Week 10 HW: Imaging and Measurement
Homework: Final Project
My project will measure several aspects related to the behavior of pure chlorophyll versus when bound to water-soluble chlorophyll-binding proteins (WSCPs). The main goal is to determine whether WSCP binding changes light sensitivity, photodegradation behavior, and its subsequent reaction with iron. For this end the following measurements will be needed:
- Important to understand how WSCPs influence the absorbance spectrum of chlorophyll and check if WSCPs are correctly folded and bond to chlorophyll
- This will indicate how the degradation of chlorophyll is carried out in pure chlorophyll vs chlorophyll-bound WSCPs
- It will also be important to understand the differences of light sensitivity in the two samples
Homework: Waters Part I — Molecular Weight
2. Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1)
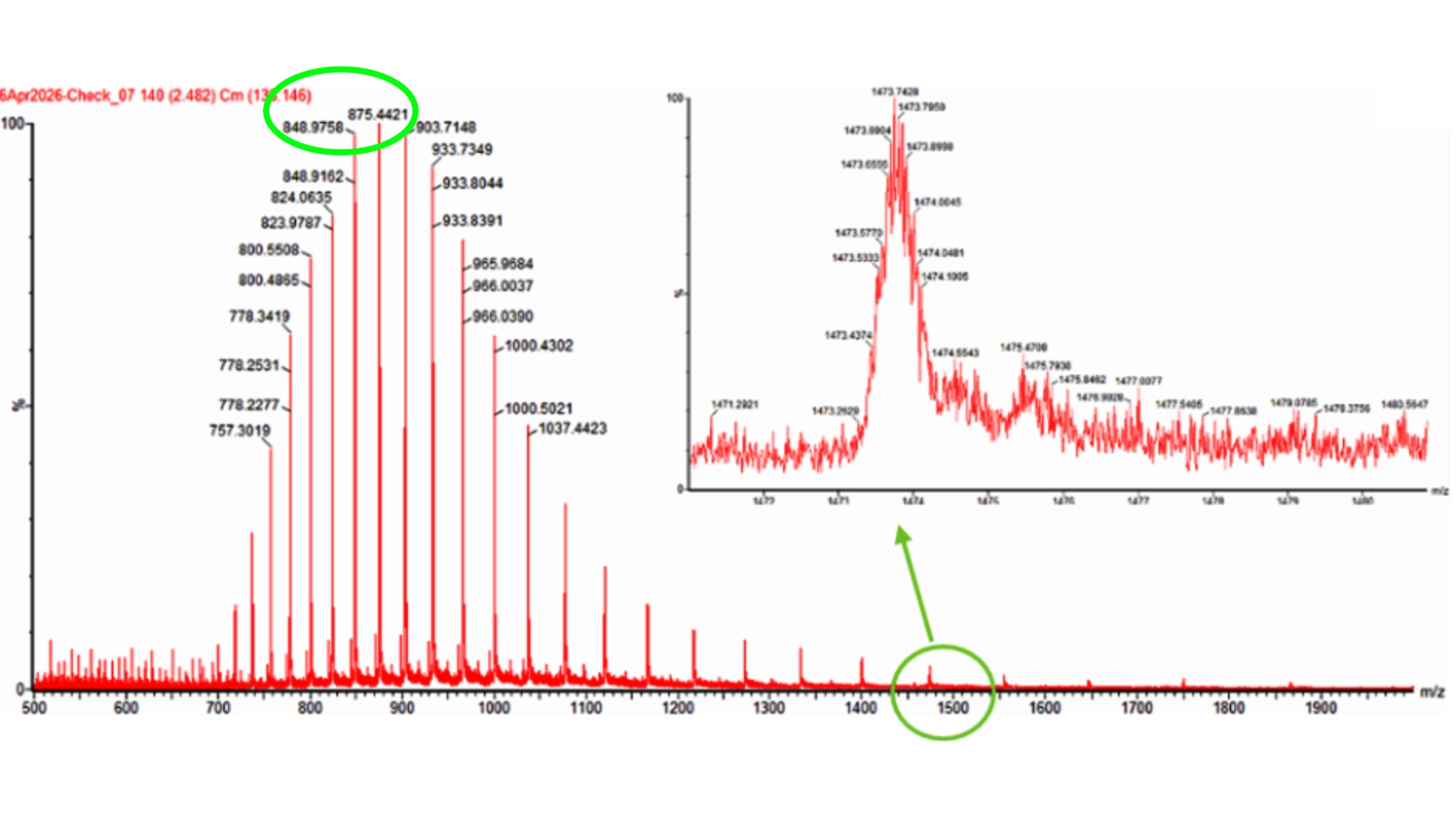 I chose to work with the two peaks circled in green
I chose to work with the two peaks circled in green
$$ \begin{aligned} z &= \frac{m/z_{n+1}}{(m/z_n - m/z_{n+1})} \ z &= \frac{848.9758}{875.4421 - 848.9758} \ z &= \frac{848.9758}{26.4663} = 32.08 \end{aligned} $$
Using the 875.4421 peak with $z = 32$:
$$ MW = 32(875.4421 - 1.0073) $$
$$ MW = 32(874.4348) $$
$$ MW \approx 27981.9\ \text{Da} $$
Check with the second peak:
$$ MW = 33(848.9758 - 1.0073) $$
$$ MW = 33(847.9685) $$
$$ MW \approx 27982\ \text{Da} $$
So the protein molecular weight is:
$$ MW \approx 27982\ \text{Da} $$
Using:
$$ \text{Accuracy} = \frac{|MW_{\text{experiment}} - MW_{\text{theory}}|}{MW_{\text{theory}}} $$
Assuming the theoretical MW of eGFP of 28006 Da, then:
$$ \text{Accuracy} = \frac{|27982 - 28006|}{28006} $$
$$ \text{Accuracy} = \frac{24}{28006} = 8.5 \times 10^{-4} $$
$$ \text{Accuracy} \approx 0.00085 $$
Homework: Waters Part III — Peptide Mapping - primary structure
2. How many peptides will be generated from tryptic digestion of eGFP?
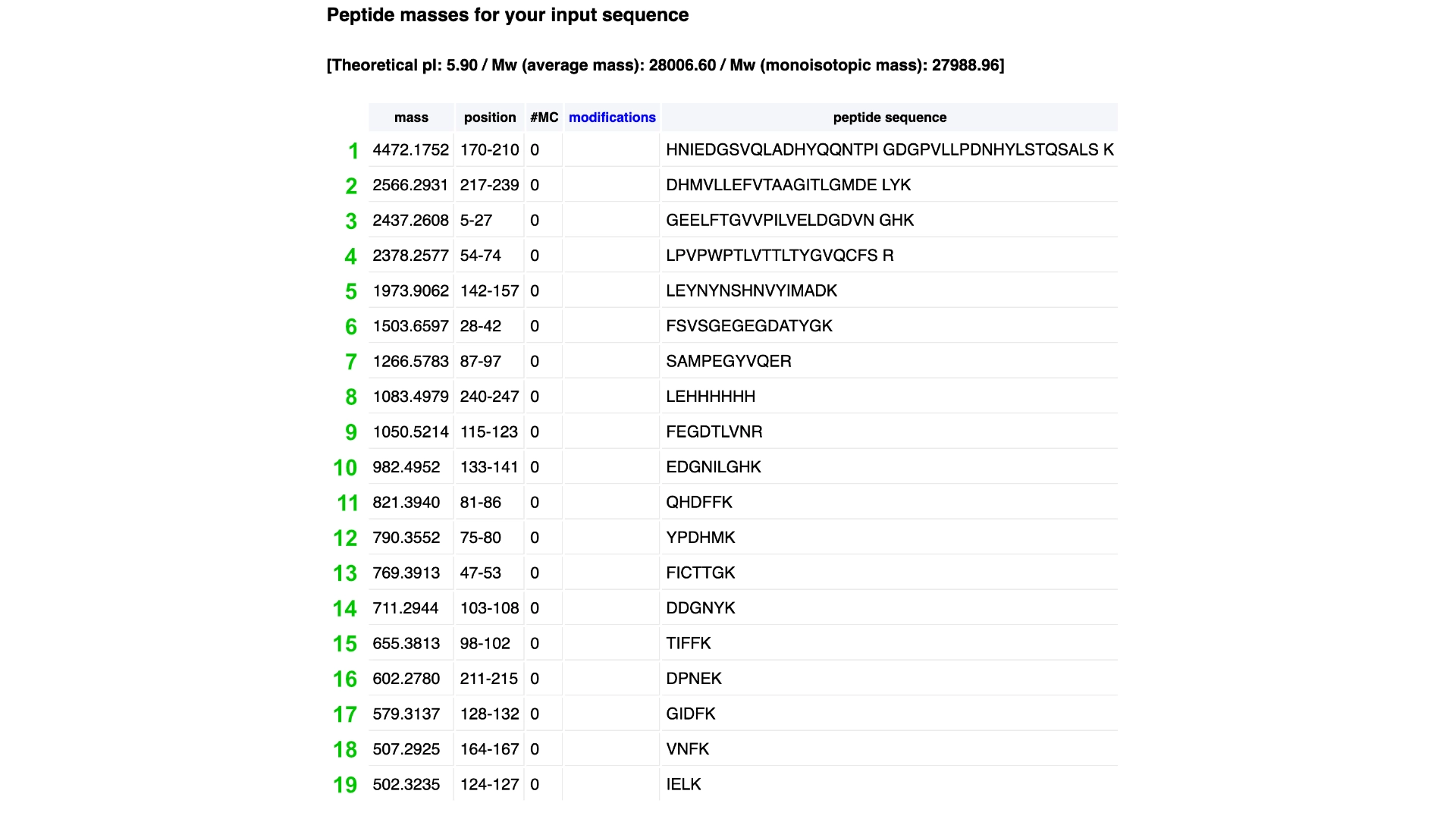 According to the Expasy PeptideMass tool, 19 peptides will be generated
According to the Expasy PeptideMass tool, 19 peptides will be generated
3. Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
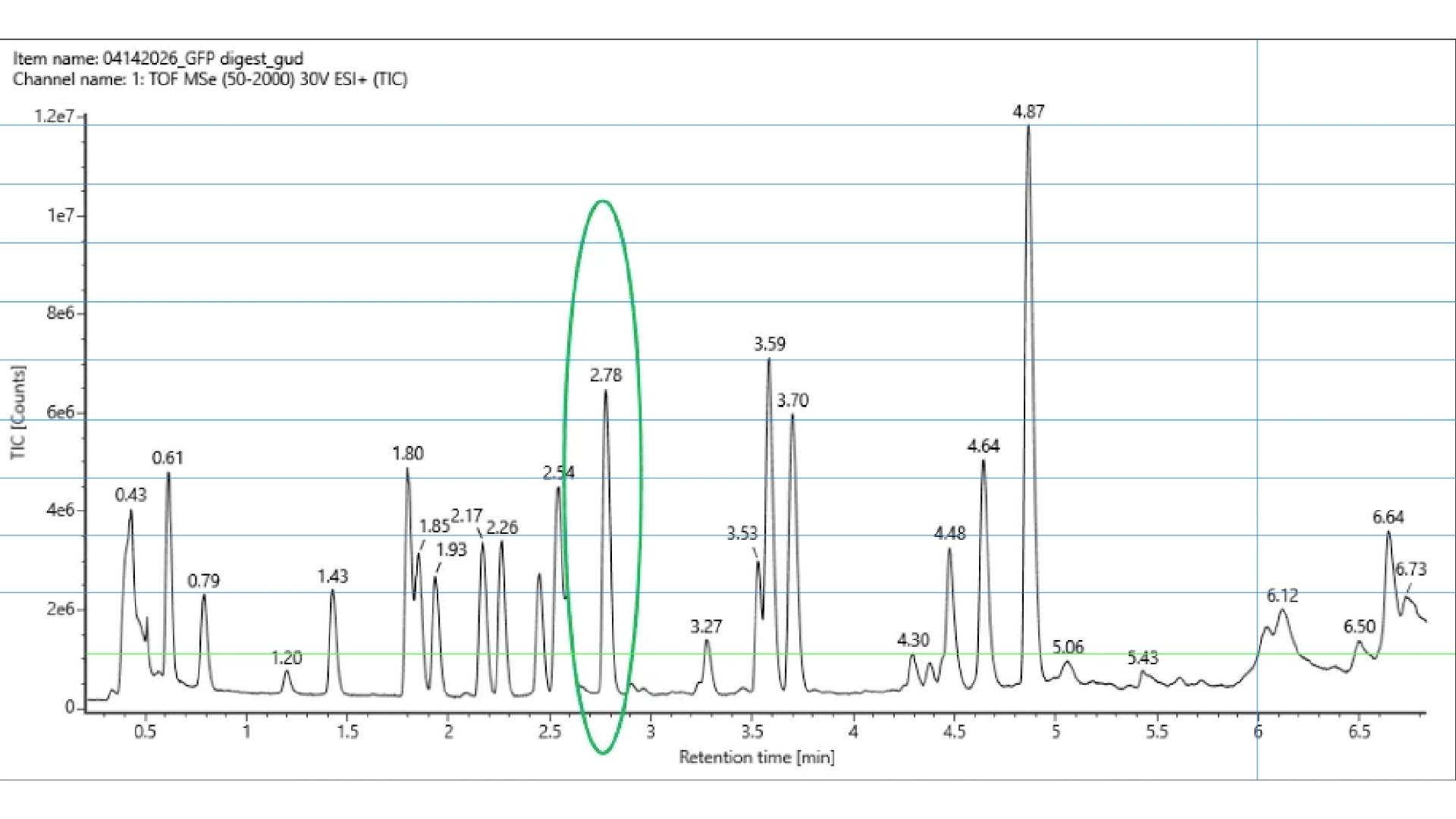
Divided the highest peak in 10
There seem to be 19, which would match the prediction
Homework: Waters Part IV — Oligomers
Using the given subunit masses: 7FU = 340 kDa 8FU = 400 kDa
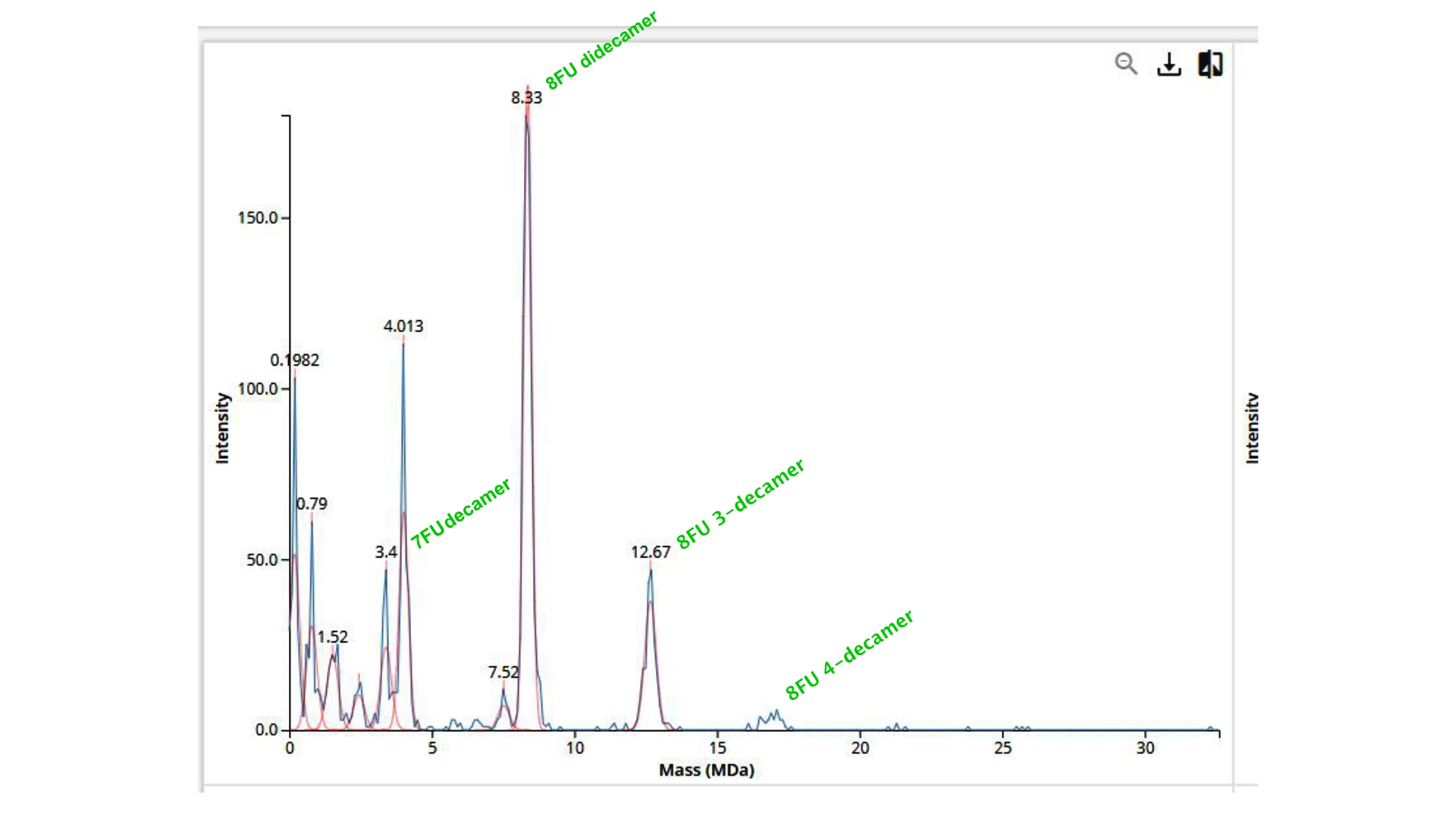
The expected oligomer masses should be:
- 7FU decamer = 10×340=3400kDa = 3.4 MDa
- 8FU didecamer = 20×400=8000kDa = 8.0 MDa
- 8FU 3-decamer = 30×400=12000kDa = 12.0 MDa
- 8FU 4-decamer = 40×400=16000kDa = 16.0 MDa — not clearly observed
Homework: Waters Part V — Did I make GFP?
| Theoretical (kDa) | Observed / Measured (kDa) | PPM Mass Error | |
|---|---|---|---|
| Molecular weight (kDa) | 28.006 | 27.982 | 857 ppm |
A 857ppm error is quite large, so I also tried to predict the molecular weight without the linker and His-tag in case that wasn’t used in the experimental version, but the results would then be even worse (Theoretical (kDa) — 26941.48) with an extremely large 38,600 ppm error, so I assume that can’t be the case.
Given the 857 ppm error and that Proteins are considered confidently identified if the mass accuracy is < 50 ppm, I assume something might be off regarding the theoretical mass probably
References
Fine tuning of chlorophyll spectra by protein-induced ring deformation