Week 2 HW: DNA Read, Write & Edit
Week 2 : Pre-HW
Professor Jacobson:
A1. DNA polymerase with proofreading has an error rate of about 1 error per 10⁶ bases (10⁻⁶). This is due to its proofreading and exonuclease activity. The human genome is about 3.2 billion base pairs. At a raw error rate of 10⁻⁶, replication would introduce thousands of errors per genome copy, which is unacceptable. Biology deals with this via multiple layers of correction, DNA Polymerase proofreading, post replication mismatch repair and other such systems.
A2. The genetic code is degenerate in nature, therefore there could be many possible DNA sequences that could encode the same protein sequence. There could be in theory, millions of DNA sequences that could encode one protein. Most of these sequences wouldn’t work due to biological constraints like Codon bias, repetitive sequences causing errors, Inhibiions in transcription/translation.
Dr. LeProust:
A1. The most common method is chemical phosphoramidite DNA synthesis. It works via:
Stepwise base addition Chemical protection/deprotection cycles Typically ~5 minutes per base addition A2. It is harder to synthesize oligos longer than ~200 nt as errors accumulate with the addition of every base. Depurination and incomplete reactions increase with time and by the time you reach ~200 nt the drops in yield and accuracy make the product unreliable.
A3. Directly synthesizing a 2000 bp gene would accumulate too many errors to be of actually use. The yield would be extremely low and the process would be expensive. We use assembly based approaches to make long genes instead via using medthods like Gibson Assemby then doing sequencing and error correction later on.
George Church:
A1. There are 10 essential amino acids that animals cannot synthesize and must get from food. They are:
Isoleucine Leucine Lysine Histidine Methionine Phenylalanine Threonine Tryptophan Valine Arginine
I wasn’t aware of the ‘Lysine Contingency’ but a quick google search revealed that it is a reference to “Jurassic Park” wherein they engineer dinosaurs so they cannot synthesize lysine and must receive it externally, acting as a biological control mechanism. In reality, all animals already lack the ability to synthesize lysine, making them inherently dependent on plants and microbes. This makes the Lysine contingency an actually real thing, but if such dependencies could be engineered then it could be used to control organisms.
Part 1: BENCHLING ADVENTURES AND GEL ART
This week’s homework was pretty daunting as it involved Benchling. Something I’d never heard of before. I just decided to follow, the steps and figure out stuff as I go.
After creating a Benchling account and logging in, I was greeted by a screen that looked so complex. A plasmid on the right, DNA sequence on the left, a lot of restriction sites. I decided to just follow the next step. After clicking on the ‘plus’ icon and selecting Import DNA/RNA sequence.
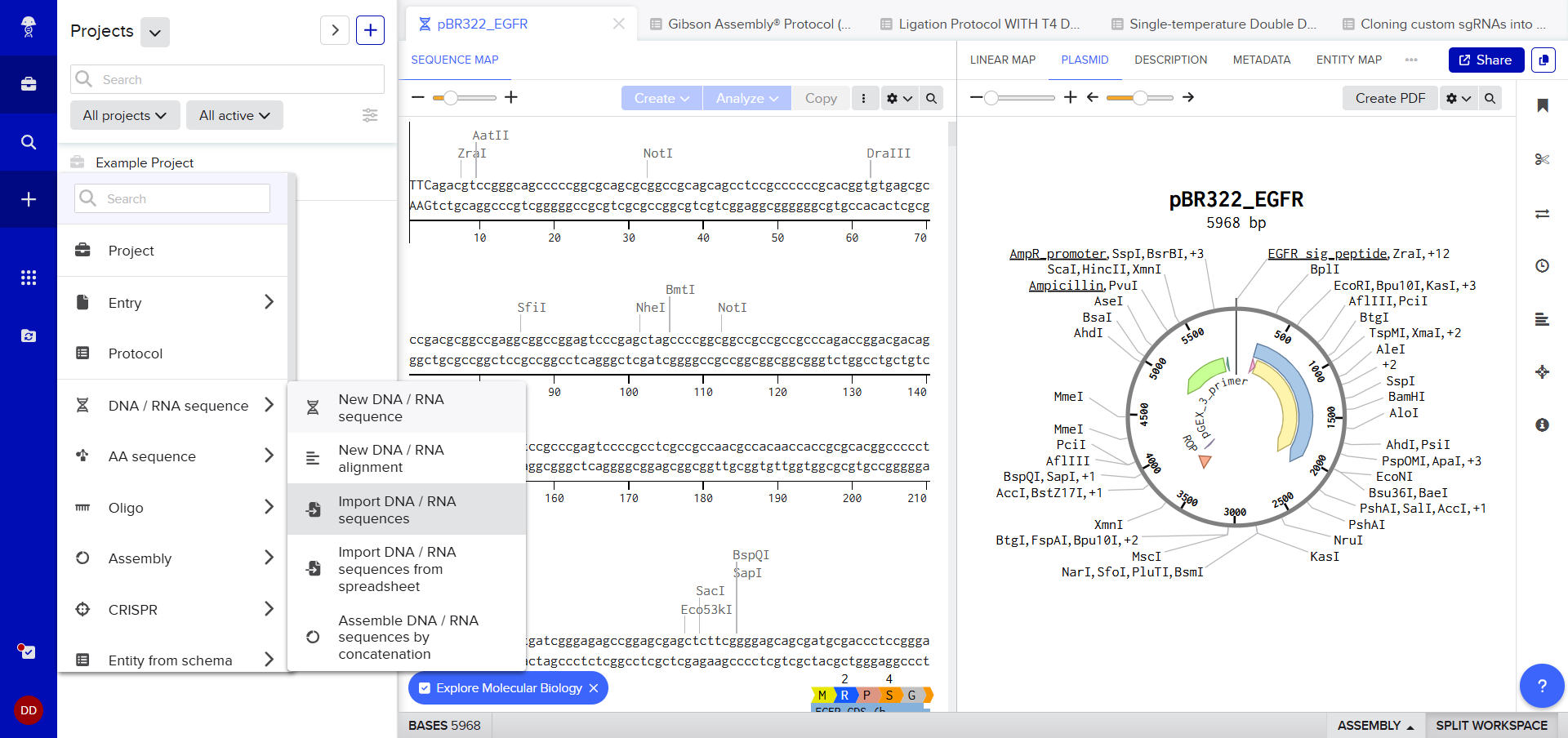
A pop up window asked me to upload the DNA Sequence, I thought I could just add the accession number or something (Something I’d used in my graduate biotechnology coursework) I wasn’t sure, so I still decided to stick to the scaffold and just follow the next step. :)
The link to lambda DNA Sequence was in the Google Doc for the homework, I opened the link and right-clicked to save the file.
I made sure that I saved with the .gb extension as I was downloading a GenBank file, it was being downloaded as a .txt file. (I didn’t want any uploading problems)
Then I just drag and dropped the .gb file to the Benchling pop-up window and the sequence started to be uploaded. (So far so good.)
I was awestruck when I saw the screen post sequence upload; I was being overwhelmed with information. Everywhere I looked, there was something new yet it seemed familiar. I then found the digest button in the side panel on the left (SCISSOR ICON).
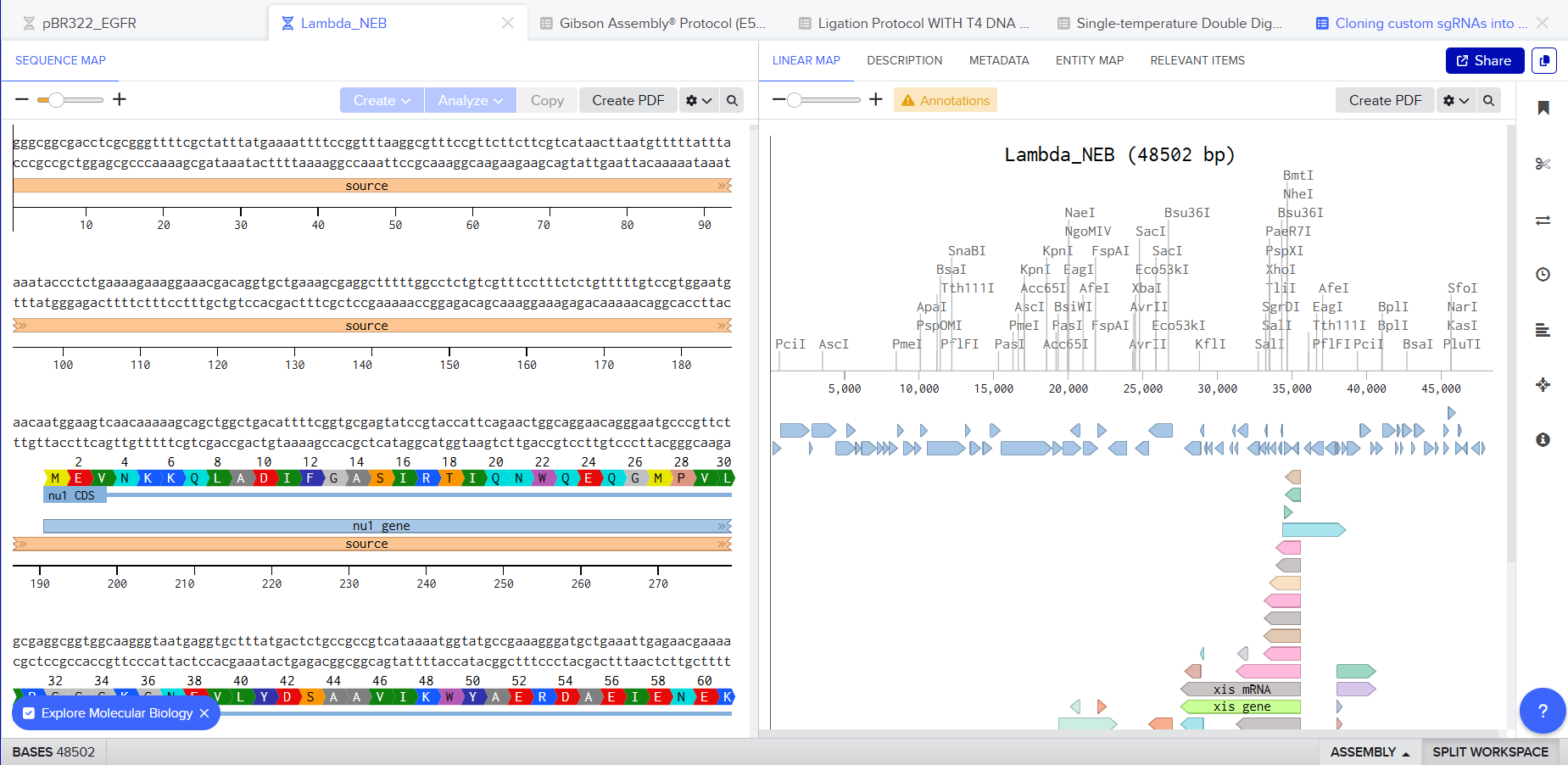
Clicking on the scissor icon, another panel for ’new digest’ opened up and it seemed intuitive. I was supposed to add the enzymes from the HW Doc, and then do an in-silico restriction of the DNA. I managed to add all the enzymes into the list and then clicked on the big blue ‘RUN DIGEST’ button.
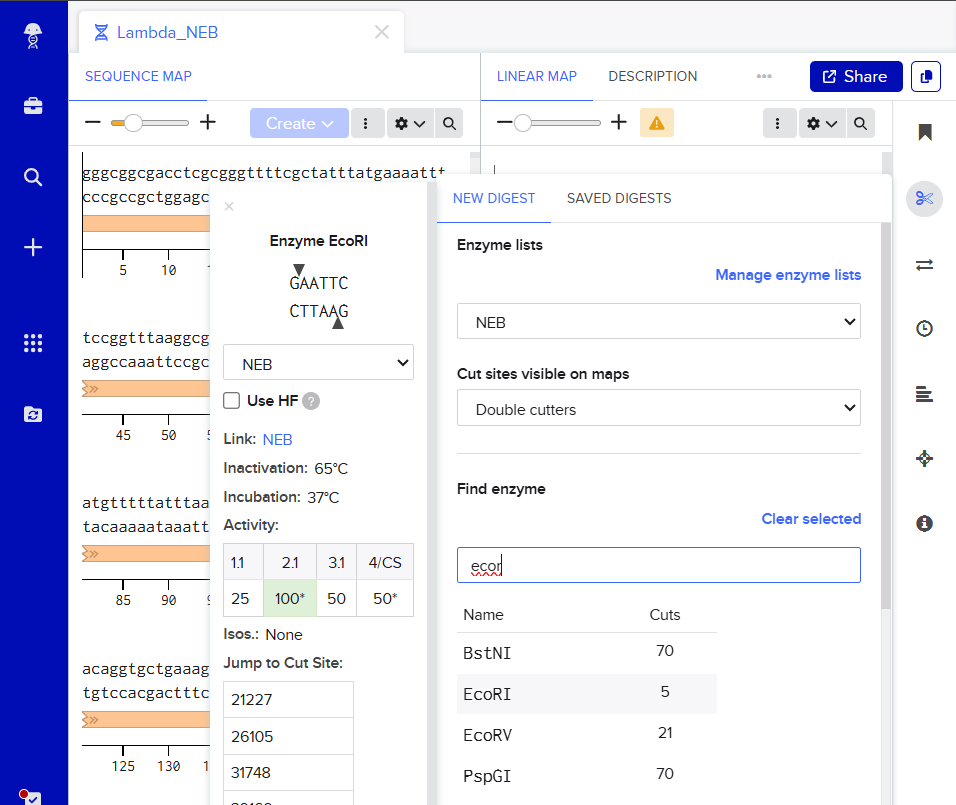
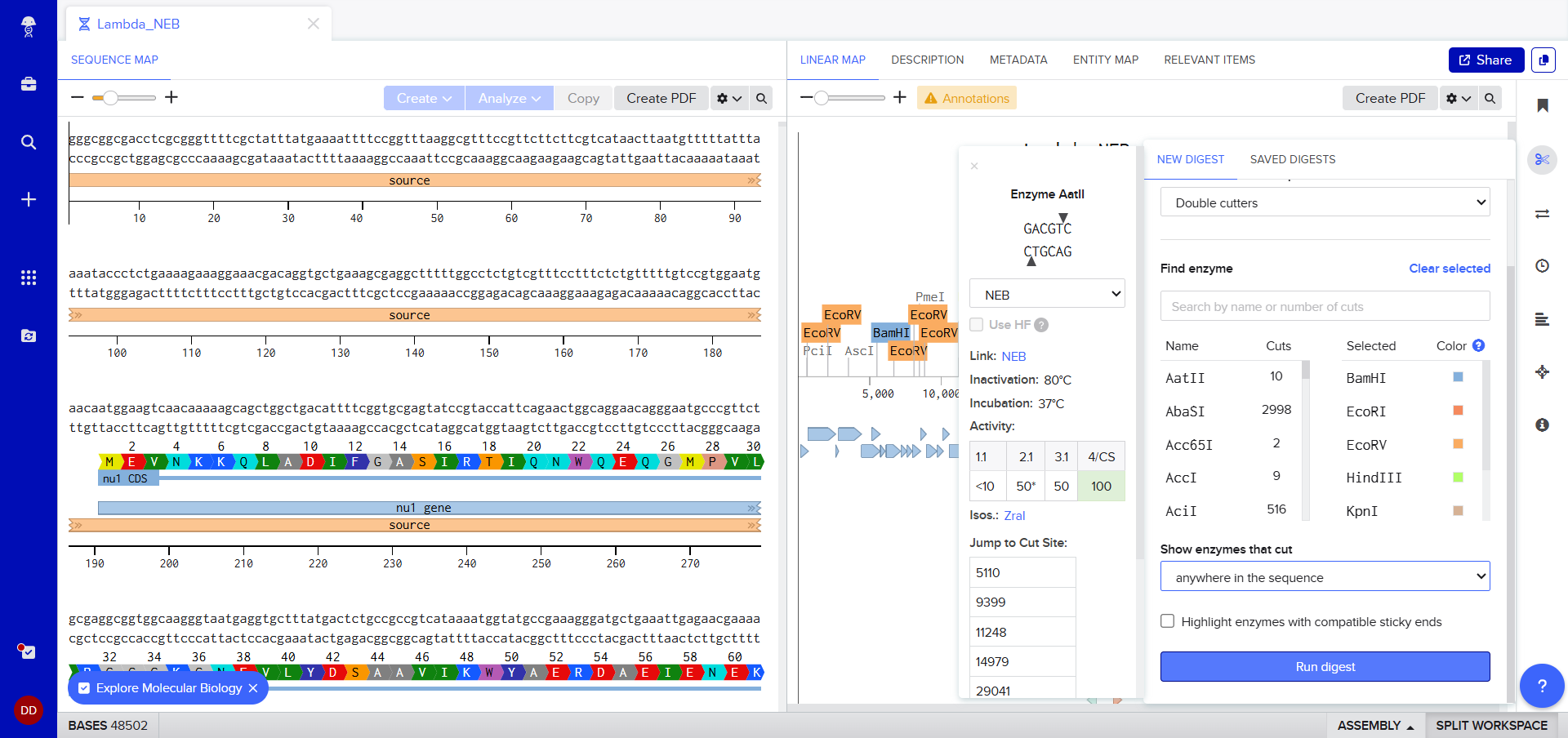
Okay so before moving ahead. I was very intimidated by Benchling and the entire homework so I had tinkered around in the whole HW Doc and I had also visited the DNA Gel Art Interface website by ‘rdonovan’ (https://rcdonovan.com/gel-art) At first I wasn’t able to understand what was happening, I only had a general idea of what this was, but there was no tutorial/tooltips, I wasn’t sure what button did what. After selecting/deselecting enzymes and pressing the arrows, I found out that this was also like Benchling’s Digest thingie but this was quicker and allowed faster tweaks. To make sure, I selected all the enzymes in DGAI (I’ll refer to rdonovan’s website this way to keep things simple) and then tried to replicate the same in Benchling.
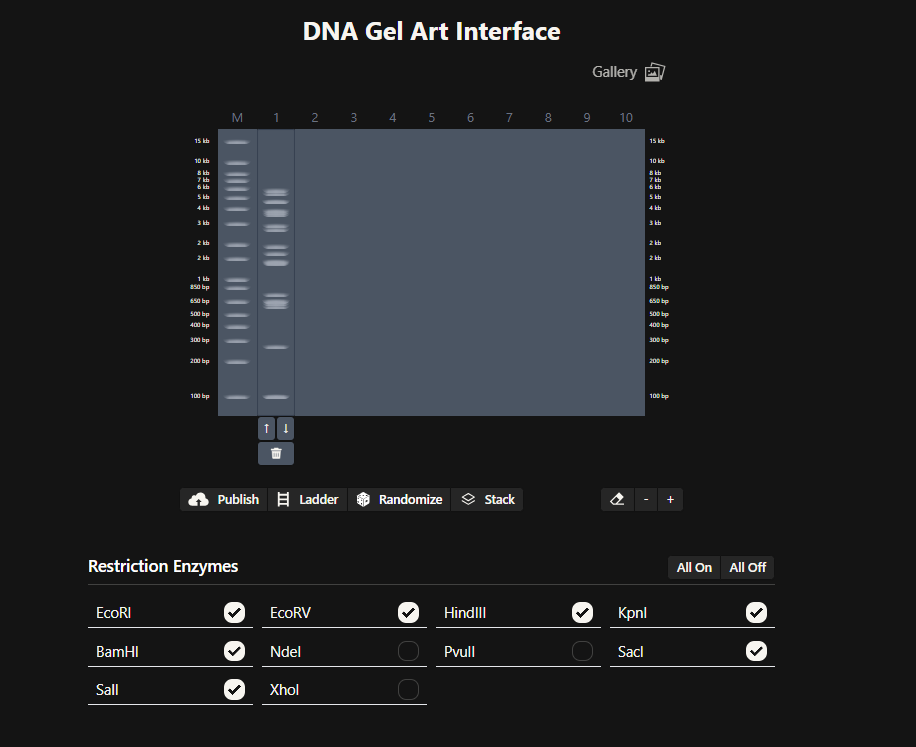
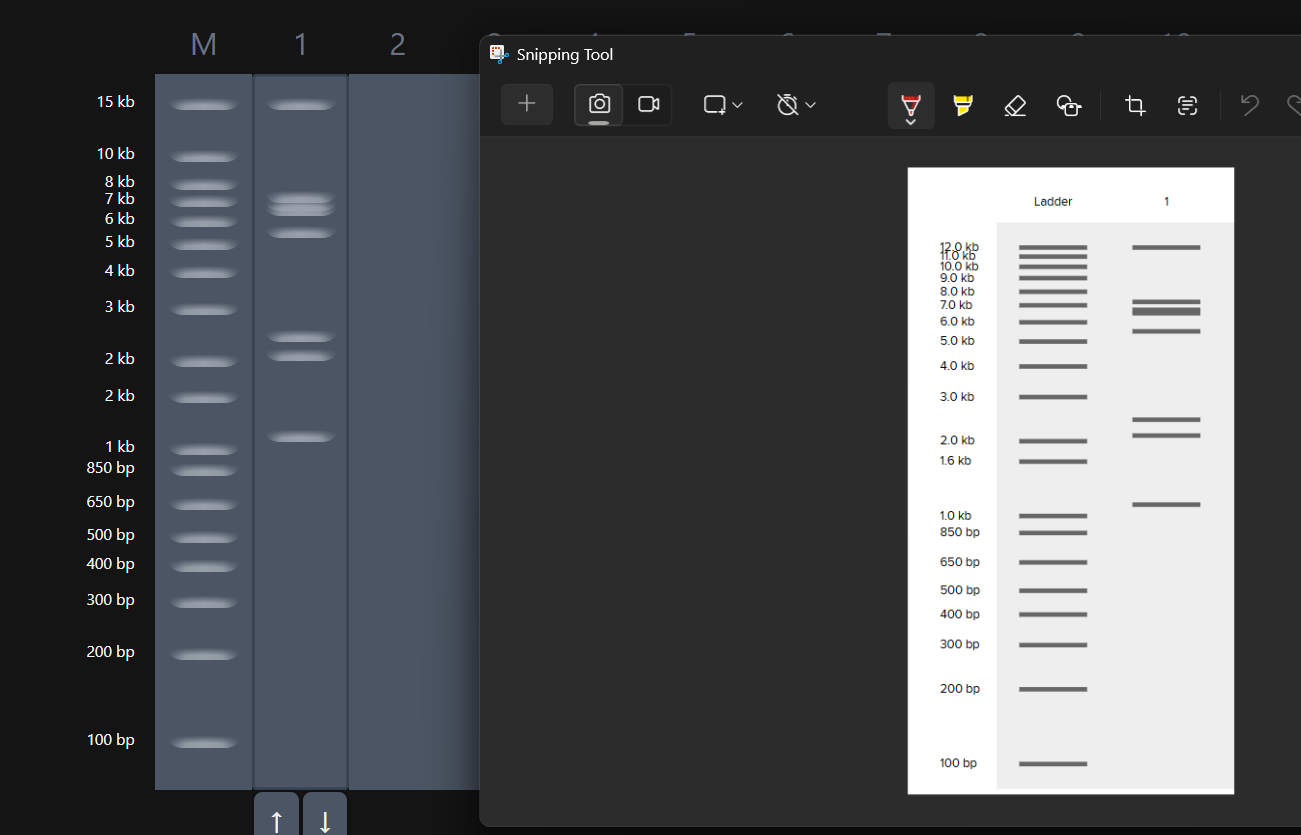
While trying to do this, I found out that the table below the enzymes in DGAI was the main thing to focus on. When I clicked on the arrows for a specific well, with a specific combination of enzymes, the table showed what enzymes were used for THAT specific result. This was I was able to find out how to replicate the DGAI gel in Benchling. For some reason, my Benchling results looked slightly different then DGAI. (side by side comparison below)
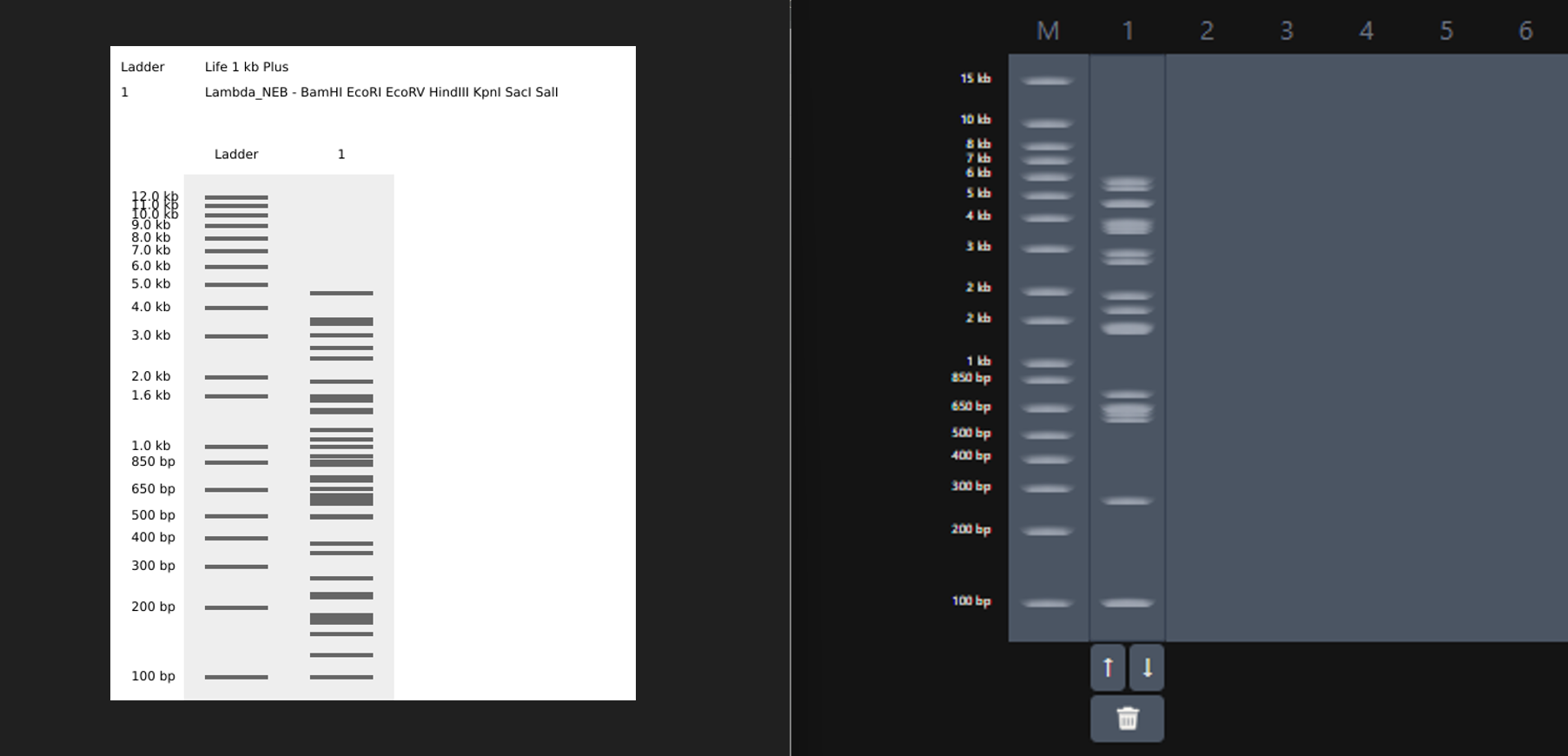
I thought maybe it was because in Benchling, it showed N/A for KpnI and SacI and in DGAI, they were selected??
I decided that to make my pattern, I’ll tinker around with different enzyme combination and see what they give me, note that down and then see what I could muster up from the patterns. I did think that maybe I could reverse engineer somehow that this combination gives these types of bands and then make a program that could somehow tell me the closest enzymes I could use to get a particular result. Like I could select the areas to keep on, like a display but all that abductive reasoning would be of no use as I wasn’t sure if it could be done. I continued with trying to make a pattern art.
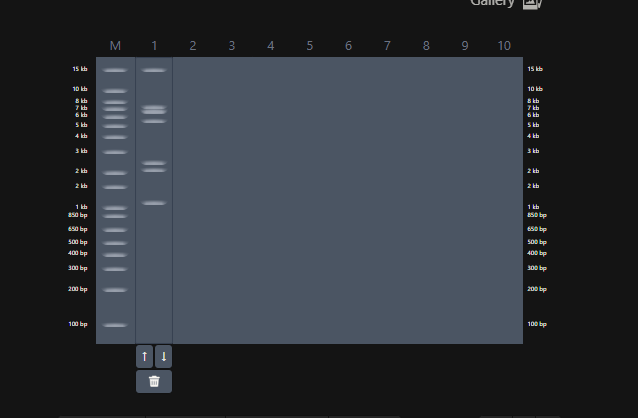
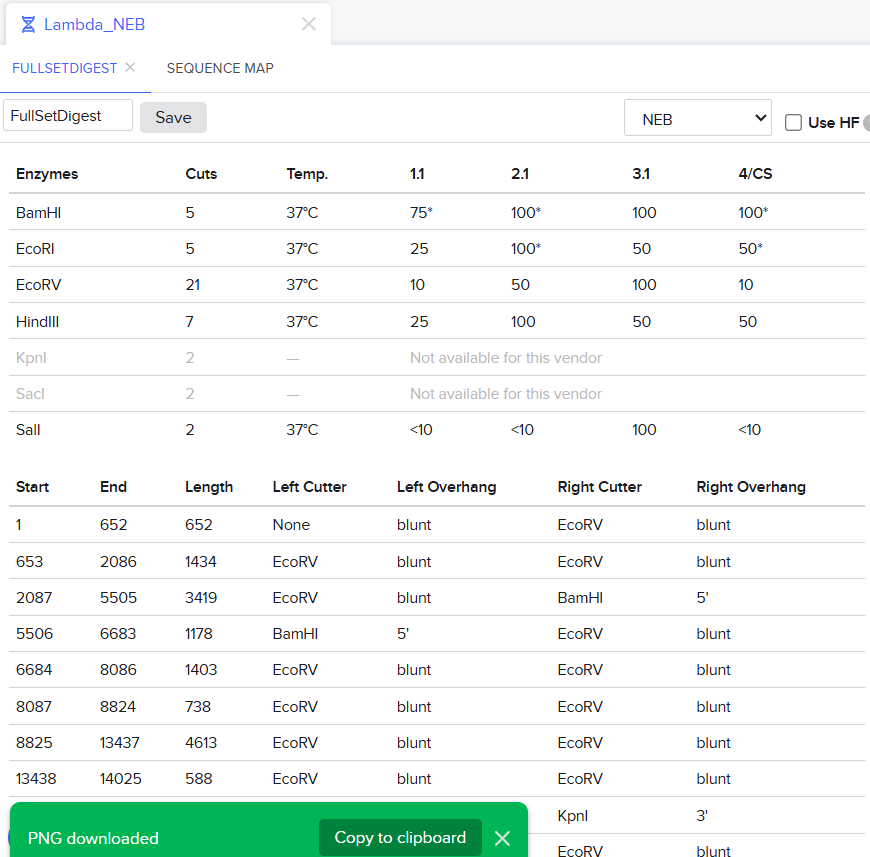
This the table that I mentioned a while ago, If you can see that in well 1, the enzymes used are BamHI and SacI. I then used the same combination in Benchling and ran a digest.
Mission Successful! I was able to get how to replicate DGAI Gels in Benchling Digest (mostly.)
Fast forward to after experimenting with multiple combos for an hour or so, I was able to make something that looked like an M. My friends said that, I do see it but to me it looked like that one cat meme (minus the whiskers and ears) in DGAI.

Then I used the table and enzymes combos from DGAI to replicate the digest in Benchling!
I had to get a bit crafty as Benchling didn’t allow an empty digest. I googled which enzyme doesn’t cut lambda DNA.
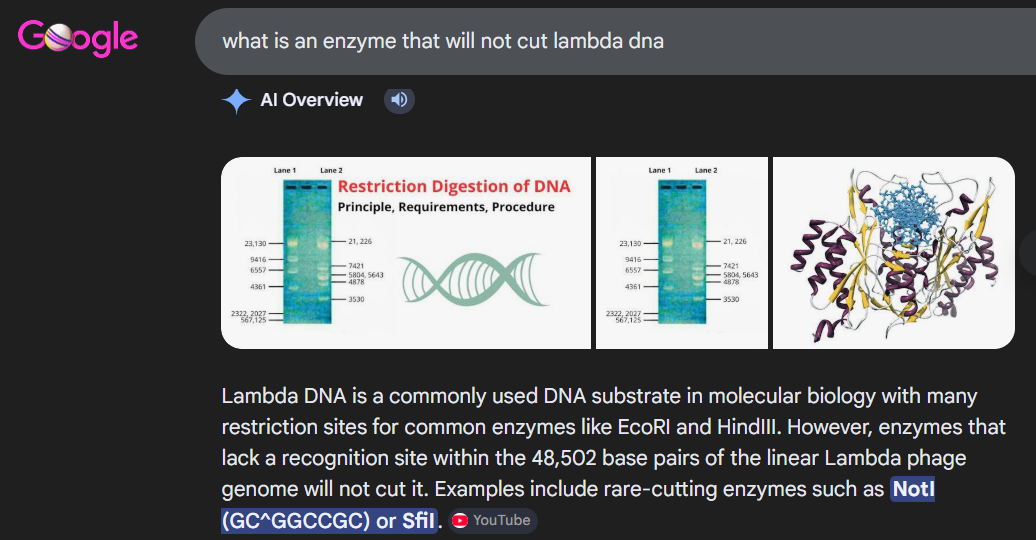
I used NotI in the digest to get an empty well. :) Mission two Successful! I was able to achieve the same output in Benchling.

It was the end but I clicked on a band in the Benchling Gel and found out that it also shows you the exact point where the cut was made and what made THAT band. I figured that if I want to refine my art further, I can maybe use this information to my advantage.
Part 3: DNA Design
3.1. Choose your protein.
The protein that I would choose is Green Fluorescent Protein. I choose GFP because it is used a lot to track other proteins, to see expression of proteins. It’s just interesting to me that it allows us to study other proteins up close.
The protein sequence for GFP (I used UniProt to get it):
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The nucleotide sequence is:
I used Bioinformatics.org Reverse Translation tool to reverse translate the AA sequence to the DNA sequence.
3.3. Codon optimization.
I would optimize the codon usage for E.coli because it grows fast, it is well-researched. The thing about GFP is that it will not be the main protein of interest but rather it’ll be used to study one. So if I have a protein that I have expressed in a certain microorganism, then I will have to optimize the codon according to that.
About why do we optimize codons, I know this! I once had a question in my mind that why is thermos thermophilus heat resistant. Why can it live in such high temperatures? I basically went on a bioinformatics quest. To answer the question so I. First, my hypothesis was that maybe it has more GC content because GC has three hydrogen bonds and just having overall more hydrogen bonds would make it more heat stable. Then to validate my hypothesis, I had to see what genes it had and I had to compare it genes with. E coli. I saw that the codons with GC were preferred more. (Codon Bias) https://www.youtube.com/watch?v=1Jrawq9fnMs&t=1791s
The reason that we optimize codons is because certain microorganisms have their own preferences of codons to use. It could be so that if the organism has a preference of a certain codon then that tRNA which is required for the protein expression is in abundance and if you pick a codon whose tRNA is not readily available in that organism, then there is a chance that because of the lack of the tRNA the protein might not be expressed. Therefore in order to increase the chances of expression we have to optimize the codons for our nucleotide sequence.
I used VectorBuilder to optimize the codons. The interface is pretty intuitive. Just paste your sequence, select the organism. (There was also an option to avoid sites for certain restriction enzymes, I think that is to avoid having the sites of the enzymes that we might work with, so that our DNA doesn’t get damaged while working with something else?)
GFP protein DNA sequence with codons optimized for E.Coli
ATGAGCAAAGGCGAAGAACTGTTTACCGGCGTGGTGCCGATTCTGGTGGAACTGGATGGCGATGTGAATGGCCATAAATTTAGCGTGAGCGGCGAAGGTGAAGGCGATGCGACCTATGGCAAACTGACCCTGAAATTTATCTGCACCACCGGTAAACTGCCGGTGCCGTGGCCGACCCTGGTGACCACCTTCAGCTACGGCGTGCAGTGTTTTAGCCGCTACCCGGATCATATGAAACAGCATGATTTTTTTAAAAGCGCGATGCCGGAAGGCTATGTGCAGGAACGCACCATTTTTTTCAAAGATGATGGCAATTACAAAACCCGTGCCGAAGTGAAATTCGAAGGCGATACCCTGGTGAATCGCATTGAACTGAAAGGCATTGATTTTAAAGAAGATGGTAACATTCTGGGCCACAAACTGGAATACAACTATAACAGCCATAACGTGTACATTATGGCGGATAAACAGAAAAATGGCATTAAAGTGAACTTTAAAATTCGCCATAACATTGAAGATGGCTCAGTGCAGCTGGCGGATCACTATCAGCAGAACACCCCGATTGGCGATGGCCCGGTTCTGCTGCCGGATAACCACTATCTGAGCACCCAGAGCGCGCTGTCGAAAGATCCGAACGAAAAACGCGATCACATGGTGCTGCTGGAATTTGTGACCGCCGCGGGCATCACCCATGGTATGGATGAACTGTATAAA
3.4. What do we do with the sequence?
The sequence can be used to chemically synthesize the DNA and then be put into a plasmid. The plasmid can then be inserted into our host organism (via electroporesis?) and then our protein can be expressed.
3.5.
- Describe how a single gene codes for multiple proteins at the transcriptional level.
A single gene can code for multiple proteins via something called ‘Alternative Splicing’. Different combinations of exons are joined together from same pre-mRNA, to create varied mRNA molecules, this allows one gene to produce multiple protein isoforms (variants).
- I aligned the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! (Using Photoshop, stitching together screenshots from Benchling)
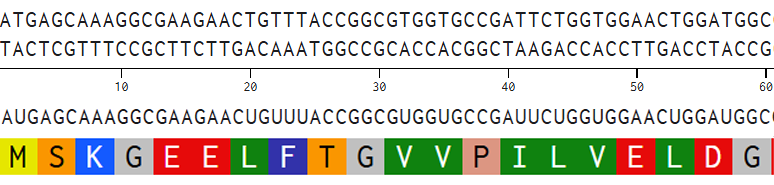
BTS: Aligning Sequences in Photoshop
Part 4 : Fake Twist DNA Synthesis Order
I just created a account using the ‘Sign Up’ Button. Pretty simple stuff really. Just add details, set a password. In the organization field I added HTGAA and Lab = 2026. I didn’t really think too much. I verified my email and I was in! I didn’t have to create a Benchling account as getting through Part 1 of the HW required using Benchling, so I already had an account.
4.2. Build Your DNA Insert Sequence
I imported the DNA Sequence into Benchling, just like from Part 1. Selected Linear topology as this is meant to be inserted into a circular vector of our choice. As I was going ahead, i realized that the exercise is already making use of GFP as an example. (well, good for me :))
I went through the sequences given in the HW document and then pasted the sequences into the Benchling file one after the other (Just the way we imported a sequence in Part 1, but here I had to copy everything one by one and then paste). I annotate the sequences based on the information in the HW document. (screenshots below on how, from the HW Doc)
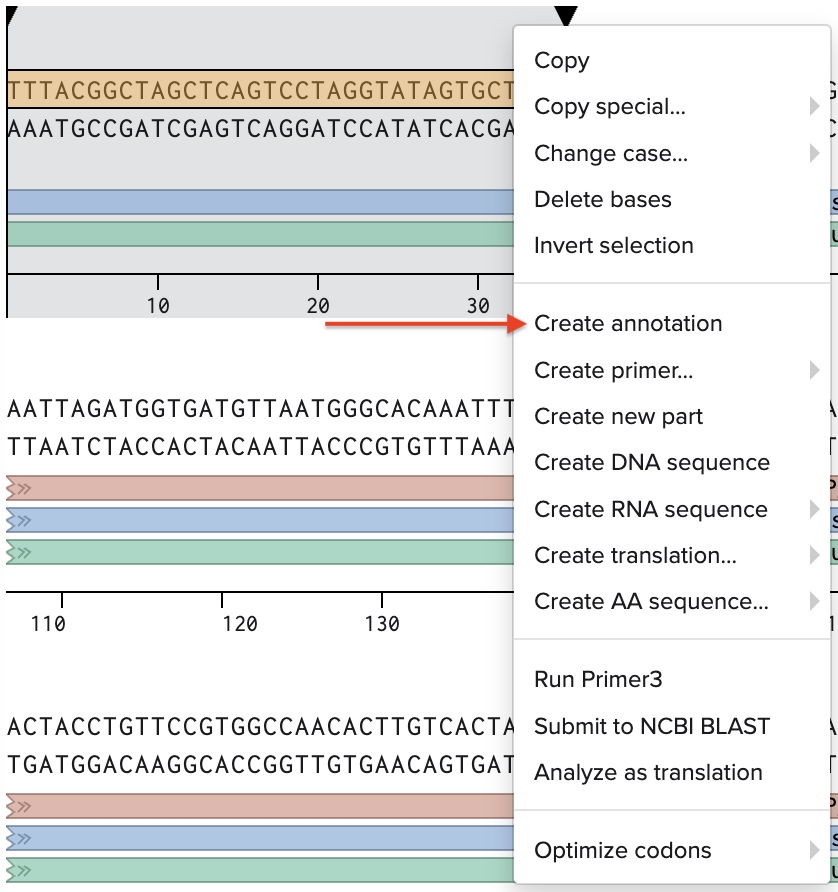
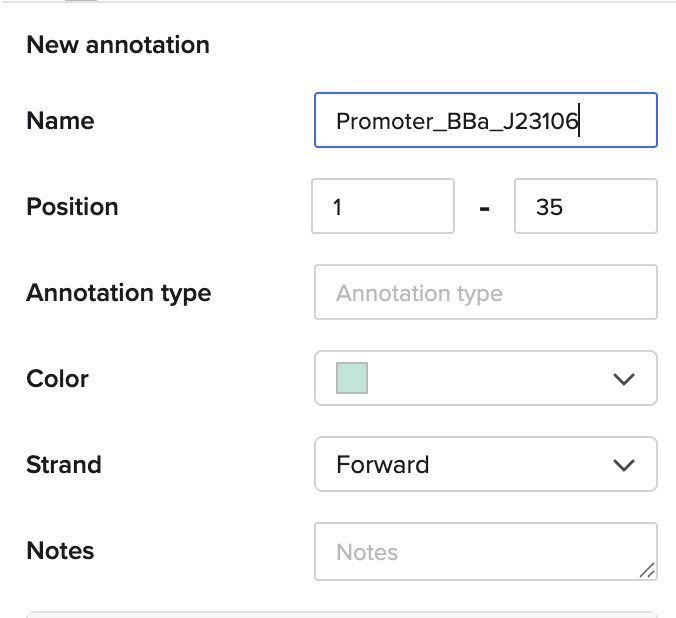
My Benchling Annotation Screenshots:
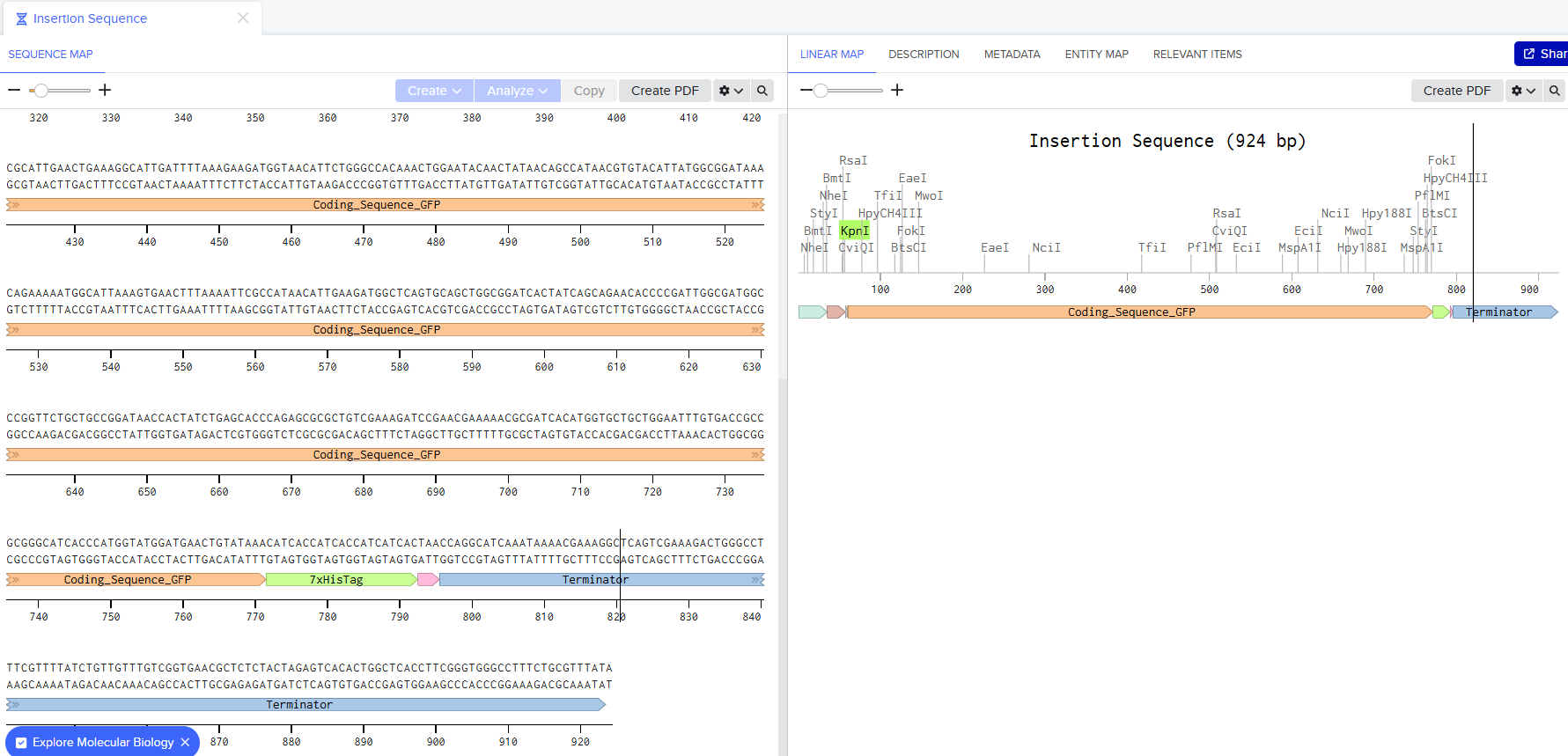
After about 15 mins, I was done with it and I turned sharing on: here’s the link to my sequence: https://benchling.com/s/seq-bXgbXvSR0KND8n5Y59IJ?m=slm-FudwlRJXB3kzUe0O5m3M
I then went and exported the sequence, by clicking on the metadata tab and then clicking on the three dots and selecting export sequence, I selected FASTA format to export.
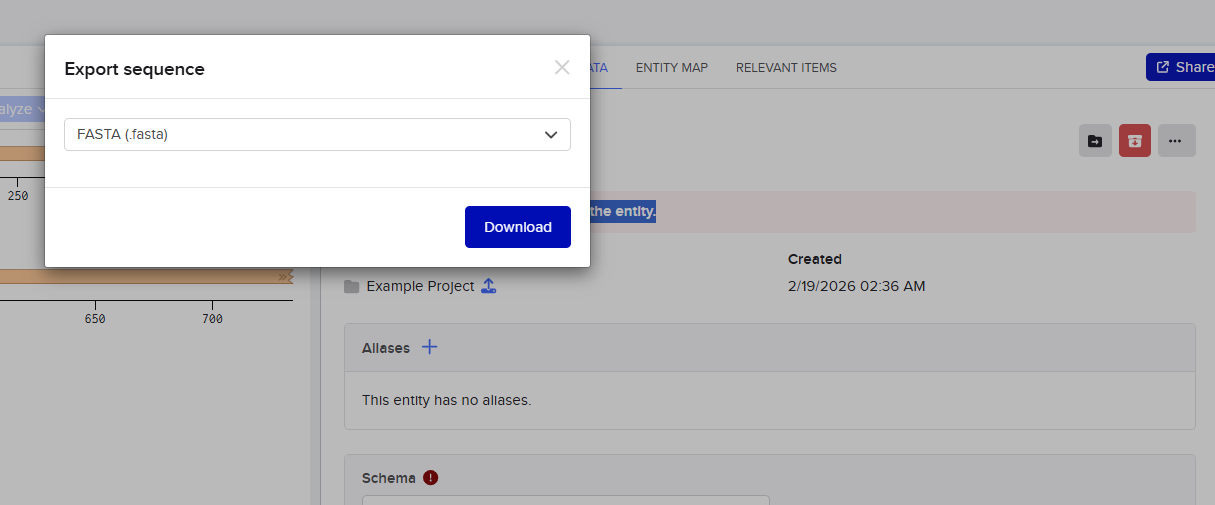
4.3. Benchling to Twist: continuing with our fake order
On the Twist E-commerce platform, I went and selected Genes -> Clonal Genes. (Screenshot from HW Doc)
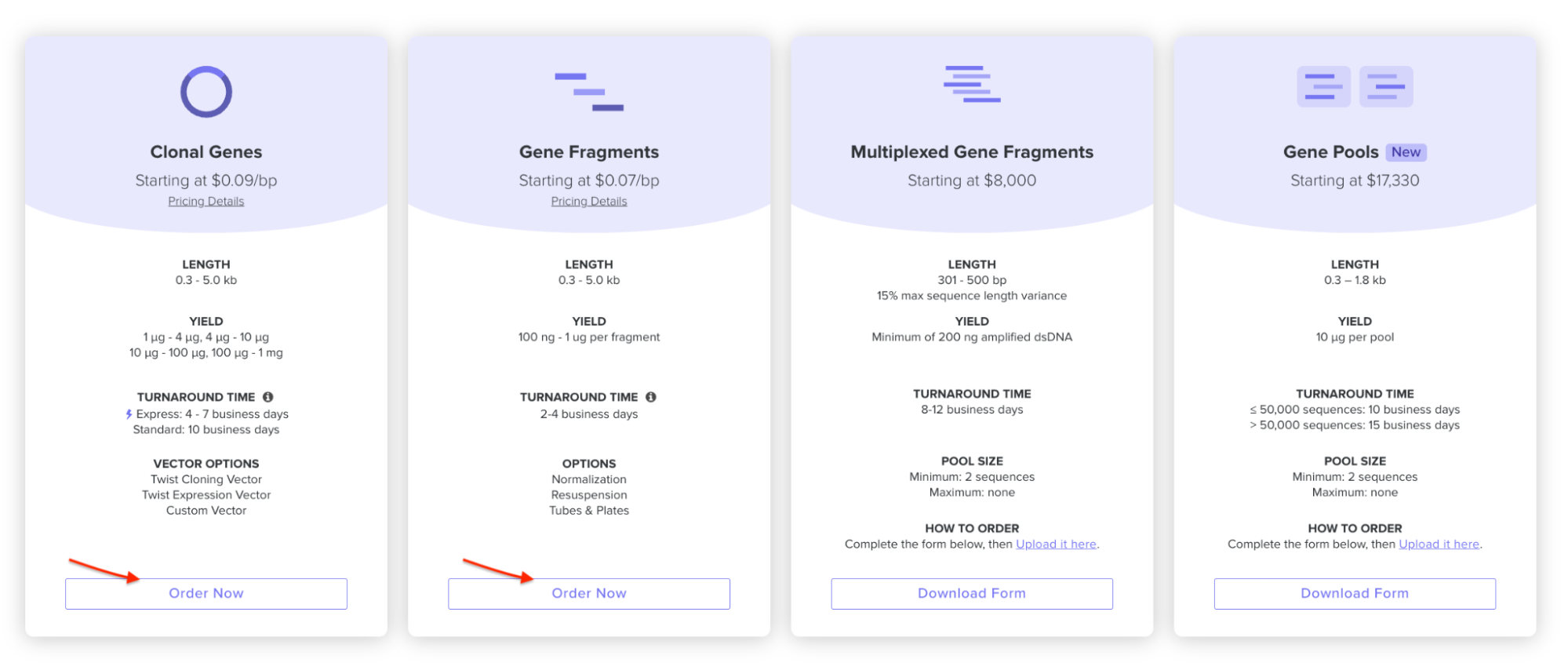 Then I had to import my sequence, I drag and dropped the FASTA file that I downloaded from Benchling.
Then I had to import my sequence, I drag and dropped the FASTA file that I downloaded from Benchling.
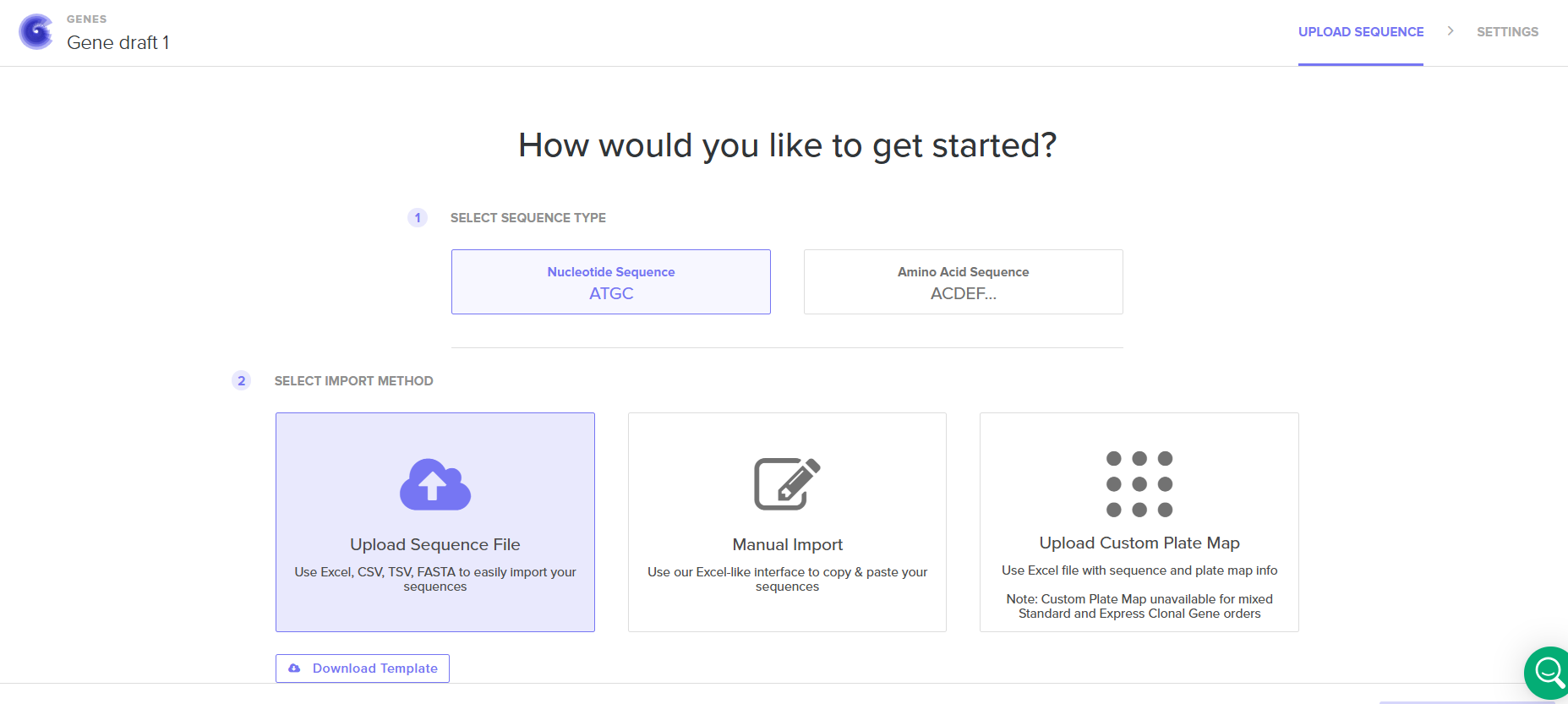
After the sequence had been uploaded successfuly, I clicked on the sequence and I saw this screen. (the twist platform also allows you to do codon optimization, niceee!)

I had to refer to the HW Doc to know what was next. Turns out I had to select a vector, I did that by clicking on select vector option on the sequence, a drop-down dialog allowed me to choose a vector in ‘Cloning’. I chose pTwist Amp High Copy based on the HW Doc.
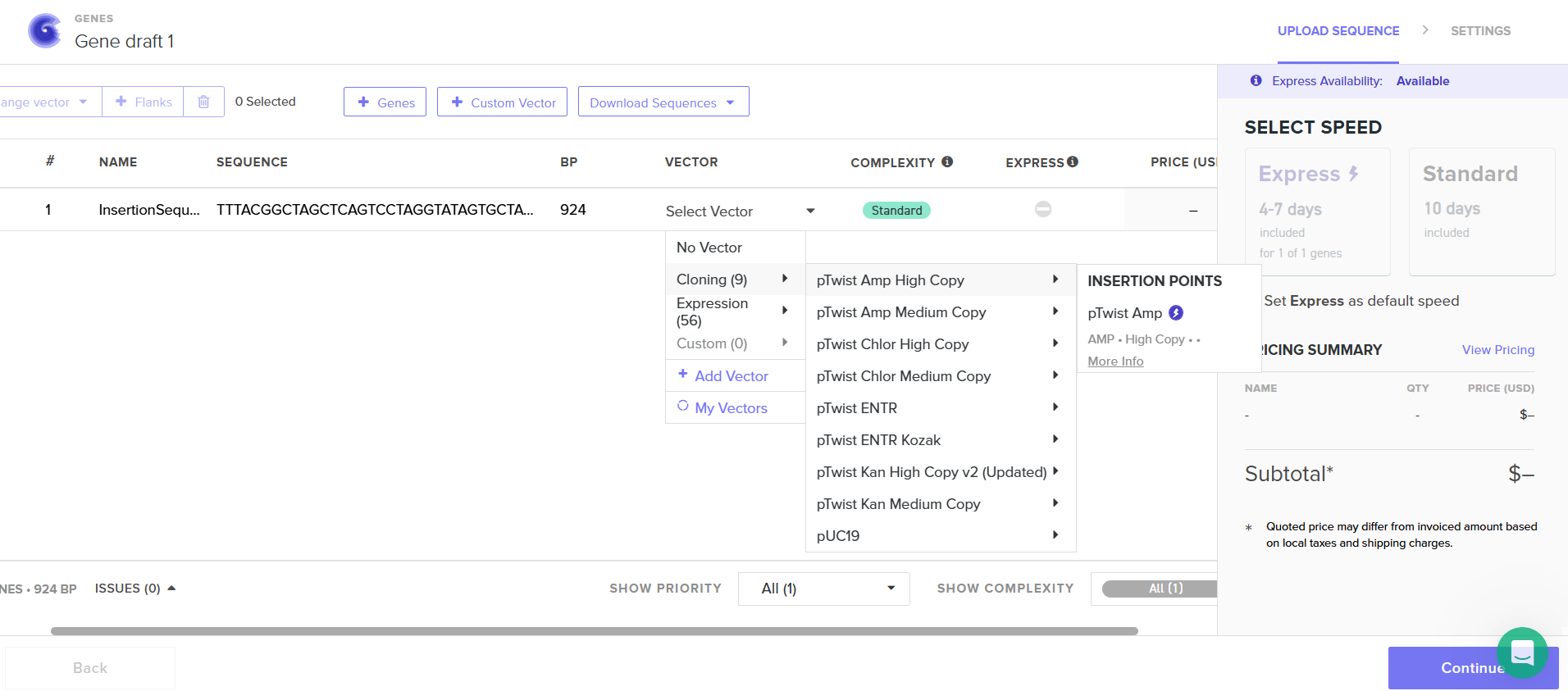
Then I clicked on my sequence again to see the ‘construct’. I pressed the ‘Show Construct’ button to view the construct and I was able to see two different tabs.
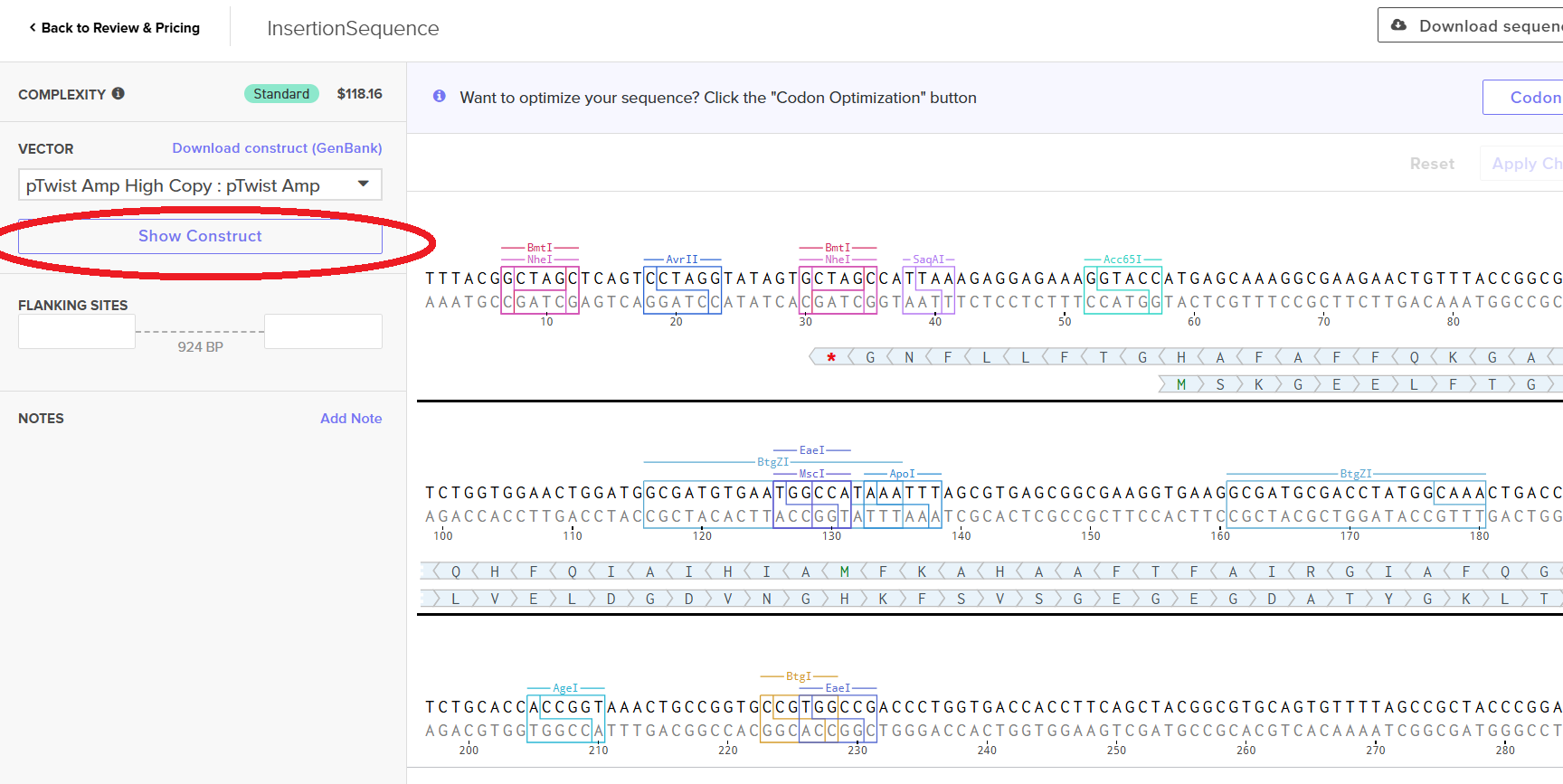
- Sequence
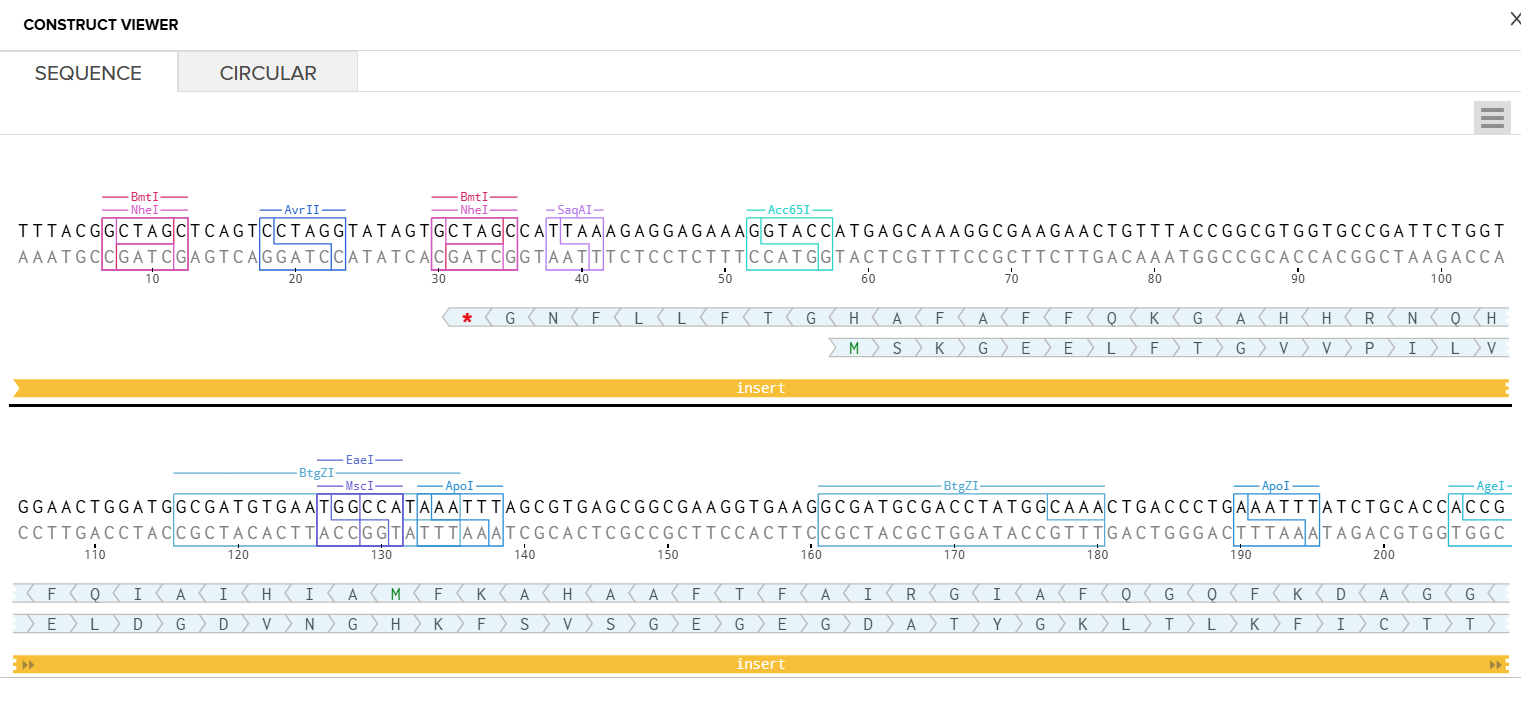
- Circular
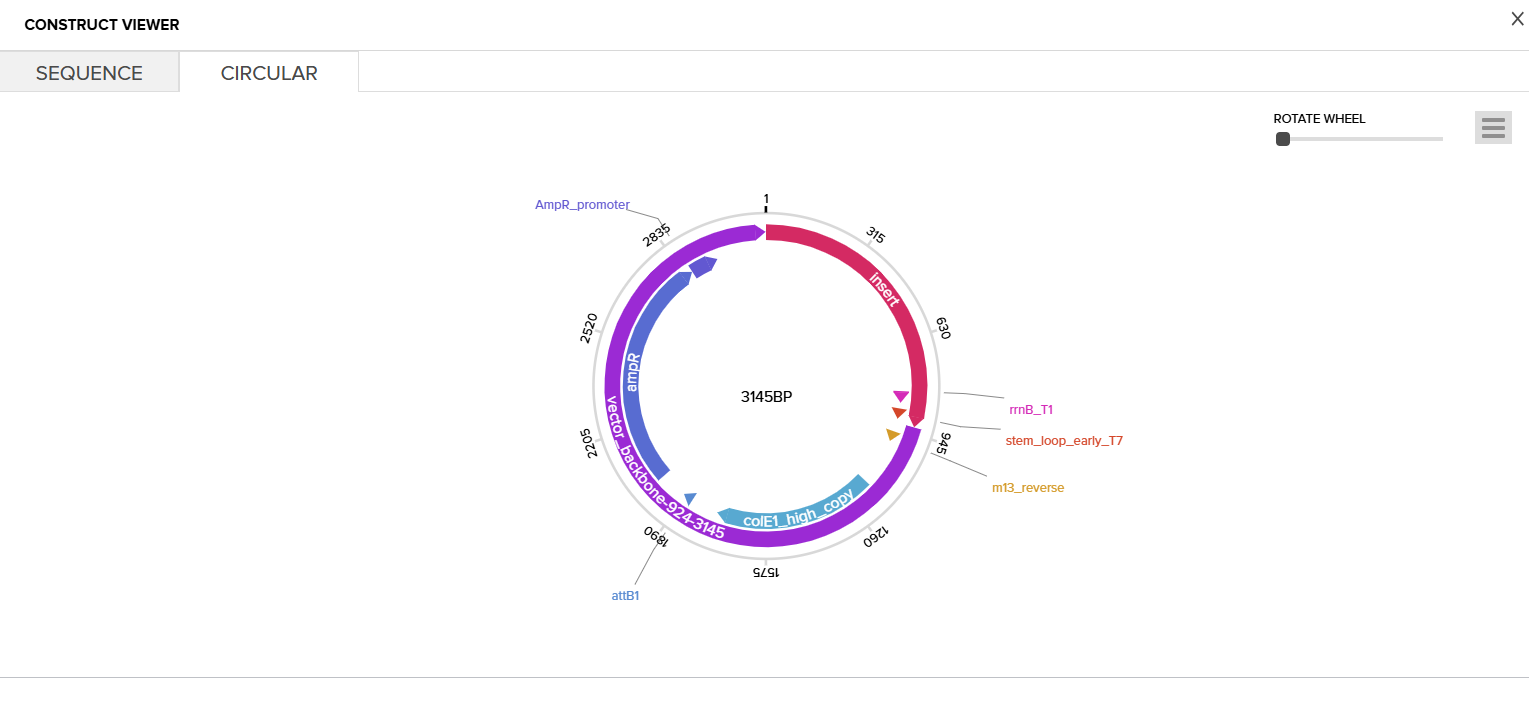
Then I clicked on the Download Construct link to download the GenBank file to my construct. (screenshot below, from HW Doc)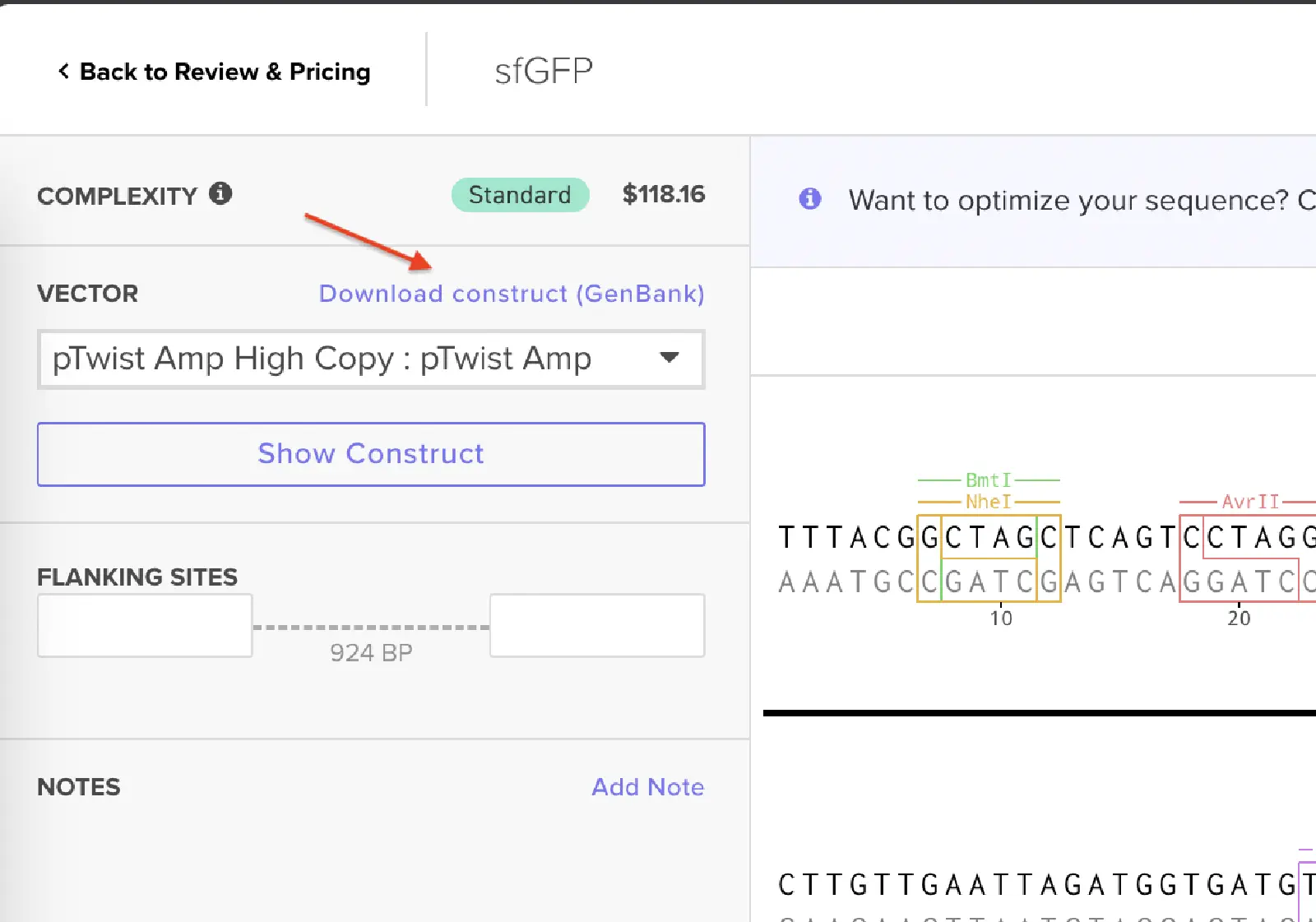 I downloaded the GenBank file of my construct and imported it to Benchling.
Part 4: Done! I built a plasmid with my own DNA of choice that is ready to insert! exhilirating feeling!
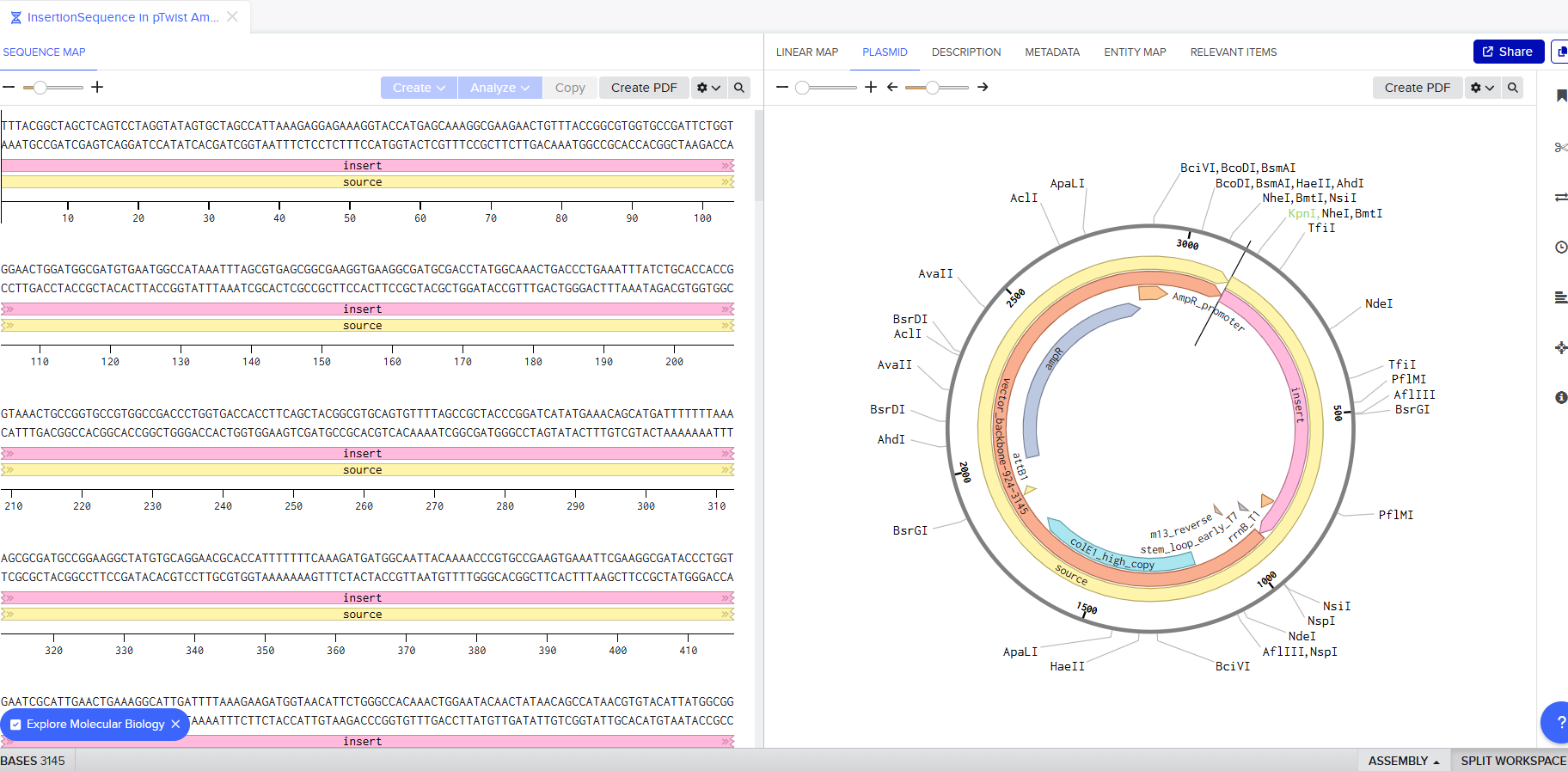
I downloaded the GenBank file of my construct and imported it to Benchling.
Part 4: Done! I built a plasmid with my own DNA of choice that is ready to insert! exhilirating feeling!
Part 5: DNA RW+
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I would want to sequence my own DNA. I’ve wanted to understand for a long while, what makes me, ‘ME’. What is my ancestry, what genes have I carried. Why am I naturally strong but fat? Why can I conserve muscle by little workout but fat just never budges? It might seem a bit small but yes I would want to read my own DNA first. (priority-wise)
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use WGS (Whole Genome Sequencing) using Illumina (Next-Gen Sequencing) as to analyze my genome, I would requite a method that covers the entire genome with high accuracy. Illumina’s NGS offers that high capability.
- Is your method first-, second- or third-generation or other? How so?
- The method is second generation as first generation methods like Sanger sequencing make use of chain-termination methods to sequence DNA and the third generation methods provide single molecule real time reading. WGS uses parallel sequencing.
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
- The input would be a DNA Sample of mine (blood/saliva). The steps to prepare would include fragmentation, (breaking the DNA down using enzymes) attaching adapters to allow primers to bind and if the sample is little, then PCR (to amplify the DNA, to make sure there’s enough to sequence)
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
- The fragments of DNA attach to the flow cell and undergo bridge amplification to create clusters of identical strands. The fluoroscently labeled reversible terminator nucleotides are added and then the polymerase adds a single matching base to the growing strand. (Thus this method is also called Sequencing by Synthesis) then a sensor captures the fluoroscent signal to identify which base was added, the terminator and dye are cleaved off, then next cycle begins.
- What is the output of your chosen sequencing technology?
- The output of this sequencing method is FASTQ files. They are like FASTA files but in FASTQ files theres also a Q (quality score) for every base, indicating how confident the machine is.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would want to synthesize the PprI gene from Deinococcus radiodurans. Ever since I heard about a bacterium, surviving Chernobyl levels of radiation. I was pretty fascinated by it. I would want it synthesize its DNA and study it further, perhaps the genes for radiation resistance can be expressed in other organisms to help them operate in radiation heavy environments.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use the phosphoramidite oligonucleotide synthesis method for DNA synthesis as to I would need accurate synthesis to create gene sequences to insert in other organisms.
- What are the essential steps of your chosen sequencing methods?
- The first step is In-Silico Design, breaking down the gene of interest into shorter chunks. Removing the chemical cap from previous base to make it reactive then add the next nucleotide to the growing chain. Blocking strands that didn’t accept the new base then oxidation to make the bases stable. Once all the short fragments are made on the silicon chip, they are released and then stitched together to form the full length genes.
- What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
- High GC sequences are difficult to synthesize because they form secondary structures and they also have high melting temperatures, which can cause synthesis to fail or introduce errors.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
This might be a bit controversial but I would want to edit my MSTN gene and try and tweak the gene for lower myostatin expression. I would want to make myself more muscular. I am aware however that the cascading effects could be unwelcome and lead to disorders. That is why this is just hypothetical.
(ii) What technology or technologies would you use to perform these DNA edits and why?
The best method I know of CRISPR. CRISPR is the most programmable and efficient method to edit specific genes.
How does your technology of choice edit DNA? What are the essential steps?
- CRISPR makes use of a Guide RNA (gRNA) that binds to the Cas9 protein and directs it to the specific DNA sequence in the MSTN gene that matches the guide. The Cas9 nuclease creates a Double-Strand Break at that precise location. The cell then repairs the breakage. If we want to add a gene we provide a template, and the cell uses homology-directed repair to copy the new sequence into the DNA.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
- We have to design the sgRNA to ensure it specifically targets the gene of interest with minimal off-target effect potential. Input materials are Cas9 nuclease, gRNA, Template DNA
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
- Off-target effects- the nuclease might accidentally cut similar sequences elsewhere and cause mutations.
- Getting the gRNA and Cas9 into the cell is difficult.
- Not all cells get edited.