<Daryoosh Afshinjoo> — HTGAA Spring 2026
About me
A highschool student from Iran, really into creative writing and biology.
A highschool student from Iran, really into creative writing and biology.
Week 1 HW: Principles and Practices
Part 1: Yeast BioShields: Engineering Yeast for Wound Protection The global problem of multi-drug-resistant bacteria (MDR) has been a widespread issue for many years now. The World Health Organization has declared this phenomenon as a major global health threat, further emphasizing the need for a solution to this problem (Renata Urban-Chmiel et al, 2022). In the pursuit of this solution, I structured my idea around a way to reduce the threat of antibiotic-resistant bacteria.
Week 2 HW: DNA read, write and edit
Part 1: Benchling & In-silico Gel Art In order to create my gel art, I first created an account in Benchling and signed into it. Then, I found and downloaded the genebank of the Lambda DNA from New England Biolabs. After that, I simply pasted the DNA sequence into my Benchling workspace, and started experimenting with the different digestions possible using the allowed enzymes.
Part 01: Python Script for Opentrons Artwork In order to create my art, I use the Automation Art Interface, and uploaded my art into the required form. Part 02: Post-Lab Questions Question one: I belive a perfect example of a paper with automated lab use would be one that is also focused on it. Therefore, I found and read the 2019 research paper “Towards a fully automated algorithm driven platform for biosystems design” by Mohammad HamediRad (https://www.nature.com/articles/s41467-019-13189-z). This paper demonstrates a fully automated laboratory platform called BioAutomata that integrates robotics with machine learning to close the entire Design-Build-Test-Learn (DBTL) cycle in synthetic biology with minimal human intervention after initial setup.
The global problem of multi-drug-resistant bacteria (MDR) has been a widespread issue for many years now. The World Health Organization has declared this phenomenon as a major global health threat, further emphasizing the need for a solution to this problem (Renata Urban-Chmiel et al, 2022). In the pursuit of this solution, I structured my idea around a way to reduce the threat of antibiotic-resistant bacteria.
My first proposal was to use GRAS bacteria as a chassis, and modify them to be receptive to the pheromones of infectious bacteria and recognize them as competitors. These modified bacteria could then be cultured and used as a natural shield for the human wounds against infections. This idea was based around the knowledge that bacteria possess a diverse range of mechanisms for inhibiting competitors, including bacteriocins, tailocins, type VI secretion systems and contact-dependent inhibition (CDI) (Booth, S.C., Smith, W.P.J. & Foster et al, 2023).

Figure 1. The dynamics of non-engineered bacteriocin-producing bacteria are complex, guaranteeing the ecological co-existence of antibiotic-resistant and antibiotic-sensitive strains. I believe that, by using yeasts as a naturally immune producer, this ecological balance may be turned in favor of eliminating resistant strains. From Booth, S.C., Smith, W.P.J. & Foster et al, 2023.
But I shifted my focus from using bacteria, be as cheap and fast to grow as they may be, to yeasts, as the former lack the ability to create more complex peptides and are endangered by the antimicrobial peptides (AMPs) they produce themselves. I plan to focus on using genetically engineered yeast within a patients gut or near an infection-vulnerable wound (such as that of a surgery, specially long ones) to eliminate antibiotic resistant bacteria.
While their growth is slightly more time-taking and expensive, yeasts hold many advantages in comparison with GRAS bacteria:
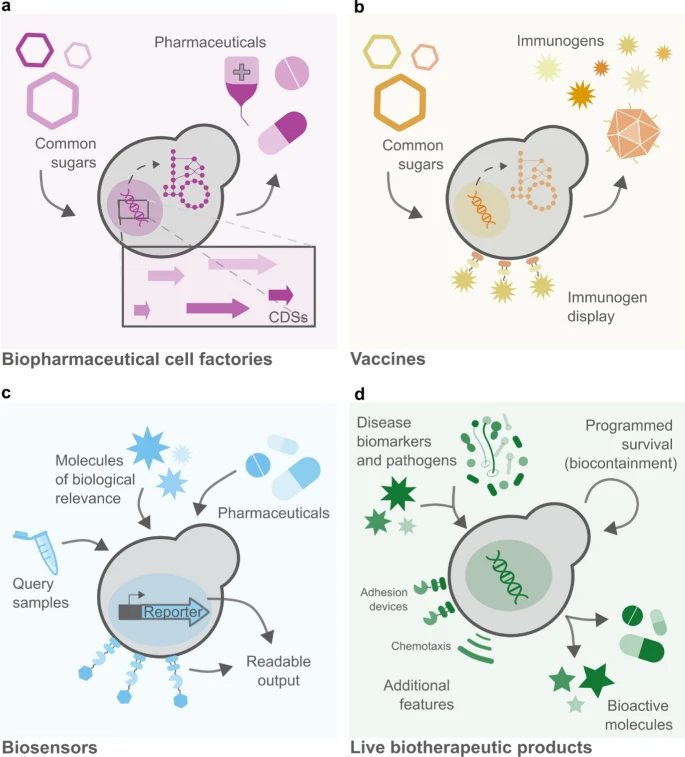
Figure 2. Different applications of the baker’s yeast in medical biotechnology. My idea relates to panel d, demonstrating yeast as a potent biotherapeutic agent. From Maneira et al., 2025.
Genetically engineered yeast can be a promising boon towards the struggle against MDRs, they can also be health risk themselves if not used carefully. For example, there has been reports of a case of bloodstream co-infection of Saccharomyces cerevisiae and Candida glabrata while using micafungin (Furuya K, Ito K, Sugiyama K, Tokuda S, Kanemoto H, Kamei K, Shimada T et al, 2023), which can cause complications regarding the use of yeast near open wounds. So, several policies are needed in order to ensure that yeast remain harmless towards patients.
For my final project and according to the aforementioned sub-goals, I suggest the following actions: A) Implementing a genetic kill switch: Actors: Researchers, Academia.
By creating a designed kill switch within the yeast, we can design the yeast to be neutralized in the case of it entering the patient’s blood.
B) Optimization of growth methods for the yeast: Actors: Researchers, Medical practitioners.
This action reduces the cost of production by increasing the yield of the yeast, using aerobic conditions and fed-batch cultivation to avoid the Crabtree effect (Gregory J. O. Martin and Sitha Chan et al, 2024).
In order to fund the bioprocess, we can encourage investors and academic institutes. This action can ensure the resources needed to fully develop the product.
| Does the option: | Action 1: Implementing a genetic kill switch | Action 2: Optimization of growth methods for the yeast | Action 3: Securing governmental or private funds |
|---|---|---|---|
| Ensuring biosafety | |||
| • By rendering the yeast incapable of causing harm to patients? | 2 | 0 | 0 |
| • By ensuring its containment and inability to spread outside its specified area of use? | 3 | 0 | 0 |
| Keeping the product affordable and accessible | |||
| • By lowering the cost of making the final yeast-based product through bioprocess optimization? | 0 | 3 | 1 |
| • By improving production capacity and upscaling the process? | 0 | 2 | 3 |
| Other considerations | |||
| • Minimizing costs and burdens to stakeholders | 1 | 3 | 2 |
| • Feasibility? | 2 | 3 | 3 |
During my research I have made some assumptions:
Answer: Even though different eukaryotic and prokaryotic DNA polymerases have different error rates, which is also subject to the environment (Balint et al., 2024) in which they function, the apporixmate error rate of all polymerases is about 1 mutation per 10,000 bases replicated. Considering the fact that the human genome is roughly 3.2 billion base pairs long, after each round of replication, one should expect to see 320,000 mutations, which is far from what is the case in the real world. This discrepancy is explained by the active role of polymerase proof-reading and error correction systems active during and after DNA is replicated by DNA polymerase.
Answer: There are 64 different codons that make up the genetic code of all living organisms, of which 61 combinations are used to code amino acids and 3 are used to stop translation. The main reason behind the existence of more than 1 codon for each of the amino acids goes back to the previous question. Even with error correction in place, there may be some mutations that pop up after each round of replication. Therefore, having more than 1 codon for certain amino acids helps preserve the function and amino acid composition of the proteins mutated genes code for.
Answer: The typical method for synthesizing DNA (chemically) is phosphoramidite synthesis (Hoose et al., 2023). As far as I could understand it, this method involves loading modified bases on a solid silicon platform, removing a protecting group from them, and binding them to another base with the protecting group still attached. See the figure below for more information.
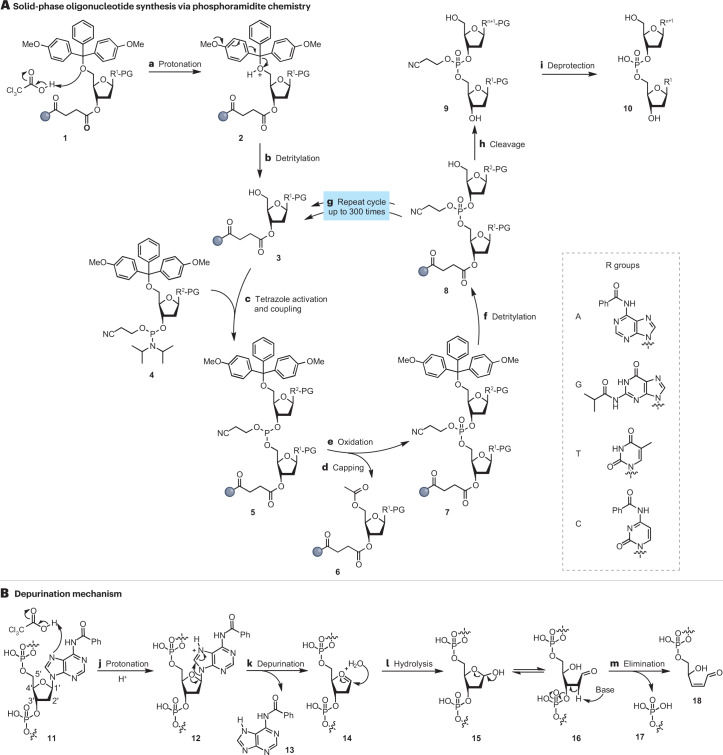
Figure 3. The phosphoramidite synthesis method. Steps h and i are important for the answer to the next question.
Answer: As it can be observed in the Figure 3, sometimes the added bases do not stay in the oligo synthesized and separate from it as an unwanted byproduct. The article introduces this as acid-catalyzed depurination, which is the reason behind the inefficiency of direct synthesis for breaking the 200nt cap.
Answer: As it was implicitly mentioned in the previous question, direct oligo synthesis using the phosphoramidite method has ‘imperfect’ cycles, meaning that at each cycle there is a chance for one or more nucleotides to break apart from the growing oligo and hinder its growth. This problem, which becomes more serious and ultimately breaks and stops oligo synthesis as the oligo grows longer, disables us from creating a 2000bp gene by direct synthesis.
Hoose et al., 2023 instead names two other methods that can be used to assemble synthesized DNA molecules like LEGO bricks: Gibson Assembly and Polymerase Cycling Assembly (see Figure 4).
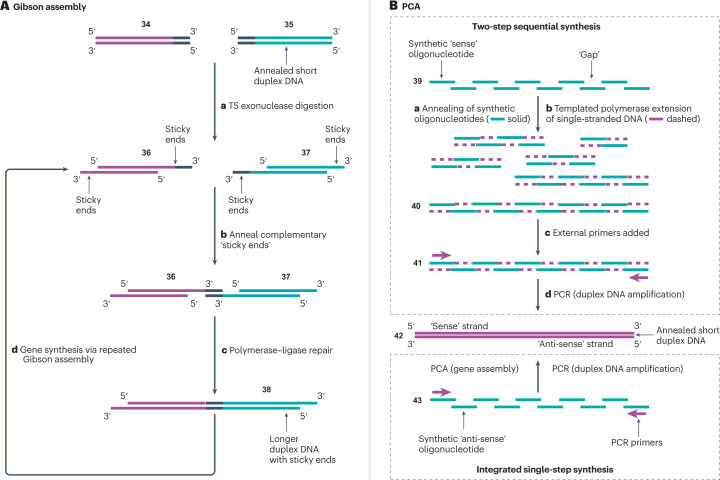
Figure 4. Two DNA assembly methods.
Answer: Cysteine, Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, Tyrosine, and Valine are considered essential amino acids for all animals (Hou et al, 2018).
Interestingly, the concept behind ‘Lysine contingency’ is similar to auxotrophy, a situation where an organism lacks the ability to make one or more compounds necessary for its growth and sustenance (usually with reference to its wild-type relatives). However, since Lysine is naturally an essential amino acid and not produced by the animal itself, the ‘Lysine contingency’ is more fiction than (feasible) reality.
I acknowledge the use of AI, particularly Gemini, for early fact checking of my raw ideas. “One thing that I was curious about: Can yeast cells produce and secrete more potent and a more diverse array of engineered/heterologous antimicrobial peptides (AMPs) than probiotic bacterial cells creating bacteriocin and other types of antimicrobial factors or AMPs?”
In order to create my gel art, I first created an account in Benchling and signed into it. Then, I found and downloaded the genebank of the Lambda DNA from New England Biolabs. After that, I simply pasted the DNA sequence into my Benchling workspace, and started experimenting with the different digestions possible using the allowed enzymes.



Eventually, I came to a design I believed to be symmetrical and graphically pleasing to look at. A nice, bored looking elephant!
 The design itself
The design itself
 What I had in mind, image created by me via Pixil
What I had in mind, image created by me via Pixil
Unfortunetly, I did not have he required lab access to complete this part. Hopefully, I could come back and revisit it once I do.
3.1. For my protein, I chose the ardA anti restriction protein, found in many strains of E. coli. My reason for choosing this protein is related to one of my original ideas for my final project. Which was to incase the phages used to eliminate antibiotic resistent bacteria, such as Salmonella enterica, with the proteins that block the restriction enzymes that protect the bacteria from these viruses. So, I found and downloaded the sequence of my protein in Unipprot.

3.2 & 3.3. Afterwards I reverse translated my protein sequence in Cusabio, and codon optimized it using Vector Builder.
 Reverse translation using Cusabio
Reverse translation using Cusabio
 Codon optimization using Vectro Builder
Codon optimization using Vectro Builder
3.4. For the production of my protien, I belive In vitro, or cell-free protein expression to be an optimal technique. As although in vitro expression is not practical for commercial large-scale recombinant protein production, it has a variety of features that make it considerably more useful and flexible for many research applications, such as labeling of proteins with stable isotopes for structural analysis and production of functional virons or toxic polypeptides.
I had a lot of trouble sigining into Twist due to the fact that I am not part of an institution or lab that. As mentioned before, I am in highschool, and was not sure if that really could be counted. Adding to that problem is the fact that my VPNs, which I need to use Twist, have been acting up. Hopefully I can get them running until the next lecture.
5.1. i. If given no limitation, I would want to read and sequnce the DNA of the naked mole rat. This creature is virtually immune to cancer and many other age-related decline, therefore managing to somehow, someday, impliment its secrets into humans in a safe and ethical way could be a wonderful advance in biology. ii. 1. To sequence the Naked Mole Rat (NMR), I would choose Pacific Biosciences (PacBio) HiFi Sequencing. Specifically, I would use the PacBio Revio system. While standard short-read sequencing (like Illumina) is great for basic spell-checking, the NMR’s “superpowers” likely lie in complex structural variations—gene duplications, insertions, and repetitive regions—that short reads simply cannot resolve. To see the full picture, we need long, unbroken reads with high accuracy. This method is classified as Third-Generation Sequencing. Third-generation (PacBio, Nanopore) sequences single molecules of DNA in real-time without the need for PCR amplification to generate the signal. It observes the DNA polymerase adding nucleotides as it happens. 2. The input is High Molecular Weight (HMW) Genomic DNA, and its essential steps are: 1- Extraction: We extract gDNA using a gentle lysis method to keep fragments massive. 2- Shearing: We use mechanical shearing (like a Megaruptor) to break the DNA into a consistent, large size range. 3- DNA Damage Repair: Enzymes are added to fix any nicks or gaps in the sugar-phosphate backbone caused by the shearing. 4- Adapter Ligation (The Critical Step): We ligate SMRTbell adapters to both ends of the double-stranded DNA. 5- Primer Annealing & Polymerase Binding: We anneal a sequencing primer to the adapter and bind the DNA polymerase enzyme to the complex. The sample is now ready for the machine. 3. The primary output is HiFi Reads.Format: Typically FASTQ or uBAM files.Characteristics:Length: Average read lengths of 15kb to 20kb (kilobases).Accuracy: Q30 or higher (99.9% accuracy).Value: This output provides the best of both worlds: the length to span difficult repetitive regions (common in complex mammal genomes) and the accuracy to detect tiny mutations (SNPs) without needing massive computational correction.
5.2. i. If, once again, given not limits, I would synthesize and refactor the entire Nitrogen Fixation (nif) gene cluster (approx. 20 genes) found in soil bacteria, optimized for expression in cereal crops like corn or wheat. This is arguably the most impactful application for global sustainability. It would eliminate the need for synthetic nitrogen fertilizers, which are a massive source of greenhouse gases and water pollution (runoff). It would trigger a second Green Revolution, allowing crops to “feed themselves.” ii. To synthesize the genetic system I described, I would use Enzymatic DNA Synthesis (EDS). 1. Enzymatic synthesis builds DNA one base at a time using a “Stop-and-Go” cycle. Firstly, A starting DNA fragment (an “initiator”) is tethered to a solid support surface in a microscopic well. Then, An engineered TdT enzyme and a specific nucleotide (A, T, C, or G) are added. TdT is a “template-independent” polymerase, meaning it can add bases without needing a guide strand. After that The excess enzymes and unused nucleotides are washed away, and mild solution is added to remove the “blocker” (the terminator) from the newly added base. This exposes the “hook” (the 3’-OH group) so the next base can be attached. 2. While EDS is the “frontier” technology of 2026, it still faces hurdles in three key areas, such as speed, accuracy, and scalability.
5.3. i. Lastly, I would edit the TP53 gene in human somatic cells, often called the “Guardian of the Genome”. In humans, we have two copies of p53. If they mutate, cancer often follows. I would edit the genome to include multiple redundant, hyper-sensitive copies of this gene, similar to what we see in elephants (who rarely get cancer despite their size). This would effectively “auto-update” our cellular security system. The moment a cell starts to turn cancerous due to UV damage or chemical exposure, the enhanced p53 would detect the break and force the cell to repair itself or safely self-destruct. ii. To perform the targeted DNA edits described, I would use a combination of CRISPR-Cas9, Base Editing, and Prime Editing. 1. The Cas9 enzyme creates a Double-Strand Break (DSB) at a precise location. The cell then tries to fix this break using Non-Homologous End Joining (NHEJ). This process is messy and often deletes or inserts random bases, which effectively “breaks” or silences the gene. These use a “deactivated” Cas9 (dCas9 or nickase) that cannot cut both strands of DNA. Instead, it is fused to other enzymes. 2. Editing requires meticulous “software design” before the biological work begins. First, we’ll identify the exact genomic coordinates. Then, we use software to create a 20-nucleotide sequence that matches the target. We must ensure a PAM sequence exists next to our target. 3. A major limitation is Mosaicism. Even with a near perfect edit, the technology might only work in about maybe 70% of the cells in a sample.
In order to create my art, I use the Automation Art Interface, and uploaded my art into the required form.

Question one: I belive a perfect example of a paper with automated lab use would be one that is also focused on it. Therefore, I found and read the 2019 research paper “Towards a fully automated algorithm driven platform for biosystems design” by Mohammad HamediRad (https://www.nature.com/articles/s41467-019-13189-z). This paper demonstrates a fully automated laboratory platform called BioAutomata that integrates robotics with machine learning to close the entire Design-Build-Test-Learn (DBTL) cycle in synthetic biology with minimal human intervention after initial setup.
Figure from the article showing the overall workflow of the BioAutomata.
As a proof-of-concept, the authors applied it to optimize the lycopene biosynthetic pathway in E. coli by fine-tuning expression levels of three genes (crtE, crtB, and crtI), exploring a combinatorial space of ~13,824 possible variants while evaluating fewer than 1% of them.
Question two: As for the integration of lab automation in my own final project, I belive the optimal choice would be the Ginkgo Nebula in biofoundries. Ginkgo Nebula is designed for exactly this: outsourcing the high-throughput “Build” phase. Instead of cloning one gene at a time, you submit a library of designs. I intend to use this automation in creation of a killswitch in my “Yeast Biosheild”.
For my final project ideas, I decided to build upon the projects I came up with for the first homework which seemed to hold great potential: 1. The use of yeast as a biosheild against bacteria, specially those with antibiotic resistence. 2. The use of moquitoes as a form of vaccination. While this idea was originally pushed aside because it has already been done before, I have recently decided to combine it with the use of gene editing to have antibodies be sprayed onto the blood the mosquito drinks as a way of stopping parasitic diseases being transferred into the saliva gland, and also improving the modified mosquitoes’ chance of reprodction through improvements such as more thirst for blood, better fly patterns and more keep sense of sight. 3. My final idea was the use of artficaial cells to replace damaged nerves and, eventually, the automation of their repair.