Week 5 HW: Protein design part 2
Part A: SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
After introducing the A4V mutation. I performed the mutation A5V based on its position in the FASTA sequence.
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using PepMLM Colab with a K value of 1, I obtained:
WRYYAVAAAHKX 8.149419
WRYYAVAAAHKX 8.149419
WRYYAVAAAHKX 8.149419
WRYYAVAAAHKX 8.149419
In comparison with SOD1-binding peptide:
FLYRWLPSRRGG with a perplexity of 5.98
The generated peptides showed perplexity scores around 8, which are higher than the example. This indicates that the model assigns lower confidence to these sequences as potential binders to the mutant SOD1 protein.
Part 2: Evaluate Binders with AlphaFold3
Due to the fact that AlphaFold does not allow the analysis of proteins with undetermined amino acids, I ran the PepMLM Colab again using a K value of 2 so that all amino acids would be fully specified.
WRYYAAGVEHKE 15.984515
WRYYVVAAAHGX 11.686807
WRYYAAGAALKX 6.211397
WRYYVVAAALKE 15.368100
And with 3:
WHYPAVGAALWE 12.713875
WRYYAVVLAHKX 10.328839
WRYYVVALAHKE 16.276417
HLYYVVGVRWKE 27.442868
So I chose the four with the lowest perplexity value:
WHYPAVGAALWE 12.713875
WRYYAAGVEHKE 15.984515
WRYYVVALAHKE 16.276417
WRYYVVAAALKE 15.368100
For the first peptide: 12.7
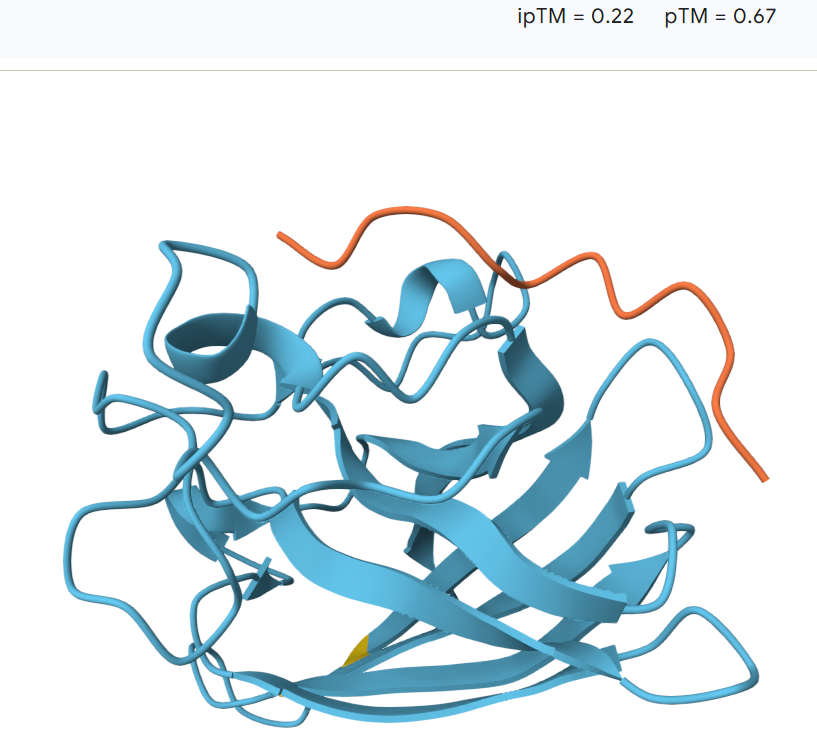
The peptide binds along the outer surface of the SOD1 structure, lying across loops adjacent to the β-barrel rather than inserting into the core of the protein. It does not localize directly near the N-terminal region where the A4V mutation is located, nor does it clearly approach the predicted dimer interface.
For the second peptide:15.9

The peptide does not localize directly near the N-terminal region where the A4V mutation resides, instead, the peptide lies primarily on the protein surface, with a portion potentially partially accommodated within a shallow surface groove. The model shows an ipTM value of 0.45 and a pTM of 0.88, indicating moderate confidence in the protein–peptide interaction while maintaining high confidence in the overall protein structure.
For the third peptide:16.2
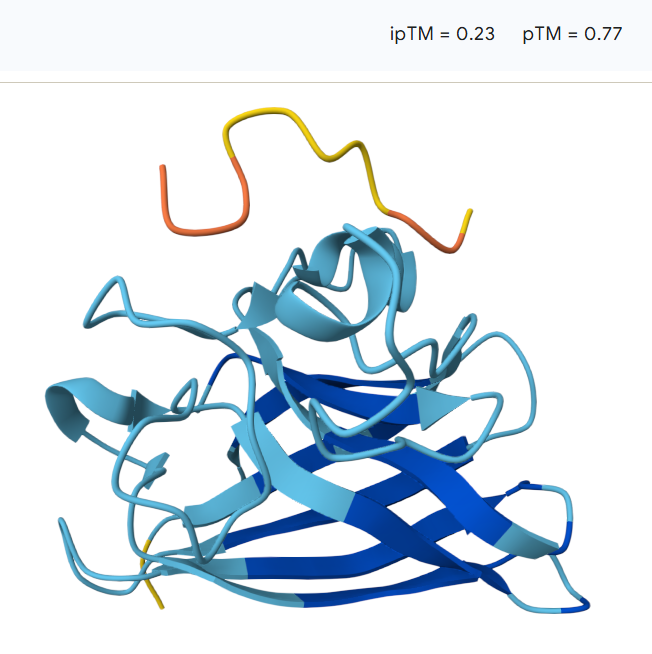
The peptide remains largely separated from the protein and appears flexible and surface-exposed, suggesting a lack of stable binding. The model shows an ipTM value of 0.23 and a pTM value of 0.77, indicating low confidence in the predicted protein–peptide interaction despite moderate confidence in the overall protein structure
The last peptide:15.3
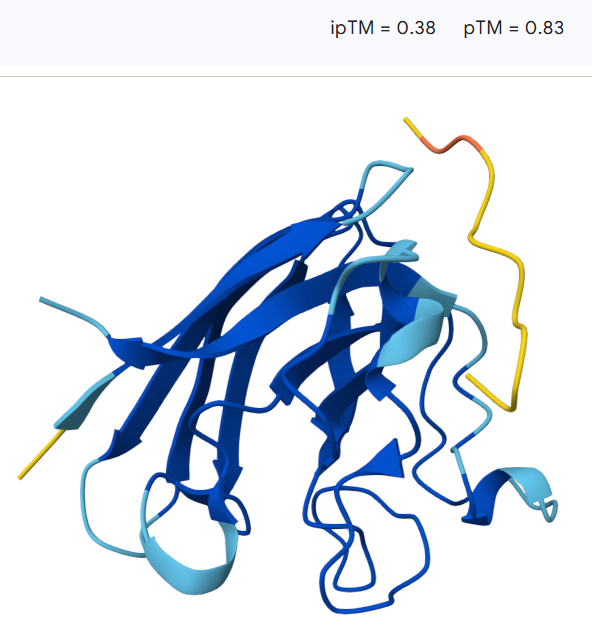
The peptide remains largely surface-bound and extended, suggesting a relatively weak interaction with the protein. The model shows an ipTM value of 0.38 and a pTM value of 0.83, indicating moderate confidence in the overall protein structure but limited confidence in the predicted protein–peptide interaction.
The predicted ipTM values range from 0.22 to 0.45, indicating generally low to moderate confidence in the protein–peptide interactions. The models with ipTM values around 0.22–0.23 suggest weak or unlikely binding, while the highest value (0.45) shows a more plausible surface interaction with the SOD1 β-barrel. However, overall the predicted interfaces remain relatively weak, and none of the PepMLM-generated peptides clearly exceed the expected binding confidence of the known SOD1 binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
The first one:
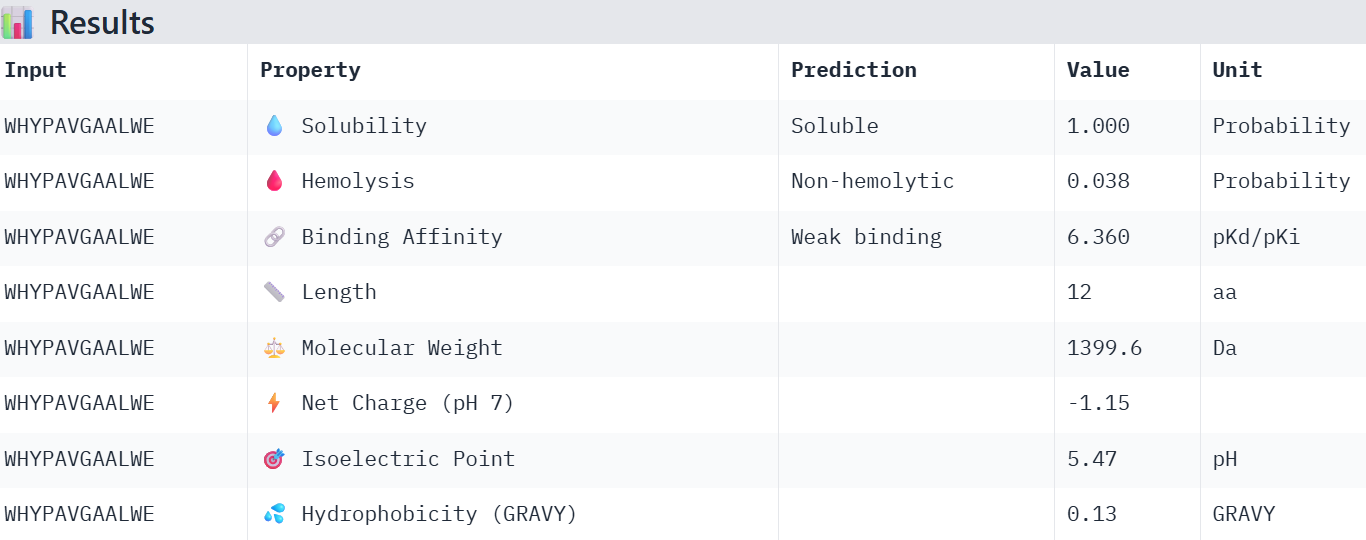
The second:
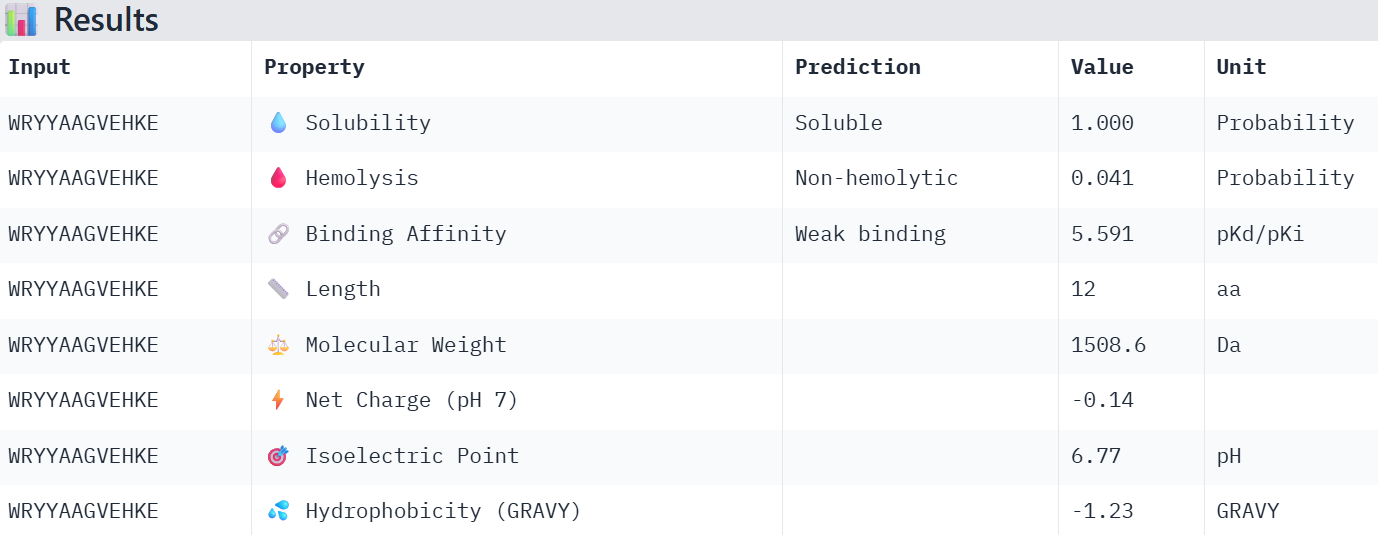
The third:
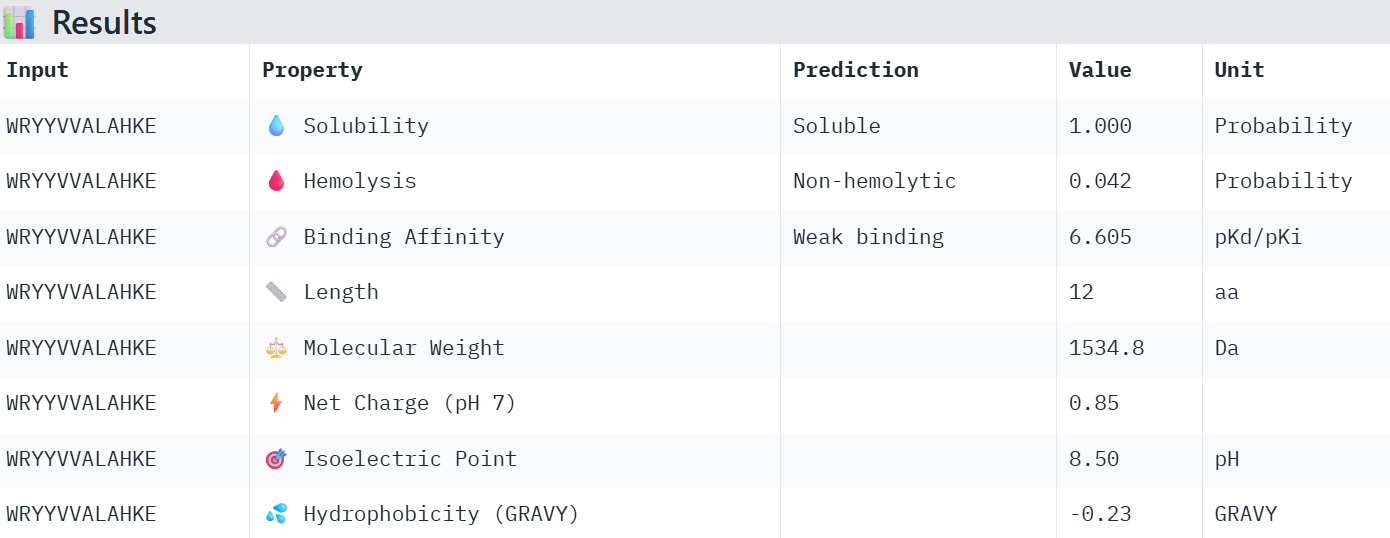
The last one:
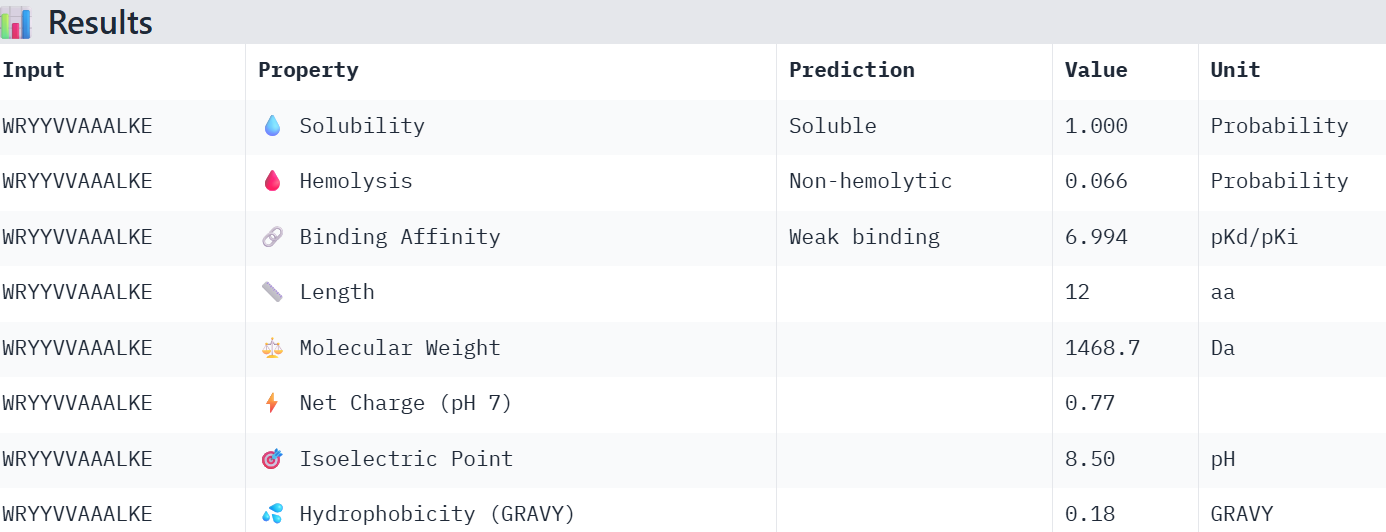
Overall, the AlphaFold models show low confidence in peptide binding (ipTM ~0.22–0.45). The peptide with the highest ipTM sits closer to the protein, but stronger ipTM doesn’t clearly match stronger predicted affinity since all peptides show weak binding. All peptides are predicted to be soluble and non-hemolytic. WRYYAAGVEHKE has the best balance because it has the strongest predicted affinity (~5.59) and good therapeutic properties. I would advance WRYYAAGVEHKE for further testing.
Part 4: Generate Optimized Peptides with moPPIt
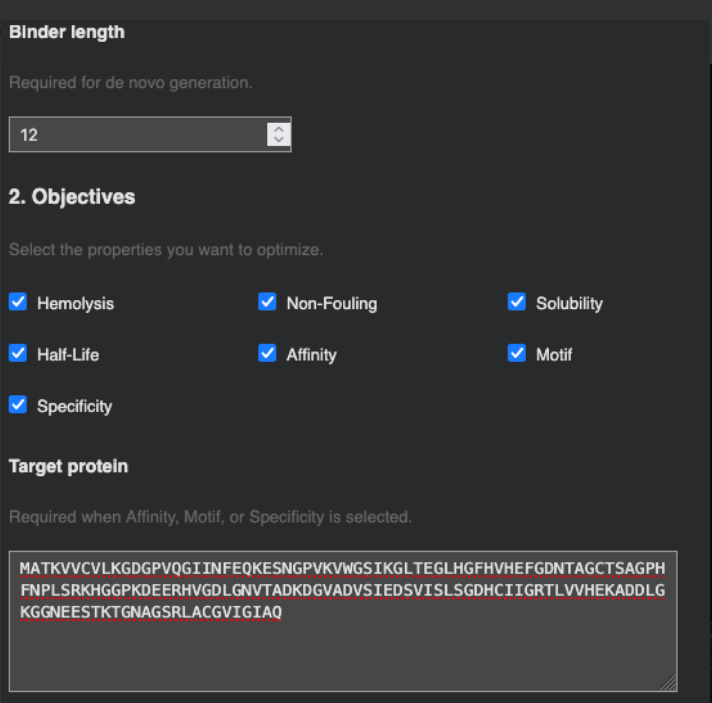
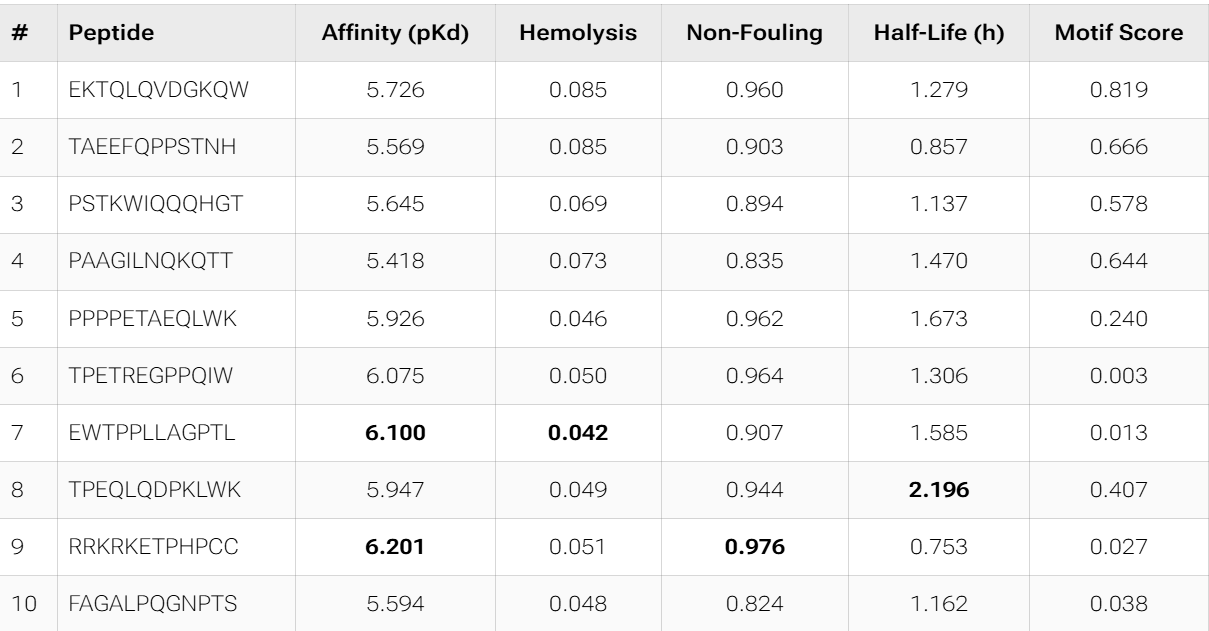
The PepMLM candidates appear more optimized from a therapeutic perspective than the moPPIt results. They consistently show excellent solubility, very low hemolysis, and more coherent physicochemical profiles, while also converging toward aromatic-rich motifs that may favor hydrophobic interactions with SOD1. In contrast, moPPIt reflects a broader multi-objective search space, with greater sequence diversity and stronger trade-offs between affinity, half-life, and motif conservation.
However, both groups still show relatively weak predicted binding affinities overall, suggesting that computational optimization is currently improving developability more effectively than absolute binding strength.
How would you evaluate these peptides before advancing them to clinical studies?
Before advancing any of these peptides toward clinical studies, I would first perform AlphaFold3 structural validation and MD simulations to confirm stable binding at the SOD1 A4V interface. From there, the most promising candidates would undergo chemical stabilization (e.g., cyclization or D-amino acid substitution), followed by in vitro aggregation inhibition assays and cell viability testing in motor neuron models.