Week 5 HW: Protein Design II
Part A: SOD1 Binder Peptide Design
Part 1:
The first step was retrieving human SOD1 sequence from Uniprot and introducing the A4V mutation. Here’s the SOD1 sequence:
Here’s the mutated SOD1 Sequence:
I used the PepMLM Colab to generate the 4 peptides of length 12 as specified in the homework. I selected the length to be 12 and I chose 4 binders as I had to generate 4 peptides. This was the result.
| Binder | Sequence | Pseudo Perplexity |
|---|---|---|
| 0 | FLYRWLPSRRGG | This is the known binder that the homework said to add in the list |
| 1 | SRWDEYTAVVAWARK | 9.686584 |
| 2 | SWYGEYTGVVAWRKK | 14.675614 |
| 3 | AHWPEYVVVVEWKKK | 20.736155 |
| 4 | SRVDEYTVRKKWARK | 15.232643 |
Part 2: Evaluate Binders with AlphaFold3
Next step was to evaluate the binders. I went to alphafoldserver.com, logged in with my google account and then I was greeted by this screen.
For each peptide, I pasted the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex. There were 5 total jobs. to be submitted.
Job 0: The Mutated SOD1 and the known binder: FLYRWLPSRRGG
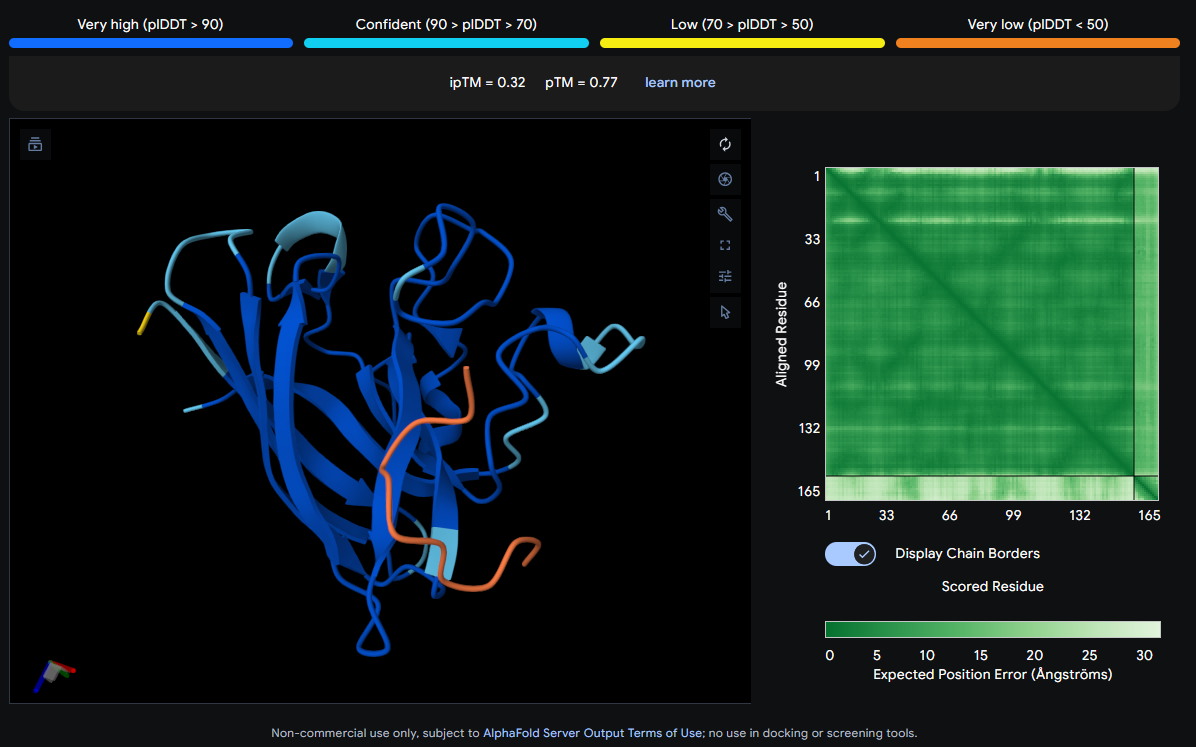
The peptide (shown in orange/red) visually wraps around one face of the β-barrel, appearing partially surface-bound. It does not appear to penetrate deeply. Its position is consistent with engagement near the loop regions connecting β-strands rather than strictly at the N-terminus where A4V (position 4) sits.
Job 2: Mutated SOD1 and Binder 1: SRWDEYTAVVAWARK
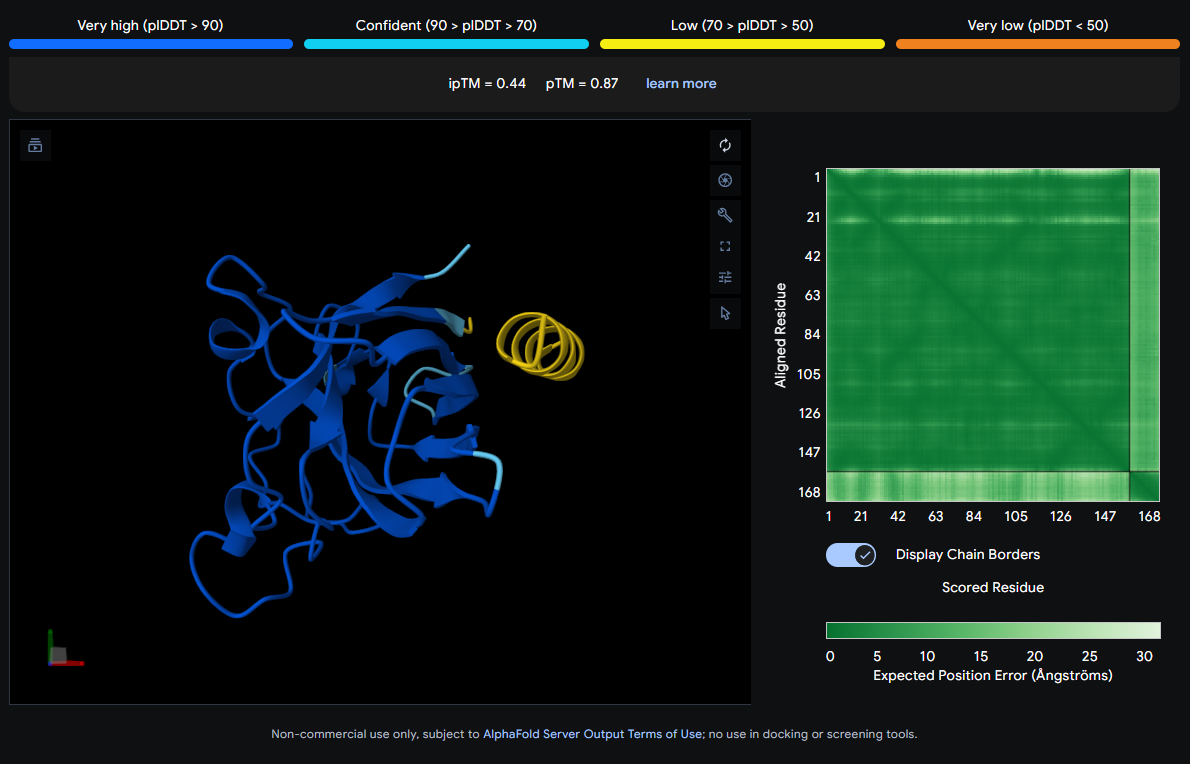
The peptide (visible as a yellow coil) appears to dock away from the main β-barrel body — positioned more distally and looking loosely tethered. It does not appear buried and likely represents a surface-level interaction, possibly near an external loop rather than the A4V mutation site directly.
Job 3: Mutated SOD1 and Binder 2: SWYGEYTGVVAWRKK
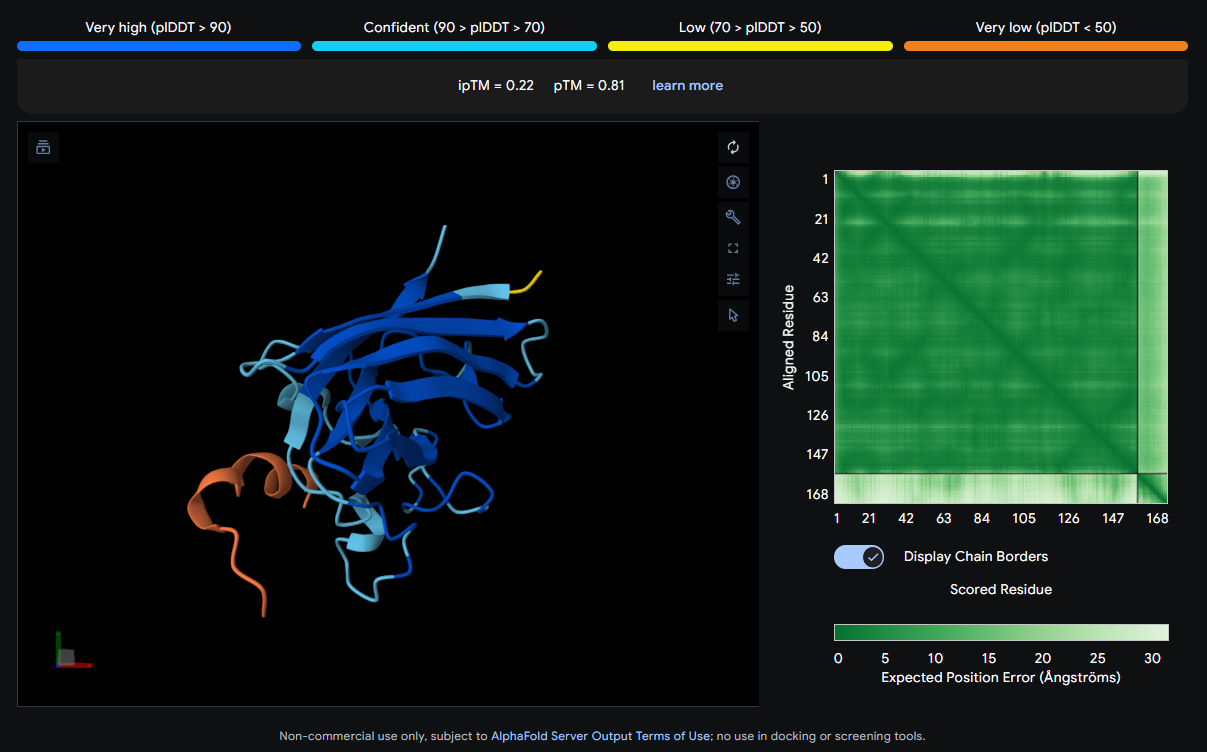
The peptide (orange/red loop) appears to contact the β-barrel on a lateral face and partially approaches what could be the dimer interface region. It sits more surface-exposed rather than buried.
Job 4: Mutated SOD1 and Binder 3: AHWPEYVVVVEWKKK
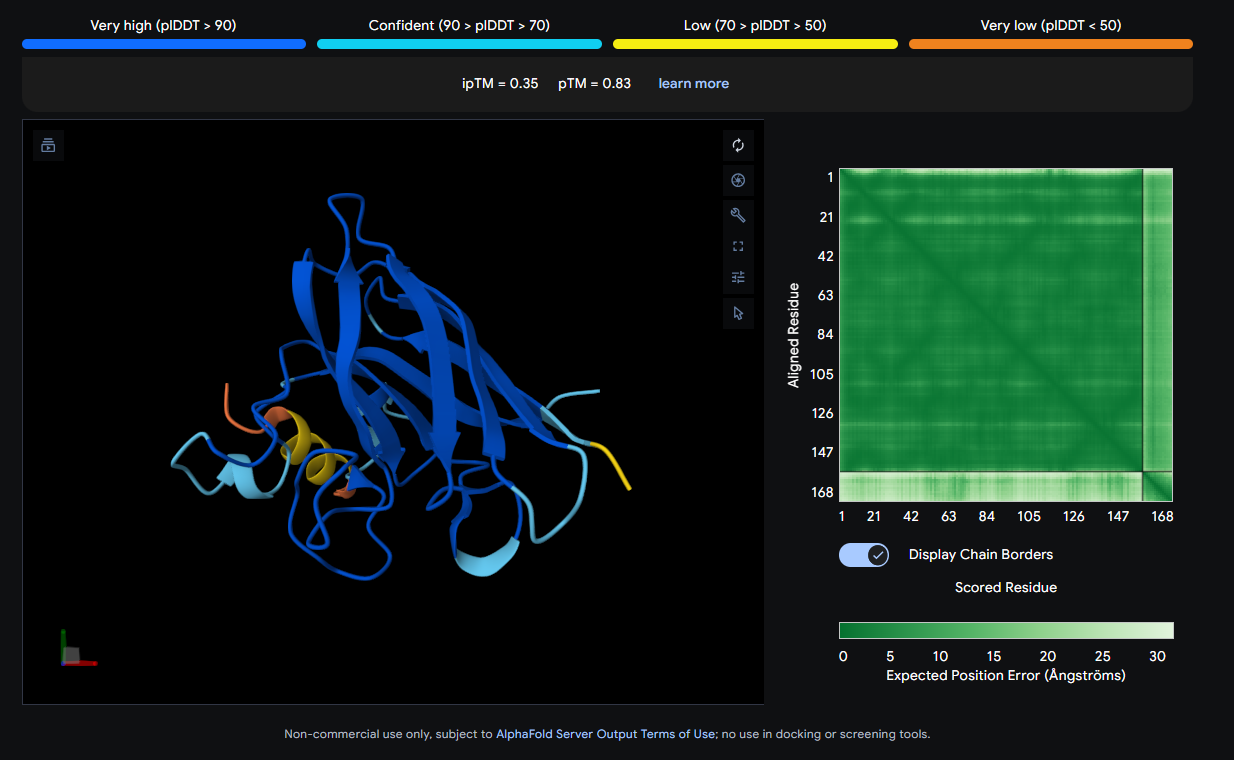
The peptide (yellow, compact) appears to bind near the front face of the β-barrel and shows relatively close association with the protein body. It could be engaging a region near the electrostatic loop or β-barrel surface, though not deeply buried.
Job 5: Mutated SOD1 and Binder 4: SRVDEYTVRKKWARK
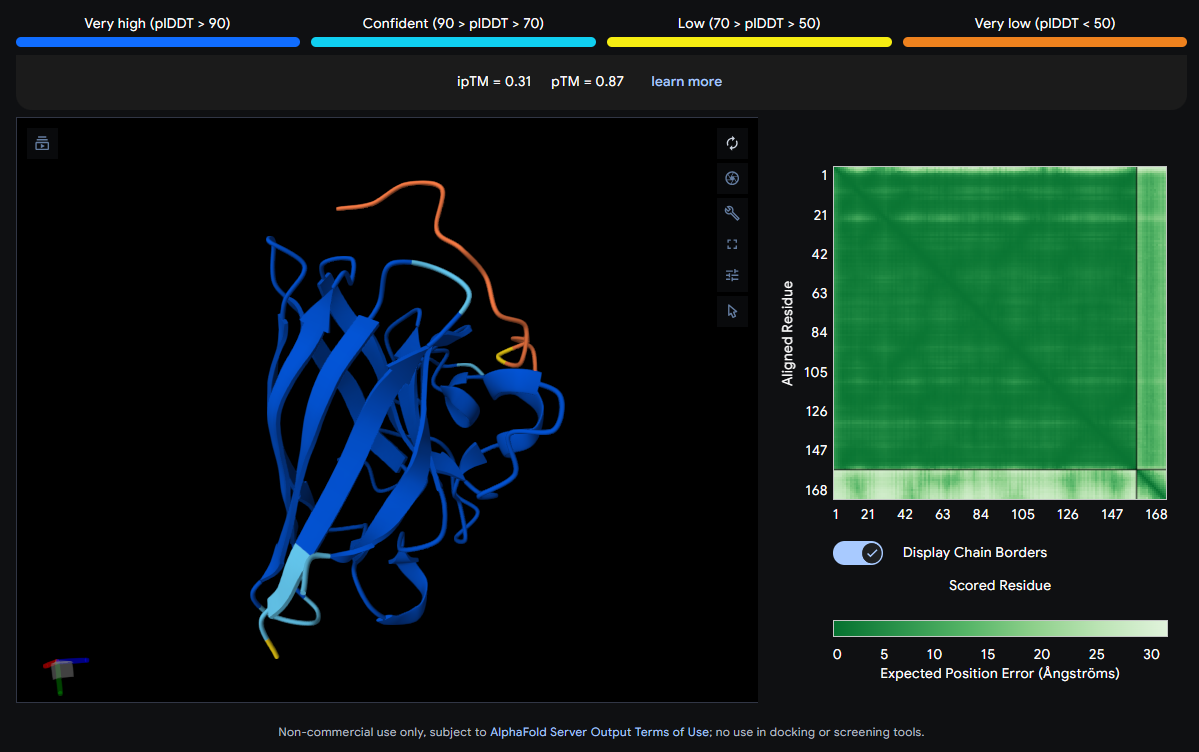
The peptide (orange/red) drapes along one edge of the SOD1 structure, appearing surface-bound. Its positioning is loosely consistent with an approach toward the N-terminal β-strand region where A4V resides, though definitive localization is limited without residue-level contact maps.
ipTM Scores and Binding Description
| Binder | Sequence | ipTM | pTM |
|---|---|---|---|
| 0 (Known) | FLYRWLPSRRGG | 0.32 | 0.77 |
| 1 | SRWDEYTAVVAWARK | 0.44 | 0.87 |
| 2 | SWYGEYTGVVAWRKK | 0.22 | 0.81 |
| 3 | AHWPEYVVVVEWKKK | 0.35 | 0.83 |
| 4 | SRVDEYTVRKKWARK | 0.31 | 0.87 |
| None of the peptides show convincing deep burial, suggesting predominantly surface-level or shallow groove engagement with the β-barrel exterior. |
The ipTM scores across all five complexes range from 0.22 to 0.44, values that collectively sit in the low-to-moderate confidence range for inter-chain interaction quality. The known binder (FLYRWLPSRRGG) achieves an ipTM of 0.32, which serves as the reference benchmark. Notably, Binder 1 (SRWDEYTAVVAWARK) is the only PepMLM-generated peptide to exceed this, reaching an ipTM of 0.44. A meaningful improvement of ~0.12 over the known binder. Binder 1 stands out as the most structurally promising among the generated candidates.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Now we had to evaluate the properties of the generated peptides, we use PeptiVerse for this.
The workflow was simple, paste the peptide seq and paste mutated SOD1 seq, check the boxes according to the homework: Predicted binding affinity, Solubility, Hemolysis probability, Net charge (pH 7), Molecular weight. Here are the results.
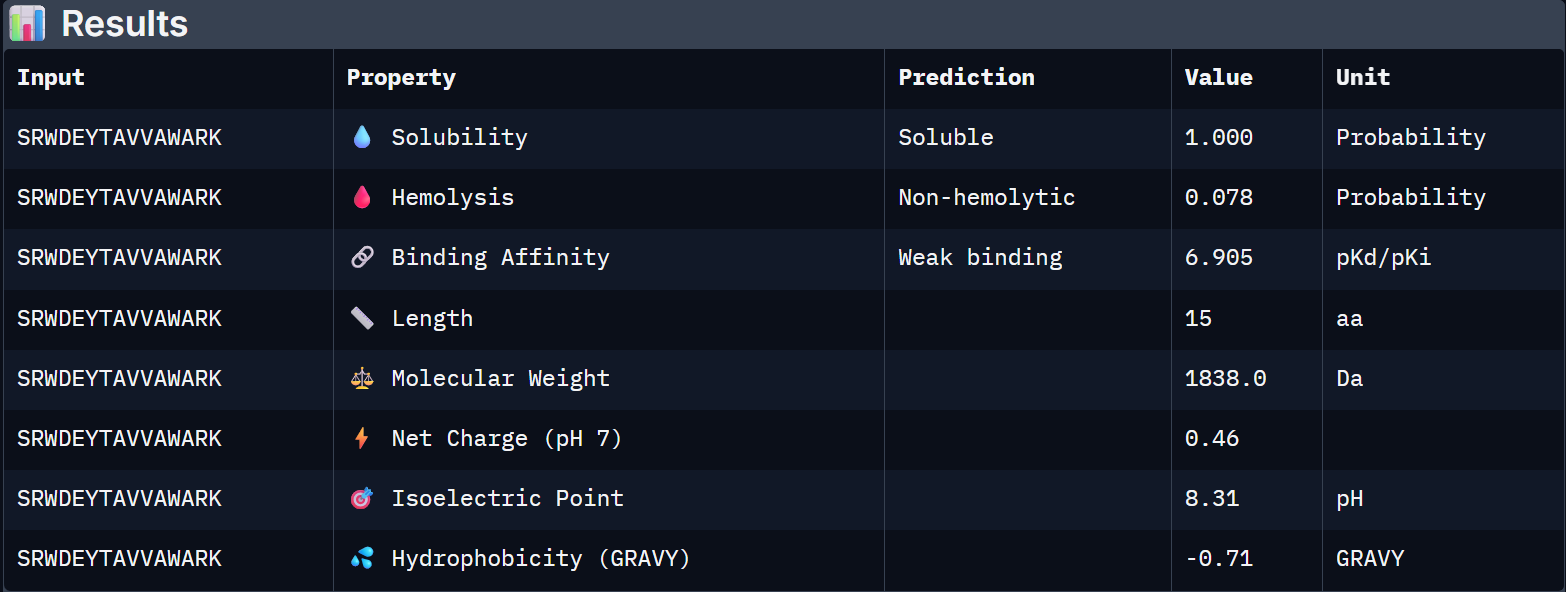
For Peptide 1: SRWDEYTAVVAWARK
For Peptide 2: SWYGEYTGVVAWRKK
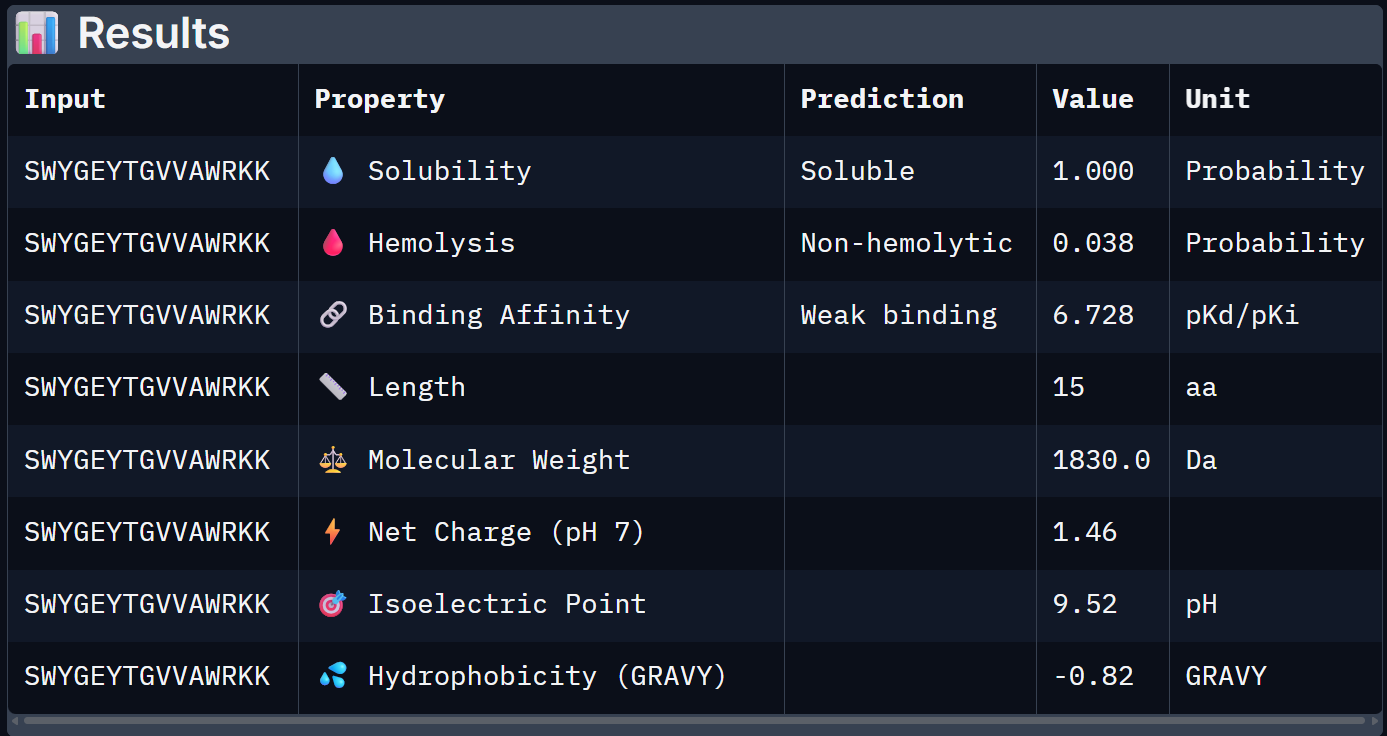
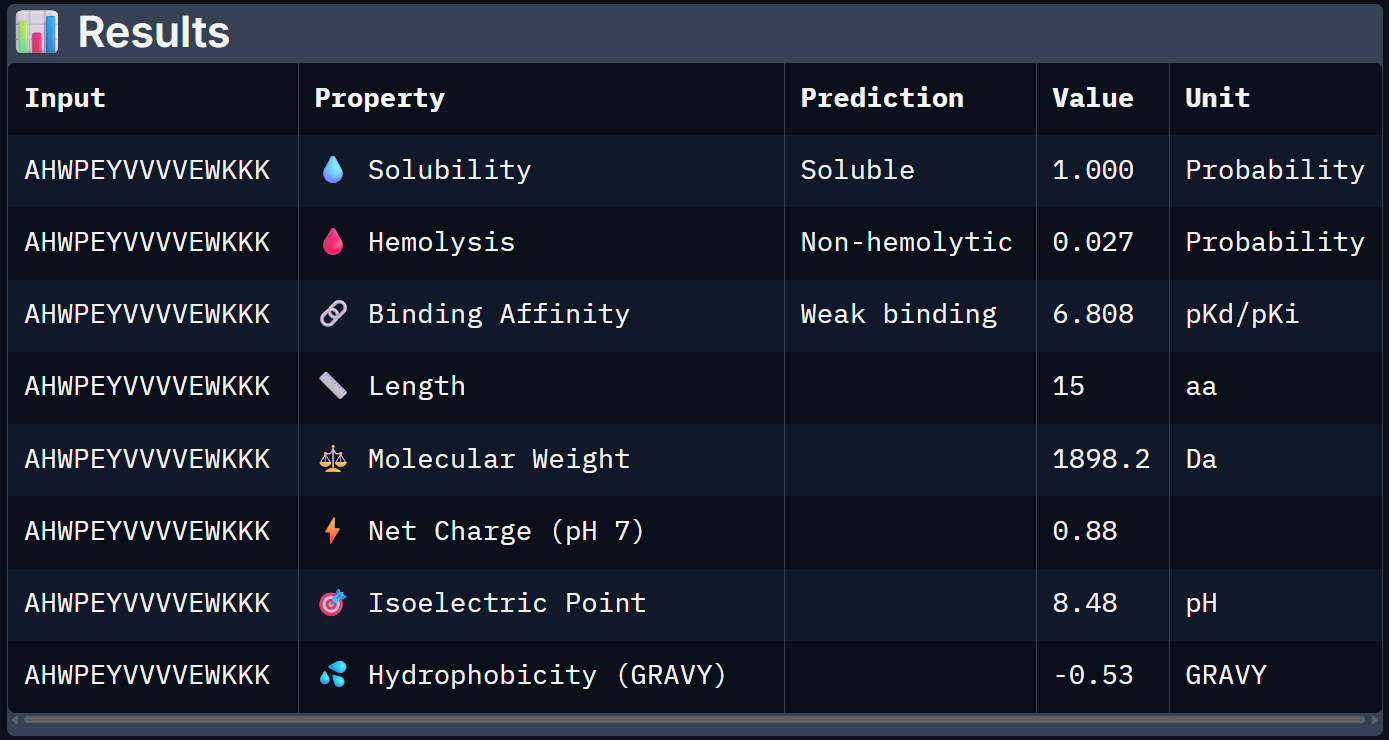
For Peptide 3: AHWPEYVVVVEWKKK
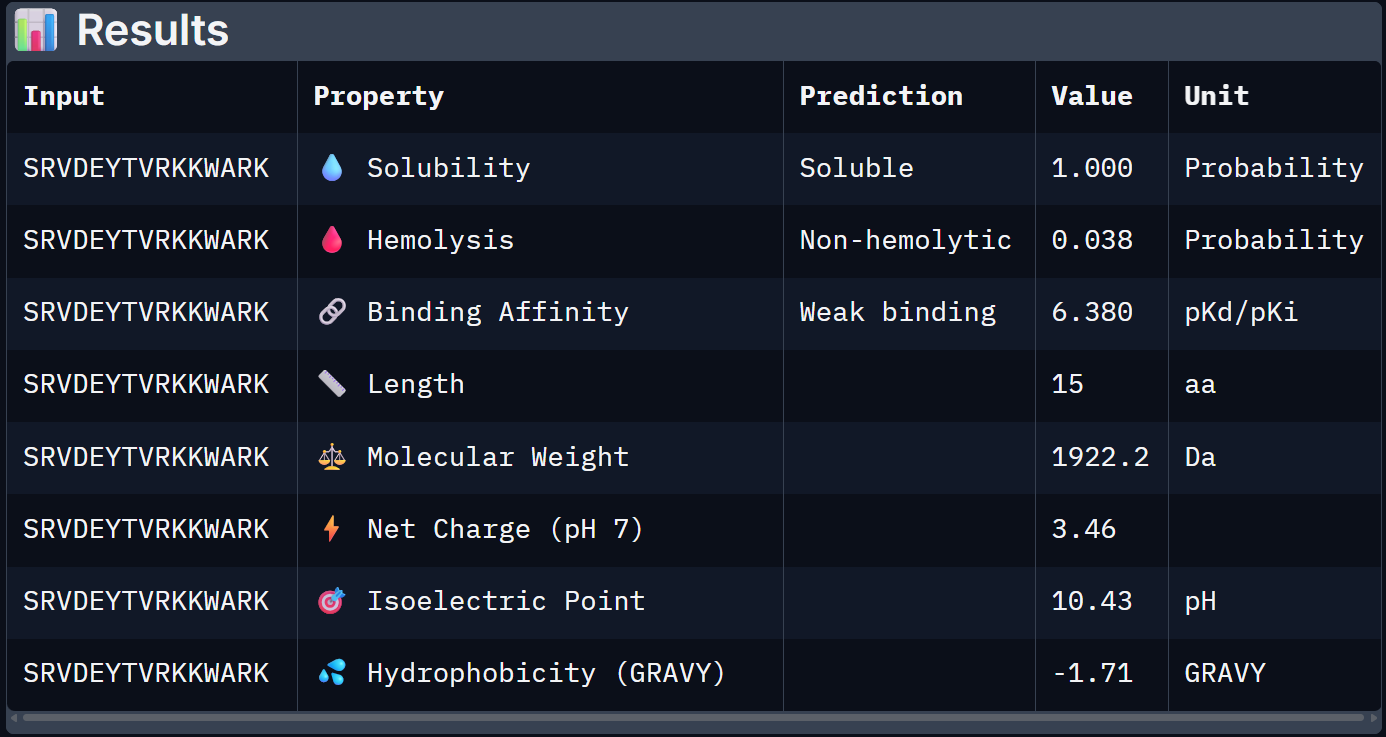
For Peptide 4: SRVDEYTVRKKWARK
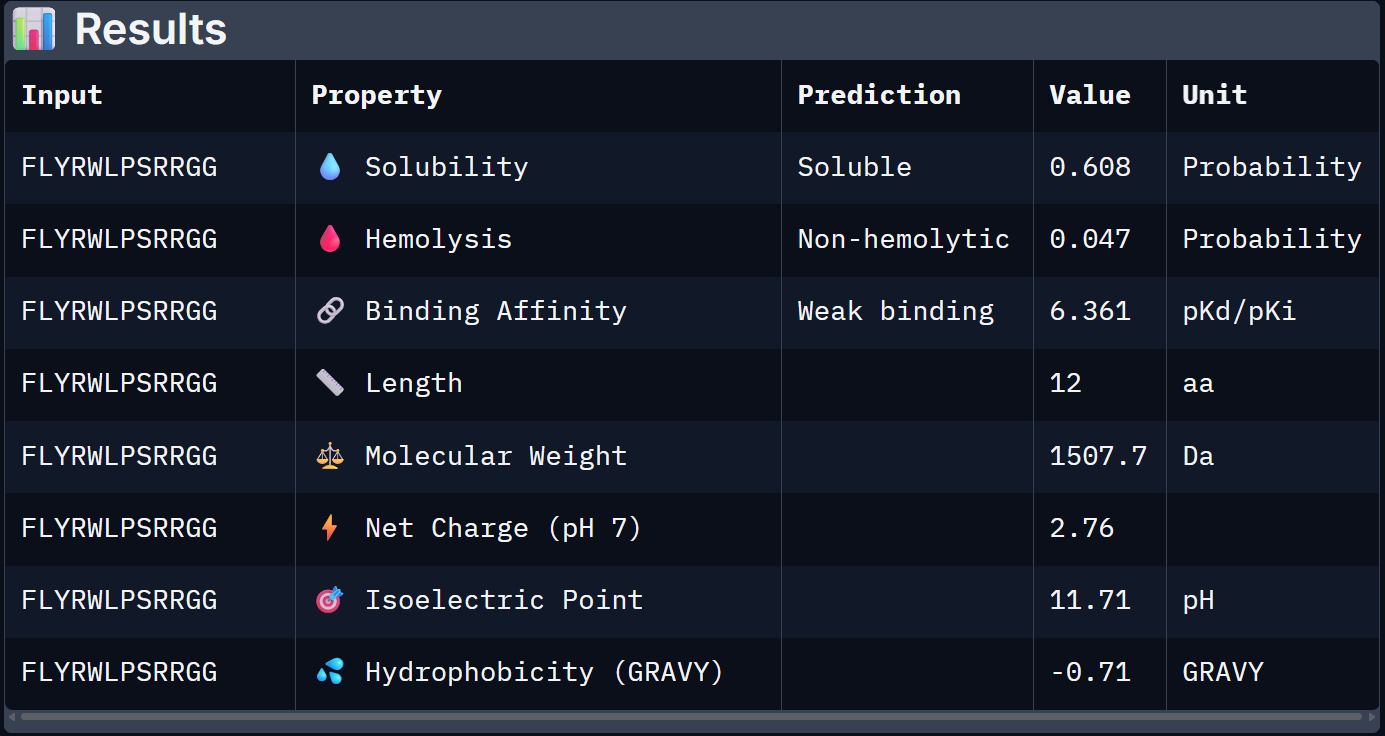
For Peptide 0, not generated but known: FLYRWLPSRRGG
| Binder | Solubility | Hemolysis Prob. | Binding Affinity (pKd) | Net Charge (pH 7) | MW (Da) |
|---|---|---|---|---|---|
| 0 – FLYRWLPSRRGG | 0.608 | 0.047 | 6.361 | +2.76 | 1507.7 |
| 1 – SRWDEYTAVVAWARK | 1.000 | 0.078 | 6.905 | +0.46 | 1838.0 |
| 2 – SWYGEYTGVVAWRKK | 1.000 | 0.038 | 6.728 | +1.46 | 1830.0 |
| 3 – AHWPEYVVVVEWKKK | 1.000 | 0.027 | 6.808 | +0.88 | 1898.2 |
| 4 – SRVDEYTVRKKWARK | 1.000 | 0.038 | 6.380 | +3.46 | 1922.2 |
When the PeptiVerse predictions are overlaid with the AlphaFold3 structural data, a reasonably coherent picture emerges. Binder 1 (SRWDEYTAVVAWARK) leads on both fronts. The highest ipTM (0.44) and the strongest predicted binding affinity (6.905 pKd), suggesting that the structural confidence in its interface correlates with a tighter predicted binding interaction. This is the clearest case of structural and biochemical agreement. Binder 3 (AHWPEYVVVVEWKKK) also performs consistently. It has a moderate ipTM of 0.35 pairs with an affinity of 6.808 pKd and the lowest hemolysis probability (0.027), making it the safest therapeutic candidate on safety metrics.
Binder 1 is the clear choice to advance. It uniquely leads on the structural confidence metric (ipTM = 0.44, the only one to exceed the known binder), has the highest predicted binding affinity (6.905 pKd), is perfectly soluble (1.000), and is non-hemolytic. Its near-neutral net charge (+0.46) is also favorable.
Part 4: Generate Optimized Peptides with moPPIt
Now for the last part, I was supposed to generate better and optimized peptides using moPPit Colab . I copied the Colab notebook. After running the first two cells, to clone the Github repo and install requirements. I found the cell with the generation setup, I chose ‘de-novo synthesis’ pasted my mutated SOD1 sequence in the target protein sequence. The target protein box opens up when you select motif/affinity guidance. I chose residues 1-10 and set the peptide length to 12. I enabled motif and affinity guidance and generated the peptides.
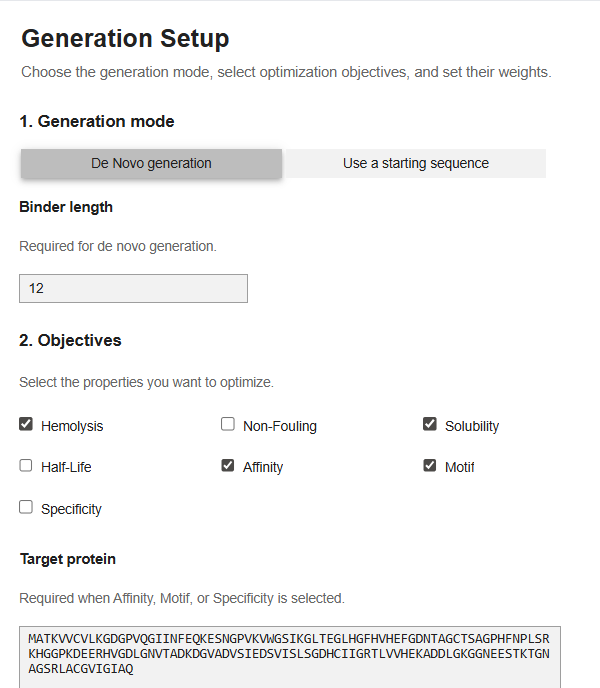
 My parameters.
My parameters.
This was the point upto which the code functioned. I tried my best to run the code but on the Colab there was always an error repeated, I tried in a new runtime, I tried all kinds of hacks using Gemini but it didn’t work. I tried finding an alternative access platform for moppit but it wasn’t available. I decided to just see the generated peptides of other people. I noticed that the moppit generated proteins are actually more optimized to the specific goal that we are trying to achieve, based on the weights, I think. (solubility, hemolysis etc.)
As about the evaluation prior to clinical studies, I would do more in-silico binding simulations, then screen the successful candidates to in-vitro binding assays like ELISA, then look for real hemolytic, cytotoxic property and then possibly move on to animal studies.
Part C L-Protein Mutants
Option 1: Mutagenesis
I used the Colab Notebook, provided in the pdf. Notebook I pasted in the L-Protein sequence and ran the notebook to get the per-substitution LLR scores and then I got a list top 20 mutations with positive score mutations.
| Position | Wild_Type_AA | Mutation_AA | LLR_Score |
|---|---|---|---|
| 50 | K | L | 2.561468 |
| 29 | C | R | 2.395427 |
| 39 | Y | L | 2.24178 |
| 29 | C | S | 2.04315 |
| 9 | S | Q | 2.014325 |
| 29 | C | Q | 1.997049 |
| 29 | C | P | 1.971029 |
| 29 | C | L | 1.960646 |
| 50 | K | I | 1.928801 |
| 53 | N | L | 1.864932 |
| 61 | E | L | 1.818098 |
| 52 | T | L | 1.813968 |
| 50 | K | F | 1.802069 |
| 29 | C | T | 1.797247 |
| 29 | C | K | 1.795878 |
| 5 | F | Q | 1.795244 |
| 5 | F | R | 1.659717 |
| 29 | C | A | 1.648656 |
| 27 | Y | R | 1.628061 |
| 22 | F | R | 1.602028 |
| 5 | F | P | 1.596891 |
| 50 | K | V | 1.594576 |
| 50 | K | S | 1.574557 |
| 5 | F | T | 1.559024 |
| 5 | F | S | 1.556417 |
| 45 | A | L | 1.539248 |
| 39 | Y | S | 1.517457 |
| 27 | Y | S | 1.497053 |
| 40 | V | L | 1.47763 |
| 27 | Y | L | 1.474637 |
| 22 | F | S | 1.423358 |
| 29 | C | E | 1.383281 |
| 39 | Y | A | 1.364999 |
| 29 | C | N | 1.362601 |
| 50 | K | A | 1.357795 |
| 29 | C | I | 1.344121 |
| 5 | F | L | 1.332615 |
| 17 | N | R | 1.323651 |
| 39 | Y | I | 1.320103 |
| 39 | Y | T | 1.302804 |
| 26 | D | R | 1.268762 |
| 29 | C | H | 1.246107 |
| 39 | Y | F | 1.245851 |
| 39 | Y | V | 1.24439 |
| 23 | K | R | 1.236555 |
| 25 | E | R | 1.22935 |
| 24 | H | R | 1.227779 |
| 50 | K | T | 1.222131 |
| 27 | Y | Q | 1.218851 |
| 27 | Y | T | 1.215567 |
| Cross-checking this with the experimental data provided along with the homework. I found out that there are only two overlapping mutations. |
- Position 29: C→R Experimental Lysis=0 LLR Score=2.3954
- . Position 50: K→I Experimental Lysis=0 LLR Score=1.9288
Now. categorizing beneficial mutations by region. I found out that
- 19 mutations improved lysis (
Lysis=1) - 63 mutations impaired lysis (
Lysis=0)
Regional Distribution
| Region | Positions | Beneficial Mutations |
|---|---|---|
| Transmembrane | 35–59 | 3 |
| Soluble | 1–34, 60–75 | 16 |
84% of beneficial mutations occur in soluble regions.
Top Beneficial Experimental Mutations
Soluble Region
| Position | Mutation | Effect |
|---|---|---|
| 13 | P→L | Improved lysis |
| 15 | S→A | Improved lysis |
| 18 | R→G | Improved lysis |
| 18 | R→I | Improved lysis |
| 19 | R→S | Improved lysis |
Transmembrane Region
| Position | Mutation | Effect |
|---|---|---|
| 44 | L→P | Improved lysis |
| 45 | A→P | Improved lysis |
| 46 | I→F | Improved lysis |
Top Language Model Predictions
| Position | Mutation | LLR Score | Region |
|---|---|---|---|
| 50 | K→L | 2.5615 | Transmembrane |
| 29 | C→R | 2.3954 | Soluble |
| 39 | Y→L | 2.2418 | Transmembrane |
| 29 | C→S | 2.0431 | Soluble |
| 9 | S→Q | 2.0143 | Soluble |
Selection of 5 Candidate Mutations
The criteria for selection was:
- Prioritized high LLR scores
- Considered experimental evidence
- Maintained regional balance (TM + soluble)
- Avoided redundant positions
- Focused on mutations supported by both datasets
Here’s a list of selected Candidate Mutations
| Mutation | Region | LLR Score | Key Rationale |
|---|---|---|---|
| K50L | Transmembrane | 2.5615 | Highest-scoring mutation; increases TM hydrophobicity and may improve membrane insertion |
| Y39L | Transmembrane | 2.2418 | Enhances hydrophobic packing within TM helix |
| C29R | Soluble | 2.3954 | High-scoring mutation that may improve oligomerization despite weak experimental support |
| S9Q | Soluble | 2.0143 | Likely enhances hydrogen bonding and N-terminal stability |
| A45P | Transmembrane | 1.5392 | Experimentally validated (Lysis=1); may improve pore geometry via helix kink formation |