Week 4: Protein Design Part I
Part A. Conceptual Questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Considering that 500 g of meat contains 20% protein, we would have 100 g of protein. After converting the protein mass to moles (assuming an average amino acid mass of 100 Da), this corresponds to approximately 6.022 × 10²³ amino acid molecules.
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because these proteins are broken down during digestion, reducing them to amino acids and peptides that are reused to synthesize our own proteins according to our genetic instructions.
- Why are there only 20 natural amino acids?
There are 20 natural amino acids because early in evolution the genetic code incorporated a set of amino acids that provided sufficient chemical diversity to build functional proteins, and once this translation system (involving tRNAs and aminoacyl-tRNA synthetases) became established, it was evolutionarily conserved and effectively “frozen,” making the addition of new amino acids highly disadvantageous.
- Can you make other non-natural amino acids? Design some new amino acids.
Yes, chemically it is possible to design new amino acids, but incorporating them biologically into proteins is difficult because they require their own tRNA, a matching aminoacyl-tRNA synthetase, and a reassigned codon. Pendiente hacer aminoacido
- Where did amino acids come from before enzymes that make them, and before life started?
Before life began, amino acids likely formed through abiotic chemical reactions driven by energy sources such as lightning, UV radiation, volcanic activity, hydrothermal systems, and possibly delivery from meteorites in a primitive earth, as demonstrated by experiments like Miller.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left-handed
- Can you discover additional helices in proteins?
Yes, additional helices are physically possible and we can design new one modifying amino acid chemistry or backbone structure.
- Why are most molecular helices right-handed?
Most molecular helices are right-handed because the chiral building blocks of life—such as L-amino acids and D-sugars—constrain backbone geometry in a way that makes right-handed helices energetically more stable and sterically favorable.
- Why do β-sheets tend to aggregate? Because their extended structure exposes backbone hydrogen bond donors and acceptors, allowing β-strands from different molecules to form stable intermolecular hydrogen bonds.
What is the driving force for β-sheet aggregation? The main driving force is the formation of intermolecular backbone hydrogen bonds, reinforced by hydrophobic interactions that stabilize the aggregate and lower the system’s free energy.
Part B. Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it. Furin is a calcium-dependent protease hat activates precursor proteins by cleaving them at specific basic amino acid recognition sequences in the Golgi apparatus and I selected it because it’s a protein I am currently interested in.
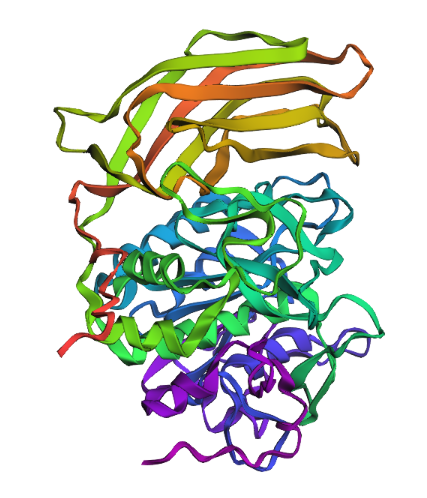
- Identify the amino acid sequence of your protein.
5JXG_1|Chain A|Furin|Homo sapiens (9606)
DVYQEPTDPKFPQQWYLSGVTQRDLNVKAAWAQGYTGHGIVVSILDDGIEKNHPDLAGNYDPGASFDVNDQDPDPQPRYTQMNDNRHGTRCAGEVAAVANNGVCGVGVAYNARIGGVRMLDGEVTDAVEARSLGLNPNHIHIYSASWGPEDDGKTVDGPARLAEEAFFRGVSQGRGGLGSIFVWASGNGGREHDSCNCDGYTNSIYTLSISSATQFGNVPWYSEACSSTLATTYSSGNQNEKQIVTTDLRQKCTESHTGTSASAPLAAGIIALTLEANKNLTWRDMQHLVVQTSKPAHLNANDWATNGVGRKVSHSYGYGLLDAGAMVALAQNWTTVAPQRKCIIDILTEPKDIGKRLEVRKTVTACLGEPNHITRLEHAQARLTLSYNRRGDLAIHLVSPMGTRSTLLAARPHDYSADGFNDWAFMTTHSWDEDPSGEWVLEIENTSEANNYGTLTKFTLVLYGTASGSLVPRGSHHHHHH
How long is it? What is the most frequent amino acid?
482, The most common amino acid is: G, which appears 47 times
How many protein sequence homologs are there for your protein?
230
Does your protein belong to any protein family?
Proprotein convertase
- Identify the structure page of your protein in RCSB
5JXG | pdb_00005jxg
When was the structure solved? Is it a good quality structure?
2016 with 1.80 Å
Are there any other molecules in the solved structure apart from protein?
Yes, Ca+2, Cl- and Na+
Does your protein belong to any structure classification family?
Serine endoprotease, hydrolase
Open the structure of your protein in any 3D molecule visualization software
Cartoon
 Ribbon
Ribbon
 Ball and stick
Ball and stick
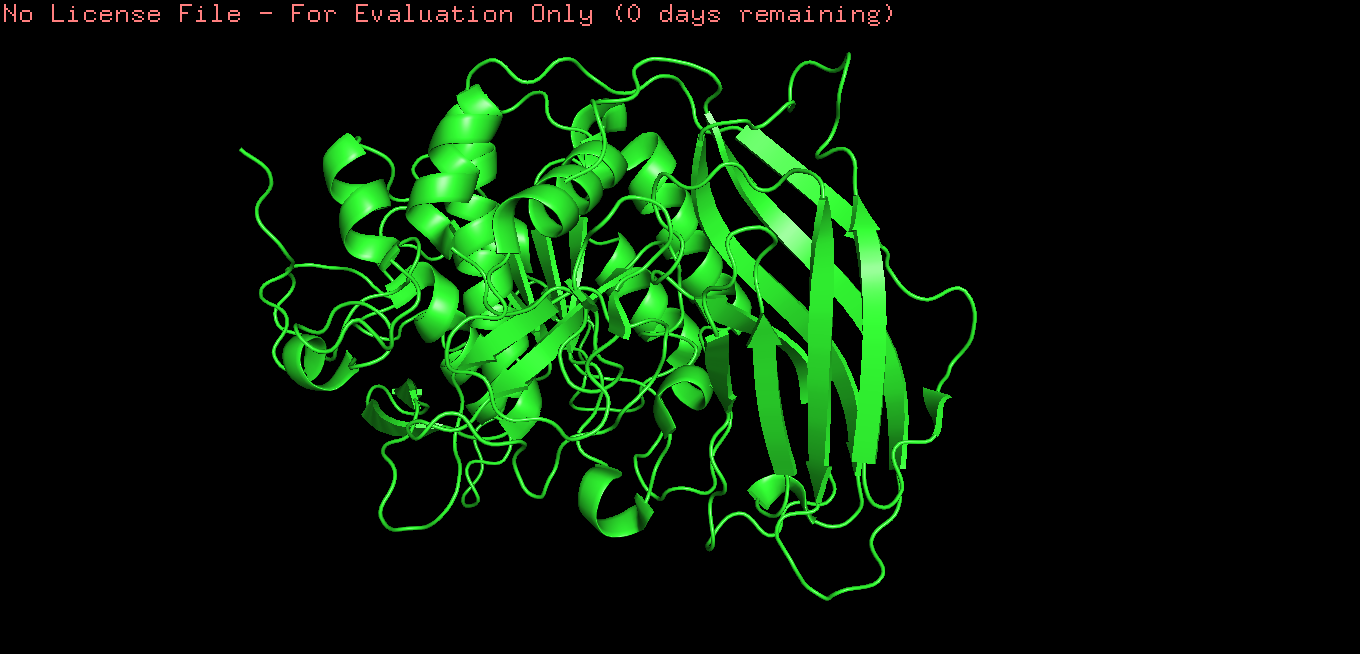
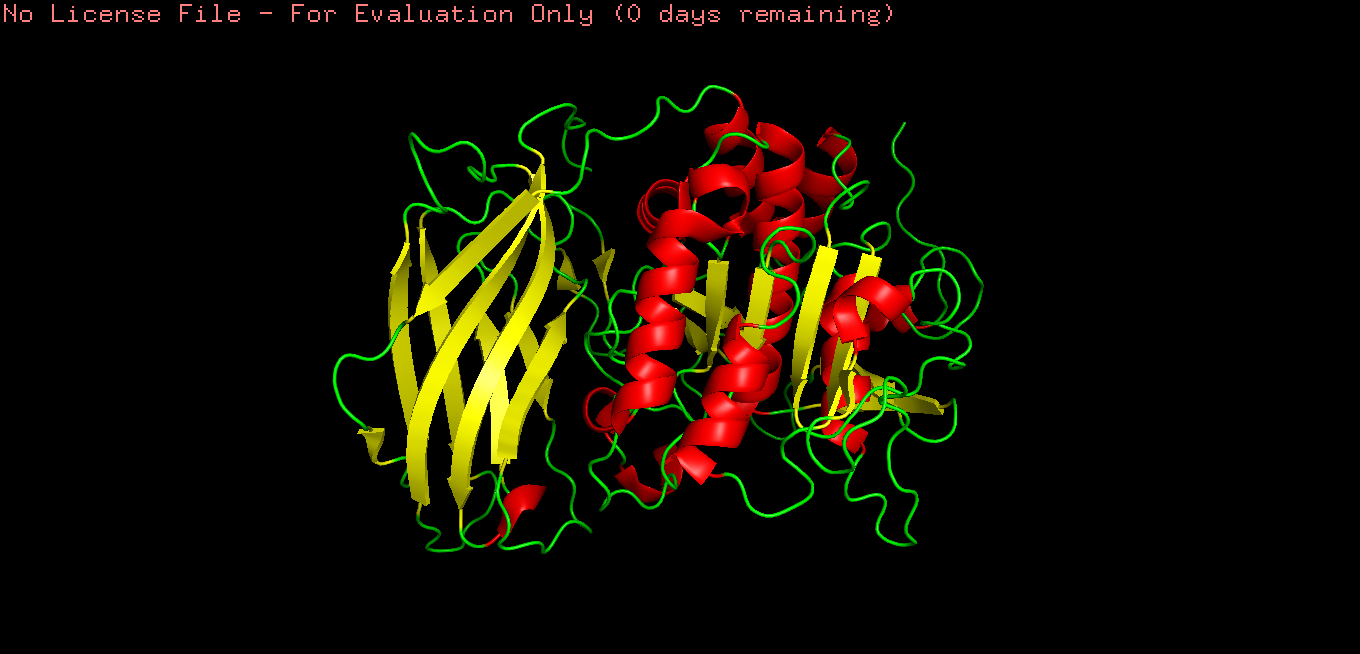
 Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by secondary structure. Does it have more helices or sheets?
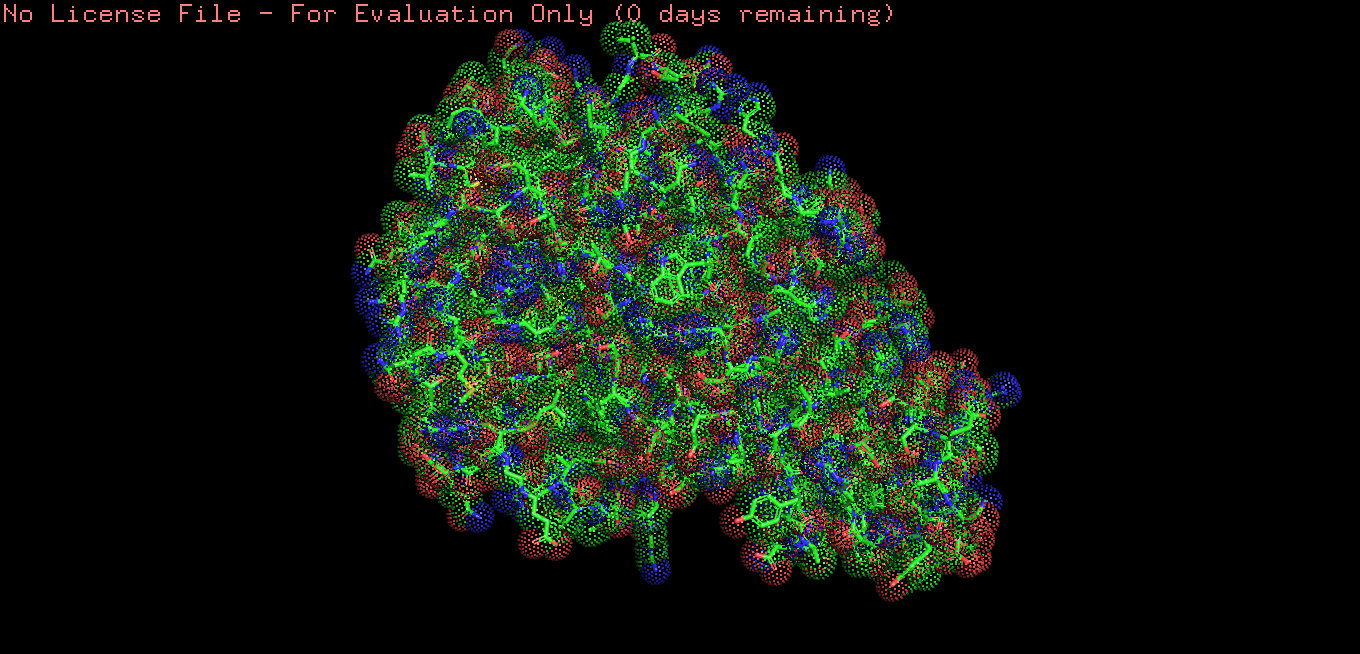
Color the protein by residue type.
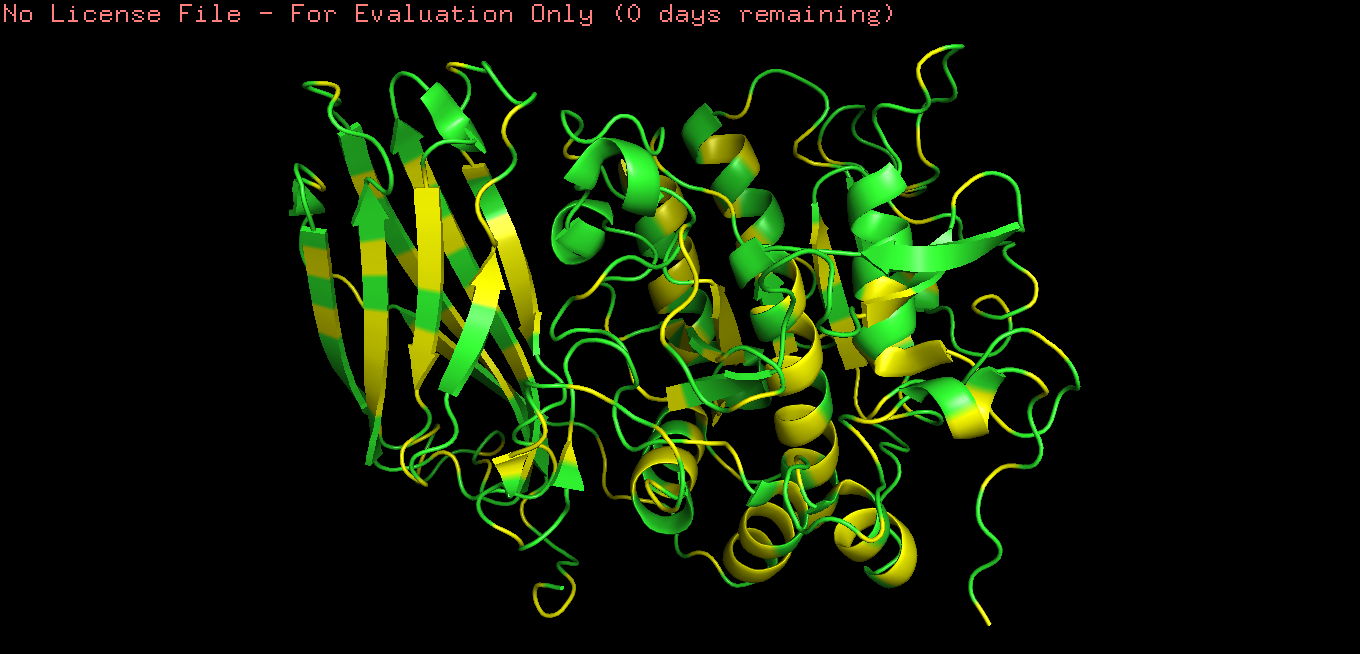
What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Hydrophobic residues are mainly buried in the protein core, while hydrophilic residues are predominantly exposed on the surface, consistent with typical folding of a soluble globular protein.
C1. Protein Language Modeling
Deep Mutational Scans
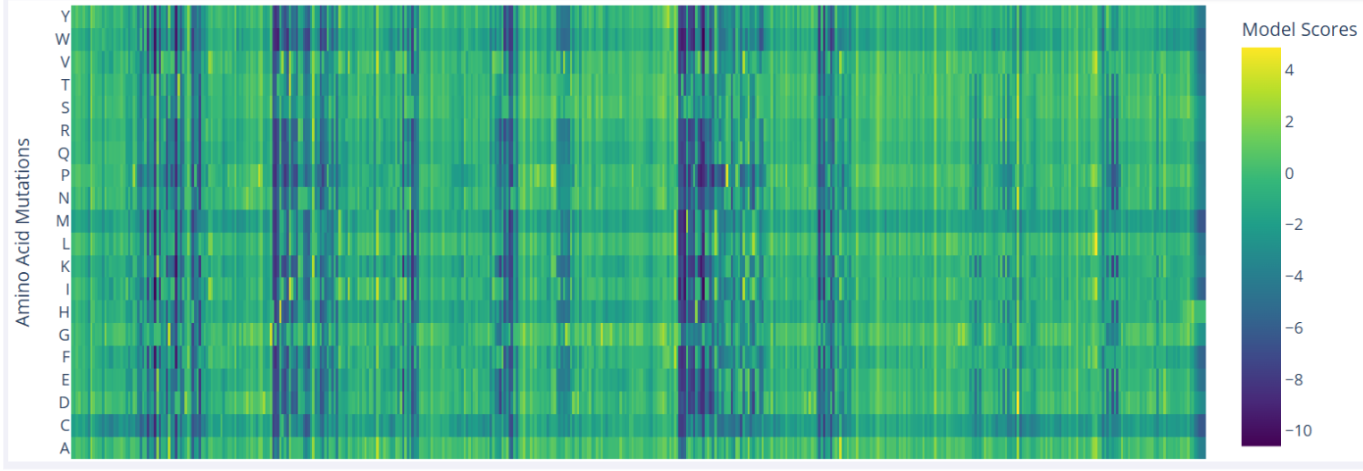
In the mutational scan of furin, one clear pattern is the presence of vertical dark bands that reflect unfavorable mutations at specific positions, which likely correspond to critical residues. For example, Ser368 is essential in the serine protease catalytic triad, and nearly all substitutions at this position are predicted to be highly deleterious.
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
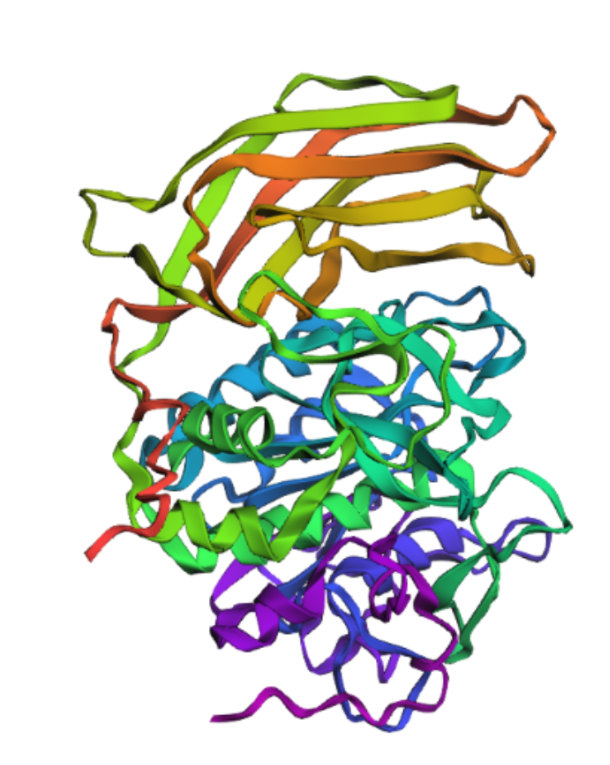
Yes, It´s very similar.
- Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
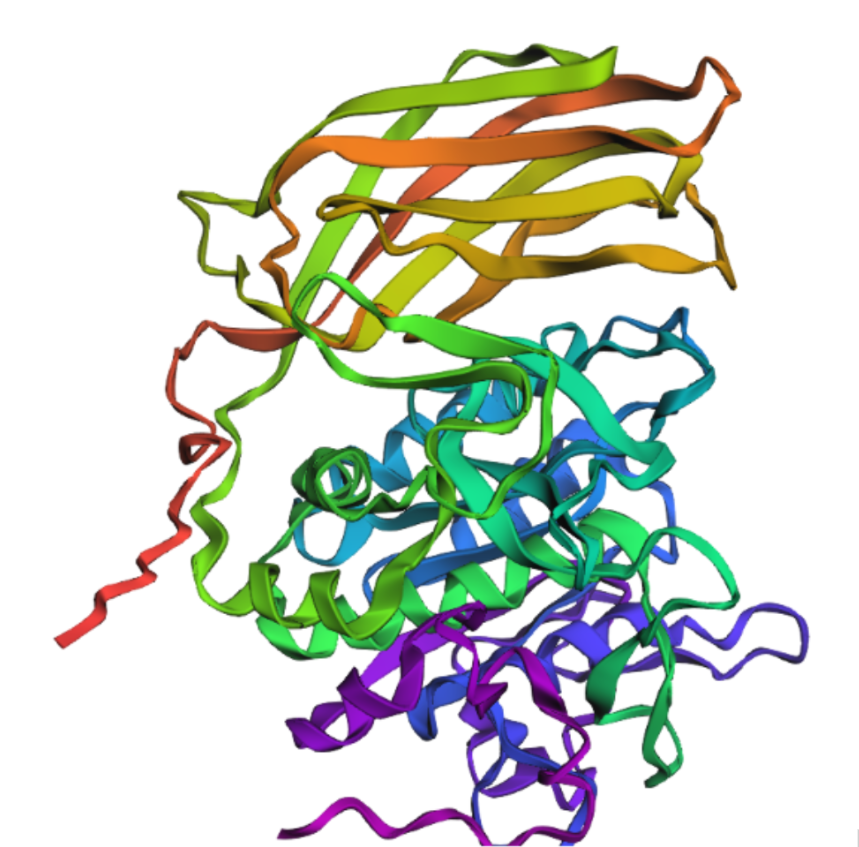
I mutated the amino acids in the active site of furin to render the protein inactive; specifically, I introduced the substitutions Ser → Ala (S368A), His → Ala (H194A), and Asp → Asn (D153N).
It’s almost the same structure, but now I changed the p-domain, the majority of the protein still remains intact mading this one resilient to mutations
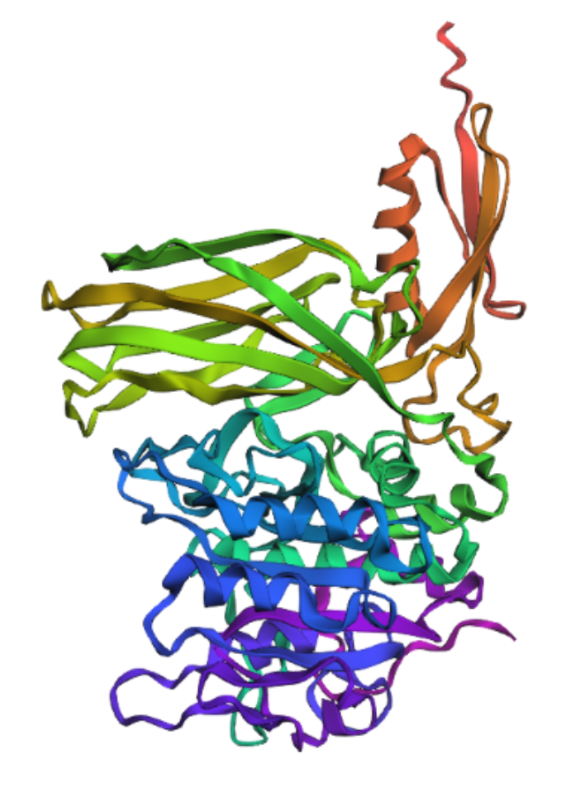
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
- Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
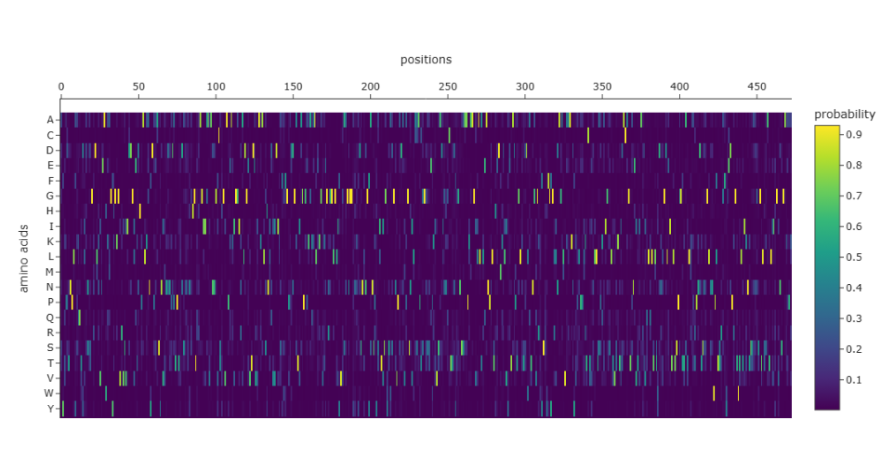
The heatmap shows that many positions in the sequence strongly favor a single amino acid, while alternative residues have very low probabilities. Suggesting that these positions are highly constrained, likely due to structural or functional requirements of the protein. Only a limited number of positions show broader amino acid tolerance, indicating potential sites where mutations might be more acceptable. This pattern is consistent with proteins that contain structurally important or catalytic regions, where mutations are less tolerated.
D. Group Brainstorm on Bacteriophage Engineering
GROUP FINAL PROJECT