Week 4 HW: Protein Design I
Part A : Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Amino Acids are protein building blocks, so whatever percent of protein the meat contains is technically the AA content. A quick google search tells me that most cooked meats contain 20%-30% protein by weight. I’ll take 25% as my number. Now, 25% of 500g is 125g. (500/4)
Amount of protein = 125g
Now, 1 AA avg. = 100 Daltons. but 1 Dalton = 1 g/mol
so 1 AA = 100g/mol. To find the amount of moles = mass / molar mass
therefore, No. of moles of amino acids = 125 / 100 = 1.25 moles
number of molecules = moles x Avogadro’s Number = 1.25 x 6.022 x 10^23
= 7.527 x 10^23 molecules of amino acids per 500 grams of meat.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The proteins from other animals are built out of the same universal building blocks and the process of digestion breaks food down into the building blocks. (catabolism) These building blocks are then used to make YOUR own proteins using YOUR DNA. (metabolism) Basically:
When humans eat cow, human body not take cow protein. Human body break down protein into free amino acids. Free amino acid used by human body to make its own protein. Free amino acid not make a human a cow or fish.
3. Why are there only 20 natural amino acids?
They’re basically evolution-wise frozen in place. Early life settled on 20 AA that were chemically diverse enough to build different functional proteins. Once the genetic code was ’locked’ There was no way that evolution could now swap it, it would break everything. 20 amino acids have enough chemical variety to accomplish the protein goal.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, it is possible to make new amino acids, labs do this using engineered tRNAs that insert a non-natural AA at the stop codon. An example of a new amino acid created by modifying the side chain is ‘fluorophenylalanine’ - it is basically a phenylalanine with a fluorine atom, making it more stable and UV trackable.
5. Where did amino acids come from before enzymes that make them, and before life started?
The Miller-Urey experiment has shown that lightning + early earth atmosphere could form amino acids spontaneously. Also amino acids have been found in meteorites like the Murchison meteorite, which could indicate that amino acids could’ve come from space. Amino acids aren’t strictly a product of life, but rather a tool life used.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Naturally, α-helixes made from L-amino acids have right handed turns. So logically if we were to use D- amino acids to make α-helixes then they should have left-handed turns.
7. Can you discover additional helices in proteins?
Skipped. (The HW said I could skip two questions so this is the first)
8. Why are most molecular helices right-handed?
Most molecular helices are right handed because the life uses L-AAs. the geometry of L-AAs favors right handed twisting when they form hydrogen bods along a backbone. It is just like the answer of Q.3, L-AAs dominated in the early life and that dominance carried over.
9.Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets have exposed hydrogen bond donors and acceptors along their edges. So when 2 β-sheets meet edge to edge, they form hydrogen bonds with each other and grow in to ordered stacks. The driving forces of this bonding are 1. Hydrogen bonding 2. Van der Waals interactions between sheets 3. Hydrophobic effect - water shoves the sheets together to get those nonpolar faces out of its way.
10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Once a protein misfolds, it can cause other copies of the same protein to misfold in the same way. In cases of misfolds, sometimes a β-strands edge can get exposed, this edge then acts like a template and causes the other proteins to misfold the same way and forms a stack, the stack keeps growing. The result is insoluble amyloid fibrils. Diseases like Alzheimer’s (Aβ plaques), Parkinson’s (α-synuclein), all involve this.
The same reason why amyloid diseases are pathological make them useful. Aggregate materials can be incredibly stable and heat resistant. They’re perfectly ordered. They are self-propagating/assembling.
11. Design a β-sheet motif that forms a well-ordered structure.
Skipped. (This is the second skipped question)
Part B : Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
I choose the protein “myoglobin”. I selected it as I know from pop sci that it is responsible for keeping muscles well-oxygenated. It is also the reason why meat is red in color. It has a higher affinity for oxygen than hemoglobin. The protein is a simple single poly peptide chain wrapped around an iron-containing heme group which holds and physically grabs an oxygen molecule. It is different from it’s well known elder cousin hemoglobin in the way that myoglobin is a tertiary monomer consisting of a single polypeptide chain while hemoglobin is a quaternary tetramer made of four polypeptide subunits.
Identify the amino acid sequence of your protein.
Here’s the amino acid sequence: MGLSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETLEKFDKFKHLKSEDEMKASEDLKKHGATVLTALGGILKKKGHHEAEIKPLAQSHATKHKIPVKYLEFISECIIQVLQSKHPGDFGADAQGAMNKALELFRKDMASNYKELGFQG
The total length of human myoglobin is 154 amino acids. The molecular weight is 17.8 kDa. The most frequent amino acid is Leucine. It appears 19 times.
Homologs :Hemoglobin α and β subunits (most famous), neuroglobin, cytoglobin, and plant leghemoglobin, they all share the same globin fold.
It belongs to the globin superfamily.
Identify the structure page of your protein in RCSB
1MBO (PDB ID for Myoglobin) was solved by S. E. Phillips in 1980, It was the first solved protein structure. It was published as “Structure and refinement of oxymyoglobin at 1.6 Å resolution.” At 1.6 Å, this is an exceptionally high-quality structure, well below the 2.70 Å “good” threshold, meaning the atomic positions are resolved with outstanding precision. This is one of the most accurately determined protein structures in the entire PDB.
Yes, There are three types of non-protein molecules are present in 1MBO beyond the protein chain itself: HEM: the heme group. The iron atom at its center is what physically binds oxygen. OXY: a molecular oxygen (O₂) molecule, bound directly to the iron atom inside the heme pocket. SO4: a sulfate ion from the crystallization buffer, sitting on the protein surface.
Yes. In SCOP, myoglobin is classified as: - Class a: All alpha proteins Fold a.1: Globin-like Superfamily a.1.1: Globin-like Family a.1.1.2: Myoglobin
Open the structure of your protein in any 3D molecule visualization software:
I just used RCSB PDB Viewer. It was online, easy and direct.
Cartoon: (Color by Amino Acid)

Ribbon: (Color by Amino Acid)

Ball and Stick (Color by Amino Acid)
When I colored the protein by secondary structure, I found out that it has more helices.
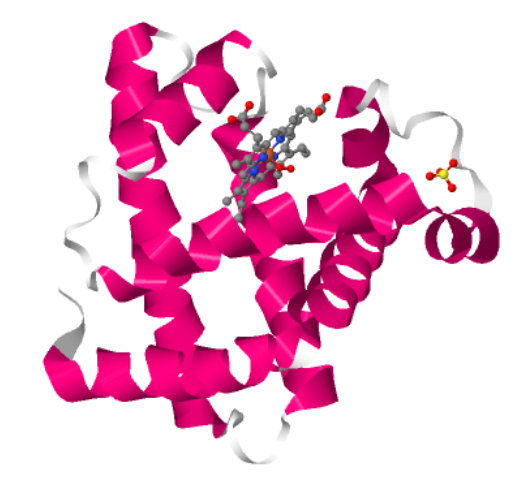
The Jsmol based viewer didn’t have an option to color by residue type but it had the option of color by hydrophobicity. Red means hydrophobic, Blue means hydrophilic.
I visualized the surface for Cavities and this is what I got.
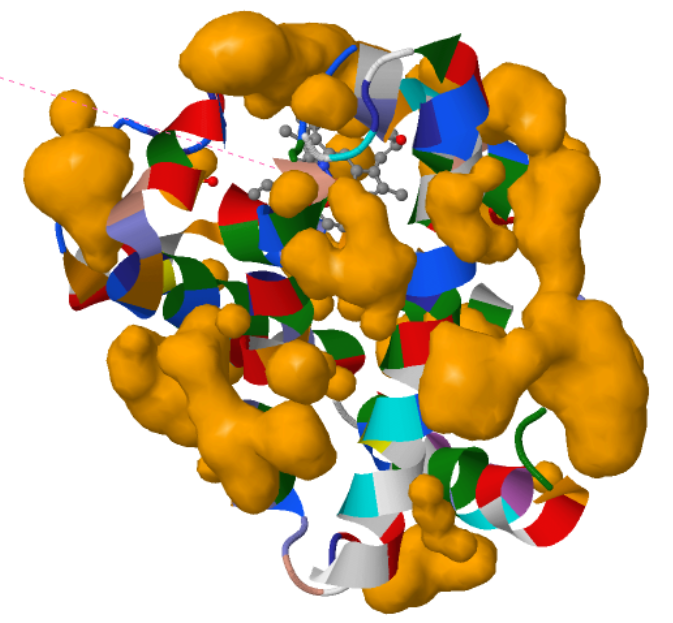 The visualization shows that the protein indeed has cavities that act as binding pockets and it makes perfect sense that a protein like myoglobin would have cavities for binding.
The visualization shows that the protein indeed has cavities that act as binding pockets and it makes perfect sense that a protein like myoglobin would have cavities for binding.
Part C : Using ML-Based Design Tools
I set up a Colab instance and copied the Protein Design Notebook from HTGAA. I chose Myoglobin again for this.
C1: Protein Language Modelling
C1.1 Deep Mutational Scans
I had to use ESM2 to generate an unsupervised deep mutational scan of my protein (myoglobin). I just opened the provided Colab notebook and replaced the protein sequence in there.
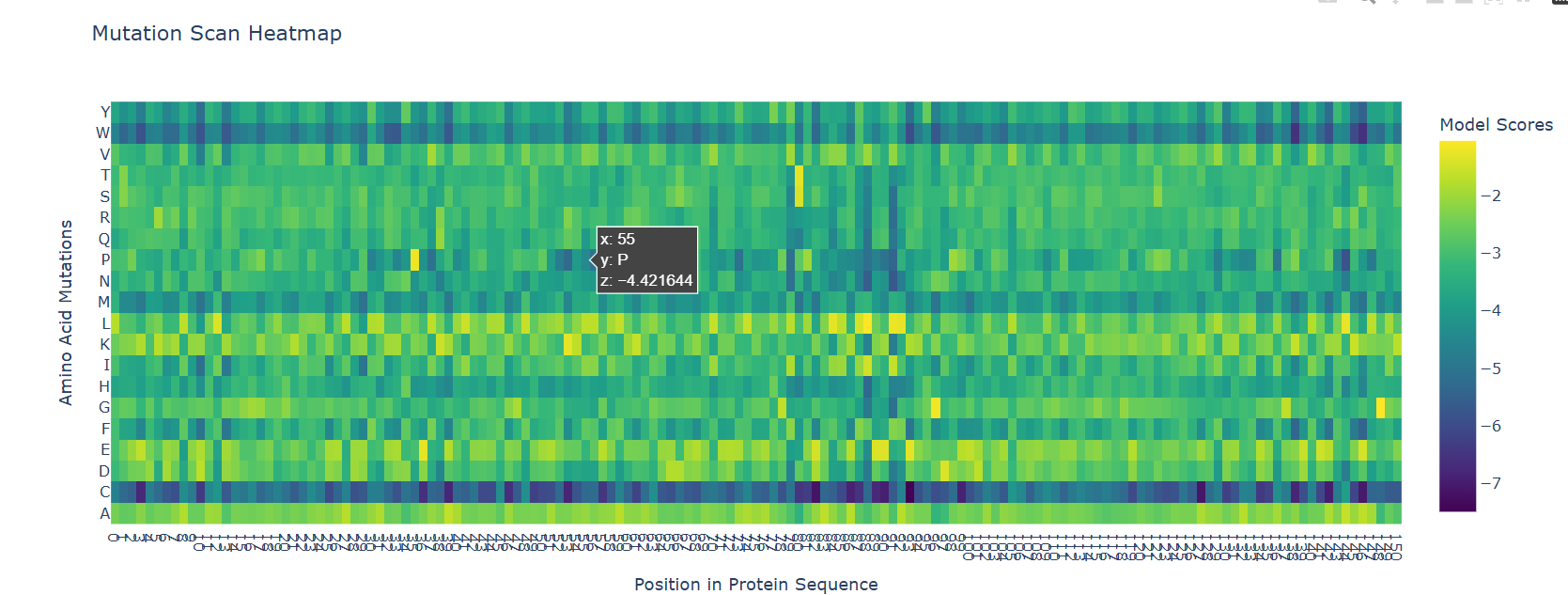
The most striking pattern visible is the bottom C row and the top W row. I do not know the reason behind the C row but as for W, tryptophan is the bulkiest amino acid and it wouldn’t fit the dense helical packaging of globin fold without causing problems to neighbouring residues.
As for the latent space analysis, I tried to use the default dataset from the notebook but it wasn’t loading correctly in the notebook. The issue was in the dataset URL itself, it was returning a corrupted FASTA file with comments. I tried to download the dataset manually but that wasn’t working either, the page didn’t open. If the code worked as expected, I would have added my myoglobin sequence to the sequences list and compared it to its neighbors. I think it would’ve been closer to other globin like proteins.
C2. Protein Folding
C2.1 Folding a protein
I tried using the given Colab Notebook for folding a protein with ESMFold, but the visualization part didn’t work. It said Py3dmol not found. I even tried to install it before that cell but even that didn’t work. I found an Web Interface for ESMFold, I decided to use that for the folding. Here’s what I got.
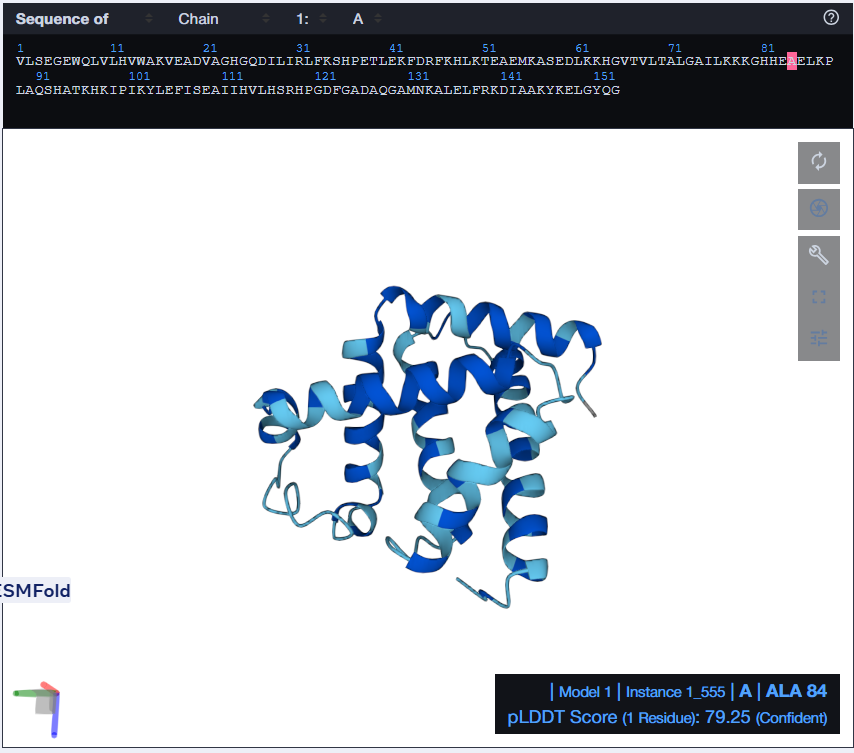
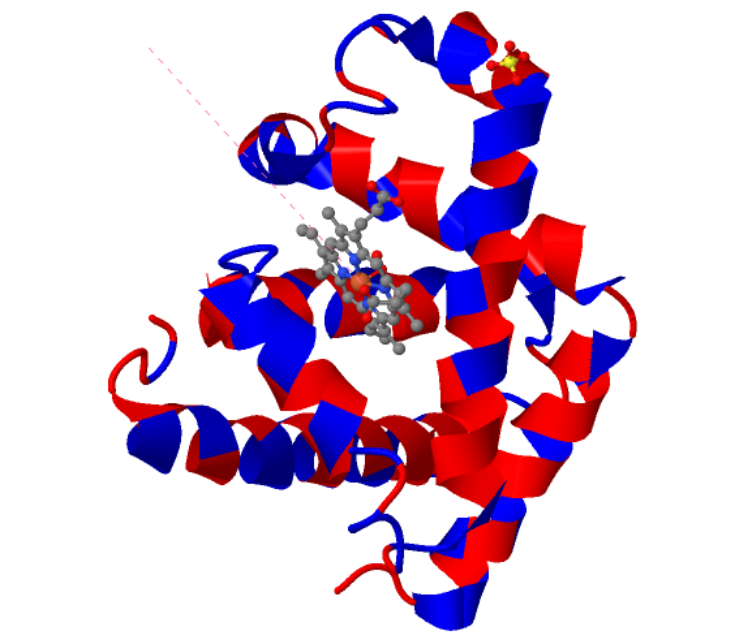
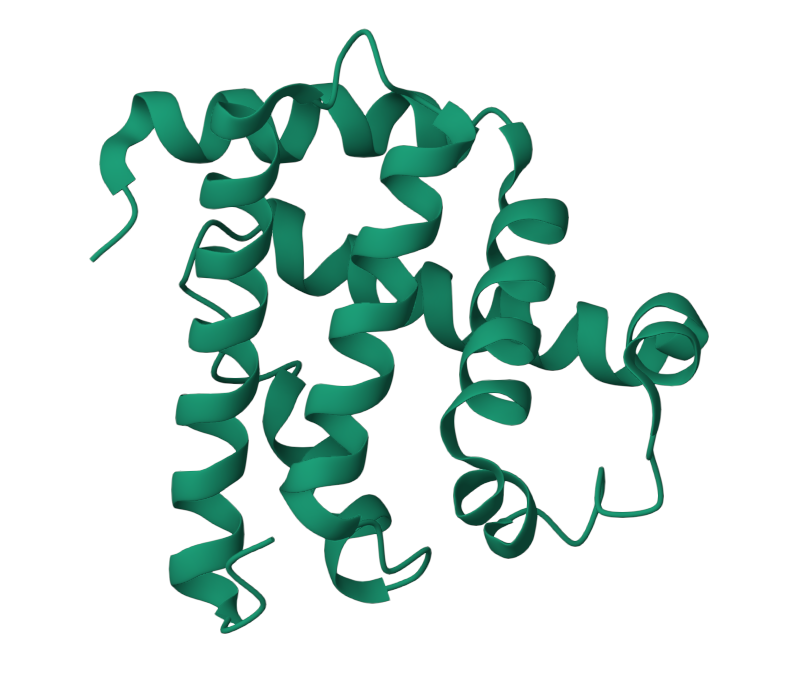 The predicted structure with ESMFold (Left) next to the original structure (right)
The predicted structure with ESMFold (Left) next to the original structure (right)
The predicted structure matches the original structure, but it isn’t very surprising as myoglobin is a well studied protein. Next up I had to try changing the sequence and see if the protein is resilient to mutations.
At first, I tried changing the K at 147th position to a W, then I folded the sequence. The overall structure remained the same but it introduced a region of low confidence into the structure. 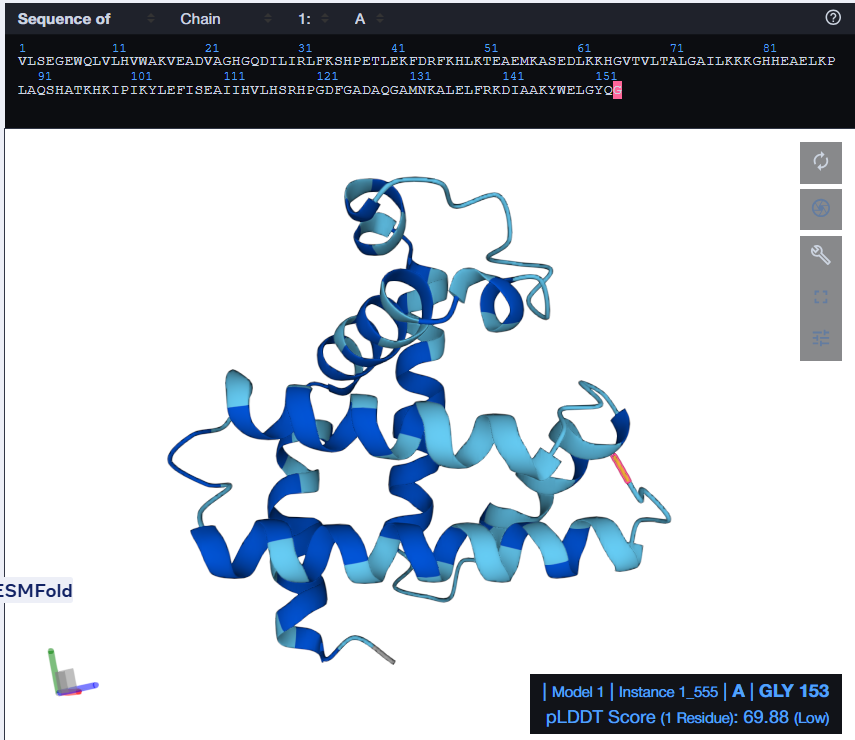
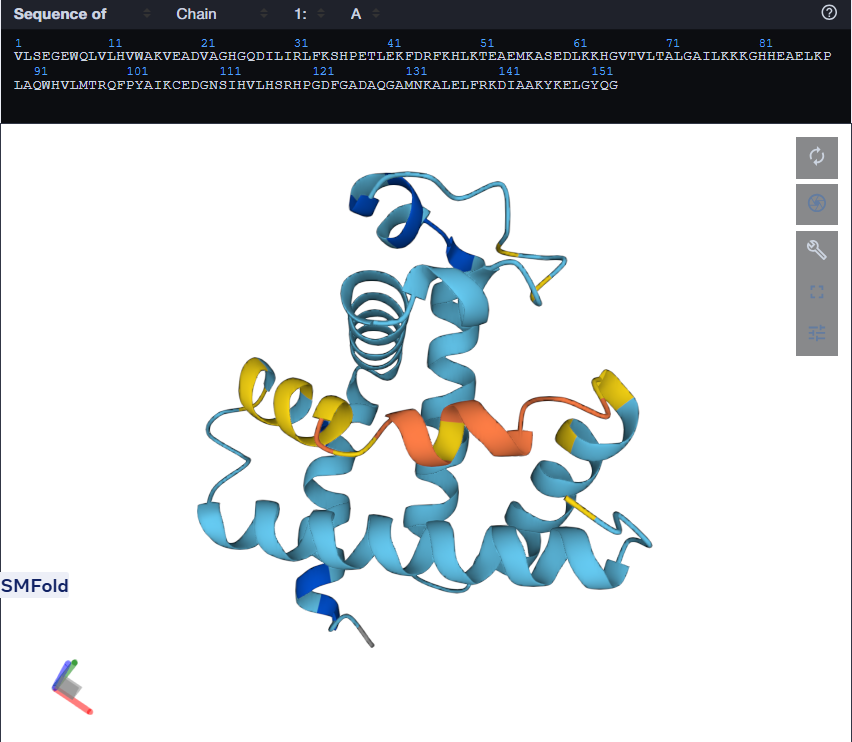 Mutant 1 (left), Mutant 2 (Right)
Mutant 1 (left), Mutant 2 (Right)
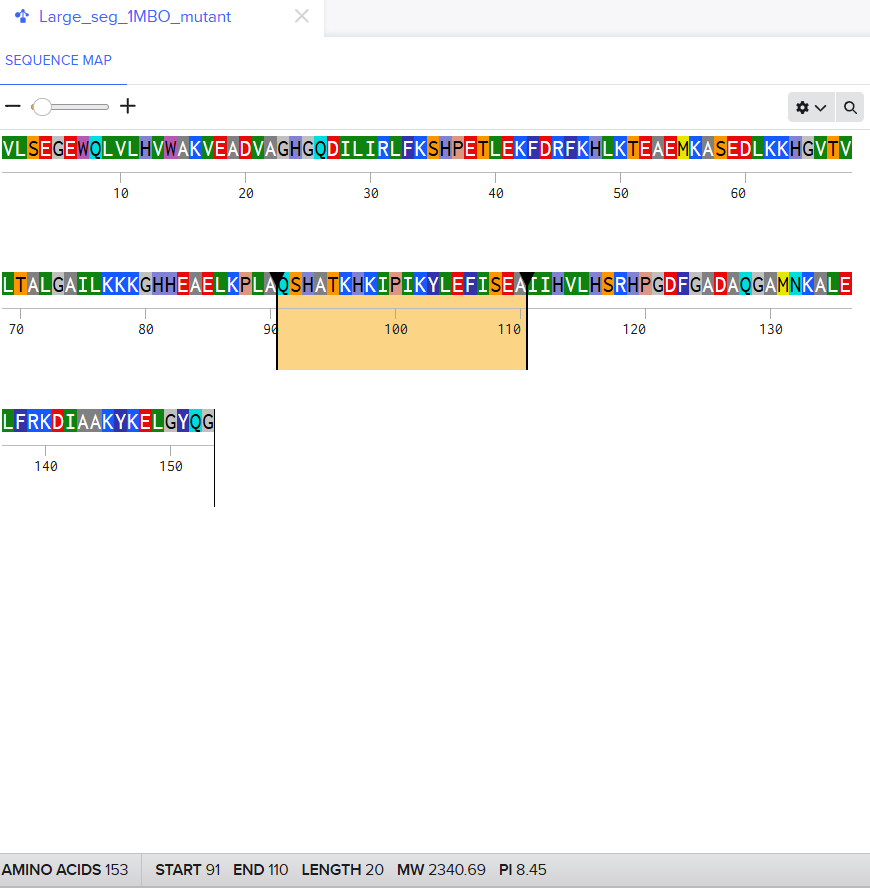
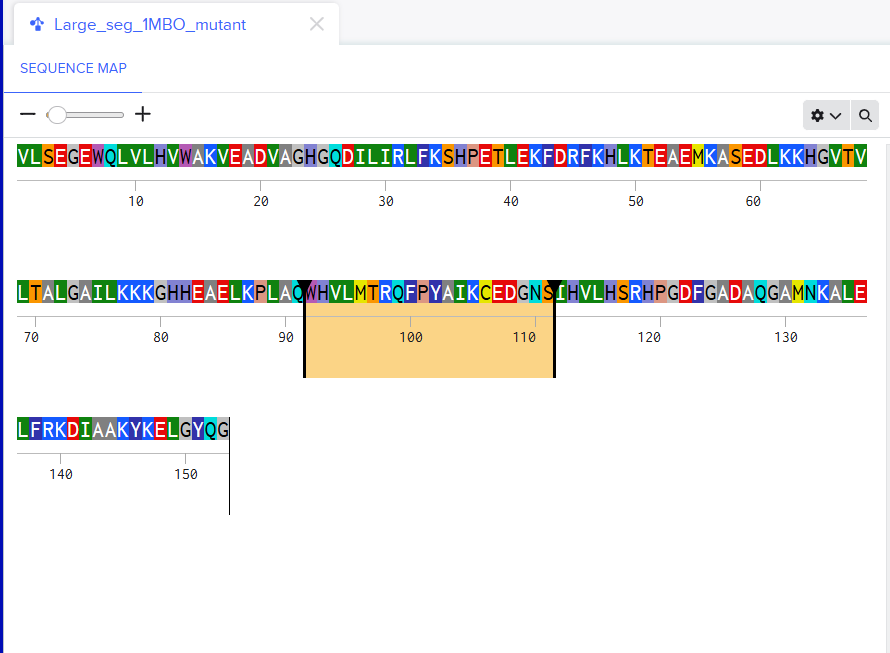
For the second mutant, changing a large segment. I switched a 20 amino acid sequence at 92-111 and replaced it with a random 20 amino acid string: W H V L M T R Q F P Y A I K C E D G N S. I lobotomized my protein. It was funny to see how a 20AA change really reduced the confidence score of the model. A lot of the structure was still conserved but the protein was certainly affected. I think the model tried to model it based on the training it had and globins are well-studied. Fun to see. I might play around more with other proteins. I used Benchling to do the mutation. (I got the random sequence via Google’s AI Mode)
C3. Protein Generation
The notebook really has it’s issues. Even MPNN wasn’t working so I had to find an alternative, I found Tamarind Bio had a online tool. I used that. There are so many tools being made. This is making me aware of my obliviousness to computational biology.
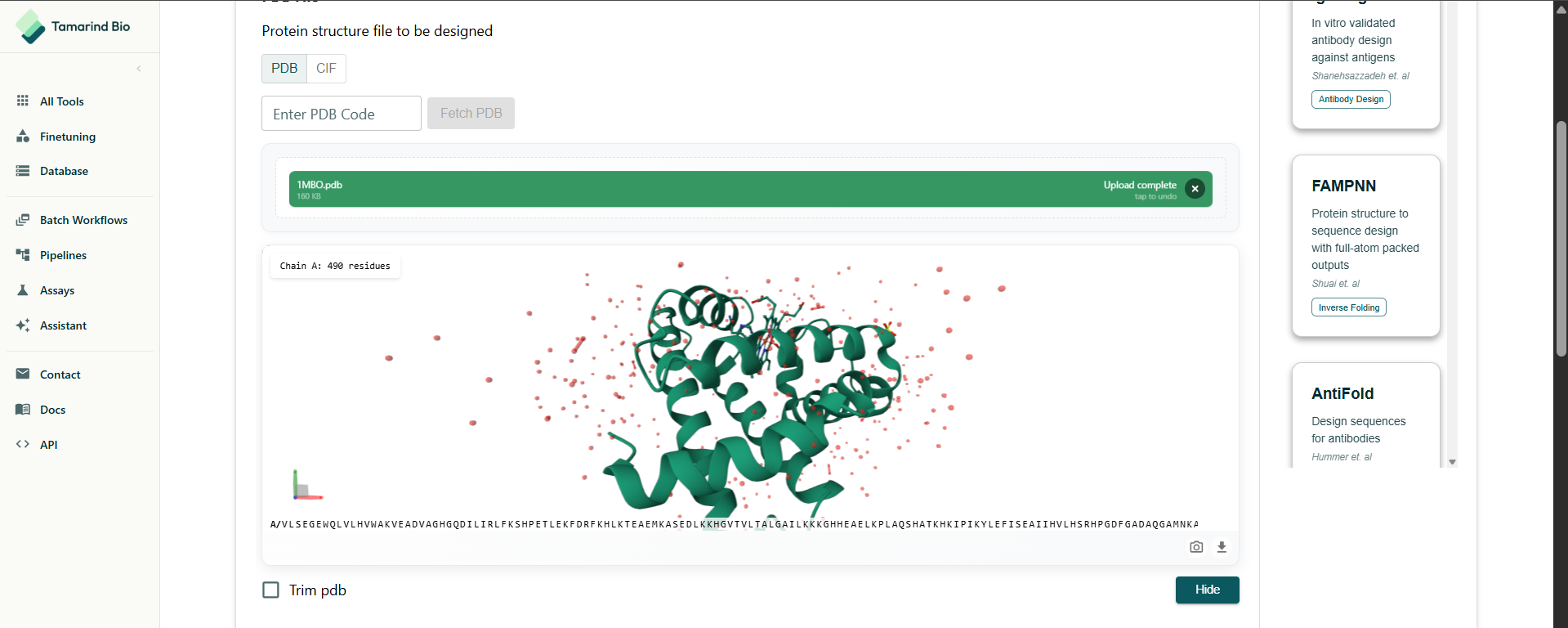
I had to select the residues to design. I decided to do a full redesign.
I got this sequence after the full redesign via ProteinMPNN:
One can tell by just looking that it is quite different from the original sequence. The length is same but the amino acids are different. But the Histindines on Position 64 and 93 have been recovered. I then proceeded to predict the structure for this via ESMFold.
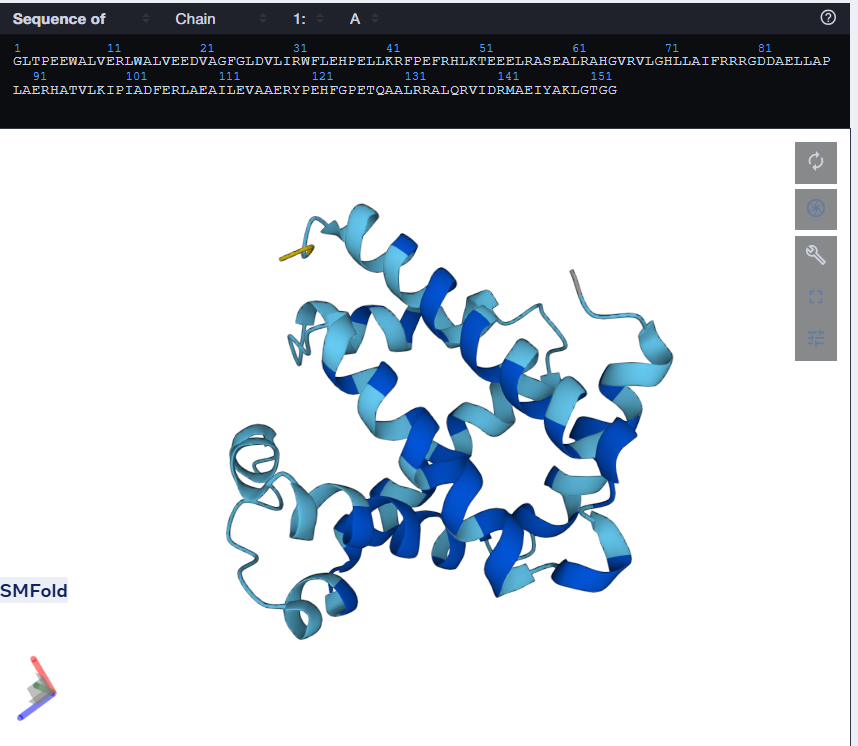
Using only the myoglobin backbone structure (153 residues, chain A), ProteinMPNN designed a new sequence with ~38–45% identity to the native protein. But, His64 and His93, the two histidines crucial for heme binding were independently recovered without any sequence information. This suggests that the geometry of the heme-binding pocket strongly constrains these positions to histidine. Other than that, a lot of surface residues were varied from the original sturcture. Predicting the structure of the redesigned sequence via ESMFold had conserved the characteristic globin helix bundle, with high-confidence pLDDT scores across the helical core. This shows that the designed sequence is structurally compatible with the myoglobin fold. Overall, ProteinMPNN generated a realistic globin-like sequence rather than simply reproducing the native one.