Week 4 HW: Protein Design part I
HW Questions:
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
- Meat is roughly 20% protein by mass, so there’s ~100g of protein in 500g of meat. Average amino acid molecular weight is ~110 Da.
- 100 g ÷ 110 g/mol ≈ 0.91 mol of amino acid residues × Avogadro’s number: 0.91 × 6.022 × 10²³ ≈ ~5.5 × 10²³ amino acid residues
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
- During digestion, proteases break down the cow/fish proteins down into their constituent free amino acids and small peptides. These are then absorbed as monomers. Your ribosomes then reassemble them according to your own mRNA instructions.
Why are there only 20 natural amino acids?
- Likely a combination of three things. First, once the genetic code co-evolved around these 20 amino acids, any change would catastrophically mis-translate the entire proteome, so those 20 were ’locked in’. Second, the 20 cover the necessary chemical space (charged, polar, hydrophobic, aromatic, etc). Third, the simplest amino acids (Gly, Ala, Asp, Glu, Val…) are exactly the ones most readily produced abiotically, so the code evolved around what was chemically accessible early on. Selenocysteine and pyrrolysine as the “21st and 22nd” amino acids show the code can expand, but only under very constrained circumstances.
Where did amino acids come from before enzymes that make them, and before life started?
- They form spontaneously from simple chemistry. Amino acids are thermodynamically reasonable products.
- The Miller-Urey experiment showed electric discharge through CH₄, NH₃, H₂O and H₂ produces Gly, Ala, Asp and others.
- Hydrothermal vents provide mineral catalysts and redox gradients. Meteorites (like Murchison) contain over 70 amino acids synthesized in space, confirming abiotic production is universal wherever C, N, O, H and energy coexist.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
- Left-handed. L-amino acids prefer φ≈−57°, ψ≈−47° → right-handed helix. D-amino acids are the mirror image → φ≈+57°, ψ≈+47° → left-handed helix.
Can you discover additional helices in proteins?
- Beyond the α-helix, several others exist.
- The 3₁₀-helix is tighter (i→i+3 H-bonds), more strained, and common at helix termini.
- The π-helix is wider (i→i+5 H-bonds) and surprisingly prevalent at functional sites — maybe 15% of proteins contain at least one π-turn.
- The polyproline II (PPII) helix has no intramolecular H-bonds at all, is left-handed, and is extremely common in disordered regions, collagen, and signaling domains (SH3 recognition).
- The collagen triple helix is three intertwined PPII-like chains stabilized by interchain H-bonds.
- New folds continue to emerge from cryo-EM and AlphaFold-era structural biology, so the catalogue is probably not closed.
Why are most molecular helices right-handed?
- Most molecular helices are right-handed because all biological amino acids are L-form, so their bond geometry naturally favors coiling clockwise when chained together.
Why do β-sheets tend to aggregate?
- Edge strands have unpaired backbone NH and C=O groups pointing outward (they’re basically unsatisfied H-bond donors and acceptors, which makes them inherently “sticky.”)
- Flat hydrophobic surfaces on sheet faces also drive stacking through the hydrophobic effect.
- Side chains interdigitate into a tight steric zipper, which is favorable in both enthalpy and entropy, and thus very hard to prevent without chaperones or proline residues.
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?\
- For many disease-associated sequences (Aβ in Alzheimer’s, α-synuclein in Parkinson’s, IAPP in type II diabetes), the amyloid state is actually thermodynamically more stable than the native fold. Stress, concentration increases, mutations, or metal ions can nucleate conversion, after which elongation proceeds rapidly like crystal growth. The resulting cross-β architecture is extraordinarily stable, resistant to heat, detergent, and proteases.
- As materials, amyloid fibrils are definitely useful. They can have a Young’s modulus of 1–20 GPa (similar to silk), high aspect ratio, and nanoscale precision. Functional amyloids already exist naturally (curli fibers in bacterial biofilms, yeast prions as regulatory switches). Proposed applications include conductive nanowires (metallized with silver or gold), hydrogels for drug delivery, tissue engineering scaffolds, and even food technology(whey protein amyloids are already used commercially as emulsifiers).
Protein Analysis and Visualization
- Insulin is a small hormone produced by the pancreas that regulates blood glucose levels. I chose it because of its fascinating history as a therapeutic protein. Decades of protein engineering have produced analogs like insulin lispro and glargine, where just one or two amino acid changes dramatically alter how the drug behaves in the body. I wanted to explore the structure underlying a protein that has been so deliberately and successfully redesigned.
- Sequence:
- GIVEQCCTSICSLYQLENYCN/FVNQHLCGSHLVEALYLVCGERGFFYTPK
- length: 51 amino acids
- most common residues are C and L, which appear 6 times each
- this protein has 228 homologs
- this specific insulin protein is in the broader insulin family
- Protein Structure Page
- The structure was released on February 24, 2009. (Note: While the very first insulin structure was solved in 1969, this version (3E7Y) is the modern high resolution reference for the native human protein).
- The quality is excellent - 1.60 Å
- There are other molecules in the solved structure of the protein, as this classic structure represents the storage form (hexamer). It contains zinc ions, chloride ions, and water molecules
- This protein is the defining member of the Insulin-like superfamily.
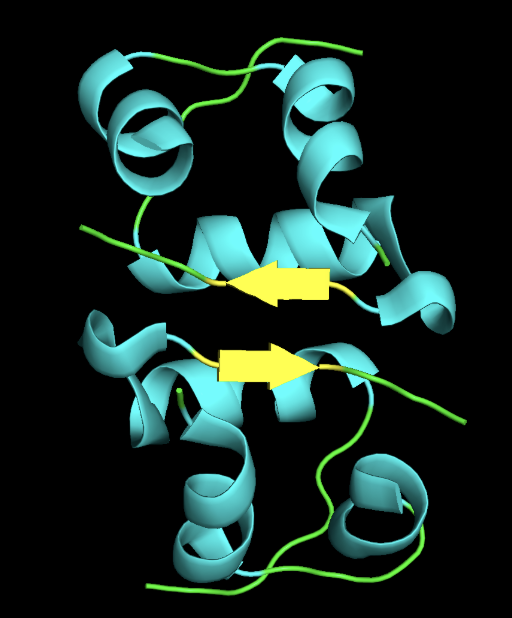
- 3D Structure
- cartoon
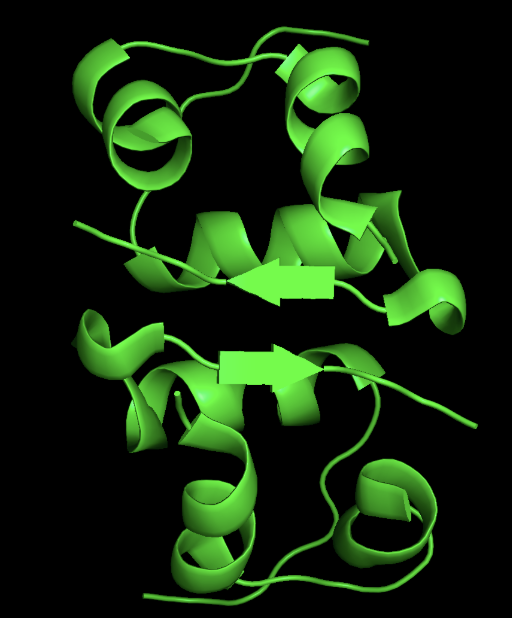
- ribbon

- sticks

- secondary structure: the protein has more helices than sheet
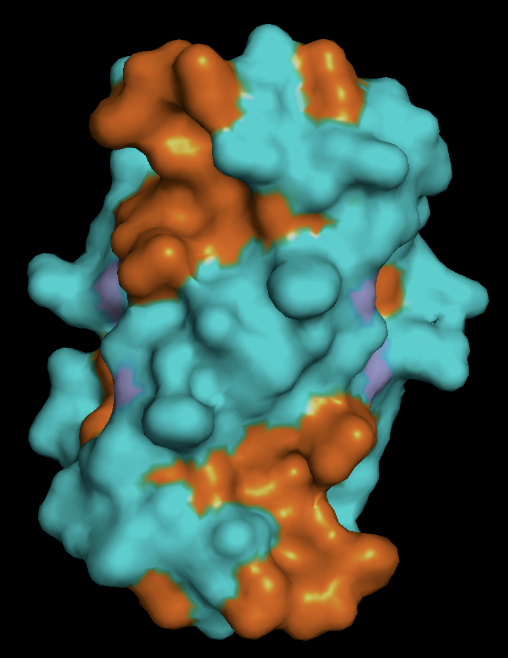
- protein surface: the distribution of hydrophobic (orange) vs hydrophillic (cyan) residues follows as expected. Most of the surface residues are hydrophillic, and the hydrophobic residues line the binding pockets
Using ML-Based Protein Design Tools
For this section, I will be using the same Insulin protein from the prior section.
GIVEQCCTSICSLYQLENYCNFVNQHLCGSHLVEALYLVCGERGFFYTPKT
PDB ID: 3E7Y
C1.1. Mutational Scan
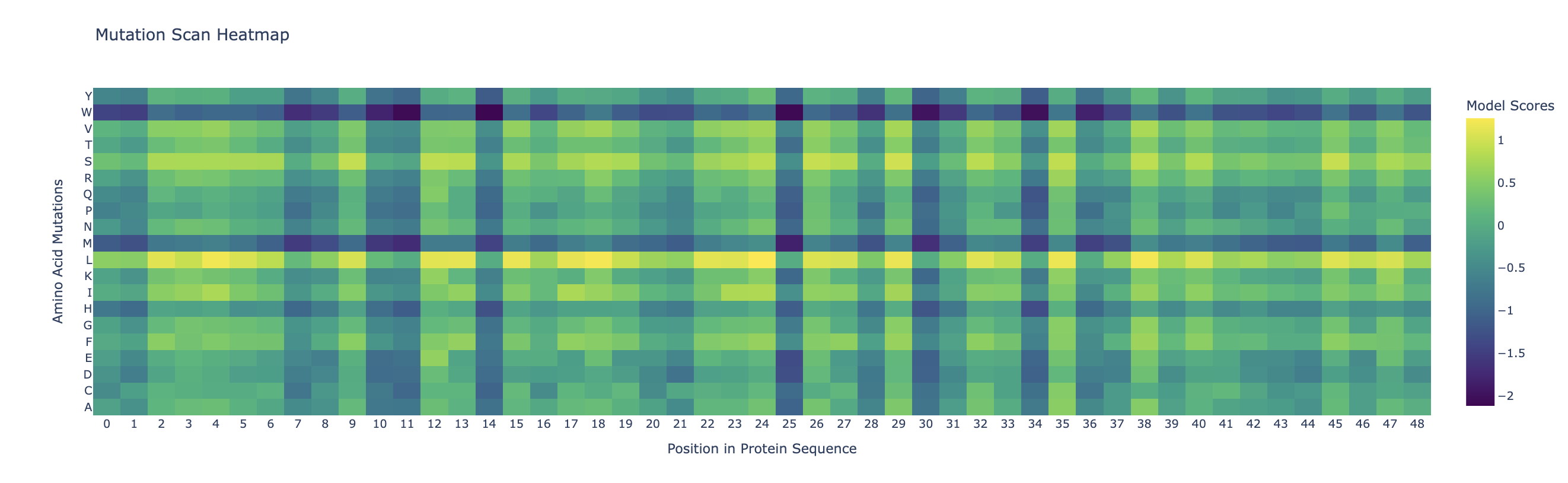
Darker columns (low probablities mean mutations are unlikely) indicate more conserved residues (wild-type is strongly preffered). Likely, these are amino acids which are most crucial to the protein’s structure and function.
C1.2. Latent Space Analysis
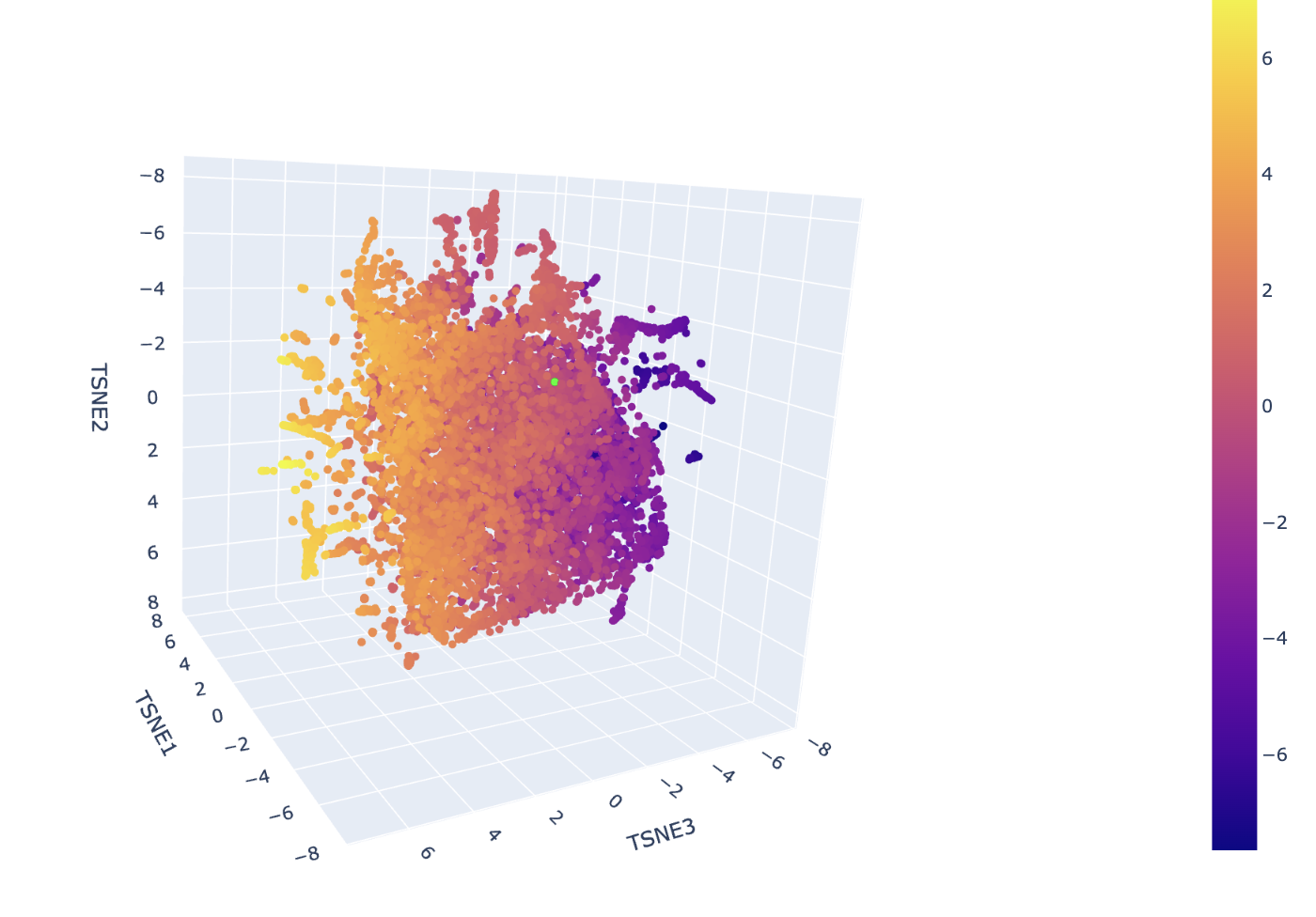
The neighborhoods in this map are determined by the high-dimensional proximity of ESM-2 embeddings, which represent the structural and evolutionary patterns of the sequences. In the ASTRAL SCOP dataset used here, these spatial clusters correspond to specific protein superfamilies and folds. Hovering over any cluster confirms that neighbors share identical or closely related SCOP classification codes, verifying that the visualization effectively approximates biological and structural similarity.
The insulin protein is represented by the lime green dot situated within a dense cluster of other signaling proteins. It is positioned in this specific neighborhood because the ESM-2 model identified its sequence signature, particularly the conserved cysteine motif, as being highly similar to the other sequences in that region. The predominant identities of its closest neighbors are other human and mammalian insulins, as well as members of the Insulin/IGF/Relaxin superfamily like IGF-1 and IGF-2. This proximity indicates that the model recognizes your protein as sharing the same disulfide-rich fold that defines the structural template for this entire cluster.
C2.1
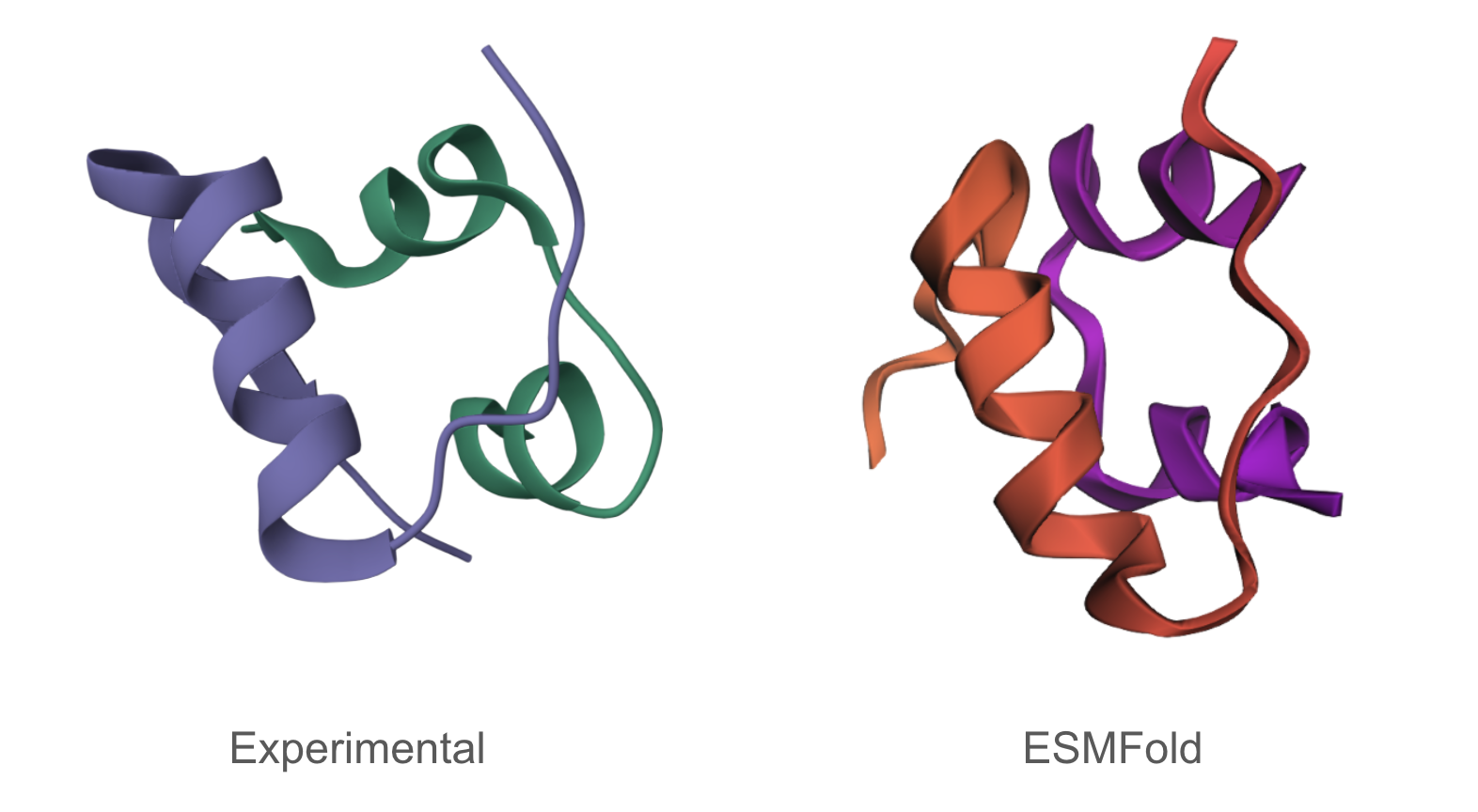
The predicted structure matches the experimentally determined structure.
C2.2
Mutating some portions had minimal impact on predicted structure, while mutations/deletions in other portions (the beginning for instance) meaningfully altered the predicted structure.
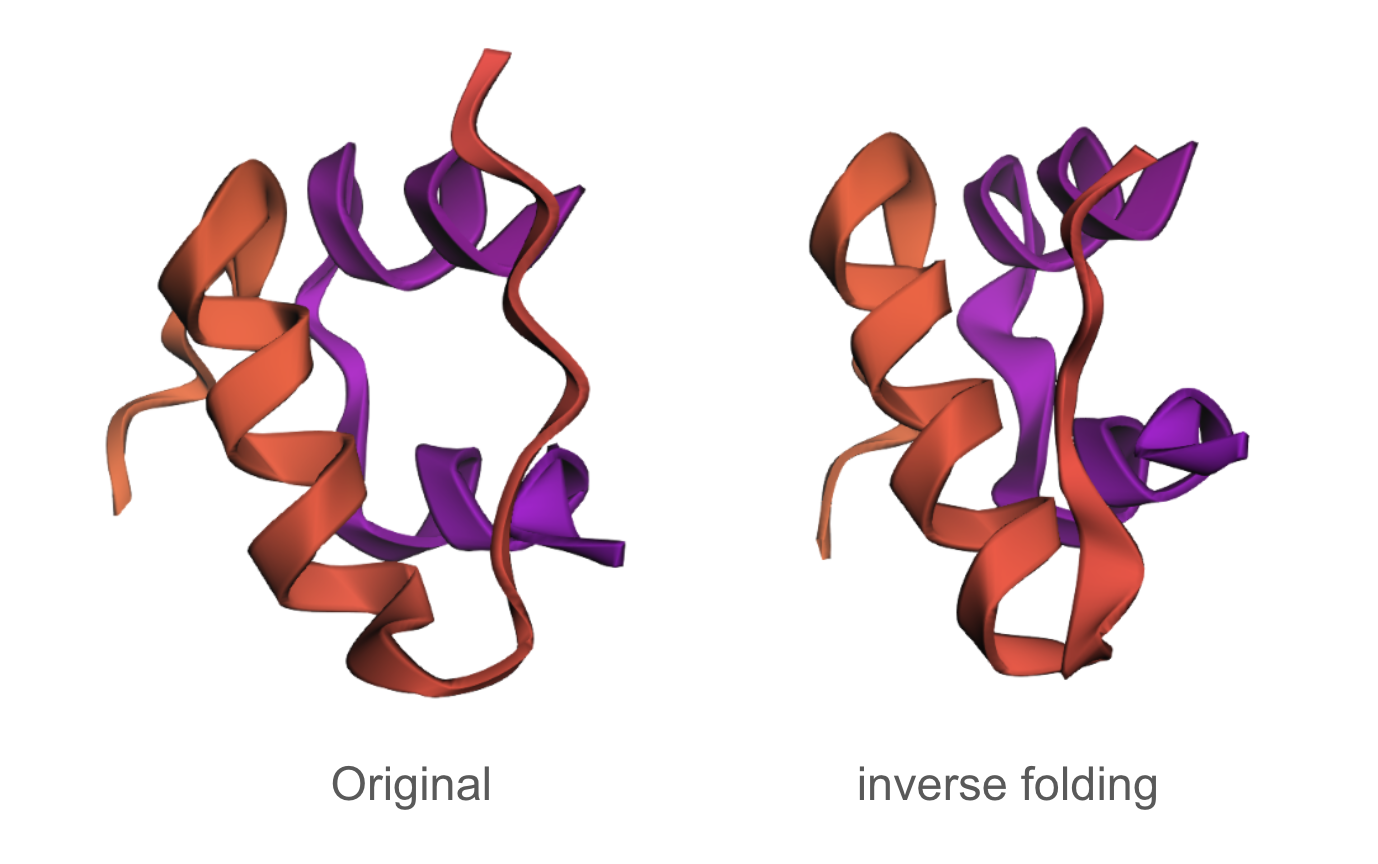
C3.1
The designed sequence obtained using inverse folding was:
GIEELCCESSCTPEELAEYCN/SSSGRYCGEELIEALAEVCGERGFTYAPP
The inverse folding analysis of the 3E7Y backbone yielded a native sequence score of 1.6521 and a ProteinMPNN-designed score of 0.9312, where the lower value for the design indicates a higher model-perceived likelihood that the new sequence will stabilize the target structure. This optimization is reflected in a sequence recovery of 0.5200, suggesting that while the model preserved 52% of the original residues to maintain the structural core, it proposed alternative mutations for the remaining 48% to improve the fit. The amino acid probability chart supports these scores by revealing that the model’s high confidence is concentrated at key positions like the cysteines, which are essential for the disulfide-rich insulin fold, while the lower scores for the redesign likely stem from the model finding more energetically favorable residues for the flexible, darker-colored regions on the heatmap.
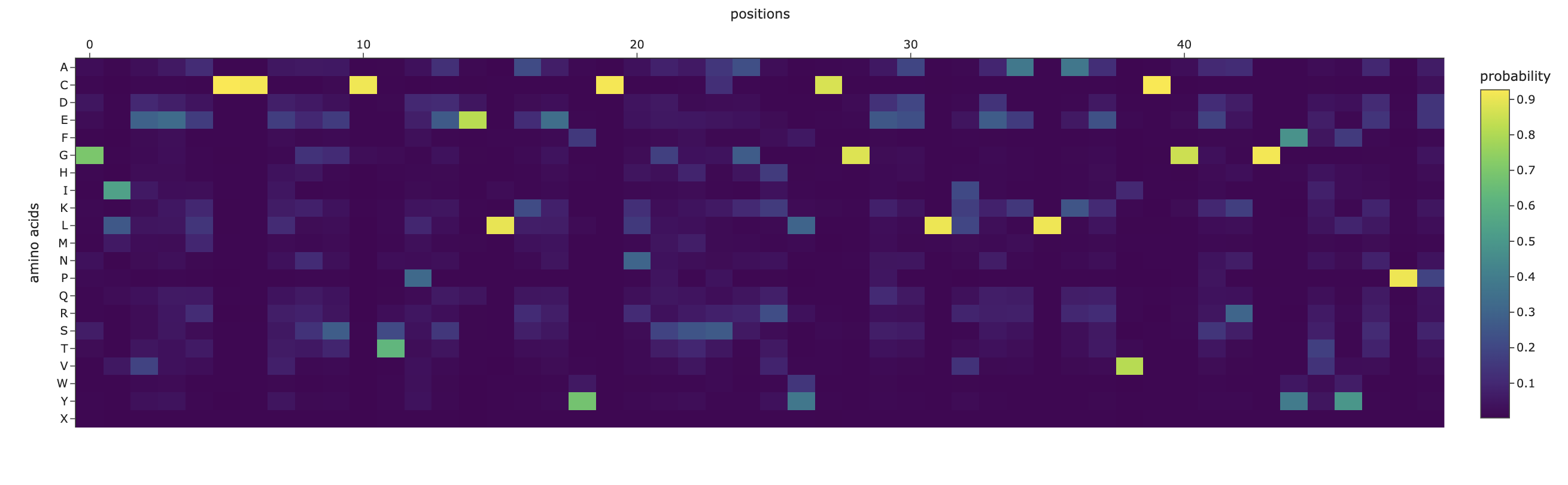
C3.2
The predicted structure of the inverse-folding result was extremely similar to the ESMFold result of the original sequence.