021-00950-3.
- ---. “Wearable Materials with Embedded Synthetic Biology Sensors for Biomolecule Detection.” Nature Biotechnology, vol. 39, 28 June 2021, pp. 1–9, www.nature.com/articles/s41587-021-00950-3, https://doi.org/10.1038/s41587-021-00950-3.
- Schaub, Theresa. “Cell-Free Protein Synthesis: A Faster, More Flexible Alternative to Traditional Expression.” Cosmo Bio USA, 2 Mar. 2026, www.cosmobiousa.com/blog/why-cell-free-protein-expression-can-be-better-than-cell-based-systems. Accessed 7 Apr. 2026.
- Steinkühler, Jan, et al. “Improving Cell-Free Expression of Model Membrane Proteins by Tuning Ribosome Cotranslational Membrane Association and Nascent Chain Aggregation.” ACS Synthetic Biology, vol. 13, no. 1, 27 Dec. 2023, pp. 129–140, https://doi.org/10.1021/acssynbio.3c00357. Accessed 11 Dec. 2025.
- ---. “Improving Cell-Free Expression of Model Membrane Proteins by Tuning Ribosome Cotranslational Membrane Association and Nascent Chain Aggregation.” ACS Synthetic Biology, vol. 13, no. 1, 27 Dec. 2023, pp. 129–140, https://doi.org/10.1021/acssynbio.3c00357. Accessed 11 Dec. 2025.
- UMBC. “CAST | Center for Advanced Sensor Technology.” Umbc.edu, 2020, cast.umbc.edu/research-new/manufacturing-of-insulin-using-cell-free-systems-cfs/.
- ---. “CAST | Center for Advanced Sensor Technology.” Umbc.edu, 2020, cast.umbc.edu/research-new/manufacturing-of-insulin-using-cell-free-systems-cfs/.Wan, Xinyi. “Synthetic Biology Enabled Cellular and Cell-Free Biosensors for Environmental Contaminants.” Ed.ac.uk, The University of Edinburgh, 6 July 2019, era.ed.ac.uk/items/eada344c-6d52-4c51-8714-73b612b1848b. Accessed 7 Apr. 2026.
- ---. “Synthetic Biology Enabled Cellular and Cell-Free Biosensors for Environmental Contaminants.” Ed.ac.uk, The University of Edinburgh, 6 July 2019, era.ed.ac.uk/items/eada344c-6d52-4c51-8714-73b612b1848b. Accessed 7 Apr. 2026.
- “What Are the Advantages of Cell-Free Protein Expression System over the Traditional in Vivo Systems? | AAT Bioquest.” Aatbio.com, 2023, www.aatbio.com/resources/faq-frequently-asked-questions/what-are-the-advantages-of-cell-free-protein-expression-system-over-the-traditional-in-vivo-systems. Accessed 7 Apr.
- 2026.Whittaker, James W. “Cell-Free Protein Synthesis: The State of the Art.” Biotechnology Letters, vol. 35, no. 2, 21 Oct. 2012, pp. 143–152, https://doi.org/10.1007/s10529-012-1075-4.
- ---. “Cell-Free Protein Synthesis: The State of the Art.” Biotechnology Letters, vol. 35, no. 2, 21 Oct. 2012, pp. 143–152, https://doi.org/10.1007/s10529-012-1075-4.“Why Use Cell-Free Protein Expression?” Cube Biotech, 2014, cube-biotech.com/our-science/cell-free-lysates/cell-free-expression/.
- “Why Use Cell-Free Protein Expression?” Cube Biotech, 2014, cube-biotech.com/our-science/cell-free-lysates/cell-free-expression/.
- Wyss Institute. “Wearable Synthetic Biology: Clothing That Can Detect Pathogens and Toxins.” YouTube, 29 June 2021, www.youtube.com/watch?v=_pHiIlNDAGk. Accessed 7 Apr. 2026.
- ---. “Wearable Synthetic Biology: Clothing That Can Detect Pathogens and Toxins.” YouTube, 29 June 2021, www.youtube.com/watch?v=_pHiIlNDAGk. Accessed 7 Apr. 2026.
- Zemella, Anne, et al. “Cell-Free Protein Synthesis: Pros and Cons of Prokaryotic and Eukaryotic Systems.” ChemBioChem, vol. 16, no. 17, 19 Oct. 2015, pp. 2420–2431, www.ncbi.nlm.nih.gov/pmc/articles/PMC4676933/, https://doi.org/10.1002/cbic.201500340.---.
Week 10 HW: Measurement Technology

Homework: Final Project
For your final project:
1. Please identify at least on6e (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
The main measurable aspect of this project is the presence or absence of the A118G polymorphism in the OPRM1 gene.
2. Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
The detection system is based on a CRISPR-Cas13 biosensor coupled to a Broccoli RNA aptamer. The elements measured include the presence of the target mutation and the resulting fluorescence signal.
When the mutant sequence (G118) is present, the crRNA guides Cas13 to specifically recognize the target RNA. This activates Cas13’s collateral cleavage activity, which degrades a blocking RNA sequence. Once the blocker is degraded, the Broccoli aptamer can fold into its active structure and bind to its fluorophore (e.g., DFHBI), producing a fluorescent signal (ON state).
In contrast, if the wild-type sequence (A118) is present, Cas13 is not activated, the blocker remains intact, and the aptamer does not fluoresce (OFF state).
Fluorescence will be measured using a fluorimeter or plate reader, allowing detection of signal presence or absence, and potentially signal intensity. Additionally, gel electrophoresis may be used to confirm RNA integrity and Cas13-mediated cleavage.
3. What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
- CRISPR-Cas13 system: Used for sequence-specific recognition of RNA and discrimination between the A118 and G118 variants. Upon recognition of the mutant sequence, Cas13 becomes activated and induces collateral RNA cleavage.
- Cell-free expression system: The biosensor will be implemented in a cell-free transcription-translation system, which allows controlled expression and interaction of the RNA components without the use of living cells. This system provides a rapid and tunable platform for biosensing.
- Fluorescent RNA aptamer (Broccoli): Functions as a reporter that emits fluorescence upon proper folding and binding to a fluorophore such as DFHBI, indicating activation of the system.
- Fluorescence detection (fluorimeter or plate reader): Used to measure the biosensor output, enabling qualitative (ON/OFF).
Homework: Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
1. Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence:
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
The calculated molecular weight of eGFP based on its amino acid sequence is approximately 28,006.60 Da (~28.0 kDa), as determined using the ExPASy Compute pI/Mw tool. This value includes the C-terminal His-tag present in the sequence.
2. Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Figure 1
Mass Spectrum of intact eGFP protein from the Waters Xevo G3 LC-MS (a mass spectrometer with 30,000 resolution) with individual charge state peaks labeled with m/z values.
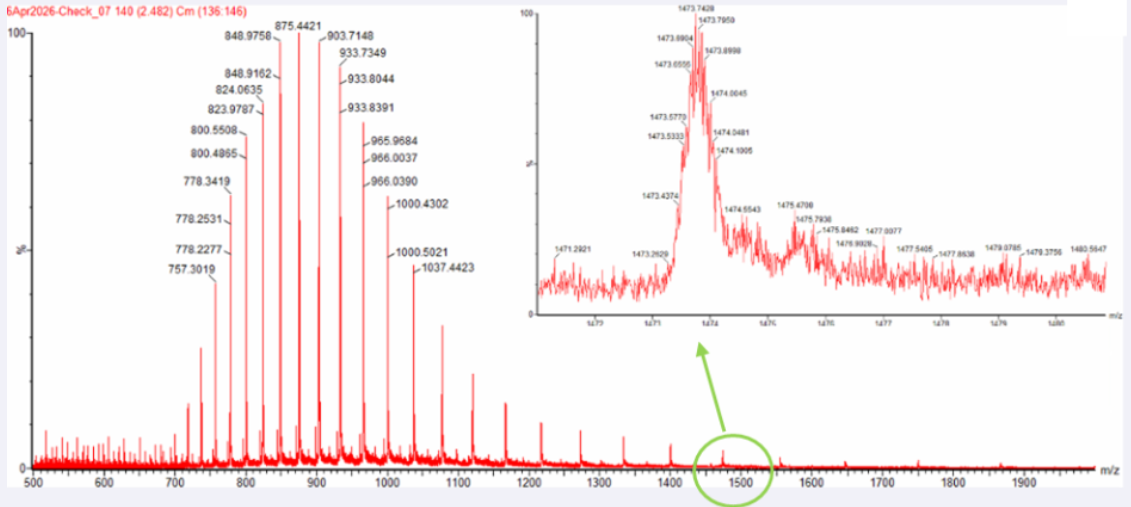
2.1 Determine z for each adjacent pair of peaks (n, n + 1) using:
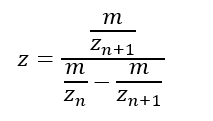
Peak 1: 875,4
Peak 2: 903,7
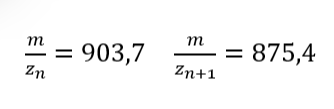
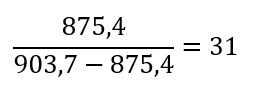
2.2 Determine the MW of the protein using the relationship between m/zn, MW, and Z

2.3 Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
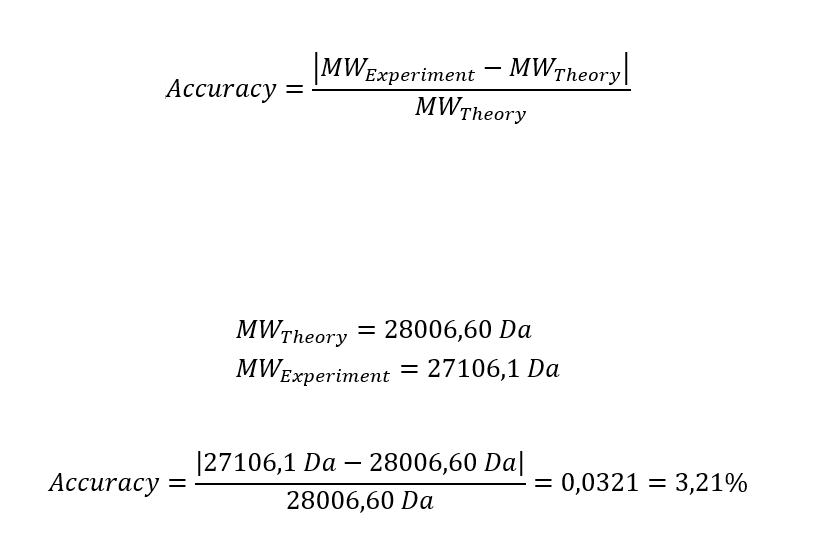
Homework: Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
1. How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
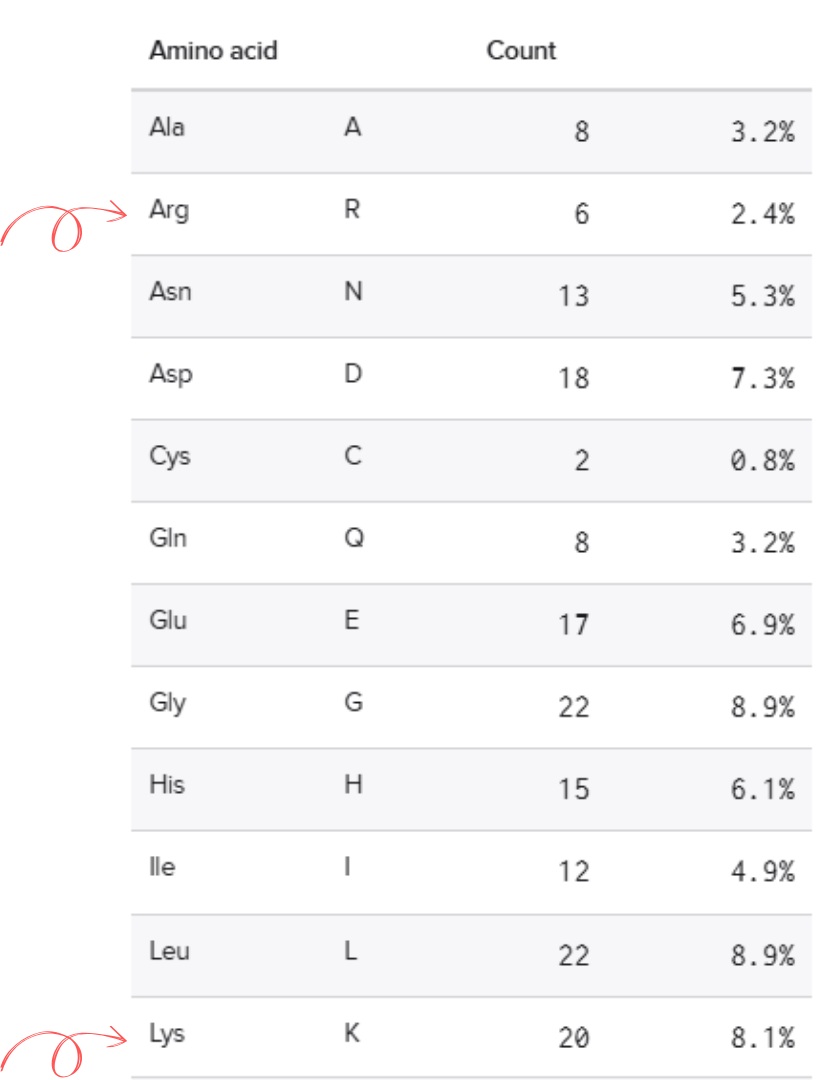
2. How many peptides will be generated from tryptic digestion of eGFP?
2.1 Navigate to https://web.expasy.org/peptide_mass/
2.2 Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
2.3 Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
Using Expasy, We obtained 19 peptides.
3. Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
 Figure 5a. Total ion chromatogram (TIC) of the eGFP peptide map. The peak at 2.78 minutes is circled, and its MS data is shown in the mass spectrum in Figure 5b, below.
Figure 5a. Total ion chromatogram (TIC) of the eGFP peptide map. The peak at 2.78 minutes is circled, and its MS data is shown in the mass spectrum in Figure 5b, below.
Approximately 17 chromatographic peaks above 10% relative abundance between 0.5 and 6 minutes.
4. Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
Assuming all the peaks are peptides, the number of peaks does not match the number of peptides predicted from question 2. This is because, in question 2, we obtain 19 peptides, but only 17 significant peaks are observed in the chromatogram. This discrepancy may be due to several factors, including co-elution of peptides, low-abundance peptides falling below the detection limit, incomplete digestion, or ionization efficiency differences between peptides, in addition to experimental noise.
5. Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide (
[M+H]+) based on its m/z and z.
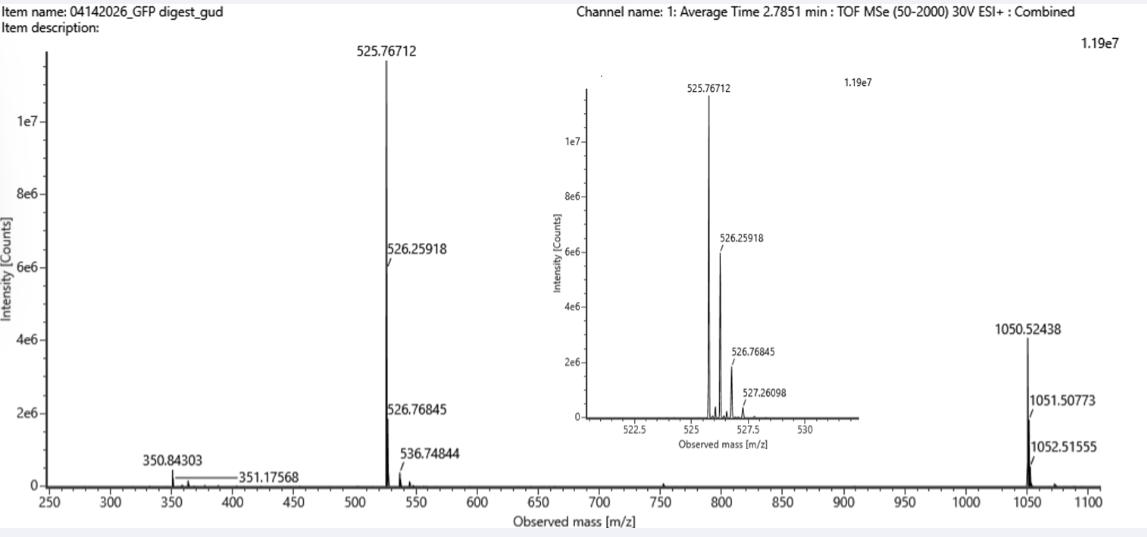 Figure 5b. Mass spectrum figure to show m/z for the chromatographic peak at 2.78 min from Figure 5a above. The inset is a zoom-in of the peak at m/z 525.76, to discern the isotope peaks.
Figure 5b. Mass spectrum figure to show m/z for the chromatographic peak at 2.78 min from Figure 5a above. The inset is a zoom-in of the peak at m/z 525.76, to discern the isotope peaks.
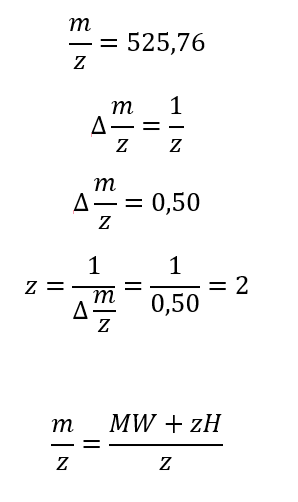

6. Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
In the table of point 2, I obtained the peptide FEGDTLVNR, with a mass of 1050.5214 Da. This peptide may correspond to the one shown in Figure 5b, whose experimental mass is 1050.5 Da.
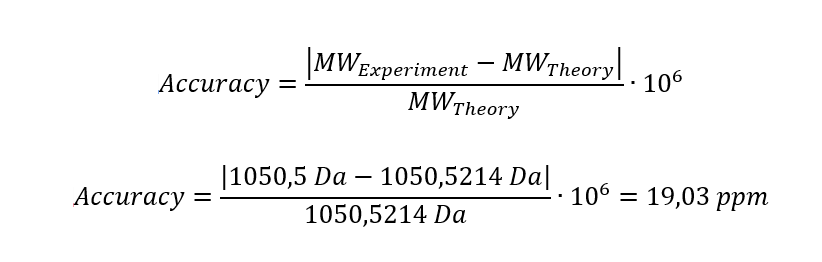
7. What is the percentage of the sequence that is confirmed by peptide mapping?
The percentage of the sequence confirmed by peptide mapping is 88%.
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution.
1. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
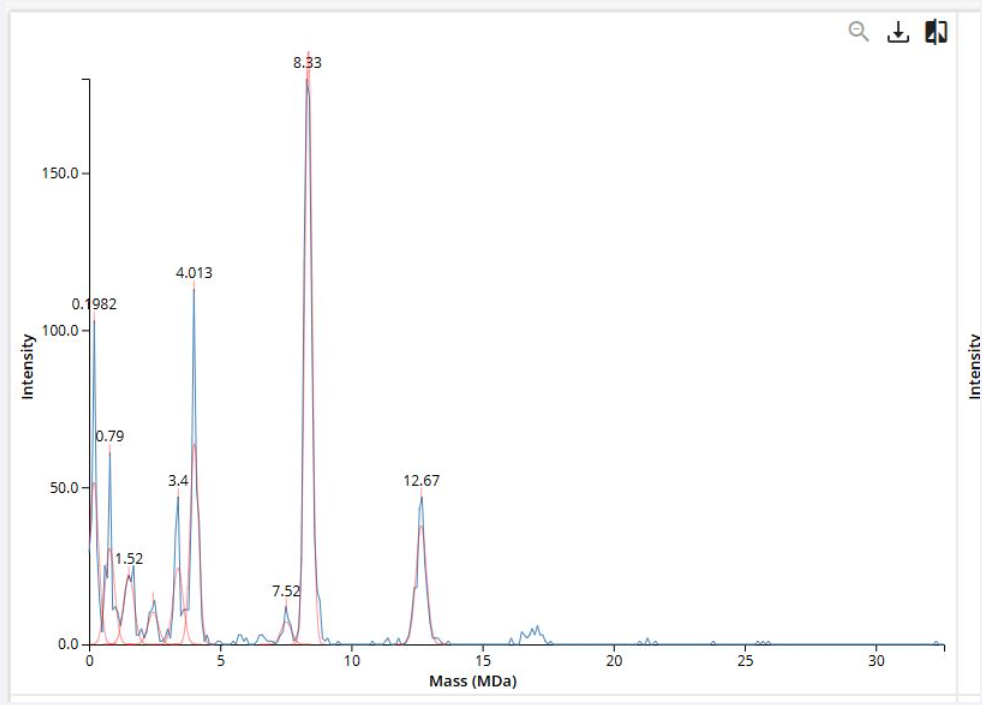 Figure 7. Mass spectrum of Keyhole Limpet Hemocyanin (KLH) acquired on the CDMS.
Figure 7. Mass spectrum of Keyhole Limpet Hemocyanin (KLH) acquired on the CDMS.
Polypeptide Subunit Name ⭢ 7FU ⭢ Subunit Mass ⭢ 340 kDa
Polypeptide Subunit Name ⭢8FU ⭢ Subunit Mass ⭢ 400 kDa
- 7FU Decamer
- 8FU Didecamer
- 8FU 3-Decamer
- 8FU 4-Decamer
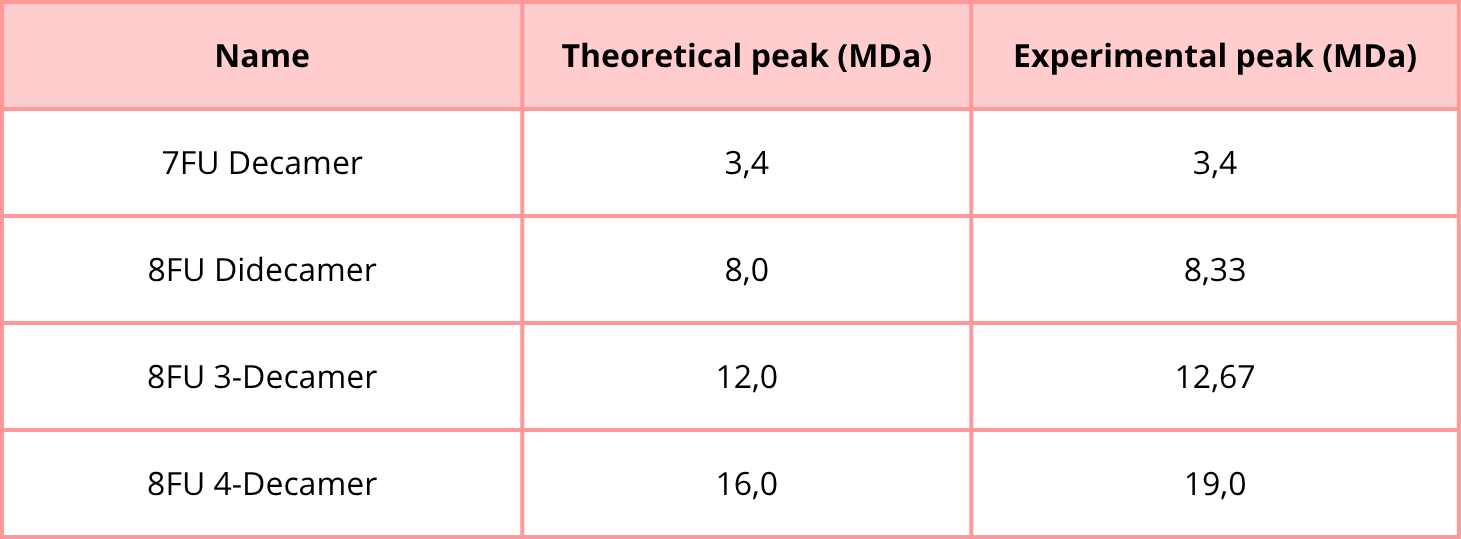
Homework: Waters Part V — Did I make GFP?
1. Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
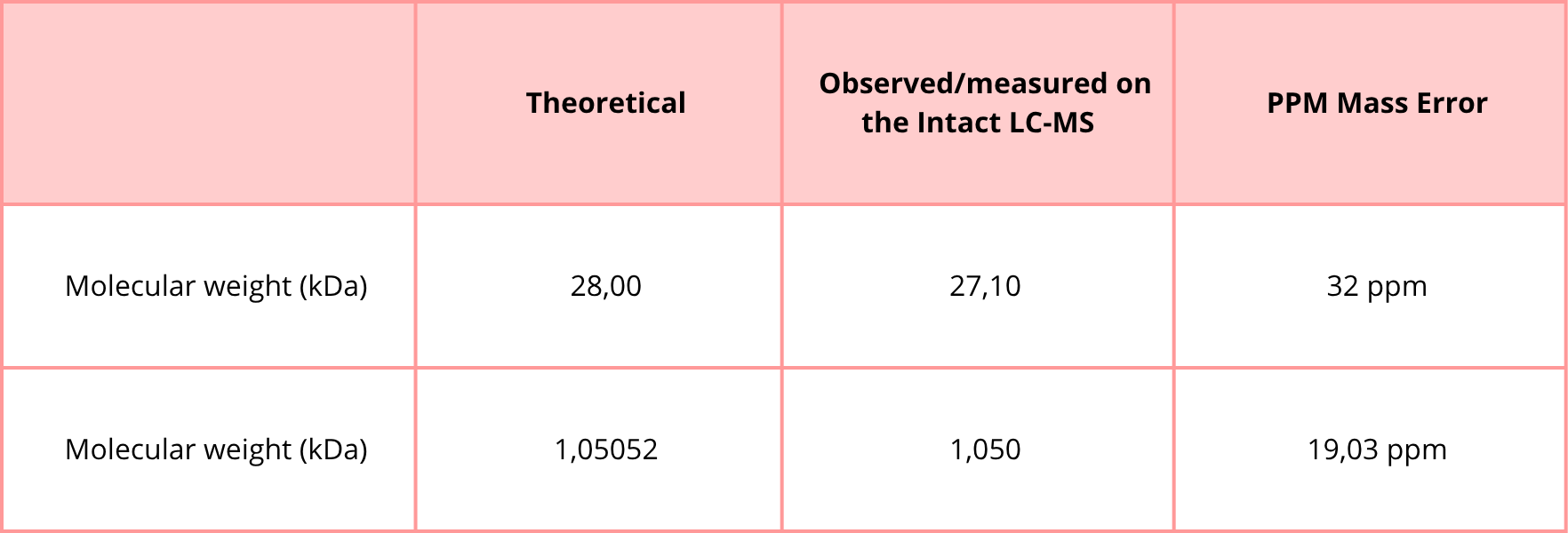
Week 11 — Bioproduction & Cloud Labs

Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
1. Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
- A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse.
- If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
Although I was not able to contribute a pixel, I noticed the suggestion about becoming a TA this fall. I think it could be an interesting opportunity, as it would allow for more involvement in collaborative projects like this one and a way to support the course community.
2. Make a note on your HTGAA webpages, including:
- What you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
- What you liked about the project, and what about this collaborative art experiment could be made better for next year.
I explored the final artwork and the collaborative process behind it.
What I liked most about the project is the idea of building a collective artwork where each participant contributes a minimal unit (a pixel), and the result collects a complex and meaningful image. I also found it engaging that the project connects science, art, and community participation.
One aspect that could be improved for next year is increasing reminders and accessibility to the participation link, since it is easy to miss the contribution window. Additionally, having a longer editing period or sending follow-up notifications could help ensure more people are able to contribute.
Overall, even without directly contributing, the project highlights the value of collaboration and creativity in scientific communities.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
1. Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
- BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
Salts/Buffer
- Potassium Glutamate
- HEPES-KOH pH 7.5
- Magnesium Glutamate
- Potassium phosphate monobasic
- Potassium phosphate dibasic
Energy / Nucleotide System
- Ribose
- Glucose
- AMP
- CMP
- GMP
- UMP
- Guanine
Translation Mix (Amino Acids)
- 17 Amino Acid Mix
- Tyrosine
- Cysteine
Additives
Backfill
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): This is a specialized lysate from an expression strain that also contains T7 RNA polymerase, allowing very efficient transcription from T7 promoters.
Potassium Glutamate: This supplies potassium ions and helps stabilize the reaction conditions needed for ribosome activity and enzyme function.
HEPES-KOH pH 7.5: This is the main buffering agent that keeps the reaction at a near-neutral pH, which is optimal for protein synthesis.
Magnesium Glutamate: This provides magnesium ions, which are essential for ribosome stability, nucleotide binding, and overall enzymatic activity.
Potassium Phosphate Monobasic: This contributes to buffering capacity and helps maintain stable chemical conditions in the reaction.
Potassium Phosphate Dibasic: This works with the monobasic form to adjust and stabilize the pH of the buffer system.
Ribose: This serves as a carbon source and helps support nucleotide regeneration pathways in the reaction.
Glucose: This provides a long-lasting energy source that helps sustain ATP regeneration and overall reaction activity.
AMP: This is a nucleotide precursor that helps maintain the adenylate pool and supports energy recycling.
CMP: This is a cytidine nucleotide precursor needed to maintain the pool of RNA building blocks.
GMP: This is a guanosine nucleotide precursor required for RNA synthesis and nucleotide balance.
UMP: This is a uridine nucleotide precursor that supports RNA production.
Guanine: This is a base precursor that can be used to help replenish guanosine nucleotide pools.
17 Amino Acid Mix: This supplies most of the amino acids required for protein synthesis.
Tyrosine: This amino acid is often added separately to fine-tune its concentration and improve solubility or balance.
Cysteine: This is added separately because it is important for protein structure and may require special handling in the mixture.
Nicotinamide: This supports redox-related reactions and can help maintain metabolic activity in the lysate.
Nuclease-Free Water This is used to complete the reaction volume while avoiding degradation of DNA or RNA by nucleases.
2. Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour optimized PEP-NTP master mix is designed for fast, high-energy protein synthesis, using PEP as a rapid ATP-regenerating substrate and preloaded NTPs to support quick transcription. In contrast, the 20-hour NMP-Ribose-Glucose master mix is built for longer reactions, relying on glucose and ribose plus NMPs as a more sustained, lower-cost energy/nucleotide regeneration system that supports extended protein production over time.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
1. Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
- sfGFP
- mRFP1
- mKO2
- mTurquoise2
- mScarlet_I
- Electra2
The amino acid sequences are shown in the HTGAA Cell-Free Benchling folder.
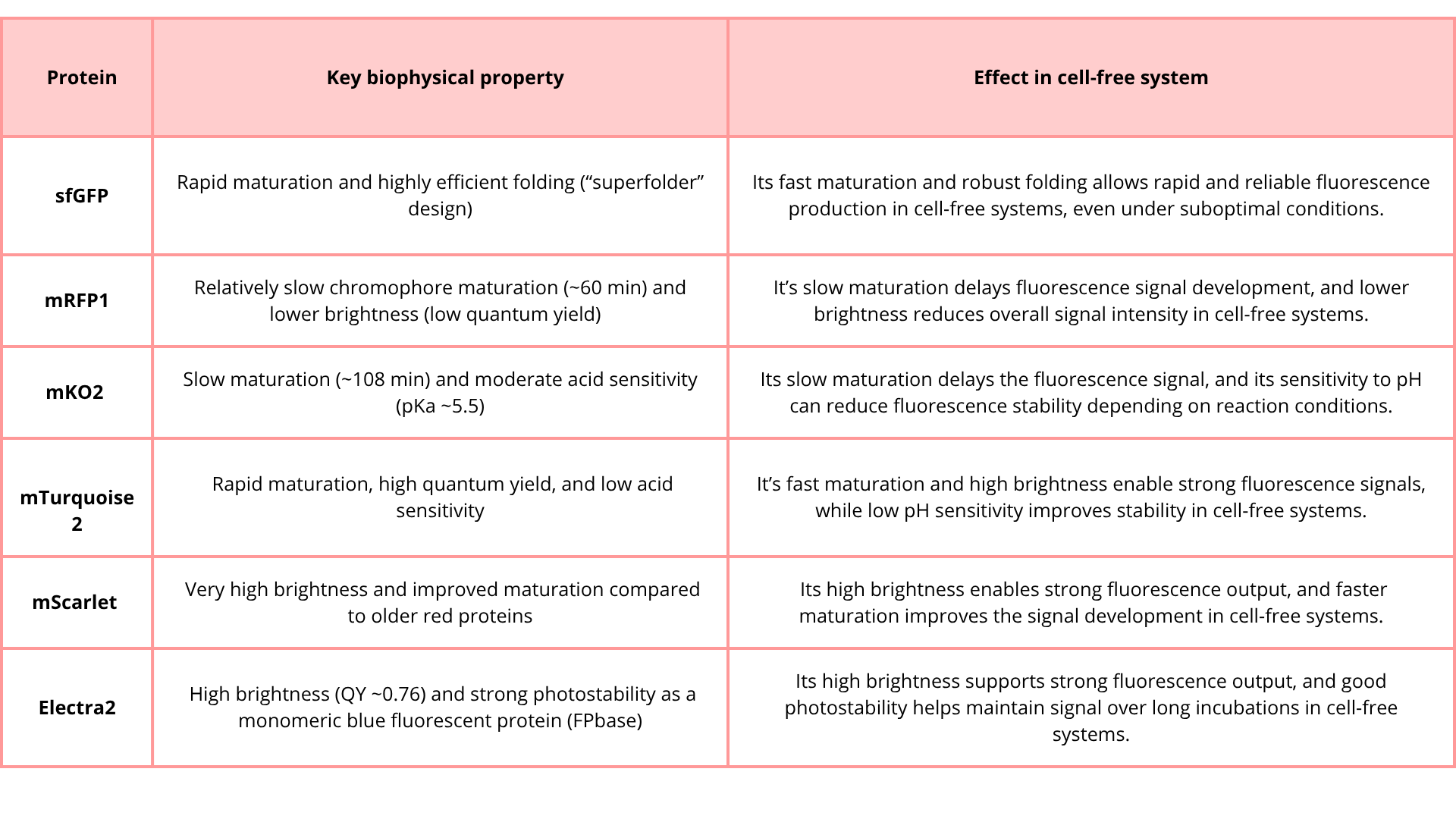
2. Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Protein: mRFP1
Hypothesis: Adding molecular chaperones (e.g., GroEL/ES or DnaK system) and increasing the energy regeneration system in the cell-free mastermix will improve folding efficiency and overall protein yield, leading to increased and more stable fluorescence signal over a 36-hour incubation period.
3. The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here
For mRFP1, I increased magnesium glutamate in the CFPS mastermix because Mg2+ is essential for efficient transcription-translation activity, ribosome stability, and formation of productive translation complexes. In cell-free systems, optimizing magnesium is important because it can directly affect protein yield, folding efficiency, and the total amount of fluorescent protein that accumulates over time, which is especially relevant for a 36-hour incubation.
My original hypothesis also included adding molecular chaperones to improve the folding of mRFP1. Chaperones would be expected to help the newly synthesized protein reach a functional conformation more efficiently, which would increase the amount of properly matured fluorescent protein and therefore improve fluorescence output.
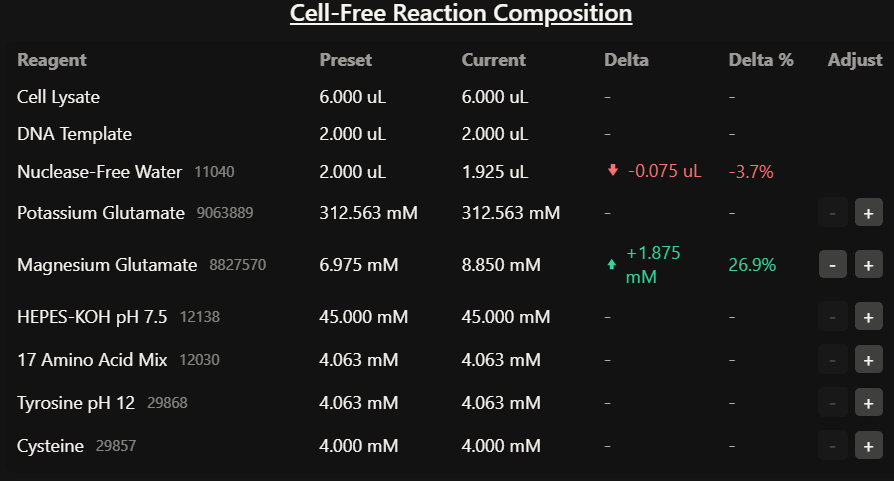

References
- Bartsch, Tabea, et al. “Cell-Free Protein Synthesis with Technical Additives – Expanding the Parameter Space of in Vitro Gene Expression.” Beilstein Journal of Organic Chemistry, vol. 20, 4 Sept. 2024, pp. 2242–2253, https://doi.org/10.3762/bjoc.20.192. Accessed 29 Nov. 2024.
- Calhoun, Kara A., and James R. Swartz. “Energizing Cell-Free Protein Synthesis with Glucose Metabolism.” Biotechnology and Bioengineering, vol. 90, no. 5, 2005, pp. 606–613, onlinelibrary.wiley.com/doi/abs/10.1002/bit.20449, https://doi.org/10.1002/bit.20449.
- “CLS Cell Lines Service GmbH.” Cytion, 2026, www.cytion.com/es/Acerca-de-Cytion/Centro-de-conocimiento/Articulos-y-novedades/Sistemas-sin-celulas-para-la-produccion-de-proteinas-Ventajas-sobre-las-celulas-vivas/. Accessed 23 Apr. 2026.
- Gregorio, Nicole E., et al. “A User’s Guide to Cell-Free Protein Synthesis.” Methods and Protocols, vol. 2, no. 1, 12 Mar. 2019, https://doi.org/10.3390/mps2010024.
- Guzman-Chavez, Fernando, et al. “Constructing Cell-Free Expression Systems for Low-Cost Access.” ACS Synthetic Biology, vol. 11, no. 3, 8 Mar. 2022, pp. 1114–1128, https://doi.org/10.1021/acssynbio.1c00342.
- Lang, Xianshengjie, et al. A Simplified and Highly Efficient Cell-Free Protein Synthesis System for Prokaryotes. 11 Dec. 2025, elifesciences.org/reviewed-preprints/109495, https://doi.org/10.7554/elife.109495.1.
- Lara, Álvaro R. “Producción de Proteínas Recombinantes En Escherichia Coli.” Revista Mexicana de Ingeniería Química, vol. 10, no. 2, 1 Aug. 2011, pp. 209–223, www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S1665-27382011000200006.
- Rubin, Harry. “Intracellular Free Mg2+ and MgATP2- in Coordinate Control of Protein Synthesis and Cell Proliferation.” Nih.gov, University of Adelaide Press, 2024, www.ncbi.nlm.nih.gov/books/NBK507263/.