Abstract Dengue rapid tests targeting NS1 protein perform inconsistently across infection stages, delaying diagnosis in the low-resource settings where the disease hits hardest. This project develops a cell-free, aptamer-based biosensor that targets EDIII — a more immunogenically distinct domain of the dengue E protein — to achieve more reliable, stage-independent detection without cold chain or laboratory infrastructure. The core system couples three integrated modules: a capture antibody and aptamer selected to bind non-competing EDIII epitopes, confirmed via protein–protein interaction modeling; an aptamer–blocker–theophylline construct engineered for low leak and EDIII-triggered release; and a cell-free circuit linked through a theophylline riboswitch to drive LacZ expression as the final colorimetric signal. Aim 2 translates this system into a portable rapid test format. Aim 3 validates diagnostic performance and expands applicability across serotypes and sample matrices
Computational Engineering of the MS2 Lysis Protein to Improve Stability, Titers, and Toxicity After reviewing the provided literature on the MS2 lysis protein (L) and discussing the project aims, our group has decided to focus on three interconnected goals:
Goal 1: Increase the stability of the L protein As the “easiest” goal, it is the most computationally tractable. A stabilized protein is less prone to degradation and misfolding, which could directly lead to higher functional titers and serve as a robust starting point for any subsequent engineering.
Subsections of Projects
Pattern-Based Rapid Diagnostic Platform for Dengue Virus Using Aptamers
Abstract
Dengue rapid tests targeting NS1 protein perform inconsistently across infection stages, delaying diagnosis in the low-resource settings where the disease hits hardest. This project develops a cell-free, aptamer-based biosensor that targets EDIII — a more immunogenically distinct domain of the dengue E protein — to achieve more reliable, stage-independent detection without cold chain or laboratory infrastructure.
The core system couples three integrated modules: a capture antibody and aptamer selected to bind non-competing EDIII epitopes, confirmed via protein–protein interaction modeling; an aptamer–blocker–theophylline construct engineered for low leak and EDIII-triggered release; and a cell-free circuit linked through a theophylline riboswitch to drive LacZ expression as the final colorimetric signal. Aim 2 translates this system into a portable rapid test format. Aim 3 validates diagnostic performance and expands applicability across serotypes and sample matrices
Project aims
Develop an integrated molecular workflow for DENV detection
‘Build a functional cell-free biosensor in silico for dengue virus detection by designing and integrating three coupled molecular modules targeting the EDIII domain of the dengue E protein’
•1.1 Select a capture antibody and DNA aptamer with non-competing EDIII binding sites, validated through protein–protein interaction modeling, to enable sandwich-style recognition without steric interference.
•1.2 Design an aptamer–blocker–theophylline construct with low basal leak and confirmed EDIII-triggered theophylline release, optimized through sequence engineering and in vitro characterization.
•1.3 Implement a cell-free transcription–translation system regulated by a theophylline riboswitch driving LacZ expression as a colorimetric output, and verify end-to-end signal activation in response to recombinant EDIII.
Design and prototype a portable rapid test format
‘Executes the experimental validation and physical integration of all three modules.’
•2.1 Synthesize aptamer and blocker for validate antibody-aptamer non-competition by competitive ELISA and assemble and test the TXTL–riboswitch–LacZ circuit, confirming theophylline-dependent colorimetric output via X-gal.
•2.2 Characterize EDIII-triggered strand displacement and theophylline release by native PAGE; integrate all three modules in solution and confirm end-to-end colorimetric output
•2.3 Lyophilize TXTL–Xgal components onto Whatman paper discs with trehalose as stabilizer; rehydrate with EDIII-spiked samples and confirm visual readout at room temperature.
Validate performance and expand diagnostic applicability
weight: 4
‘If fully realized, this platform could redefine point-of-care dengue diagnostics by replacing traditional tests with a programmable, aptamer-driven biosensor accessible to anyone, anywhere’
•3.1 Evaluate sensitivity, specificity, and limit of detection against commercial NS1 tests and RT-PCR across infection stages
•3.2 Assess adaptability across serotypes and evaluate extension to other flaviviruses, enabling a single platform to support both individual diagnosis and community-level outbreak surveillance.
•3.3 Pharmacy-available, self-administered, room-temperature stable, and readable by eye. No clinic, no equipment; just accurate early diagnosis in the hands of patients who need it most and sensitivity, specificity, and limit of detection against commercial NS1 tests and RT-PCR across infection stages
decentralized data for real-time epidemiological response.
Chen et al. (2015) reported an DNA aptamer (S15), capable of binding the envelope protein domain III (EDIII) of dengue virus with high affinity, selected by SELEX against recombinant DENV-2 ED3. The aptamer adopts a G-quadruplex structure, with both the quadruplex fold and the 5′ sequence region essential for binding activity, and NMR titration experiments mapped its binding site to a highly conserved loop between the βA and βB strands of EDIII. Critically, S15 demonstrated neutralization activity against all four DENV serotypes, confirming that EDIII represents a serotype-conserved and immunogenically accessible target that a single aptamer can engage with broad cross-serotype coverage. This work was important because establishes the molecular rationale for targeting EDIII with a DNA aptamer in my project, directly informing the recognition module design of the aptamer–blocker–theophylline construct developed in Aim 1.
Rapid, Low-Cost Detection of Zika Virus Using Programmable Biomolecular Components
Pardee et al. (2016) established a landmark proof of concept for freeze-dried, cell-free (FDCF) diagnostic systems by embedding Zika virus–responsive toehold switch sensors into lyophilized paper discs that could be rehydrated with patient sample and read colorimetrically within one hour at room temperature. The system required no cold chain, no laboratory infrastructure, and no trained personnel, demonstrating that the full transcriptional and translational machinery of an E. coli TXTL system could survive lyophilization and retain functional activity after ambient storage. The paper further showed that toehold switch RNA sensors could be coupled to reporter genes — including LacZ — to produce visually interpretable colorimetric outputs, and that specificity could be maintained against closely related viral sequences including dengue. This work is the foundational precedent for the cell-free module of this project and validates the core architectural assumption that a toehold switch–LacZ TXTL circuit embedded on paper is a feasible and field-ready diagnostic format.
Novelty and Innovation
Existing dengue rapid diagnostics are architecturally static: they rely primarily on NS1 antigen detection or antibody-based serological targets, and cannot be reprogrammed when viral variants emerge or when a new pathogen requires detection. This project replaces that fixed architecture with a modular, programmable system in which each component: the aptamer, the blocker strand, the riboswitch, and the reporter — can be independently redesigned and reordered as a synthetic DNA sequence, enabling rapid adaptation without rebuilding the platform from scratch. Using multivalent AuNP-mediated upstream amplification of theophylline release seeks to transform a linear 1:1 detection stoichiometry into a signal-amplified cascade that pushes sensitivity toward the picomolar range without adding instrumentation. More broadly, the project expands the synthetic biology toolkit for diagnostics by demonstrating that a toehold-mediated strand displacement mechanism can serve as a chemical transducer between a protein recognition event and a nucleic acid–regulated gene expression circuit, as conceptual bridge between protein biosensing and programmable cell-free gene regulation that has broad applicability beyond dengue.
Why This Project Matters
Dengue is a systemic viral infection transmitted between humans by Aedes mosquitoes, with an estimated 390 million infections occurring annually across tropical and subtropical regions, placing an enormous burden on health systems across Latin America, Southeast Asia, and sub-Saharan Africa; regions where laboratory infrastructure is scarce and cold chain logistics are unreliable (Bhatt et al., 2013). In 2024 alone, over 14 million cases and nearly 9,500 deaths were recorded globally, representing a 12-fold increase compared to 2014, underscoring the accelerating urgency of this diagnostic gap (Haider et al., 2025). Current NS1 rapid tests achieve only 50–80% sensitivity, perform significantly worse in secondary infections, and lose reliability after day 7 of illness, meaning that a significant proportion of dengue patients in the highest-burden settings receive no actionable diagnosis at the moment when clinical management decisions matter most. By designing explicitly for pharmacy availability, room-temperature stability, self-administration, and a cost target below existing rapid tests, this project addresses not just a technical gap but a structural inequity in who gets access to accurate diagnosis. If successful, the platform would enable decentralized, real-time epidemiological surveillance through georeferenced positive result reporting, generating outbreak data from communities that currently fall entirely outside formal health surveillance networks, transforming individual diagnostic events into population-level public health intelligence. Beyond dengue, the modular aptamer–blocker–riboswitch–TXTL architecture is inherently adaptable: swapping the aptamer and adjusting the blocker sequence could redirect the platform toward other flaviviruses sharing structural homology with DENV.
Ethical Implications
My approach integrates several core bioethical principles into its foundational design. The principle of justice is central: dengue disproportionately burdens low-income populations in tropical regions who are systematically underserved by existing diagnostic markets, and deliberately designing a low-cost, pharmacy-available, self-administered test is an ethical act of redistributive access, ensuring that diagnostic innovation reaches those with the greatest need rather than those with the greatest purchasing power. The principle of beneficence governs the research itself: all experimental work involving human serum samples must proceed under IRB-approved protocols with full informed consent, de-identification of all samples, and adherence to BSL-2 biosafety standards. The georeferenced outbreak surveillance component raises privacy concerns that must be governed by the principle of non-maleficence: aggregating location data from self-administered tests could inadvertently expose individuals or communities to stigmatization, insurance discrimination, or government surveillance if not designed with strict anonymization, voluntary participation, and transparent data governance from the outset.
Concretely, several measures are required to ensure this project remains ethical throughout its development and deployment. IRB approval must be secured before any human sample is handled, and community engagement with populations in target endemic regions must occur before deployment, so that affected communities are stakeholders in the design of the tool rather than passive recipients of it. A critical uncertainty is whether the platform’s sensitivity in real patient serum will be sufficient to avoid false negatives, which carry their own harm: a patient who tests negative and does not seek care when in fact infected. To mitigate this, performance claims must be rigorously benchmarked against RT-PCR across all infection stages before any public deployment, and clear communication of the test’s limitations must accompany any over-the-counter format. An alternative to self-administration would be community health worker–administered testing, which adds a layer of human oversight and counseling but reduces accessibility, the tension between oversight and access is itself an ethical design decision that should be resolved in dialogue with the communities the platform is intended to serve.
Experimental design, techniques, tools and technology.
Retrieve the crystal structure of DENV EDIII from the Protein Data Bank (PDB) and identify an available antibody co-crystal data.
Select a anti-EDIII antibody (in my case 4E11), map its binding footprint on EDIII using published structural data (PDB).
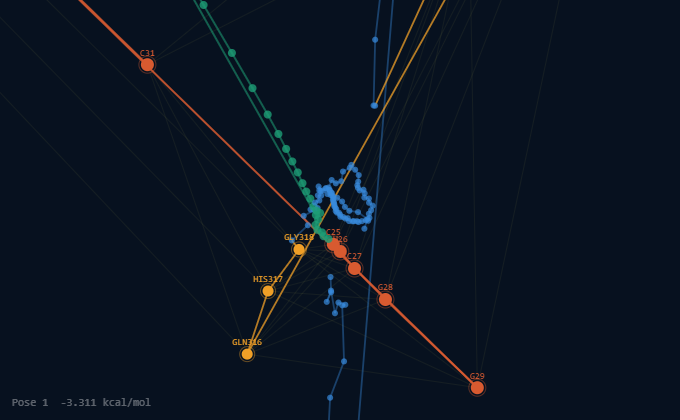
Identify an aptamer para EDIII and design 3D structure (RNA composer) and dock them computationally against the identified EDIII patch using Alphafold or Autodock.
Using pyMOL predicts binding affinity and position from the antibody and aptamer to ensure that they don’t overlap and compete for the union site
Design a blocker strand partially complementary to the aptamer’s binding region, such that hybridization sequesters the aptamer in the absence of EDIII; incorporate a theophylline-conjugated toehold overhang to enable downstream riboswitch activation upon EDIII-triggered strand displacement.
Use NUPACK or mfold to model secondary structures of the aptamer–blocker duplex, optimizing for low basal leak (ΔG favoring duplex), fast displacement kinetics upon EDIII binding, and minimal off-target folding.
Test the functionality of the strand displacement and blocker strand using the Asimov Kernel to validate the following design:
Estado ON:
A se une a EDIII → B se desplaza → T se libera
3 — Cell-Free Circuit Assembly and End-to-End Validation (WEEK 3)
Design a linear DNA construct encoding a theophylline riboswitch upstream of the lacZ reporter gene, using the canonical E. coli theophylline aptazyme sequence (Jenison et al.) as the regulatory element; optimize ribosome binding site (RBS) strength using the RBS Calculator (Salis Lab) to maximize dynamic range.
Prepare cell-free TXTL reactions using a commercial E. coli-based TXTL system; titrate theophylline concentration to establish dose-response curve for LacZ expression using X-gal colorimetric assay
Integrate all three modules: add recombinant EDIII to the aptamer–blocker construct, collect released theophylline, introduce it into the TXTL reaction, and measure LacZ output colorimetrically.
1 — Validate antibody-aptamer non-competition. Assemble and test the TXTL–riboswitch–LacZ circuit, confirming theophylline-dependent colorimetric output via X-gal. (WEEK 4/5)
Synthesize the DNA aptamer and blocker strand (Twist Bioscience or IDT); resuspend in TE buffer and verify integrity by agarose gel electrophoresis.
Incubate aptamer, capture antibody, and recombinant EDIII simultaneously; run competitive ELISA to confirm that aptamer and antibody bind non-competing EDIII epitopes.
Prepare the riboswitch–lacZ construct as a linear gBlock; verify sequence by Sanger sequencing.
Set up TXTL reactions with the construct and X-gal as substrate; titrate theophylline and record colorimetric output at 2 hours to confirm theophylline-dependent LacZ activation.
2 — Characterize EDIII-triggered strand displacement and theophylline release by native PAGE; integrate all three modules in solution and confirm end-to-end colorimetric output (WEEK 5/6)
Incubate the aptamer–blocker construct with increasing concentrations of recombinant EDIII at room temperature
Run native PAGE to visualize blocker displacement: the duplex band should decrease and a free aptamer band should appear as EDIII concentration increases.
Quantify released theophylline by absorbance or fluorescence according to the conjugation method used
Transfer the displacement reaction supernatant directly into the TXTL–X-gal reaction and monitor color development to confirm end-to-end signal activation
Run negative controls: scrambled aptamer, buffer without EDIII, and unrelated antigen
3 — Lyophilize TXTL–Xgal components onto Whatman paper discs with trehalose as stabilizer; rehydrate with EDIII-spiked samples and confirm visual readout at room temperature. (WEEK 6/7)
Mix TXTL components with the riboswitch–lacZ construct, X-gal, and trehalose; spot 2–5 µL per Whatman No. 1 paper disc.
Lyophilize discs using a benchtop lyophilizer; store at room temperature in sealed bags with desiccant.
Rehydrate each disc with 5–10 µL of human serum spiked with recombinant EDIII at varying concentrations.
Incubate at room temperature and record colorimetric output visually and by portable spectrophotometry.
Compare lyophilized discs against fresh reactions to assess activity retention after lyophilization.
1 — Evaluate sensitivity, specificity, and limit of detection against commercial NS1 tests and RT-PCR across infection stages (WEEK 8 - 12)
LOD determination: Prepare serial dilutions of recombinant EDIII in dengue-negative human serum (blood samples); run each concentration on lyophilized discs and record the lowest concentration producing a visible colorimetric signal. Compare against a standard curve generated by spectrophotometry.
Test the platform against a panel of potential cross-reactants including other flaviviruses antigens at equivalent concentrations for confirm absence of colorimetric output in all non-DENV samples.
Obtain samples from patients at early (days 1–3), acute (days 4–7), and late (days 8+) infection stages; run each sample on the lyophilized disc platform alongside a commercial NS1 rapid test (e.g., SD Bioline Dengue NS1) and RT-PCR to generate side-by-side sensitivity and specificity data.
Calculate sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) for each infection stage; construct ROC curves to compare diagnostic performance across platforms
2 — Assess adaptability across serotypes and evaluate extension to other flaviviruses, enabling a single platform to support both individual diagnosis and community-level outbreak surveillance (WEEK 12 - 30)
Express and purify recombinant EDIII from all four DENV serotypes; verify identity and purity by SDS-PAGE and Western blot using serotype-specific antibodies.
Test the existing aptamer against EDIII from each serotype by BLI or competitive ELISA to map cross-reactivity; if binding drops significantly for any serotype, design and order serotype-specific aptamer variants.
Spot aptamer variants for each serotype in spatially distinct zones on a single paper disc; rehydrate with DENV1–4 spiked samples and assess whether zone-specific colorimetric output enables serotype discrimination visually.
Test platform performance against Zika and West Nile EDIII to assess structural homology-driven cross-detection; evaluate whether riboswitch variants or aptamer modifications can tune selectivity for surveillance applications targeting multiple co-circulating flaviviruses.
Recruit volunteer participants, provide them the lyophilized disc test with a one-page illustrated instruction sheet only; record time-to-result, error rate, and ability to correctly interpret colorimetric output without assistance.
Calculate per-test material cost including paper disc, lyophilized TXTL components, aptamer, and packaging; benchmark against commercial NS1 rapid tests and RT-PCR to confirm economic accessibility for pharmacy distribution.
Store lyophilized discs at 37°C and 60% relative humidity for 4 weeks to simulate tropical field conditions; test activity retention weekly by rehydrating with EDIII-spiked samples and comparing colorimetric output to freshly prepared discs.
Design a data capture tool (QR code printed on the disc packaging) linking to a georeferenced reporting form, allowing positive results self-reported from any location to feed into a real-time outbreak map, enabling decentralized surveillance without requiring clinic visits or laboratory confirmation.
Identify the regulatory framework applicable in target endemic countries; map the evidence package required for approval as an over-the-counter diagnostic and outline the validation study design needed to meet those standards.
Techniques Checklist
☑ DNA Construct Design
☑ Databases (RCSB PDB, NCBI, NUPACK)
☑ Protein Design
☑ Use of Benchling
☑ Cell-Free Systems
☑ Primer Design or Selection
Technique Deep-Dives
DNA Construct Design:
The DNA design is a linear construct encoding a theophylline-responsive riboswitch upstream of a T7 promoter–driven lacZ reporter gene. The full sequence — including the validated Jenison et al. aptazyme, the lacZ coding sequence codon-optimized for E. coli cell-free expression, and a T7 terminator were assembled in silico using Benchling andwould be verified for OFF/ON structural states using NUPACK and ViennaRNA before synthesis. The construct will be ordered as a linear gBlock from Twist Bioscience, requiring no cloning, as linear DNA is compatible with TXTL reactions, thereby simplifying the workflow. In parallel, the aptamer–blocker–theophylline module will also be designed as synthetic DNA oligos with a precisely tuned toehold region (6–10 nt) and blocker GC content optimized computationally to minimize leak in the OFF state while ensuring fast strand displacement kinetics upon EDIII binding.
Cell-Free Systems:
The cell-free (TXTL) system is the core signal-generating engine of the biosensor, chosen because it operates without living cells, making it compatible with lyophilization and room-temperature storage, essential for a field-deployable diagnostic. Using a commercial E. coli–based TXTL kit, the riboswitch–lacZ construct will be added directly to the cell-free reaction alongside X-gal as colorimetric substrate, and theophylline will be titrated across a concentration range to establish a dose-response curve defining the minimum activation threshold. Once validated in solution, the complete TXTL mixture — including the gBlock, substrate, and trehalose as a cryoprotectant — will be spotted onto Whatman No. 1 paper discs and lyophilized using a benchtop lyophilizer, producing a dry, stable disc that reactivates upon addition of patient sample. This freeze-dried cell-free format eliminates cold chain requirements entirely and enables single-step operation: the patient adds sample, and a visible color change within 60 minutes or less indicates a positive dengue result.
In the EDIII and aptamer S15G3 interactions, binding interface involves the region of EDIII: residues 311–322 (the β-A/BC region) , suggesting stacking interactions with the RNA bases. On the aptamer side, the most engaged nucleotides are located in the 3′ stem-loop (C18–U26, G28–C31), which is consistent with RNA aptamers that use hairpin structures for protein recognition.
In the EDIIand antibody 4E11 interactions, involve the region: residues 20–90 that forms part of the antibody-binding epitope. These contacts likely contribute to specific antigen recognition and may involve exposed β-sheet surfaces characteristic of ED3. Aromatic and charged residues within this region may participate in hydrogen bonding, hydrophobic, and electrostatic interactions that stabilize the complex.
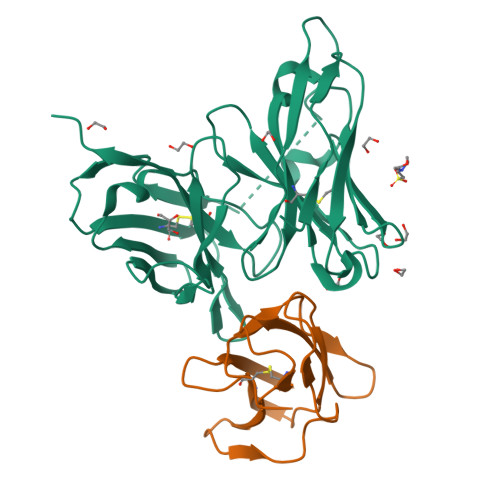
Overall, these results suggest that the capture antibody (4E11) and the aptamer (S15G3) occupy distinct binding sites on the ED3 domain. This ensures that neither molecule interferes with the binding activity of the other; instead, they provide a dual-recognition mechanism that is expected to enhance both target capture and molecular recognition
Aspects to validate
The design of a linear DNA construct encoding a theophylline-responsive riboswitch upstream of a lacZ reporter gene, representing the core signal-generating module of the biosensor. Confirme that this construct produces theophylline-dependent LacZ expression in a cell-free TXTL system is the critical proof-of-concept that underpins the entire downstream architecture, if the riboswitch–LacZ circuit does not activate reliably in response to theophylline, no other module in the system has diagnostic value.
Detailed Protocol
Retrieve the validated theophylline aptazyme sequence (Jenison et al., 1994) and the E. coli lacZ coding sequence from NCBI (GenBank accession V00296); assemble the full construct in Benchling in the following order: T7 promoter → theophylline riboswitch → RBS → lacZ → T7 terminator.
Verify OFF and ON secondary structures of the riboswitch computationally using NUPACK: confirm that in the absence of theophylline the RBS is sequestered within a stable stem-loop, and that theophylline binding shifts the equilibrium toward an open conformation exposing the RBS.
Design the construct as a linear gBlock (2,500–3,000 bp) and submit the sequence to Twist Bioscience for synthesis; upon arrival, resuspend in TE buffer to 10 ng/µL stock and verify size by 1% agarose gel electrophoresis.
Prepare a theophylline titration series in nuclease-free water: 0, 0.1, 0.5, 1, 2, 5, and 10 mM theophylline stocks.
Set up TXTL reactions on ice using myTXTL master mix following manufacturer protocol; add gBlock and X-gal as colorimetric substrate to each reaction; add theophylline at each concentration across reactions; include a no-DNA negative control and a constitutive lacZ positive control.
Load reactions into a 96-well plate; incubate in a plate reader and measure absorbance at 570 nm every 10 minutes for 3 hours.
If an Opentrons OT-2 robot is available, program the liquid handling steps as a Python protocol to ensure reproducibility and minimize pipetting error across replicates; run in triplicate for each theophylline concentration.
Plot absorbance at 570 nm versus time for each theophylline concentration; calculate fold-activation relative to the 0 mM control at the 120-minute timepoint; generate a Hill-function dose-response curve fitting signal vs. log[theophylline].
Synthetic Biology Techniques Utilized
The validation integrates multiple synthetic biology techniques. DNA construct design was performed entirely in silico using Benchling for sequence assembly and NUPACK for thermodynamic structural prediction of riboswitch OFF/ON states, ensuring the construct was computationally validated before synthesis. The construct would be ordered as a linear gBlock through Twist Bioscience, applying the principle that linear DNA is directly compatible with cell-free TXTL systems and requires no cloning, reducing turnaround time and eliminates transformation-related failure modes. Cell-free reaction setup and theophylline titration were performed using standard pipetting technique under the myTXTL manufacturer protocol, with colorimetric LacZ output measured via X-gal absorbance for visual and quantitative signal readout. Where available, an Opentrons OT-2 liquid handling robot would be programmed in Python to automate the reaction assembly, ensuring consistent volumes across all replicates and reducing human pipetting variability as a confounding source of error in the dose-response data.
Additional information
References
• Bhatt, S., Gething, P. W., Brady, O. J., Messina, J. P., Farlow, A. W., Moyes, C. L., Drake, J. M., Brownstein, J. S., Hoen, A. G., Sankoh, O., Myers, M. F., George, D. B., Jaenisch, T., Wint, G. R. W., Simmons, C. P., Scott, T. W., Farrar, J. J., & Hay, S. I. (2013). The global distribution and burden of dengue. Nature, 496(7446), 504-507. https://doi.org/10.1038/nature12060
• Chen, H., Hsiao, W., Lee, H., Wu, S., & Cheng, J. (2015). Selection and Characterization of DNA Aptamers Targeting All Four Serotypes of Dengue Viruses. PLoS ONE, 10(6), e0131240. https://doi.org/10.1371/journal.pone.0131240
• Haider, N., Hasan, M. N., Onyango, J., Billah, M., Khan, S., Papakonstantinou, D., Paudyal, P., & Asaduzzaman, M. (2025). Global dengue epidemic worsens with record 14 million cases and 9000 deaths reported in 2024. International Journal Of Infectious Diseases, 158, 107940. https://doi.org/10.1016/j.ijid.2025.107940
• Pardee, K., Green, A. A., Takahashi, M. K., Braff, D., Lambert, G., Lee, J. W., Ferrante, T., Ma, D., Donghia, N., Fan, M., Daringer, N. M., Bosch, I., Dudley, D. M., O’Connor, D. H., Gehrke, L., & Collins, J. J. (2016). Rapid, Low-Cost Detection of Zika Virus Using Programmable Biomolecular Components. Cell, 165(5), 1255-1266. https://doi.org/10.1016/j.cell.2016.04.059
• Tricou, V., Vu, H. T., Quynh, N. V., Nguyen, C. V., Tran, H. T., Farrar, J., Wills, B., & Simmons, C. P. (2010b). Comparison of two dengue NS1 rapid tests for sensitivity, specificity and relationship to viraemia and antibody responses. BMC Infectious Diseases, 10(1), 142. https://doi.org/10.1186/1471-2334-10-142
Group Final Project
Computational Engineering of the MS2 Lysis Protein to Improve Stability, Titers, and Toxicity
After reviewing the provided literature on the MS2 lysis protein (L) and discussing the project aims, our group has decided to focus on three interconnected goals:
Goal 1: Increase the stability of the L protein
As the “easiest” goal, it is the most computationally tractable. A stabilized protein is less prone to degradation and misfolding, which could directly lead to higher functional titers and serve as a robust starting point for any subsequent engineering.
Goal 2: Increase bacteriophage titers through improved lysis efficiency.
Phage therapy relies on high phage titers for effective bacterial killing and scalable manufacturing, but phage production can be limited by inefficient lysis or poor coordination between phage replication and host destruction. Improving the efficiency and timing of host cell lysis can therefore directly increase the number of phage particles released per infected cell.
The MS2 L protein is a small 75–amino acid membrane protein that triggers bacterial lysis and is essential for the release of new phage particles. In the paper Mutational analysis of the MS2 lysis protein L, it is described how MS2 L functions as a single-gene lysis protein that disrupts bacterial cell envelope integrity without classical enzymatic activity. Additionally, L interacts with the host chaperone DnaJ, which modulates its activity and timing of lysis. In MS2 Lysis of Escherichia coli Depends on Host Chaperone DnaJ it is shown that lysis timing strongly affects the number of virions produced before the host cell bursts, meaning that engineering improved L variants may increase overall phage titers.
Goal 3: Increase the toxicity of the lysis protein.
This proposal addresses the subproblem of increasing the toxicity of the L lysis protein from Bacteriophage MS2. Instead of random mutagenesis, toxicity will be approached as a multi-factor optimization problem involving structural stability, membrane insertion, oligomerization efficiency, and expression kinetics in Escherichia coli. The objective is to design L variants that enhance membrane disruption while maintaining proper folding and stability.
E. coli chaperone DnaJ.
Additionally, we will explore disrupting the interaction between the L protein and the E. coli chaperone DnaJ.
The reading “Identification MS2 lysis protein dependency on DnaJ” establishes this interaction as critical for function. By computationally predicting and then disrupting this interface, we can test its necessity and potentially create a DnaJ-independent lysis mechanism, offering a new avenue for controlling lysis timing.
Together, these three goals form a coherent strategy: stabilizing the L protein may improve its folding and expression, which can increase functional titers, while further engineering of membrane disruption and host interactions may increase toxicity and lysis efficiency.
Proposed Computational Tools and Approaches
Proposed Tools and Approaches We will build a computational pipeline using the tools introduced in recitation and the provided resources. The key steps and tools are:
Step 1: Structural Modeling of the L Protein
Tool: AlphaFold2 (via ColabFold for ease of use).
Why: No high-resolution experimental structure of the full-length MS2 L protein exists. A reliable 3D model is the absolute foundation for all downstream analysis, allowing us to visualize which parts are structured vs. disordered.
Step 2: Modeling the L-DnaJ Complex
Tool: AlphaFold-Multimer.
Why: To disrupt the interaction, we first need to know where it occurs. AlphaFold-Multimer is the current state-of-the-art for predicting protein-protein complexes and will generate a testable model of the L protein bound to E. coli DnaJ.
Step 3: In Silico Mutagenesis for Stability
Tool: Rosetta (or FoldX). Specifically, the ddg_monomer application for predicting changes in folding free energy (ΔΔG).
Why: These tools are parameterized using vast amounts of experimental data on protein stability. They can systematically mutate each residue in our L protein model and predict whether the change (e.g., A->V) makes the protein more stable (negative ΔΔG) or less stable (positive ΔΔG).
Step 4: Visualizing and Selecting Interface Mutations
Tool: PyMOL and the HTGAA Protein Engineering Tools spreadsheet.
Why: We will use PyMOL to visually inspect the predicted L-DnaJ complex from Step 2 and select residues at the interface. We will then use the spreadsheet to check the conservation of those residues and manually design mutations (e.g., swapping a large hydrophobic residue for a charged one) predicted to break the interaction.
Protein Language Models (PLMs)
Protein language models such as ESM or ProtBERT will be used to perform in silico mutagenesis on the MS2 L protein sequence. These models can suggest mutations that preserve structural and functional constraints learned from large protein datasets.
This approach allows us to generate multiple candidate mutations across the L protein, avoid mutations likely to disrupt folding, and explore sequence space beyond naturally occurring variants
AlphaFold Structure Prediction
Each candidate L variant will be analyzed using AlphaFold to predict protein structure and membrane topology. Since the C-terminal transmembrane region is essential for lytic activity, structural prediction will help identify mutations that preserve this functional domain.
Structural predictions will also help identify:
misfolded variants
mutations that destabilize the transmembrane region
variants that may alter oligomerization or membrane insertion
Interaction Modeling with Host Proteins
Because MS2 L interacts with the DnaJ chaperone, which affects lysis timing, candidate variants can be evaluated using AlphaFold-Multimer to predict changes in the L–DnaJ interaction.
This could help identify variants that:
maintain necessary folding assistance
reduce excessive dependency on host chaperones
improve robustness of lysis across physiological conditions
Proposed Computational Strategy
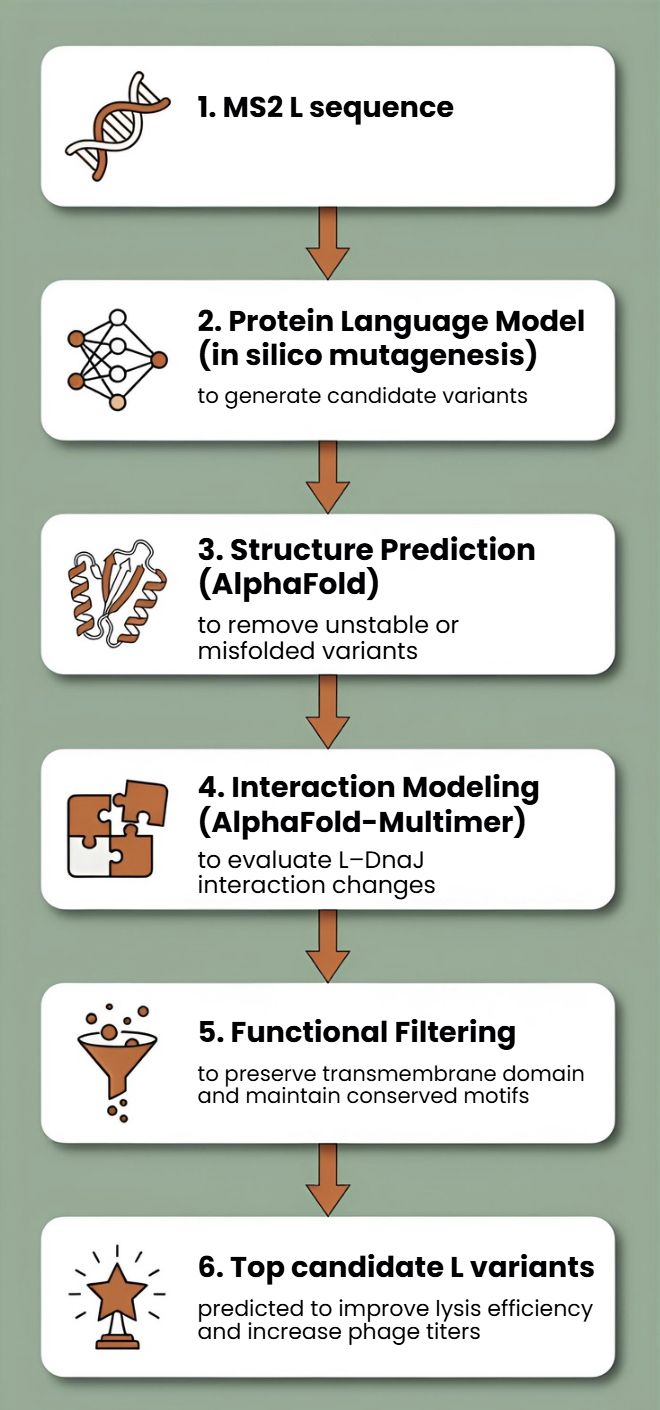
First, protein language models (e.g., ESM-2, ProtT5) will be used to perform directed in silico mutagenesis. These models capture evolutionary constraints and residue interactions, enabling the generation of structurally plausible variants while identifying mutation-tolerant and functionally critical positions. This step efficiently reduces the combinatorial search space.
Second, predicted variants will be structurally evaluated using AlphaFold2 for monomer folding and AlphaFold - Multimer to assess oligomerization and interaction with host factors such as DnaJ.
Third, membrane compatibility will be analyzed using membrane-aware modeling (RosettaMP) and selected molecular dynamics simulations.
Fourth, ΔΔG prediction tools (e.g., FoldX, Rosetta energy functions) will filter out destabilizing mutations.
In parallel, codon optimization algorithms will redesign selected variants for improved expression in E. coli, as toxicity depends on both structure and intracellular concentration.
Potential Pitfalls
Pitfall 1: Dynamic Regions and Model Quality
The L protein is small and likely has flexible/disordered regions, especially in its N-terminal domain.
Pitfall 2: Stability vs. Function Trade-off
A mutation that makes the protein more stable in its monomeric state might prevent it from undergoing the necessary conformational changes to oligomerize and form a pore in the membrane.
Pitfall 3: Lack of Membrane Context
Our stability predictions (Rosetta) are performed in a virtual “aqueous” environment and do not account for the energetic complexity of the lipid bilayer.
Limited biological data: There is still limited structural and mechanistic knowledge about MS2 L.
Cellular context not captured computationally Protein modeling tools may not fully capture membrane environment.
One limitation is the scarcity of quantitative datasets linking specific mutations to measured lysis kinetics.
L-Protein Mutants
To generate the first two mutations in the L protein of bacteriophage MS2 within the transmembrane region, I selected the top candidates predicted by the Python models and the spreadsheet analysis for that region. I applied the same approach to the soluble region, ensuring that all mutations were introduced at amino acid positions with less constrained mutability.