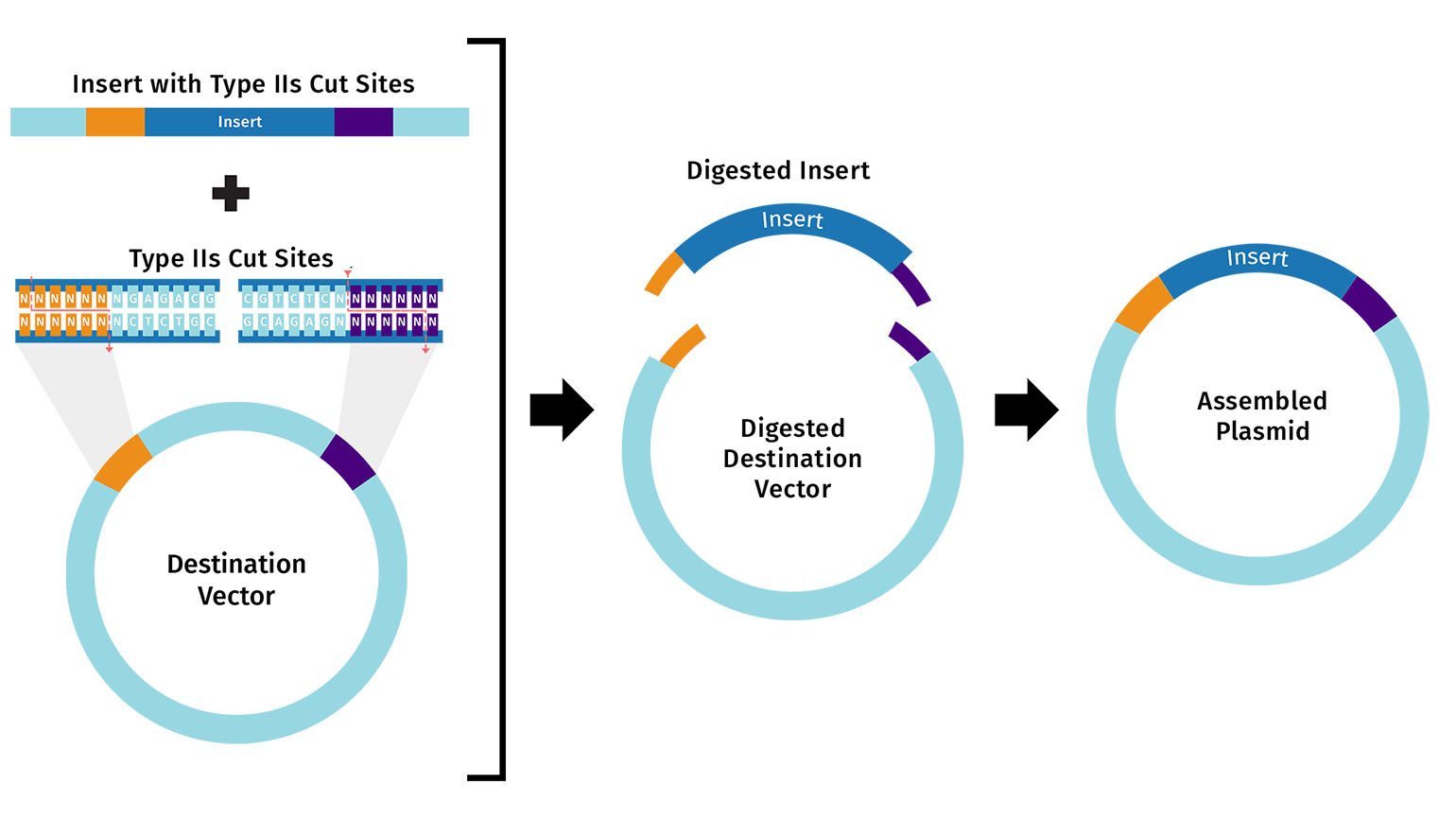First, describe a biological engineering application or tool you want to develop and why
Pattern-Based Rapid Diagnostic Platform for Dengue Virus: A rapid diagnostic platform for dengue virus (DENV) that integrates innate immune recognition, molecular recognition, and biosensor engineering to address key limitations of existing diagnostic methods. The proposed system combines mannose-binding lectin for the recognition of viral glycoproteins, dengue-specific aptamers targeting conserved regions of viral proteins, and signal transduction through a portable biosensor to enable rapid readout. This approach is motivated by the fact that current dengue diagnostics are often expensive and exhibit reduced sensitivity and reliability in dengue-endemic regions, particularly in countries like mine (Colombia), where prior flavivirus exposure compromises serological test performance and access to reliable diagnostics is limited by public healthcare infrastructure (Terenteva et al., 2025).
Homework: Final Project Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. Expression of the Reporter Gene LacZ
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements β-galactosidase hydrolyzes chromogenic substrates, producing a colored product that can be visually detected or quantified spectrophotometrically. LacZ will be measured using a colorimetric assay with ONPG, enabling both spectrophotometric quantification and visual detection
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Make a note on your HTGAA webpages including:
What you contributed to the community bioart project
I made part of the 2026 on the upper left plate
What you liked about the project, and what about this collaborative art experiment could be made better for next year.
Opentrons Artwork opentrons-art.rcdonovan.com/?id=oevp91e27i3m061
Post-Lab Questions Find and describe a published paper that utilizes the Opentrons This article combines an open‑source liquid‑handling robot (Opentrons OT‑One‑S Hood) with four interchangeable modules that perform magnetic‑bead DNA isolation, isothermal recombinase polymerase amplification (RPA) of the ctrA gene, exonuclease digestion to generate single‑stranded DNA, and detection on a paper‑based vertical‑flow microarray (VFM) using anti‑biotin gold nanoparticles for colorimetric read‑out.
Part A. Conceptual Questions How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Considering that 500 g of meat contains 20% protein, we would have 100 g of protein. After converting the protein mass to moles (assuming an average amino acid mass of 100 Da), this corresponds to approximately 6.022 × 10²³ amino acid molecules.
Assignment: DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Phusion High-Fidelity DNA polymerase: enzyme responsible for synthesizing new DNA strands while possessing activity, which reduces errors during replication.
dNTPs (deoxynucleotide triphosphates): Nucleotide substrates incorporated by DNA polymerase into the elongating DNA strand during synthesis
reaction buffer: contain compounds as Tris-HCl that maintains the correct pH and salts like KCl which help stabilize primer binding and enzyme activity
Part A: SOD1 Binder Peptide Design Part 1: Generate Binders with PepMLM After introducing the A4V mutation. I performed the mutation A5V based on its position in the FASTA sequence.
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using PepMLM Colab with a K value of 1, I obtained:
Assignment Part 1: Intracellular Artificial Neural Networks What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Instead of relying on ON/OFF logic, they process information in a continuous and distributed way, closer to how real cells behave. This allows them to integrate multiple signals with different intensities and avoid the exponential complexity that arises when scaling Boolean circuits. In addition, IANNs are inherently more flexible, since their behavior can be tuned by adjusting interaction strengths rather than completely redesigning the system, enabling more complex and nonlinear decision-making. Overall, they provide a more efficient, scalable, and biologically realistic framework for intracellular computation.
Assignment Part A: General and Lecturer-Specific Questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. You can control everything directly. You can adjust pH, temperature, ion concentrations, and cofactors and even add or remove components during the reaction. Also, it’s faster since you don’t need cell growth or maintenance.
Subsections of Homework
Week 1 HW: Principles and Practices
First, describe a biological engineering application or tool you want to develop and why
Pattern-Based Rapid Diagnostic Platform for Dengue Virus: A rapid diagnostic platform for dengue virus (DENV) that integrates innate immune recognition, molecular recognition, and biosensor engineering to address key limitations of existing diagnostic methods. The proposed system combines mannose-binding lectin for the recognition of viral glycoproteins, dengue-specific aptamers targeting conserved regions of viral proteins, and signal transduction through a portable biosensor to enable rapid readout. This approach is motivated by the fact that current dengue diagnostics are often expensive and exhibit reduced sensitivity and reliability in dengue-endemic regions, particularly in countries like mine (Colombia), where prior flavivirus exposure compromises serological test performance and access to reliable diagnostics is limited by public healthcare infrastructure (Terenteva et al., 2025).
2.Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
GOAL 1:
Ensure that diagnostic accuracy supports appropriate clinical decision-making, minimizing the risk of misdiagnosis, delayed care, and public health mismanagement.
Subgoals:
Diagnostic Reliability: Standards validated in endemic populations and ongoing monitoring of false positives and false negatives.
Prevention of clinical misinterpretation: Support test results with clear and accessible interpretive guidance.
GOAL 2:
Ensure to promote equitable access and global health justice in the development and use of the diagnostic technology.
Subgoals:
Affordability and Accessibility: Promote public–private collaboration for the deployment of dengue diagnostics in high-burden countries, without dependence on specialized infrastructure.
Prevent Technological Exclusion: Ensure that the diagnostic tool is usable in decentralized healthcare settings, such as rural clinics and community health centers
3.Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”)
Establish context-specific validation requirements for diagnostic deployment:
The objective of this action is to prevent clinical harm caused by diagnostic failures that persist under current practices in endemic settings, particularly false negatives in secondary dengue infections and false positives due to flavivirus cross-reactivity.
To achieve this, regulatory agencies and public health institutions should require that rapid dengue diagnostics be validated directly in endemic communities—especially among people with prior flavivirus exposure—before they’re approved or rolled out. This means shifting approval pathways so they rely on real‑world performance data, not just controlled lab studies or trials in non‑endemic settings.
Assuming that regulators can review performance data in the specific contexts where diagnostics will be used, that accuracy can vary across different populations, and that manufacturers will adapt their designs to meet these requirements. Even so, a “false success” could occur if compliance is limited to minimal testing in some endemic populations. True success would mean diagnostics that work reliably across diverse populations, helping to reduce misdiagnosis and support appropriate clinical decisions.
Ensuring Transparency and Open Validation in Diagnostic Development:
The purpose of this action is to promote responsible innovation and build trust by ensuring that the limitations of diagnostics are openly documented and shared before large-scale deployment. By making failure modes, cross-reactivity profiles, and other constraints visible early, developers, regulators, and clinicians can make better-informed decisions and reduce risks to patients and public health.
Funding agencies and scientific journals should require transparent reporting of assay limitations as part of the evaluation and publication process. Shared validation datasets could be established. Incentives—such as eligibility for specific funding programs, prioritized review, or formal recognition—can encourage participation from researchers and companies while improving the overall reliability and comparability of diagnostic technologies.
This approach assumes that increased transparency improves diagnostic quality and that researchers and companies will share performance data to reduce failure across the sector. Potential failure points include limited industry participation due to intellectual property concerns or competitive pressures. Success is defined by establishing routine, independent testing of diagnostic performance claims, creating a standard expectation of reliability prior to clinical adoption.
4.Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
–>
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
X
• By helping respond
X
Foster Lab Safety
• By preventing incident
X
• By helping respond
X
Protect the environment
• By preventing incidents
X
• By helping respond
X
Other considerations
• Minimizing costs and burdens to stakeholders
X
• Feasibility?
X
• Not impede research
X
• Promote constructive applications
X
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties
Based on the above, the following actions would be prioritized:
Establish context-specific validation requirements for dengue diagnostics.
This action has the highest priority because it directly ensures diagnostic reliability in endemic populations. Requiring validation in communities with prior flavivirus exposure reduces false negatives, false positives, and clinical mismanagement. It provides the regulatory foundation necessary for safe, equitable deployment. Without this step, large-scale implementation could compromise patient care and public health decision-making.
Ensure transparency and open reporting of diagnostic limitations.
This action strengthens accountability and trust by requiring disclosure of performance data, cross-reactivity profiles, and failure modes. However, its effectiveness depends on clear validation standards. Properly designed, it improves long-term reliability and supports informed clinical use, while balancing industry participation and innovation incentives.
Ethical concerns that arose, especially any that were new to you.
This week, I learned that developing a diagnostic platform is more than just a technical or experimental challenge; it is also a matter of governance and biosecurity—areas that were largely new to me. Previously, I focused mainly on protocol design, molecular mechanisms, and performance metrics. In class and doing the homework, I began to understand that every diagnostic tool exists within a broader regulatory, ethical, and public health framework that determines how it is validated, deployed, and monitored in real-world settings.
One key ethical concern that emerged for me is that diagnostic errors are not just laboratory inaccuracies—they can produce systemic harm. Poor validation, lack of transparency, or weak oversight can lead to misdiagnosis, inequitable access, and loss of public trust.
To address these issues, some governance actions that I think should be required are context‑specific validation before any large‑scale deployment, establishing clear transparency standards to ensure diagnostic limitations are openly reported, and implementing post‑market monitoring to quickly identify and respond to performance gaps.
References
Terenteva, S., Golani-Zaidie, L., Avivi, S., Lustig, Y., Indenbaum, V., Koren, R., Hoa, T. M., Tuyen, T. T. K., Huyen, M. T., Hoan, N. M., Hoi, L. T., Trung, N. V., Schwartz, E., & Danielli, A. (2025). Sensitivity and Cross-Reactivity analysis of Serotype-Specific Anti-NS1 serological assays for dengue virus using optical modulation biosensing. Biosensors, 15(7), 453. https://doi.org/10.3390/bios15070453
WEEK 2 LECTURE PREP
Questions from Professor Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate of replicative DNA polymerases is roughly 10⁻⁵ errors per nucleotide incorporated. With proofreading (3’→5’ exonuclease activity), this improves to about 10⁻⁷, and after post-replicative mismatch repair, the final error rate drops to approximately 10⁻⁹ to 10⁻¹⁰ per base per replication cycle.
Comparing that to the human genome, which is about 3 × 10⁹ base pairs per haploid. If replication occurred at 10⁻⁵ error frequency with no correction, that would mean tens of thousands of mutations per cell division, incompatible with genomic stability. Even at 10⁻⁷, you would expect hundreds of mutations per division.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
An average 400 amino acids long. Because of the degeneration of the genetic code, most amino acids are specified by multiple synonymous codons.
In practice, most of these sequences do not function equivalently because:
1. Codon usage bias
2. mRNA secondary structure
3. GC content constraints
4. Regulatory elements within coding regions
Questions from Dr. LeProust
What’s the most commonly used method for oligo synthesis currently?
The most common method for oligo synthesis is solid-phase phosphoramidite chemistry. Nucleotides are added stepwise in the 3’→5’ direction on a solid support, with cyclic coupling, capping, oxidation, and deprotection steps.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Because each coupling step is not 100% efficient. The yield drops with length. At 99% efficiency per step, a 200-mer has only a few full-length products. Beyond that, truncated products dominate, and purification becomes inefficient.
Why can’t you make a 2000bp gene via direct oligo synthesis?
Because the cumulative stepwise loss would make the full-length product essentially nonexistent. Long genes are built by assembling shorter oligos not by single continuous chemical synthesis.
Question from George Church
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids in animals are: histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine, and arginine. These cannot be synthesized de novo and must be obtained from the diet.
Respect “Lysine Contingency”:
Lysine is not the only essential amino acid; it is one of several indispensable amino acids. This means that engineering a specific dependence on lysine is conceptually no different from creating dependence on any other essential amino acid. Its practical value does not lie in biochemical uniqueness, but in its controllability: environmental availability can be tightly regulated, making it a useful strategy.
Source: ChatGPT (Open AI)
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Expression of the Reporter Gene LacZ
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements
β-galactosidase hydrolyzes chromogenic substrates, producing a colored product that can be visually detected or quantified spectrophotometrically.
LacZ will be measured using a colorimetric assay with ONPG, enabling both spectrophotometric quantification and visual detection
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail
Colorimetric β-Galactosidase Assay
To quantify the final output of the biosensor—LacZ expression—which reflects the presence of the DENV pathogen
Homework: Waters Part I — Molecular Weight
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight?
The molecular weight of eGFP with His-purification tag and a linker is 5.90 / 28006.60
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Peaks: 933.7848 and 903.7844
Determine z for each adjacent pair of peaks (n,n +1)
z=30,1
Determine the MW of the protein using the relationship: (Peak 1 and Z)
MW=27983,3
Cal culate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1
Accuracy=8,3x10(4)
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
No, the charge state cannot be determined from the enlarged peak, because the peak does not show adjacent peaks or sufficient isotopic resolution to infer the charge
Homework: Waters Part II — Secondary/Tertiary structure
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
Native proteins retain their folded, 3D structure, stabilized by interactions such as hydrogen bonding, hydrophobic interactions, and salt bridges.
Denatured proteins have lost this folded structure due to disruption of these interactions. The protein becomes unfolded or extended.
When a protein unfolds: Lys, Arg, His become exposed, the protein accept more protons during electrospray ionization (ESI) and this leads to higher charge states (larger z values)
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800 M/Z? What is the charge state? How can you tell?
It’s approximately. z=10
Homework: Waters Part III — Peptide Mapping - primary structure
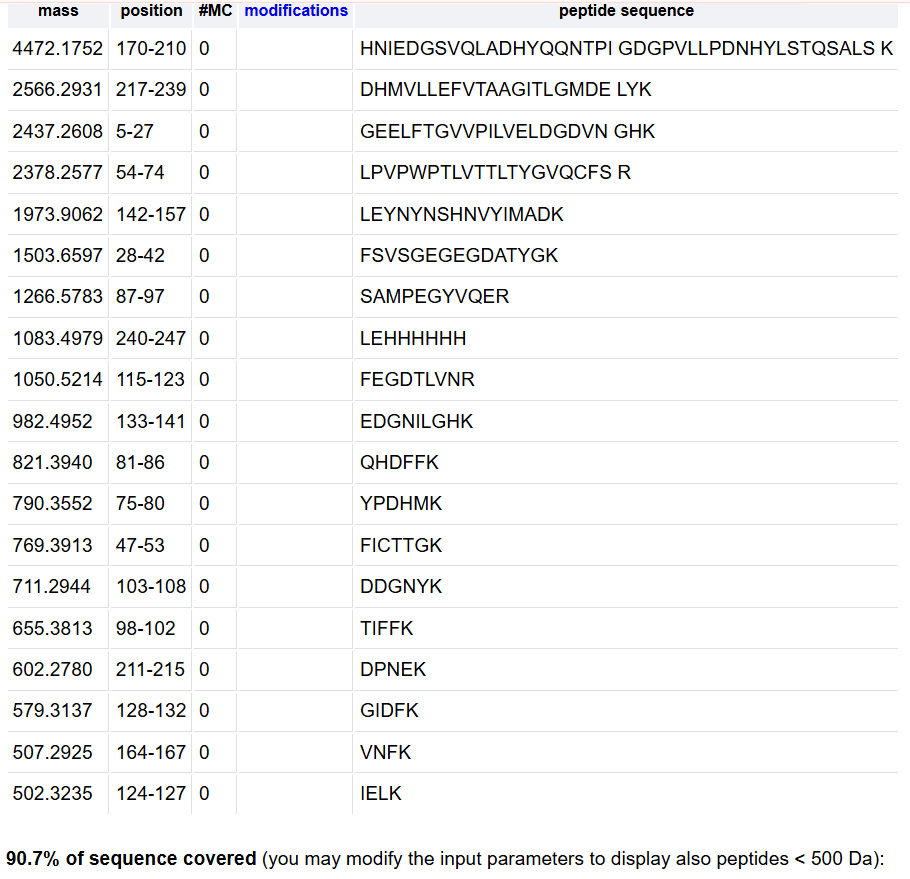
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above.
26
How many peptides will be generated from tryptic digestion of eGFP?
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance
21
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
Not, there are fewer
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state).
The dominant peak cluster is centered at m/z≈525.76
charge state: z=2+
Neutral peptide mass: ≈1049.5Da
Mass of [M + H]+:1050,5 m/z
dentify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement?
Peptide: DLGEEYVQAFK (GFP)
Mt= 1049.53 Da
Mexp= 1049.52 Da
Error= 9.5 ppm
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
88%
Homework: Waters Part IV — Oligomers
Identify where the following oligomeric species are on the spectrum shown below from the CDMS
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Make a note on your HTGAA webpages including:
What you contributed to the community bioart project
I made part of the 2026 on the upper left plate
What you liked about the project, and what about this collaborative art experiment could be made better for next year.
I liked the idea of working with hundreds of people on a single collaborative project that can express the essence of all the participants. What could be improved next year would be to add more colors.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): Supplies a high-yield transcription system via T7 RNA polymerase, and the Star strain reduces RNase activity for increased mRNA stability.
Salts/Buffer
Potassium Glutamate: Maintains ionic strength and mimics the intracellular environment more effectively than chloride salts, enhancing translation efficiency.
HEPES-KOH pH 7.5: Buffers the reaction mixture at an optimal pH for enzymatic activity.
Magnesium Glutamate: Serves as a necessary cofactor for ribosome function, tRNA binding, and RNA polymerase activity.
Potassium phosphate monobasic: Along with its dibasic form, helps maintain pH and supplies phosphate for energy regeneration.
Potassium phosphate dibasic: Works with the monobasic form to buffer the reaction and contribute to the phosphate pool.
Energy / Nucleotide System
Ribose and glucose: Act as energy sources and carbon backbones to fuel metabolic pathways that regenerate ATP and GTP.
AMP, CMP, GMP y UMP: Are converted to the corresponding nucleotide triphosphates (ATP, CTP, GTP, UTP) to serve as substrates for transcription and energy metabolism.
Guanine: Can be salvaged to produce GTP, which is critical for translation initiation and elongation.
Translation Mix (Amino Acids)
17 Amino Acid Mix: Supplies the common amino acids required for protein synthesis; typically excludes tyrosine and cysteine to allow controlled addition
Tyrosine and cysteine: Added separately to prevent chemical modification or precipitation that can occur during long-term storage of the full amino acid mix.
Additives
Nicotinamide: Helps regenerate NAD⁺ and inhibits certain proteases, thereby improving reaction longevity and yield.
Backfill
Nuclease Free Water: Adjusts the final volume of the reaction to achieve the desired concentrations of all components, while ensuring no contaminating nucleases degrade mRNA.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The main difference is the energy and nucleotide supply strategy. The 1-hour uses pre-supplied NTPs for immediate, rapid energy regeneration, enabling fast protein synthesis. In contrast, the 20-hour uses simple precursors that are metabolically converted into NTPs over time, which supports sustained, long-term protein production.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP: Robust and rapid folding even without chaperone assistance, however, its chromophore maturation still requires molecular oxygen, meaning anaerobic or oxygen-depleted CFPS reactions will show reduced fluorescence despite proper translation. sfGFP’s high resistance to aggregation makes it ideal for CFPS, but its sensitivity to acidic pH (below ~6.5) can quench fluorescence, requiring careful buffer maintenance during extended incubations
mRFP1: Slow maturation time due to its obligate requirement for chromophore oxidation and dehydration, which limits its utility in short reactions. Exhibits some sensitivity to oligomerization at high concentrations, lacking membrane compartments, this can lead to solubility issues or altered spectral properties compared to monomeric red FPs.
mKO2: Rapid maturation rate due to its efficient chromophore formation. Like all GFP-derived fluorescent proteins, mKO2 requires molecular oxygen for chromophore oxidation, meaning that oxygen depletion in extended cell-free reactions can limit final fluorescence yield despite active protein synthesis.
mTurquoise2: High quantum yield and efficient maturation, but its chromophore is sensitive to acidic pH (pKa ~5.1), meaning that any drop in pH during extended cell-free reactions can quench fluorescence readout. Additionally, it requires proper oxidation of its chromophore, making it dependent on adequate oxygen levels in the reaction mix for full fluorescent signal development.
mScarlet_I: Accelerated maturation, which is beneficial for the systems because it allows rapid fluorescence development within short reaction times. Its red chromophore requires precise oxidative maturation conditions, and any oxygen limitation or redox imbalance in the lysate can reduce final fluorescence yield. but, it exhibits excellent monomeric behavior and pH stability, making it less sensitive to mild acidification.
Electra2: High photostability, but its chromophore maturation is relatively slow and oxygen-dependent, requiring sufficient dissolved oxygen in the reaction for complete fluorescent signal development. It has lower quantum yields and are more prone to acid sensitivity, meaning that pH drops during extended incubations
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Limitation: Its chromophore maturation requires molecular oxygen, and extended 36-hour incubations lead to oxygen depletion and pH acidification, which can quench fluorescence even if the protein remains folded.
Adjust:
Long-term ATP regeneration and buffer against acidification:
Glucose increase from 6.9 mM to 15–20 mM
Ribose increase from 77.4 mM to 100 mM
HEPES-KOH pH 7.5 increase from 45 mM to 80 mM
Protect the oxidizing chromophore from peroxide damage:
Catalase add 100 U/mL
‘‘This combined adjustment is expected to yield stable sfGFP fluorescence for up to 36 hours, with at 1.5–2× higher endpoint signal compared to the standard 20-hour master mix, by preventing both energy collapse and oxidative stress during prolonged incubation’’
Mail not received
Unable to do without data
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
Because several codons encode the same amino acid, but the frequency at which these codons are used varies among organisms. Each species has preferences for particular codons, a phenomenon known as codon usage bias.
When expressing a gene from one organism in a different host without prior codon optimization, translation efficiency can be reduced, leading to lower protein production or even affecting proper protein folding.
Which organism have I chosen to optimize the codon sequence for, and why?
I chose to optimize the sequence for Escherichia coli because it is one of the most widely used systems for recombinant protein production. It is easy to cultivate, grows quickly, and is cost‑effective. In addition, there are well‑established genetic tools that enable efficient protein expression when codons are adapted to its natural codon bias, and it is the organism I am most familiar with.
You have a sequence! Now what?:
What technologies could be used to produce this protein from your DNA?
Recombinant gene expression in bacteria, yeasts, or cell‑free systems
Cell-dependent transcription and translation
The gene is inserted into a plasmid and then introduced into a host organism.
The process begins with cloning, where the codon‑optimized gene is inserted into a plasmid. During transformation or transfection, the plasmid is delivered into the host cell. Inside the cell, the host RNA polymerase recognizes the promoter and transcribes the DNA into mRNA. This mRNA is then read by ribosomes, which synthesize the protein by assembling amino acids according to the codon sequence. Finally, the newly synthesized protein folds and, if necessary, undergoes further modifications.
MBL for be used as an initial viral capture molecule due to its affinity for high-mannose glycans present on the viral envelope protein.
What technology or technologies would you use to perform sequencing on your DNA and why?
To sequence DNA, I would use Illumina sequencing for its high accuracy and cost-effectiveness, since my idea is targeted gene analysis and Illumina technology provides low error rates and high throughput, making it ideal for detecting deletions.
Is your method first-, second or third-generation or other? How so?
Second generation, because it performs massive sequencing in parallel, use clonal amplification (bridge amplification) before reading and is necessary perform the amplification before sequencing
What is your input? How do you prepare your input? List the essential steps.
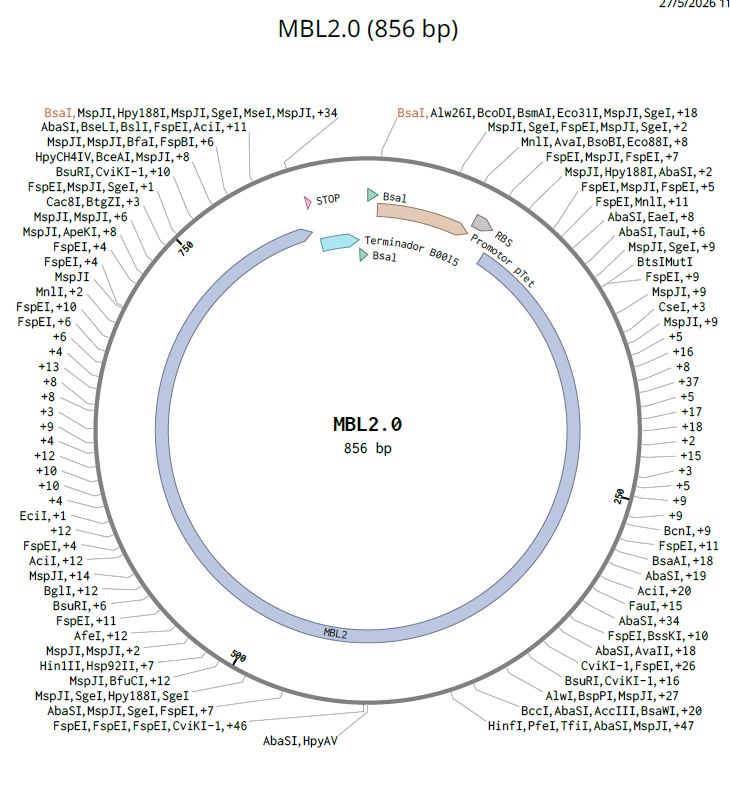
Input: MBL2
Preparation: Using linear synthetic DNA MBL2 sequence
End repair.
A-tailing.
Adapter ligation for Illumina-compatible adapters
PCR enrichment of adapter-ligated fragments.
Cleanup and quantification.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample?
Essential steps:
Flow Cell Binding
MBL2 gene fragments with adapters are loaded onto the flow cell.
The fragments hybridize to complementary oligonucleotides immobilized on the surface.
Bridge Amplification
Each fragment is locally amplified, forming clonal clusters, generating many identical copies of each fragment, increasing the detectable signal during sequencing.
Sequencing by Synthesis
Fluorescently labeled nucleotides with reversible terminators.
In each cycle:
• A labeled nucleotide (A, T, C, or G) is added.
• Only one nucleotide is incorporated per cycle.
• A camera detects the emitted fluorescence.
• The system records the corresponding color.
• The fluorophore and chemical blocking group are then removed.
• The cycle is repeated.
Base calling performed through:
Optical detection of the fluorescent signal in each cluster.
Each color corresponds to a specific base (A, T, C, or G), the software converts the light signal into a nucleotide sequence, and a quality score is assigned to each base.
What is the output of your chosen sequencing technology?
Millions of short reads. Generated in FASTQ format files, with the nucleotide sequence of each read and a quality score assigned to every base.
WRITE
What DNA would you want to synthesize (e.g., write) and why?
would synthesize a codon-optimized version of the MBL2 coding sequence (CDS) to produce recombinant MBL protein for use as a viral capture molecule in a diagnostic assay.
What technology or technologies would you use to perform this DNA synthesis and why?
Array-based or column-based phosphoramidite DNA synthesis. Because it is a method with high sequence fidelity and control over sequence design
What are the essential steps of your chosen sequencing methods?
a) Solid-phase attachment
b) Deprotection
c) Coupling
d) Capping
e) Oxidation
f) Cleavage and deprotection
g) Sequence verification
What are the limitations of your sequencing method in terms of speed, accuracy, scalability?
In terms of accuracy, each coupling step is not 100% efficient, so errors can accumulate as the sequence length increases. Regarding length, synthesis is limited to about 150–200 base pairs per oligonucleotide, meaning longer genes must be assembled from multiple overlapping fragments. In terms of speed, synthesis is relatively fast for short oligos, but full gene synthesis requires extra assembly and validation steps,increasing turnaround time. Concerning scalability, array-the individual yield per oligo may be lower compared to column-based synthesis. Finally, cost increases with more complex constructs due to the need for assembly and error correction.
EDIT
What DNA would you want to edit and why?
MBL2 gene, which encodes Mannose-binding lectin, to enhance its expression in individuals with naturally low serum MBL levels. Increased MBL expression could potentially improve early immune recognition of viral pathogens
What kinds of edits might you want to make to DNA? Why?
Precise regulatory modification rather than altering the protein-coding sequence. Introducing promoter variants associated with higher transcriptional activity could increase protein levels without changing protein structure, minimizing unintended functional consequences while enhancing host defense.
What technology or technologies would you use to perform these DNA edits and why?
CRISPR-Cas9 with Homology-Directed Repair, because the precise control over the type of genetic modification and flexibility depending on whether the goal is a single-base change or a larger regulatory insertion.
How does your technology of choice edit DNA? What are the essential steps?
Using a guide RNA (gRNA) to direct the Cas9 nuclease to a specific genomic sequence.
Target recognition
DNA cleavage
DNA repair
What preparation do you need to do and what is the input for the editing?
Design a specific guide RNA targeting for MBL2, verifying minimal off-target sites using bioinformatic tools.
Inputs: Cas9 protein, Guide RNA, delivery system and target cell
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Efficiency:
HDR efficiency is often low in non-dividing cells, and editing rates depend on the cell type and genomic context.
Precision:
Potential off-target edits if the guide RNA is not highly specific and NHEJ repair may introduce unintended insertions or deletions.
Week 2 HW: Lab Automation
Opentrons Artwork
opentrons-art.rcdonovan.com/?id=oevp91e27i3m061
Post-Lab Questions
Find and describe a published paper that utilizes the Opentrons
This article combines an open‑source liquid‑handling robot (Opentrons OT‑One‑S Hood) with four interchangeable modules that perform magnetic‑bead DNA isolation, isothermal recombinase polymerase amplification (RPA) of the ctrA gene, exonuclease digestion to generate single‑stranded DNA, and detection on a paper‑based vertical‑flow microarray (VFM) using anti‑biotin gold nanoparticles for colorimetric read‑out.
Magnetic Dynabeads capture pathogen DNA from cerebrospinal fluid or water samples, after which the beads are washed and the DNA is released into the amplification mix 3. The RPA reaction runs at 37 °C, using a biotin‑labelled forward primer and a 5′‑phosphate reverse primer; the resulting double‑stranded amplicons are digested by Lambda exonuclease, which removes the phosphorylated strand and leaves a biotin‑tagged single strand for hybridisation. The single‑stranded amplicons are applied to nitrocellulose VFM spots that contain capture probes for ctrA; anti‑biotin gold nanoparticles bind the biotin tag, and a signal‑enhancement solution produces a visible colour change at positive spots.
ecause all steps are scripted on the Opentrons platform, the robot performs liquid handling automatically, reducing hands‑on time to 110 min for eight samples—about 18 % faster than manual processing—and lowering consumable cost to roughly USD 16 per sample. The open‑source hardware and standard lab consumables make the system adaptable to other pathogens and suitable for deployment in low‑resource settings.
Write a description about what you intend to do with automation tools for your final project
For my final project, I intend to develop a low-cost diagnostic test for the detection of Dengue virus (DENV), optimized for use in low-resource settings. The system would integrate:
A lateral-flow or paper-based detection platform
Automation in the creation and production of the test
The goal is to reduce human error, increase reproducibility, and lower production costs while maintaining diagnostic sensitivity and specificity.
Final Project Ideas
Week 4: Protein Design Part I
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Considering that 500 g of meat contains 20% protein, we would have 100 g of protein. After converting the protein mass to moles (assuming an average amino acid mass of 100 Da), this corresponds to approximately 6.022 × 10²³ amino acid molecules.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because these proteins are broken down during digestion, reducing them to amino acids and peptides that are reused to synthesize our own proteins according to our genetic instructions.
Why are there only 20 natural amino acids?
There are 20 natural amino acids because early in evolution the genetic code incorporated a set of amino acids that provided sufficient chemical diversity to build functional proteins, and once this translation system (involving tRNAs and aminoacyl-tRNA synthetases) became established, it was evolutionarily conserved and effectively “frozen,” making the addition of new amino acids highly disadvantageous.
Can you make other non-natural amino acids? Design some new amino acids.
Yes, chemically it is possible to design new amino acids, but incorporating them biologically into proteins is difficult because they require their own tRNA, a matching aminoacyl-tRNA synthetase, and a reassigned codon.
Pendiente hacer aminoacido
Where did amino acids come from before enzymes that make them, and before life started?
Before life began, amino acids likely formed through abiotic chemical reactions driven by energy sources such as lightning, UV radiation, volcanic activity, hydrothermal systems, and possibly delivery from meteorites in a primitive earth, as demonstrated by experiments like Miller.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left-handed
Can you discover additional helices in proteins?
Yes, additional helices are physically possible and we can design new one modifying amino acid chemistry or backbone structure.
Why are most molecular helices right-handed?
Most molecular helices are right-handed because the chiral building blocks of life—such as L-amino acids and D-sugars—constrain backbone geometry in a way that makes right-handed helices energetically more stable and sterically favorable.
Why do β-sheets tend to aggregate?
Because their extended structure exposes backbone hydrogen bond donors and acceptors, allowing β-strands from different molecules to form stable intermolecular hydrogen bonds.
What is the driving force for β-sheet aggregation?
The main driving force is the formation of intermolecular backbone hydrogen bonds, reinforced by hydrophobic interactions that stabilize the aggregate and lower the system’s free energy.
Part B. Protein Analysis and Visualization
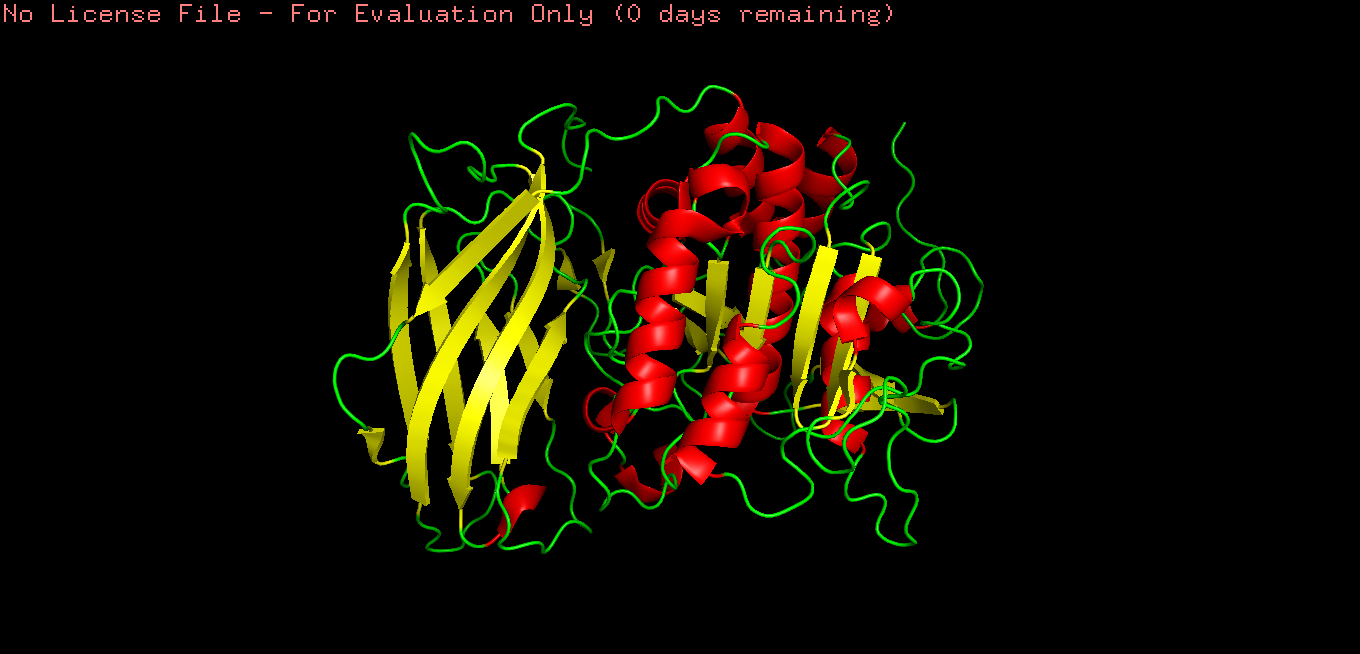
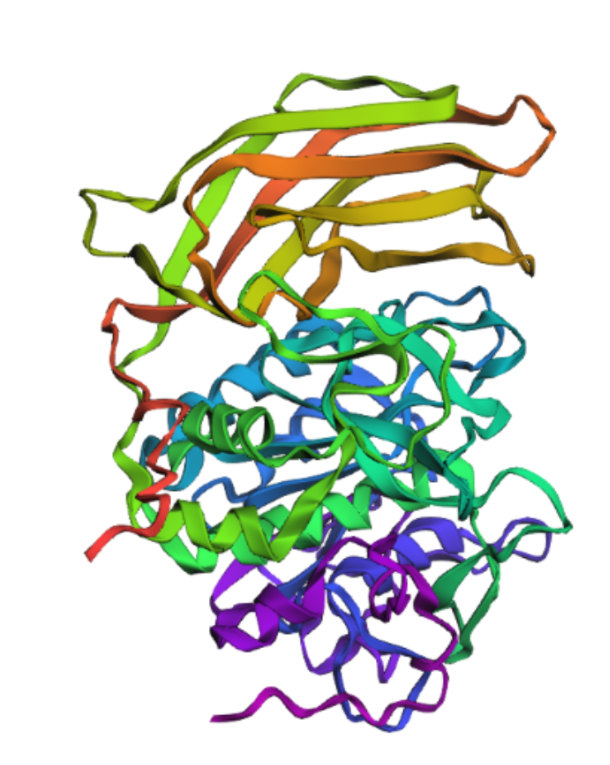
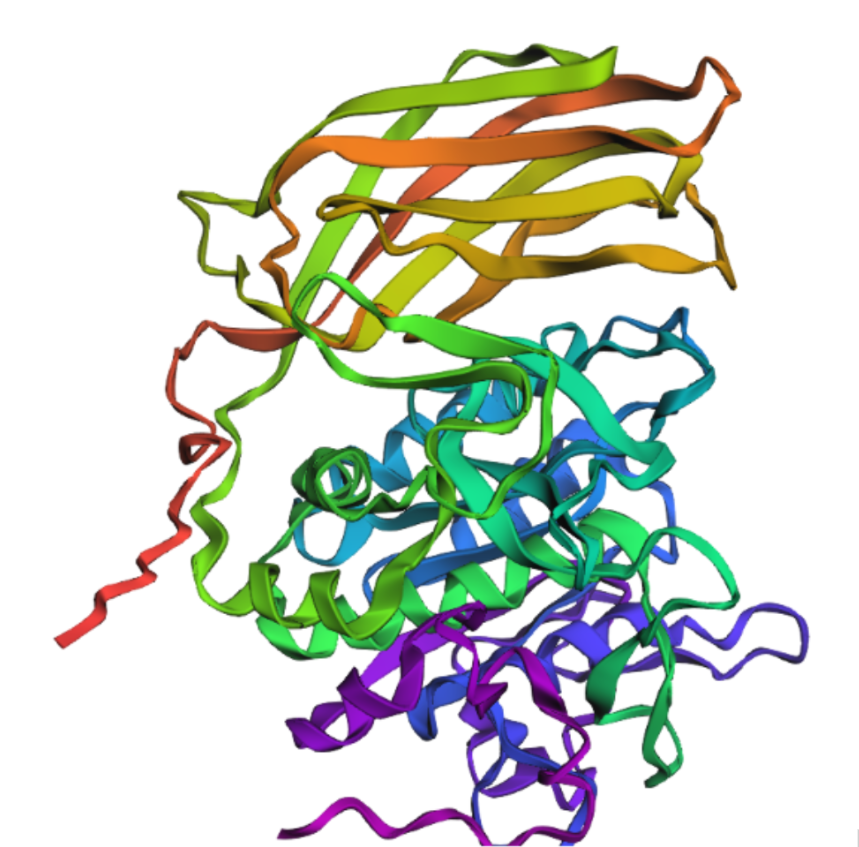
Briefly describe the protein you selected and why you selected it.
Furin is a calcium-dependent protease hat activates precursor proteins by cleaving them at specific basic amino acid recognition sequences in the Golgi apparatus and I selected it because it’s a protein I am currently interested in.
How long is it? What is the most frequent amino acid?
482, The most common amino acid is: G, which appears 47 times
How many protein sequence homologs are there for your protein?
230
Does your protein belong to any protein family?
Proprotein convertase
Identify the structure page of your protein in RCSB
5JXG | pdb_00005jxg
When was the structure solved? Is it a good quality structure?
2016 with 1.80 Å
Are there any other molecules in the solved structure apart from protein?
Yes, Ca+2, Cl- and Na+
Does your protein belong to any structure classification family?
Serine endoprotease, hydrolase
Open the structure of your protein in any 3D molecule visualization software
Cartoon
Ribbon
Ball and stick
Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by residue type.
What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Hydrophobic residues are mainly buried in the protein core, while hydrophilic residues are predominantly exposed on the surface, consistent with typical folding of a soluble globular protein.
C1. Protein Language Modeling
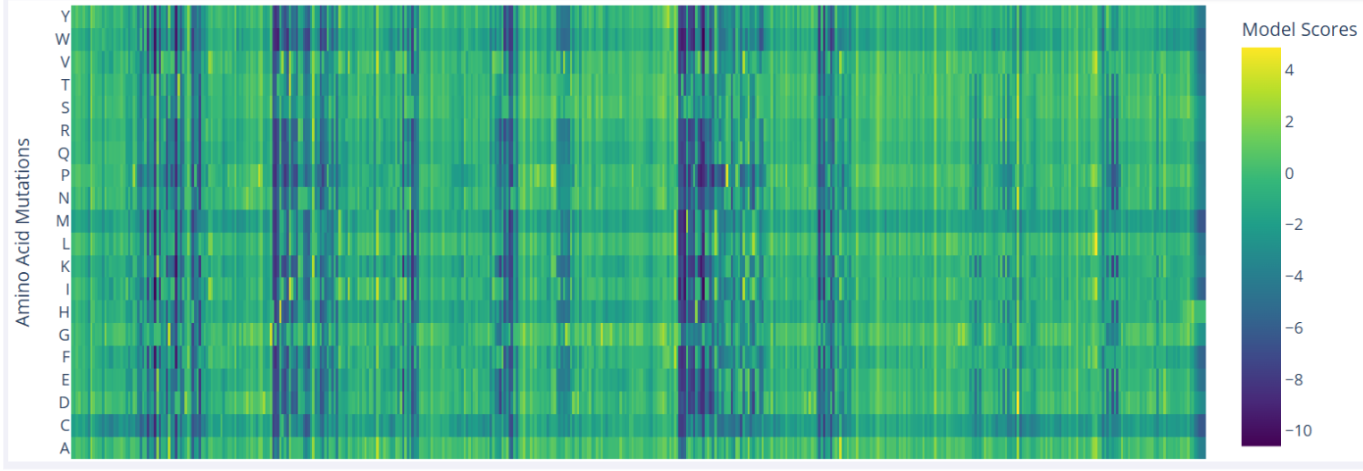
Deep Mutational Scans
In the mutational scan of furin, one clear pattern is the presence of vertical dark bands that reflect unfavorable mutations at specific positions, which likely correspond to critical residues. For example, Ser368 is essential in the serine protease catalytic triad, and nearly all substitutions at this position are predicted to be highly deleterious.
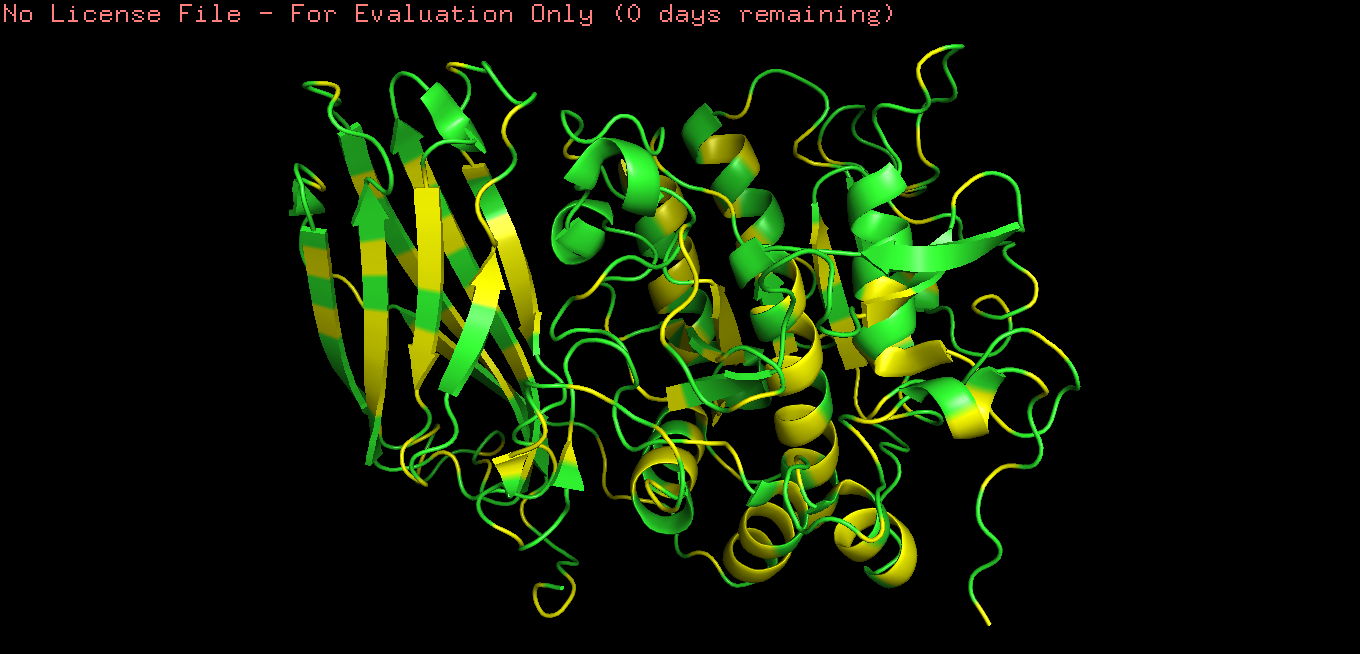
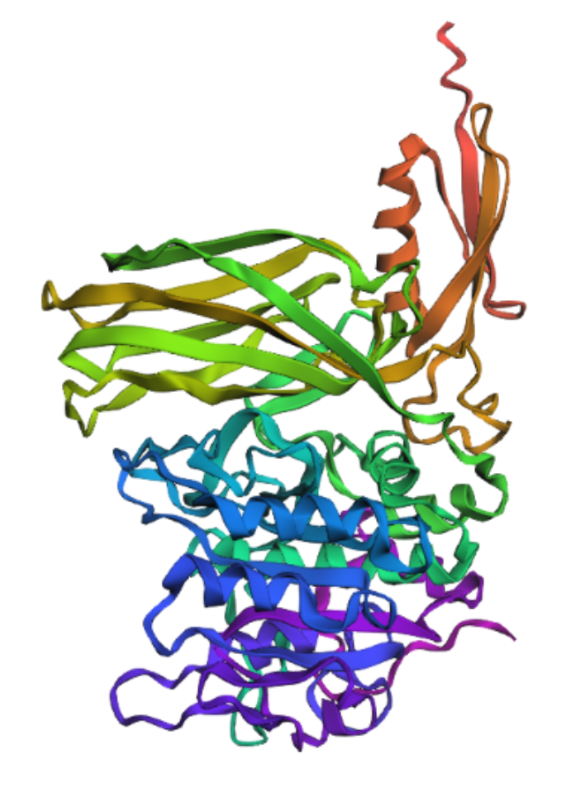
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Yes, It´s very similar.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
I mutated the amino acids in the active site of furin to render the protein inactive; specifically, I introduced the substitutions Ser → Ala (S368A), His → Ala (H194A), and Asp → Asn (D153N).
It’s almost the same structure, but now I changed the p-domain, the majority of the protein still remains intact mading this one resilient to mutations
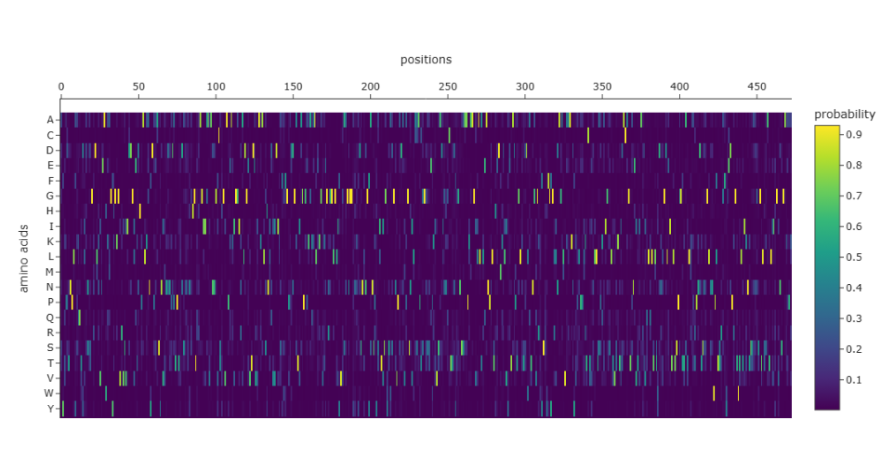
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
The heatmap shows that many positions in the sequence strongly favor a single amino acid, while alternative residues have very low probabilities. Suggesting that these positions are highly constrained, likely due to structural or functional requirements of the protein. Only a limited number of positions show broader amino acid tolerance, indicating potential sites where mutations might be more acceptable. This pattern is consistent with proteins that contain structurally important or catalytic regions, where mutations are less tolerated.
D. Group Brainstorm on Bacteriophage Engineering
GROUP FINAL PROJECT
Week 5 HW: Genetic circuits part 1
Assignment: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity DNA polymerase: enzyme responsible for synthesizing new DNA strands while possessing activity, which reduces errors during replication.
dNTPs (deoxynucleotide triphosphates): Nucleotide substrates incorporated by DNA polymerase into the elongating DNA strand during synthesis
reaction buffer: contain compounds as Tris-HCl that maintains the correct pH and salts like KCl which help stabilize primer binding and enzyme activity
MgCl₂: Essential cofactor required for DNA polymerase catalytic activity.
What are some factors that determine primer annealing temperature during PCR?
Tm Range: reflects the temperature at which half of the primer–template duplex dissociates, it depends largely on the primer nucleotide composition, particularly the GC content.
Primer length: As longer primers higher melting temperatures due to more base-pair interactions with the template.
Secondary structures: May require adjustment of the annealing temperature.
Reaction conditions: can alter primer–template stability and thus influence the optimal annealing temperature.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR and restriction digests serve different purposes: PCR amplifies a specific DNA fragment using primers and an enzyme like Taq polymerase, making it ideal when you need a lot of a precise sequence or start with very little DNA. In contrast, restriction enzymes such as EcoRI cut DNA at specific sites, which is useful for cloning or checking constructs. So basically, PCR = amplify, restriction digest = cut, and they’re often used together.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Primers should be designed to add some bp of overlapping homologous sequences between the insert and the vector. After PCR or digestion, fragments are verified by gel electrophoresis to confirm the correct size and purity, and the overlap regions should be checked in silico, for example by calculating the predicted digest in Benchling to verify the expected band sizes.
How does the plasmid DNA enter the E. coli cells during transformation?
Heat shock: Generate pores in bacterial cell wall with an abrupt temperature change
Electroporation: Generate pores in bacterial cell wall with high electrical voltage
In both methods the cells are shocked causing the cell membrane to “open up”
Describe another assembly method in detail
Golden Gate Assembly
Allows the assembly of multiple DNA fragments in a single reaction, using a Type IIS restriction enzyme, which cuts DNA outside of its recognition sequence to create specific overhangs. DNA fragments and the vector are designed so that these overhangs are complementary, ensuring that the fragments assemble in the correct order. During the reaction, the restriction enzyme cuts the DNA while T4 DNA Ligase simultaneously ligates the compatible ends. Because the restriction sites are removed after ligation, the final assembled plasmid cannot be cut again, which improves assembly efficiency. This method is widely used for assembling multiple fragments quickly and accurately in a one-pot reaction.
Assignment: Asimov Kernel
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
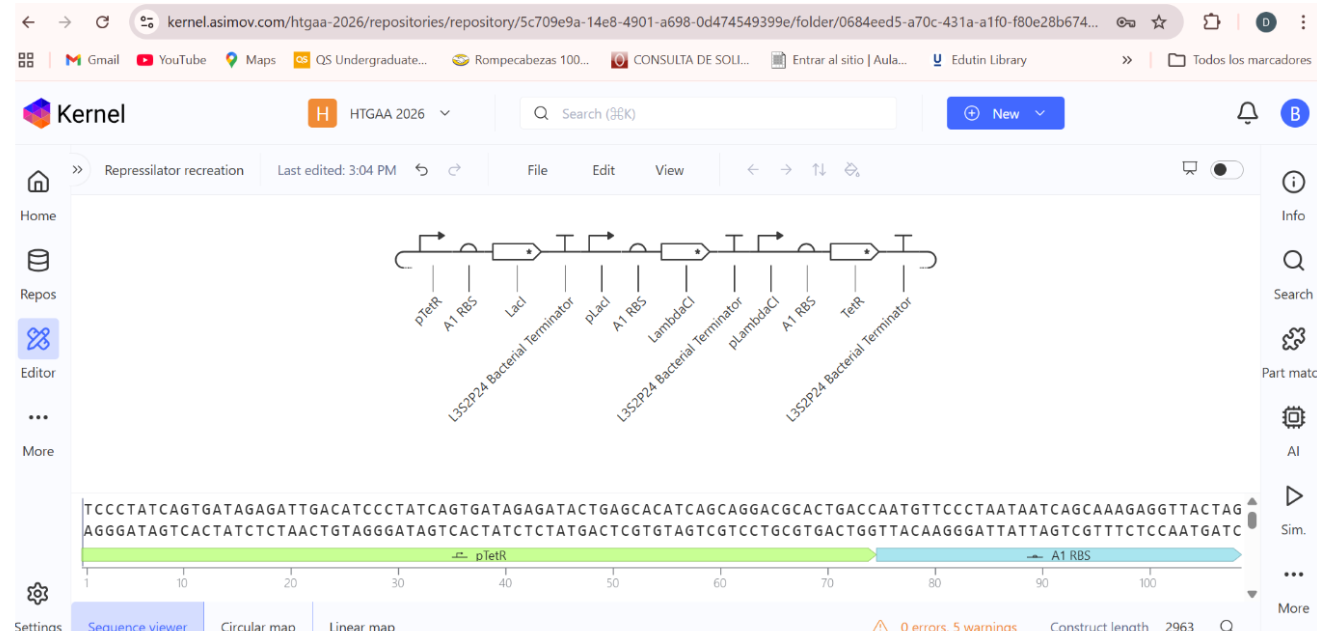
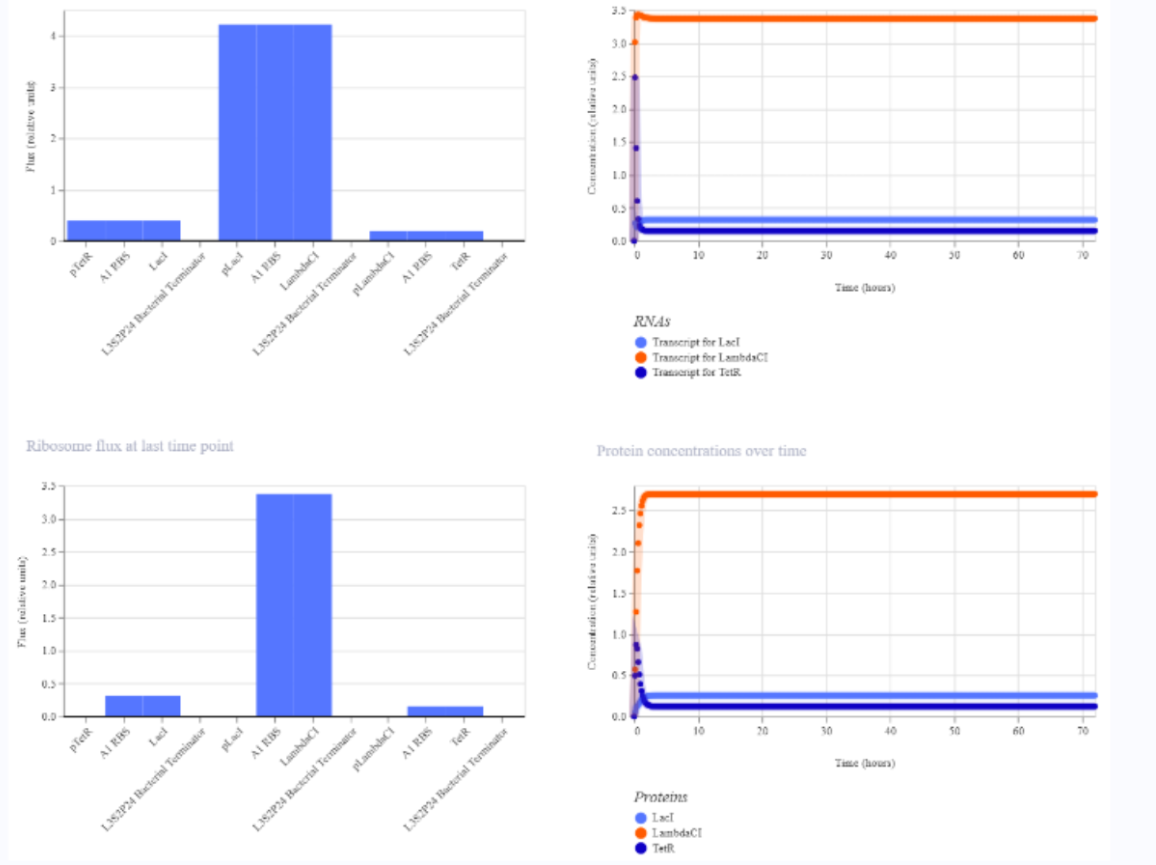
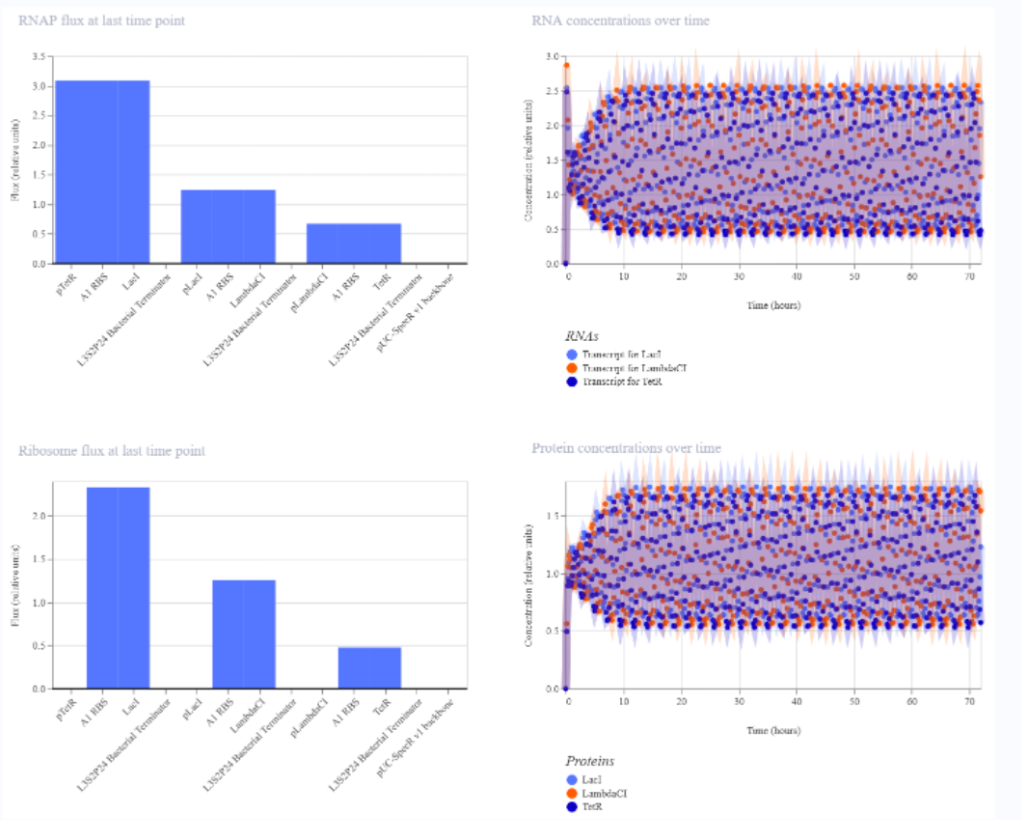
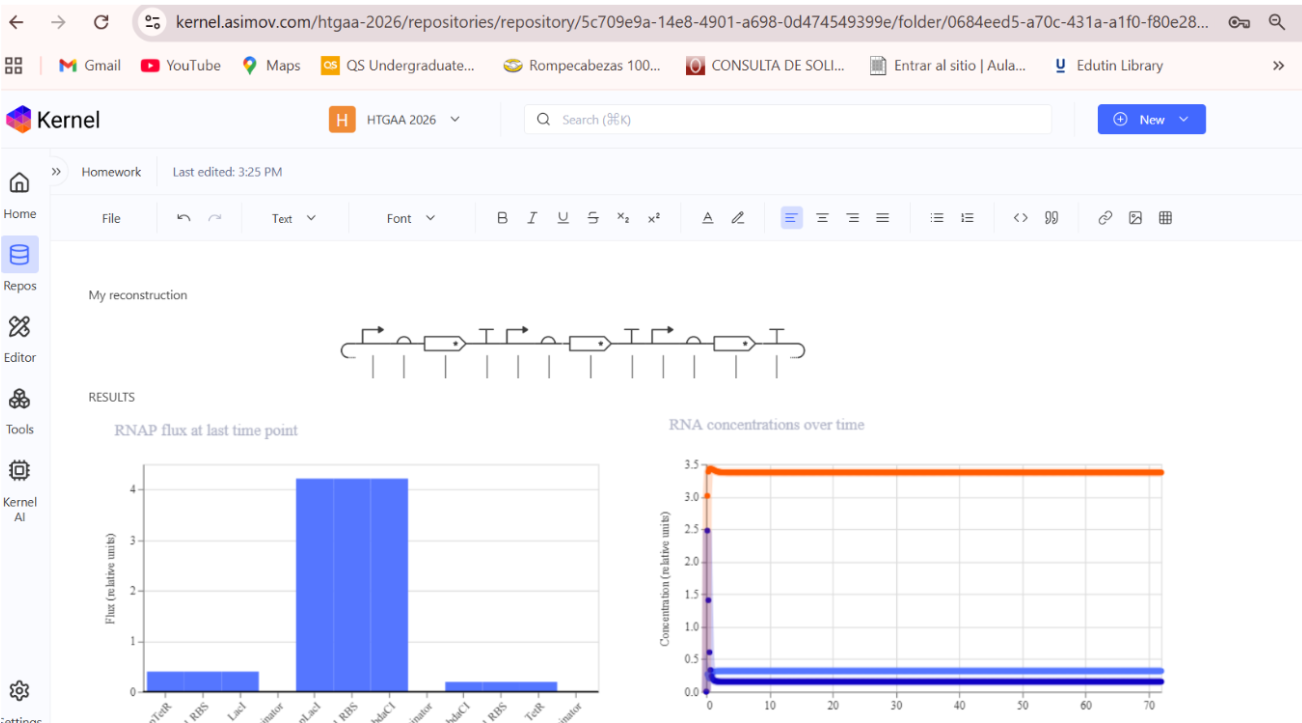
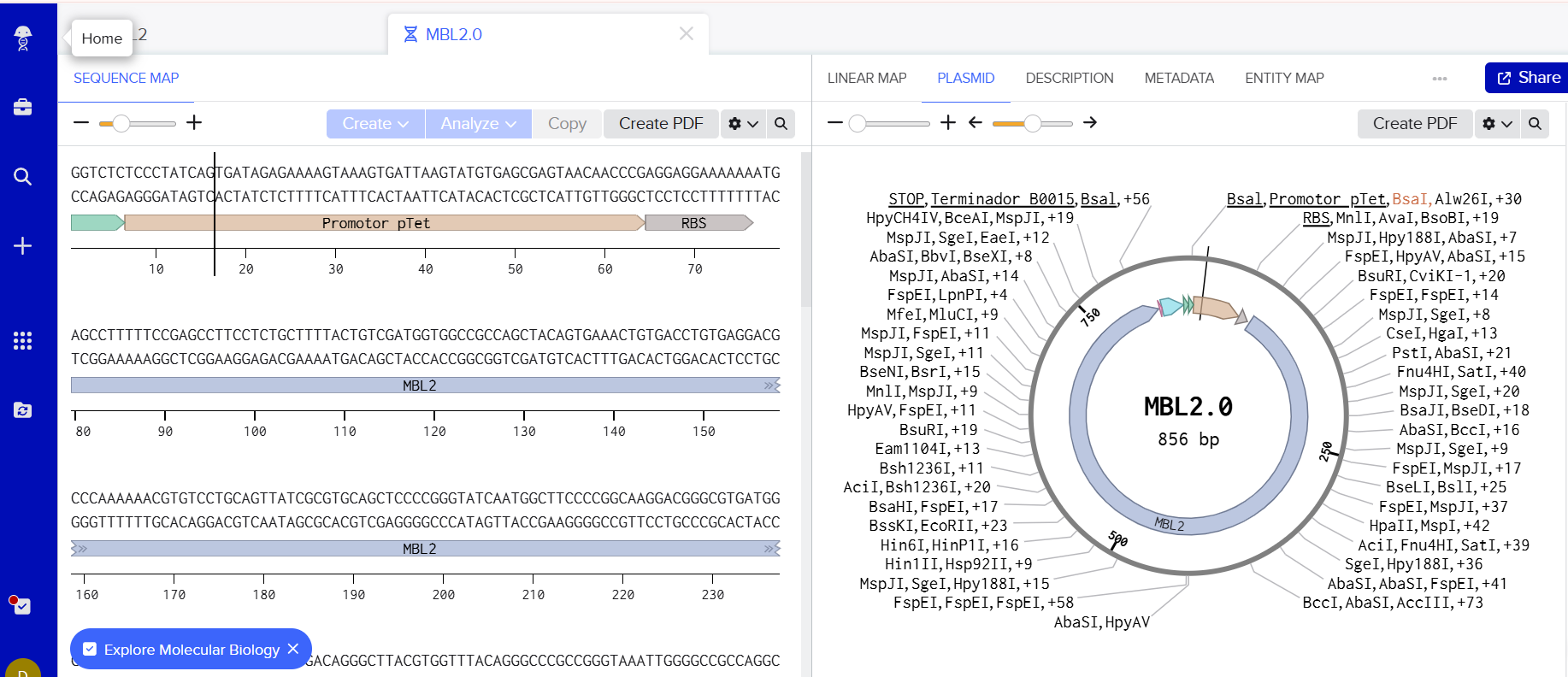
a) A dengue-responsive genetic sensor based on a derepression mechanism. An inducible promoter continuously drives the expression of the repressor TetR, which binds to the Tet-regulated promoter (PTet) and blocks transcription of the reporter gene (sfGFP) under normal conditions, keeping fluorescence at basal levels. When dengue is present, its biomarker (E), represented as an inducible promotor, is recognized by an aptamer that functionally inhibits TetR. This inhibition prevents TetR from binding to PTet, thereby relieving repression and allowing transcription and translation of sfGFP. As a result, GFP fluorescence increases in the presence of NS1, meaning the construct converts dengue detection into a measurable fluorescent output, with low signal in the absence of the ligand and high signal when it is present.
b) Three expression cassettes assembled in a single construct. Together, these cassettes implement a threshold-detection circuit that produces GFP only when the dengue-derived ligand is present above a sufficient concentration.
The circuit integrates two constitutively expressed repressors (TetR and LacI) and a dual-repressed reporter cassette, allowing the system to behave as a biological logical gate sensitive to ligand concentration. Correct functionality relies on specifically chosen promoters for each cassette, as the use of pLtetO‑1 in Cassette 2.
C) Genetic construct designed as a ligand-responsive transcriptional amplifier based on TetR–pTet regulation. The system operates through double negation and feedback, such that an external ligand indirectly activates gene expression by inhibiting a transcriptional repressor.
Final project
Week 5 HW: Protein design part 2
Part A: SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
After introducing the A4V mutation. I performed the mutation A5V based on its position in the FASTA sequence.
Using PepMLM Colab with a K value of 1, I obtained:
WRYYAVAAAHKX 8.149419
WRYYAVAAAHKX 8.149419
WRYYAVAAAHKX 8.149419
WRYYAVAAAHKX 8.149419
In comparison with SOD1-binding peptide:
FLYRWLPSRRGG with a perplexity of 5.98
The generated peptides showed perplexity scores around 8, which are higher than the example. This indicates that the model assigns lower confidence to these sequences as potential binders to the mutant SOD1 protein.
Part 2: Evaluate Binders with AlphaFold3
Due to the fact that AlphaFold does not allow the analysis of proteins with undetermined amino acids, I ran the PepMLM Colab again using a K value of 2 so that all amino acids would be fully specified.
WRYYAAGVEHKE 15.984515
WRYYVVAAAHGX 11.686807
WRYYAAGAALKX 6.211397
WRYYVVAAALKE 15.368100
And with 3:
WHYPAVGAALWE 12.713875
WRYYAVVLAHKX 10.328839
WRYYVVALAHKE 16.276417
HLYYVVGVRWKE 27.442868
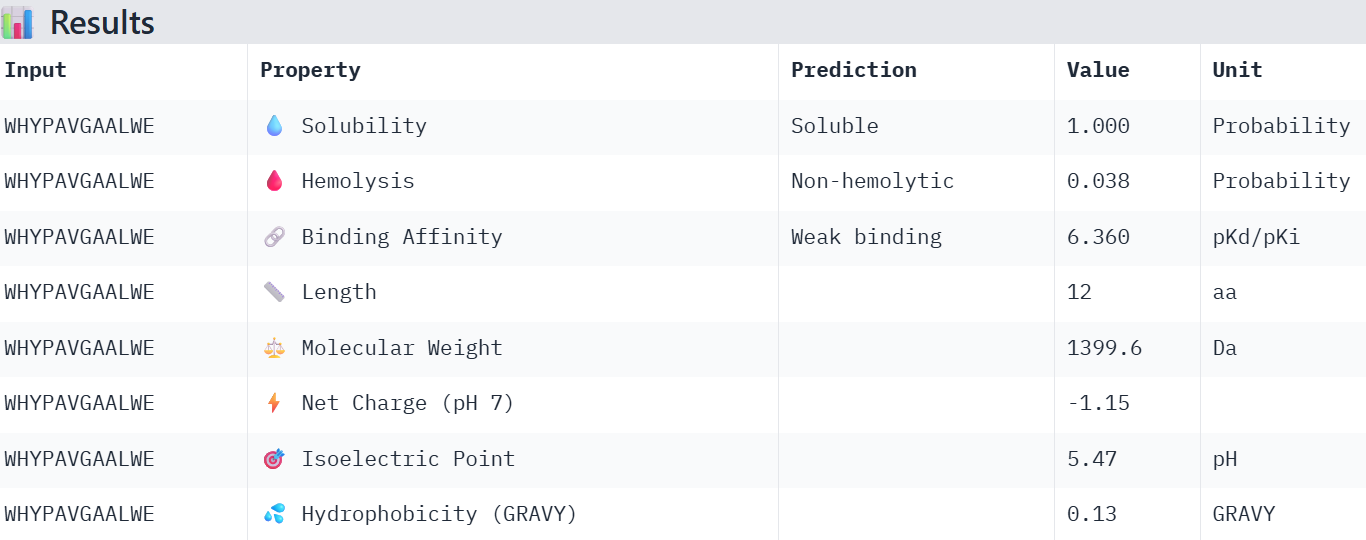
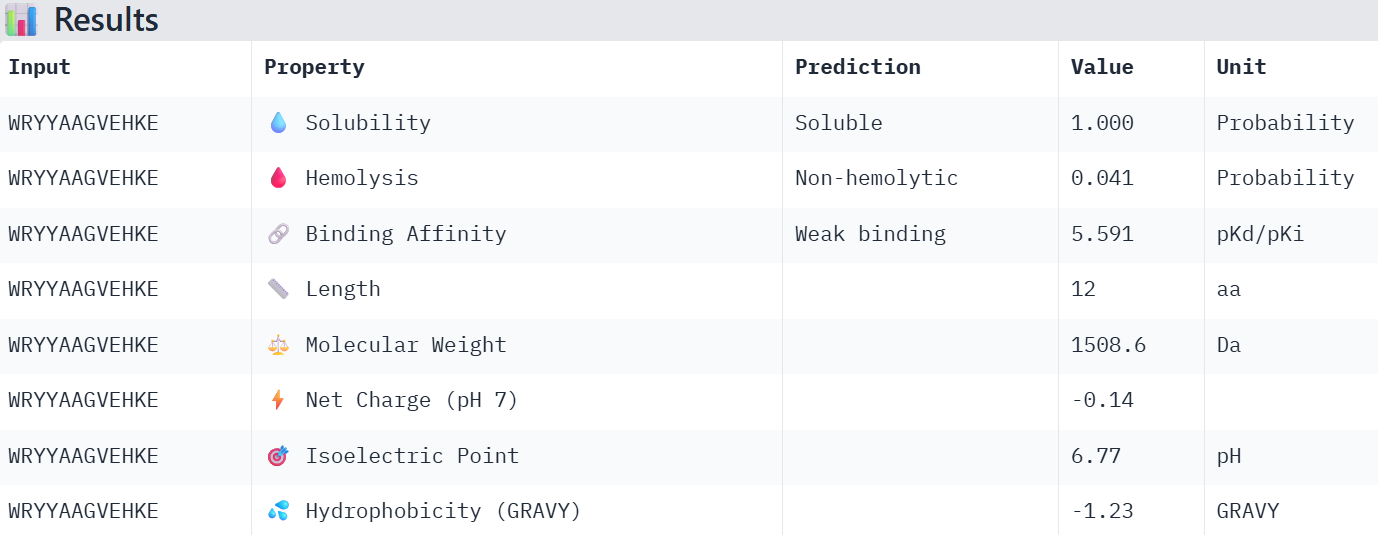
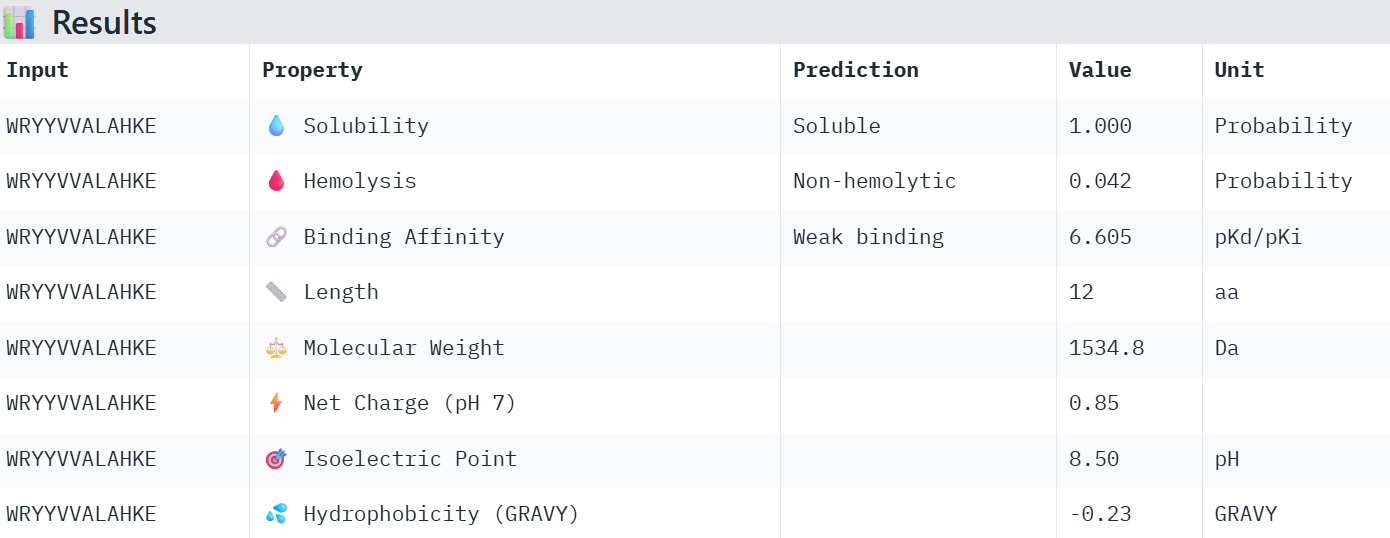
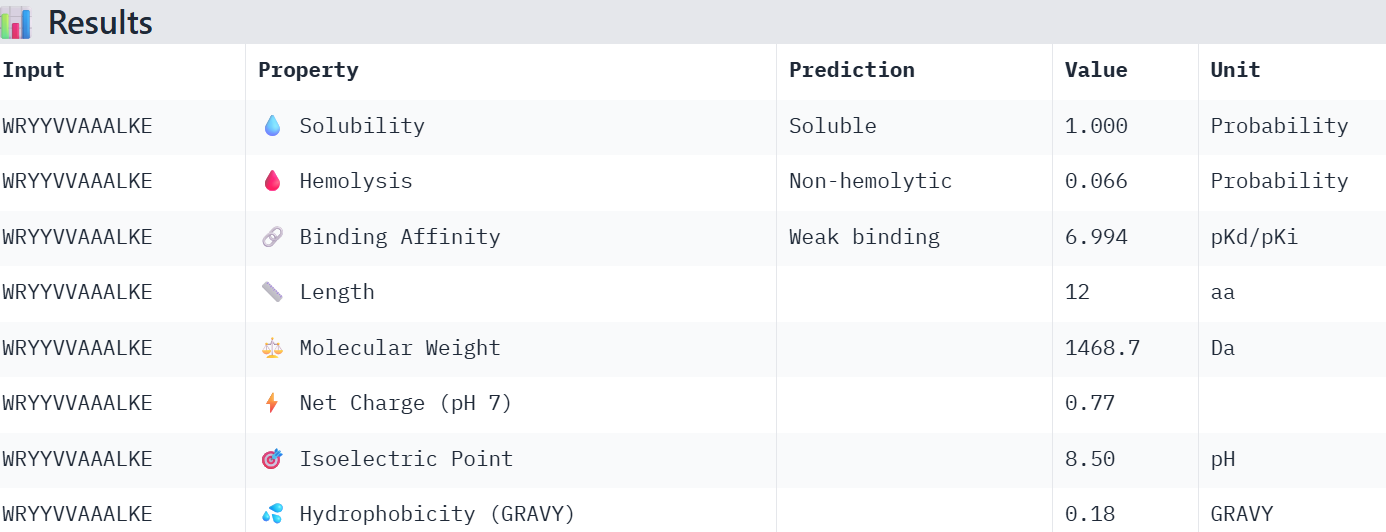
So I chose the four with the lowest perplexity value:
WHYPAVGAALWE 12.713875
WRYYAAGVEHKE 15.984515
WRYYVVALAHKE 16.276417
WRYYVVAAALKE 15.368100
For the first peptide: 12.7
The peptide binds along the outer surface of the SOD1 structure, lying across loops adjacent to the β-barrel rather than inserting into the core of the protein. It does not localize directly near the N-terminal region where the A4V mutation is located, nor does it clearly approach the predicted dimer interface.
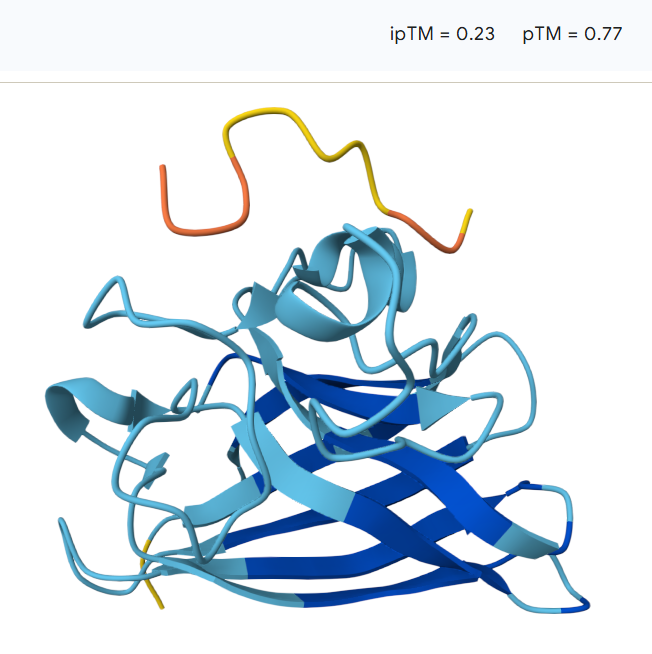
For the second peptide:15.9
The peptide does not localize directly near the N-terminal region where the A4V mutation resides, instead, the peptide lies primarily on the protein surface, with a portion potentially partially accommodated within a shallow surface groove. The model shows an ipTM value of 0.45 and a pTM of 0.88, indicating moderate confidence in the protein–peptide interaction while maintaining high confidence in the overall protein structure.
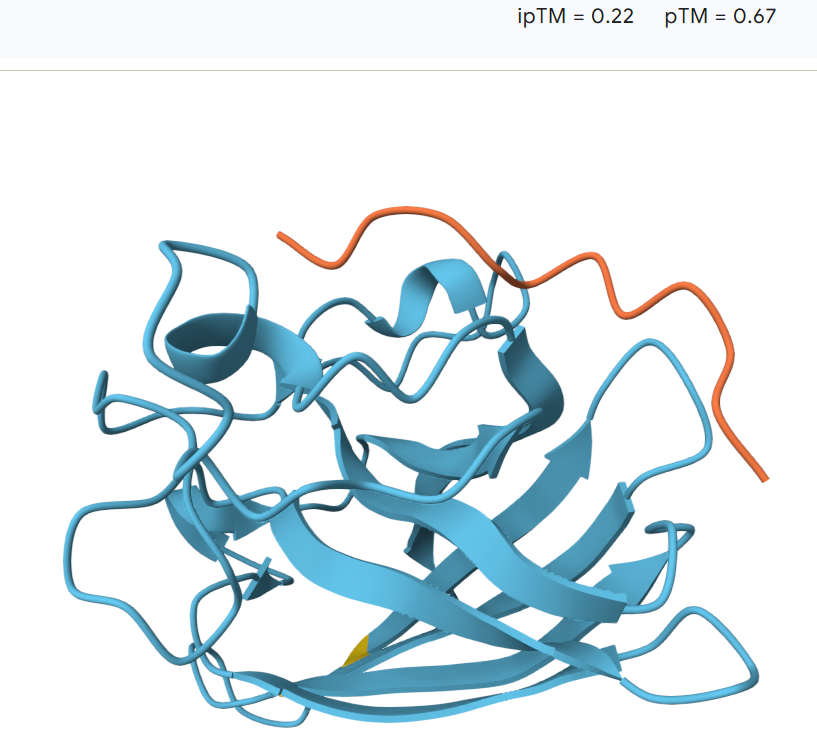
For the third peptide:16.2
The peptide remains largely separated from the protein and appears flexible and surface-exposed, suggesting a lack of stable binding. The model shows an ipTM value of 0.23 and a pTM value of 0.77, indicating low confidence in the predicted protein–peptide interaction despite moderate confidence in the overall protein structure
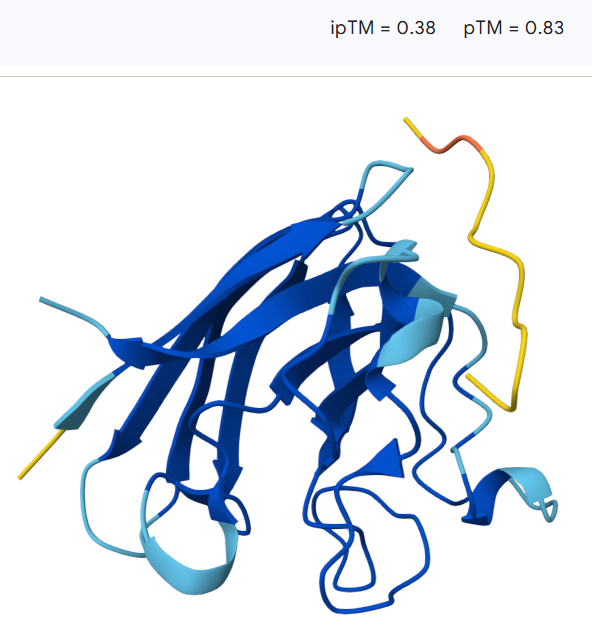
The last peptide:15.3
The peptide remains largely surface-bound and extended, suggesting a relatively weak interaction with the protein. The model shows an ipTM value of 0.38 and a pTM value of 0.83, indicating moderate confidence in the overall protein structure but limited confidence in the predicted protein–peptide interaction.
The predicted ipTM values range from 0.22 to 0.45, indicating generally low to moderate confidence in the protein–peptide interactions. The models with ipTM values around 0.22–0.23 suggest weak or unlikely binding, while the highest value (0.45) shows a more plausible surface interaction with the SOD1 β-barrel. However, overall the predicted interfaces remain relatively weak, and none of the PepMLM-generated peptides clearly exceed the expected binding confidence of the known SOD1 binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
The first one:
The second:
The third:
The last one:
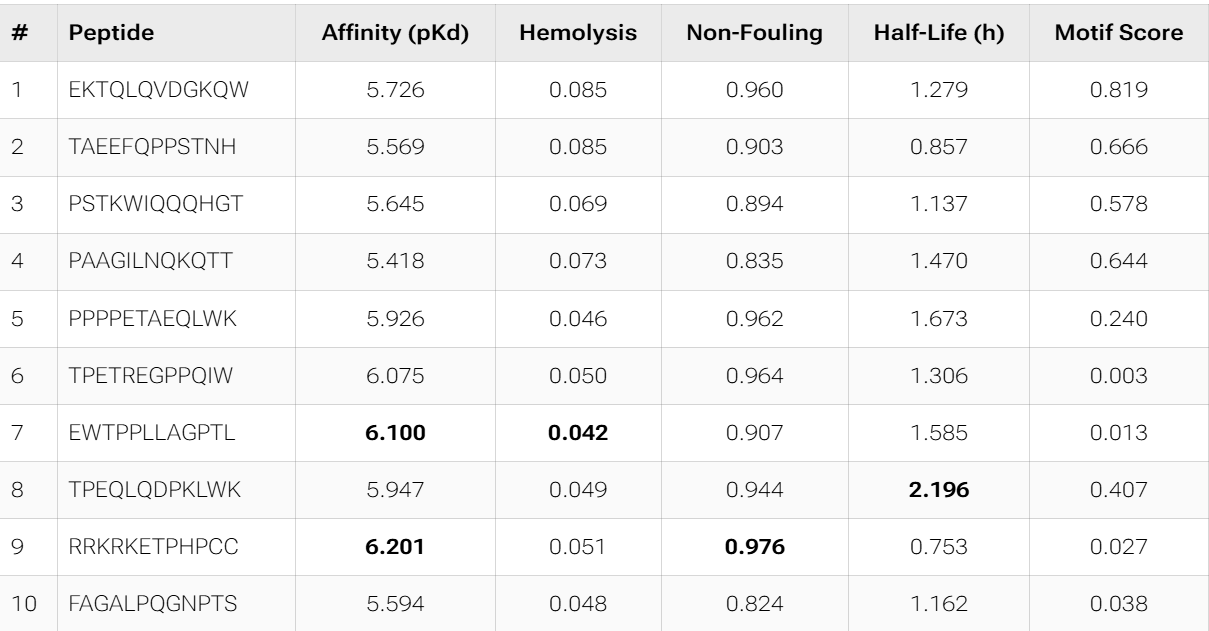
Overall, the AlphaFold models show low confidence in peptide binding (ipTM ~0.22–0.45). The peptide with the highest ipTM sits closer to the protein, but stronger ipTM doesn’t clearly match stronger predicted affinity since all peptides show weak binding. All peptides are predicted to be soluble and non-hemolytic. WRYYAAGVEHKE has the best balance because it has the strongest predicted affinity (~5.59) and good therapeutic properties. I would advance WRYYAAGVEHKE for further testing.
Part 4: Generate Optimized Peptides with moPPIt
The PepMLM candidates appear more optimized from a therapeutic perspective than the moPPIt results. They consistently show excellent solubility, very low hemolysis, and more coherent physicochemical profiles, while also converging toward aromatic-rich motifs that may favor hydrophobic interactions with SOD1. In contrast, moPPIt reflects a broader multi-objective search space, with greater sequence diversity and stronger trade-offs between affinity, half-life, and motif conservation.
However, both groups still show relatively weak predicted binding affinities overall, suggesting that computational optimization is currently improving developability more effectively than absolute binding strength.
How would you evaluate these peptides before advancing them to clinical studies?
Before advancing any of these peptides toward clinical studies, I would first perform AlphaFold3 structural validation and MD simulations to confirm stable binding at the SOD1 A4V interface. From there, the most promising candidates would undergo chemical stabilization (e.g., cyclization or D-amino acid substitution), followed by in vitro aggregation inhibition assays and cell viability testing in motor neuron models.
Week 7: Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Instead of relying on ON/OFF logic, they process information in a continuous and distributed way, closer to how real cells behave. This allows them to integrate multiple signals with different intensities and avoid the exponential complexity that arises when scaling Boolean circuits. In addition, IANNs are inherently more flexible, since their behavior can be tuned by adjusting interaction strengths rather than completely redesigning the system, enabling more complex and nonlinear decision-making. Overall, they provide a more efficient, scalable, and biologically realistic framework for intracellular computation.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
A useful application of an IANN would be an intracellular cancer classifier that integrates multiple biomarkers, such as oncogene expression, tumor suppressor loss, and metabolic changes, as continuous inputs. Instead of a simple Boolean response, it would compute a weighted, nonlinear decision and trigger outputs like pro-apoptotic gene expression only when the overall profile strongly indicates malignancy, improving specificity. However, limitations include difficulty in tuning interaction strengths, cellular noise, crosstalk with native pathways, and the challenge of reliably calibrating the system in vivo.
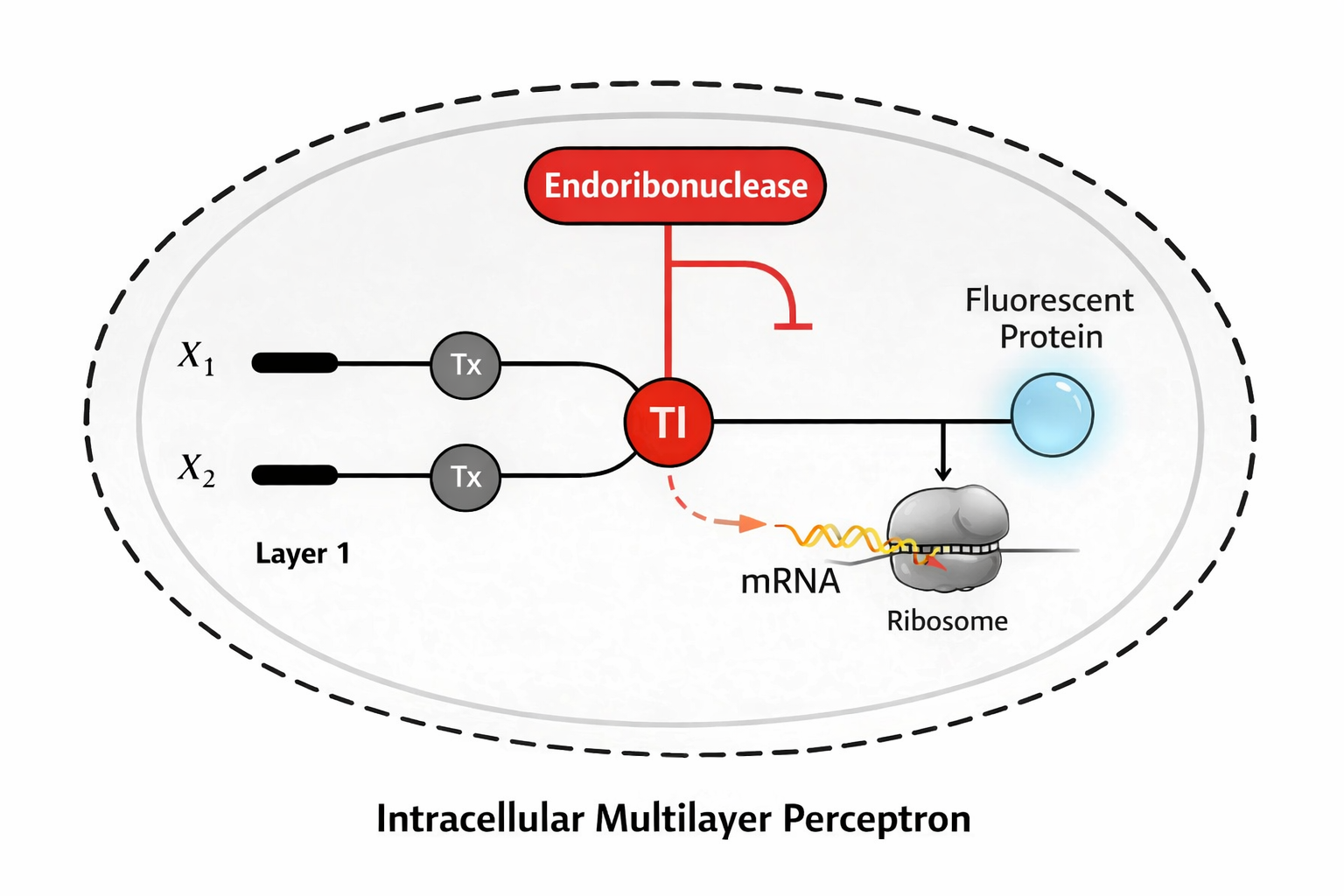
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Derived from mycelium:
Based packaging: Used as a substitute for polystyrene foam in protective packaging.
Leather: Used in fashion as vegan leather alternatives.
Construction materials: Bricks, insulation panels, and composites used in architecture for lightweight, biodegradable building components
They are advantageous because are biodegradable, renewable, and can be grown with low energy input using agricultural waste, making them much more sustainable than petroleum-based plastics or animal-derived materials.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
For the biodegradation of environmental pollutants, especially industrial textile waste, since many fungi already have strong natural decomposing abilities. For example, Trametes versicolor can help reduce textile contamination because it produces enzymes like laccases and peroxidases that break down synthetic dyes. This capability could be enhanced by overexpressing ligninolytic enzymes (laccases, manganese peroxidases, lignin peroxidases), coupling their expression to inducible systems that activate in the presence of pollutants, and improving tolerance to harsh industrial conditions such as extreme pH, temperature, and heavy metals. Additionally, optimizing secretion pathways would increase extracellular enzyme release and overall degradation efficiency, making the process more effective for wastewater treatment.
For the biodegradation of environmental pollutants, especially industrial textile waste, since many fungi already have strong natural decomposing abilities. For example, Trametes versicolor can help reduce textile contamination because it produces enzymes like laccases and peroxidases that break down synthetic dyes. This capability could be enhanced by overexpressing ligninolytic enzymes (laccases, manganese peroxidases, lignin peroxidases), coupling their expression to inducible systems that activate in the presence of pollutants, and improving tolerance to harsh industrial conditions such as extreme pH, temperature, and heavy metals. Additionally, optimizing secretion pathways would increase extracellular enzyme release and overall degradation efficiency, making the process more effective for wastewater treatment.
Advantages over bacteria for synthetic biology:
They are eukaryotic organisms that can perform complex post-translational modifications and proper protein folding required for many eukaryotic proteins.
They have a strong natural capacity for protein secretion, which simplifies production and purification of recombinant proteins.
More complex genetic regulation and larger gene constructs, making them suitable for expressing multi-step pathways.
Their cellular organization and scalability in industrial fermentation further make them attractive platforms for producing pharmaceuticals, enzymes, and biomaterials, although they are typically slower growing and less genetically tractable than bacteria.
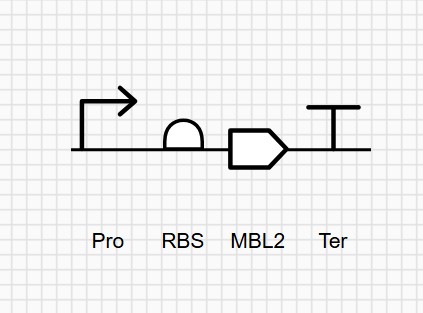
Part 3: First DNA Twist Order
This insert is designed to be cloned into the pUC19 plasmid for expression in E. coli. The construct includes a pTet inducible promoter, allowing controlled expression of the gene of interest.
Week 9: Cell-Free Systems
Assignment Part A: General and Lecturer-Specific Questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
You can control everything directly. You can adjust pH, temperature, ion concentrations, and cofactors and even add or remove components during the reaction. Also, it’s faster since you don’t need cell growth or maintenance.
In terms of control, you can precisely tune the system and avoid cellular regulation that might limit expression.
Cases where it’s more beneficial:
Expression of toxic proteins, because they would kill the cells
Rapid protein production for screening experiments, testing many variants quickly
Describe the main components of a cell-free expression system and explain the role of each component.
Cell extract: Provides the machinery (ribosomes, tRNAs, enzymes) for transcription and translation
DNA or mRNA template: Contains the gene for the protein to be expressed
Amino acids: Building blocks for the protein
Energy system (ATP, GTP): Powers transcription and translation
Buffers and salts (Mg²⁺, K⁺): Maintain optimal conditions for the reaction
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Because energy systems are consumed very quickly during transcription and translation. If it runs out, protein synthesis stops.
To maintain ATP levels, it’s possible to use phosphoenolpyruvate (PEP). It works by transferring a phosphate group to ADP, continuously regenerating ATP during the reaction.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic systems are faster, cheaper, and give high yields, but they lack complex post-translational modifications.
Eukaryotic systems are slower and more expensive, but they allow proper folding and modifications like glycosylation.
Examples:
Prokaryotic: GFP, because it’s simple and doesn’t need modifications
Eukaryotic: an antibody, because it requires correct folding and post-translational modifications to function properly
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
To mimic the membrane: Add liposomes or nanodiscs
Keep the protein soluble: Use mild detergents
Adjust conditions like Mg²⁺, temperature, and protein concentration
Include chaperones to help folding of the protein
Challenges and how to adress:
Misfolding: Membrane proteins don’t fold well without lipids
→ Solution: add liposomes or nanodiscs + chaperones
Aggregation: Hydrophobic regions stick together
→ Solution: use mild detergents and lower expression rate (e.g., lower temperature)
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Poor DNA/template quality or design
→ Troubleshooting: use a stronger promoter, optimize codons, check DNA purity
Energy depletion
→ Troubleshooting: improve the energy regeneration system (e.g., add PEP or increase substrates)
Protein misfolding or degradation
→ Troubleshooting: add chaperones, lower temperature, optimize reaction conditions
Assignment Part A: question from Kate Adamala
Pick a function and describe it.
What would your synthetic cell do?
It would act as a diagnostic biosensor for the detection of DENV. Its primary function is to transduce the presence of the viral E protein into a detectable signal, utilizing a two-stage recognition system:
External Sensor: A specific antibody and aptamer recognizes the DENV E protein, this recognition event triggers the release of theophylline, which acts as a messenger molecule.
Internal Sensor (Synthetic cell): The theophylline binds to a theophylline riboswitch, inducing a conformational change that exposes the ribosome binding site (RBS), allowing the translation of the LacZ reporter gene, generating a detectable signal.
What is the input and what is the output? (synthetic cell)
Input: Theophylline released by the external sensor
Output: Expression of beta-galactosidase (LacZ)
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Yes.
This process can be carried out using only a cell-free Tx/Tl system. In this simplified setup, theophylline diffuses freely to activate LacZ expression without the need for encapsulation.
Could this function be realized by genetically modified natural cell?
It’s possible, but a cell-free system is more economical and streamlined than traditional cell-based methods for this purpose.
Describe the desired outcome of your synthetic cell operation.
The primary objective is to develop a sensitive, specific, user-friendly, and cost-effective biosensor for DENV detection.
BUT the desired outcome of the synthetic cell operation is the expression of beta-galactosidase (LacZ) in the presence of theophylline
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
Phospholipid liposomes and polymersomes to ensure membrane stability, permeability, and biocompatibility
What would you encapsulate inside? Enzymes, small molecules.
Genetic circuit, E. coli extract for Tx/Tl machinery and chromogenic substrates for β-galactosidase
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason?
E. coli
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
Utilizing an alpha-hemolysin channel to ensure the diffusion of theophylline and ONPG (the substrate for beta-galactosidase)
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
System functionality is quantified through beta-galactosidase-mediated hydrolysis of ONPG
Assignment Part A: question from Peter Nguyen
FirstIdea
Write a one-sentence summary pitch sentence describing your concept
Walls that use cell-free photosynthetic systems integrated into the material to capture CO₂ from the air and transform it locally into oxygen during daily sun exposure.
How will the idea work, in more detail? Write 3-4 sentences or more.
The building’s exterior walls incorporate modular layers containing cell-free systems based on artificial or reconstituted photosynthetic pathways. These modules are activated by sunlight and controlled ambient humidity, triggering biochemical reactions that capture CO₂ from the air and release oxygen as a byproduct
What societal challenge or market need will this address?
The urgent need to reduce CO₂ in dense urban environments, it proposes an architecture that not only minimizes its footprint but also actively acts as environmental infrastructure, which is especially relevant in cities with high pollution and constant solar exposure.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Activation is controlled by micro-encapsulation, which regulates water ingress, preventing unwanted continuous activation.
For stability, stabilizers and housed in replaceable cartridges within the façade, allowing for periodic maintenance.
By acknowledging the one-time or limited-cycle nature of these reactions, the idea is treat them as consumable layers—similar to filters—integrated into a building renewal cycle without compromising its main structure.
SecondIdea
Write a one-sentence summary pitch sentence describing your concept
An eco-friendly, ’living’ window coating utilizing freeze-dried cell-free systems to express UV-active chromoproteins that create avian-visible warning patterns while remaining perfectly transparent to the human eye.
How will the idea work, in more detail? Write 3-4 sentences or more.
A cell-free platform embedded into a transparent, porous biopolymer matrix applied to glass surfaces. When activated, the machinery expresses specialized UV-chromoproteins or enzymes that produce pigments with high absorbance/reflectance in the $300\text{–}400\text{ nm}$ range, which falls within the tetrachromatic visual spectrum of birds. These biological “inks” are arranged in specific geometric patterns that birds recognize as solid obstacles, triggering an avoidance response. Because humans lack UV photoreceptors and the proteins do not scatter visible light, the window appears clear to us while appearing “solid” or patterned to birds.
What societal challenge or market need will this address?
Every year, it is estimated that up to one billion birds die just in the United States alone due to collisions with glass windows, making it a leading cause of avian mortality and biodiversity loss. While UV-reflective stickers and “fritted” glass exist, they are aesthetically unpleasing, or lose effectiveness over time. There is a significant market need for sustainable, “invisible” retrofitting solutions for residential and commercial skyscrapers that can protect migratory species without altering architectural aesthetics
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Stability: The cell-free machinery is freeze-dried within a protective matrix of trehalose or synthetic polymersomes, allowing it to remain dormant and stable during transport and installation.
Activation: The system is designed for hydro-activation; ambient humidity or rain triggers the controlled release of water into the micro-compartments, initiating protein synthesis precisely when the risk of collision (often higher in overcast/rainy weather) is present.
Longevity: To address the one-time use limit, the system would incorporate genetic circuits for protein stability and “slow-release” mechanisms where the chromoproteins are cross-linked to the matrix, ensuring the UV signal persists for several months before a simple, biodegradable “recharge” spray is required.
Assignment Part A: question from Ally Huang
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
High‑energy heavy ions (HZE radiation) are a major hazard in deep‑space missions and are known to cause complex DNA and protein damage. Understanding how radiation accelerates molecular aging is essential for astronaut health, long‑duration missions, and space habitation. Cell‑free systems provide a lightweight, non‑living platform to directly study radiation‑induced molecular damage without confounding cellular repair mechanisms. This makes them ideal for spaceflight experiments, where resources are limited and biological containment is critical. Studying molecular aging in space has direct implications for human longevity, cancer risk, and the stability of biological systems beyond Earth.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Green fluorescent protein (GFP)–encoding DNA and expressed GFP protein as molecular reporters of radiation‑induced damage and aging.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
GFP is a well‑characterized protein whose fluorescence is sensitive to errors in DNA transcription, protein folding, and structural integrity. Damage from HZE radiation may reduce protein yield, alter folding efficiency, or degrade fluorescence intensity. Using GFP in a BioBits® cell‑free system allows direct measurement of radiation‑induced molecular aging without cellular repair or replication. Differences in expression level or fluorescence provide a clear molecular readout of how space radiation affects fundamental biological processes.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Exposure to HZE radiation causes measurable molecular aging in cell‑free systems, leading to decreased protein expression efficiency and reduced fluorescence intensity.
Cell‑free reactions lack DNA repair mechanisms and protein turnover, making them highly sensitive indicators of cumulative radiation damage. If HZE radiation accelerates molecular aging, irradiated GFP‑encoding DNA or protein synthesis machinery will produce less functional GFP compared to non‑irradiated controls. By comparing fluorescence output, this experiment will isolate the direct effects of space radiation on molecular stability and function, independent of living cells.
Outline your experimental plan—identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Freeze‑dried BioBits® reactions containing GFP DNA will be exposed to HZE radiation and compared to Earth‑based and flight non‑irradiated controls. Reactions will be rehydrated simultaneously and incubated under identical conditions. GFP fluorescence intensity will be measured using the P51 Molecular Fluorescence Viewer. Controls include unexposed freeze‑dried reactions and reactions expressing pre‑folded GFP. Data will consist of fluorescence intensity and expression consistency across conditions.