Week 4 HW: Protein Design Part1
Part A. Conceptual Questions
Part B: Protein Analysis and Visualization
3. Identify the structure page of your protein in RCSB
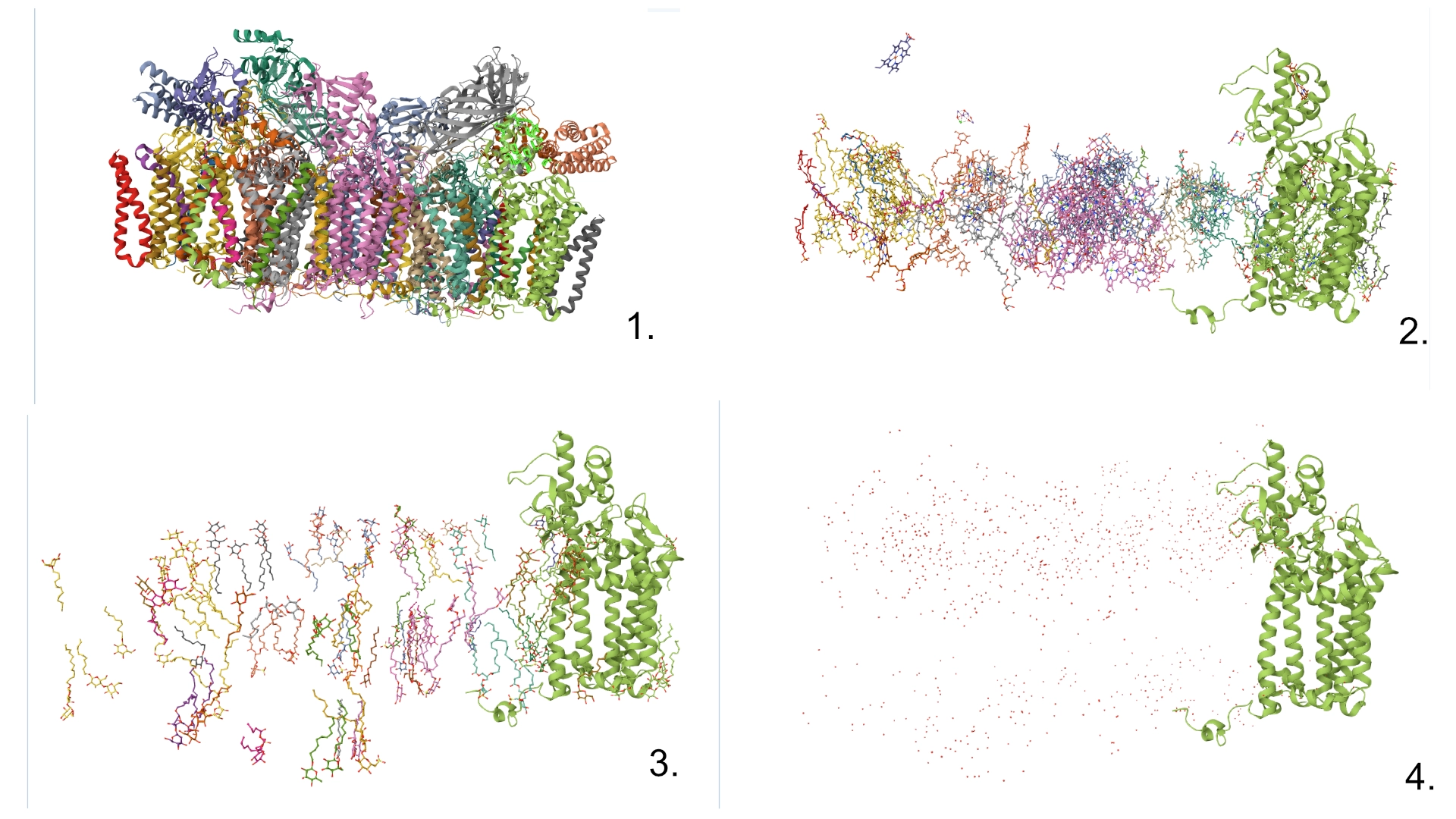
- First solved in 2020 with 2.58 resolution, version of 2021 is the one with best resolution 1.93
- Yes, there are present the other proteins(1.) that constitute Photosystem II as well as ligands(2.), sugars(3.), ions and water molecules(4.)
4. Open the structure of your protein in any 3D molecule visualization software
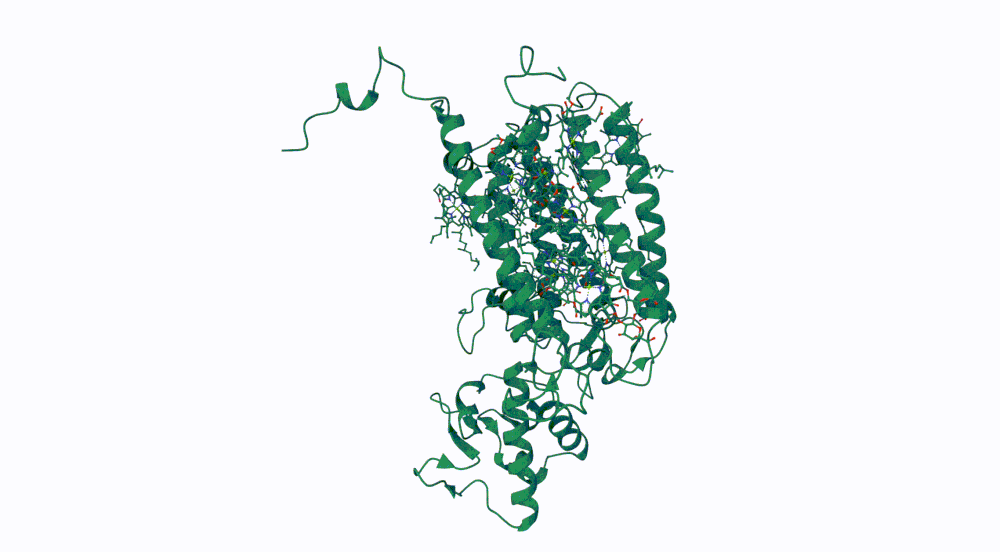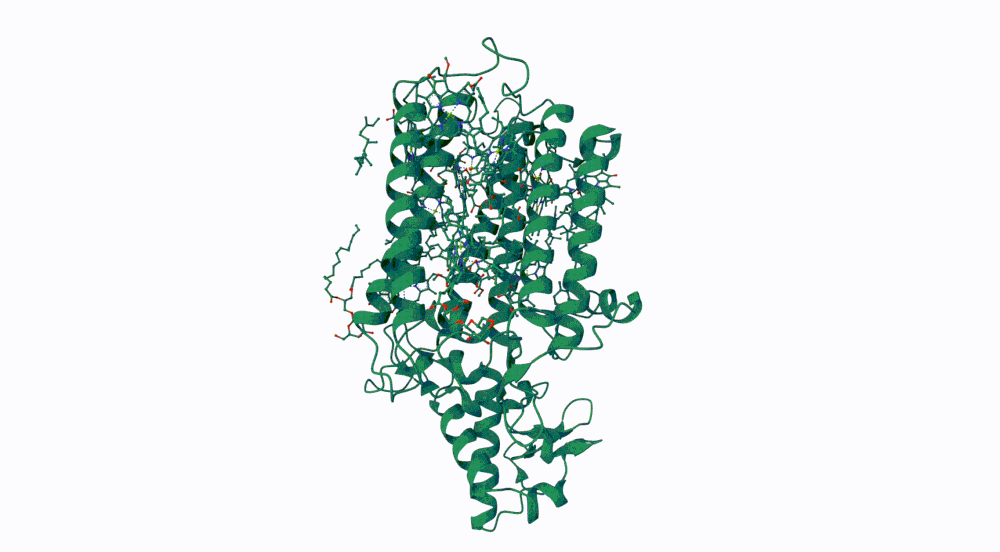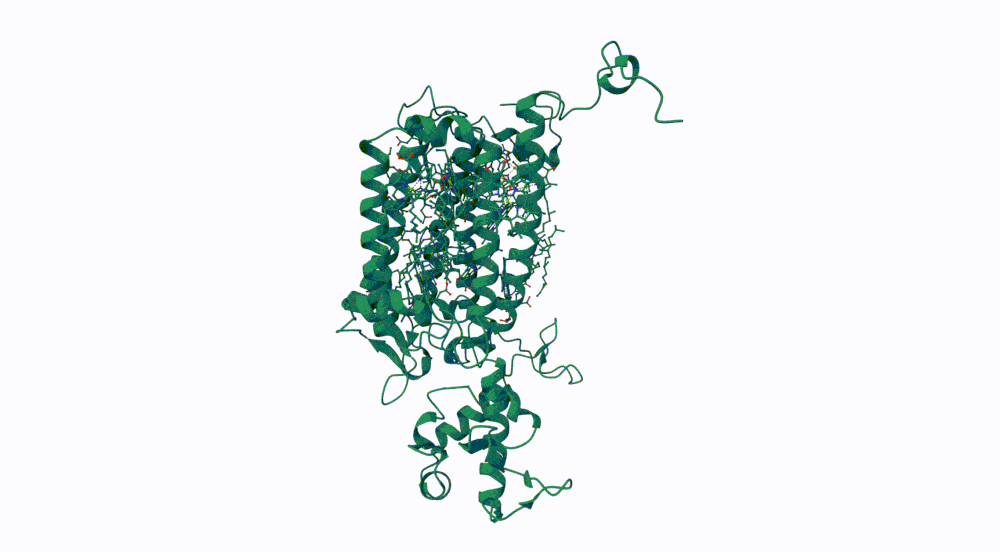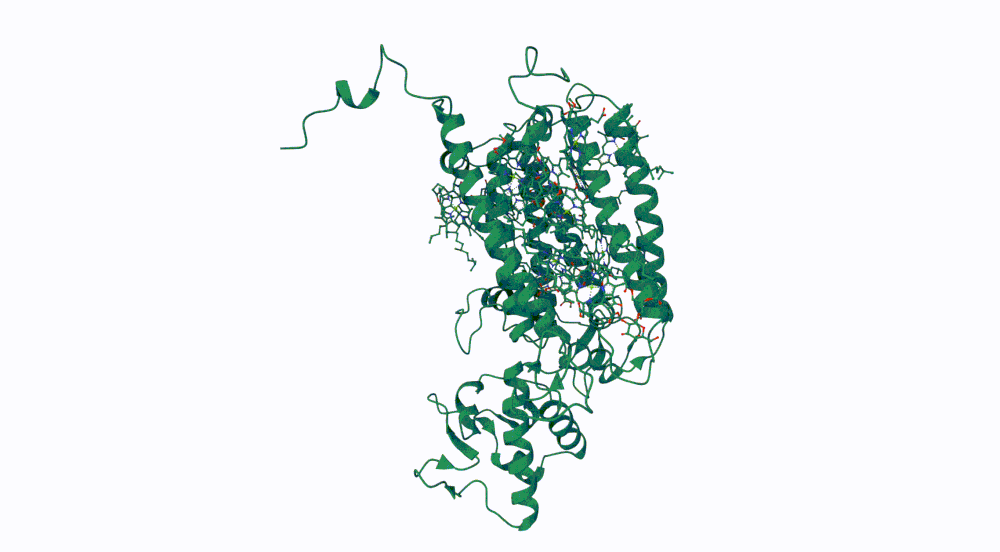
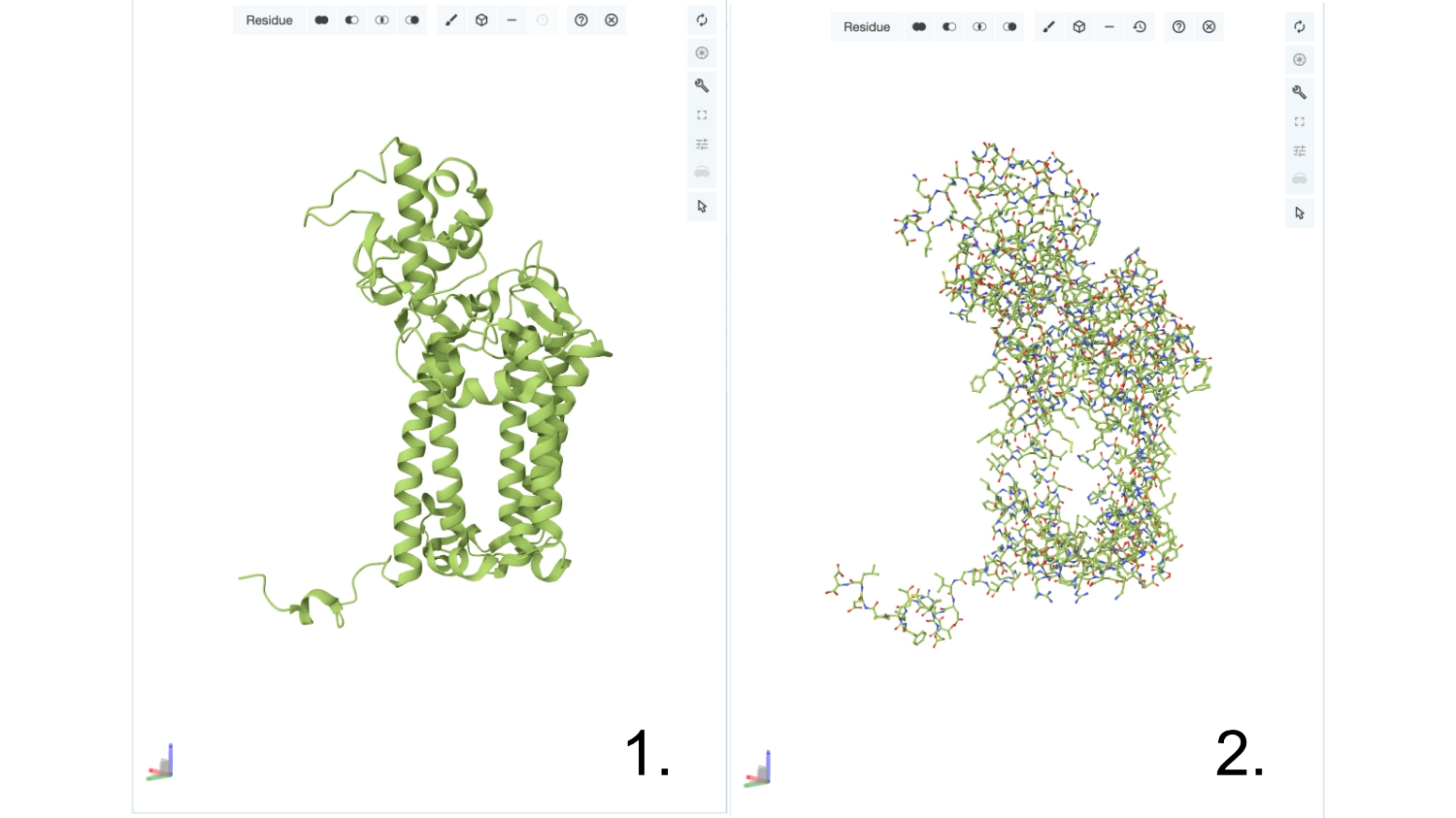
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Color the protein by secondary structure. Does it have more helices or sheets? Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
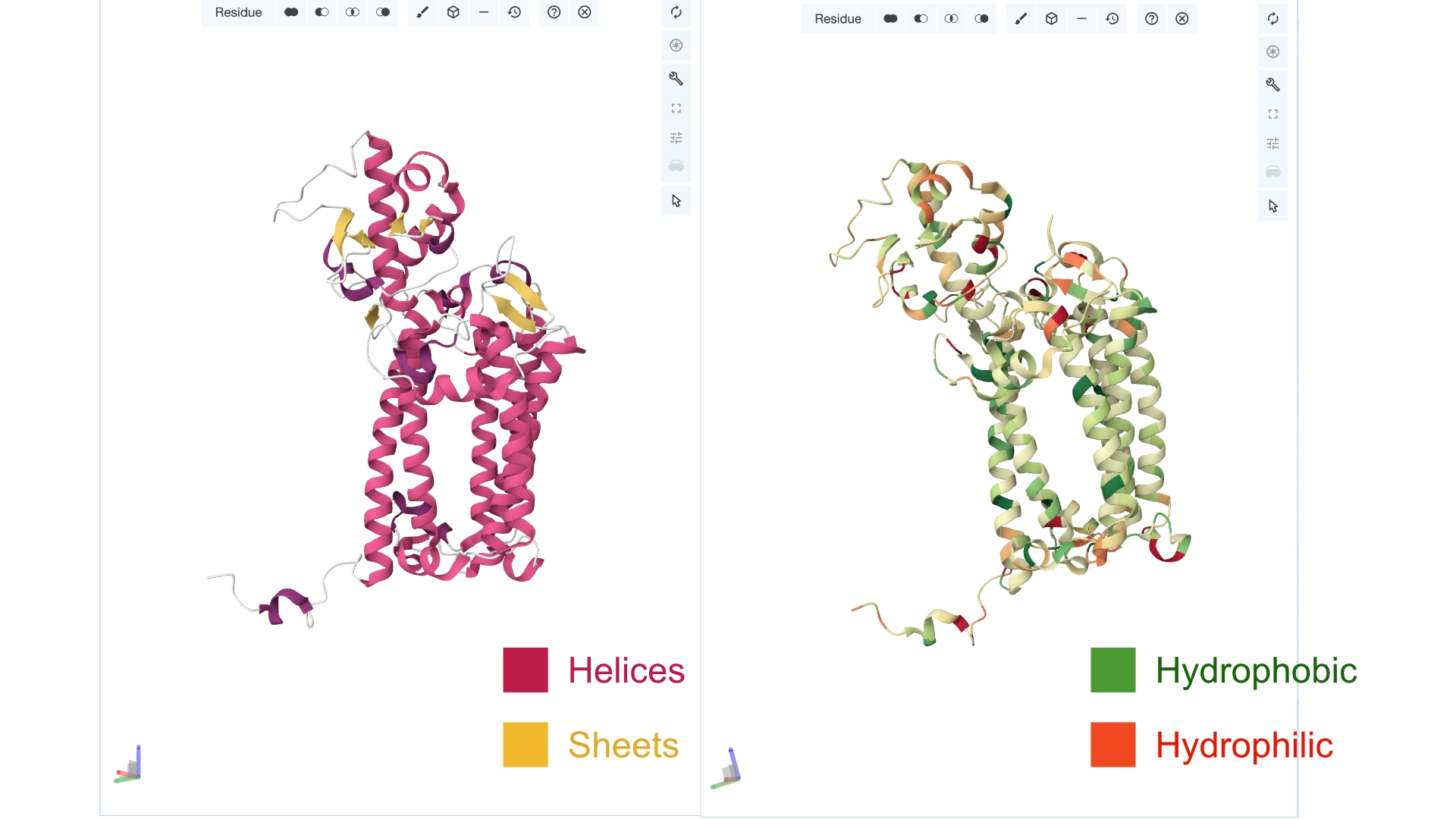
More helices, Hydrophobic
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
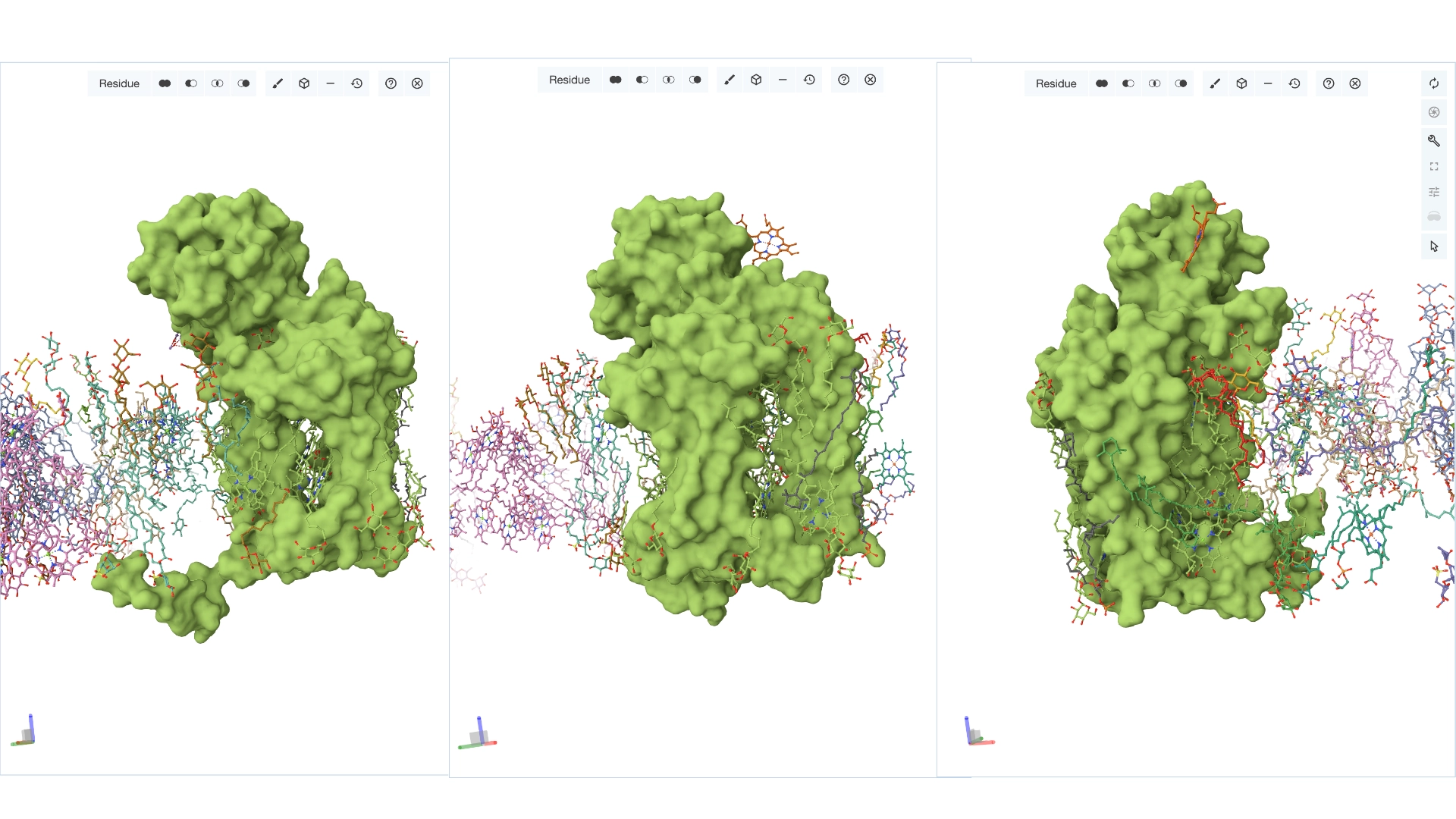
Yes it has several binding pockets. I turned on the ligands and sugars visualization to try to understand the binding pockets better
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Documentation
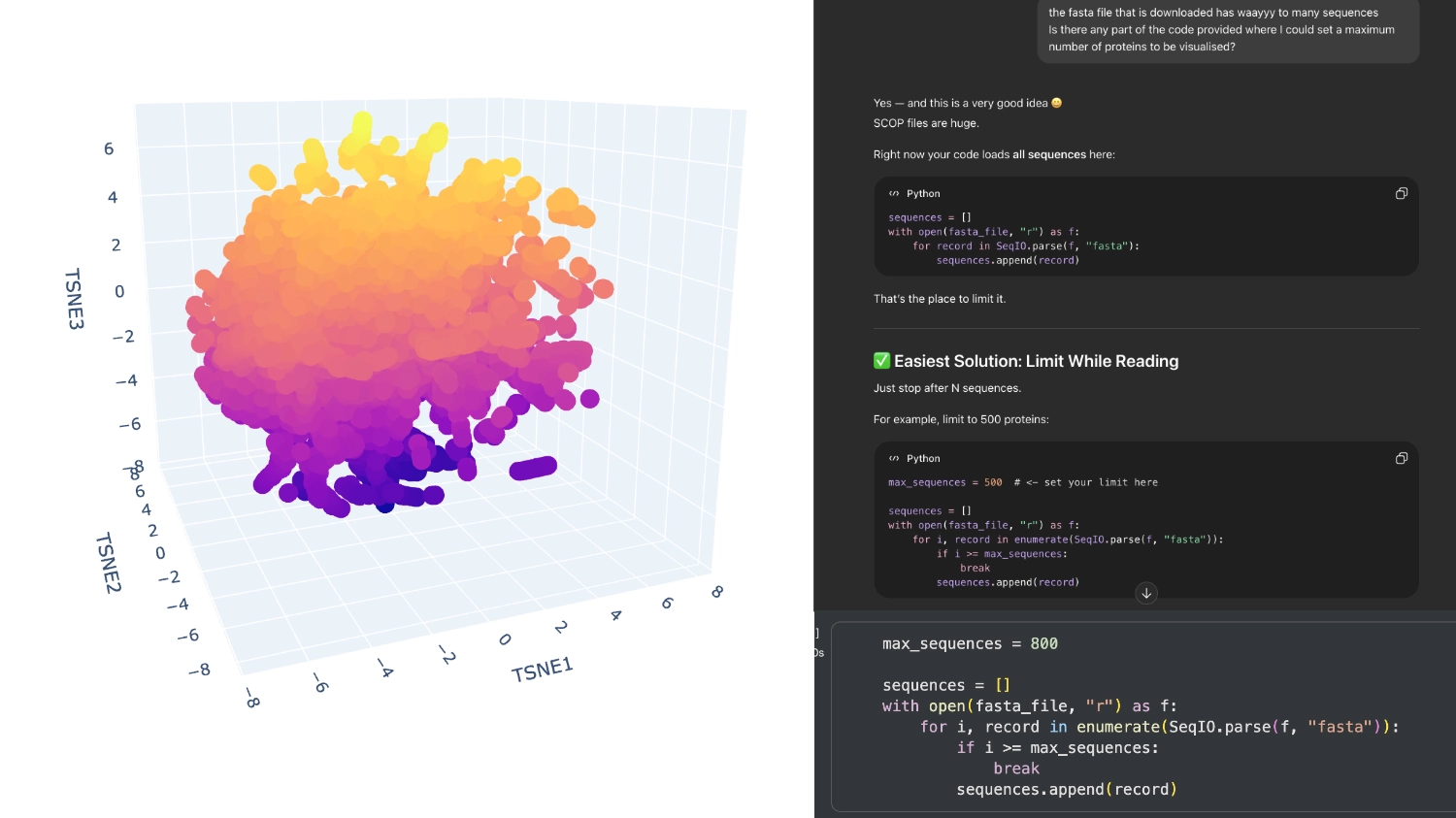
When I first ran the code there were way too many proteins in the visualization, then I understood that the fasta file being loaded had around 15 thousand sequences which were being rendered. So I asked Chat GPT (in order to not break the whole code by experimenting) where I could limit the amout of pronteins that were being embeded to a more manageable array.
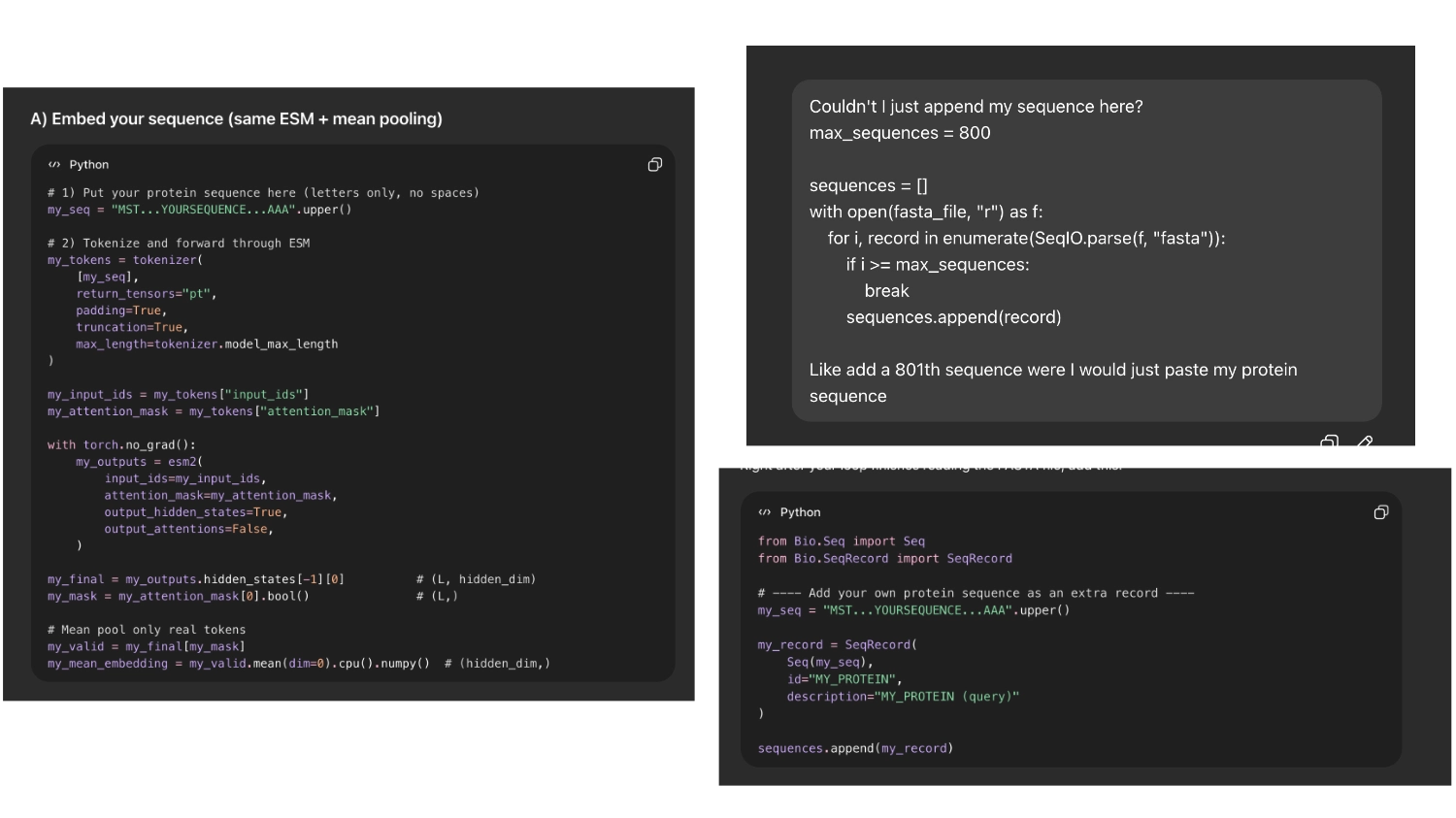
Then I asked chat gpt how I could insert my protein and it first tried to code it into a token. But it seemed easier and less risky If I could append my sequence to the array of sequences imported so I asked it to do that
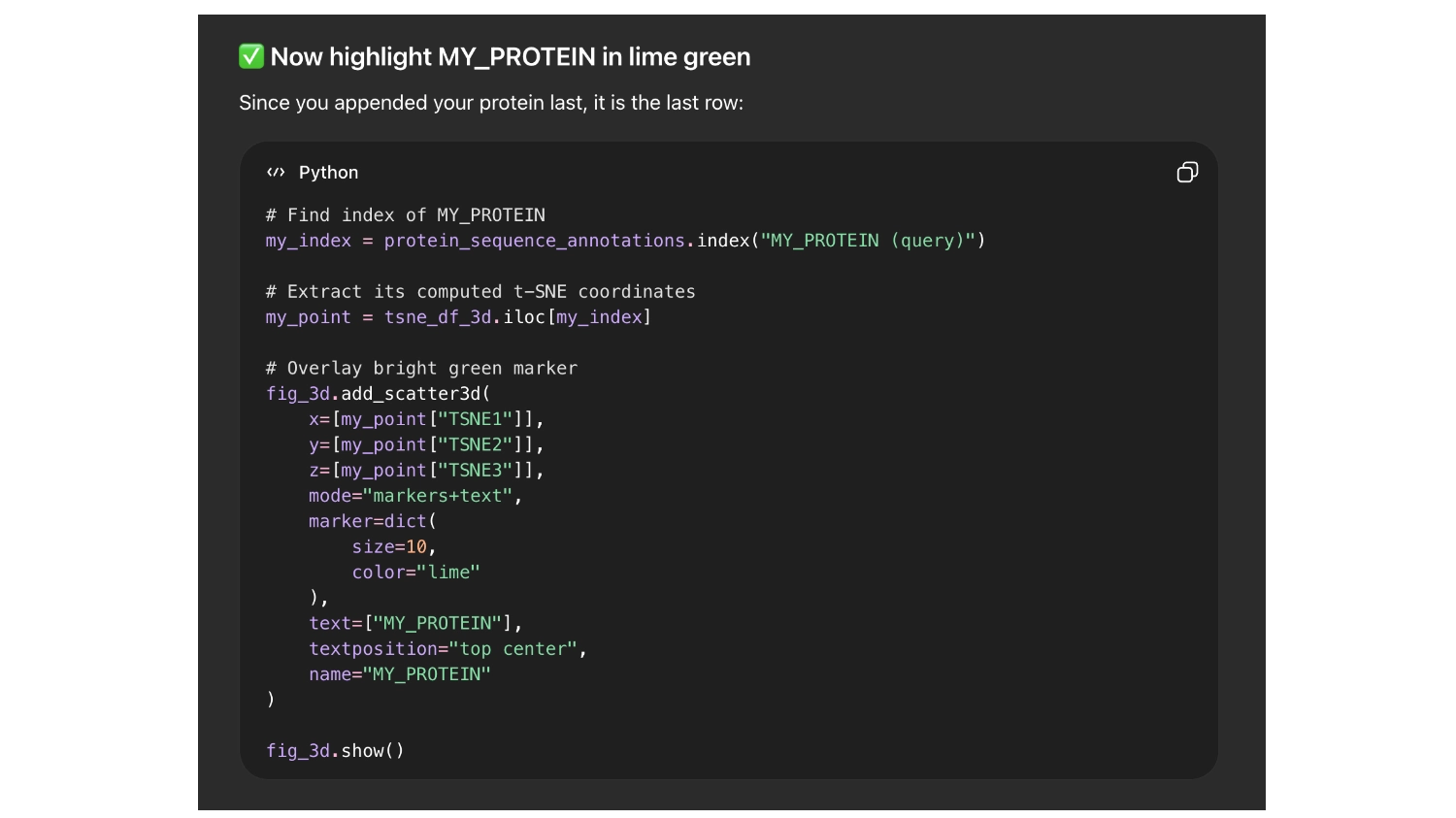
So it would be easier to find my protein I asked chat GPT to label it with the name ““My protein”” and color it bright green
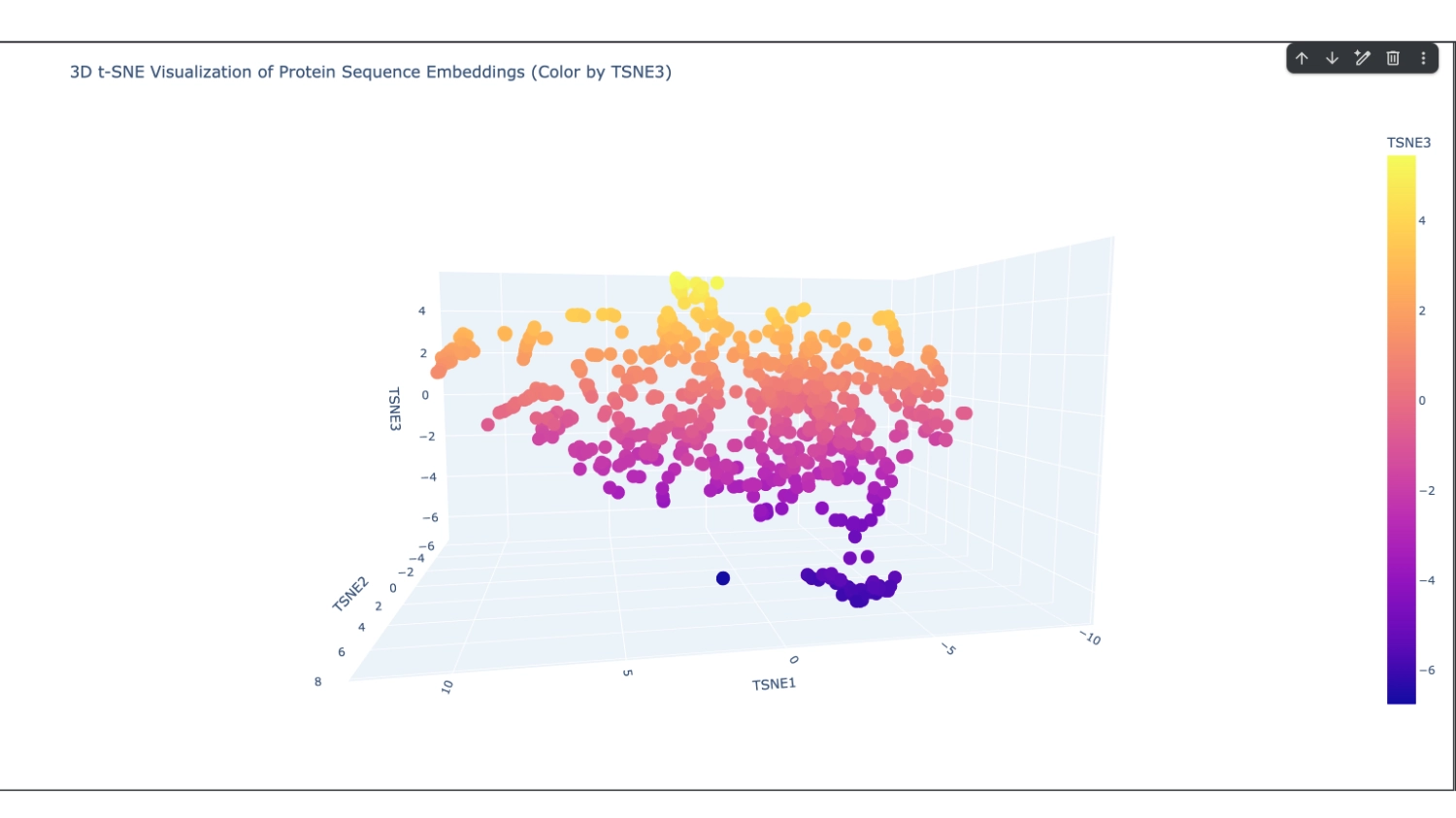
I set it to 800 which seemed to have a good amount of variation for me to understand what was happening. After that, I increased the array to 8000 because I didn’t have too many direct neighbours to my protein
1. Deep Mutational Scans
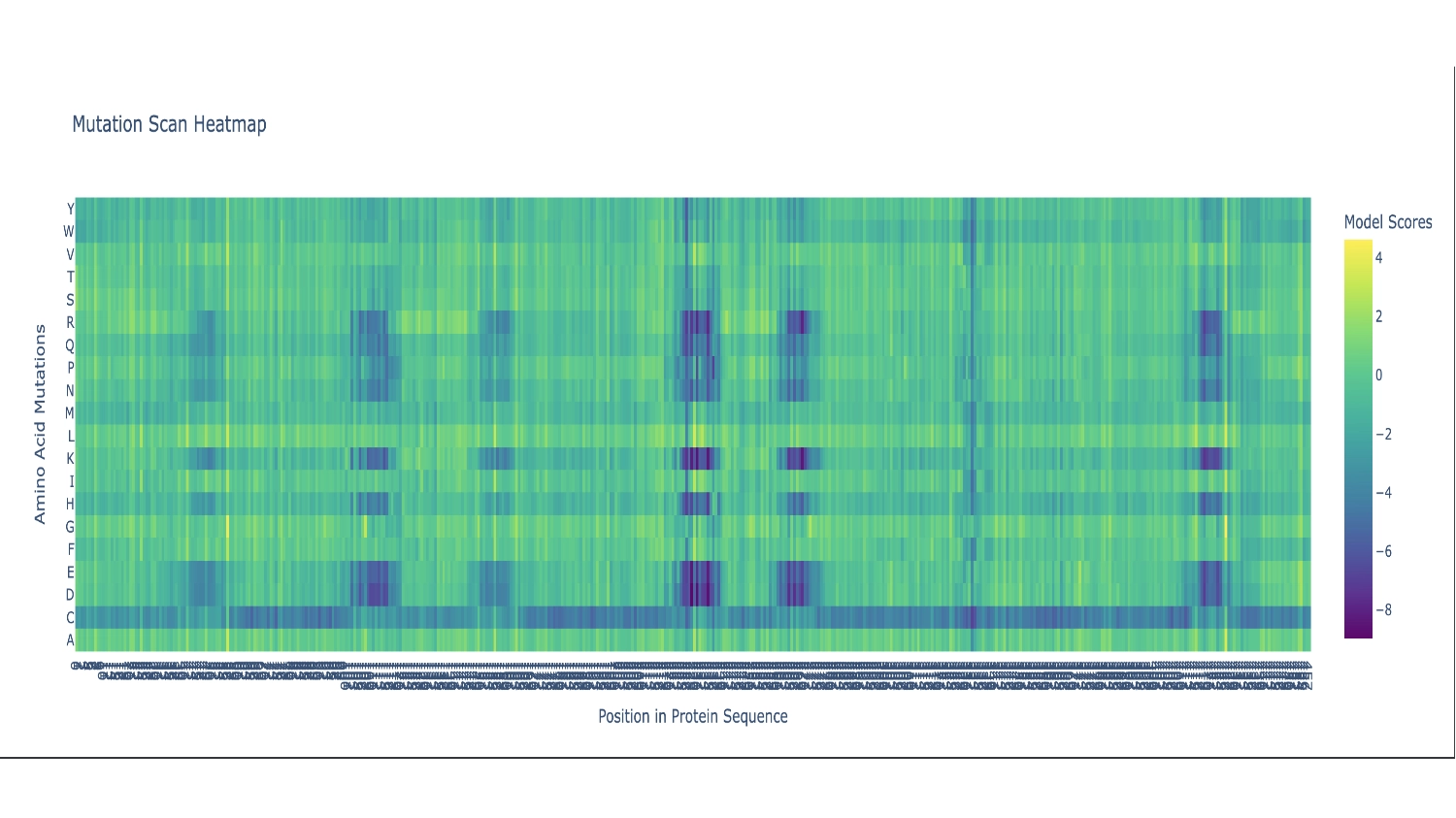
There are two primarily noticable patterns. The first one is the Cystine residue which mostly seems like a bad fit throughout the whole sequence, persumably because the chemistry of this amino acid would disrupt the function of this protein in most places— the actual protein sequence only contains 2 Cystines. The second pattern is the vertical blue columns throughout the sequence that indicate parts of the protein sequence which are propably very functionaly critic zones, thus toleratig very little exchange of residues. In these columns there are some interuptions for certain amino acids which seem to be fairly well accepted along the whole sequence which include: G (the most comon AA), and L, F, V and I which are all hidrophobic, since the protein as a whole is mainly hidrophobic this makes sence.
2. Latent Space Analysis
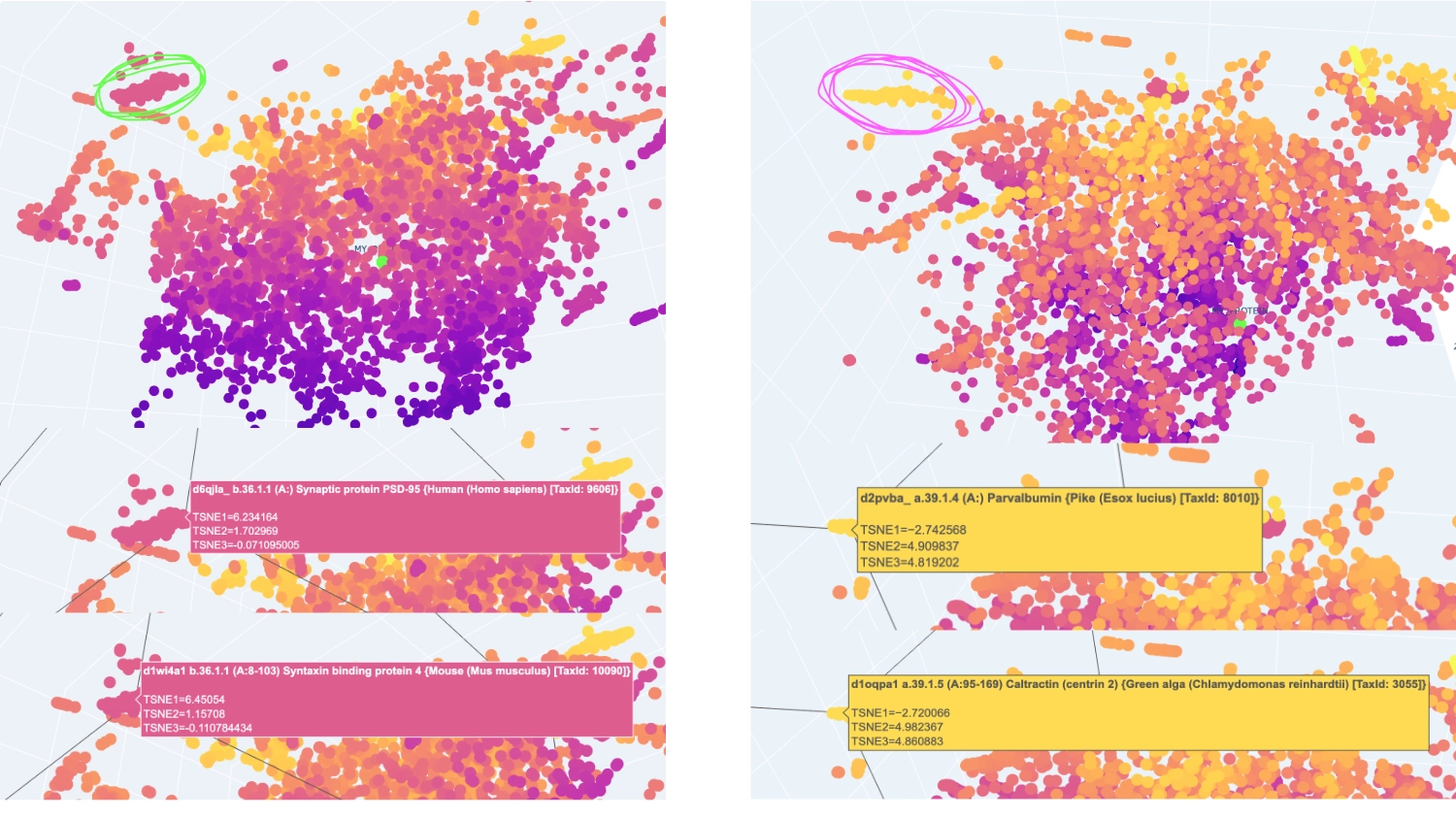
I analysed 2 well identifiable distinct neighborhoods. The one selected in green has several proteins related to neural function, while the one in pink has several calcium-binding proteins that serve different functions.
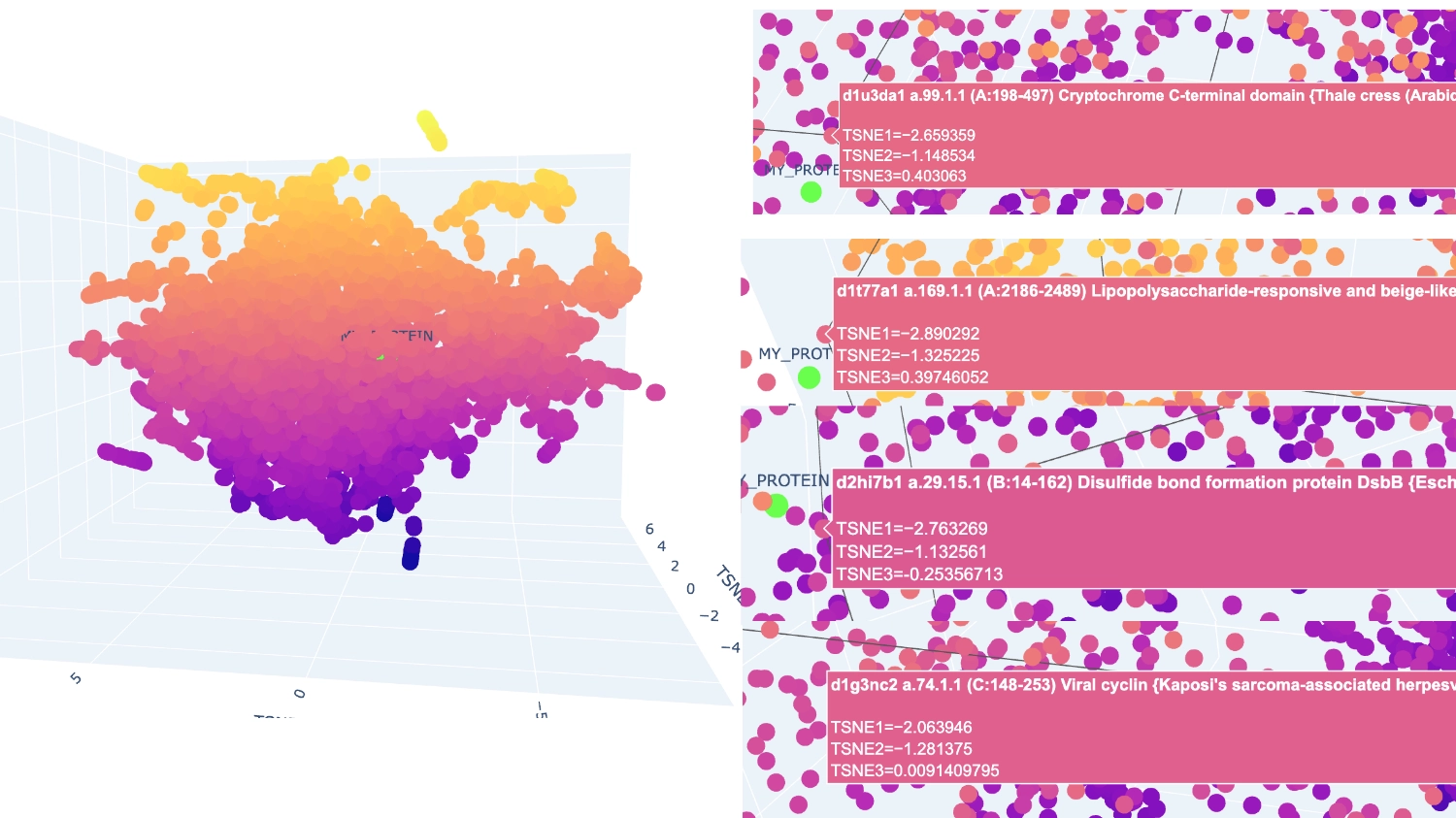
The closest and most interesting neighbour that I could identify as aproximate in function was a Cryptochrome C-terminal domain which is a flavoprotein blue light-sensing photoreceptors found in plants, animals, and microorganisms that regulate circadian rhythms and developmental processes Most other neighbours seemed to be related by being membrane-related proteins or also being rich in helices like Disulfide bond formation protein, E. coli (membrane protein with transmembrane helices), Cytochrome c peroxidase (predominantly alpha-helical) and Viral nucleoproteins (helix-rich folds).
C2. Protein Folding
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
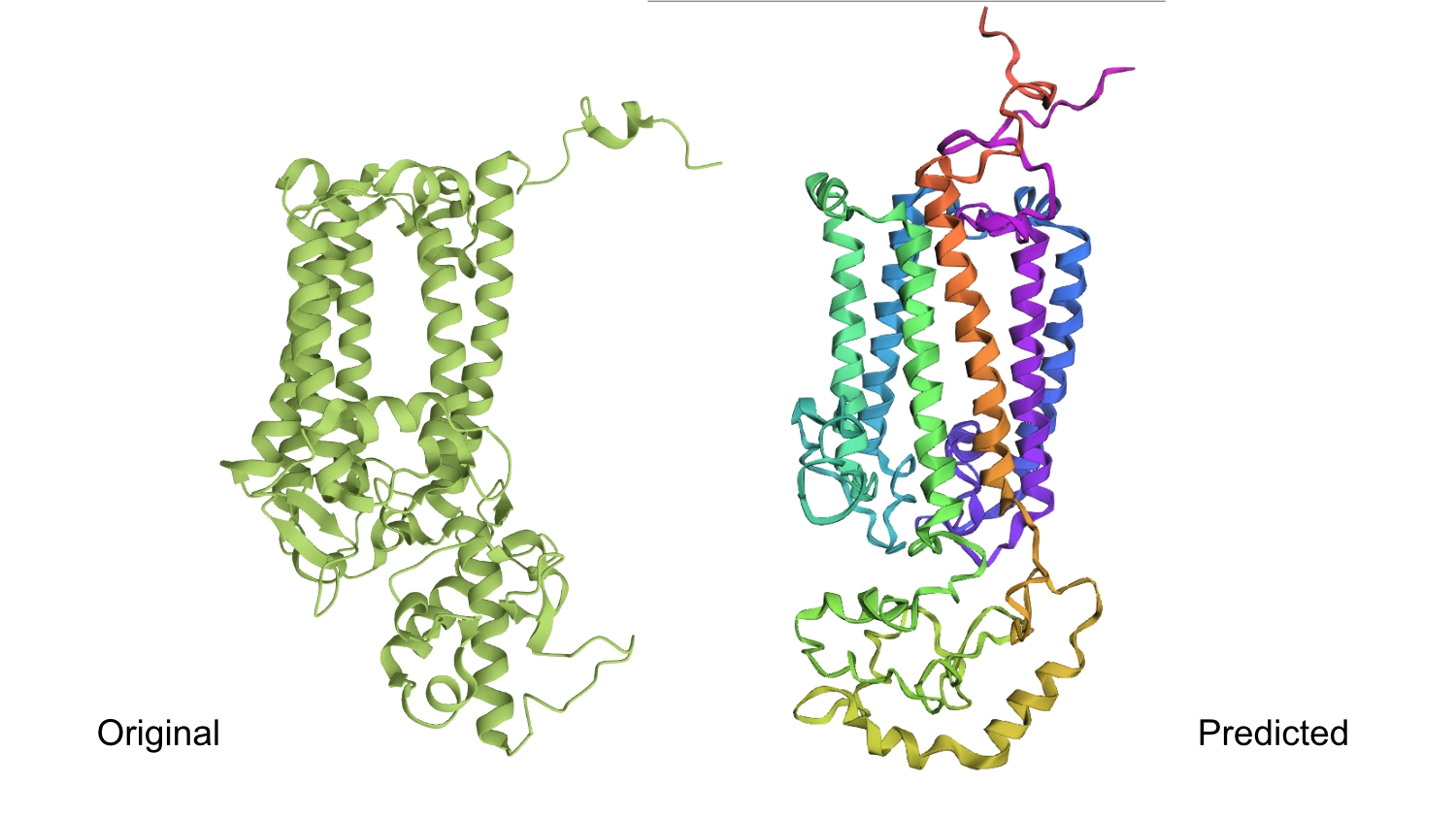
The predicted structure seems slightly different regarding the helices on the bottom part than the actual protein.
2. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
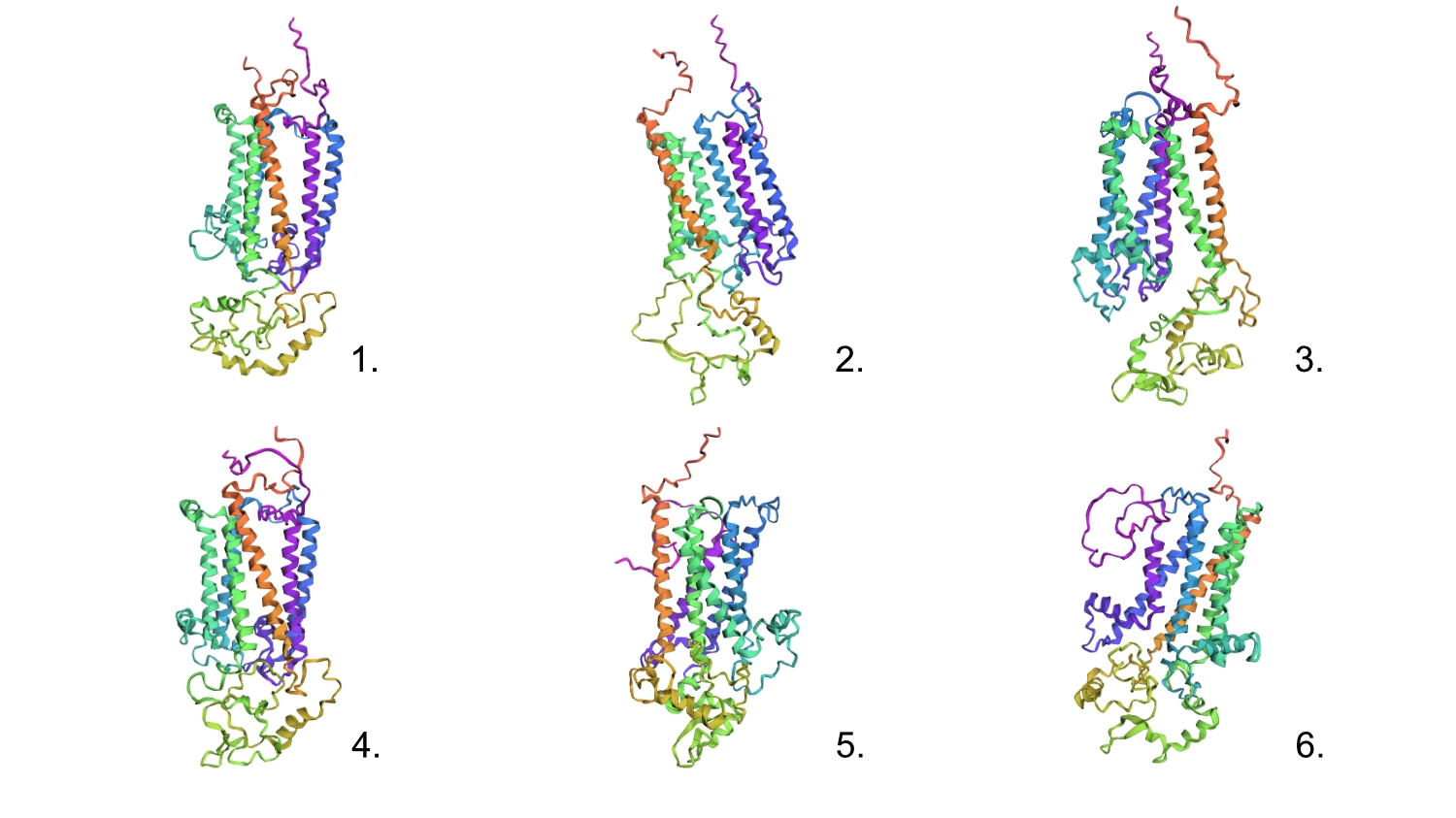
1.First I changed a random base for a G since it is one of the most well accepted residues and the protein seemed to maintain its shape.
2.After that, I changed 4 residues to Cs (the least accepted residue) and nothing seemed to change too much in the conformation.
3.Changed 4 sets of 6 to Cs
4.Changed 1 random set of 20 to Cs
5.Deleted 4 random residues — one of the six helixes partially uncoiled and smaller horizontal helices formed
6.Deleted 10 random residues — seems to have aggravated the latter step but still resembles the original protein
All these changes were cumulative so this protein seems relatively resilient to mutations
C3. Protein Generation
Documentation
The first problem I ran into was that I couldn’t download the file for the 3D structure of CP43 protein alone, as it is part of photosystem II, all the proteins involved were also included in the in the files. I tried several ways through the RCBS website, but with no success. And so, I resorted to Chat GPT that spat out a python script to run on my computer terminal in order to download the correct file, and it worked.
Once I had the right file (checked it by opening it on the RCBS site), with both the backbone and all ligands to that specific protein. I tried to run the inverse folding code on the Collab notebook but with no success and couldn’t find a way to import hat was missing so I tried to use an online tool for inverse folding from neurosnap.ai and it seemed to work just fine!
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
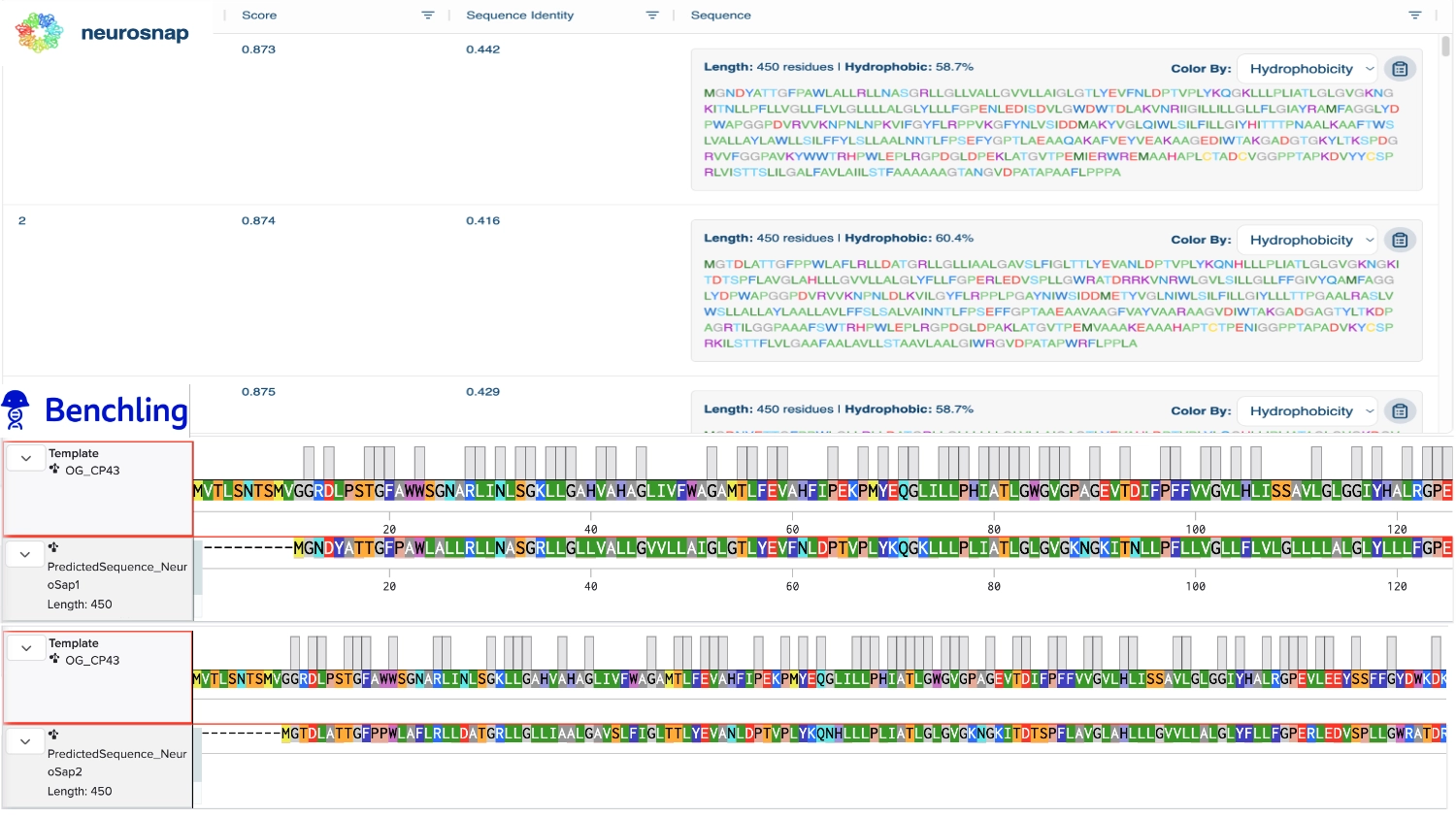
Then compared some of the predicted sequences to the original one by performing alignments on Benchling to see what residues were used on the predictions and how similar they were to the original. As observed on the heatmap from earlier the C residues were kept at a minimum and the most used residues fluctuated between the best accepted ones like L and G. It was also interesting to observe that between predicted sequences there were similar areas that have the same residues in common as the original, which must be the most crutial for that type of folding. Also, the predicted sequences have -10 residues at the very beggining, you can see it from the benchling screenshot, and which I interpret as a N-terminus or C-terminus that the inverse folding model didn’t replicate since they probably weren’t solved in the 3D structure.
2. Input this sequence into ESMFold and compare the predicted structure to your original.
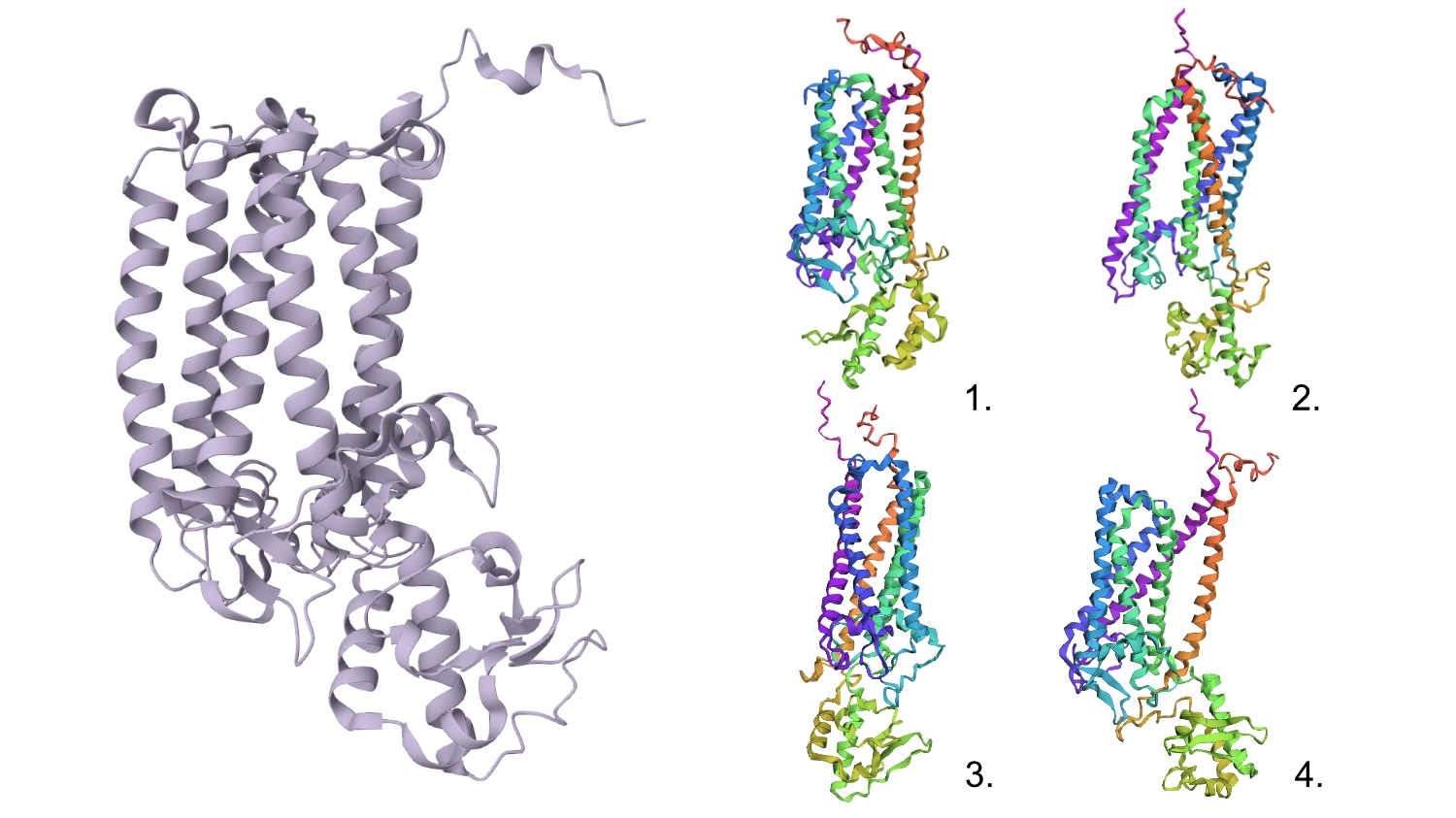
Regarding the 3D structure, the new predicted proteins were similar to the original, however, the main differences rested on the width of the 6 helices barrel which in wider on the original CP43 and the same helices are not as well organized, straight and vertical as the original.
Part D. Group Brainstorm on Bacteriophage Engineering
This was the result of my initial research for the group phage project. My group has setup a shared docs and we are working in the goal of stabilizing the L protein.
