Week 2 HW: DNA Read, Write & Edit
.webp) (squinting might help)
(squinting might help)
Part 1: Benchling & In-silico Gel Art
Documentation

First of all I started by making a digest with a single enzyme at a time.
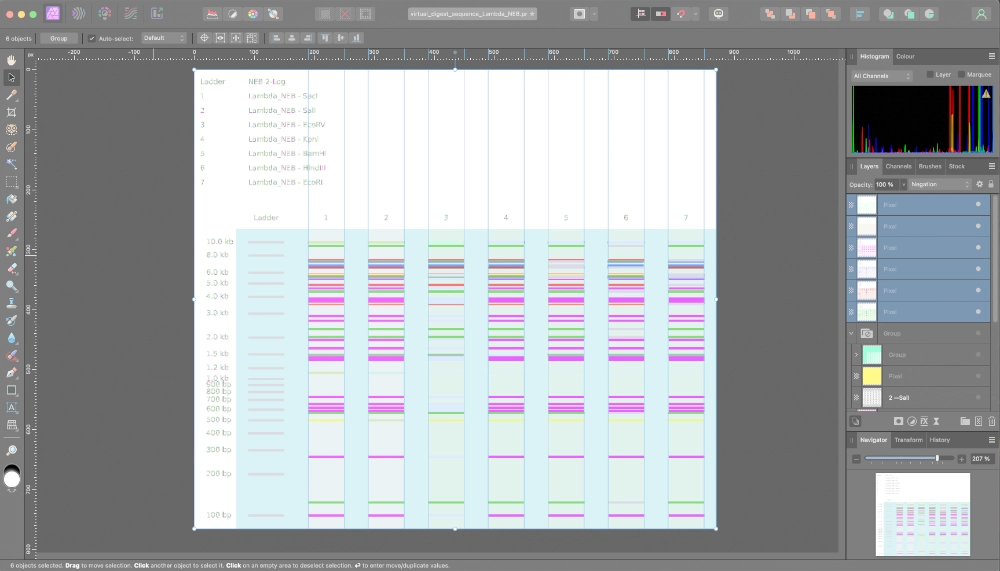
Then tried to color code the result of every enzyme and superimpose them on top of each other so to create a “grid” were I would make my design. I soon understood it would be way too confusing, plus, that the result of using a combination of enzymes doesn’t necessarily correspond to the superimposition of the lines created by each enzyme separately.
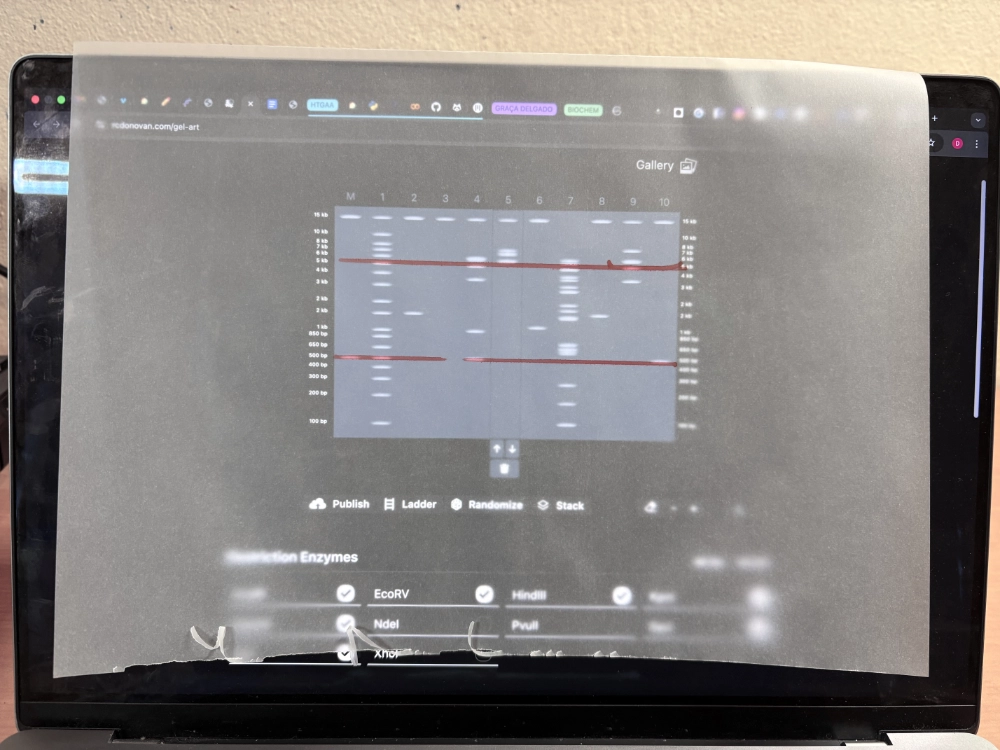
Then I started using Ronan’s website to iterate on combinations of enzymes + using some unconventional techniques.
Final Result — “HTGAA”
.png)
For the cover image I just edited out some of the space between lines of the same letter to make it more perceptible
- This was a fun exercise that allowed me to visually understand the logic of enzyme’s digests and pay attention to some details I might haved overlooked otherwise.
Part 3: DNA Design Challenge
3.1. Choose your protein
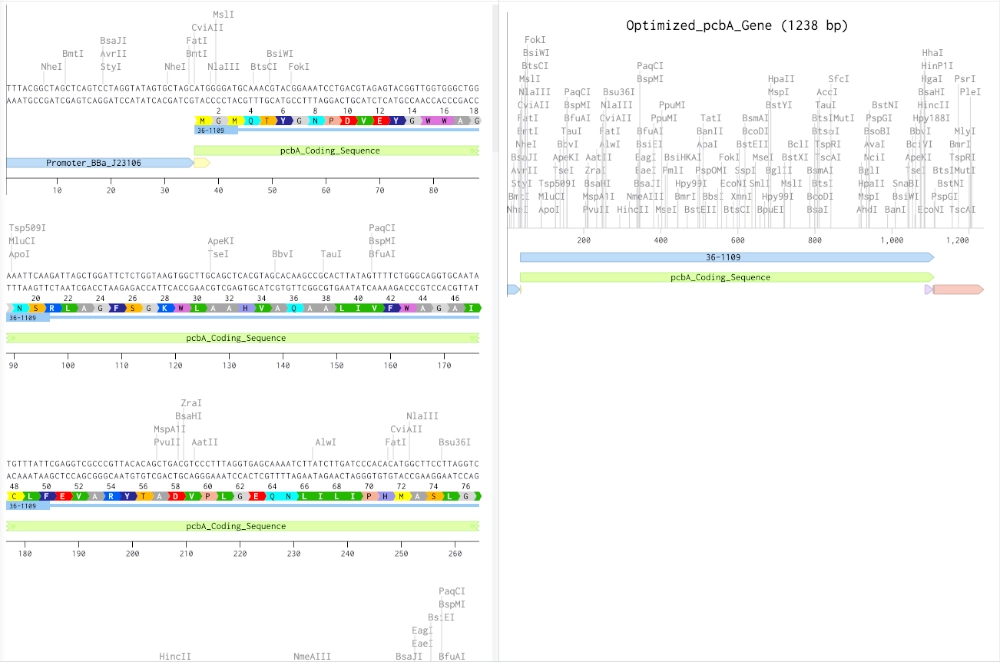
The protein I’d like to work with is the prochlorophyte chlorophyll-binding (Pcb) protein which is the light-harvesting protein (LHP) in prokaryotes that uses only chlorophyll as their photosensitive pigment. A modified version of this protein could be used to efficiently absorb light causing degradation of chlorophyll a, b and d molecules into porphyrin-type derivatives — that can be used to bind iron and create photographic images in a cell-free system.
(from NCBI chlorophyll a/b binding protein [Prochloron didemni])
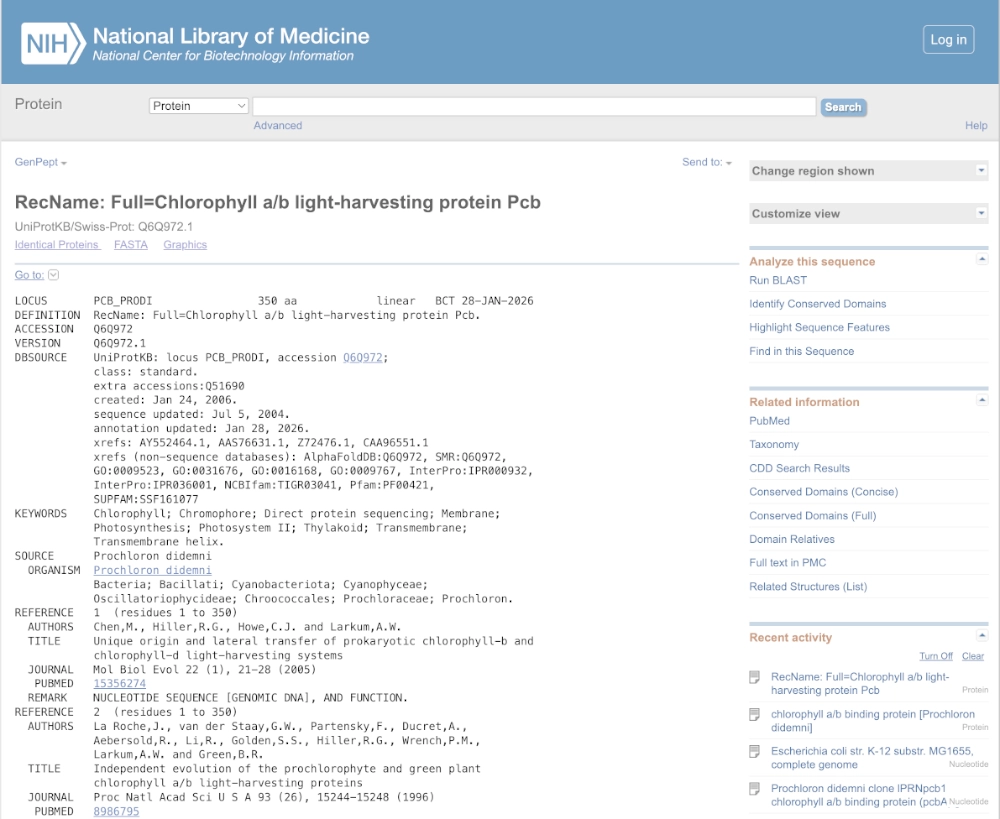
3.2. Reverse Translate — AA to DNA
(converted using bioinformatics.org)
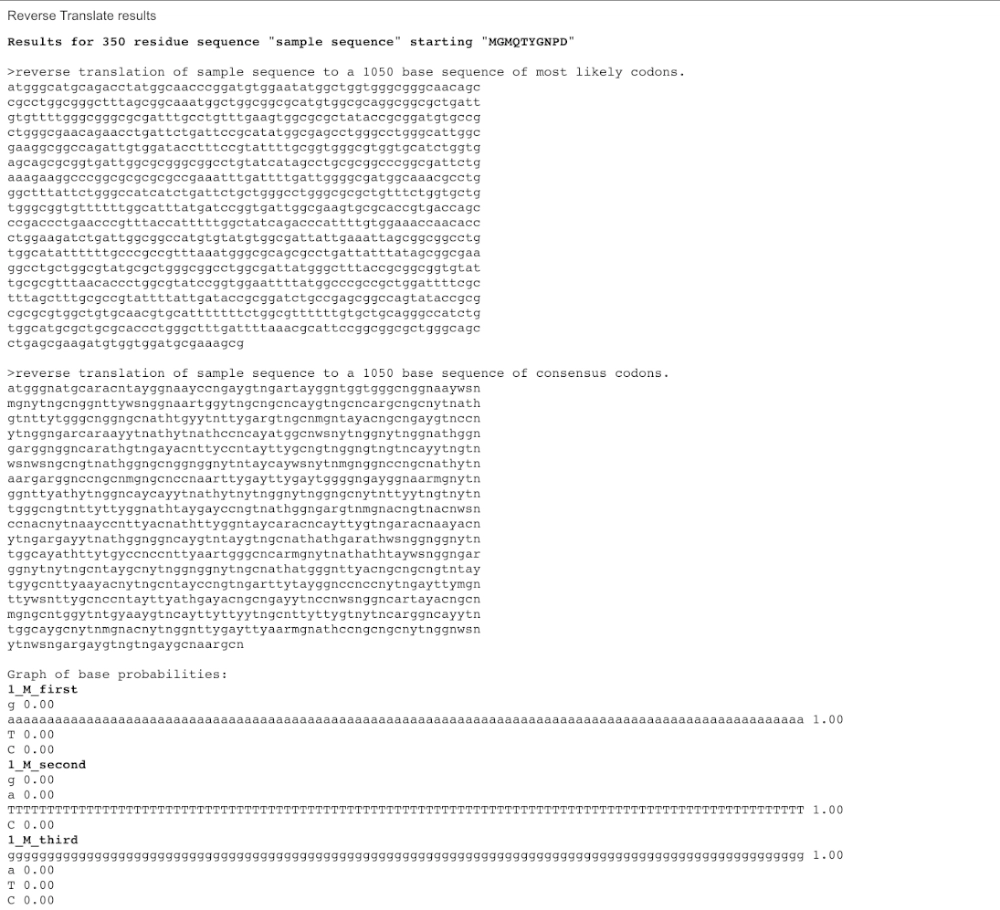
3.3. Codon optimization.
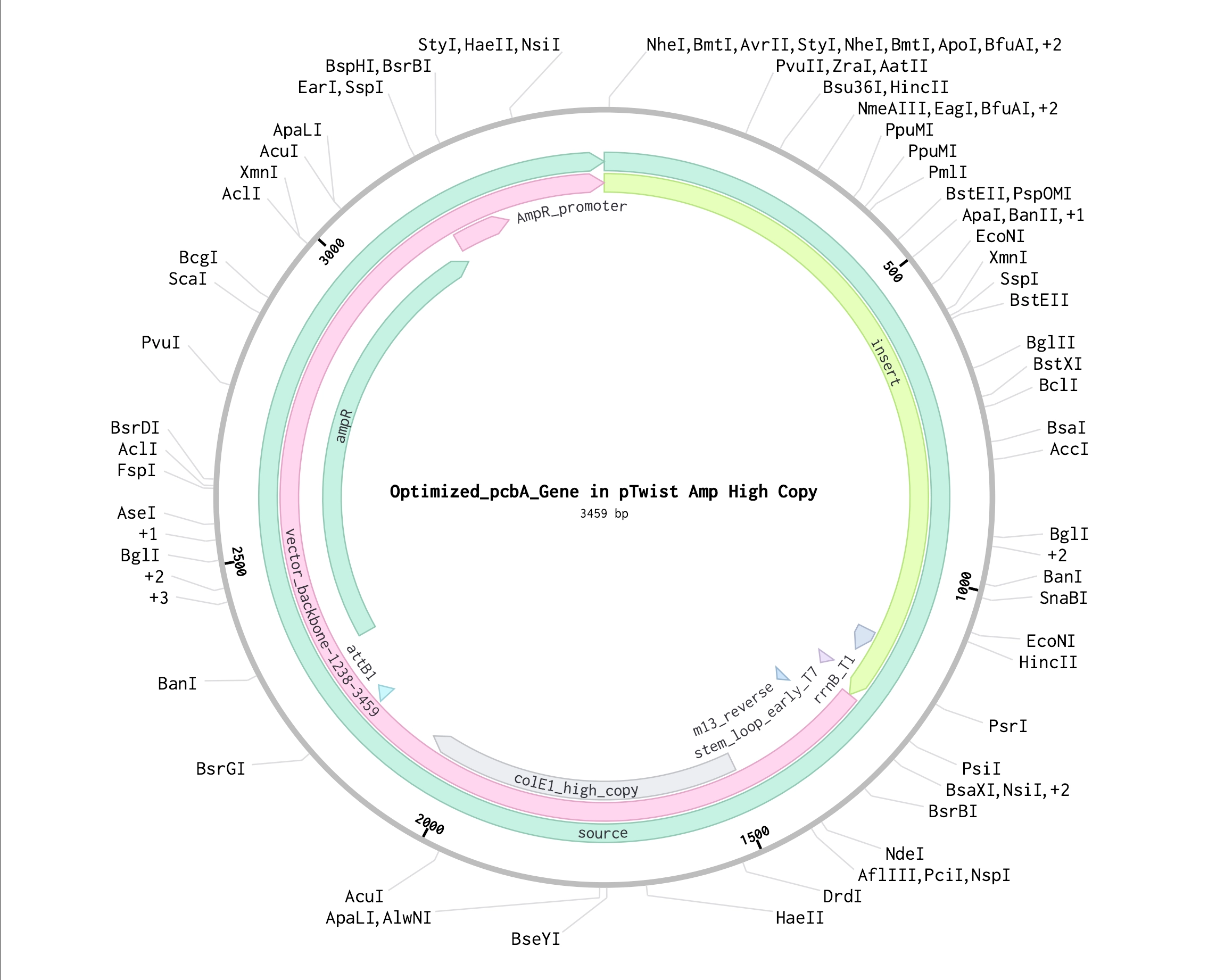
Codon optimization is an important process due to different organisms having and producing different amino acids in different proportions. So, if a gene codes for a rare amino acid, it might slow the translation process and therefore the folding of the protein and might even render the protein non-functional. In this case, the gene should be optimized for e. coli which is probably the best choice since the primary objective is to express a protein that is going to be used in a cell-free system and it is the simplest organism to work with. For this codon optimization I avoided Type IIS enzyme recognition sites for BsaI, BsmBI, and BbsI — these are some enzymes that are useful for ligation with plasmid backone.
(made with Codon Optimization Tool | Twist Bioscience)
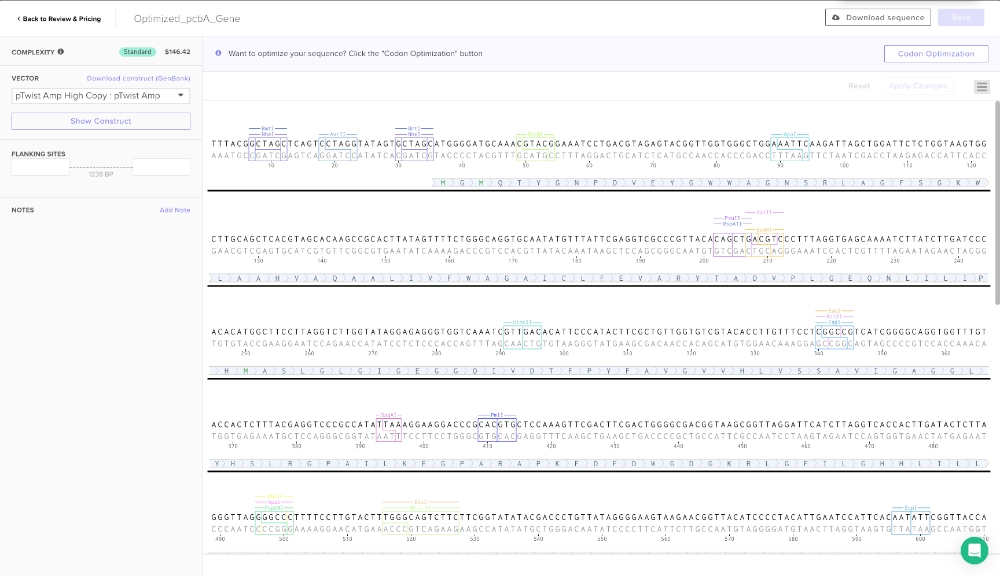
3.5. How does it work in nature/biological systems?
1. Describe how a single gene codes for multiple proteins at the transcriptional level.
In biological systems, a single gene can code for multiple proteins at the transcriptional level through the process of alternative splicing (in eukaryotes)— a process where different combinations of exons from the same pre-mRNA molecule are joined together. This process happens inside the nucleus during the processing of pre-mRNA, leading to the synthesis of multiple protein isoforms, which are related forms of the same protein, but with different structural or functional properties. Another process that allows for a single gene to code for multiple proteins (both in eukaryotes and prokaryotes) is the action of alternative promoter genes that create different initiation sites, affecting which exons are included in transcription.
2. Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!!
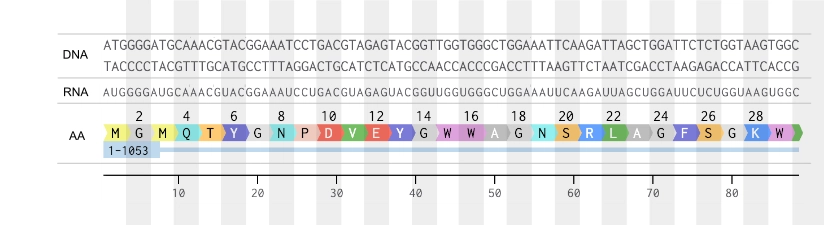
Part 4: Prepare a Twist DNA Synthesis Order