Week 5 HW: Protein Design Part2
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
Documentation
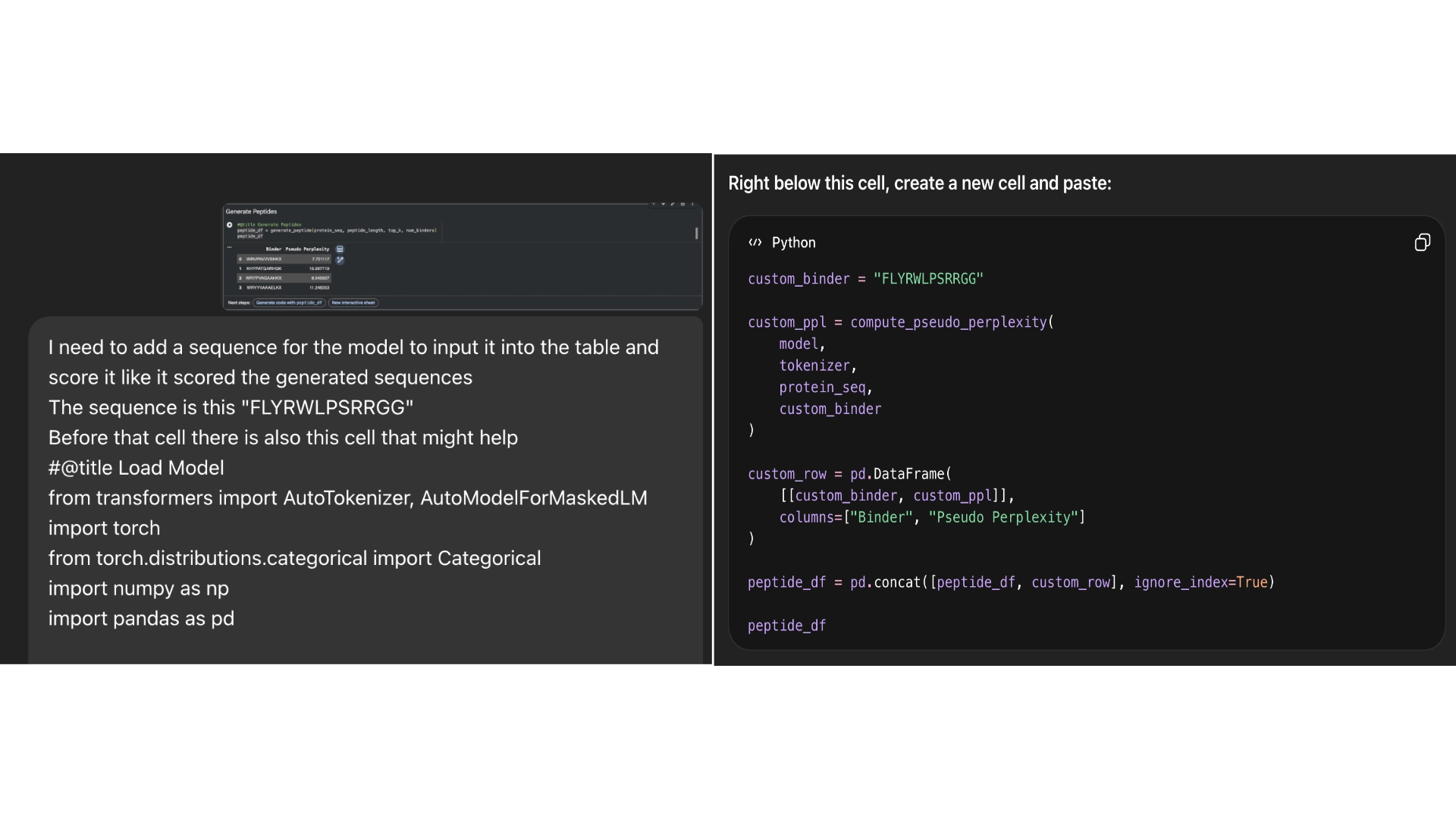 After generating the 4 sequences I resorted to Chat GPT to generate code to help me input the known sequence into the model so that PepMLM would grade its confidence in the known binder as well.
After generating the 4 sequences I resorted to Chat GPT to generate code to help me input the known sequence into the model so that PepMLM would grade its confidence in the known binder as well.
Part 2: Evaluate Binders with AlphaFold3
3. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
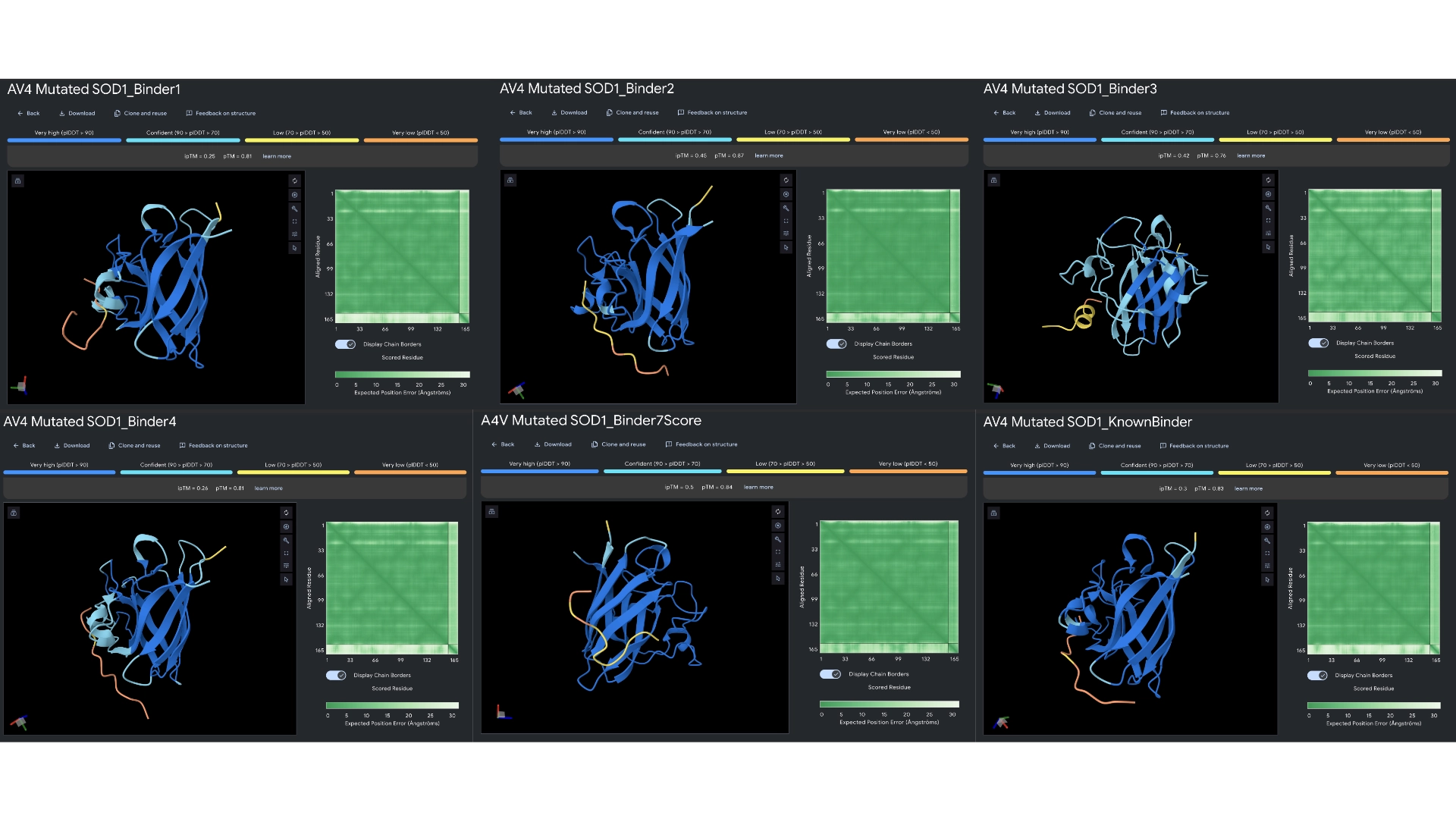
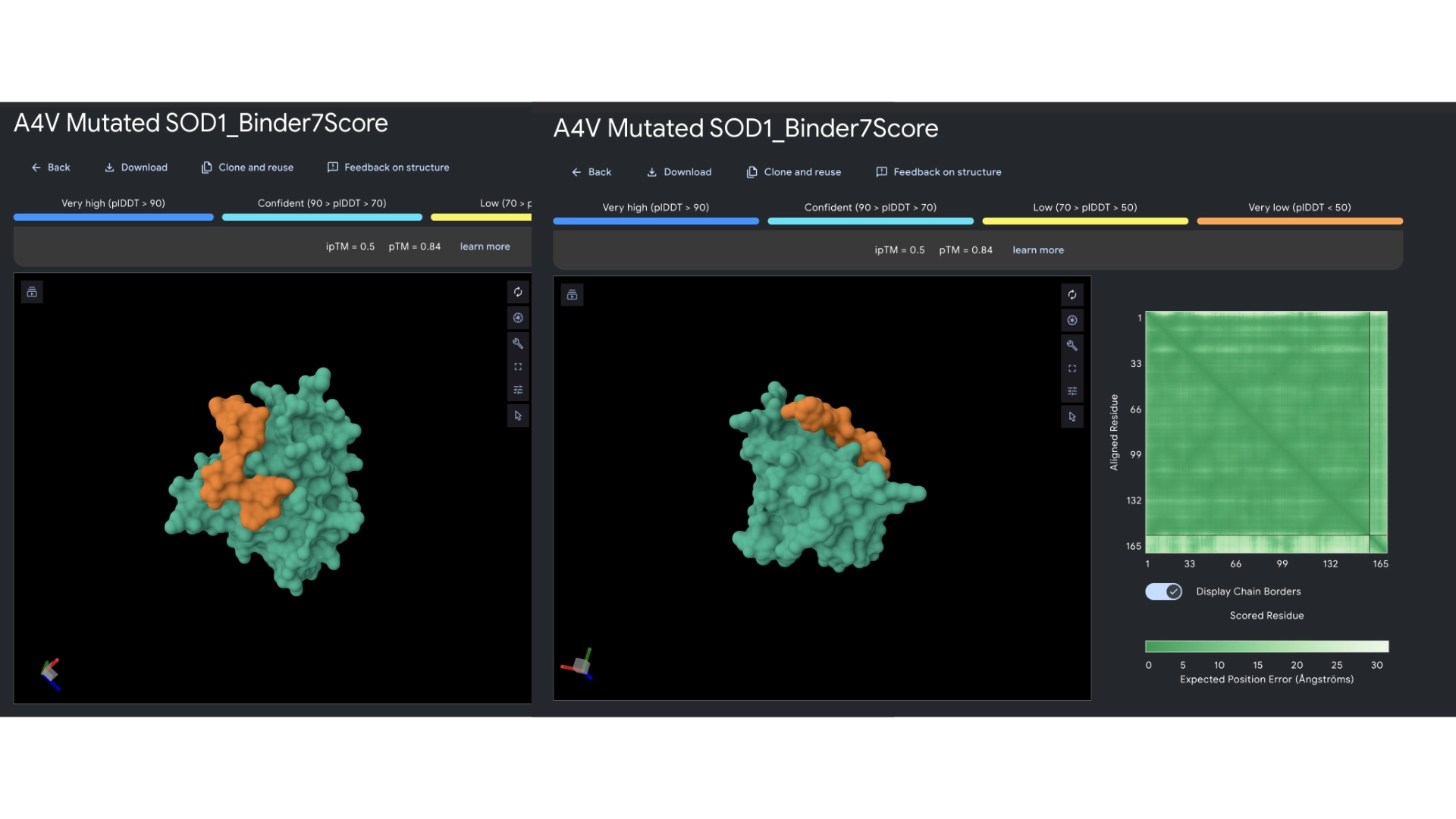 All the pepties I visualized bonded to the barrel region. The one I was able to generate which had the best perplexity score (of 7) and the best ipTM (of 5) bonded slightly closer to the N-Terminal, but still on the barrel region. I included a molecular surface visualization for that one so it could be better analized. It seems to bind at the surface, doesn’t seem to be partially burried.
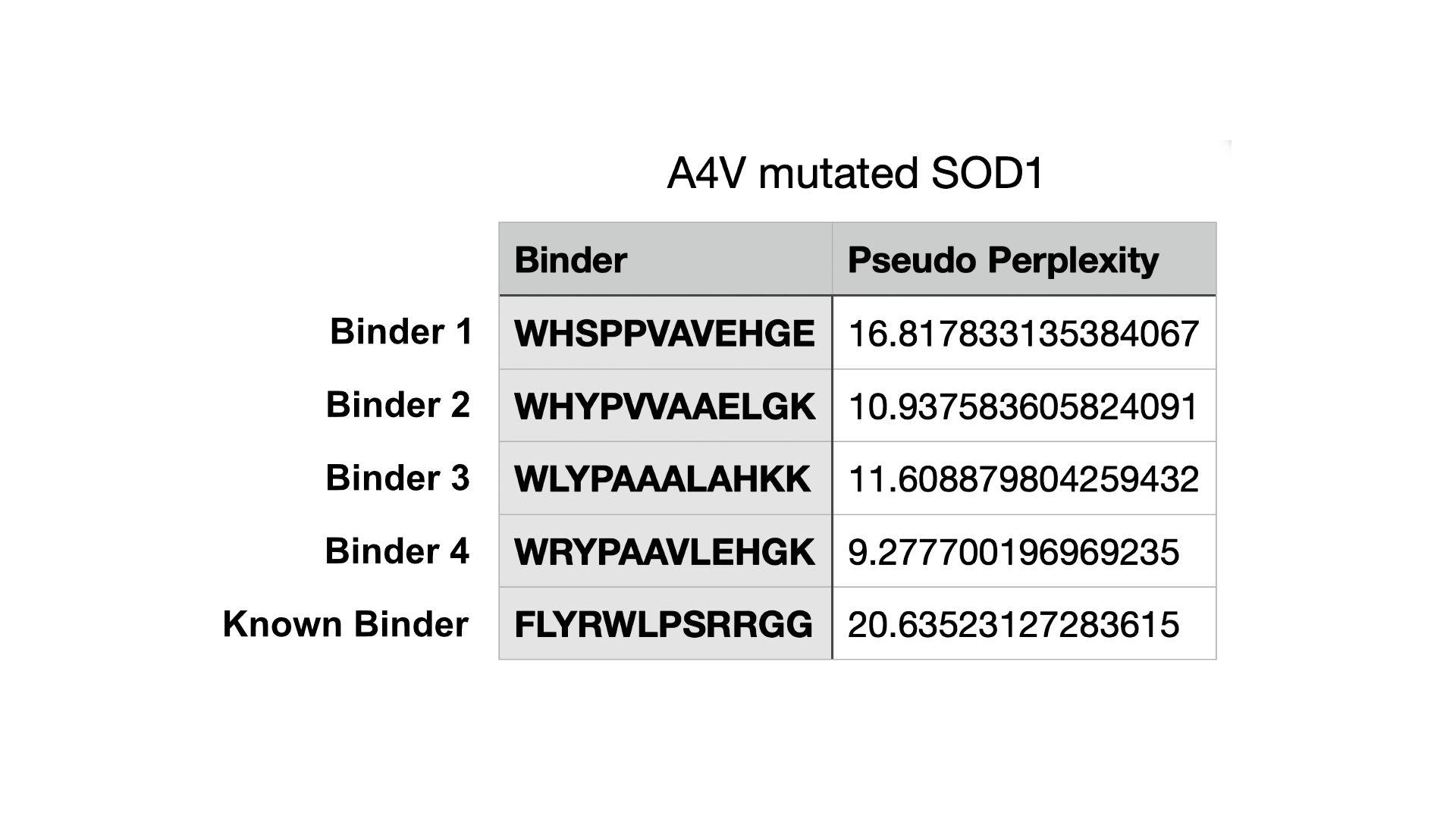
All the pepties I visualized bonded to the barrel region. The one I was able to generate which had the best perplexity score (of 7) and the best ipTM (of 5) bonded slightly closer to the N-Terminal, but still on the barrel region. I included a molecular surface visualization for that one so it could be better analized. It seems to bind at the surface, doesn’t seem to be partially burried.
The known binder got a perplexity score of 20 and a ipTM score of 0.3, so I’m not sure how indicative these scores are being of the actual binding abilities of these peptides
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
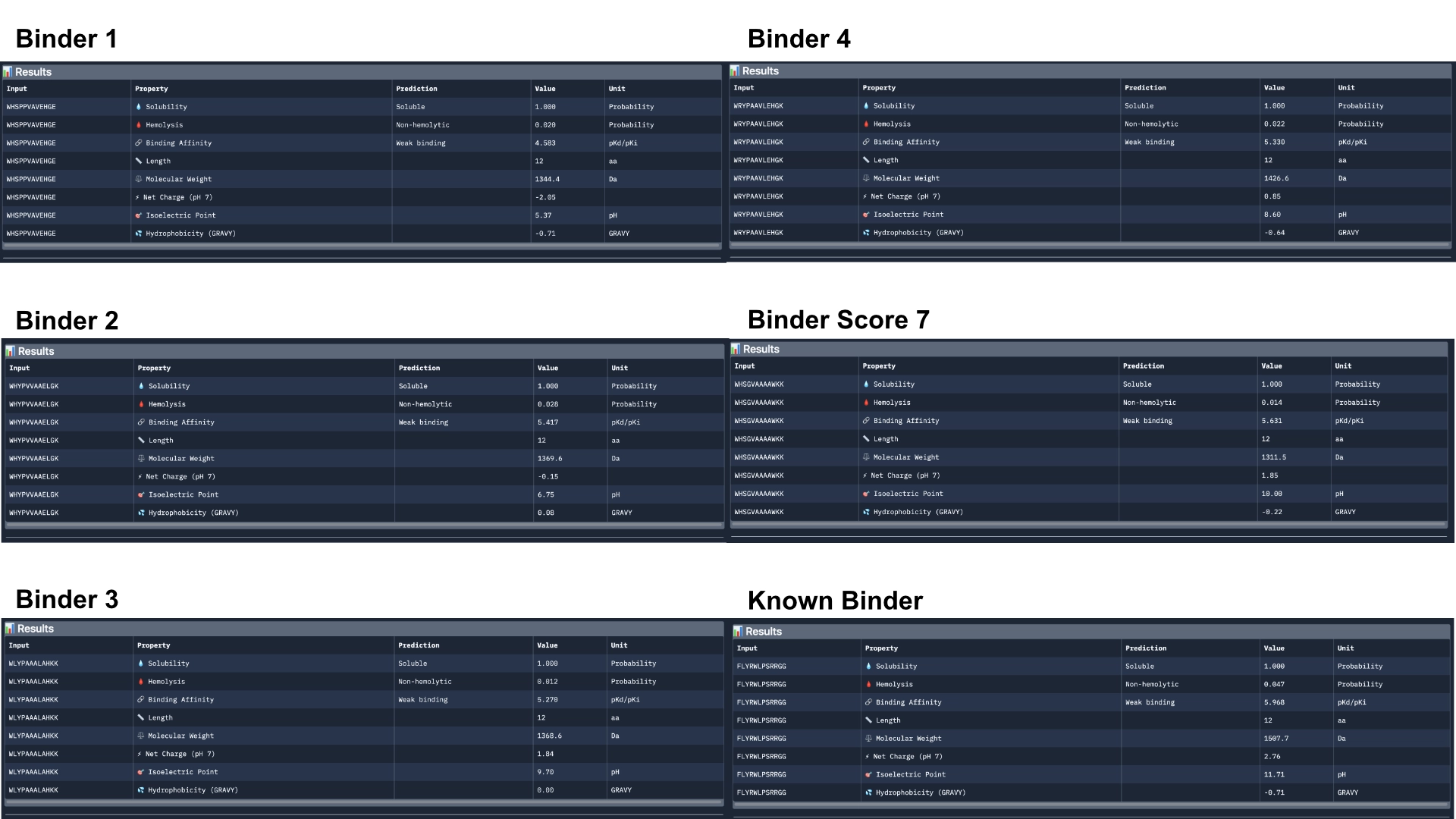
Overall, Peptiverse considers all generated binder and known binder as weak binders, the known binder having the highest score of 5.968, followed by the binder which had the highest ipTM score, with a binding affinity 5.631. However, binders 2 and 3, which surpassed the known binder in ipTM score, were not attributed better binding affinitty, having the scores 5.417 and 5.270 respectively. All predicted binders had good therapeutic properties scores and none were non-soluble nor hemolytic. The best overall in balancing all aspects was the known binder with 5.968 of predicted binding and 0.047 of hemolytic probability. The best predicted binder, althought, it has a slightly lower hemolytic probability of 0.014, that might not balance out the 0.337 difference in binding affinity (5.631). However, this would probably be the best to advance with, even though it might not exceed the qualities of the already known binder.
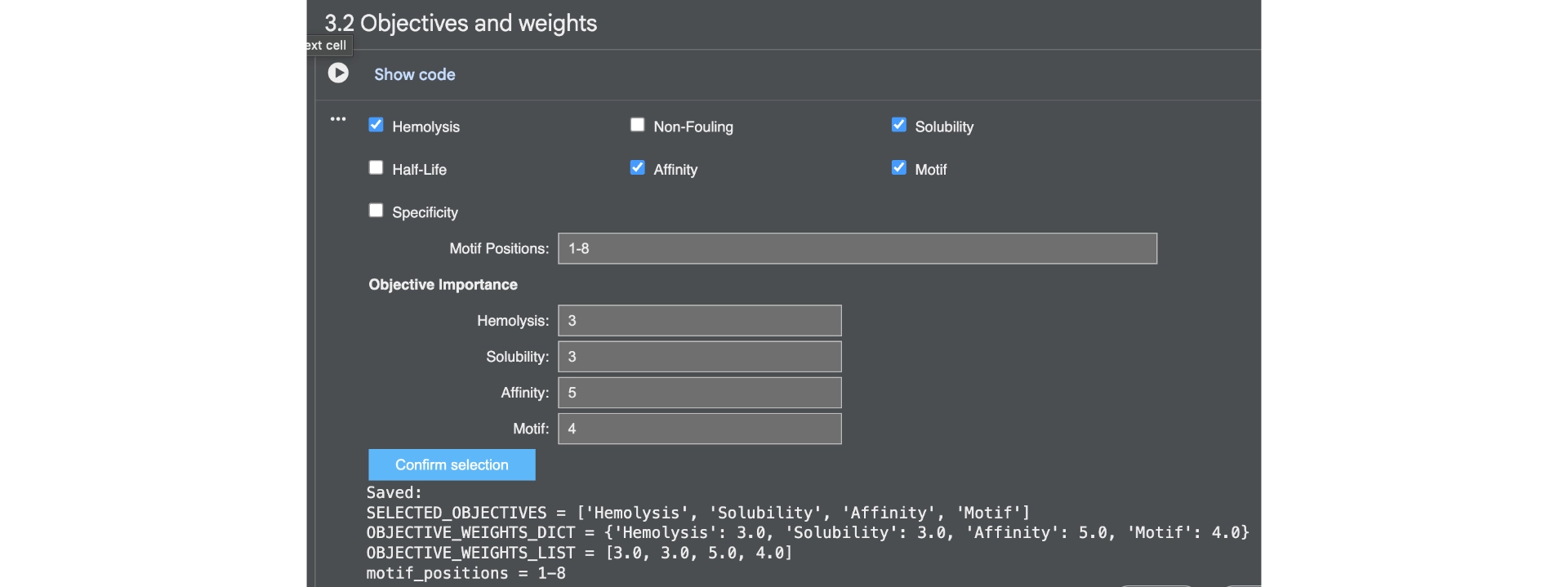
Part 4: Generate Optimized Peptides with moPPIt
First of all I generated 3 peptides for the A4V mutated SOD1 with the only parameters being affinity and motif whith the following weights:
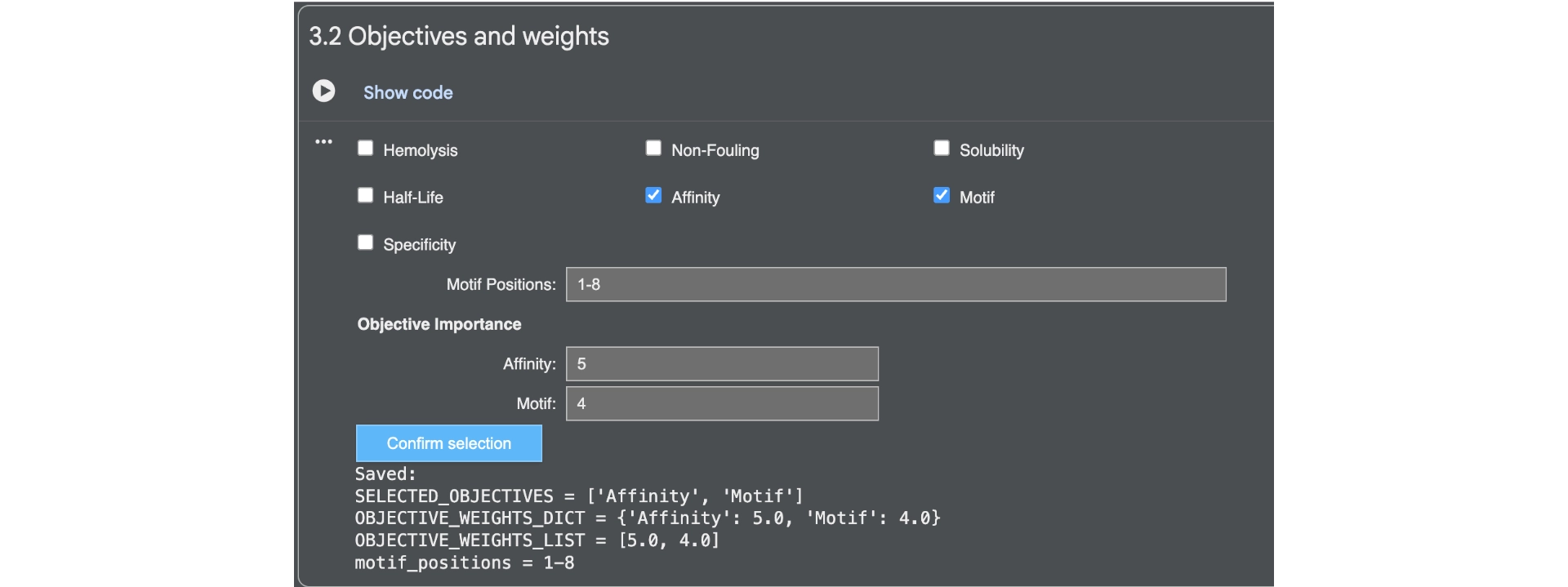
Then analysed the resulting peptides in PeptiVerse.
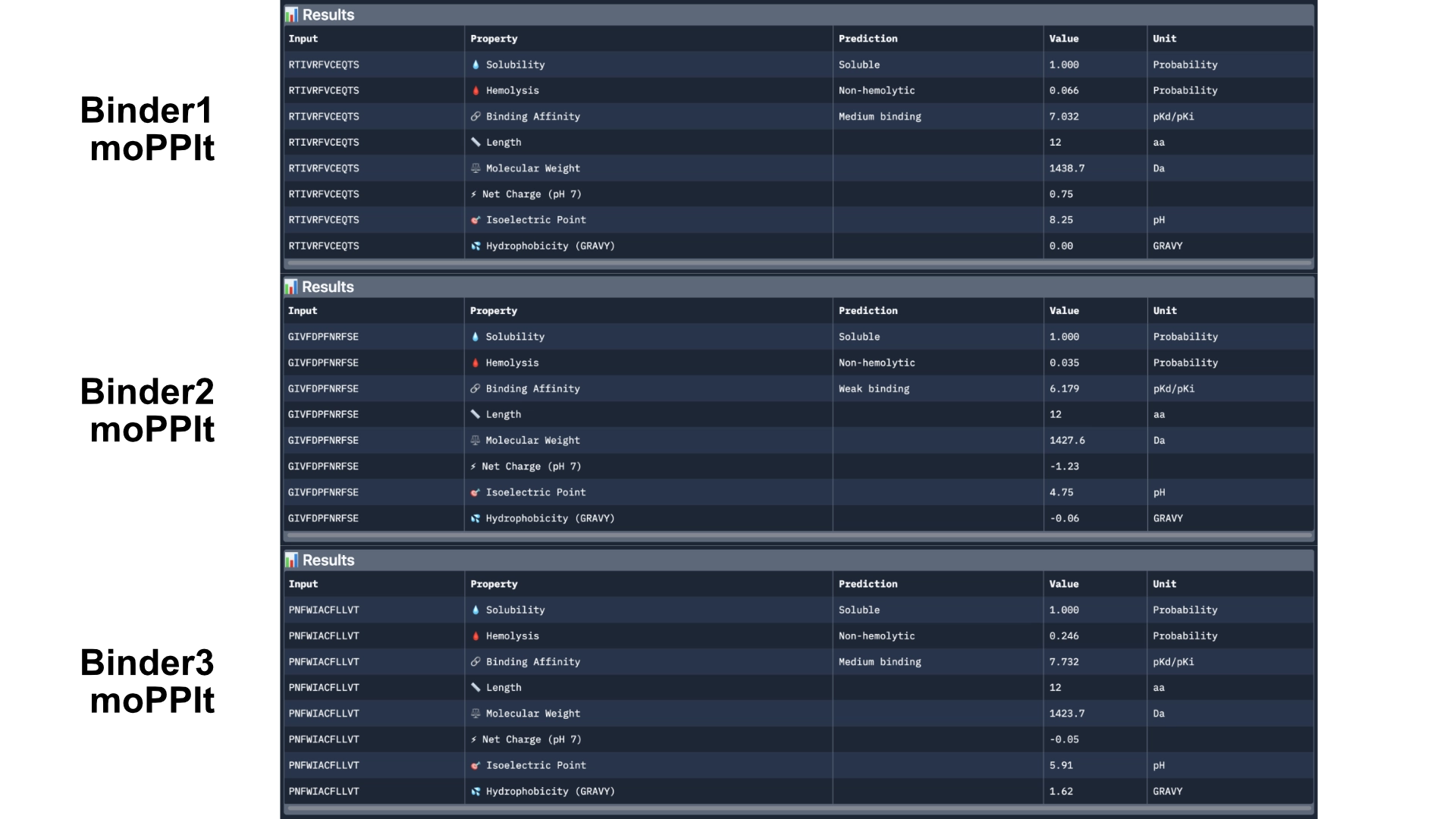
Comparing these to the PepMLM ones, there already was a good improvement as they were all soluble and non-hemolytic and had better affinities with two of them reaching medium binding instead of the previous weak binding levels.
Then generated other 3 peptides with non-hemolytic and solubility objectives to watch how they would differ from the previous ones.
Then analysed with PeptiVerse again.
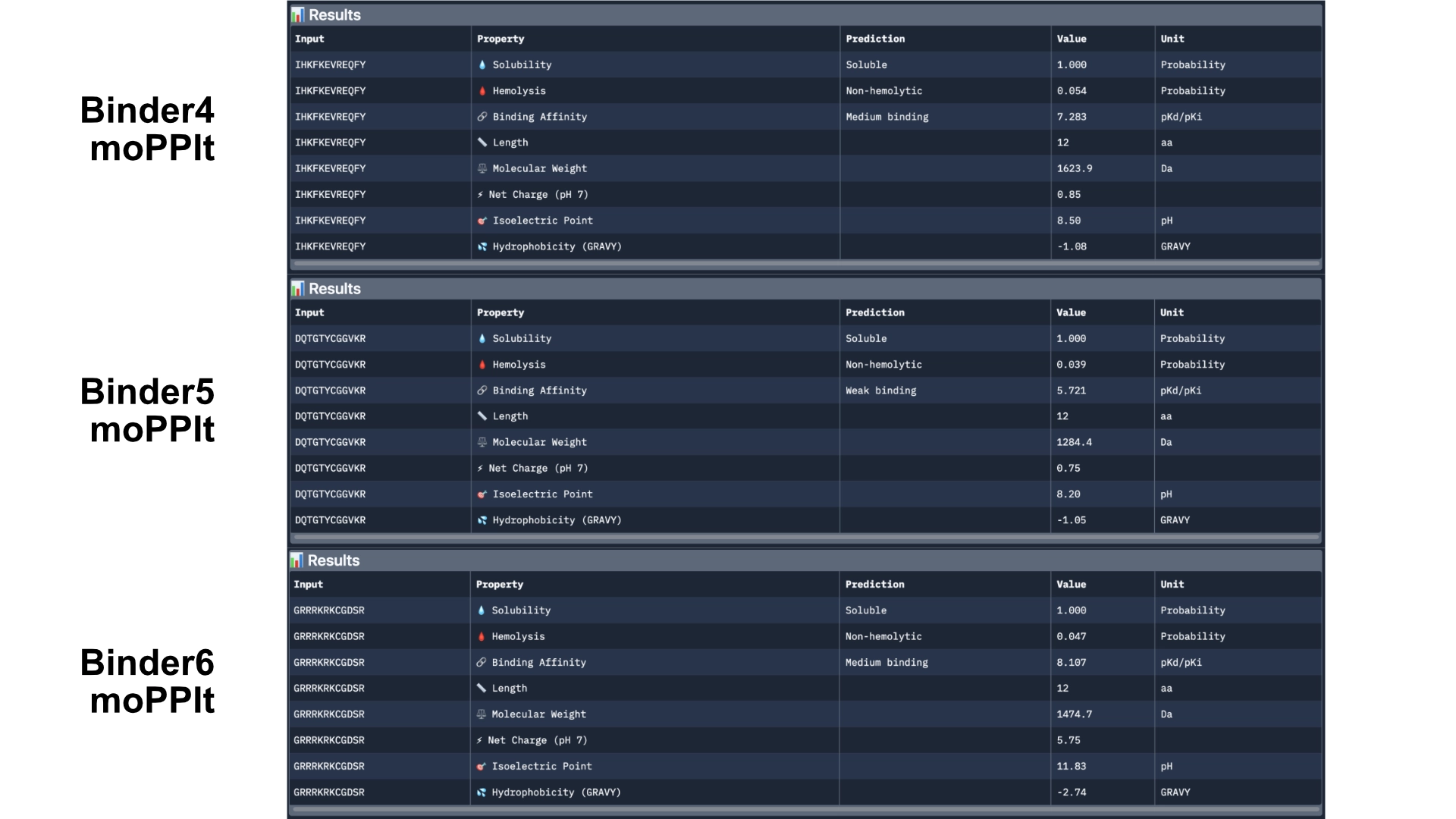
The results came back with low homlytic probabilities, soluble and with good affinity
In orther to reach some more conclusions I fed the best 2 peptides— Binders 1 and 6— to Alphafold to see if they would bind closer to the N-terminus which was the motif I had input and see if the ipTM score had gotten any better.
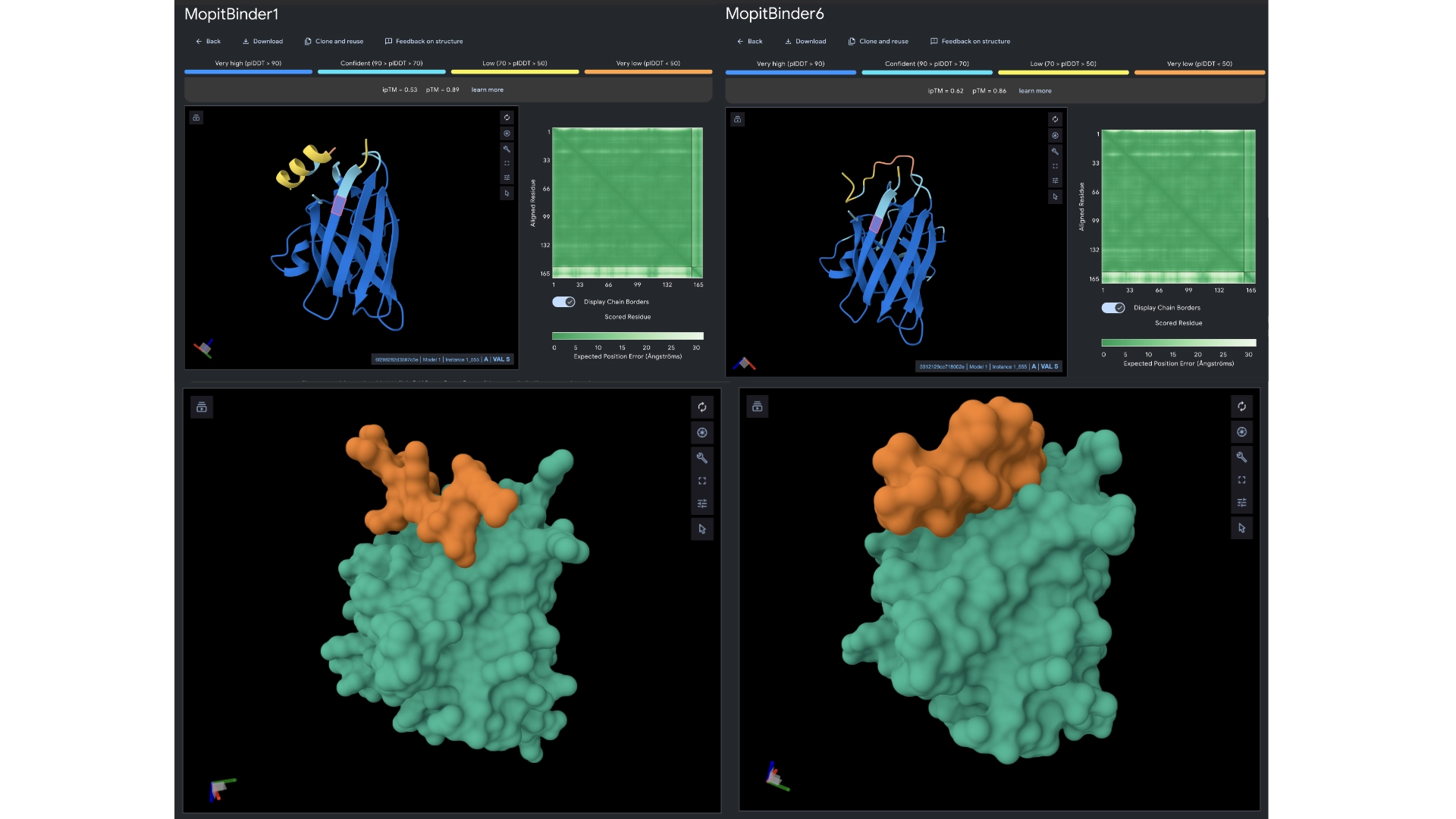
The ipTM scores came back the highest yet, with binder 6 reaching 0.62. They did bind closer to the N-terminus and not to the barrel region. Binder 1 did form a helix in the visualization and this might be interesting since a helical structure is more stable and could be further improved to have really high binding affinity. Then ran them against more therapeutic parameters— non-fouling, halflife and permeability— to see how these would hold up even not having been optimized for those purposes.
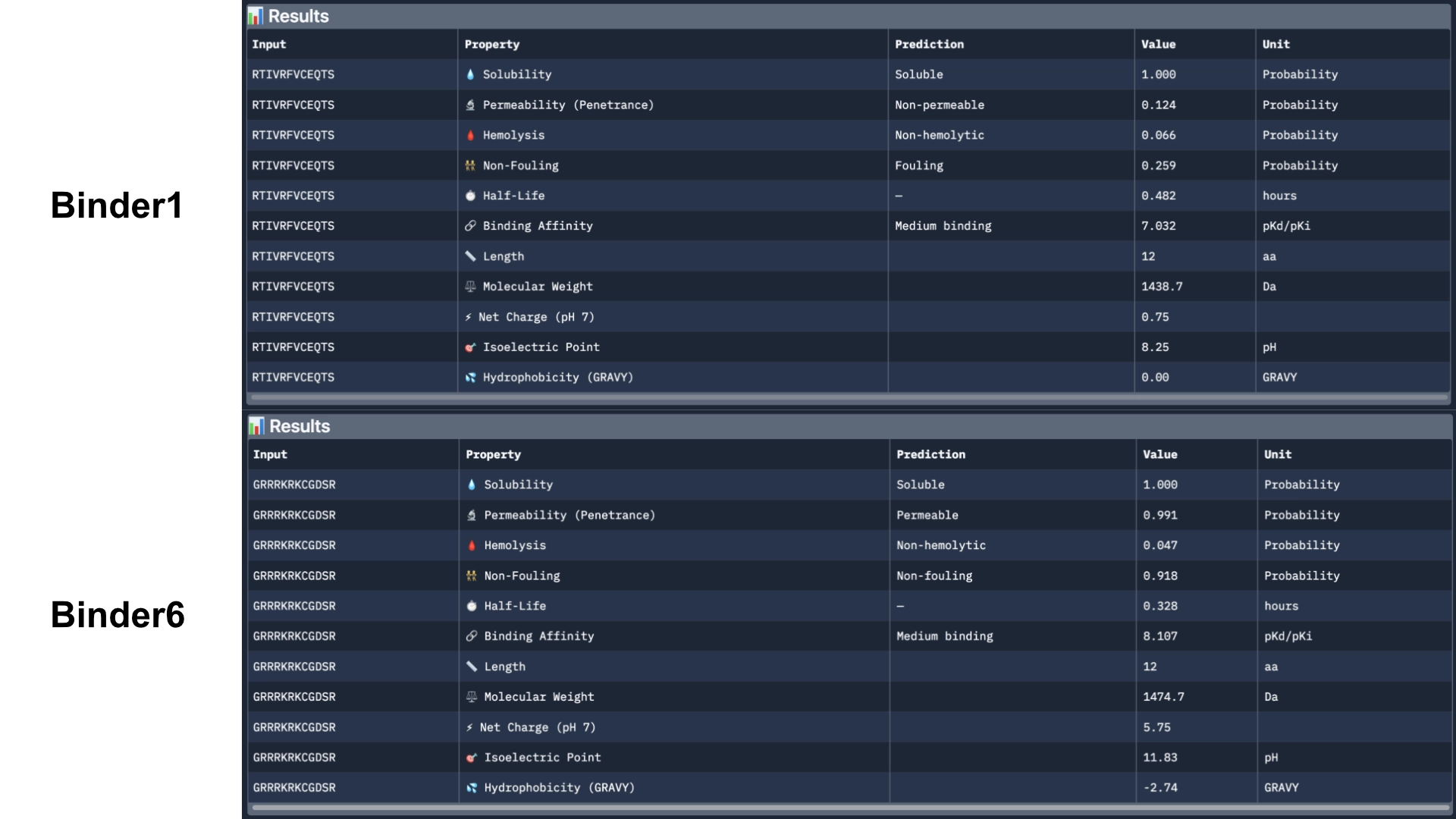
Binder 1 did come back as fouling and non-permeable. However, Binder 6 came back as permeable, non-fouling and with a half-life of 0.328 (which might be on the lower side). I would further optimize these 2 best peptides for the different theurapeutic qualities where they are weakest— fouling and permeability and half-life for binder 1 and half-life for binder 6— using a MOG-DMF model and run them against those parimeters using PeptiVerse.
Part C: Group Project: L-Protein Mutants
Documentation
First of all, I used Chat GPT to number the residue sequence in the heatmap for me to compare the residues more easily
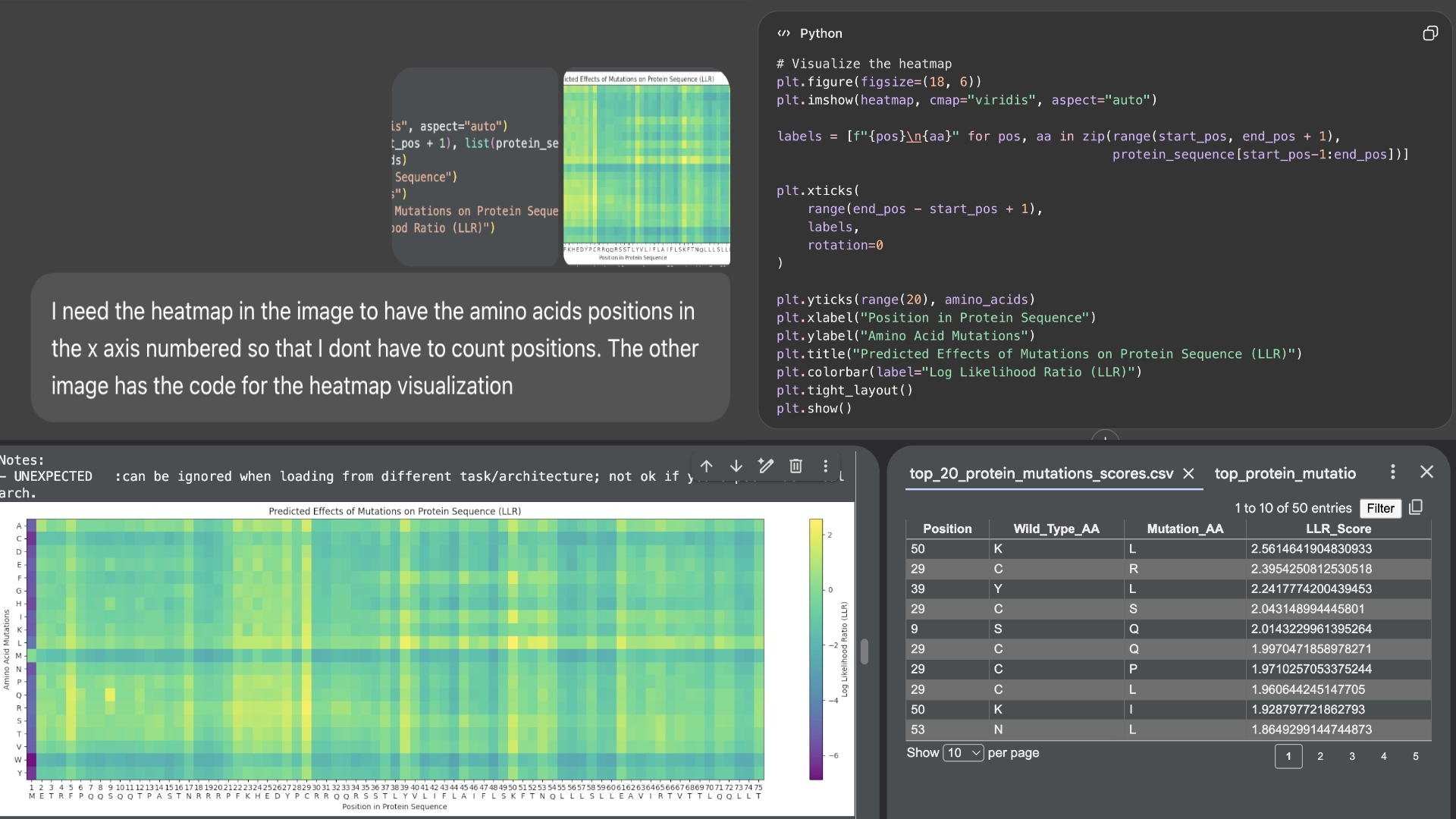
I started by making 2 mutants with several mutations to the hydrophilic domain, would be interesting to make it more stable and not dependent on DnaJ, by making changes in the positions best scored by the LLR and confirming them with the experimental data.
Mutant 1
6 best LLR scored residues in hydrophilic zone (no changes to transmembrane zone)
- 29 C->S (C->R was the best according to LLR score but was negative for lysis on the experimental data)
- 39 Y->L
- 9 S->Q
- 5 F->Q
- 27 Y->R
- 22 F ->R
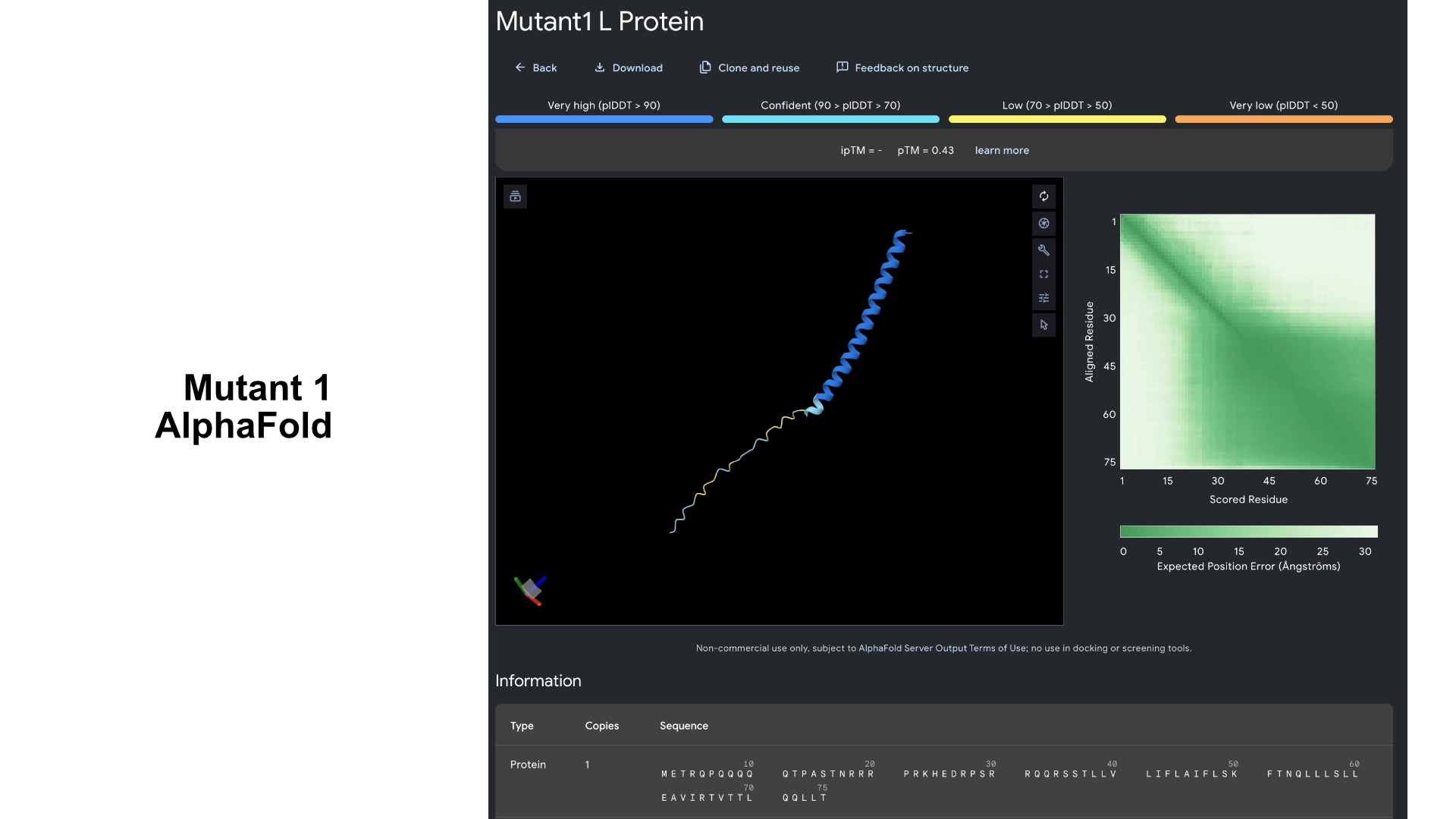
Then I visualized it in Alpha fold to see what had changed. While the hydrophilic region in the original L protein had low accuracy in the visualization and wasn’t uniform, this Mutant 1 had better values of accuracy and more uniformity in the hydrophilic tail.
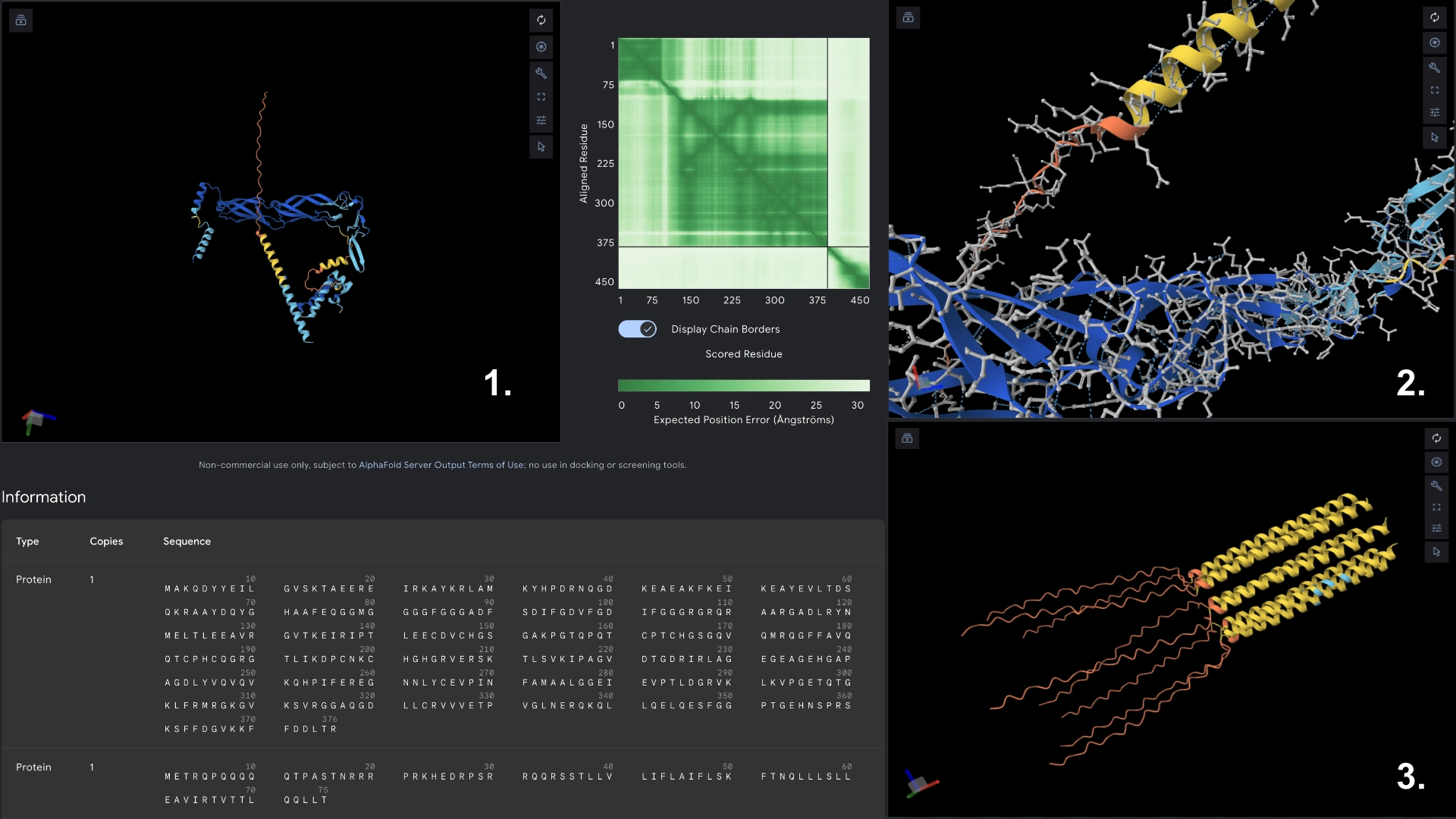
- Redered interaction with DnaJ
- Detail where we can see no bonds between DnaJ and L protein Mutant
- Multimer made up of 8 Mutant L proteins. The aggregation seems close to what would be expected of wild type L Protein
Mutant 2
17 best residues in hydrophilic zone (no changes to transmembrane zone) Added to the previous best LLR scored residues the best mutant for other 11 residues always double checking with experimental data sheet
- 39 Y->L
- 9 S->Q
- 5 F->Q
- 27 Y->R
- 22 F ->R
- 17 N->R
- 26 D->R
- 23 K->R
- 2 E -> A
- 6 P -> Q
- 12 T -> Q
- 24 H -> R
- 25 E -> R
- 32 Q -> R
- 33 Q -> R
- 37 T -> L
- 14 A -> S
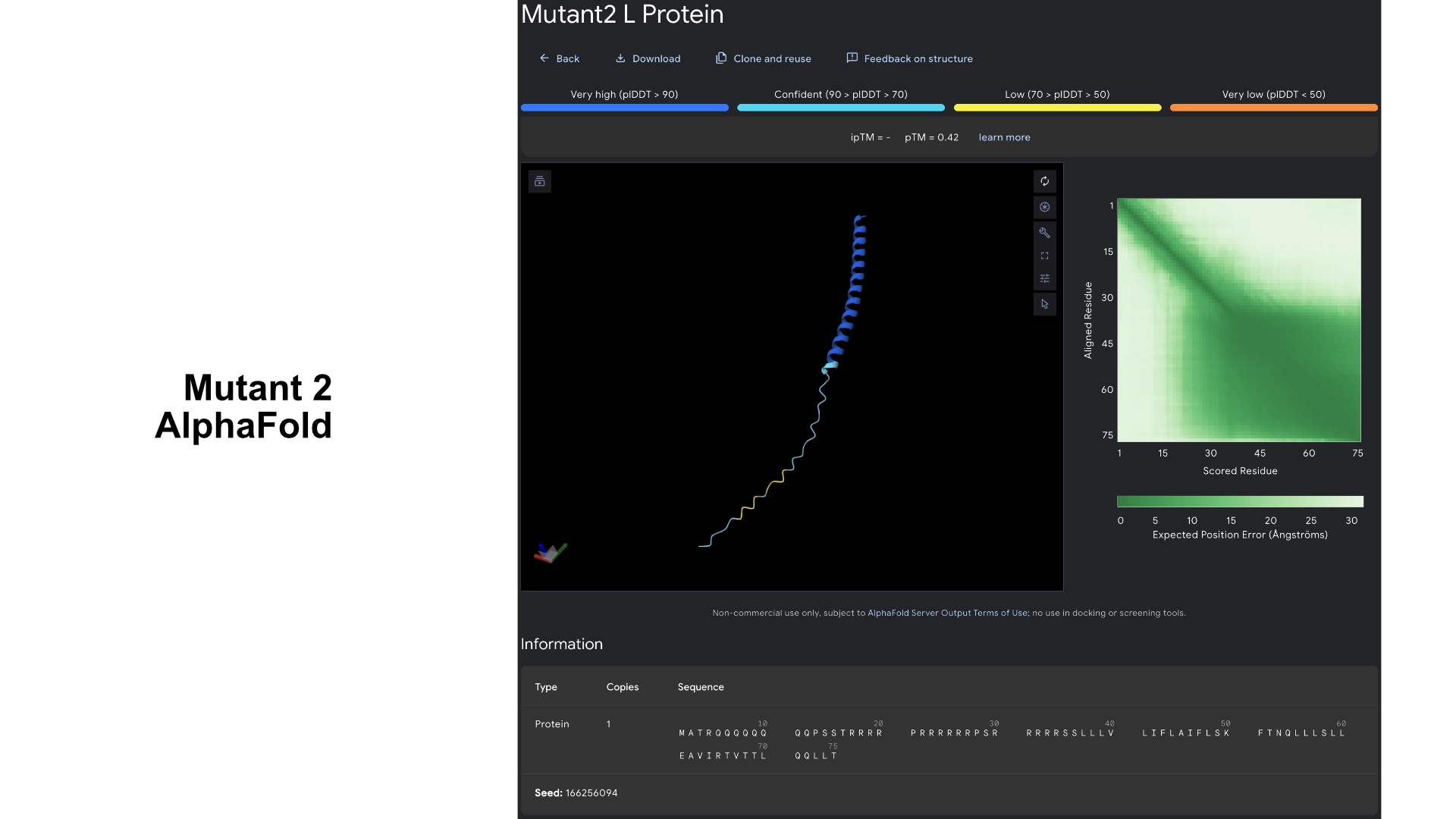
This version had slightly better scoring in regards to prediction confidence and was slightly more uniform, but no significant changes in visualization
- Redered interaction with DnaJ
- Detail where we can see no bonds between DnaJ and L protein Mutant
- Multimer made up of 8 Mutant L proteins. The aggregation seems close to what would be expected of wild type L Protein
Mutant 3
5 best LLR scored residues in transmembrane region always double checking with experimental data sheet (no changes to hydrophylic zone)
- 50 K -> L
- 53 N -> L
- 61 E -> L
- 52 T -> L
- 45 A -> L
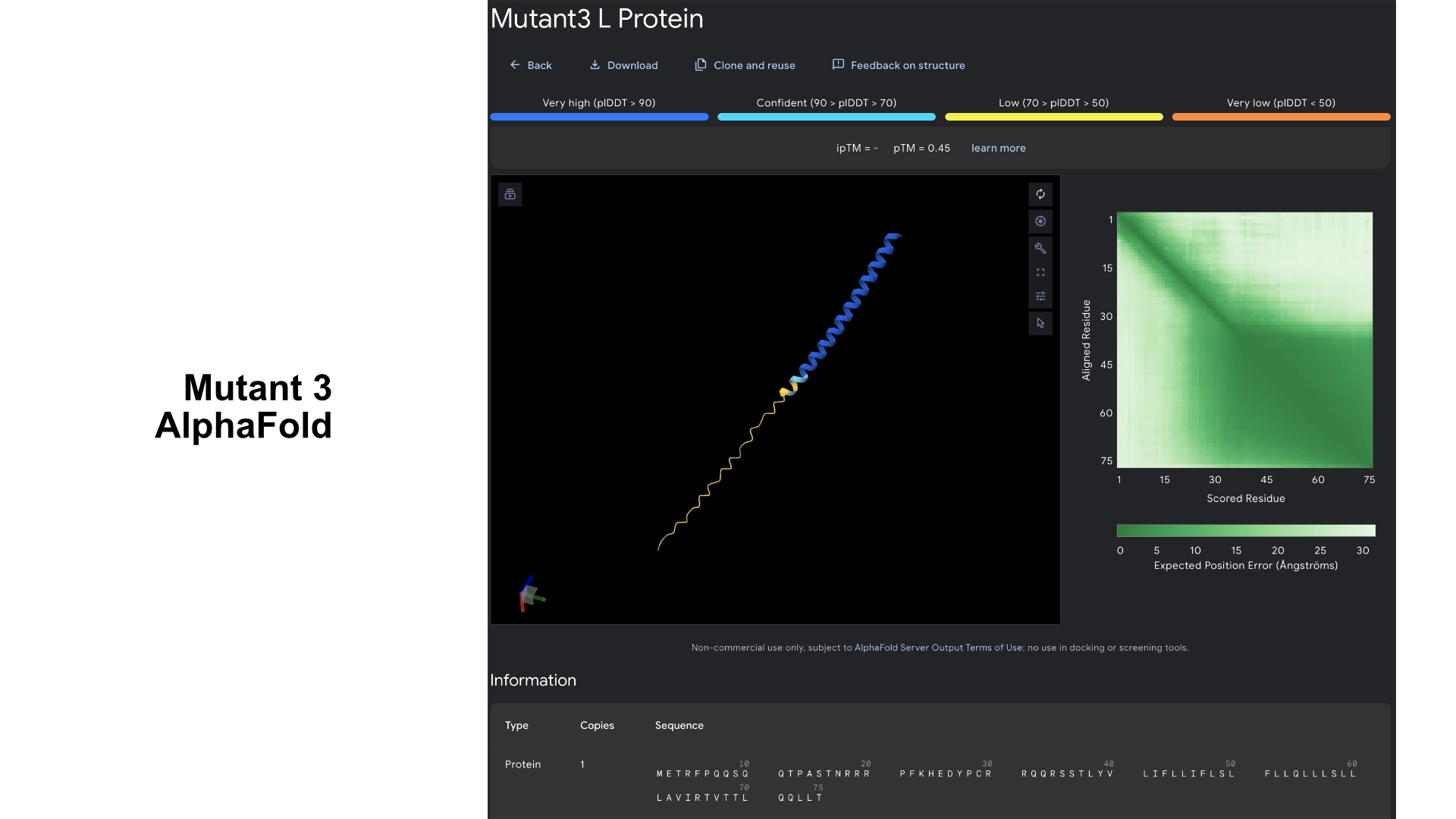
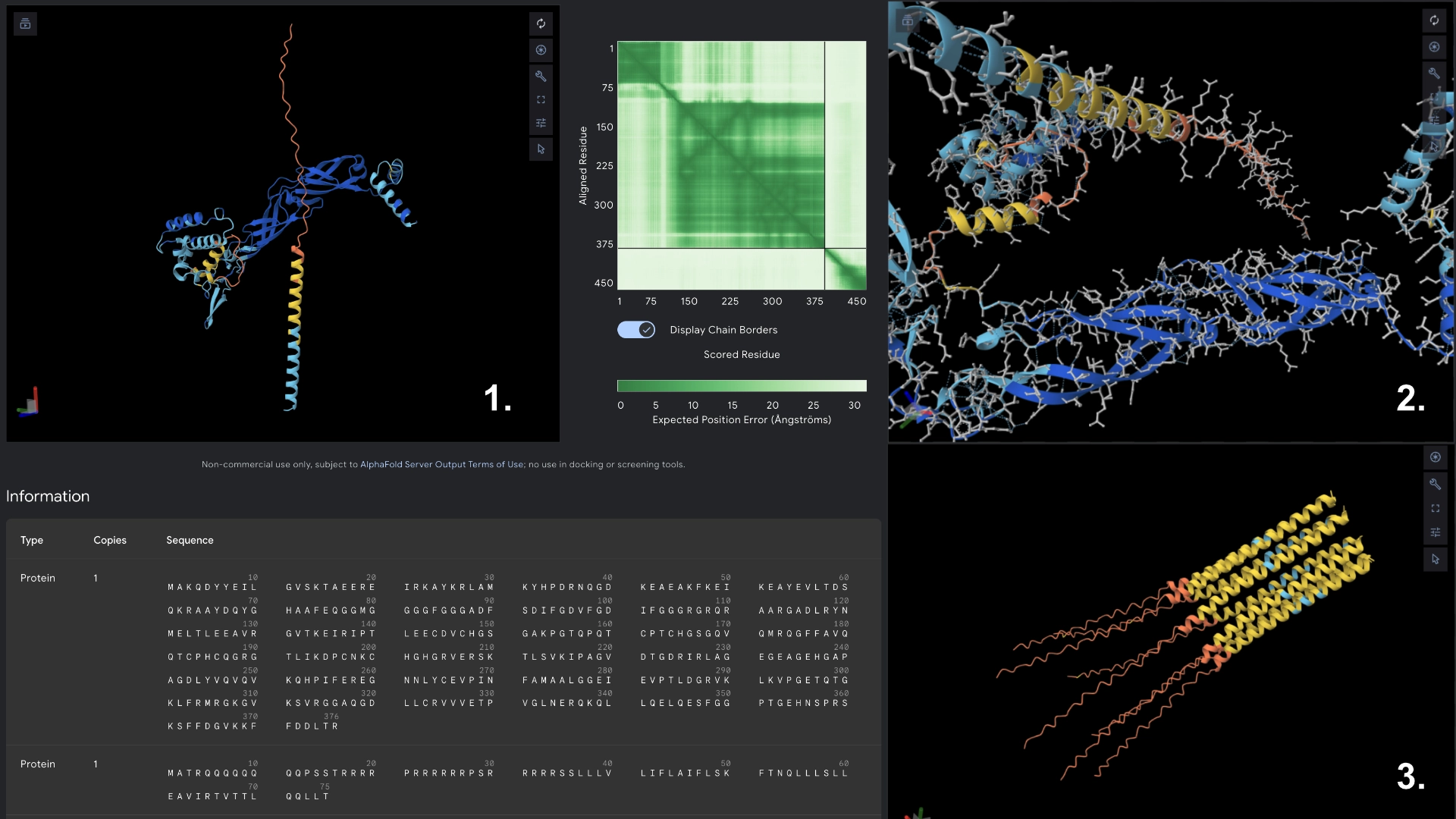
These residues correspond to the areas in the heatmap where mutation seems to be more accepted. As the transmembrane region already has a well defined structure and good prediction confidence in the original protein, no significant changes were noticeable in the visualization.
- Redered interaction with DnaJ
- Detail where we can see no bonds between DnaJ and L protein Mutant
- Multimer made up of 8 Mutant L proteins. The aggregation here changed a lot, having a circular conformation, but with the 8th monomer at the center, and the hydrophylic tails folded inwards at the end
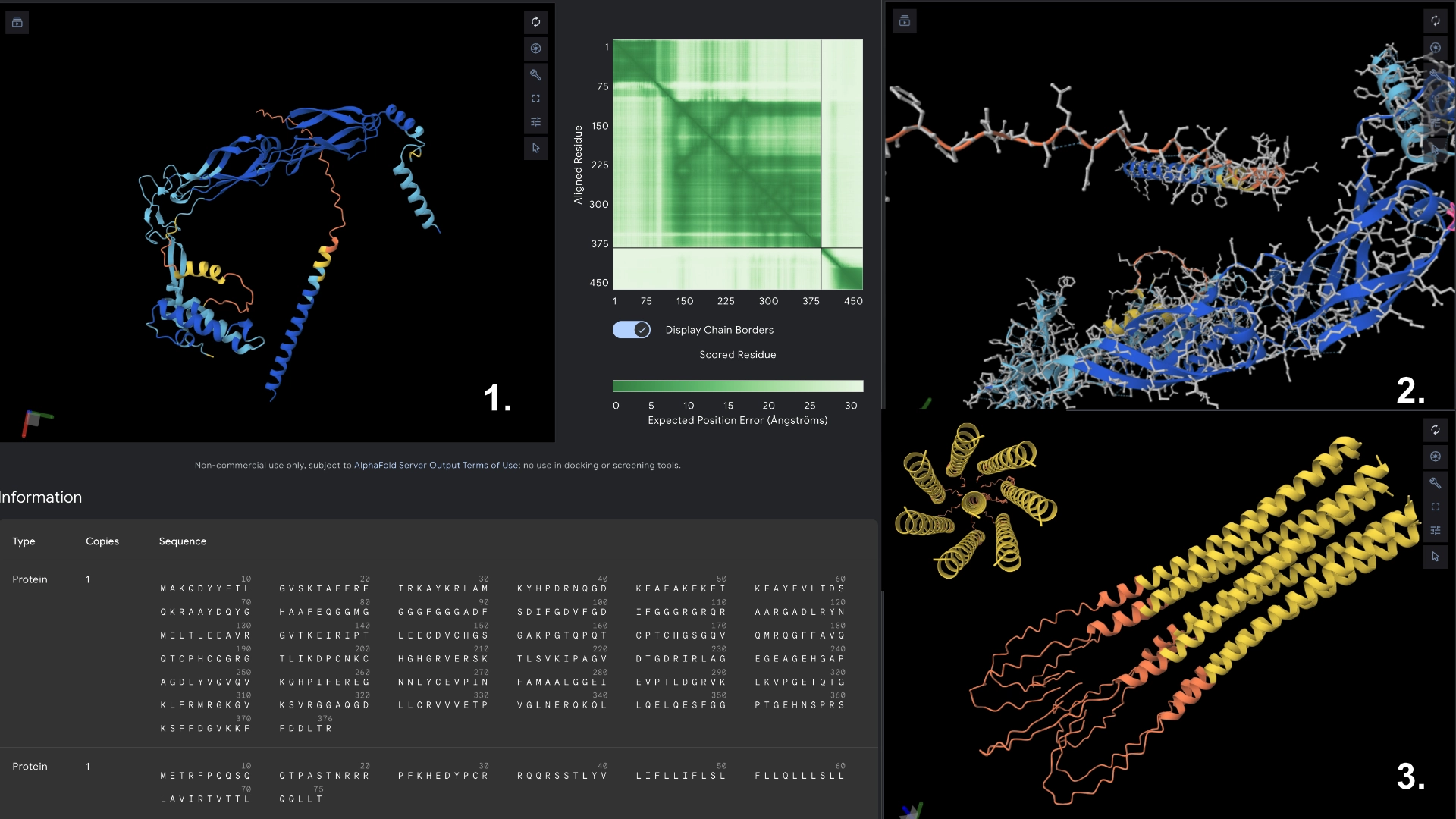
Mutant 4
Added the known positive mutations for lysis from experimental data to mutant 3 (no changes to hydrophylic zone)
- 50 K -> L
- 53 N -> L
- 61 E -> L
- 52 T -> L
- 45 A -> P
- 44 L -> P
- 46 I -> F
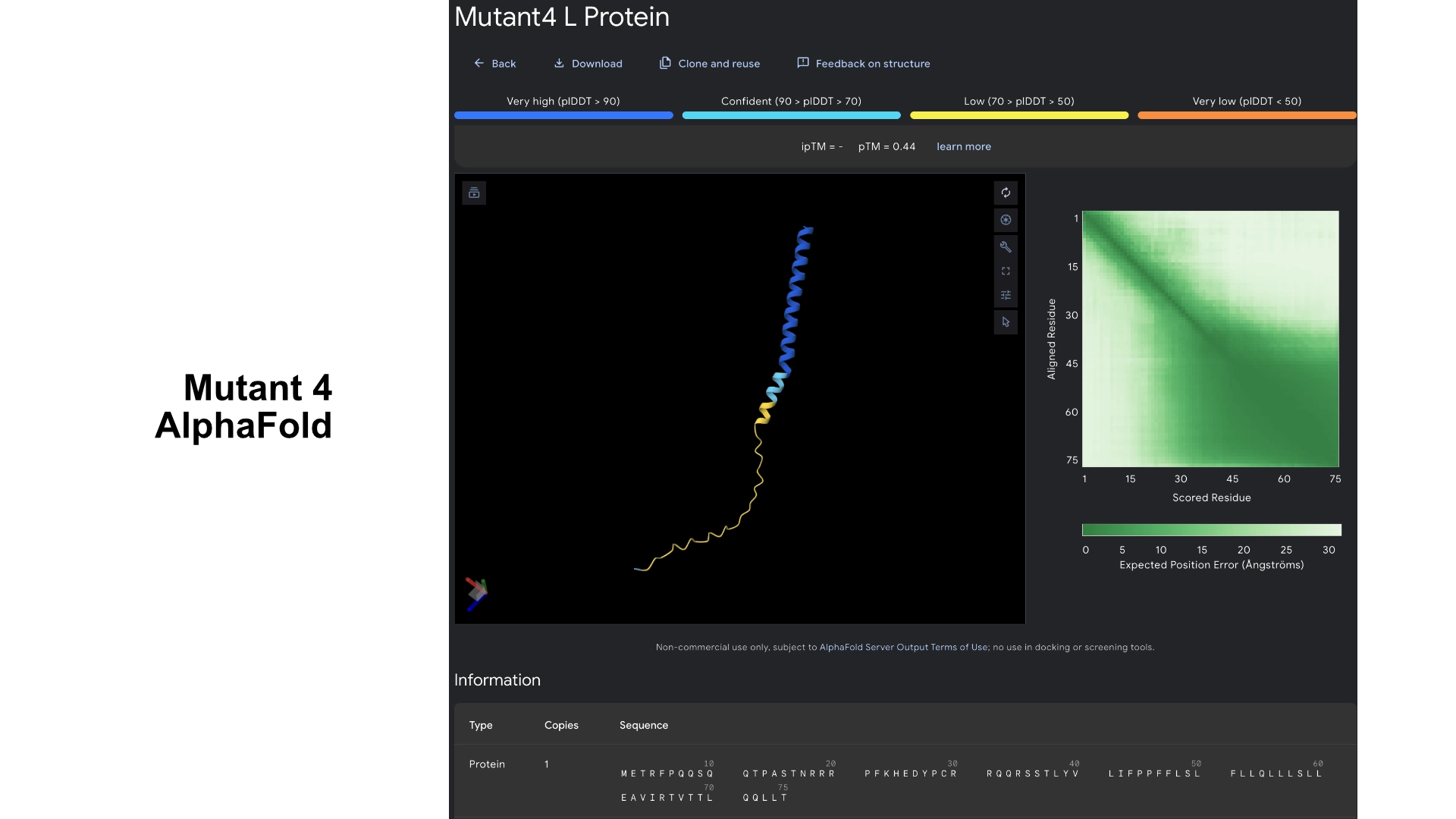
For this version I thought that maybe if the transmembrane region has a mix of better residues for Lysis (from experimental sheet) combined with better structure from best scored residues in the less conserved areas it could have an overall benefit for Lysis and anchoring to the membrane. It was interesting to see that most changes to residues in the transmembrane region in the experimental data resulted in negative impact on lysis, and that the residues that came back positive for lysis in experimental data don’t necessarily correspond to the structural scoring of the LLR
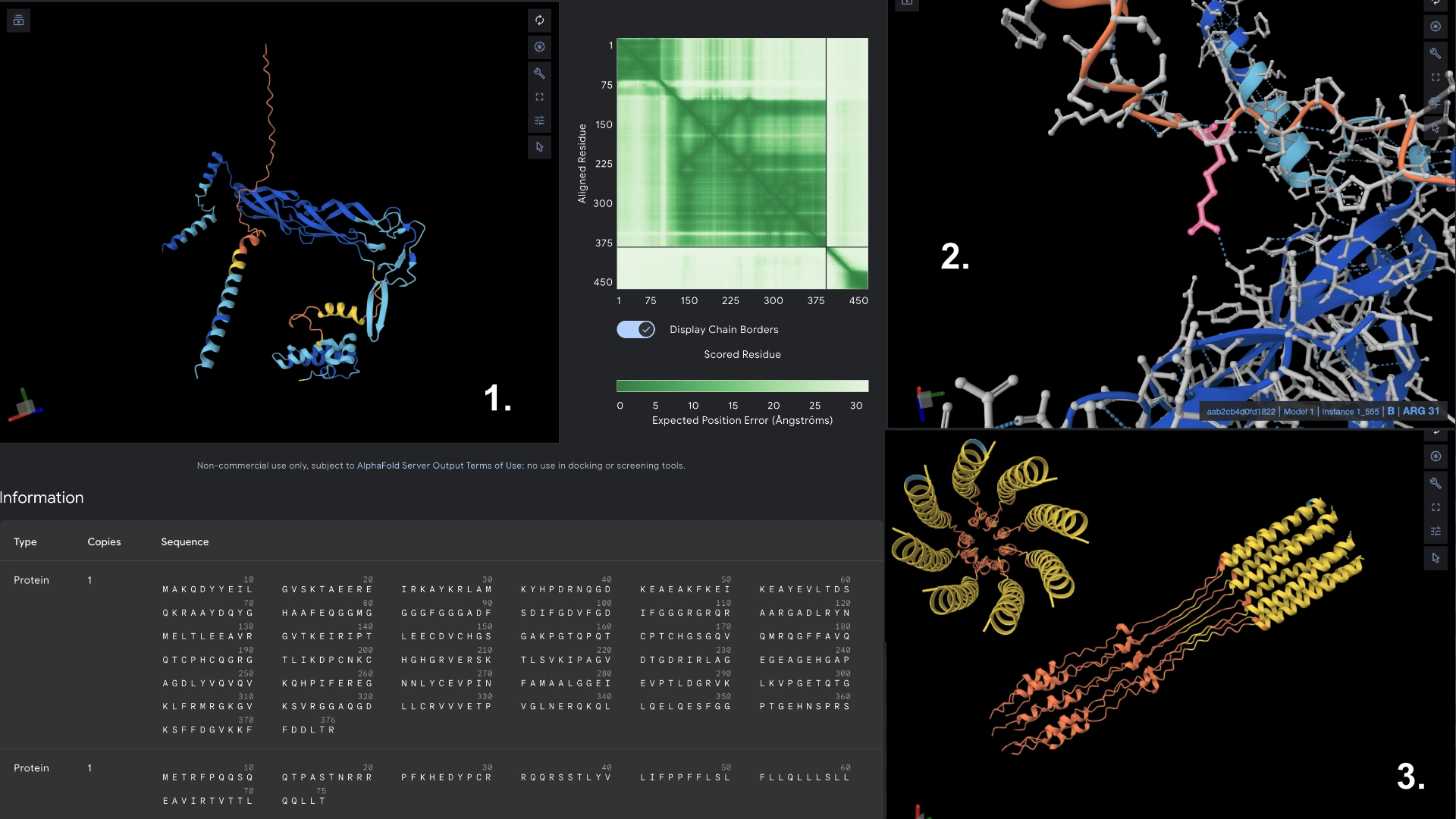
- Redered interaction with DnaJ
- Detail where we can see 1 bond to DnaJ on the residue 31 of Mutant 4
- Multimer made up of 8 Mutant L proteins. The aggregation here changed even more, uncoilig some of the transmembrane portions. It was interesting to understand how mutating a protein might not affect it’s individual folding but can affect a lot its interactions with other proteins, and the low tolerance of the transmembrane zone to mutations.
Mutant 5
Combined mutant 2 and 4
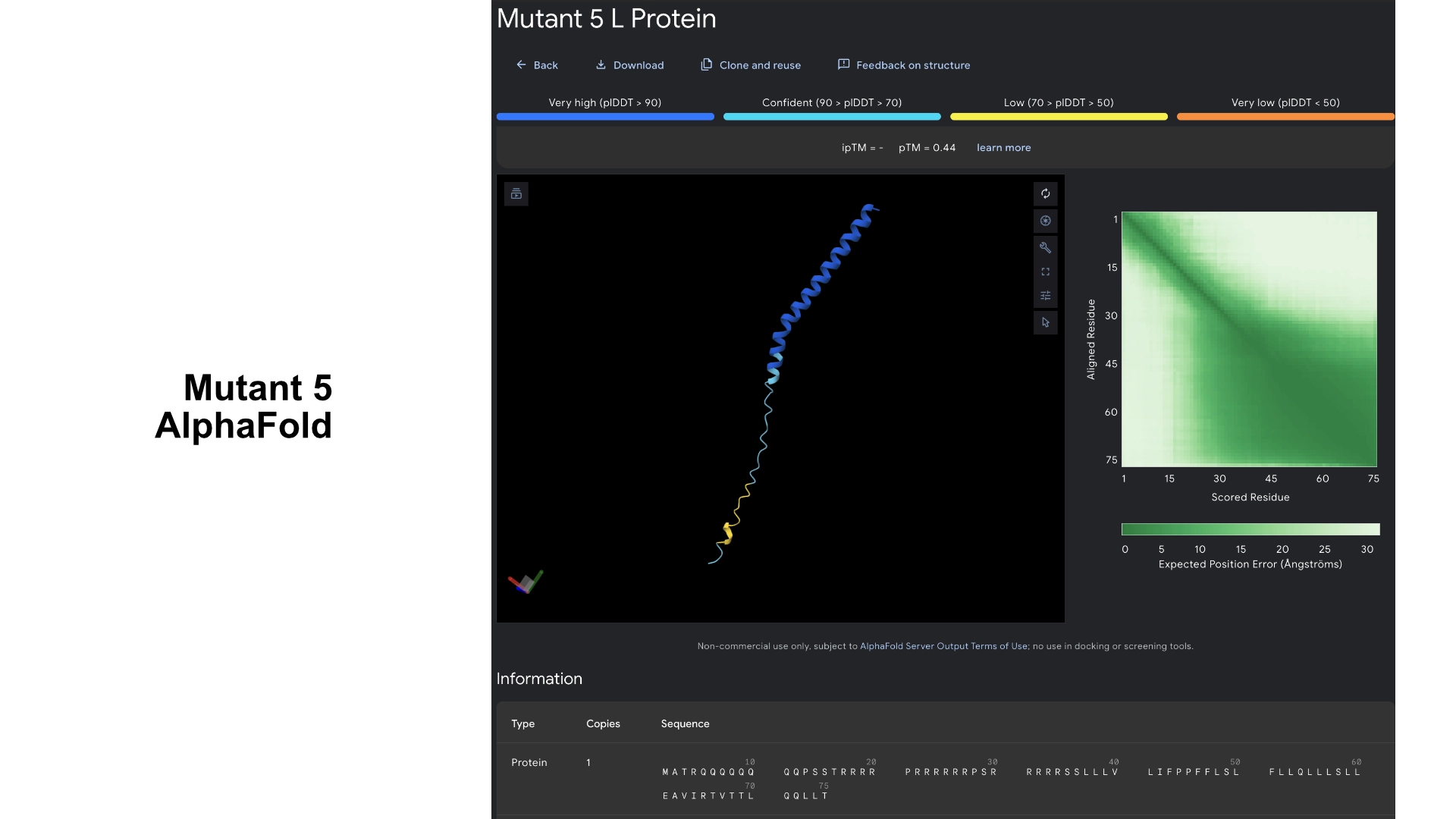
It was interesting to observe that in the visualization this version formed a small helix segment in the hydrophilic region
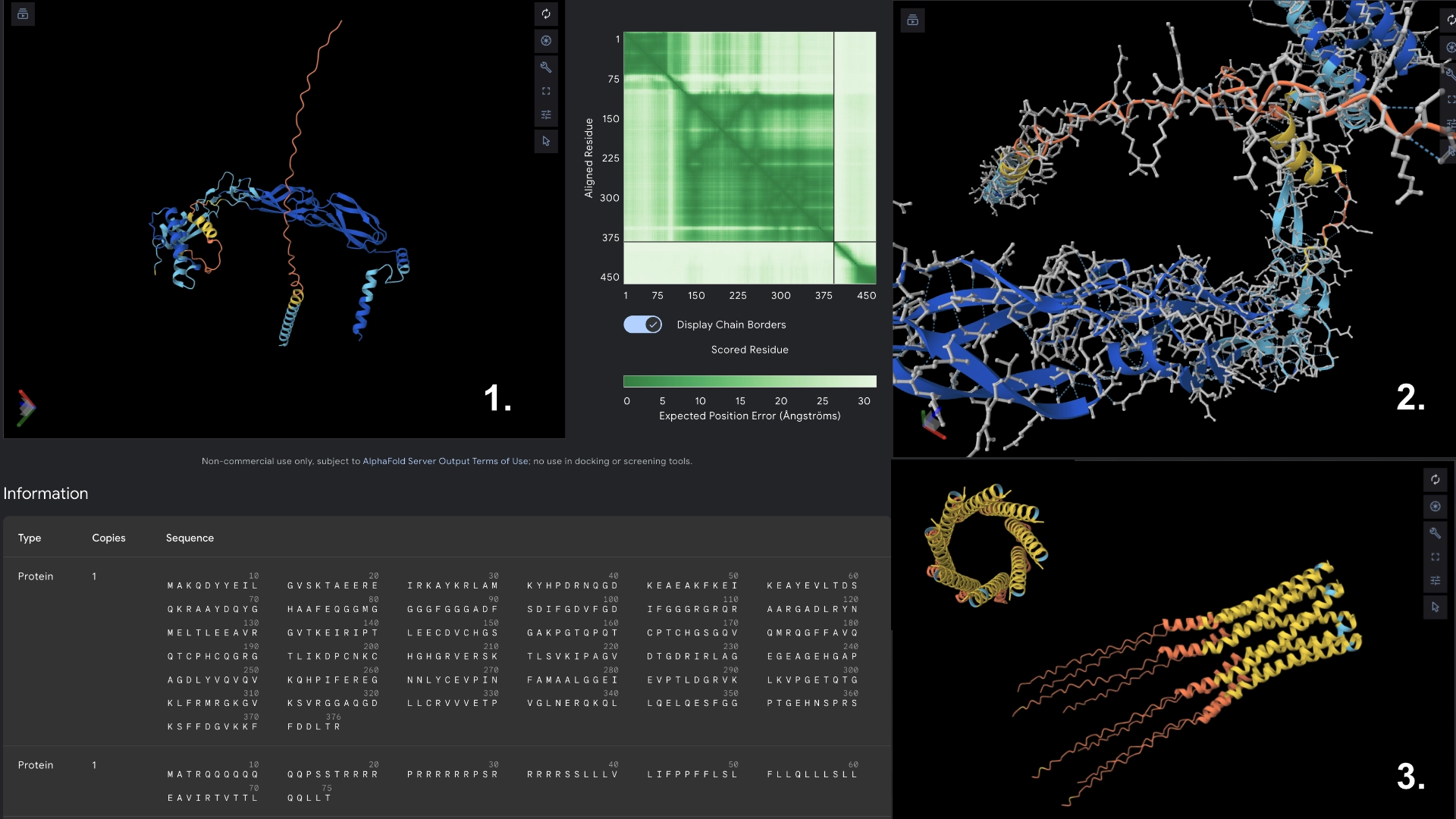
- Redered interaction with DnaJ
- Detail where we can see no bonds between DnaJ and L protein Mutant
- Multimer made up of 8 Mutant L proteins. The aggregation seems close to what would be expected of wild type L Protein, although the monomers bend slightly outwards