First, describe a biological engineering application or tool you want to develop and why. My plan for the final project is a synthetic membrane that has Mesenchymal Stem Cells Microvesicules (which have scientifically proven regenerative and other positive properties) intercalating inbetween the membrane’s layers. Which could be used for burn wounds and/or donation organs preservation while in transportation.
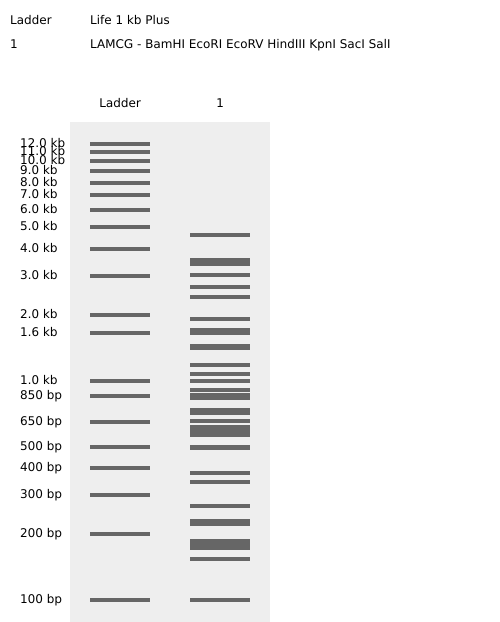
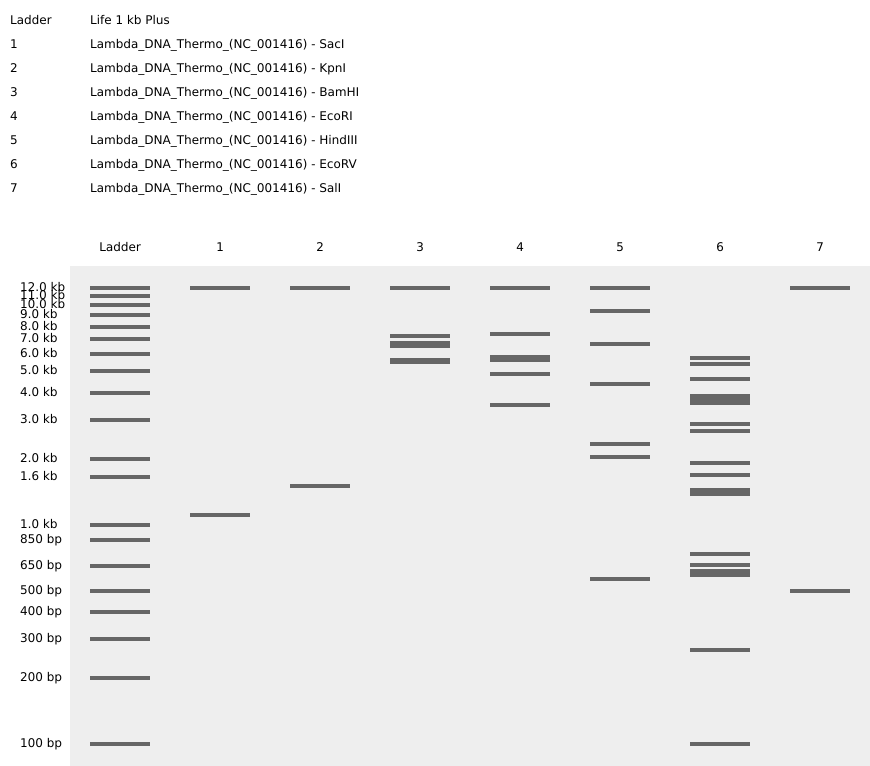
Part 1: Benchling & In-silico Gel Art: Simulate Restriction Enzyme Digestion with the following Enzymes: Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. I tried to make a pattern that looked like a staircase going down by using logic but couldn’t quite seem to get it right…
Part 1: Python Script for Opentrons Artwork I used Ronan’s provided GUI and a nice little pixelart of a kidney to try out the design and figure out coordinates. Python coding was new and quite… surprising. But using the toad.py provided by the USFQ node I figured out how to get the code to work. This is a kidney art that i’ll try to print. Hopefully it actually resembles something!
Part A. Conceptual Questions How many molecules of amino acids do you take with a piece of 500 grams of meat? Roughly 3 x 10^24 individual amino acid molecules.
1- Why do humans eat beef but do not become a cow, eat fish but do not become fish? Because digestion is a disassembly line. We completely chop up their proteins into individual amino acid building blocks, then use our own DNA blueprints to rebuild them into human.
Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM The human SOD1 sequence (UniProt P00441) was retrieved and the A4V mutation introduced by substituting alanine for valine at residue 4:
Uniprot (P00441) Sequence: MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Mutated (A4V) Sequence: MATVKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Assignment: DNA Assembly 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Phusion High-Fidelity PCR Master Mix Components:
Phusion DNA Polymerase: A high-accuracy enzyme with 3′→5′ exonuclease (proofreading) activity that minimizes mutations during amplification dNTPs (Deoxynucleotide Triphosphates): The nucleotide building blocks (A, T, C, G) used to synthesize the new DNA strand.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) 1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Intracellular Artificial Neural Networks (IANNs) transcend the limitations of binary Boolean logic by enabling analog and graded signal processing within a cell. While traditional genetic gates are restricted to “ON/OFF” states, IANNs can integrate multiple continuous environmental inputs and assign them specific “weights,” allowing the cell to make nuanced decisions based on a threshold of combined signals. This analog capability is particularly superior for pattern recognition and processing complex biomarkers, as it mimics natural biological decision-making more closely than rigid digital circuits. Furthermore, IANNs can often achieve high levels of computational complexity with fewer genetic parts, as they leverage the inherent non-linearities of biochemical reactions as natural “activation functions,” thereby reducing the metabolic burden on the host organism compared to massive, multi-gate Boolean architectures.
General homework questions 1- Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. In traditional in vivo (inside the cell) methods, the cell’s own survival is the priority. In CFPS, your protein is the priority. You can add non-natural amino acids, chaperones, or labeling molecules directly to the mix without worrying about transport across a cell membrane. You can monitor and tweak variables like pH, temperature, and redox potential in real-time. If a protein is lethal to a living cell (e.g., a pore-forming toxin), CFPS is the only way to produce it because there is no “cell” to kill. You can go from a linear DNA template (PCR product) to a protein in hours, whereas cell production requires days for cloning and transformation.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork I missed the date to contribute :c Will try my best to become a TA next year!
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction. Component Roles Component Category and their role in the Reaction:
Subsections of Homework
Week 1 HW: Principles and Practices
1. First, describe a biological engineering application or tool you want to develop and why.
My plan for the final project is a synthetic membrane that has Mesenchymal Stem Cells Microvesicules (which have scientifically proven regenerative and other positive properties) intercalating inbetween the membrane’s layers. Which could be used for burn wounds and/or donation organs preservation while in transportation.
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
I can’t seem to see much possibility for malfeasance after proper testing. But the two biggest questions that arise are: Reproductibility and Universal (or almost) access to the treatment.
A. Reproductibility: This step is essential to make sure that the design is simple enough to be able to be reproduced by as many labs as possible even in the harshest conditions.
Sub goals:
Testing and optimizing the design so it uses the simplest technology possible.
Trialing in several conditions and labs across the world.
B. Ensuring universal access: This step is to make sure that, after proper testing, the therapy is accessible by the general population and not just a select few.
Sub goals:
Collaboration with public health systems globally.
Ensuring no malicious patenting of the design can be made.
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Action 1: Creation of a bioethics and biosafety commitee;
Purpose: Making sure that every material is non-harmful and is in fact composed of what it is expected to. Example: Making sure that the MV’s are indeed microvesicules and that their content is beneficial to health.
Design: Proper lab testing protocols such as Flow Cytometry, Electron Microscopy and such; And parameters for consistency in the design.
Risk of failure and “success”: Failure: the output is not as expected and the consistency of the therapy can’t be guaranteed. Success: the design is properly tested and may be applied to patients willing to test.
Action 2: Implementation of proper trials and validation;
Purpose: Testing the therapeutic with willing and acknowledging subjects to ensure that the effects are real, beneficial and consistent.
Design: Double blind studies.
Risk of failure and “success”: Failure: the therapeutics has no effect or is harmful to whoever is subjected. Success: the effects are, in fact, benign and consistend independent of characteristics of the patient.
Action 3: Preemptive adoption of a framework for dealings with regulatory organs;
Purpose: Making sure that, after the testing is done, the therapy can be introduced to as many patients as possible while being in concordance with local regulations.
Design: Keeping proper documentation of all design steps, dialoguing with regulatory organs to plan proper introduction to new patients.
Risk of failure and “success”: Failure: the design is proper and has effects but it can’t be introduced due to not fitting regulations. Success: the design is introduced properly to more populations.
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
1
n/a
n/a
• By helping respond
1
n/a
n/a
Foster Lab Safety
• By preventing incident
2
n/a
n/a
• By helping respond
2
n/a
n/a
Protect the environment
• By preventing incidents
1
1
n/a
• By helping respond
1
3
n/a
Other considerations
• Minimizing costs and burdens to stakeholders
2
3
1
• Feasibility?
1
2
1
• Not impede research
1
2
1
• Promote constructive applications
3
1
1
5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties
I would prioritize making sure that international and national regulations do not become an obstacle for the application of the therapy. While it may seem idealist to aim for it, I believe in universal healthcare and the feasability of more accessible therapies.
6. Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
Mainly the use of creatures (animals and others) that may not consent to their usage in research. For governance actions, regulations and societies and maybe even the general public should make sure that the subjects of the research are treated in the most humane way possible.
Week 2 lecture preparation
Professor Jacobson:
1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
1:10⁶. The human genome consists of about 3.2 billion base pairs. The discrepancy is dealt with using proofreading mechanisms and other corrective measures.
2. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
According to uniprot the average human protein consists of about 400 aminoacids. Most AA’s have multiple codons which encode them (being redundant). That makes it so that there are more possibilities of DNA coding than humanly possible to consider. In practice, some of the reasons why they wouldn’t work are: codon usage bias (some codons are preferred due to optimal functioning), formation of CPG islands promoting methylation and thus leaving the DNA useless. Other reasons include structural integrity and such…
Dr. LeProust:
3. What’s the most commonly used method for oligo synthesis currently?
Phosphoramidite chemistry on solid-phase synthesizers
4. Why is it difficult to make oligos longer than 200nt via direct synthesis?
Could not find this in the slides, had to make use of the emergencial AI resource through the use of Deepseek AI (Where I just made the same question as the homework). Apparently, it is because the efficiency of coupling is not 100%, accumulating various errors and unwanted reactions after said length.
5. Why can’t you make a 2000bp gene via direct oligo synthesis?
Reffering to the answer above… The actual yield would be too low. Deepseek said that that’s the reason why “synthetic biology relies on assembly methods rather than direct synthesis for genes and long constructs.”
Mr. George Church:
6. [(Advanced students)] Given the one paragraph abstracts for these real 2026 grant programs sketch a response to one of them or devise one of your own:
I chose the BoSS – BioStabilization Systems program from Arpa-H.
My response towards this problem would be to employ AI to find more stable molecular structures of existing or even of new proposed drugs. Simulating responses towards a multitude of different conditions, and trying to predict different environmental parameter threshholds that are supported by the molecules.
Week 2 HW: DNA Read Write and Edit
Part 1: Benchling & In-silico Gel Art:
Simulate Restriction Enzyme Digestion with the following Enzymes:
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
I tried to make a pattern that looked like a staircase going down by using logic but couldn’t quite seem to get it right…
Part 3: DNA Design Challenge:
Protein Choice: AFP III (Antifreeze Protein Type III)
I chose AFP III because it has an incredible practical application: allowing organisms to survive in sub-zero temperatures. In biotechnology, it is used to create smoother ice cream textures (by preventing large ice crystals) and in the preservation of organs for transplant. It is a fascinating example of how nature solves extreme physical challenges through molecular engineering.
Protein Sequence (UniProt P05140):sp|P05140|ANP3_MACAM Antifreeze protein type 3 NQASVVANQLIPINTALTLVMMKAEVVTPMGIPAEEIPNLVGMQVNRAVPLGTTLMPDMV KNY
Codon Optimization
In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
This answer takes back to last homework, due to codon bias, using a sequence that’s not ideal for your chosen organism, you may have some problems with codons used tipically by the species that produces said protein naturally. I picked E.Coli because it is the standart for easy cultivation.
I ran VectorBuilder’s tool with some uncertainty but it seemed good because it was the only one that said ‘free’ on the google search.
This is the optimized sequence:
AACCAGGCGAGCGTGGTGGCGAATCAGCTGATTCCGATTAATACCGCCCTGACCCTGGTGATGATGAAAGCCGAAGTGGTGACCCCGATGGGCATTCCGGCGGAAGAAATTCCGAACCTGGTGGGCATGCAGGTGAATCGCGCGGTGCCGCTGGGCACCACCCTGATGCCGGATATGGTGAAAAATTAT
You have a sequence, now what?
Cell-Dependent Expression (In Vivo): The most common method is to transform E. coli cells with the plasmid. When an inducer (like IPTG) is added to the growth media, the bacteria’s internal machinery (RNA Polymerase and Ribosomes) begins to transcribe the DNA into mRNA and translate that mRNA into the physical AFP III protein.
Cell-Free Protein Synthesis (In Vitro): Alternatively, one could use a “cell-free” extract. This involves mixing the DNA directly with a “molecular soup” containing ribosomes, enzymes, and amino acids in a test tube.
Part 4: Prepare a Twist DNA Synthesis Order:
I made an account and twist doesn’t seem to allow me to use the website, saying I need to contact a distributor…
I checked with a fellow brazilian colleague and he seems to be having the same issue, maybe it’s a regional block?
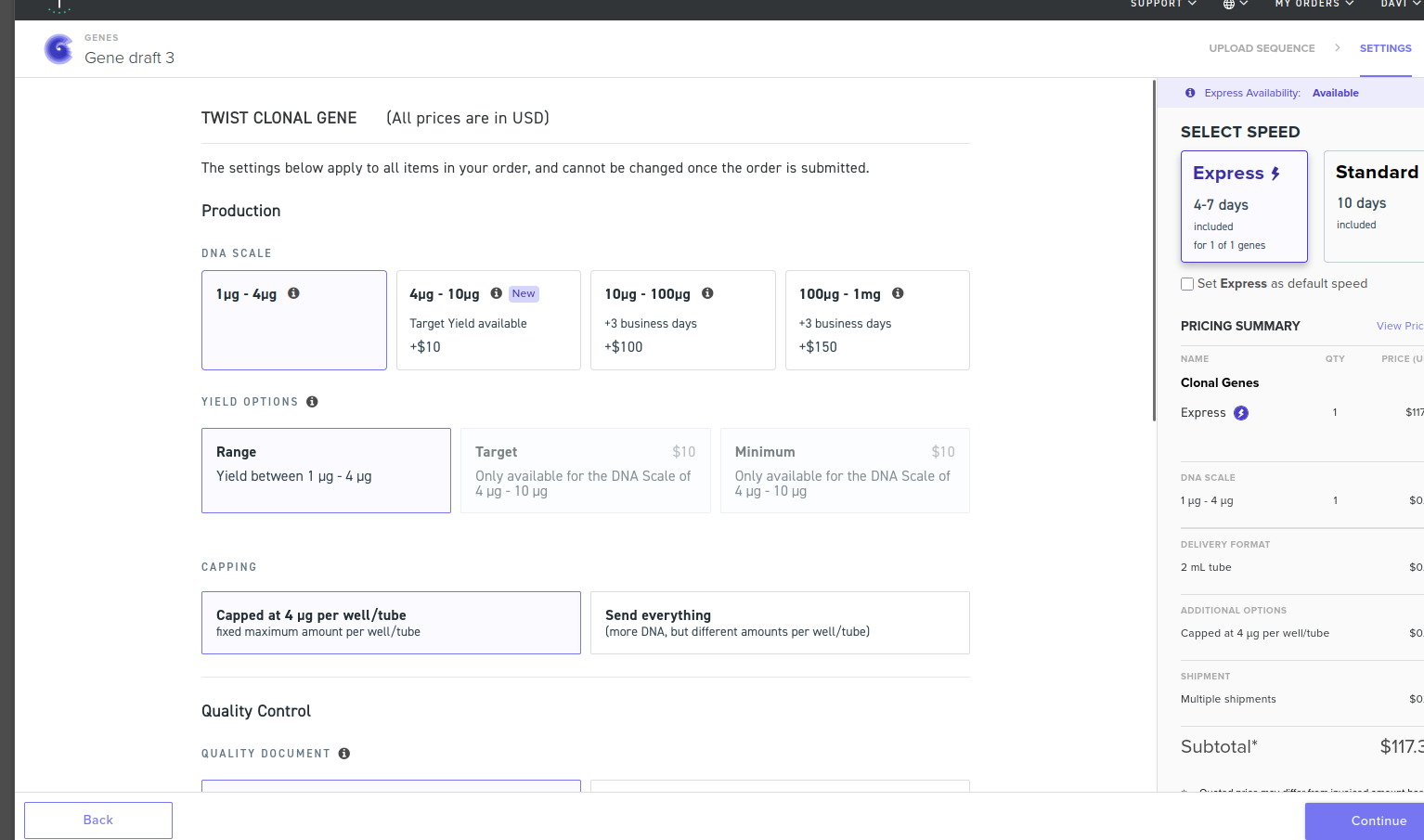
05/25/2026 - I just did this part because of the final project, so I’ll just post the cronological order of processess and hope for the best…
First I optimized the sequence using idtDNA Codon Optimization tool to optimize the sequence.
I also ran digestion to see the restriction sites which showed 0 cuts for the parts that seemed possibly problematic.
Then, I exported as FASTA, and proceeded to try to use twist once again.
I uploaded the FASTA to twist and there were a LOT of warnings and one error, proceeded to try and optimize the sequence.
I had to have BT1311 promoter replaced with synthetic Bacteroides P1 promoter (Whitaker et al. 2017) for synthesis compatibility, retaining equivalent constitutive expression activity. Changed it in benchling and redid everything…
Still with issues… to finish the homework will change the ClyA portion to sfGFP + a HisTag…
For the Twist ordering exercise, sfGFP was used as a placeholder CDS to demonstrate cassette design workflow. The actual therapeutic construct (ClyA-P2) presents synthesis challenges due to coiled-coil repeat regions in ClyA, which would require either: (1) gene fragmentation and assembly, (2) use of a truncated ClyA transmembrane domain only, or (3) a custom synthesis provider tolerant of complex sequences.
Here’s the final order screen (I hope this constitutes as a done homework…)
Part 5: DNA R/W/E:
In graduation I learned a ton about mangroves and their importance, so, to sequence the mangrove microbiome, I’d target the DNA in the sediment and root zones of these coastal forests. Mangroves are massive “blue carbon” sinks, and by reading the genetic material of the microbes living there, we can find hidden enzymes that fix nutrients or even break down plastics in salty water. This helps us understand how these forests fight climate change and gives us new “bio-tools” for cleaning up the environment.
I would use Illumina Sequencing for this because it’s the best for handling the complex mix of species found in soil. This is a second-generation technology that uses “sequencing by synthesis” to read millions of DNA fragments at once. First, I’d extract the DNA, break it into tiny pieces, and attach “adapters” so they can stick to a glass flow cell. Inside the machine, the DNA is copied into clusters, and as fluorescently labeled bases are added, they flash a specific color for each letter (A,T,C, or G). A camera captures these flashes, and the software translates them into digital FASTQ files that we can piece together like a giant puzzle.
For the “DNA Write” part, I’d synthesize a caffeine biosynthetic pathway to put into yeast. This would let us “brew” caffeine in a lab without needing huge coffee or tea plantations, saving a lot of land and water. I’d use silicon-based synthesis (like Twist Bioscience), which uses a silicon chip to build thousands of DNA strands at once using a chemical process called phosphoramidite chemistry. We’d print short pieces of the caffeine genes and then stitch them together into a full circuit. The main catch is that it’s hard to write very long or repetitive sequences, but it’s incredibly fast and scalable for these kinds of metabolic projects.
Finally, I’d use CRISPR-Cas9 to edit a banana or tomato so it grows with the caffeine pathway built right in. The idea is to create a “Caffeine Fruit” for a natural, healthy morning energy boost. CRISPR works like a molecular GPS and scissors; a guide RNA leads the Cas9 enzyme to a specific spot in the plant’s DNA to make a cut. By providing a “donor template” with our caffeine genes, the plant’s own repair system pastes the new instructions into its genome. We’d use a “shuttle” like Agrobacterium to get the CRISPR tools into the plant cells. It’s a powerful method, though it can sometimes be slow to grow a full plant from an edited cell, and we have to be careful that the enzyme doesn’t cut the DNA in the wrong place.
Week 3 HW: Lab Automation
Part 1: Python Script for Opentrons Artwork
I used Ronan’s provided GUI and a nice little pixelart of a kidney to try out the design and figure out coordinates.
Python coding was new and quite… surprising. But using the toad.py provided by the USFQ node I figured out how to get the code to work.
This is a kidney art that i’ll try to print. Hopefully it actually resembles something!
This is a small test of the gui and code, it reads nefrologia (nephrology in portuguese) and has a small heart.
Part 2: Post lab
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
https://www.nature.com/articles/s44172-025-00575-3 is the paper I picked because it initially uses opentrons for a small scale production of a living cell hydrogel. I found it quite alike my proposed final project product so it may serve as a nice starting point to figure out fabrication. Although the paper uses their product for drug trialing instead of what I proposed.
I reckon the best use of automation for my idea is to help with the fabrication with properly placing the scaffolds and the microvesicules in between them.
Part 3: Final project ideas
I uploaded my ideas to the slide deck. Here is a copy of my slide and ideas.
Week 4 HW: Protein design P1
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat?
Roughly 3 x 10^24 individual amino acid molecules.
1- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because digestion is a disassembly line. We completely chop up their proteins into individual amino acid building blocks, then use our own DNA blueprints to rebuild them into human.
2- Why are there only 20 natural amino acids?
It’s a perfect sweet spot. Those 20 give us enough chemical variety to build everything life needs, but using more would make the system too complicated and error-prone.
3- Can you make other non-natural amino acids? Design some new amino acids.
Yes, one could for example make a glowing one by attaching a light-emitting molecule
4- Where did amino acids come from before enzymes that make them, and before life started?
They were probably cosmic cooking experiments. Lightning and UV rays zapped simple gases and water in the early Earth’s atmosphere, spontaneously creating them.
5- If you make an α-helix using D-amino acids, what handedness would you expect?
You’d get a left-handed helix. They’re mirror images, so using the mirror-image building blocks flips the spiral direction.
7- Can you discover additional helices in proteins?
Yes. We’re still finding new ones. The π-helix (a wider, rarer spiral) and the 310-helix (a tighter, stubby one) are two examples already discovered.
8- Why are most molecular helices right-handed?
It’s probably a historical accident. Once early life settled on right-handed sugars and left-handed amino acids, their interactions locked everything into a right-handed spiral trend.
9- Why do β-sheets tend to aggregate? What is the driving force?
Their edges are like sticky Velcro. They have exposed backbone “claws” (called hydrogen bond donors and acceptors) that desperately want to latch onto another sheet’s identical claws to hide from water.
Part B: Protein Analysis and Visualization
Renin I picked renin because it’s kind of a trend here on my lab. It’s basically an enzyme that regulates blood pressure and volume.
The sequence I got from uniprot(listed as: P00797 · RENI_HUMAN): MDGWRRMPRWGLLLLLWGSCTFGLPTDTTTFKRIFLKRMPSIRESLKERGVDMARLGPEWSQPMKRLTLGNTTSSVILTNYMDTQYYGEIGIGTPPQTFKVVFDTGSSNVWVPSSKCSRLYTACVYHKLFDASDSSSYKHNGTELTLRYSTGTVSGFLSQDIITVGGITVTQMFGEVTEMPALPFMLAEFDGVVGMGFIEQAIGRVTPIFDNIISQGVLKEDVFSFYYNRDSENSQSLGGQIVLGGSDPQHYEGNFHYINLIKTGVWQIQMKGVSVGSSTLLCEDGCLALVDTGASYISGSTSSIEKLMEALGAKKRLFDYVVKCNEGPTLPDISFHLGGKEYTLTSADYVFQESYSSKKLCTLAIHAMDIPPPTGPTWALGATFIRKFYTEFDRRNNRIGFALAR It has 406 AA’s and the most frequent is Leucina (L) with 34 appearances. It has 432 homologs.
The structure for it was deposited at 1992-02-05. And it has a resolution of 2.5Å. It belongs to the: pepsin-like family and it has a N-acetyl-D-glucosamine apart of the molecule.
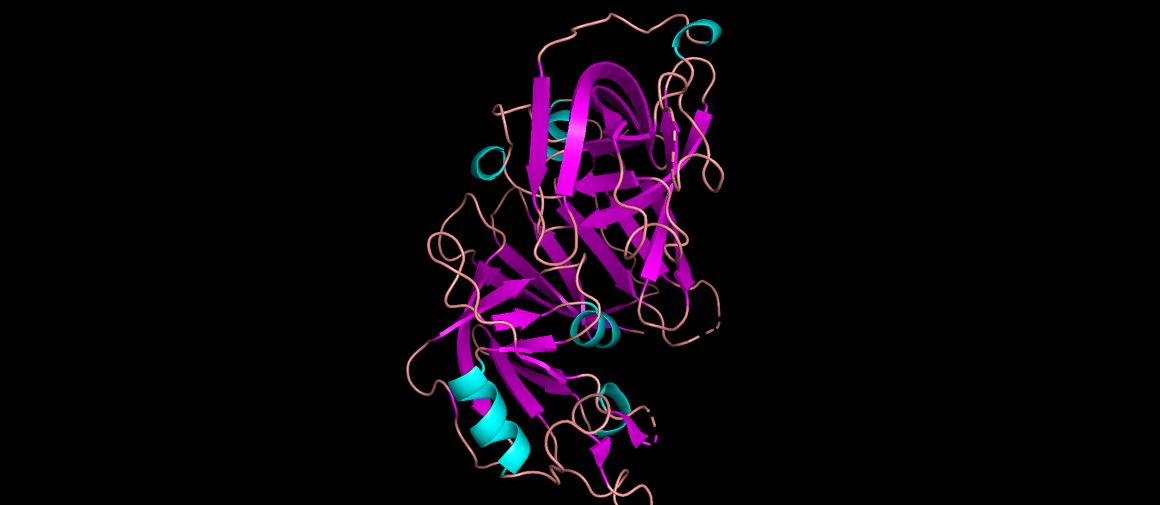
Renin as cartoon, colored by secondary structure… It mainly has beta sheets.
Renin as ribbons, colored by residues. Code:
#Hydrophobic (Ala, Val, Ile, Leu, Met, Phe, Trp, Pro, Tyr) color yellow, resn ALA+VAL+ILE+LEU+MET+PHE+TRP+PRO+TYR #Positive charges (Arg, Lys, His) color blue, resn ARG+LYS+HIS #Negative charges (Asp, Glu) color red, resn ASP+GLU #Polar (Ser, Thr, Asn, Gln, Cys) color magenta, resn SER+THR+ASN+GLN+CYS #Glycines (often left as a special case) color green, resn GLY
Renin as ball and sticks, still colored by residues.
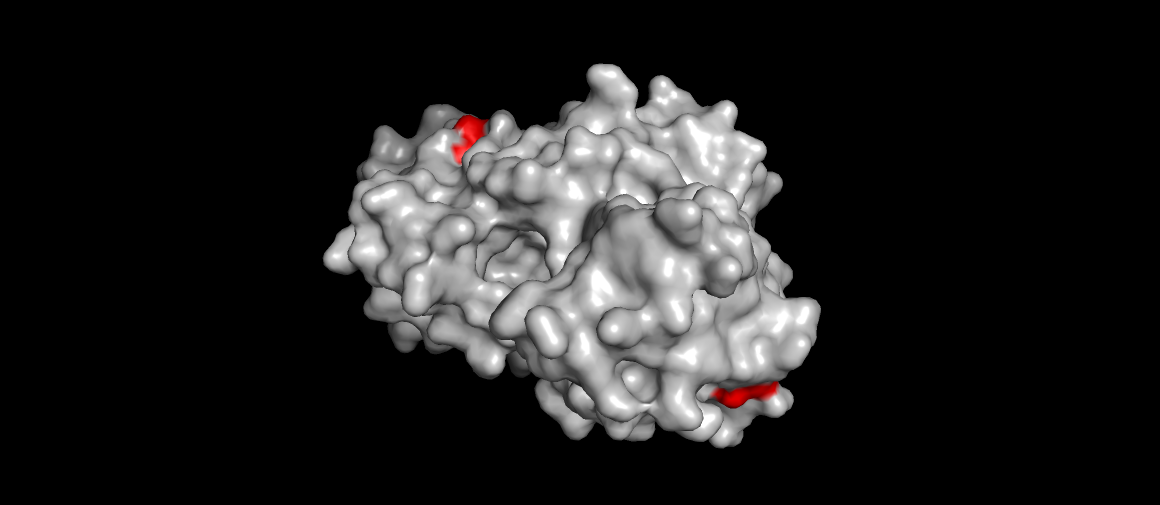
Renin surface view, catalytic aspartases (main mechanism) highlighted. Also the site were the drug Aliskiren (the direct renin inhibitor) binds…
Part C: Using ML-Based Protein Design Tools
The protein chosen for this analysis is the IgV domain of human KIM-1/TIM-1
(PDB: 5DZO), a membrane receptor involved in kidney injury, viral entry (Ebola,
SARS-CoV-2), and immune regulation. The structure was solved at 1.30 Å resolution
by X-ray diffraction and contains 107 residues with a canonical immunoglobulin
beta-sandwich fold.
C1. Protein Language Modeling
Deep Mutational Scan
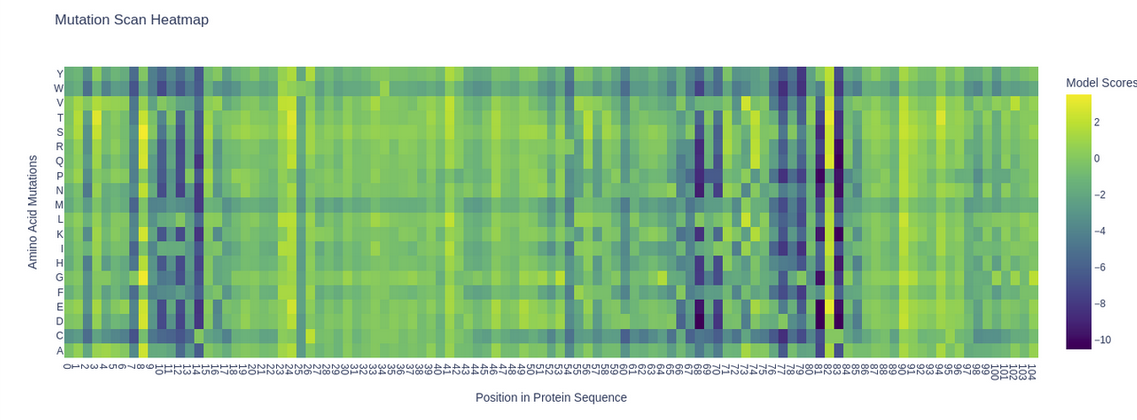
The ESM2 model (esm2_t6_8M_UR50D) was used to generate an unsupervised deep
mutational scan of the KIM-1 IgV domain. Log Likelihood Ratio (LLR) scores were
computed for all possible single amino acid substitutions at each position.
A clear pattern emerges: several positions show deep purple columns across nearly
all substitutions, indicating highly conserved residues that the model considers
essential. Notably, the cysteine row (C) is almost entirely purple throughout the
sequence, reflecting the model’s strong penalization of introducing cysteines at
non-cysteine positions — consistent with the known disulfide bonds (C15–C53) that
stabilize the IgV fold. A striking example is position 53 (C53), where any
substitution receives strongly negative scores, highlighting its structural
importance in the beta-sandwich framework.
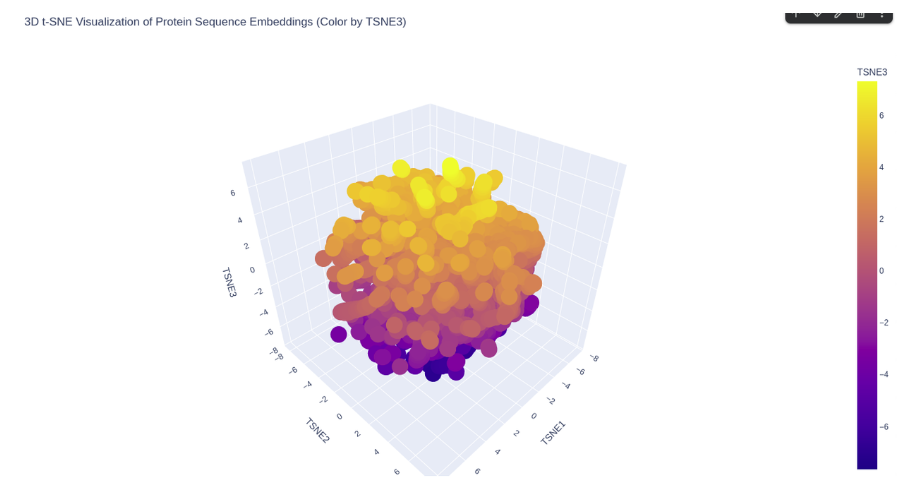
Latent Space Analysis
The ESM2 embeddings of 500 randomly sampled SCOP proteins were projected into 2D
using t-SNE.
ESM2 latent space of SCOP proteins.
KIM-1 occupies a peripheral region of the latent space, somewhat isolated from
the main cluster of SCOP proteins. This is consistent with its nature as a
membrane-associated immunoglobulin domain — a combination less represented in the
SCOP dataset, which is dominated by soluble globular proteins. Its nearest
neighbors in the embedding are likely other immunoglobulin-like or beta-sandwich
domains.
C2. Protein Folding
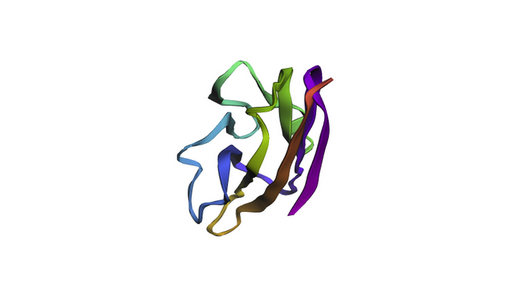
The KIM-1 sequence was folded using ESMFold directly in the notebook.
ESMFold prediction of KIM-1 IgV domain. The beta-sandwich architecture is
clearly visible, consistent with the experimental 5DZO crystal structure.
The predicted structure recapitulates the canonical IgV topology, with the
characteristic two-layer beta-sheet sandwich. The overall fold matches the
experimental structure well, as expected for a well-conserved immunoglobulin
domain. Based on the ESM2 mutational scan, positions with strongly negative LLR
scores (such as C15, C53, and several buried hydrophobic residues) are predicted
to be structurally intolerant to substitution — mutations at these sites would
likely destabilize the fold. In contrast, surface-exposed loop regions show more
permissive LLR profiles, suggesting greater resilience to mutation.
C3. Inverse Folding with ProteinMPNN
The backbone of 5DZO was used as input to ProteinMPNN to propose alternative
sequences compatible with the KIM-1 structure.
Amino acid probability heatmap: The ProteinMPNN probability matrix shows that
structurally critical positions — particularly the cysteines involved in disulfide
bonds — are strongly constrained, with near-exclusive probability mass on the
original residue. Surface-exposed and loop positions show broader distributions,
allowing more sequence diversity.
The generated sequences achieve ~50% sequence recovery while maintaining lower
scores than the native (indicating higher structural compatibility with the
backbone). Key structural residues such as the disulfide-forming cysteines are
preserved in both designs, while surface loops show substantial sequence
variation. This demonstrates that the KIM-1 IgV backbone is structurally robust —
it can accommodate diverse sequences while maintaining its fold, which is a
hallmark of the immunoglobulin superfamily.
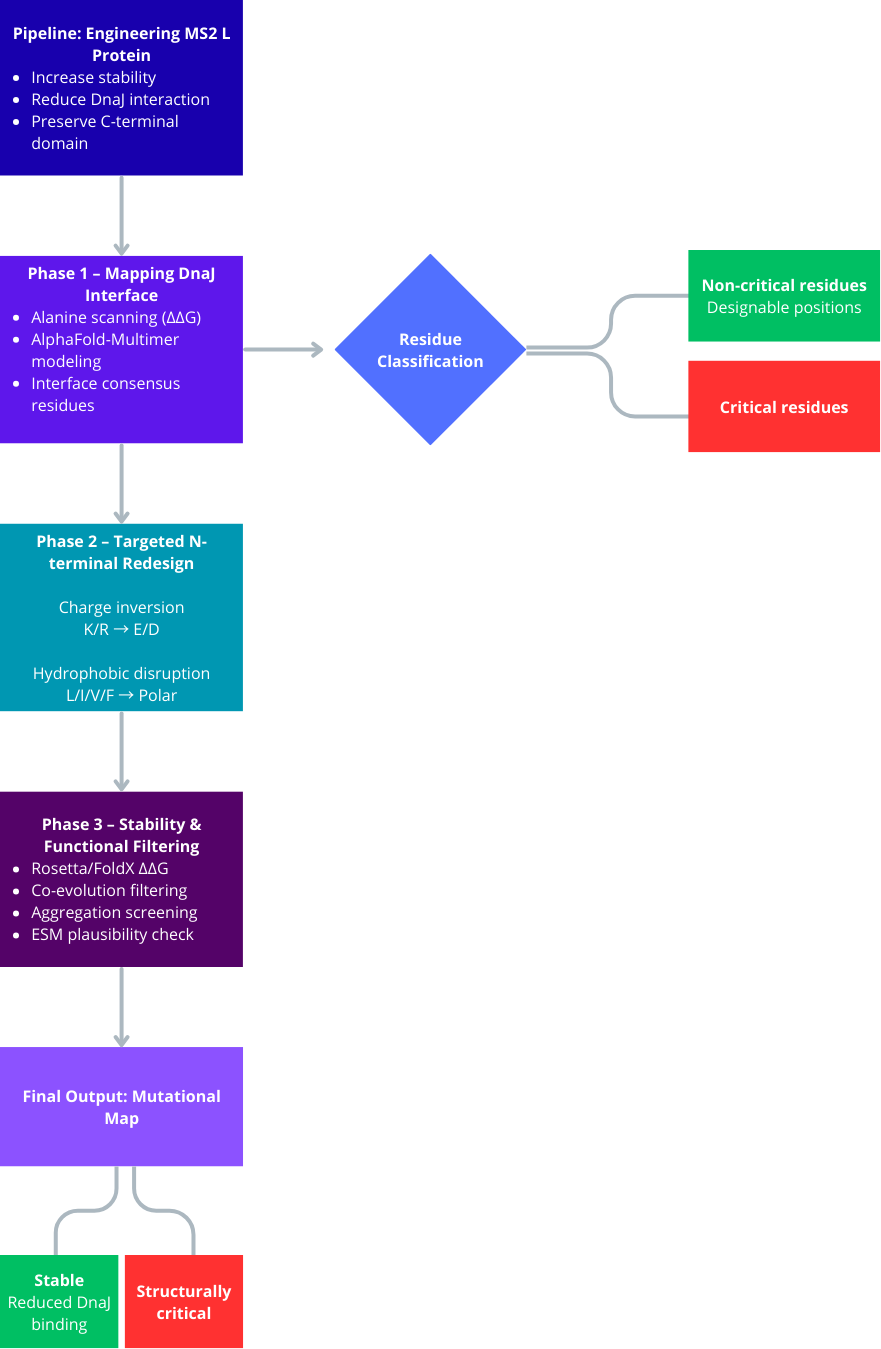
Part D. Group Brainstorm on Bacteriophage Engineering
Project Objective
Engineer the L protein of the MS2 phage to increase structural stability.
Disrupt or reduce its interaction with the bacterial chaperone DnaJ.
Preserve the C-terminal lysis domain to maintain lytic function.
Avoid mutations that interfere with structurally or evolutionarily coupled residues.
Phase 1: Mapping the DnaJ Interaction Interface
Since the exact binding interface between the L protein and DnaJ is unknown, the first step is to identify it computationally rather than introducing arbitrary mutations.
Use AlphaFold-Multimer to model the complex between L protein and DnaJ.
Generate multiple structural predictions and select the top-ranked models.
Identify consensus interface residues that consistently appear in the predicted binding interface.
Perform in silico alanine scanning of the N-terminal residues in the complex to determine which residues significantly contribute to binding energy (ΔΔG).
Analyze whether the N-terminal region resembles known DnaJ-binding motifs, typically hydrophobic residues flanked by basic amino acids.
This phase defines which residues are critical for interaction and should not be mutated randomly.
Phase 2: Targeted N-Terminal Redesign
Instead of deleting regions or performing extensive random substitutions, introduce controlled chemical modifications to disrupt interaction while preserving structural stability.
Focus on charge inversion strategies:
Basic residues (K, R) → Acidic residues (E, D)
Acidic residues (E, D) → Basic residues (K, R)
Disrupt hydrophobic interaction patches:
Hydrophobic residues (L, I, V, F) → Polar residues (S, T, N, Q)
Aromatic residues (F, Y, W) → Aliphatic or small residues
Generate a graded library of variants:
Minor charge modifications
Moderate interface perturbations
Strong hydrophobic disruption
This creates a Pareto front of variants balancing reduced DnaJ interaction and preserved protein stability.
Phase 3: Stability and Functional Filtering
To ensure that redesigned variants remain structurally viable and functionally relevant:
Use Rosetta or FoldX to calculate ΔΔG and verify that mutations do not destabilize the overall protein fold.
Confirm that mutations in the N-terminal region do not propagate structural stress toward the C-terminal lysis domain.
Identify residue pairs that co-evolved between the N-terminal and C-terminal regions.
Avoid mutating co-evolved residues independently to prevent functional disruption.
Evaluate aggregation propensity using tools such as Aggrescan3D to ensure that mutations do not create exposed hydrophobic patches leading to cytoplasmic aggregation.
Assess sequence plausibility using protein language models such as ESM to filter out unlikely or non-natural variants.
Key Limitations
The DnaJ binding mode may be transient or dynamic, reducing AlphaFold-Multimer accuracy.
Protein language model scores do not guarantee in vivo functionality.
Intrinsically disordered regions may not be accurately modeled.
Computational predictions must ultimately be validated experimentally.
Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
The human SOD1 sequence (UniProt P00441) was retrieved and the A4V mutation
introduced by substituting alanine for valine at residue 4:
Four peptides of length 12 were generated using PepMLM-650M conditioned on the
mutant sequence, and the known binder FLYRWLPSRRGG was added for comparison.
Pseudo-perplexity scores were computed for all peptides using the
compute_pseudo_perplexity function from the PepMLM Colab notebook. Lower
perplexity indicates higher model confidence in the peptide as a binder.
Index
Binder
Pseudo Perplexity
1
KHYPVVAAELKA
10.79
2
WHYYAAALAHKA
14.46
3
WHVVAAAVRWKE
20.09
4
FLYRWLPSRRGG (known binder)
20.59
Part 2: Evaluate Binders with AlphaFold3
The four PepMLM-generated peptides and the known binder FLYRWLPSRRGG were submitted to the AlphaFold3 server as separate chains paired with the A4V mutant SOD1 sequence. The resulting ipTM scores were: KHYPVVAAELKA (0.48), WHYYAAALAHKA (0.46), WHVVAAAVRWKE (0.37), and FLYRWLPSRRGG (0.32). Notably, all three top PepMLM-generated peptides exceeded the known binder in structural confidence. KHYPVVAAELKA and WHYYAAALAHKA appeared to engage the surface near the β-barrel region, while WHVVAAAVRWKE showed a more peripheral interaction. None of the peptides achieved ipTM values above 0.5, which is consistent with the inherent challenge of modeling short peptide-protein complexes, but the PepMLM candidates consistently outperformed the reference binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
PeptiVerse predictions revealed that all four peptides are soluble and non-hemolytic, with binding affinity scores ranging from 5.89 to 6.70 pKd (all classified as weak binding). There was no strong correlation between ipTM and predicted affinity: WHVVAAAVRWKE had the highest pKd (6.70) despite a lower ipTM (0.37), while KHYPVVAAELKA combined the best ipTM (0.48) with acceptable affinity (5.89). The known binder FLYRWLPSRRGG showed the lowest solubility probability (0.61) and highest net charge (+2.76), suggesting potential pharmacological liabilities despite its experimental validation.
Selected candidate: KHYPVVAAELKA. This peptide ranked first in structural confidence (ipTM 0.48), first in PepMLM perplexity (10.79), is completely soluble (1.00), non-hemolytic (0.07), and carries a near-neutral charge (+0.85), making it the most promising candidate across all evaluated dimensions.
Part 4: Generate Optimized Peptides with moPPIt
Using moPPIt with affinity and motif guidance targeting residues 1–10 of the A4V mutant SOD1 (the N-terminal region destabilized by the mutation), 10 peptides of length 12 were generated. The results revealed a notable trade-off between affinity and motif scores. CVYCCVDGCVWV achieved the highest predicted affinity (pKd 8.12), crossing the threshold for moderate binding, but had a low motif score (0.39), suggesting it does not engage the target region specifically. In contrast, DTPPCYAPVICY balanced strong motif engagement (0.730) with reasonable affinity (6.81). Compared to PepMLM peptides, moPPIt candidates are richer in cysteines, likely forming disulfide bonds for structural stability, and show higher affinity scores overall — reflecting the benefit of multi-objective guided optimization over unconditional sampling.
Before advancing any of these peptides to clinical studies, further evaluation would include: (1) experimental binding validation via SPR or ITC; (2) AlphaFold3 structural modeling of the moPPIt candidates; (3) stability and proteolysis assays; (4) cell-based toxicity screens; and (5) in vivo pharmacokinetic profiling.
Part B: MS2 Phage Lysis Protein Engineering (Option 1: Mutagenesis)
Overview
The MS2 lysis protein (L-protein, UniProtKB P03609) was analyzed using the ESM2
protein language model to predict the effect of single amino acid substitutions
across all positions. The sequence used was:
Soluble N-terminal domain (residues 1–37): responsible for interaction with
the E. coli chaperone DnaJ
Transmembrane domain (residues 38–75): responsible for membrane integration
and lysis activity
ESM2 Scoring and Correlation with Experimental Data
The ESM2 notebook generated Log Likelihood Ratio (LLR) scores for all possible
single amino acid substitutions at every position. A positive LLR score indicates
that the model considers the mutation more favorable than the wild-type residue
in that context.
Crossreferencing the ESM2 scores with the experimental L-protein mutant dataset
revealed a weak but consistent trend: mutants with confirmed lysis activity
(Lysis=1) had a mean ESM2 score of -0.156, compared to -0.407 for
non-functional mutants (Lysis=0). This suggests that ESM2 captures some
evolutionary signal relevant to protein function, but is insufficient as a
standalone predictor. A notable exception was C29R (score +2.40), which the
model ranked highly but showed no lysis activity experimentally — highlighting
the limitations of sequence-only models for membrane proteins.
Proposed Mutations
Five mutations were selected based on positive ESM2 scores, avoidance of
experimentally confirmed loss-of-function positions, and biological rationale:
#
Mutation
Region
ESM2 Score
Rationale
1
F5Q
Soluble
+1.80
Position not highly conserved; Q substitution predicted favorable by ESM2 and may reduce hydrophobic burial in the soluble domain
2
C29S
Soluble
+2.04
Free cysteines can form aberrant disulfide bonds destabilizing the protein; serine is a conservative, polar replacement with high ESM2 confidence
3
Y39L
Transmembrane
+2.24
Tyrosine is poorly suited for membrane-buried positions; leucine increases hydrophobicity and favors stable TM helix formation
4
A45L
Transmembrane
+1.54
Leucine substitution at this position increases hydrophobic packing within the TM helix, potentially improving membrane integration
5
E61L
Transmembrane
+1.82
Glutamate is a charged residue unfavorable in transmembrane regions; leucine substitution improves the hydrophobic character of the helix
Mutations 1 and 2 target the soluble domain to potentially alter DnaJ-independent
folding. Mutations 3, 4, and 5 target the transmembrane domain to improve
membrane integration efficiency, which could accelerate lysis kinetics and reduce
the window for E. coli to acquire resistance.
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix Components:
Phusion DNA Polymerase: A high-accuracy enzyme with 3′→5′ exonuclease (proofreading) activity that minimizes mutations during amplification
dNTPs (Deoxynucleotide Triphosphates): The nucleotide building blocks (A, T, C, G) used to synthesize the new DNA strand.
Mg2+ (Magnesium Ions): A critical cofactor for polymerase activity. It stabilizes the interaction between the enzyme and the DNA.
Buffer System: Maintains optimal pH and ionic strength to ensure enzyme stability and efficient primer binding.
2. What are some factors that determine primer annealing temperature during PCR?
Melting Temperature (Tm), GC content, Lenght of the primer, and Salt concentration.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Protocol: PCR uses thermal cycling (heating and cooling) to amplify specific sequences exponentially. Restriction digests involve incubating DNA with specific enzymes at a constant temperature (usually 37∘C) to “cut” the DNA at recognition sites.
Comparison: PCR is preferable when you need to amplify a specific gene from a small amount of template or add custom “overhangs” for assembly. Restriction digests are preferable for verifying plasmid maps (diagnostic digests) or using “sticky-end” cloning when appropriate sites are already present.
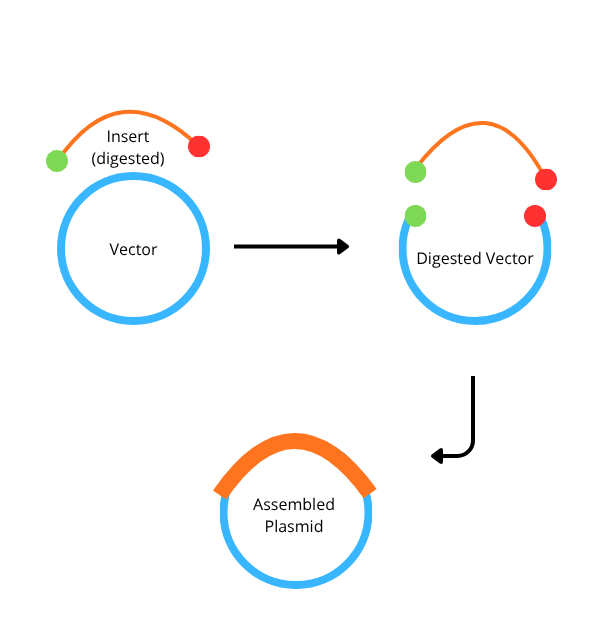
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure success, you must design your fragments with overlapping homologous ends (usually 20−40 bp long). This is achieved by designing PCR primers where the 5′ end of the primer matches the end of the adjacent fragment. Additionally, you must ensure the fragments are free of template DNA (via DpnI digestion) and that the overlaps do not contain stable secondary structures that could interfere with the exonuclease step of the Gibson reaction.
5. How does the plasmid DNA enter the E. coli cells during transformation?
In chemically competent E. coli, the cells are treated with divalent cations (like Ca2+) to neutralize the negative charges of the DNA and the cell membrane. During Heat Shock (moving from ice to 42∘C), a thermal gradient is created that opens temporary pores or “adhesion zones” in the membrane, allowing the plasmid DNA to be pulled into the cell.
6. Describe another assembly method in detail (such as Golden Gate Assembly)
Golden Gate Assembly (GGA) is a molecular cloning method that allows for the simultaneous, one-pot assembly of multiple DNA fragments using Type IIS restriction enzymes and T4 DNA ligase. Unlike traditional restriction enzymes, Type IIS enzymes (such as BsaI) cut at a specific distance away from their non-palindromic recognition sites, creating unique 4-base pair overhangs. These custom overhangs are designed so that the recognition sites are removed from the final ligated product, making the reaction essentially irreversible and driving it toward the desired construct. This “scarless” nature allows for the seamless joining of various biological parts, such as promoters, genes, and terminators, in a predefined order. Because the enzyme and ligase work in a single cycle of temperature shifts, GGA is significantly faster and more efficient than traditional multi-step cloning. Consequently, it has become a standard tool in synthetic biology for building modular systems and large library collections.
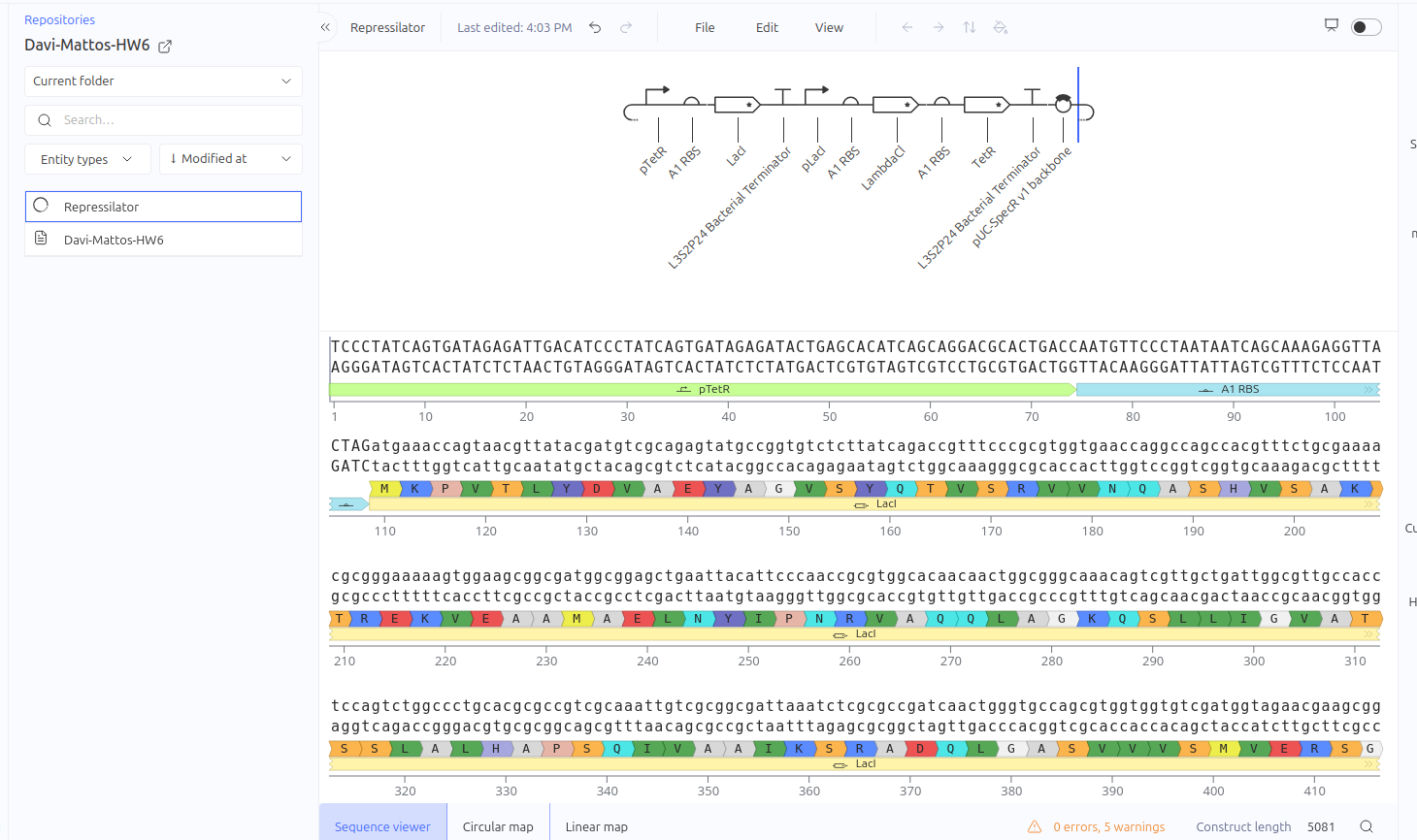
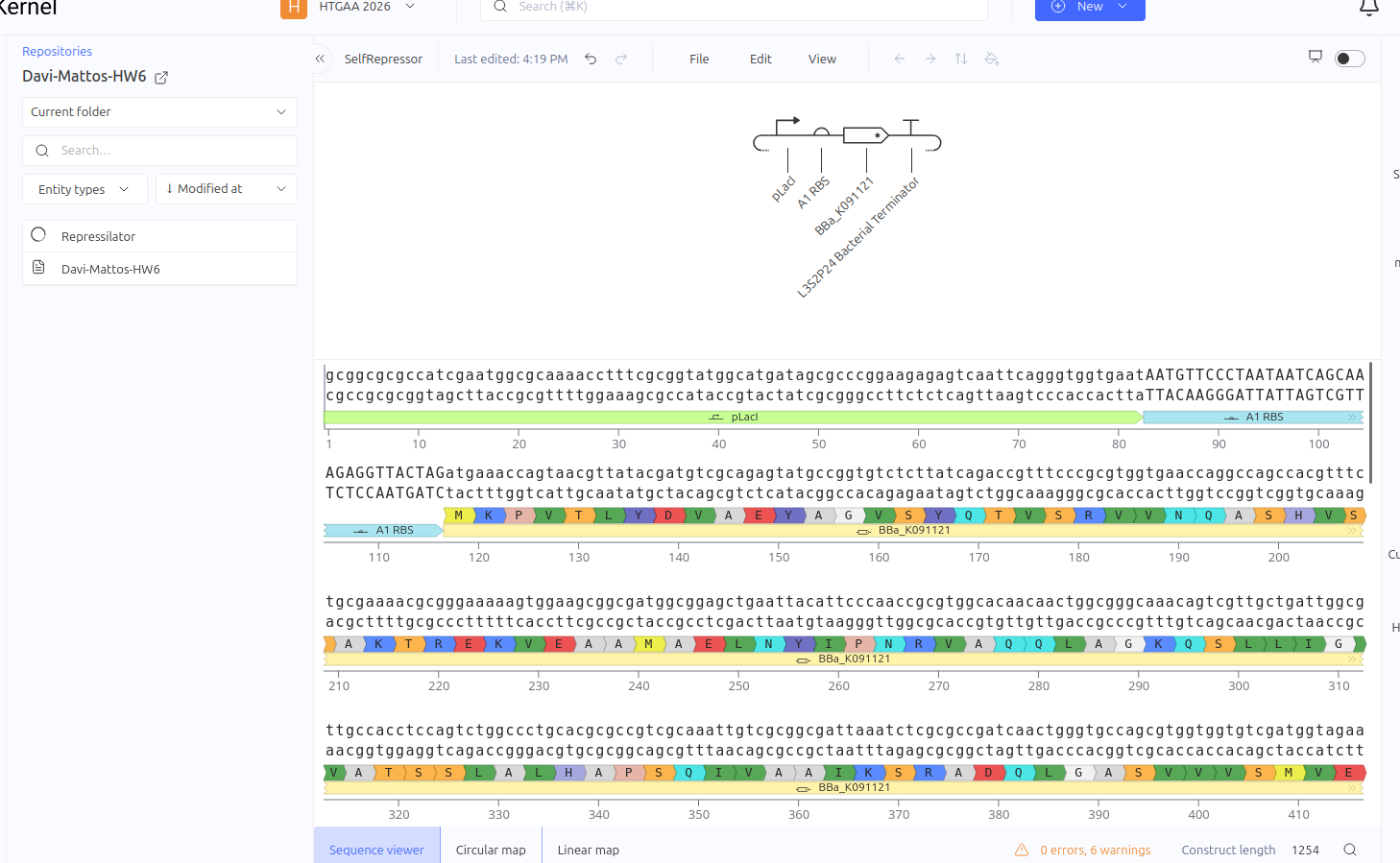
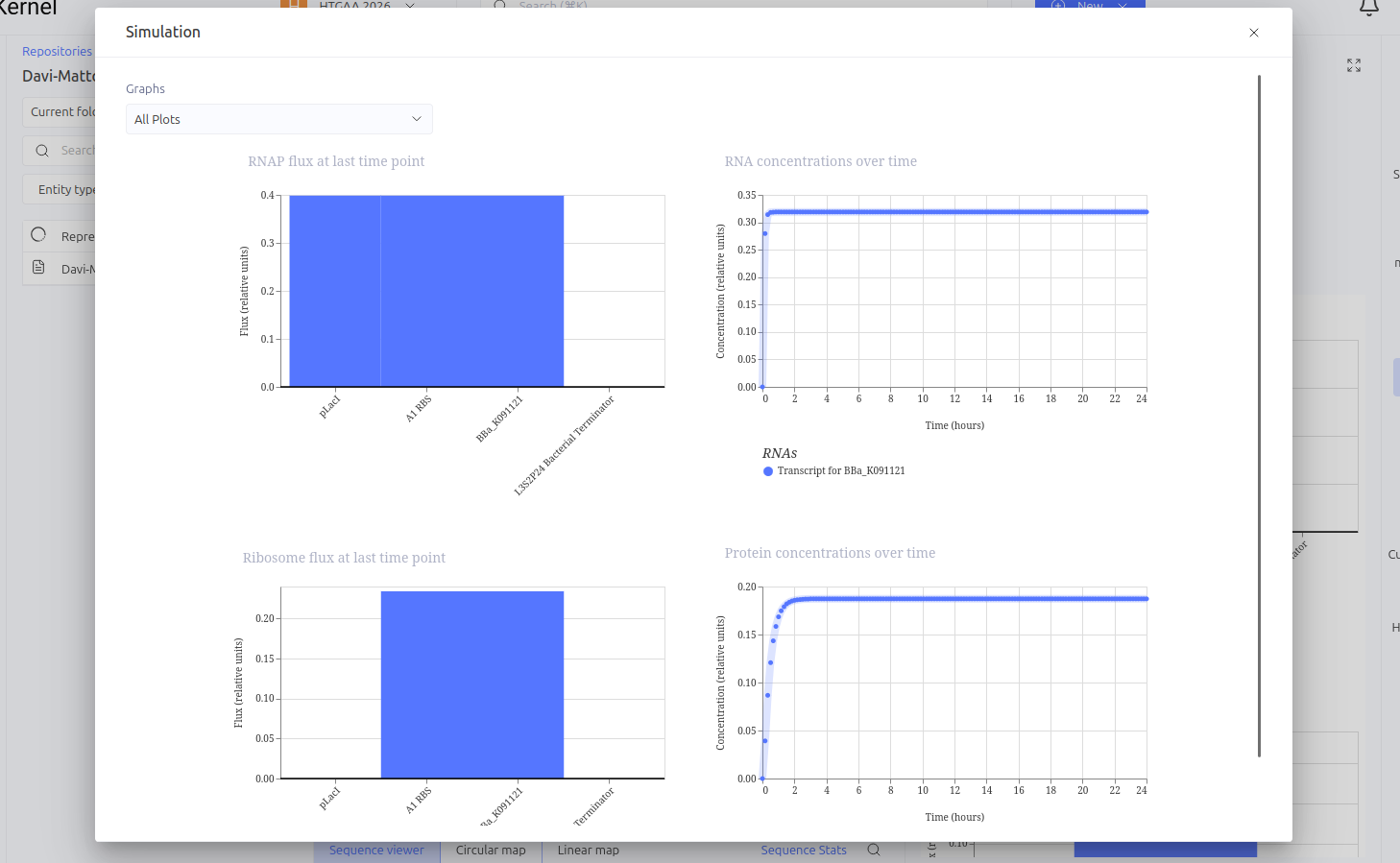
This circuit was designed as a simple negative feedback loop. The pLacI promoter initiates the transcription of the LacI gene. As the LacI protein is produced and its concentration increases, it binds to its own promoter (pLacI), blocking RNA polymerase and inhibiting its own production. This mechanism is fundamental for cellular homeostasis, allowing the protein to reach a steady state more quickly and in a controlled manner, preventing the waste of cellular resources.
Expected vs. Observed Results: In the simulator, it was observed that the protein concentration does not grow indefinitely; instead, it reaches a plateau (steady state) within a short time. Initially, a construction error occurred by using two promoters in series, resulting in a lack of translation. After correcting the sequence to Promoter -> RBS -> CDS (LacI), the self-regulating behavior was confirmed by the graphs."
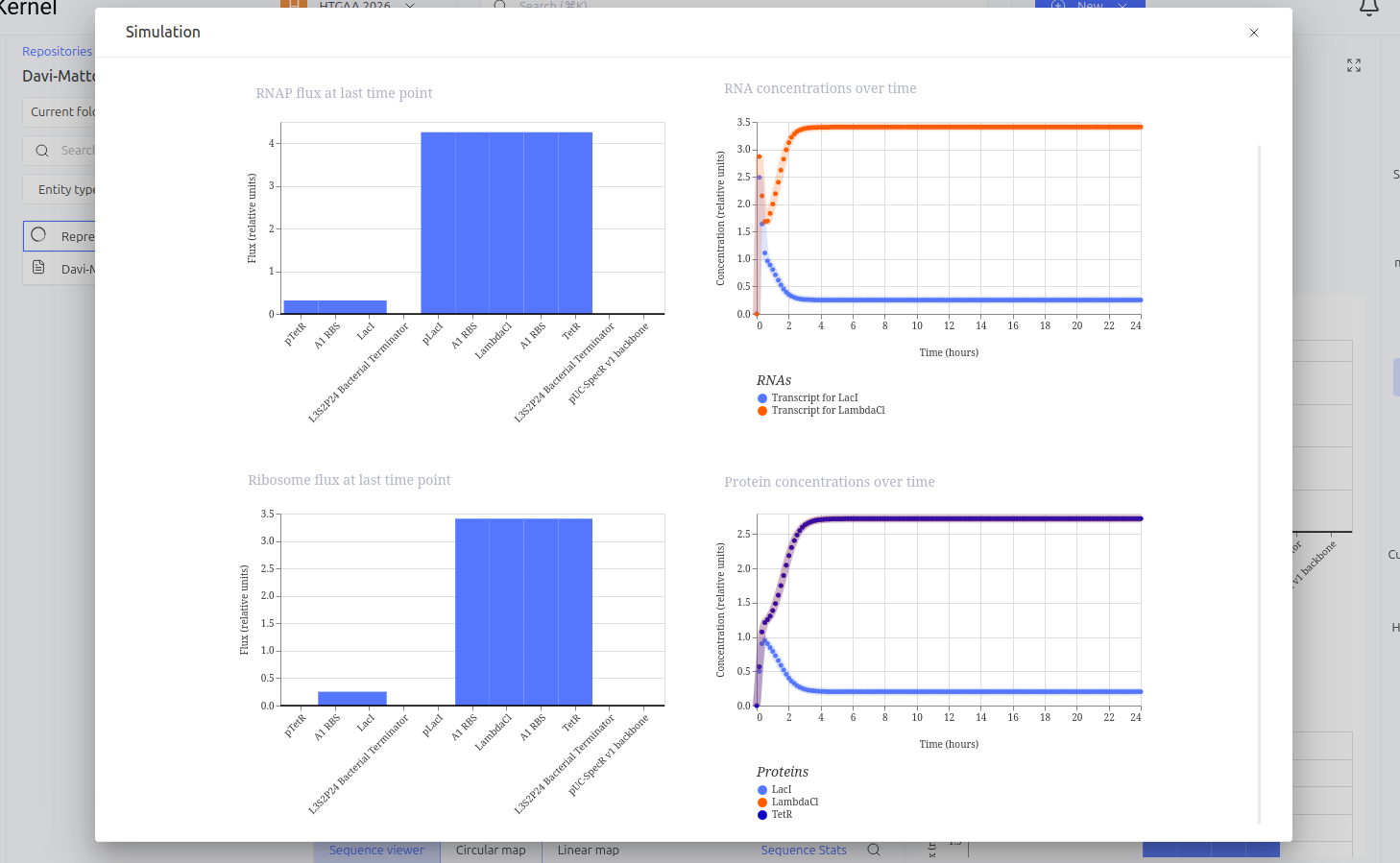
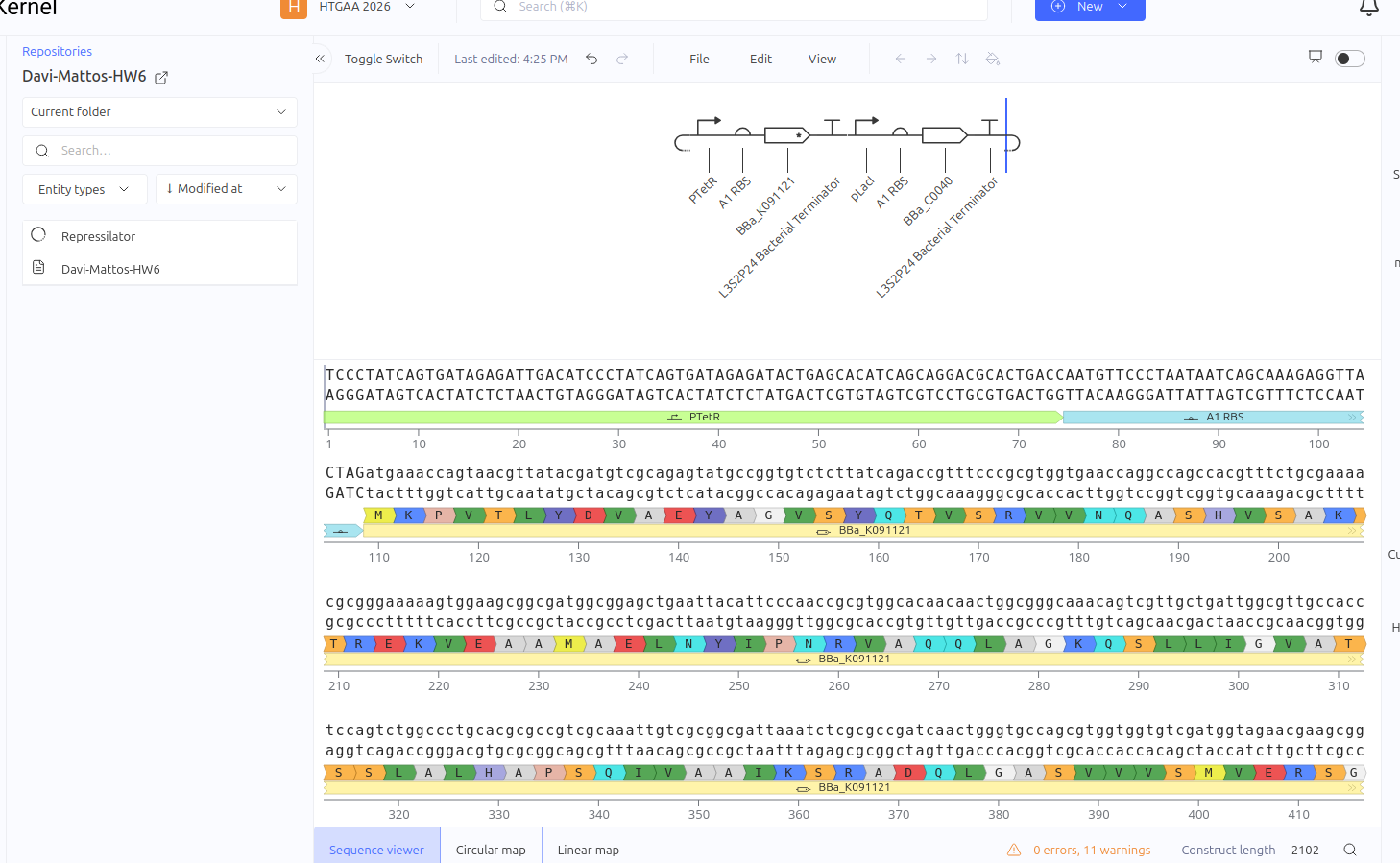
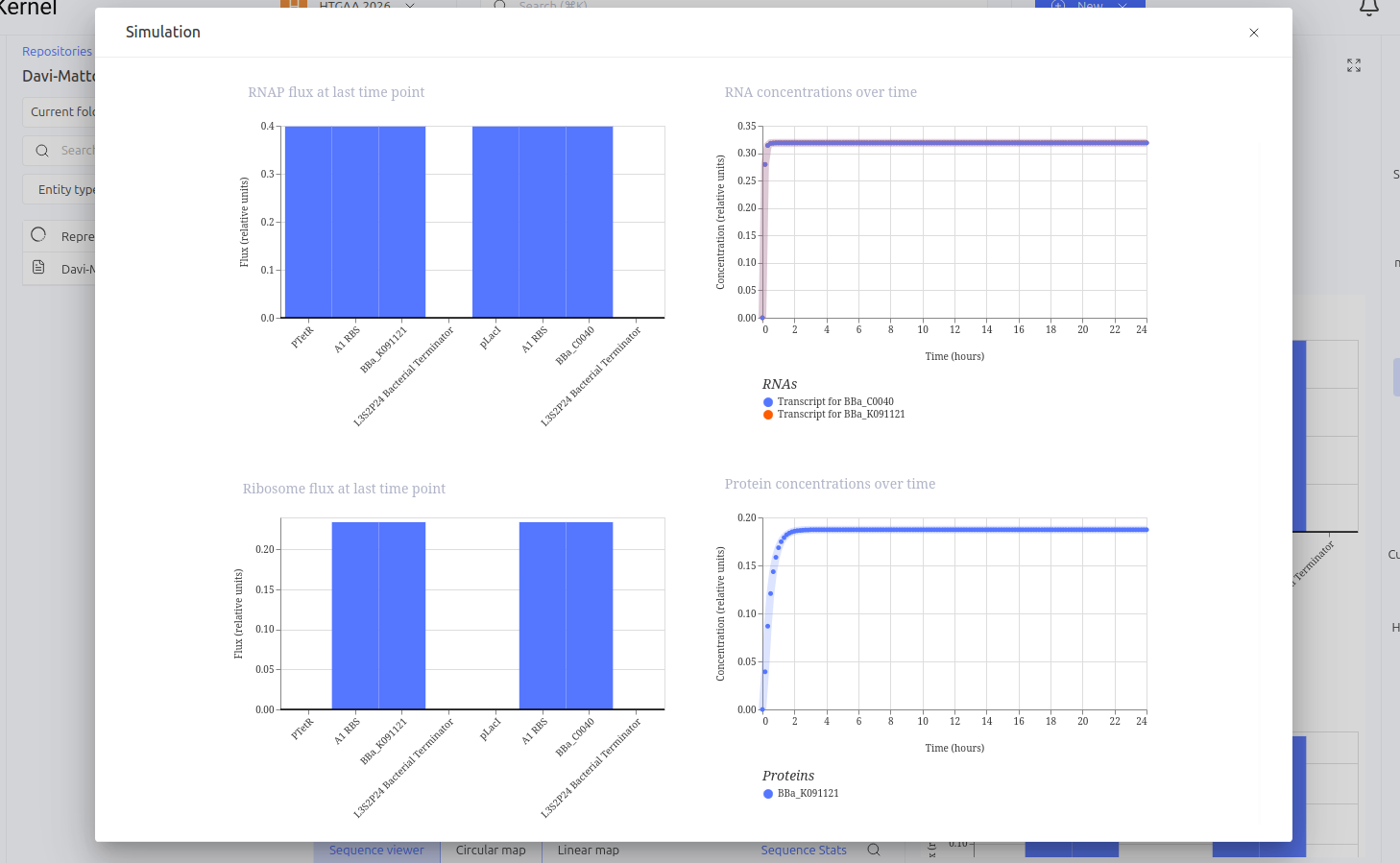
The Toggle Switch is a circuit composed of two repressors that mutually inhibit each other’s expression (LacI and TetR). The design consists of two cassettes: the first where the pTetR promoter controls the expression of LacI, and the second where the pLacI promoter controls the expression of TetR. This arrangement creates a bistability system, functioning as a biological memory that can flip between two stable states: (1) High LacI / Low TetR or (2) High TetR / Low LacI.
Expected vs. Observed Results: Simulation results showed the system ‘choosing’ one of the stable states. The LacI protein (BBa_K091121) dominated the system, keeping the expression of TetR (BBa_C0040) repressed and close to zero throughout the simulated time. This demonstrates the circuit’s ability to maintain a stable state even in the absence of external inducers.
Construct 3: Repression Cascade (Inverter / NOT Gate)
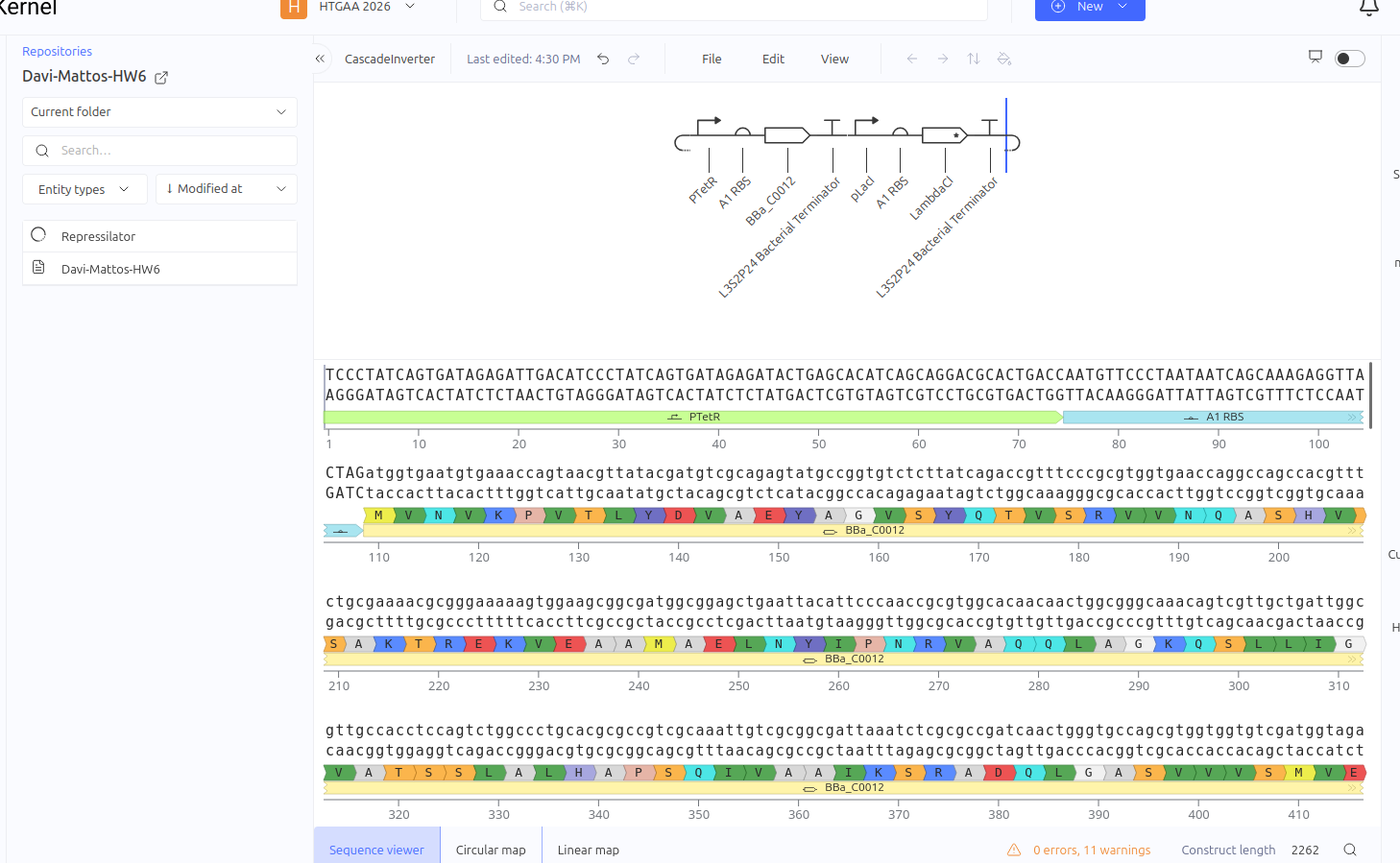
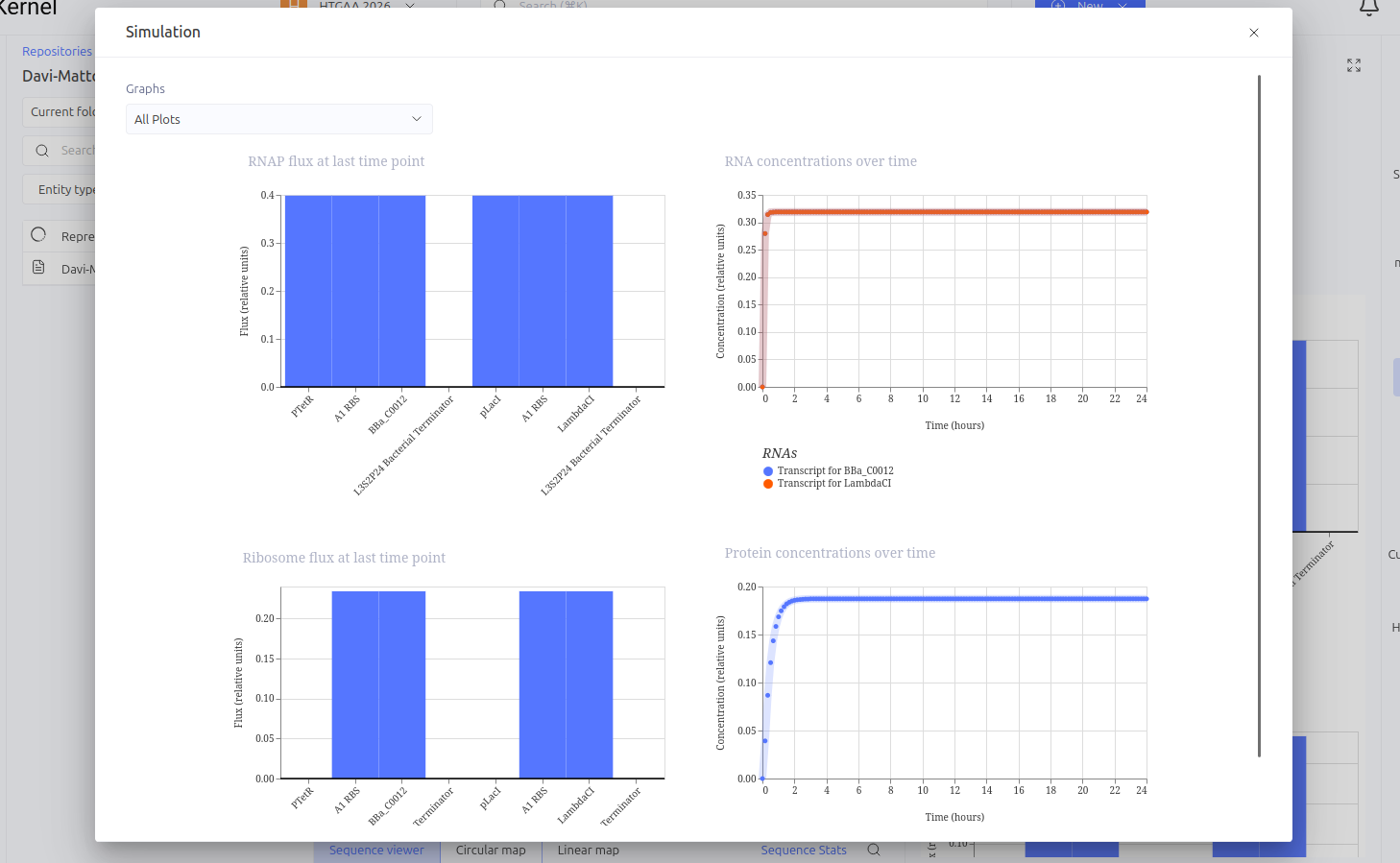
This circuit functions as a linear repression cascade, simulating a NOT logic gate. The first stage uses the pTetR promoter to express the LacI gene constitutively (in the absence of TetR). The second stage contains the LambdaCI gene under the control of the pLacI promoter. The system’s logic dictates that the presence/activation of the first stage necessarily results in the shutdown of the second stage.
Expected vs. Observed Results: As expected, the simulator showed high levels of RNA and protein for the first gene (LacI), while the second gene (LambdaCI) showed only a brief initial transient peak of RNA before being completely repressed. The ribosome flux for the second gene dropped drastically, confirming that the biological signal inversion was successful.
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
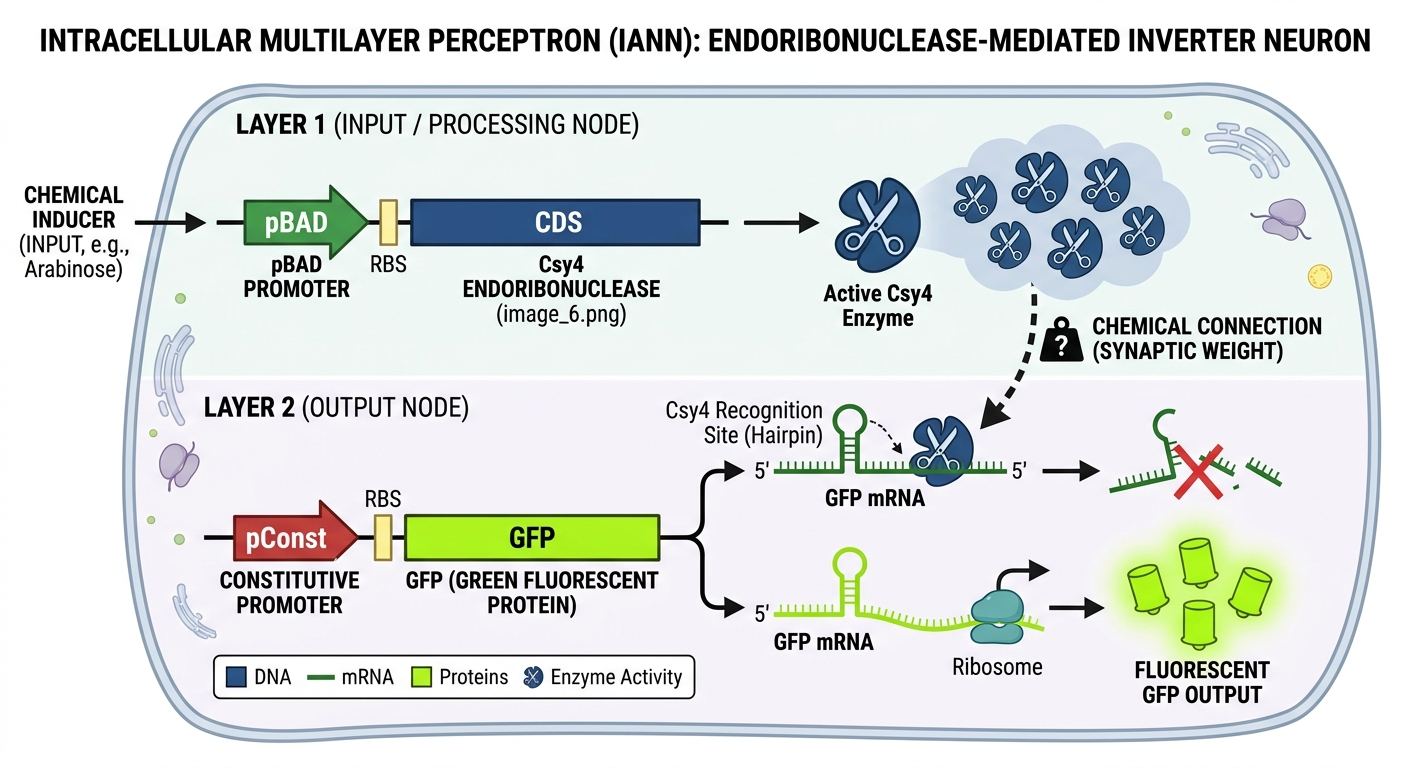
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Intracellular Artificial Neural Networks (IANNs) transcend the limitations of binary Boolean logic by enabling analog and graded signal processing within a cell. While traditional genetic gates are restricted to “ON/OFF” states, IANNs can integrate multiple continuous environmental inputs and assign them specific “weights,” allowing the cell to make nuanced decisions based on a threshold of combined signals. This analog capability is particularly superior for pattern recognition and processing complex biomarkers, as it mimics natural biological decision-making more closely than rigid digital circuits. Furthermore, IANNs can often achieve high levels of computational complexity with fewer genetic parts, as they leverage the inherent non-linearities of biochemical reactions as natural “activation functions,” thereby reducing the metabolic burden on the host organism compared to massive, multi-gate Boolean architectures.
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
A highly useful application for an IANN is a “smart” diagnostic classifier designed to trigger programmed cell death (apoptosis) only upon detecting a specific signature of multiple microRNA (miRNA) biomarkers associated with a particular cancer subtype. In this system, the IANN inputs would be the varying concentrations of intracellular miRNAs, which are weighted based on their diagnostic significance; the output would be the expression of a pro-apoptotic protein like Bax once the weighted sum of inputs exceeds a safety threshold. However, this application faces significant limitations, such as “leaky” expression where the circuit might trigger accidentally due to molecular noise, potentially killing healthy cells. Additionally, the metabolic load required to maintain the synthetic “neurons” can stress the cell, and the current scarcity of orthogonal (non-interfering) biological parts makes it difficult to scale these networks into deep, multi-layer architectures without cross-talk.
3. Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
I generated this diagram with Gemini.
This diagram illustrates a two-layer biological computer where a cell “calculates” a response to its environment. In Layer 1, the cell senses an external input (like a chemical inducer), which acts as a switch to produce Csy4 endoribonuclease enzymes (the “scissors”). These enzymes represent the processed signal traveling to the next stage. The efficiency of this enzyme’s action acts as a synaptic weight, determining how strongly the signal is passed forward. In Layer 2, the cell is constantly trying to produce a Green Fluorescent Protein (GFP), but the “scissors” from the first layer recognize and cut the GFP’s instruction manual (the mRNA). This results in an inversion logic: if the input is high, the scissors destroy the output, and the cell stays dark; if the input is low, the scissors aren’t made, and the cell glows bright green.
Assignment Part 2: Fungal Materials
1. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials, primarily mycelium composites, are grown by inoculating agricultural waste (like hemp or wood chips) with fungal spores. The mycelium acts as a natural biological glue, binding the substrate into solid shapes. Key examples include biodegradable packaging (an alternative to Styrofoam), myco-bricks for sustainable construction insulation, and fungal leather (like Mylo) used in the fashion industry as a vegan alternative to animal hides.
The primary advantage of fungal materials is their sustainability; they are carbon-negative, fire-resistant, and fully compostable, whereas traditional plastics and leathers rely on petrochemicals or high-energy livestock farming. However, they face disadvantages in structural consistency and water resistance. While a plastic brick has standardized strength, a grown myco-brick can vary based on the “diet” of the fungi, and many fungal materials tend to absorb moisture, which can lead to premature degradation if not properly sealed.
2. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
We can genetically engineer fungi to create “living materials” with functional properties. For example, engineering a fungal brick to be self-healing would allow the material to remain dormant until a crack appears, at which point moisture triggers the fungi to grow and “fill” the gap. Other goals include bioluminescent architecture for zero-electricity lighting or “sensing” materials that change color when they detect toxic pollutants in the air through embedded genetic biosensors.
Fungi offer several biological advantages over bacteria for material science. As multicellular eukaryotes, they grow in long, branching filaments called hyphae, allowing them to create massive, physically interconnected 3D structures that single-celled bacteria cannot form. Additionally, fungi are “professional secretors”; they are naturally evolved to pump huge amounts of enzymes into their environment, making them superior to bacteria for industrial-scale production of complex proteins and the breakdown of tough environmental waste.
Assignment Part 3: First DNA Twist Order
Week 9 HW: Cell-Free Systems
General homework questions
1- Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
In traditional in vivo (inside the cell) methods, the cell’s own survival is the priority. In CFPS, your protein is the priority. You can add non-natural amino acids, chaperones, or labeling molecules directly to the mix without worrying about transport across a cell membrane. You can monitor and tweak variables like pH, temperature, and redox potential in real-time. If a protein is lethal to a living cell (e.g., a pore-forming toxin), CFPS is the only way to produce it because there is no “cell” to kill. You can go from a linear DNA template (PCR product) to a protein in hours, whereas cell production requires days for cloning and transformation.
2- Describe the main components of a cell-free expression system and explain the role of each component.
Main Components and Roles
Crude Extract: This is the “machinery.” It contains ribosomes, aminoacyl-tRNA synthetases, and translation factors from a lysed cell (like E. coli or yeast).
Energy Solution: Contains ATP and GTP to fuel the reaction, plus an energy regeneration source (like Phosphoenolpyruvate).
Amino Acids: The building blocks for the protein chain.
Cofactors and Salts: Magnesium (Mg2+) and Potassium (K+) are critical for ribosome stability and function.
DNA Template: The “blueprint” (usually a plasmid or PCR fragment) encoding your target protein.
3- Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Translation is energy-expensive. For every amino acid added to a chain, multiple high-energy phosphate bonds (ATP/GTP) are consumed. Without regeneration, ATP levels drop rapidly, and inhibitory byproducts (like inorganic phosphate) build up, stopping the reaction in minutes. You can add Creatine Phosphate along with the enzyme Creatine Kinase. This enzyme “recharges” spent ADP back into ATP by transferring a phosphate group from the creatine phosphate, keeping the “battery” full for several hours.
4- Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic vs. Eukaryotic Systems
Prokaryotic (e.g., E. coli): Fast, high yield, and cheap. It lacks complex “post-translational modifications” (PTM) like glycosylation.
Protein to produce: GFP (Green Fluorescent Protein). It’s a simple, robust protein that doesn’t need complex folding or sugar tags to function.
Eukaryotic (e.g., CHO or Wheat Germ): Slower and more expensive, but capable of complex folding and adding sugar groups (PTMs).
Protein to produce: Human Erythropoietin (EPO). This protein requires specific glycosylation patterns to be biologically active in humans, which only eukaryotic machinery can provide.
5- How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Membrane proteins are “greasy” (hydrophobic). If produced in a watery cell-free mix, they will clump together (aggregate) and become useless because they have no “home” (lipid bilayer) to sit in. We can maybe add Nanodiscs or Liposomes. Include synthetic lipid bilayers in the reaction. As the protein is synthesized, it can insert directly into these membranes, maintaining its native shape and/or use mild detergents to shield the hydrophobic parts of the protein until it can be purified.
6- Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Possible Reason
Troubleshooting Strategy
Template Degradation
Check the DNA quality on a gel. Use RNase inhibitors to prevent the “blueprint” (mRNA) from being destroyed by stray enzymes in the extract.
Magnesium Imbalance
Ribosomes are very sensitive to Mg2+ Run a Magnesium titration experiment, testing small increments of concentration to find the “sweet spot” for your specific protein.
Codon Bias
If the DNA uses “rare” codons that the extract doesn’t have many tRNAs for, the ribosome will stall. Supplement the mix with extra tRNAs (e.g., using a specialized “RIL” extract) to speed up translation.
Homework question from Kate Adamala
Filamentous bulking is a nightmare for wastewater treatment plant operators; it occurs when long, thread-like bacteria (like Microthrix parvicella) overgrow, preventing the “floc” (sludge) from settling properly. This results in poor water quality and sludge carryover.
Using the Synthetic Minimal Cell (SMC) framework, we can design a targeted “Seek and Destroy” sentinel to combat this issue without the risks of broad-spectrum biocides like chlorine.
Function and Logic
The Function: A “Filament-Specific Lysis Sentinel.” It detects high concentrations of specific metabolites or signal molecules (like long-chain fatty acids) used by bulking filaments and responds by releasing specialized enzymes to break them down.
Input/Output:
Input: Oleic Acid (a common substrate and signal for M. parvicella).
Output: Chitinase or Lysozyme (enzymes that degrade the cell walls of specific filamentous bacteria).
Why Encapsulate? In the turbulent environment of a wastewater tank, enzymes would be diluted instantly. Encapsulation allows the SMC to protect its “payload” until it is in the heart of a sludge floc where the concentration of filaments is highest.
Natural Cell Alternative? Using a genetically modified bacterium (GMM) in wastewater is legally and ethically difficult because they can “escape” into the environment. An SMC is a non-living, non-replicating machine that “runs out of batteries” and disappears after its task is done.
Design of Components
Membrane: POPC and Palmitic Acid. Including a fatty acid in the membrane helps the SMC “blend in” and adhere to the lipid-loving filamentous bacteria.
Internal Contents: * E. coli cell-free Tx/Tl system.
ATP/GTP and an energy regeneration system (Creatine Phosphate).
Plasmids containing the sensor-actuator circuit.
Organism Source: Bacterial (E. coli). It provides the most robust and rapid protein production for enzymatic payloads.
Communication: Small fatty acids are permeable to the liposome membrane. The output enzyme (Chitinase) requires a pore to exit.
Experimental Details
The “Sensor” Gene: fadR (a fatty acid-responsive regulator). In its default state, FadR represses a promoter. When Oleic Acid (the input) enters the SMC, it binds FadR, releasing the repression.
The “Pore” Gene: α-hemolysin (αHL). This gene is placed under the control of a FadR-repressed promoter. Presence of filament-related lipids → FadR releases → αHL is expressed → Membrane becomes porous.
The “Actuator” Gene: ChiA (Chitinase). This enzyme specifically degrades the complex polysaccharides in certain filamentous cell walls. It is constitutively expressed but remains trapped until the αHL pore opens.
Measurements:
Sludge Volume Index (SVI): In a lab-scale bioreactor, measure the settling rate of the sludge before and after adding the SMCs. A decrease in SVI indicates a successful reduction in bulking.
Microscopy: Use Gram staining or FISH (Fluorescence In Situ Hybridization) to visually observe the physical degradation of the long filaments after SMC treatment.
Desired Outcome:
The SMCs are added to the “Return Activated Sludge” (RAS) line. They float into the aeration tank and become trapped within the tangled filaments. Once they sense the high lipid concentration of the filaments, they “fire,” releasing a concentrated burst of Chitinase directly onto the target. This breaks the filaments into smaller pieces, allowing the healthy sludge flocs to settle normally and restoring the plant’s efficiency without harming the beneficial “floc-forming” bacteria.
Homework question from Peter Nguyen
Carbon monoxide (CO) is often called the “silent killer” because it is colorless and odorless. Integrating a cell-free biosensor into heating
systems or textiles (like curtains near a furnace) could provide a life-saving, zero-electricity backup to traditional electronic detectors.
The Pitch:
A bio-synthetic “smart vent” filter that detects dangerous carbon monoxide levels from faulty heaters and undergoes a rapid, irreversible color change to provide a visible emergency alert before toxic levels are reached.
The Concept:
The core of this system is the CooA protein, a natural carbon monoxide sensor found in bacteria like Rhodospirillum rubrum. In our cell-free system, freeze-dried E. coli extract is engineered with the coo promoter system. When CO gas is present, it binds to the CooA transcription factor, causing a structural shift that “turns on” the expression of a highly concentrated red chromoprotein (like amilCP). This system would be embedded in a porous, breathable mesh placed over heating vents or furnace enclosures. As the heater pushes air through the filter, any CO present rehydrates the encapsulated cell-free “pellets” (utilizing the ambient humidity or a small, integrated moisture-release bead), triggering the rapid production of the red pigment.
Societal Challenge and Market Need:
Carbon monoxide poisoning causes thousands of hospitalizations annually, often due to faulty space heaters or furnaces during power outages (e.g., during winter storms). Electronic detectors are effective but rely on batteries or grid power, which can fail. This biological sensor acts as a fail-safe, passive indicator. It requires no electricity and can be integrated directly into low-cost household materials like vent filters, curtains, or even “stickers” placed on the side of a water heater, making high-level safety accessible to low-income households or in off-grid disaster scenarios.
Addressing Limitations:
Activation: To solve the rehydration problem, the cell-free pellets can be co-packaged with deliquescent salts (which pull moisture from the air) or a specialized hydrogel that maintains a specific “ready-state” water activity without allowing the reaction to start prematurely.
Stability: Carbon monoxide is a highly stable gas, and the CooA protein is remarkably robust. By using trehalose-based freeze-drying, the biological machinery can remain shelf-stable for 1–2 years inside the filter packaging.
One-time Use: In this context, one-time use is a safety advantage. Once the filter turns red, it serves as a permanent, “latched” record that a CO event occurred. Even if the gas clears, the red stain remains, forcing the user to acknowledge the danger and call a technician to repair the faulty heater before replacing the sens
Homework question from Ally Huang
Background: The Challenge
Deep-space missions expose astronauts to high-LET (Linear Energy Transfer) radiation, which triggers chronic oxidative stress and neuroinflammation. This damage threatens long-term cognitive function and mission safety. Currently, we lack real-time, lightweight tools to monitor neurological health in orbit. Understanding how the blood-brain barrier (BBB) integrity shifts and how specific neuro-protective proteins respond is vital for humanity to become a multi-planetary species. This research is scientifically significant because it explores the limits of biological resilience in extreme environments, offering insights into neurodegenerative diseases on Earth.
The Target
Our target is the Glial Fibrillary Acidic Protein (GFAP) and its encoding mRNA. GFAP is a hallmark clinical biomarker for astrocyte activation and brain injury.
Relation to Space Biology
In microgravity, “fluid shifts” increase intracranial pressure, potentially stressing astrocytes—the brain’s support cells. Combined with cosmic radiation, this causes astrocytes to overexpress GFAP as they become reactive. Monitoring GFAP mRNA levels provides a “real-time” look at neural stress before physical symptoms manifest. By using cell-free systems to detect these specific transcripts from a blood or saliva sample, we can quantify neuro-environmental stress without the need for complex, heavy lab equipment like traditional immunoassays, making it an ideal diagnostic approach for resource-constrained spacecraft.
Hypothesis and Research Goal
Hypothesis: We hypothesize that BioBits® cell-free pellets can be engineered into a rapid, “just-add-water” diagnostic tool to quantify GFAP mRNA levels using a fluorescence-based riboswitch, and that these levels will be significantly higher in samples exposed to simulated cosmic radiation.
Goal: Our goal is to validate a modular detection system using the BioBits® kit. By creating a specialized RNA sensor (a “toehold switch”) that only triggers the production of a fluorescent protein when it binds to GFAP mRNA, we can create a visual “red-light/green-light” safety test for astronaut neural health. This reasoning is based on the high sensitivity of cell-free systems to small RNA concentrations and the portability of fluorescence viewers, providing a low-mass alternative to terrestrial diagnostic labs.
5. Experimental Plan
We will test three samples:
Synthetic GFAP mRNA (positive control).
Scrambled RNA (negative control).
Astronaut-derived RNA (simulated via irradiated human cell culture extracts).
Samples will be added to BioBits® pellets containing the GFAP toehold-switch DNA. If GFAP mRNA is present, the BioBits® machinery will produce a fluorescent protein. We will use the miniPCR® for precise incubation temperatures (37°C) and the P51 Molecular Fluorescence Viewer to record light intensity. Data will consist of fluorescence brightness, quantified against a standard curve to determine mRNA concentration.
Homework Part B: Individual Final Project
Week 11 HW: Bioproduction and Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I missed the date to contribute :c Will try my best to become a TA next year!
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
Component Roles
Component Category and their role in the Reaction:
E. coli Lysate : Provides the essential molecular “machinery,” including ribosomes and translation factors; the Star mutation in RNase E prevents mRNA degradation to increase yields, while the DE3 lysogen provides T7 RNA Polymerase for high-level transcription.
Salts / Buffers: HEPES-KOH maintains a stable pH (~7.5) necessary for enzyme function; Potassium and Magnesium salts act as essential cofactors that stabilize the ribosome structure and facilitate the assembly of the translation complex.
Energy / Nucleotide System: Ribose and Glucose serve as metabolic precursors to regenerate energy; the NMPs (AMP, CMP, GMP, UMP) are phosphorylated into the high-energy triphosphates (ATPs, etc.) required for fueling both transcription and translation.
Translation Mix: Provides the 20 building blocks (amino acids) required to assemble the protein chain; Tyrosine and Cysteine are often added separately or in excess because they have lower solubility or are more easily degraded in extract.
Additives: Nicotinamide (part of NAD) acts as a crucial co-factor for redox reactions, helping maintain metabolic flow and preventing the depletion of energy substrates during the reaction.
Backfill: Nuclease-Free Water is used to reach the final reaction volume without introducing enzymes that would destroy the DNA template or mRNA.
Master Mix Comparisons
The 1-hour PEP-NTP mix is designed for speed and “burst” production, utilizing the high-energy substrate Phosphoenolpyruvate (PEP) and pre-supplied nucleotide triphosphates to reach peak yields rapidly before the energy is exhausted. In contrast, the 20-hour NMP-Ribose-Glucose mix is optimized for longevity; it relies on slower internal metabolic pathways to regenerate energy and nucleotides from cheaper, more stable precursors, resulting in a lower initial expression rate but sustained production over a much longer period.
Bonus: Transcription without GMP?
Transcription can occur even if GMP (Guanosine Monophosphate) is missing because the system contains Guanine and the necessary enzymes within the E. coli lysate to perform salvage pathways. Specifically, the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT) can convert the Guanine base into GMP by attaching a ribose-phosphate group. Once GMP is formed, it is further phosphorylated by kinases in the lysate into GTP, which is the actual substrate used by RNA polymerase to build the RNA chain.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
Biophysical & Functional Properties
sfGFP (superfolder GFP): This protein is engineered for extremely fast folding and high thermodynamic stability, which allows it to reach peak fluorescence quickly in cell-free systems even when ribosomes are limited.
mRFP1 (monomeric Red Fluorescent Protein): This protein has a relatively slow maturation time (the time it takes for the internal chromophore to chemically “bond” after folding), which can lead to a significant lag between protein production and visible color.
mKO2 (monomeric Kusabira Orange 2): It is highly sensitive to pH levels; if the cell-free reaction becomes too acidic due to metabolic byproduct accumulation (like lactic acid), the orange fluorescence will dim significantly.
mTurquoise2: This variant has a very high quantum yield (brightness) but requires specific “gate-keeping” residues to stay folded; it is particularly sensitive to the concentration of molecular chaperones present in the lysate.
mScarlet-I: While it is the brightest monomeric red protein, its chromophore formation is highly oxygen-dependent, meaning it may underperform in thick hydrogels or deep “wells” where oxygen diffusion is restricted.
Electra2: Designed for rapid “photo-activation” or high-speed maturation, its primary constraint in cell-free systems is its tendency to aggregate if the translation rate is too high, requiring a balanced synthesis speed to stay soluble.
Optimization Hypothesis
Protein: mScarlet-I
Reagents: Potassium Phosphate (Buffer) and Nicotinamide (Additives)
Hypothesis: By increasing the concentration of Potassium Phosphate and adding a secondary redox-active Nicotinamide/NAD+ cocktail, we can stabilize the pH and maintain metabolic flow for a full 36-hour run.
Reasoning:
In long-term (36-hour) incubations, the primary threat to mScarlet-I is the “stalling” of chromophore maturation. This maturation is an oxidative process. As the reaction progresses, the environment often becomes more acidic (reducing brightness) and oxygen is depleted.
Increasing the Phosphate buffer prevents the pH drop that would otherwise quench the red signal.
Optimizing the Nicotinamide levels helps maintain the metabolic “engine” that handles oxygen-consuming byproducts, ensuring that available dissolved oxygen is directed toward the mScarlet-I chromophore formation rather than just being sucked up by secondary metabolic stress.
Expected Effect: This should result in a deeper, more saturated red hue that continues to intensify throughout the 36-hour window rather than plateauing at hour 8.