Week 4 HW: Protein design P1
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? Roughly 3 x 10^24 individual amino acid molecules.
1- Why do humans eat beef but do not become a cow, eat fish but do not become fish? Because digestion is a disassembly line. We completely chop up their proteins into individual amino acid building blocks, then use our own DNA blueprints to rebuild them into human.
2- Why are there only 20 natural amino acids? It’s a perfect sweet spot. Those 20 give us enough chemical variety to build everything life needs, but using more would make the system too complicated and error-prone.
3- Can you make other non-natural amino acids? Design some new amino acids. Yes, one could for example make a glowing one by attaching a light-emitting molecule
4- Where did amino acids come from before enzymes that make them, and before life started? They were probably cosmic cooking experiments. Lightning and UV rays zapped simple gases and water in the early Earth’s atmosphere, spontaneously creating them.
5- If you make an α-helix using D-amino acids, what handedness would you expect? You’d get a left-handed helix. They’re mirror images, so using the mirror-image building blocks flips the spiral direction.
7- Can you discover additional helices in proteins? Yes. We’re still finding new ones. The π-helix (a wider, rarer spiral) and the 310-helix (a tighter, stubby one) are two examples already discovered.
8- Why are most molecular helices right-handed? It’s probably a historical accident. Once early life settled on right-handed sugars and left-handed amino acids, their interactions locked everything into a right-handed spiral trend.
9- Why do β-sheets tend to aggregate? What is the driving force? Their edges are like sticky Velcro. They have exposed backbone “claws” (called hydrogen bond donors and acceptors) that desperately want to latch onto another sheet’s identical claws to hide from water.
Part B: Protein Analysis and Visualization
Renin I picked renin because it’s kind of a trend here on my lab. It’s basically an enzyme that regulates blood pressure and volume.
The sequence I got from uniprot(listed as: P00797 · RENI_HUMAN): MDGWRRMPRWGLLLLLWGSCTFGLPTDTTTFKRIFLKRMPSIRESLKERGVDMARLGPEWSQPMKRLTLGNTTSSVILTNYMDTQYYGEIGIGTPPQTFKVVFDTGSSNVWVPSSKCSRLYTACVYHKLFDASDSSSYKHNGTELTLRYSTGTVSGFLSQDIITVGGITVTQMFGEVTEMPALPFMLAEFDGVVGMGFIEQAIGRVTPIFDNIISQGVLKEDVFSFYYNRDSENSQSLGGQIVLGGSDPQHYEGNFHYINLIKTGVWQIQMKGVSVGSSTLLCEDGCLALVDTGASYISGSTSSIEKLMEALGAKKRLFDYVVKCNEGPTLPDISFHLGGKEYTLTSADYVFQESYSSKKLCTLAIHAMDIPPPTGPTWALGATFIRKFYTEFDRRNNRIGFALAR It has 406 AA’s and the most frequent is Leucina (L) with 34 appearances. It has 432 homologs.
The structure for it was deposited at 1992-02-05. And it has a resolution of 2.5Å. It belongs to the: pepsin-like family and it has a N-acetyl-D-glucosamine apart of the molecule.
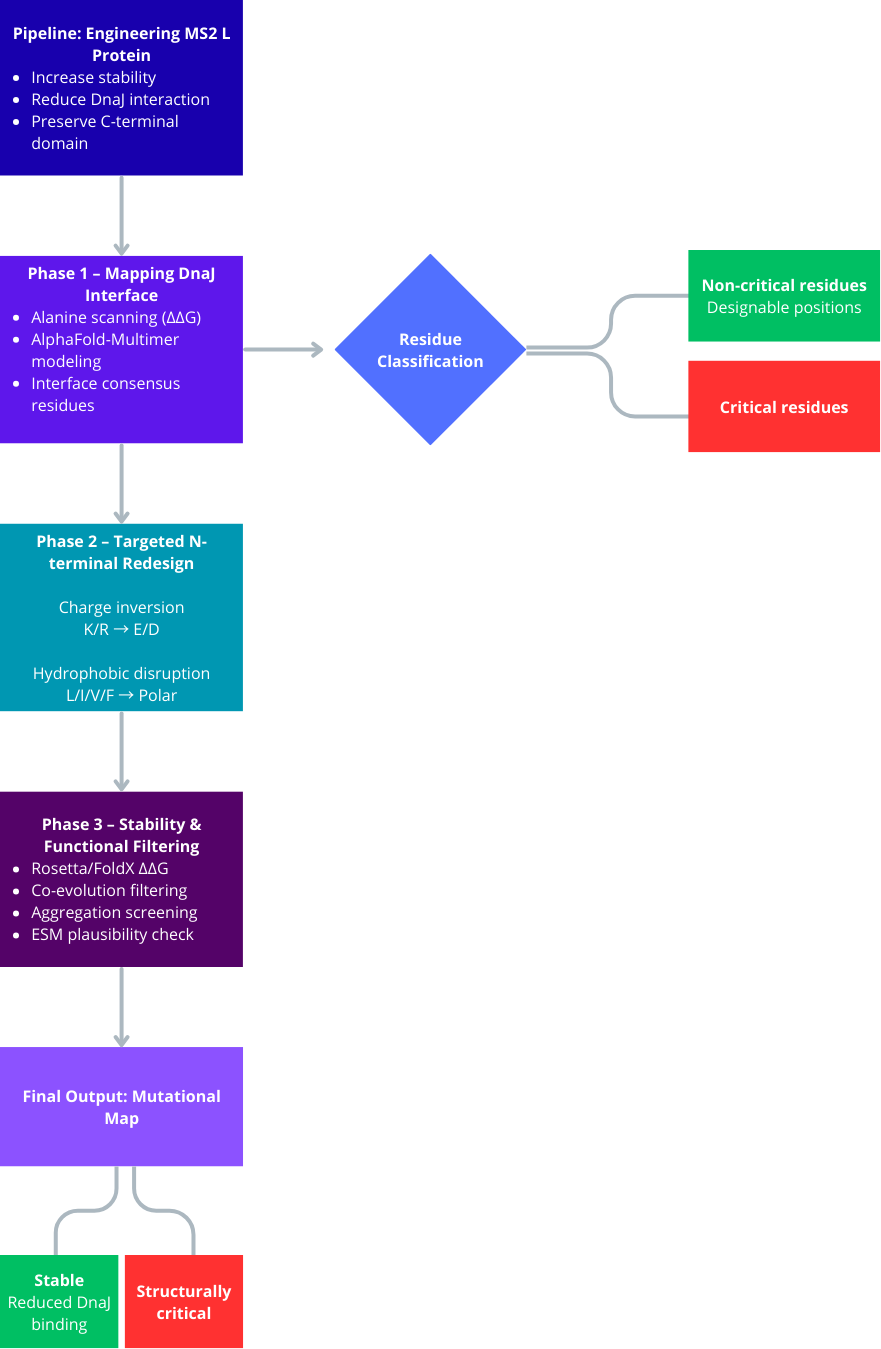
Renin as cartoon, colored by secondary structure… It mainly has beta sheets.

Renin as ribbons, colored by residues. Code:
#Hydrophobic (Ala, Val, Ile, Leu, Met, Phe, Trp, Pro, Tyr) color yellow, resn ALA+VAL+ILE+LEU+MET+PHE+TRP+PRO+TYR #Positive charges (Arg, Lys, His) color blue, resn ARG+LYS+HIS #Negative charges (Asp, Glu) color red, resn ASP+GLU #Polar (Ser, Thr, Asn, Gln, Cys) color magenta, resn SER+THR+ASN+GLN+CYS #Glycines (often left as a special case) color green, resn GLY

Renin as ball and sticks, still colored by residues.
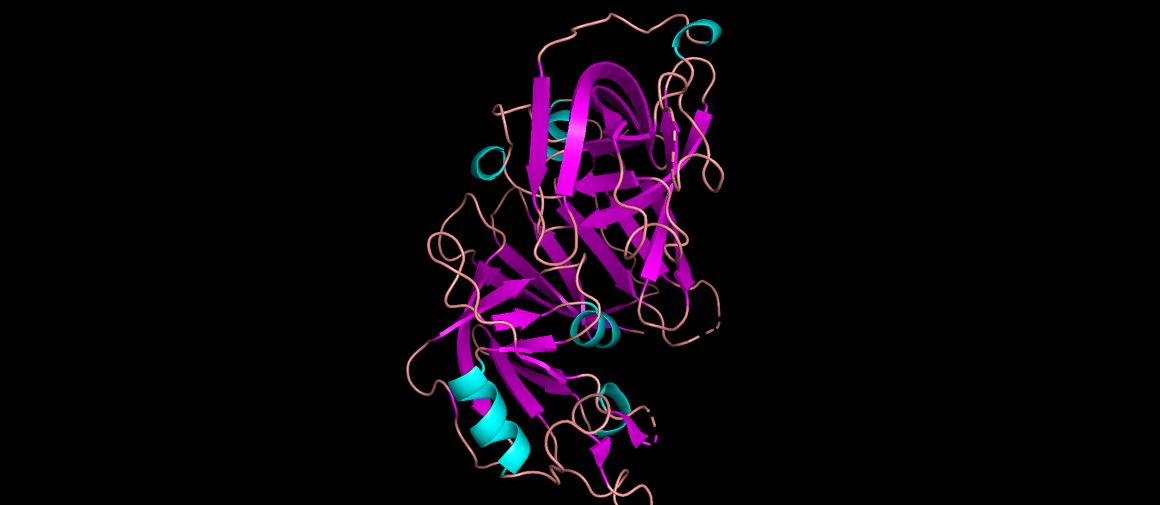
Renin surface view, catalytic aspartases (main mechanism) highlighted. Also the site were the drug Aliskiren (the direct renin inhibitor) binds…
Part C: Using ML-Based Protein Design Tools
The protein chosen for this analysis is the IgV domain of human KIM-1/TIM-1 (PDB: 5DZO), a membrane receptor involved in kidney injury, viral entry (Ebola, SARS-CoV-2), and immune regulation. The structure was solved at 1.30 Å resolution by X-ray diffraction and contains 107 residues with a canonical immunoglobulin beta-sandwich fold.
C1. Protein Language Modeling
Deep Mutational Scan
The ESM2 model (esm2_t6_8M_UR50D) was used to generate an unsupervised deep mutational scan of the KIM-1 IgV domain. Log Likelihood Ratio (LLR) scores were computed for all possible single amino acid substitutions at each position.
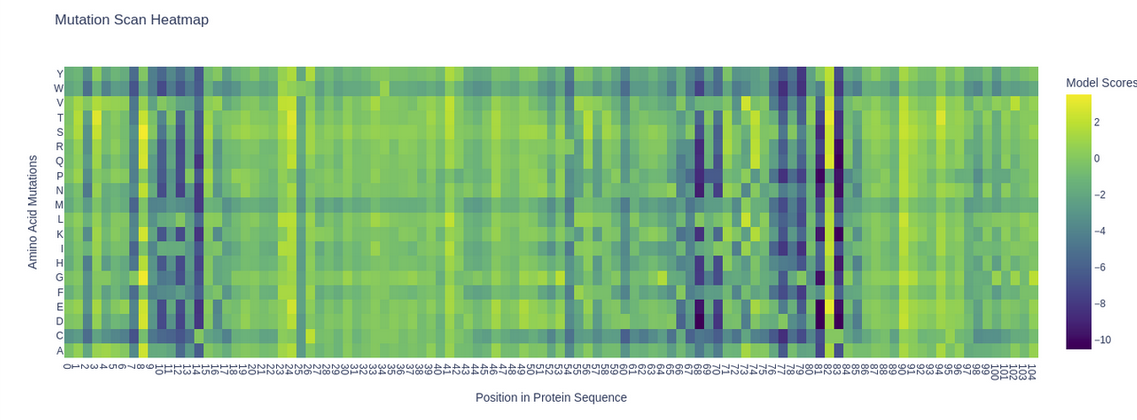
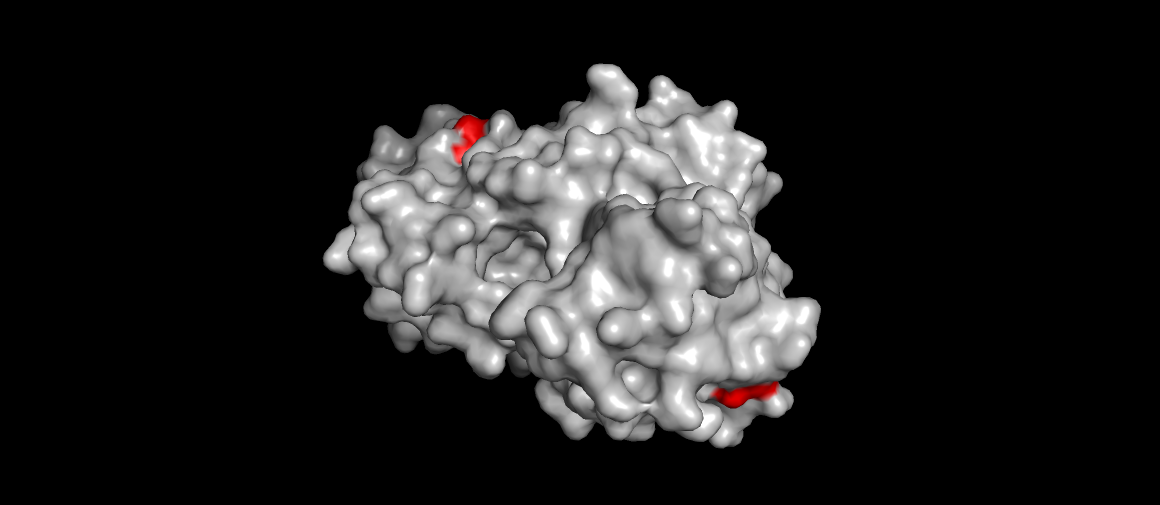
Mutation scan heatmap of KIM-1 (5DZO). Yellow = favorable substitutions (positive LLR), purple = highly deleterious substitutions (negative LLR).
A clear pattern emerges: several positions show deep purple columns across nearly all substitutions, indicating highly conserved residues that the model considers essential. Notably, the cysteine row (C) is almost entirely purple throughout the sequence, reflecting the model’s strong penalization of introducing cysteines at non-cysteine positions — consistent with the known disulfide bonds (C15–C53) that stabilize the IgV fold. A striking example is position 53 (C53), where any substitution receives strongly negative scores, highlighting its structural importance in the beta-sandwich framework.
Latent Space Analysis
The ESM2 embeddings of 500 randomly sampled SCOP proteins were projected into 2D using t-SNE.
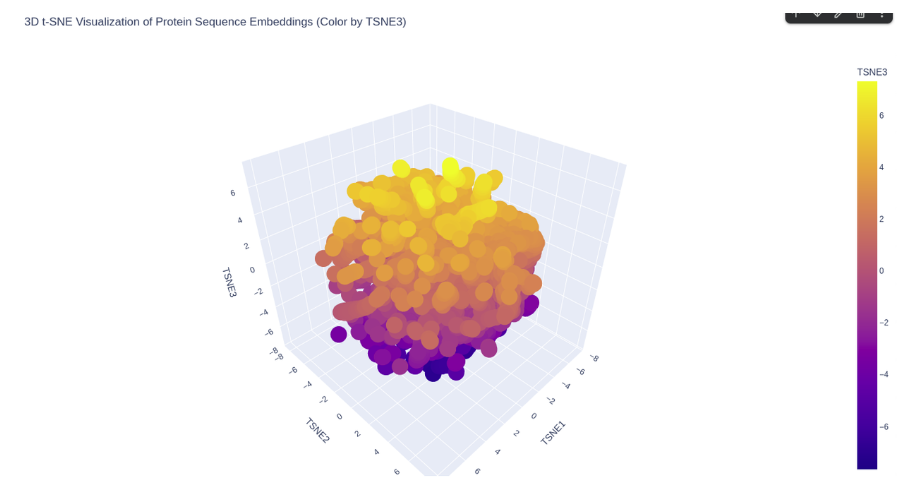
ESM2 latent space of SCOP proteins.
KIM-1 occupies a peripheral region of the latent space, somewhat isolated from the main cluster of SCOP proteins. This is consistent with its nature as a membrane-associated immunoglobulin domain — a combination less represented in the SCOP dataset, which is dominated by soluble globular proteins. Its nearest neighbors in the embedding are likely other immunoglobulin-like or beta-sandwich domains.
C2. Protein Folding
The KIM-1 sequence was folded using ESMFold directly in the notebook.
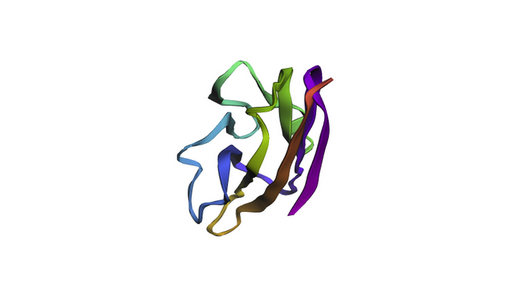
ESMFold prediction of KIM-1 IgV domain. The beta-sandwich architecture is clearly visible, consistent with the experimental 5DZO crystal structure.
The predicted structure recapitulates the canonical IgV topology, with the characteristic two-layer beta-sheet sandwich. The overall fold matches the experimental structure well, as expected for a well-conserved immunoglobulin domain. Based on the ESM2 mutational scan, positions with strongly negative LLR scores (such as C15, C53, and several buried hydrophobic residues) are predicted to be structurally intolerant to substitution — mutations at these sites would likely destabilize the fold. In contrast, surface-exposed loop regions show more permissive LLR profiles, suggesting greater resilience to mutation.
C3. Inverse Folding with ProteinMPNN
The backbone of 5DZO was used as input to ProteinMPNN to propose alternative sequences compatible with the KIM-1 structure.
Amino acid probability heatmap: The ProteinMPNN probability matrix shows that structurally critical positions — particularly the cysteines involved in disulfide bonds — are strongly constrained, with near-exclusive probability mass on the original residue. Surface-exposed and loop positions show broader distributions, allowing more sequence diversity.
Generated sequences (T=0.1):
| Sequence | Score | Recovery | |
|---|---|---|---|
| Native | MVKVGGEAGPSVTLPCHYSGAVTSMCWNRGSCSLFTCQNGIVWTNGTHVTYRKDTRYKLLGDLSRRDVSLTIENTAVSDSGVYCCRVEHRGWFNDMKITVSLEIVPP | 1.4549 | — |
| Sample 1 | MVKVGGVEGPPVTLPCTYSGEVAPFCWNRGPCSESVCPNAIVRSDGTKVTWREDPRYVVEGDLSKNDYSLTIRDTRVEDSGLYCCQVLRSGEDDNVKILIELTVTPA | 0.7971 | 55.1% |
| Sample 2 | VIEVGGVEGEPVTLPCSYSGPVRPFCWNKGPCSEEWCPNEIVRSDGTSVLWTADPRYVVEGDLSKNDYSLTIKDTKEDSGYYCCQVKRDGEDDDTKTLIKLTITPK | 0.7997 | 49.5% |
The generated sequences achieve ~50% sequence recovery while maintaining lower scores than the native (indicating higher structural compatibility with the backbone). Key structural residues such as the disulfide-forming cysteines are preserved in both designs, while surface loops show substantial sequence variation. This demonstrates that the KIM-1 IgV backbone is structurally robust — it can accommodate diverse sequences while maintaining its fold, which is a hallmark of the immunoglobulin superfamily.
Part D. Group Brainstorm on Bacteriophage Engineering
Project Objective
- Engineer the L protein of the MS2 phage to increase structural stability.
- Disrupt or reduce its interaction with the bacterial chaperone DnaJ.
- Preserve the C-terminal lysis domain to maintain lytic function.
- Avoid mutations that interfere with structurally or evolutionarily coupled residues.
Phase 1: Mapping the DnaJ Interaction Interface
Since the exact binding interface between the L protein and DnaJ is unknown, the first step is to identify it computationally rather than introducing arbitrary mutations.
- Use AlphaFold-Multimer to model the complex between L protein and DnaJ.
- Generate multiple structural predictions and select the top-ranked models.
- Identify consensus interface residues that consistently appear in the predicted binding interface.
- Perform in silico alanine scanning of the N-terminal residues in the complex to determine which residues significantly contribute to binding energy (ΔΔG).
- Analyze whether the N-terminal region resembles known DnaJ-binding motifs, typically hydrophobic residues flanked by basic amino acids.
This phase defines which residues are critical for interaction and should not be mutated randomly.
Phase 2: Targeted N-Terminal Redesign
Instead of deleting regions or performing extensive random substitutions, introduce controlled chemical modifications to disrupt interaction while preserving structural stability.
Focus on charge inversion strategies:
- Basic residues (K, R) → Acidic residues (E, D)
- Acidic residues (E, D) → Basic residues (K, R)
Disrupt hydrophobic interaction patches:
- Hydrophobic residues (L, I, V, F) → Polar residues (S, T, N, Q)
- Aromatic residues (F, Y, W) → Aliphatic or small residues
Generate a graded library of variants:
- Minor charge modifications
- Moderate interface perturbations
- Strong hydrophobic disruption
This creates a Pareto front of variants balancing reduced DnaJ interaction and preserved protein stability.
Phase 3: Stability and Functional Filtering
To ensure that redesigned variants remain structurally viable and functionally relevant:
Use Rosetta or FoldX to calculate ΔΔG and verify that mutations do not destabilize the overall protein fold.
Confirm that mutations in the N-terminal region do not propagate structural stress toward the C-terminal lysis domain.
Perform co-evolutionary analysis (e.g., EVcouplings):
- Identify residue pairs that co-evolved between the N-terminal and C-terminal regions.
- Avoid mutating co-evolved residues independently to prevent functional disruption.
Evaluate aggregation propensity using tools such as Aggrescan3D to ensure that mutations do not create exposed hydrophobic patches leading to cytoplasmic aggregation.
Assess sequence plausibility using protein language models such as ESM to filter out unlikely or non-natural variants.
Key Limitations
- The DnaJ binding mode may be transient or dynamic, reducing AlphaFold-Multimer accuracy.
- Protein language model scores do not guarantee in vivo functionality.
- Intrinsically disordered regions may not be accurately modeled.
- Computational predictions must ultimately be validated experimentally.