Week 5 HW: Protein Design part II
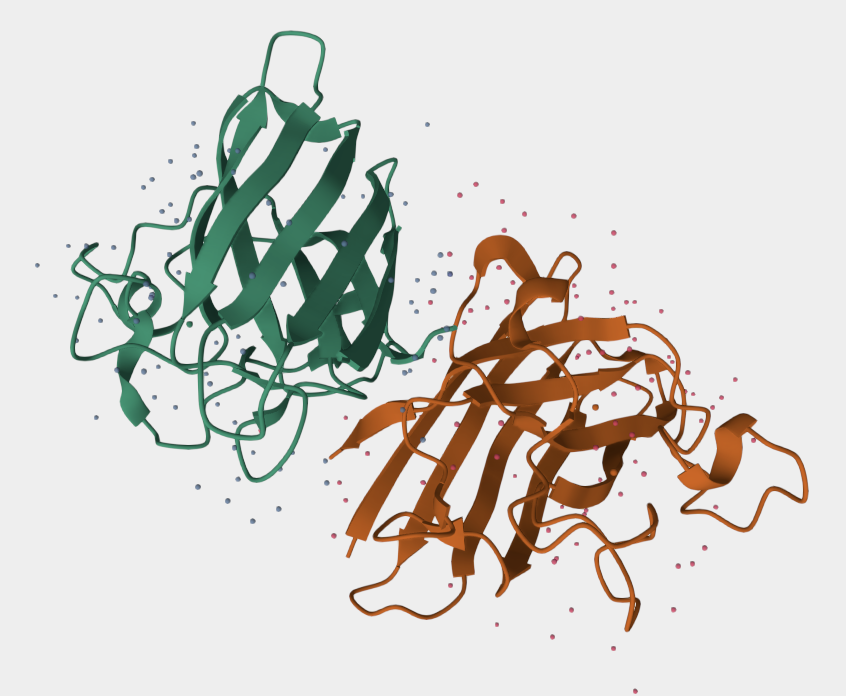
Part A: SOD1 Binder Peptide Design
sequences, scores, structure, and properties for all peptides
PepMLM binder generation
- Perplexity scores for known and generated peptides:
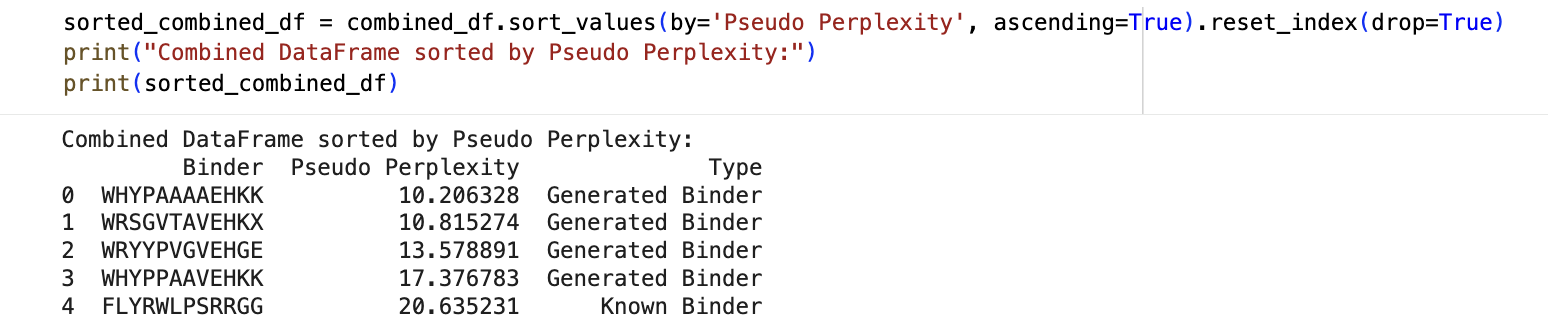
Alphafold binder evaluation
- ipTM Values and Comparison to Known Binder
The ipTM values across all peptides are low, ranging from 0.27 to 0.43, and none exceed 0.5 — the general threshold for confident protein-peptide interaction prediction. Notably, two PepMLM-generated peptides (Sequence_0 at 0.40 and Sequence_1 at 0.43) actually exceed the known binder (Sequence_4 at 0.32), suggesting the model produced candidates with comparable or slightly better predicted interface confidence. However, all predictions share the same binding
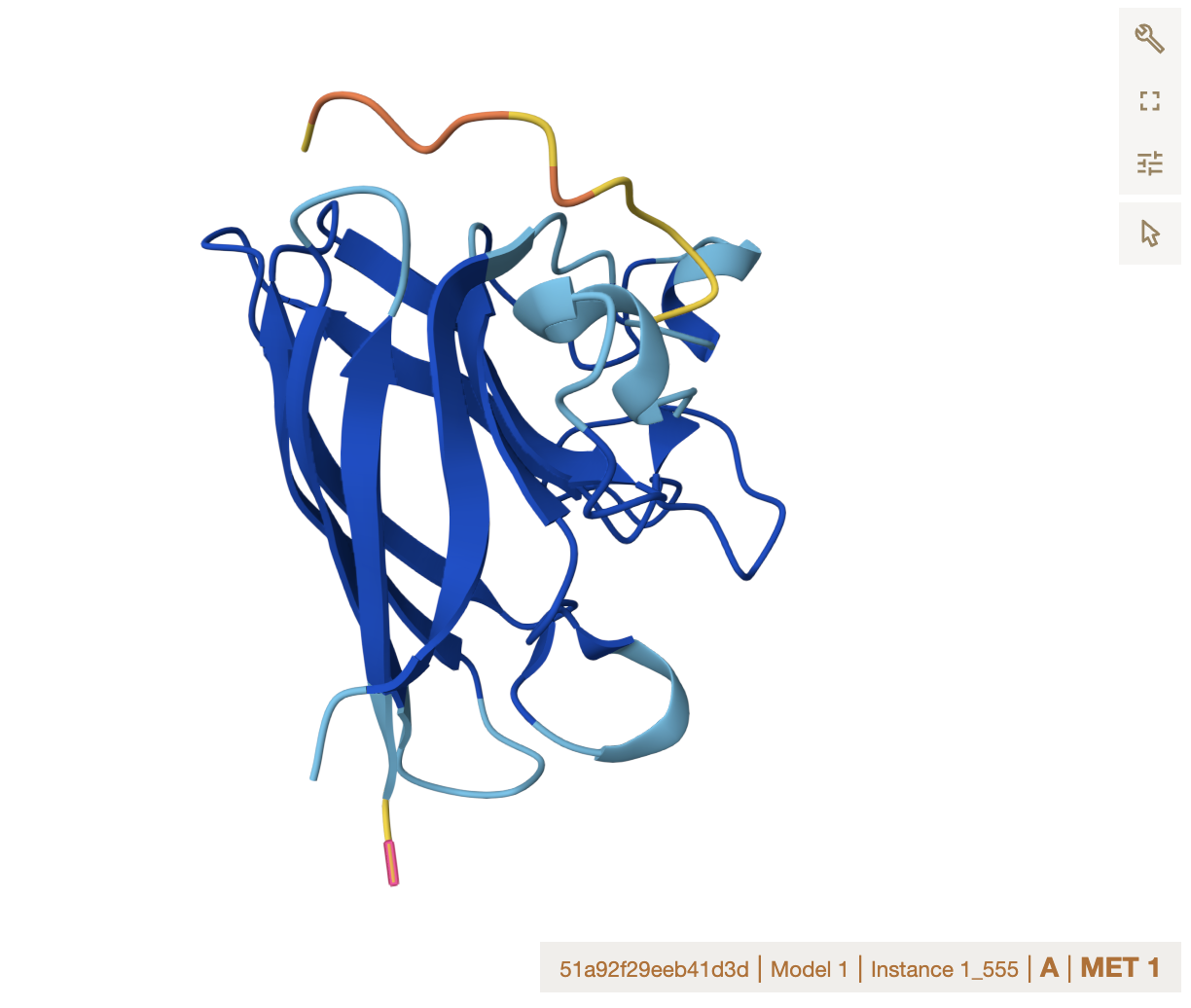
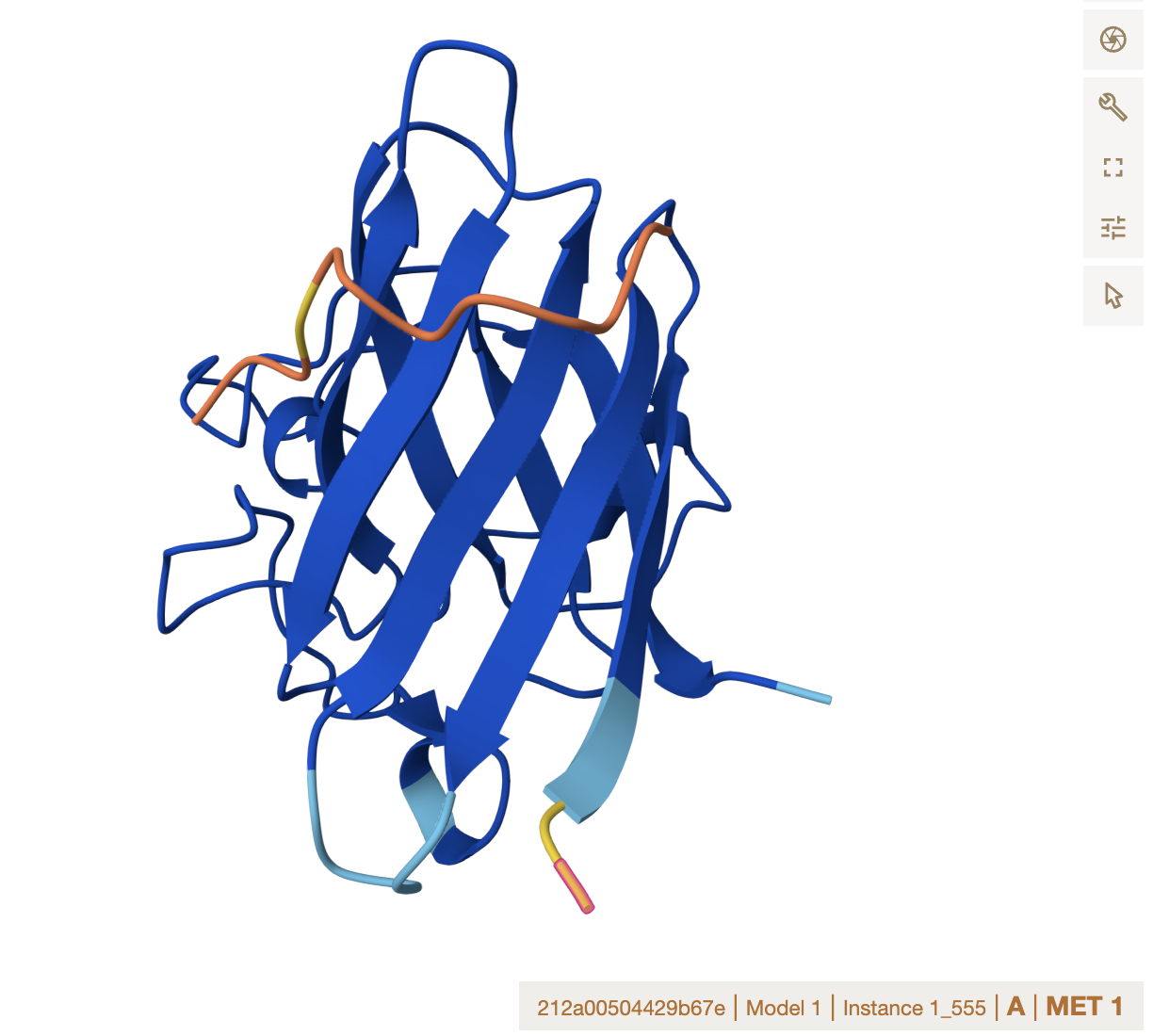
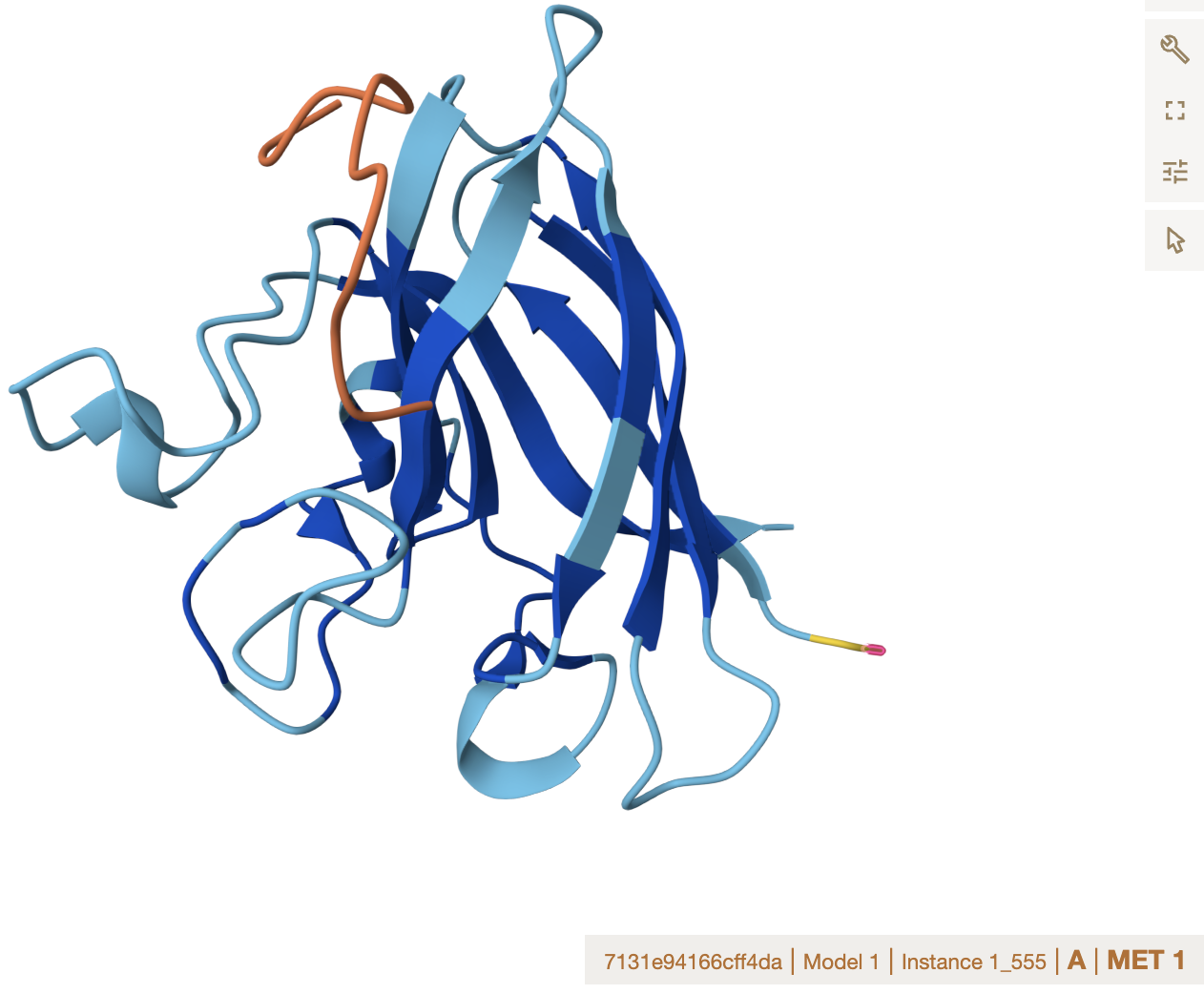
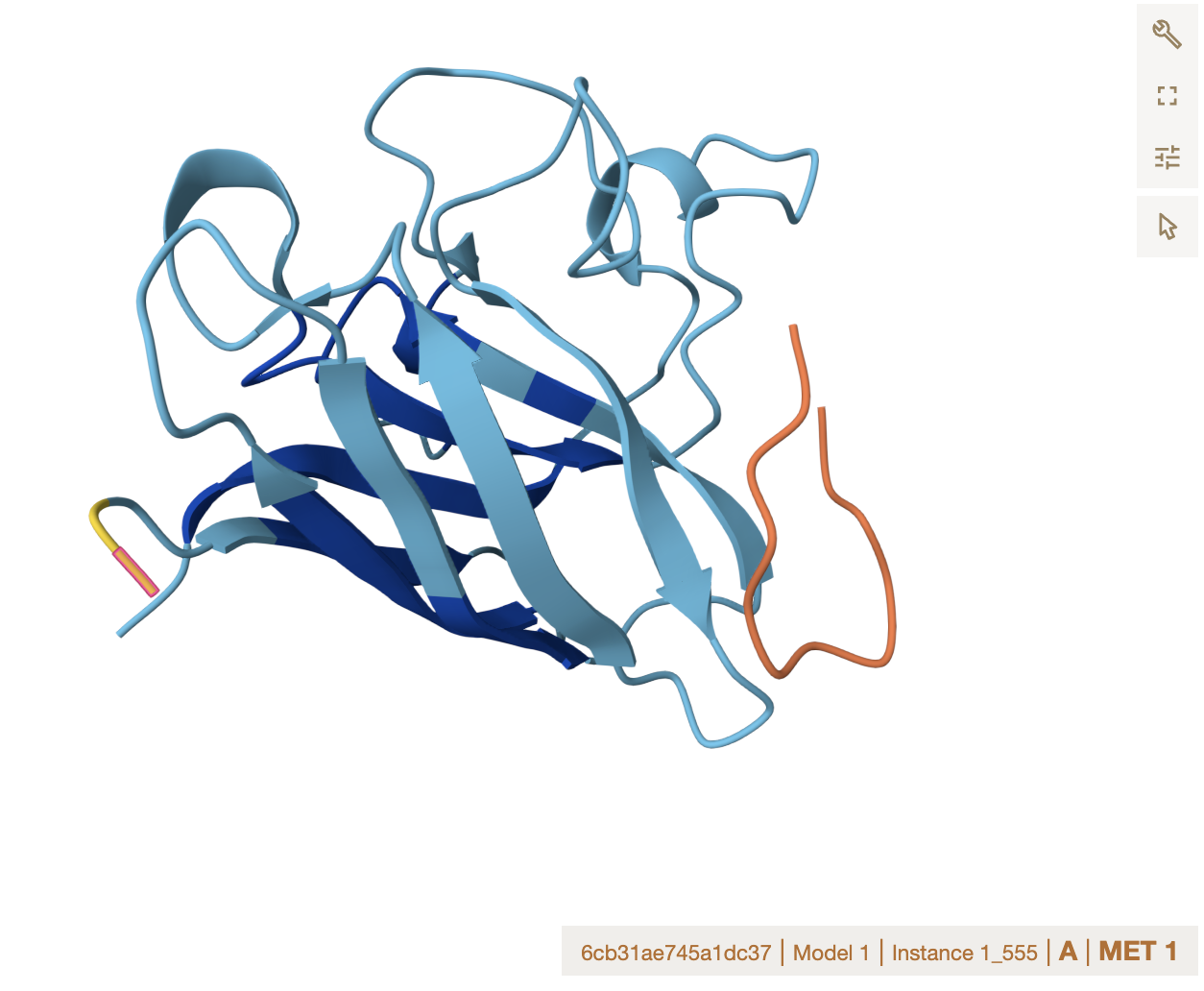
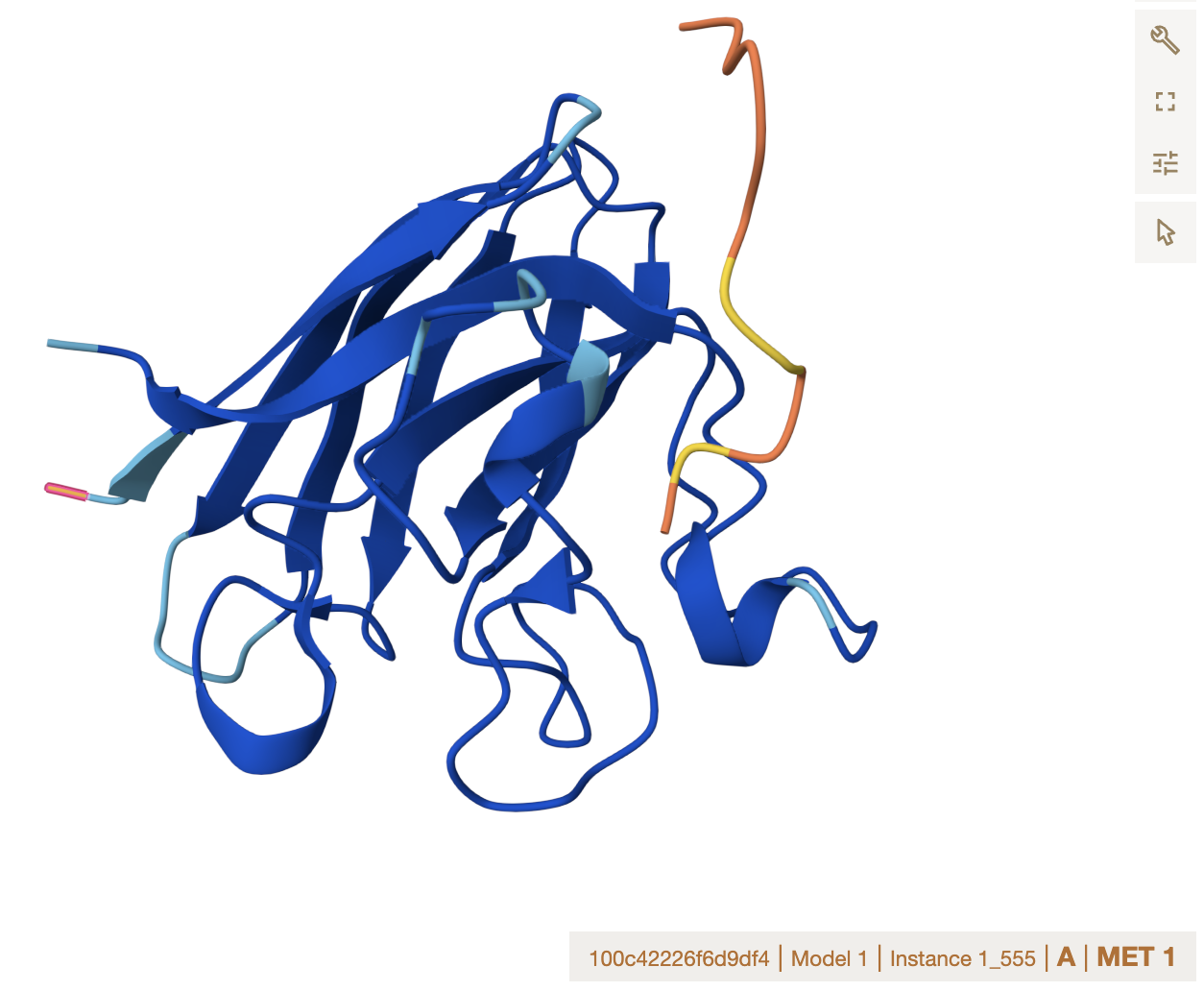
Location note: surface-exposed, near the dimer interface, and not near the A4V mutation site.
Important caveat: none of these peptides, including the known binder, appear to engage the mutation directly, which limits their utility for direct mutant stabilization.
Peptiverse property evaluation
- Peptiverse vs. AlphaFold3 Structural Predictions
There is no consistent correlation between ipTM and predicted binding affinity. Sequence_1 has the highest ipTM (0.43) but one of the weaker affinities (5.183 pKd/pKi), while the known binder has a modest ipTM (0.32) yet the strongest predicted affinity (5.968 pKd/pKi) — suggesting the two metrics are capturing different aspects of binding. Encouragingly, all peptides are fully soluble (P=1.00) and non-hemolytic, presenting clean safety profiles. The known binder is the closest to a hemolysis concern (P=0.047) and carries a high positive charge (+2.76), raising off-target interaction risk. Sequence_0 best balances the available metrics — strong ipTM among generated candidates (0.40), highest predicted affinity of the PepMLM sequences (5.624 pKd/pKi), full solubility, low hemolysis risk (P=0.034), and near-neutral charge (−1.14).
Peptide to Advance: Sequence_0 (WRYYPVGVEHGE)
I would advance Sequence_0. It offers the best combination of structural confidence, predicted affinity, and therapeutic safety among the generated candidates, and compares favorably to the known binder on hemolysis and charge. Crucially, like all candidates here, it binds near the dimer interface rather than the mutation site directly — meaning the likely mechanism of action would be dimer stabilization rather than direct rescue of the A4V destabilization. This is still therapeutically meaningful, since SOD1 A4V toxicity is closely linked to aberrant monomerization and aggregation, and stabilizing the dimer interface could slow that cascade. That mechanistic framing should be explicitly tested in follow-up biochemical and cell-based assays, but Sequence_0 represents the strongest starting point for that line of investigation.