Hallo there, I’m Diogo, a interdisciplinary artist interested in symbiosis and more-than-human relationships. Currently researching the chlorophyll molecule 🌿
1. First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
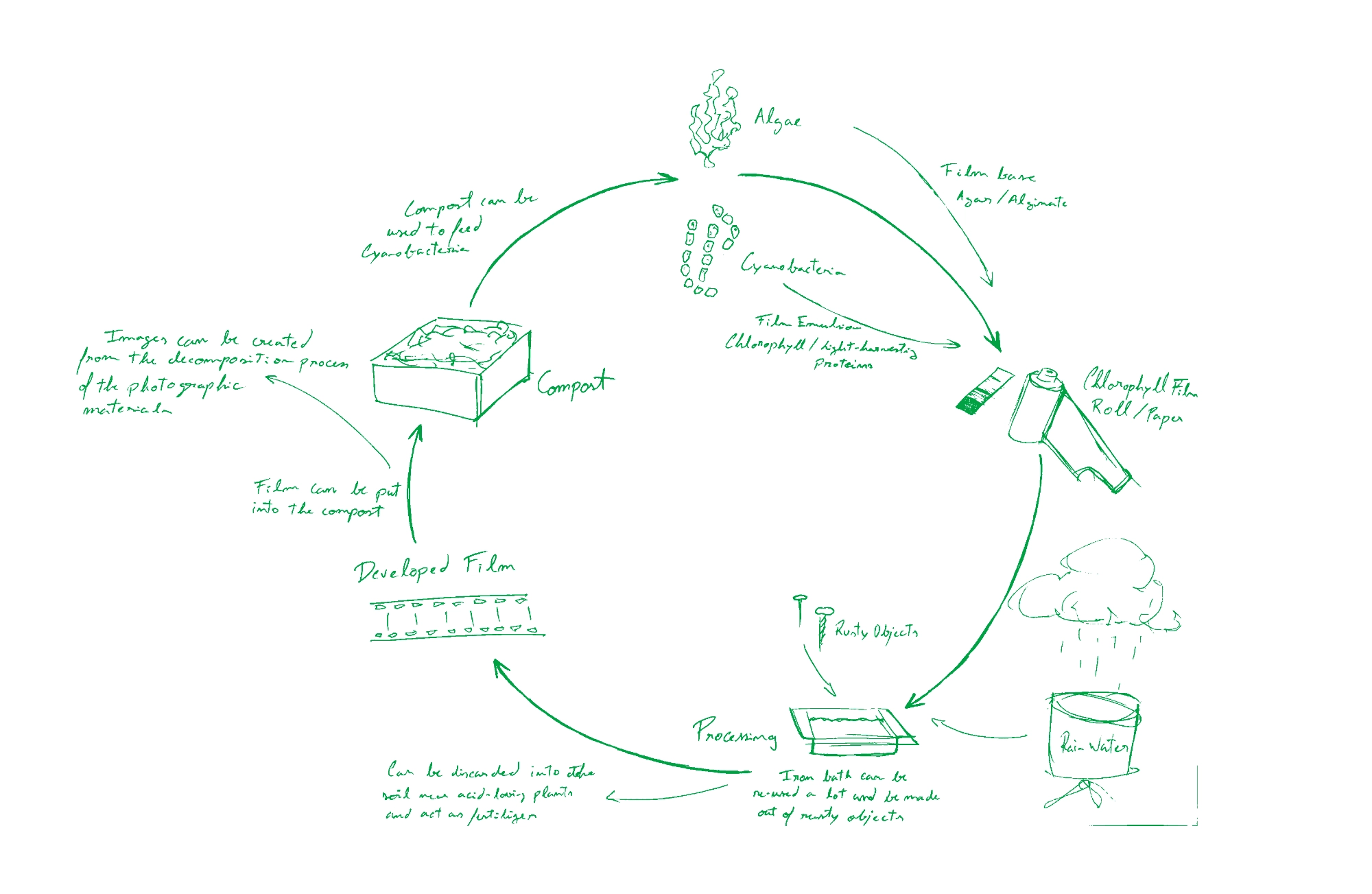
Analog photography has been experiencing a growing revival and with it a growing ecological concern, specially regarding the impacts of its “magical” component — silver halides. Much of the movement of trying to address the environmental impact of analogue film has fallen on individual artists and researchers, by trying to mitigate the consequences of silver. However, despite the efforts of exploring plant-based developers, and darkroom procedures to prevent damaging disposal of silver contaminated solutions, (extremely toxic for the environment affecting primarily microbial life) we are still left with the need to use this toxic metal in lack of any other option for analog camera photography.
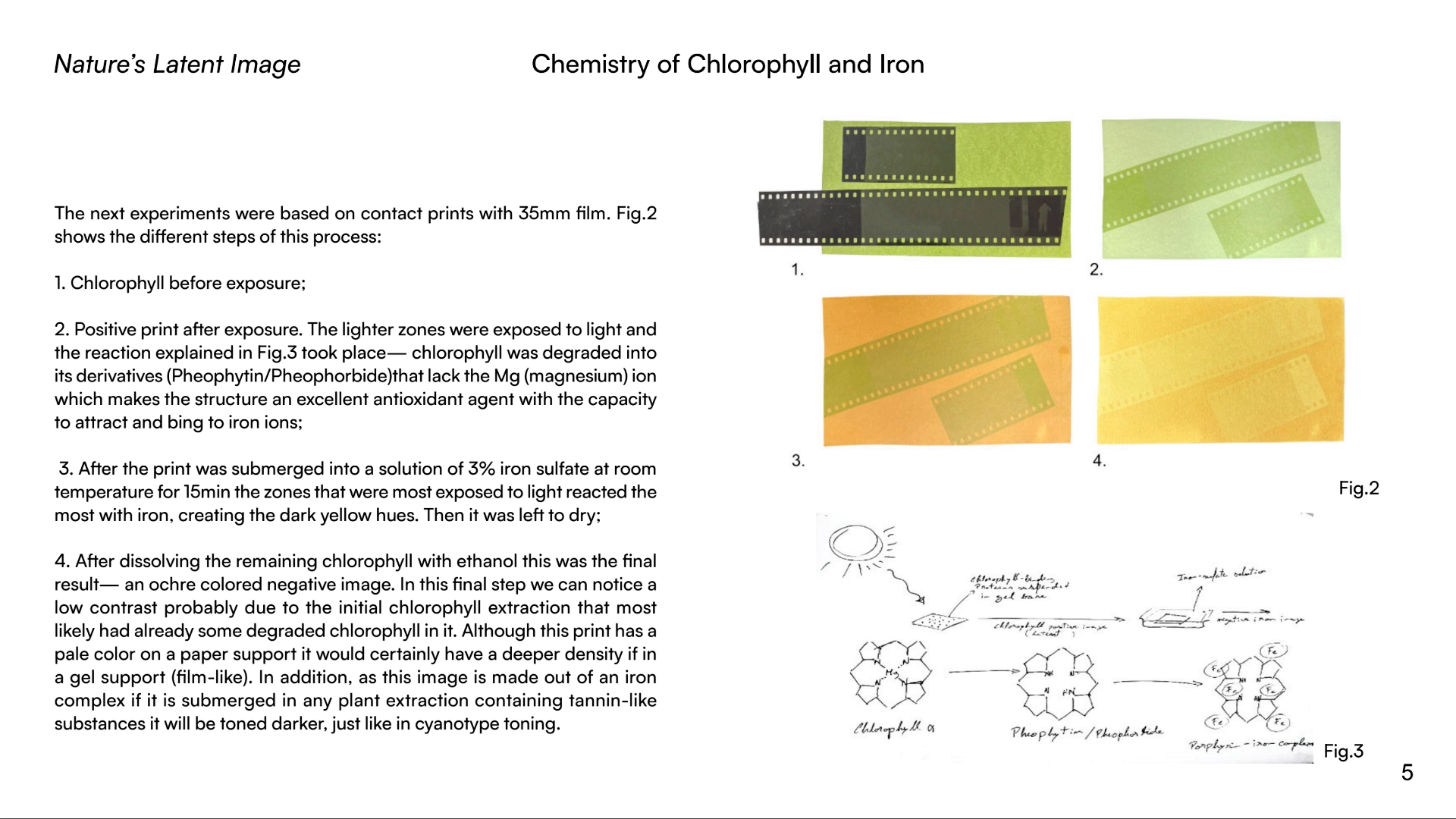
Based on chlorophyll’s photosensitive effectiveness, my research is focused on exploring this molecule as an alternative substance to silver. As of now, I have reached the conclusion that there is a potential in this molecule due to the process of degradation that occurs when chlorophyll is exposed to light outside a living cell — it can demetallate into a porphyrin-type structure that is able to chelate iron, therefore, creating a negative image formed by iron complexes. This hypothesis uses chlorophyll as the photosensitizer and iron as the density builder in order to obtain an image. By developing the image with iron and creating porphyrin-iron type complexes it’s possible to confer a permanent image formation — archival quality to be tested.
For this effect I would like to explore the possibility of engenineering bacteria that could produce a modified version of chlorophyll (that could be organized into supramolecular structures) for optimal photographic application, or an adapted version light-harvesting chlorophyll proteins (LHCPs). The use of bacteria for this effect would ensure a renewable efficient way of producing a photographic emulsion at industrial level.
In a more speculative note, there has also been a growing interest in the experimental photography community to use SCOBY membranes as photographic printing support. It would be interesting if the bacteria could be designed to form a chlorophyll layer at the surface of the cellulose membrane in order to grow photographic “paper”.
Observed: Chlorophyll photodegradation, porphyrin demetallation, iron chelation
Speculative: supramolecular organization of chlorophyll, engineered LHCPs for photographic purposes, SCOBY-grown photographic paper
This intersection between biotechnology and the foundation of an artistic medium can incentivize the much-needed discussion around the role of art when confronted with technological advances and the revision of artistic practices. Specially in the context of ecological artistic practices, there is an interesting space to explore the limits of what is considered ethical in order to make the most out of other-than-human interactions and the creation of symbiotic links through biotechnology.
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
Although this project is primarily focused on a material design/engineering point of view and the possible development of a new photographic process it does pose ethical questions both at practical and conceptual levels. Specially if entertaining the idea of a photographic SCOBY, that could be passed from one enthusiast to another like it happens today with kombucha cultures. Still, even if we just contemplate the possibility of a genetic modification derived chlorophyll film, that would no longer contain living cells, there could be some implications at conceptual levels regarding people that are developing ecological practices. Taking this into account, some governance/policy goals that could make this project come to life in a safe and ethical manner are:
Transparency regarding the modifications
Have open documentation of genetic modifications in the cells used and processes of production of the film, to allow for an informed ethical evaluation by the users.
The same would apply to a SCOBY plus clearly stating what living cells would that culture contain.
Esurance of biosafety
Utilization of bacteria that present low biosafety hazard risk, both for human handling and eventual environmental release.
Create clear protocols of disposal and deactivation of the cultures.
Understand the impact of a modified culture that could be grown and passed from one person to another in an amateur context
Use of a “kill switch” – nutrient without which the SCOBY culture couldn’t survive
Environmental sustainability
Understanding the life cycle of the engineered material and create clear protocols for sustainable use
Design the materials that compose the film to ensure biodegradability, like substituting the gelatine used in current films for algae derived gels and using bioplastic as film base.
Avoid “greenwashing” through a narrative of sustainability without being sure of the extent of the possible impacts
Pedagogy and discussion
Generate open discussions about ethical use of synthetic biology and offer workshops on the use of this technology
Use as teaching tool to contribute to a more distributed knowledge about biotechnology and how it can be used for creation and evolution. The SCOBY could be a great opportunity to demystify synthetic biology.
Preventing misuse or misinterpretation
While trying to democratize the knowledge about synthetic biology the take measures to prevent the public notion of biological = harmless
Preventing unregulated bio-modification
Thinking about these subjects made me understand the difficult role of defining where should be the limit in making synthetic biology more accessible to the public and made me eager to start dialogues around the role of the arts in play in this. I still think it is important to show how humans can relate with other species at different levels and through symbiosis (whatever form it takes) evolve
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Action 1(most plausible): Disclosure standard for bio-engineered materials
Actors: Academic and Scientific researchers, Art institutions, Funding institutions
Purpose:
Regarding the case of the chlorophyll film which could be produced through GM bacteria, but the final product wouldn’t contain any viable »
cells. This, by Portuguese and EU law wouldn’t have the need of any labeling regarding its origin of production. As a result, users and audiences are typically unaware of the biological engineering involved in the production process.
Proposal: Introduce a voluntary disclosure standard for this type of product outside the food and feed context that are derived from GMO production processes but are themselves non-living, clarifying their origin.
Design:
Develop a standardized disclosure label or documentation stating:
That the material was produced using genetically modified microorganisms under contained conditions
The procedure of production: which type of organism was used and in which material it contributed to
That the final product contains no living organisms or viable genetic material
Adoption driven by:
Research institutions
Art schools, Museums and Galleries
Funding agencies — transparency statements
Assumptions:
It’s expected that transparency of production methods increases trust rather than fear
Artists, researchers and institutions are willing to adopt ethical commitments
The public will be receptive to the difference between process-based and product-based genetic modification when explained clearly
Risks of Failure & Sucess:
Failure:
Low adoption due to lack of incentives
Misinterpretation of disclosure as associated risk
Success:
Voluntary disclosure could become an informal requirement
May unintentionally reinforce the idea that GMO-derived products are Inherently suspect
Action 2 (still plausible): Offering demonstrations in contained conditions
Actors: Researchers; Selected Laboratories (could be biolabs); Experimental Photography Organizations; Public — artists, enthusiasts
Purpose:
Regarding both the chlorophyll film and the SCOBY.
Proposal: Through the reach of international photography organizations arrange in collaboration with biolabs demonstrations of the production of both the film and the live photographic SCOBY in contained conditions.
Design:
According to biosafety levels of both the needed GMOs and the laboratories it should be possible to realize demonstrations of the chlorophyll film production using modified bacteria and the growing of photographic SCOBY membranes since these wouldn’t leave biosafety areas.
These demonstrations could include the following:
The protocols for extracting the modified chlorophyll from the bacteria and turning it into photographic emulsion
Developing and processing chlorophyll film
Overview the safe and sustainable disposal of the film and chemicals used
The protocols used to grow the photographic SCOBY membrane such as feeding, processing the grown membrane, print an image on it and develop it.
This would be a great opportunity to be able to understand the opinion of the artistic community regarding the use of synthetic biology. >And if it seems justifiable for this end.
Assumptions:
By sharing the production protocols of a new analog photography technology artists might be more interested to build upon it and feel more confident about biotechnology
The interaction with a living GM SCOBY would largely contribute for the demystification of synthetic biology
The public interested in both traditional analog photography and experimental photography would be available to understand more about a new and ecological way of using film
Risks of Failure & “Success”Failure:
Lack of adherence due to preconceived ideas about GMOs and ethical collision against ecological practices
Success:
Increase of concerns about GMOs due to the demonstration being restricted to biosafe infrastructures
Action 3 (least plausible): Framework for release and sharing of GM SCOBY
Actors: Portuguese and EU regulatory bodies; Research centers; Community Labs
Purpose:
Under Portuguese and EU regulation, the deliberate release of genetically modified organisms into the environment — including sharing living cultures outside contained laboratory conditions — is heavily restricted and prohibited without formal authorization. Informal circulation of living GMOs through artistic or DIY communities is not legally accommodated.
Proposal: Establish a formal regulatory framework that would allow, under strict conditions, the deliberate release and downstream sharing of that genetically modified SCOBY, used for artistic or photographic purposes.
Design:
Develop a dedicated authorization pathway under existing GMO delierate release legislations adapted for non-agricultural, non-food, artistic uses
Requirements would include:
Environmental risk acessment
Proof of containent or ecological self-limitation
Monitoring and reporting obligations
Clear disposal protocols
Oversighted by the Portuguese regulator (APA), possibly coordinated at the EU level
Participation could imply institutional support from an university/research center, community biolabs for approval prior to sharing or release
Assumptions:
The environmental risks of a modified SCOBY can be sufficiently predicted and controlled
Regulators would be willing to differenciate cultural/artistic uses from agricultural and and commercial uses
That downstream user would comply with handling, propagation and disposal protocols
That a legal framework would reduce informal or illegal dissemination
Risks of Failure & “Success”Failure:
High admnistrative and financial burden could make the framework unusable
Difficulty in ensuring compliance once organisms start circulating
Public opposition to deliberate release of GMO undermines feasibility
Success:
Normalising the release of GMO for artistic porpuses could endanger biosafety norms
Authorized release could be interpreted as being biologically harmless and contribute to reckless use
Aproval could legitimize risky practices under the context of art
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Disclosure Standard for bio-engineered materials
Offering demonstrations in contained conditions
Creation of a framework for downstream sharing of GM SCOBY
Esurance of biosafety
1
2
3
Transparency regarding the modifications
1
1
2
Environmental sustainability
2
1
n/a
Pedagogy and discussion
n/a
1
2
Preventing misuse or misinterpretation
2
1
3
Other considerations
• Minimizing costs and burdens to stakeholders
1
2
3
• Feasibility?
1
2
3
• Not impede research
1
1
1
• Promote constructive applications
1
1
2
5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Disclosure standard for bio-engineered materials
This action would receive the most priority since having a disclosure that allows the consumer to make an informed ethical decision about the technology offered is essential and creates an opportunity to broaden the perception of the range of synthetic biology use.
Offering demonstrations in contained conditions
I consider this action the most interesting to accomplish the main two objectives of increasing proximity and dissemination of biotechnology in the arts and incentivize research on the topic of this project. However, it would imply more difficulties due to the need for living GM cultures, that not being authorized to leave biosafe areas, would need to be reproduced in every lab the demonstrations took place.
Homework Questions from Professor Jacobson
1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The initial insertion of nucleotides by polymerases incurs in an error once every 10000 to 100000. When including the effect of exonuclease proofreading domain, accuracy is increased by 100- to 1000- fold, making the final error rate one per 106 107 nucleotides. The haploid human genome is roughly 3 billion base pairs and a diploid cell (before division), this is 6 x 10^9 base pairs — If the polymerase only had its intrinsic proofreading ability error rate, a single cell division would result in roughly 30 to 600 errors per replication. This would be an unsustainable rate of mutation for a multicellular organism.
Thus, biology employs a multi-tiered, highly efficient repair system to ensure high fidelity, resulting in an overall mutation rate of less than one mutation per genome per cell division by: Proofreading (Immediate Correction), Mismatch Repair (Post-Replication Repair), Redundancy and Non-coding DNA and Low-Fidelity Backup — In cases of severe DNA damage, the cell uses specialized, “error-prone” polymerases (translesion synthesis) to skip over damage to prevent cell death, allowing for a temporary increase in mutations, but saving the cell.
2. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Most amino acids are coded by 2-6 codons, therefore, for an average human protein—roughly 300 to 500 amino acids long—the number of potential DNA sequences is astronomical. So most of these potential genetic codes will not produce a functional protein due to: Codon Usage Bias & Translation Speed: there are preferred codons to improve speed; Co-translational Folding Errors: the folding is coordinated with speed of translation, so if the speed is slowed down by the use of rare codons the proteins might not fold properly; mRNA Stability and Structure; Splicing Errors:in eukaryotes, the coding sequence (exons) is interrupted by non-coding sequences (introns). Eukaryotic DNA sequences contain “hidden” splicing signals that tell the cell where to cut and join RNA. A different coding sequence might accidentally introduce or destroy these sites, resulting in an improperly spliced mRNA; Regulatory Site Disruption: DNA regions often contain dual information: coding for a protein and containing regulatory signals (e.g., enhancers, transcription factor binding sites). Changing the DNA code to a synonym might destroy a crucial regulatory element, meaning the protein is simply never produced.
Homework Questions from Dr. LeProust
1. What’s the most commonly used method for oligo synthesis currently?
It’s the solid phase chemichal synthesis — Phosphoramidite Method.
2. Why is it difficult to make oligos longer than 200nt via direct synthesis?
Even with most highly optimized protocols, each step of the chemical synthesis cycle is not 100% efficient (99% — 99.5%). As the sequence lenght increases. The effect of these small cumulative losses in a 200 nt segment can reduce the final yield to < 30%.
3. Why can’t you make a 2000bp gene via direct oligo synthesis?
Due to the accuracy limitations, chemical synthesis is mostly limited to 500 bp. Making a gene longer than that implies the posterior ligation of the several smaller diferent fragments to ensure a viable gene, otherwise, the errors would accumulate into a non-working gene.
Homework Question from George Church
1. What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency"?
Being the 10 essential amino acids in animals (PVT TIM HALL): Phenylalanine, Valine, Tryptophan, Threonine, Isoleucine, Methionine, Histidine, Arginine, Leucine and Lysine — the “Lysine Contingency” would be lacking the main component of a biological kill switch which is dependant on a substance that any given organism wouldn’t be able to get outside controlled systems. If Lysine is already an essential amino acid that all animals, and presumably dinosaurs, need to find through their diet, then it can’t be considered a contingency since the dinosaurs could find it anywhere outside the island through eating plants or other animals. For this amino acid contingency to be functional the GM dinosaur would need to be dependant on some kind of completely synthetic amino acid that could not be substituted by any naturally occurring one.
First of all I started by making a digest with a single enzyme at a time.
Then tried to color code the result of every enzyme and superimpose them on top of each other so to create a “grid” were I would make my design. I soon understood it would be way too confusing, plus, that the result of using a combination of enzymes doesn’t necessarily correspond to the superimposition of the lines created by each enzyme separately.
Then I started using Ronan’s website to iterate on combinations of enzymes + using some unconventional techniques.
Final Result — “HTGAA”
For the cover image I just edited out some of the space between lines of the same letter to make it more perceptible
This was a fun exercise that allowed me to visually understand the logic of enzyme’s digests and pay attention to some details I might haved overlooked otherwise.
Part 3: DNA Design Challenge
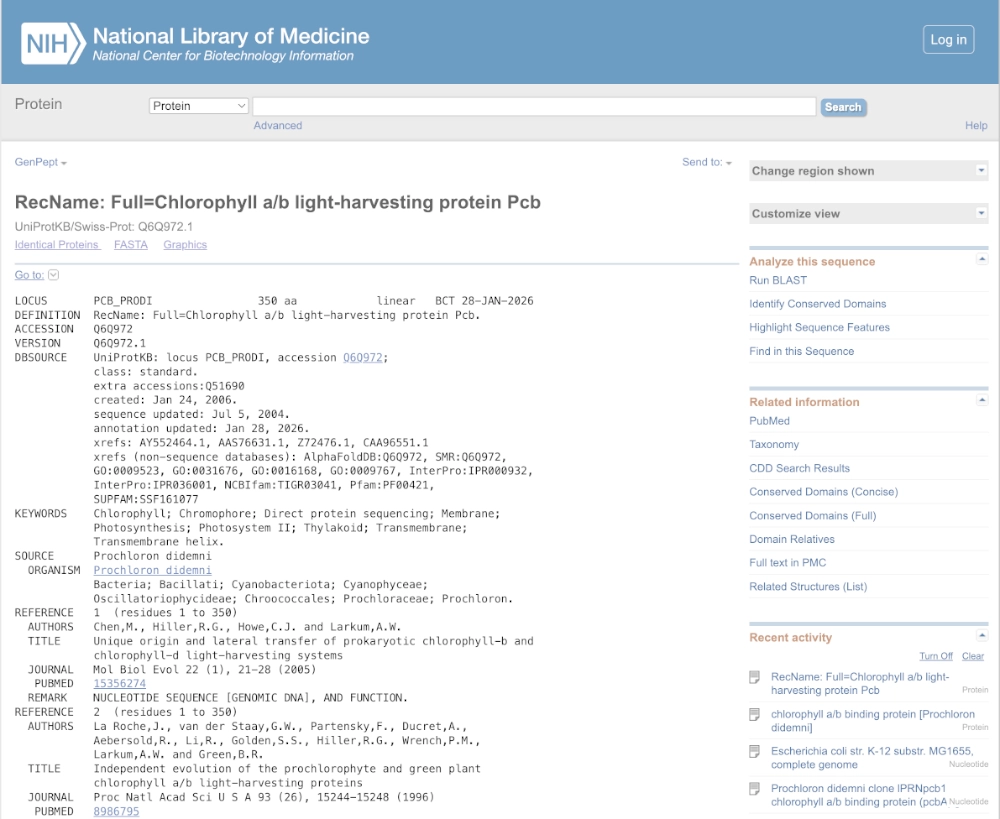
3.1. Choose your protein
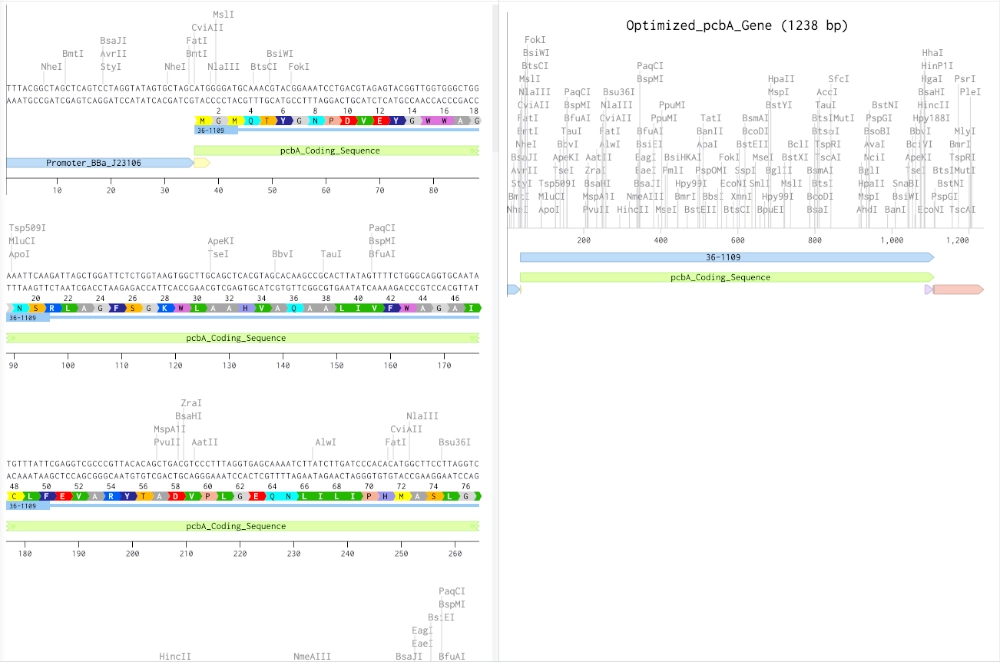
The protein I’d like to work with is the prochlorophyte chlorophyll-binding (Pcb) protein which is the light-harvesting protein (LHP) in prokaryotes that uses only chlorophyll as their photosensitive pigment. A modified version of this protein could be used to efficiently absorb light causing degradation of chlorophyll a, b and d molecules into porphyrin-type derivatives — that can be used to bind iron and create photographic images in a cell-free system.
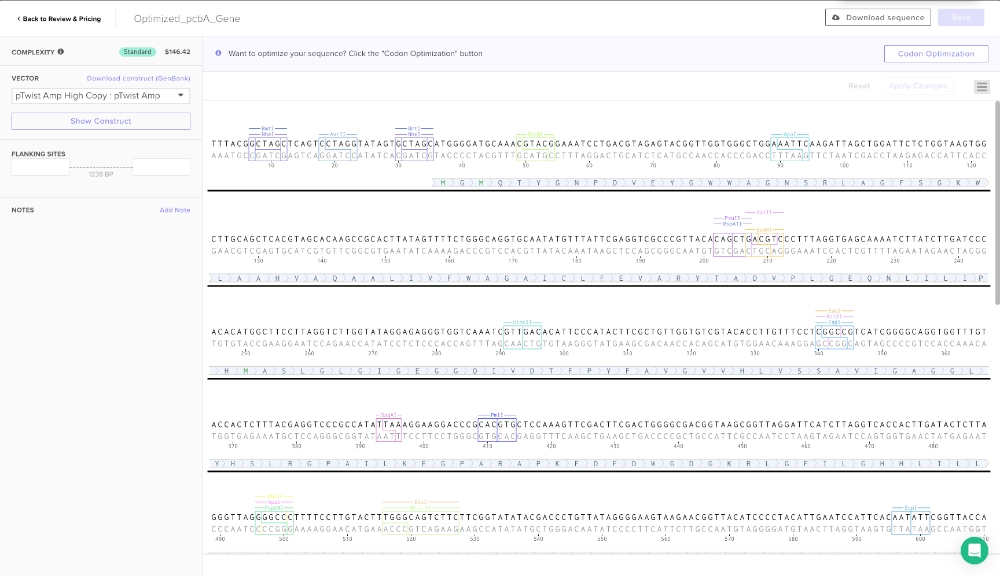
Codon optimization is an important process due to different organisms having and producing different amino acids in different proportions. So, if a gene codes for a rare amino acid, it might slow the translation process and therefore the folding of the protein and might even render the protein non-functional. In this case, the gene should be optimized for e. coli which is probably the best choice since the primary objective is to express a protein that is going to be used in a cell-free system and it is the simplest organism to work with. For this codon optimization I avoided Type IIS enzyme recognition sites for BsaI, BsmBI, and BbsI — these are some enzymes that are useful for ligation with plasmid backone.
(made with Codon Optimization Tool | Twist Bioscience)
3.4. You have a sequence! Now what?
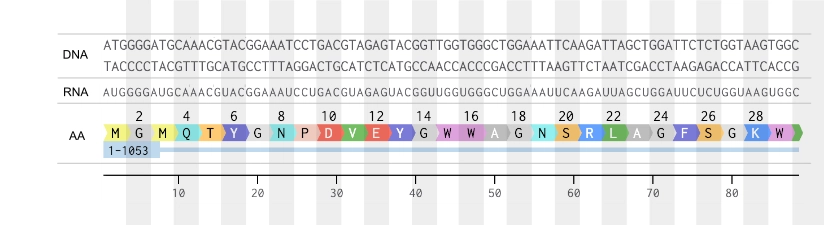
This gene sequence could be synthesized through chemical synthesis on silicon chips and assembled into a vector backbone— bacterial plasmid— then, put into e. coli. This allows for the use of the bacteria’s own cellular machinery to first transcribe this DNA sequence into mRNA (which would be identical to this coding DNA strand, except for “U”), and finally the bacteria’s ribosomes would translate the resulting mRNA into the final amino acid sequence. This AA sequence would be the pcbA protein in its apoprotein form— since e. coli lacks the machinery to produce chlorophyll molecules that play as a co-factor in the folding of this protein— which would be later combined with chlorophyll extract to render it functional.
3.5. How does it work in nature/biological systems?
1. Describe how a single gene codes for multiple proteins at the transcriptional level.
In biological systems, a single gene can code for multiple proteins at the transcriptional level through the process of alternative splicing (in eukaryotes)— a process where different combinations of exons from the same pre-mRNA molecule are joined together. This process happens inside the nucleus during the processing of pre-mRNA, leading to the synthesis of multiple protein isoforms, which are related forms of the same protein, but with different structural or functional properties. Another process that allows for a single gene to code for multiple proteins (both in eukaryotes and prokaryotes) is the action of alternative promoter genes that create different initiation sites, affecting which exons are included in transcription.
2. Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!!
Part 4: Prepare a Twist DNA Synthesis Order
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
The DNA I’m interested in sequencing and further understanding is cyanobacteria’s genes for the production of Chlorophyll LHP (Light-Harvesting proteins) which is the nature’s way of organizing chlorophyll molecules in order to get the most light absorption out of them. This biological way of organizing light sensitive pigments could be the answer to a new generation of analog photography media and can also be used to engineer more efficient solar cells to produce energy.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
For this type of application, the most adequate sequencing technology would probably be Sanger sequencing using a device like Sanger-ABI, a 1st generation technology that has been around since 1977 but would be more than enough for things like reading single protein coding sequences. This would be a small-scale project needing only to analyze relatively small nucleotide sequences, it wouldn’t demand the comparison of more complex genes like comparing/analyzing whole genomes.
For this method I would make a DNA extraction from cyanobacteria cells and purify it, followed by designing primers specific for the sequence I want to analyze and amplify it through PCR, then remove excess primers and dNTPs. Next step would be to perform a cycle sequencing reaction “chain terminator PCR” using single primers, DNA polymerases, dNTPs and fluorescently labeled ddNTPs— these fluorescent ddNTPs act as chain terminators, stopping synthesis randomly at every possible length to create labeled fragments. Clean up residual dye labeled ddNTPs to prevent noise during read and submit these tagged fragments to Sanger-ABI capillary electrophoresis, which separates the fragments by length and then makes the read by exciting the label of each fragment and detecting the color emitted— then using software to translate the fluorescence signals into a chromatogram, revealing the sequence of the DNA sample.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would need to synthesize a bacterial plasmid (for e. coli) with the insert for the Chlorophyll LHP gene, in this case the PcbA protein. The application for this would be to develop a way to keep these proteins functioning in a cell free system in order to create a novel biomaterial that would serve as photographic emulsion for analog film.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Probably the most affordable and effective way to synthesize this kind of DNA would be the through clonal gene chip-based chemical synthesis and assembly of the DNA sequence into a plasmid vector through Golden Gate Assembly using type IIS restriction enzymes and T4 DNA ligase. Due to the small-scale nature of this project and standard difficulty of synthesizing this kind of DNA, I don’t think there would be significant limitations speed, accuracy and scalability wise.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
For the objective of this project the DNA that would be interesting to edit would range from the genes coding for the chlorophyll synthesis pathway— in order to develop a modified version of chlorophyl that would be more optimized for photographic purposes— and the sequences coding for the LHP if there is the need to modify the natural occurring proteins, either by decreasing the protection these proteins confer against chlorophyll degradation or by potentially improving on their ability to maximize chlorophyll’s light absorption qualities.
(ii) What technology or technologies would you use to perform these DNA edits and why?
CRISPR-based genome editing would probably be the best choice for this purpose since Chlorophyll biosynthesis involves multiple genes, often with regulatory fine-tuning rather than simple on/off behavior. CRISPR systems allow gene-specific, locus-specific edits, making them well suited for altering enzyme functionality in the chlorophyll synthesis pathway; modifying regulatory regions that affect pigment ratios (e.g. chlorophyll a, b or d) and engineering specific amino-acid changes in LHPs.
CRISPR edits DNA by using an RNA guide to bring a DNA-cutting enzyme (nuclease) to a specific genomic site which is cut and where the cell’s own DNA-repair machinery makes the final change utilizing a DNA template (single stranded or double stranded for larger edits) which is delivered together with the Cas9 enzyme.
For this end, the first steps would be to define an objective precisely like make chlorophyll more sensitive to light or more prone to degradation under certain conditions, or reduce photoprotective quenching of the LHP and identify genes, regulatory regions or domains that are relevant for those functions. After that, decide what type of edit strategy is needed for a specific site (gene knockout, single or few nucleotide changes or sequence replacement) and design a guide RNA to bind to the target DNA of that specific site, and if a sequence rewrite is needed, design the DNA template for that repair which must have homologous endings that match those surrounding the cut site.
To perform the actual edit the gRNA, Cas9 nuclease and template DNA are combined into a plasmid vector or ribonucleoprotein and introduced into the cells via heat shock, electroporation or lipofection.
The limitations of this method might include off-target effects by binding and editing similar but not the exact sites; in a cell culture it might happen that not all cells be edited resulting in a mixture of edited and unedited cells, which can make it difficult to achieve a uniform result, and the HDR might have a low efficiency compared to the non-homologous end joining (NHEJ) pathway, not resulting in the intended edit.
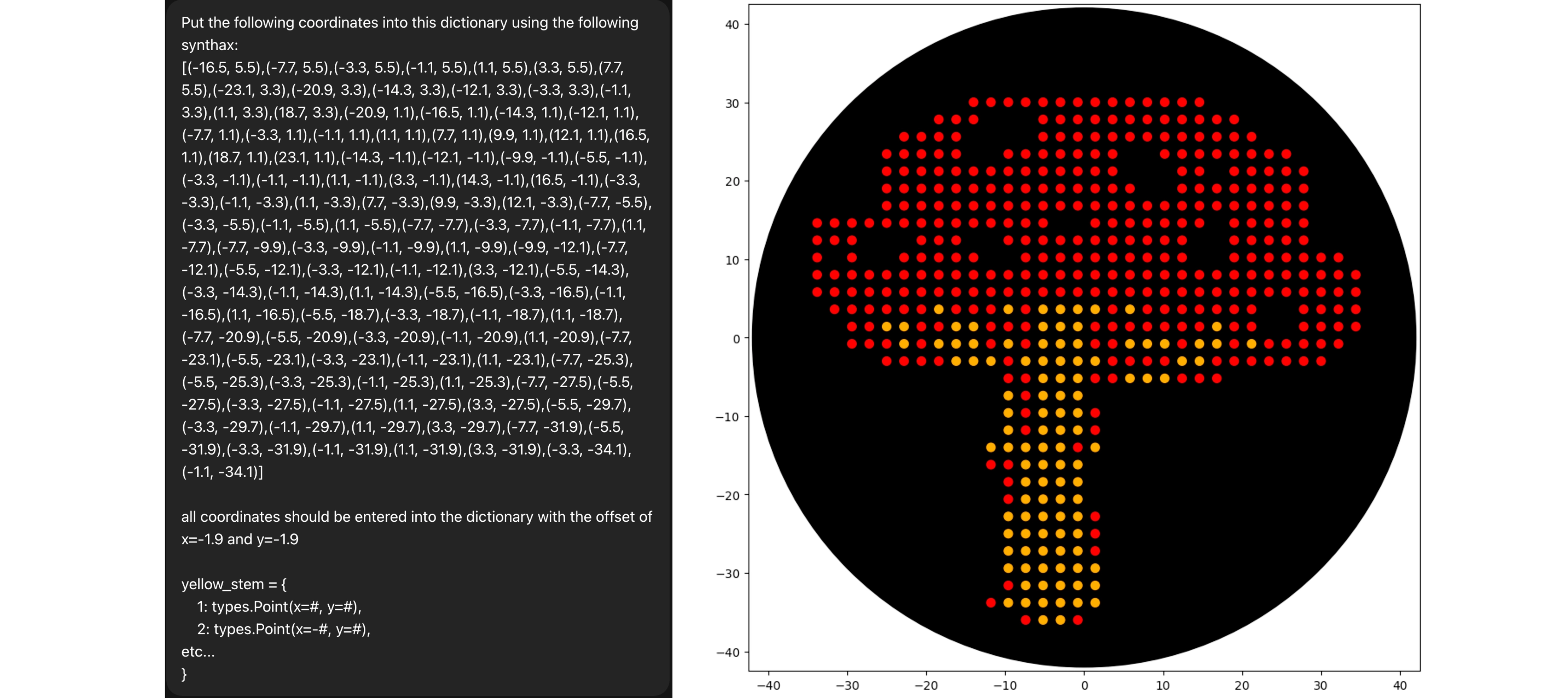
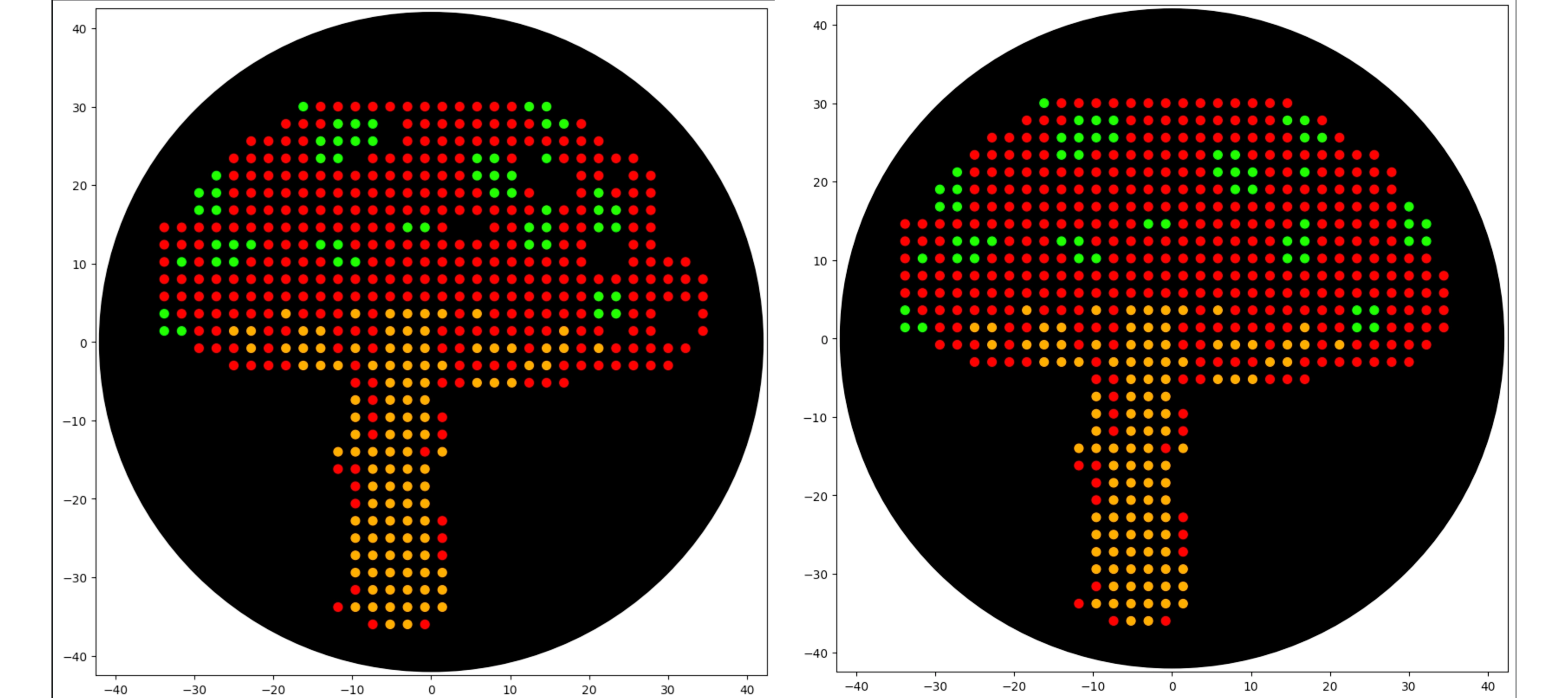
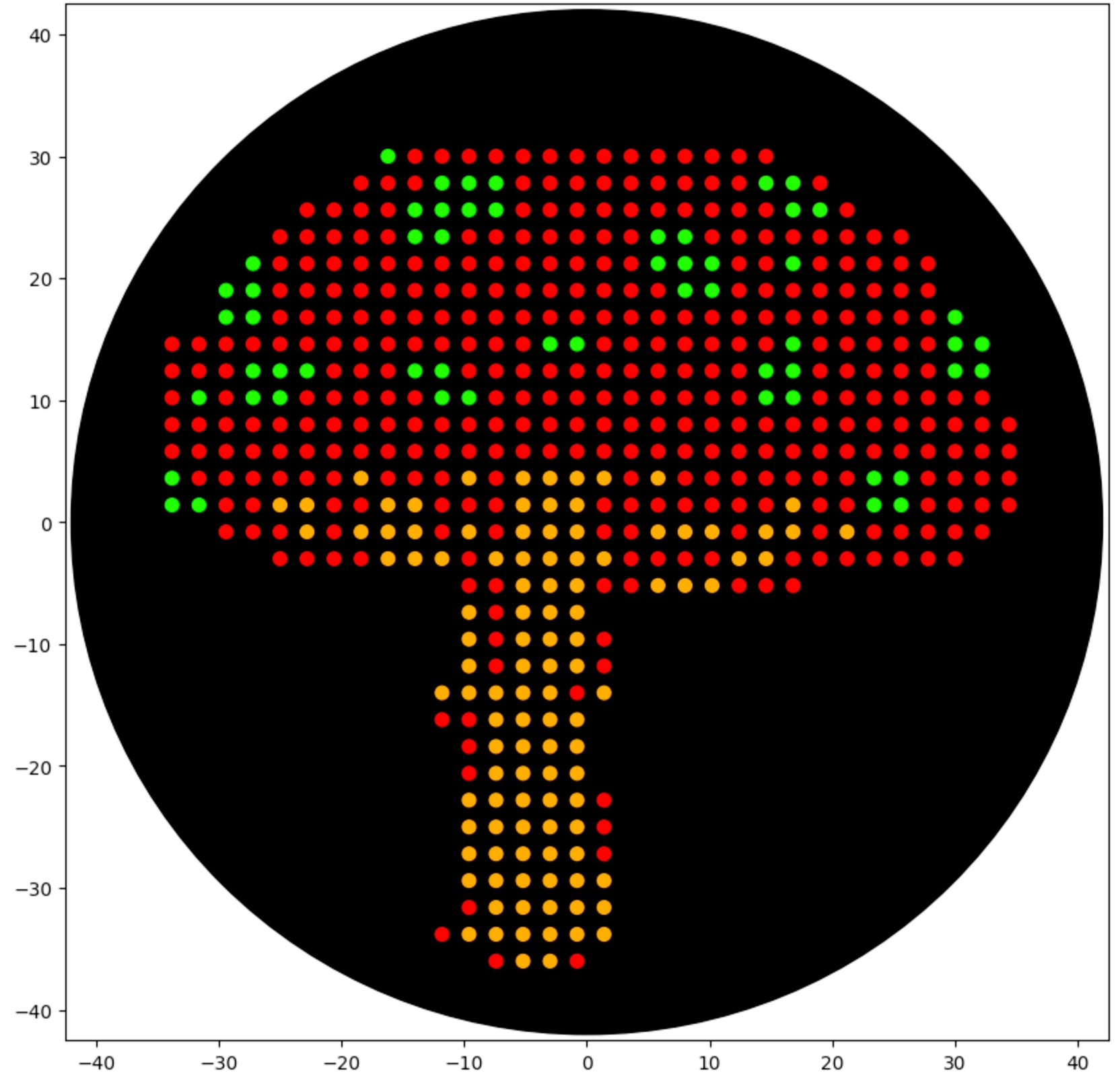
First of all I used opentrons-art.rcdonovan.com to generate a base design with an image from “vecteezy.com” and then modified it manually to reach the final design
Then created the zones in red with the following logic (adapting from Example 5 and 7):
1
2
3
4
5
6
7
8
9
10
11
12
13
14
foriinred:ifi%40==0:## Every 40 drops (not 20 because .5 drops), including at i == 0 for the start## Aspirate the smaller value between pipette_20ul max_volume and how much volume is still needed given that each drop is a .5 droppipette_20ul.aspirate(min(pipette_20ul.max_volume,(red.stop-i)*0.5),location_of_color('Red'))dispense_and_detach(pipette_20ul,.5,cursor)cursor=cursor.move(types.Point(y=0,x=2.2))## Here I start printing the red part of the cap in a side to side printer style movement — in an attempt to not have to enter every single coordinate and jumping the areas for other colors ifi==13:cursor=cursor.move(types.Point(y=-2.2,x=2.2))ifi>13:cursor=cursor.move(types.Point(y=0,x=-4.4))ifi==25:cursor=cursor.move(types.Point(y=0,x=-2.2*3))
Used Chat GPT to write all the coordinates using a “template dictionary” I wrote, and then adapted the logic I used before to work with the dictionary instead of a range:
1
2
3
4
5
6
7
8
9
10
11
dots=list(yellow_stem.values())foriinrange(len(dots)):ifi%40==0:## same logic of refill but adapted to the yellow_stem dictionarypipette_20ul.aspirate(min(pipette_20ul.max_volume,(len(dots)-i)*0.5),location_of_color('Orange'))cursor=center_location.move(dots[i])dispense_and_detach(pipette_20ul,0.5,cursor)pipette_20ul.drop_tip()
Repeated the same process for the green spots, but because I made some mistake in the red dots the coordinates from the green weren’t matching as they should, so I solved that by manually adjusting the red dots to the placement of the green spots
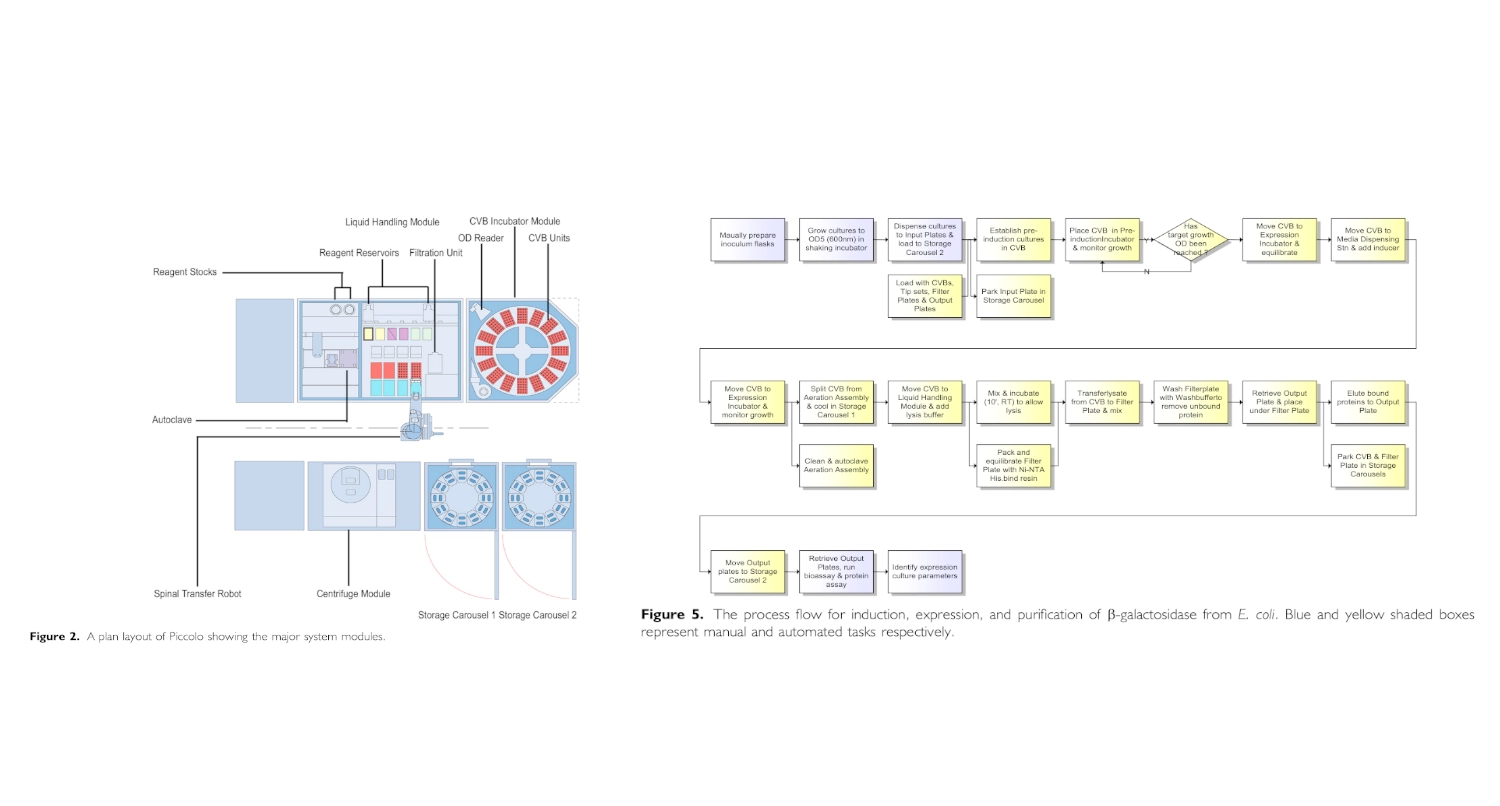
I found and was interested in two articles: The first one “Automation and Optimization of Protein Expression and Purification on a Novel Robotic Platform” published by Journal of Laboratory Automation (October 2006) that describes an automated robotic system for expression and purification of recombinant proteins grown both in E. coli and other bacterial cells and eukaryotic cells.
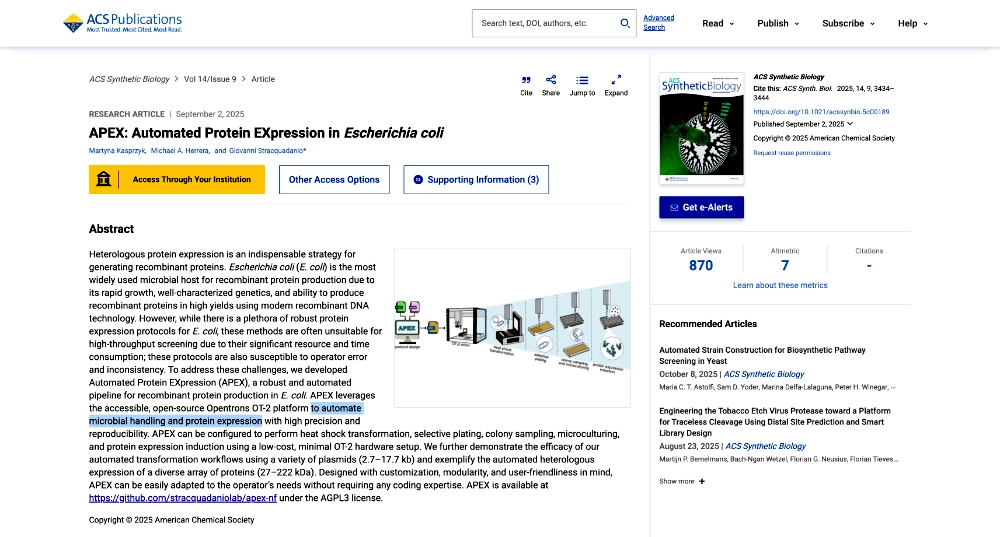
The second “APEX: Automated Protein EXpression in Escherichia coli” published by ACS Synthetic Biology (September 2, 2025) describes an automated pipeline for recombinant protein production in E. coli, leveraging the open-source Opentrons OT-2 platform to handle microbe culturing and protein expression.
Automation and Optimization of Protein Expression and Purification on a Novel Robotic Platform published by Journal of Laboratory Automation (October 2006)
Overview:
This paper describes the development of a robotic system designed to automate the process of recombinant protein production and purification. Protein expression optimization is traditionally labor-intensive, requiring repeated manual adjustments to growth conditions, induction timing, and purification steps. The authors introduce a robotic platform capable of coordinating bacterial culture growth, induction, cell harvesting, lysis, and affinity purification within a same workflow.
A key innovation of the platform is its ability to conduct parallel experiments that test different expression conditions in a controlled and automated manner. Instead of performing expression trials sequentially, the robotic system enables simultaneous evaluation of variables such as induction timing and culture density. The workflow integrates liquid handling, incubation, and affinity purification into a continuous process, reducing manual intervention and variability. By linking culture monitoring with automated downstream purification, the system demonstrates how laboratory automation can streamline workflows that are typically fragmented across separate instruments and manual steps.
Findings:
The study demonstrates that automation in parallel significantly increases experimental throughput and improves the efficiency of identifying optimal protein expression conditions. Compared with traditional manual workflows, the automated approach reduced hands-on time and enabled rapid exploration of a large experimental space. Overall, the findings support the use of integrated robotic systems to accelerate protein production workflows and reduce bottlenecks in research requiring purified recombinant proteins.
(Chat GPT was used to assist in the summarization of this paper)
Although this paper might be outdated since it was published 20 years ago it helped me better understand automation of experimentation in living cells.
APEX: Automated Protein EXpression in Escherichia coli published by ACS Synthetic Biology (September 2, 2025)
Overview:
This paper presents APEX (Automated Protein EXpression), an end-to-end automation pipeline designed to streamline recombinant protein production in E. coli using the open-source Opentrons OT-2 platform to automate microbial handling and protein expression. Protein expression workflows are traditionally labor-intensive and prone to variability due to repeated manual steps such as heat shock transformation, plating, colony picking, culturing, and induction. APEX integrates these processes into four modular automated protocols: heat shock transformation; selective plating; colony sampling and microculturing; and protein expression. The system is designed to operate on a minimal OT-2 configuration, requiring only the thermocycler module and standard pipettes, making automation accessible to smaller laboratories without specialized robotics infrastructure.
A defining feature of APEX is its emphasis on reproducibility and usability. Rather than requiring programming expertise, experiments are configured using spreadsheet-based input files (JSON and CSV), which are processed through a Nextflow computational pipeline to automatically generate robot-ready Python protocols and user documentation. The workflow also includes automated spotting and colony sampling (illustrated in Figure 2).
Findings:
The authors validated APEX across multiple experimental scenarios and compared its performance to manual workflows. Transformation efficiency remained comparable to manual methods even when transformation volumes were miniaturized, and the expected decrease in efficiency with increasing plasmid size was observed in both automated and manual conditions. Automated colony sampling methods were tested under varying colony densities, with a spiral sampling strategy demonstrating improved robustness. Finally, the complete automated workflow successfully expressed soluble proteins spanning a wide molecular weight range (29 kDa to 222 kDa), with results comparable to manual processing. These results demonstrate that APEX maintains reliability while increasing throughput and reducing hands-on time.
(Chat GPT was used to assist in the summarization of this paper)
Write a description about what you intend to do with automation tools for your final project.
The development on photographic emulsions has an added difficulty of them being light-sensitive. So having an automated workflow to produce iterations of different possibilities would largely make the process more efficient
Automation for culturing of e coli and synthesis of an array of modified and non-modified chlorophyll binding proteins.
Lipid-induced folding of those different proteins by combining with chlorophyll extract
Maybe further along the research process the expression of these proteins could be done with cyanobacteria that already have the metabolic pathways for production of chlorophyll which would facilitate the scalability of the project.
Combination of the different final proteins with an agarose base to allow dispersion onto a base surface
3d printed holder for the base supports for the chlorophyll protein emulsion to be dispersed on
Drying of the emulsion in dark conditions
During a testing phase, there might be a better way of testing these protein complexes for light sensitivity and for reactivity towards iron once exposed to light without having to create and emulsion and disperse it onto a base support. This could eventually be achieved through biosensing? — detecting if, once exposed to light, the chlorophyll attached to the proteins degrades into the right derivatives that are good chelating agents for iron
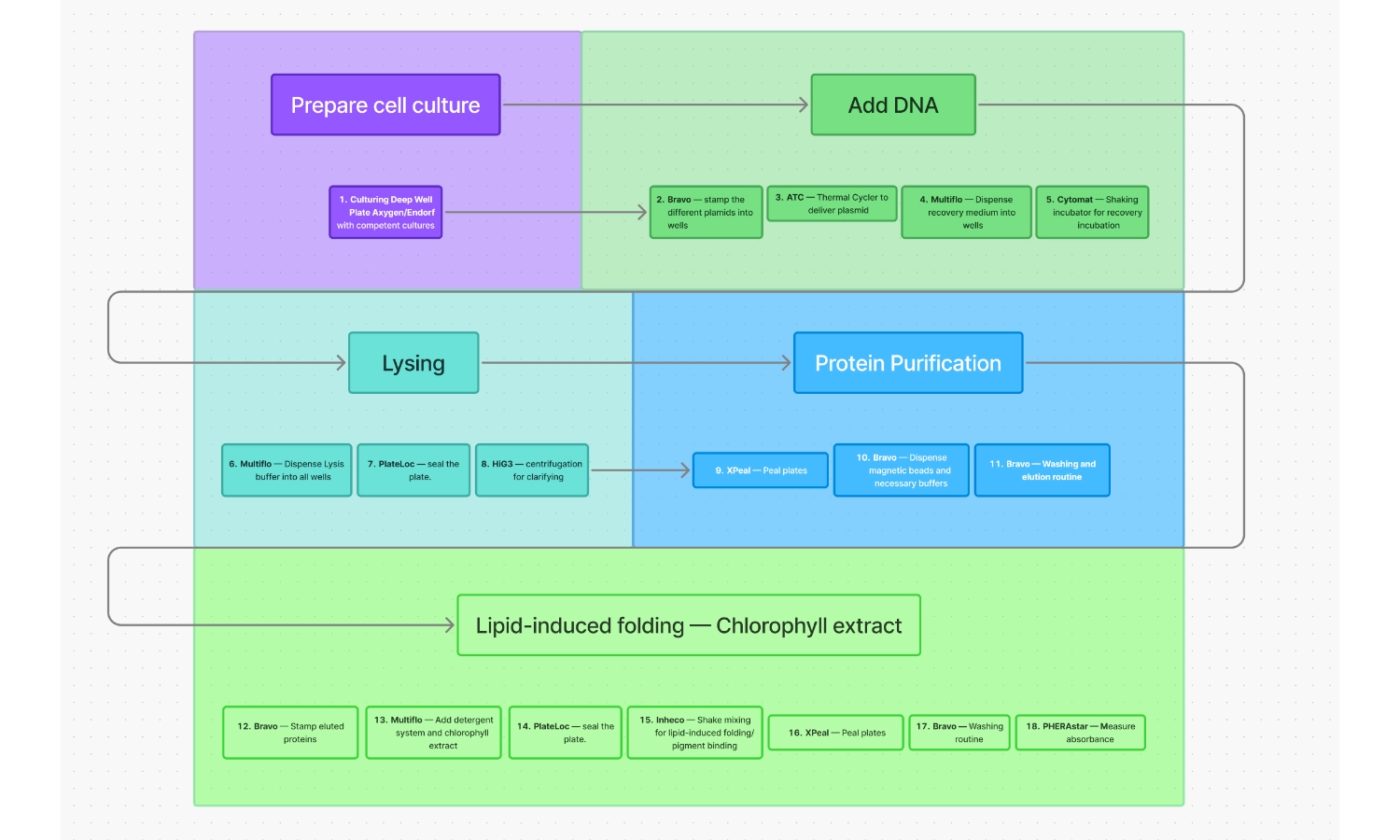
For the purpose of this exercise, I tried to create a comprehensible workflow for the following operations that I think would be essential for this project:
Workflow for expression of chlorophyll-binding proteins
Culturing Deep Well Plate Axygen/Endorf with competent cultures
Bravo — Stamp the different plasmids into wells
ATC — Thermal Cycler to deliver plasmid via thermo shock
Multiflo — Dispense recovery medium into wells
Cytomat — Shaking incubator for recovery incubation
Multiflo — Dispense Lysis buffer into all wells
PlateLoc — Seal the plate
HiG3 — Centrifugation for clarifying
XPeal — Peal plates
Bravo — Dispense magnetic beads and necessary buffers
Bravo — Washing and elution routine
Bravo — Stamp eluted proteins
Multiflo — Add detergent system and chlorophyll extract
PlateLoc — Seal the plate
Inheco — Shake mixing for lipid-induced folding/pigment binding
XPeal — Peal plates
Bravo — Washing routine
PHERAstar — Measure absorbance
After completing this part of the homework, I realized that CFPS might be a better bet, using an automated system, for the expression and testing of this particular kind of protein since the open system nature of this method would allow the direct addition of chlorophyll into the reaction mixture and allow for instant protein folding. Further along the progression of this project, when the chemistry of the proteins is optimized, living cells — like cyanobacteria — could be used to express them in larger quantities
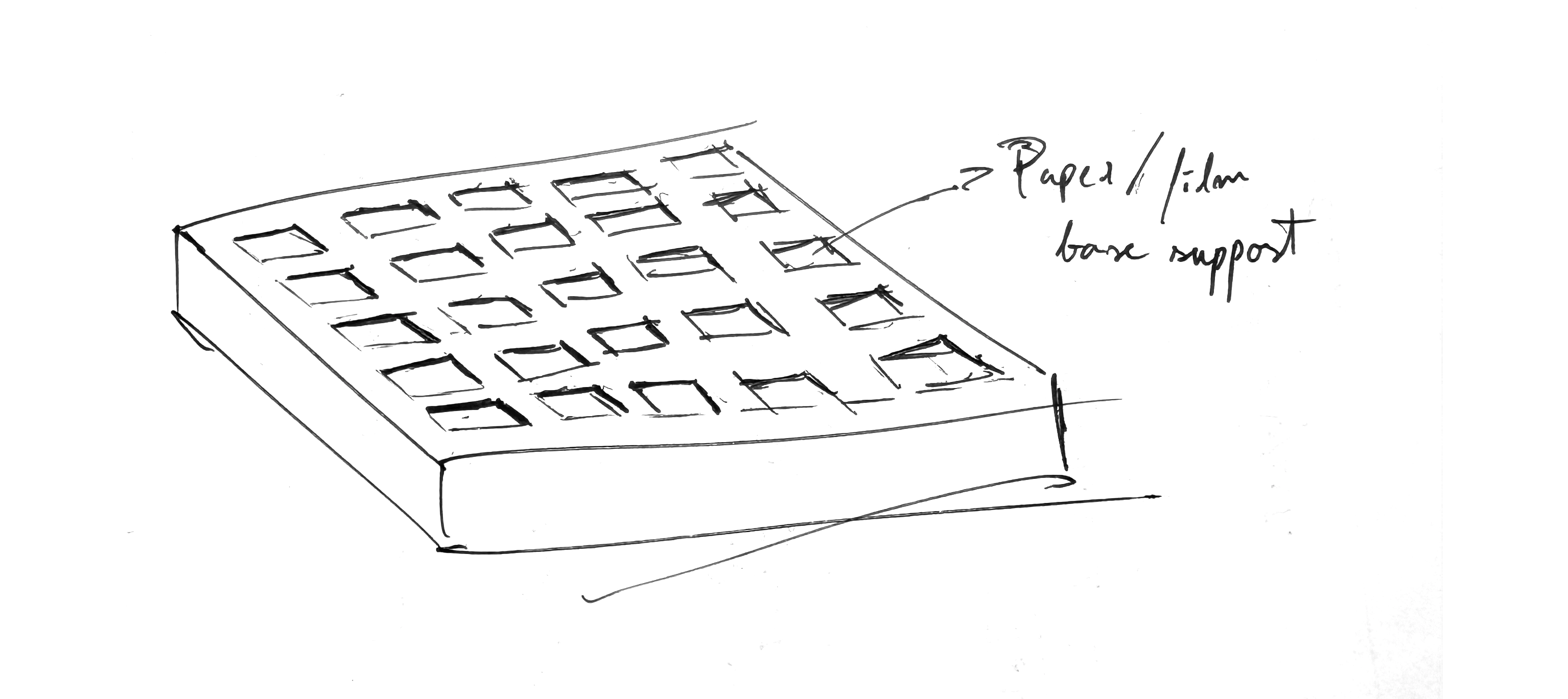
3D printing of a holder for base supports for the chlorophyll emulsion
For the chlorophyll proteins (suspended in some kind of gelling agent like agar) to be dispersed on, so field tests could be performed with pinhole cameras (for the testing of an array of photographic emulsion iterations). Bellow a quick sketch of what it could be.
1. How many molecules of amino acids do you take with a piece of 500 grams of meat?
If we assume that 100g of meat has on average ≈ 26g of protein then 500g would have 130g.
100 Daltons per amino acid is ≈ to 100g|mol of amino acids.
So if we get how many moles are present in 130g of protein and multiply that by the Avogadro’s number we get the number of molecules of amino acids present.
N = number of molecules
N = 130g/100g|mol x (6.022x10^23molecules|mol)
N ≈ 7.83 x 10^23 molecules
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When an animal eats any other being the digestion system breaks down and processes the food into particles our cells can use to function. In the case of proteins, when we consume any other being we are repurposing the amino acids that being contains and turning them into the combinations we need to make our own proteins. We don’t start using the same proteins other beings use. Our DNA contains the code to build human proteins that repurpose and rearrange the building blocks that compose what we eat.
3. Why are there only 20 natural amino acids?
I assume the reason there are only 20 natural amino acids is that those molecules were the ones that ended up being easier to produce through metabolic pathways, were the most stable and had enough differences between them to perform all the needed interactions. Other iterations of amino acids that appeared throughout evolution must have stopped been produced because they didn’t fit these parameters one way or another.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, we can synthesize new non-natural amino acids through chemical synthesis or enzyme-based reactions
5. Where did amino acids come from before enzymes that make them, and before life started?
Given that amino acids are just molecules— combinations of atoms, they can organize spontaneously through chemical reactions under the right conditions, without any type of biological intervention.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
A left handedness would be expected, since what can be observed from natural occurring amino acids that build proteins, which are left-handed, is the formation of right-handed alpha helices because this clockwise twisting promotes less clashes between the side chains and the peptide backbone. So if the amino acids were right-handed the opposite would probably happen.
7. Can you discover additional helices in proteins?
Yes, there are some other types of protein helices, though they are less common than alpha.
There are 3-10 helices, often found at the ends of helices. They are tighter, with 3 residues per turn and (instead of the 3.6 found in alpha), and often observed in membrane proteins acting as an intermediate conformation in the unfolding/folding of alpha helices.
Also, Pi-helices, which are wider with aprox. 4.4 residues per turn, inserted inside alpha-helices, appearing as a bulge. Often found in functionally important regions of proteins, such as active sites or ion-binding sites, providing increased conformational flexibility.
8. Why are most molecular helices right-handed?
If we assume the context of biology then most helices are either DNA/RNA or proteins, hand both the building blocks of these types of substances confer a final right-handed helical formation. DNA/RNA— the building blocks of these structures have a certain bias, specially the sugars D-deoxyribose/D-ribose which force the backbone to twist to the right in order to minimize strain. And proteins— which are made up of L-amino acids which also have a preferable twisting bias to the right.
9. Why do β-sheets tend to aggregate?
Beta-sheets tend to aggregate because their structures leave hydrogen-bonding edges exposed. While helices stabilize themselves by having hydrogen bonds stabilizing the coil from the inside, beta sheets leave C=O from the backbone and N=H groups free, and so they tend to aggregate with other nearby sheets or fold into themselves in a structure known as steric zipper.
10. Why do many amyloid diseases form β-sheets?
The beta-sheet formation is the more energetically favorable conformation for satisfying the backbone hydrogen bonds, so whenever a protein misfolds the probability of it’s backbone folding into a default of beta-sheet formation is high. The problem of beta-sheets is that they self-aggregate and so they can form a cascading effect of by attracting other proteins backbone with the exposed hydrogen bonds, which can stack into amyloid fibrils and can accumulate and disrupt cell and organ function.
Beta-sheets form highly ordered and stable molecular structures providing rigidity while allowing for flexibility, resulting in high tensile strength, as observed in natural fibers like silk or collagen. So, amyloid-like fibrils which follow a cross-beta motif generally unbranched and form chains into a continuous, repeating, ribbon-like core are perfect for a strong fiber-like material.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
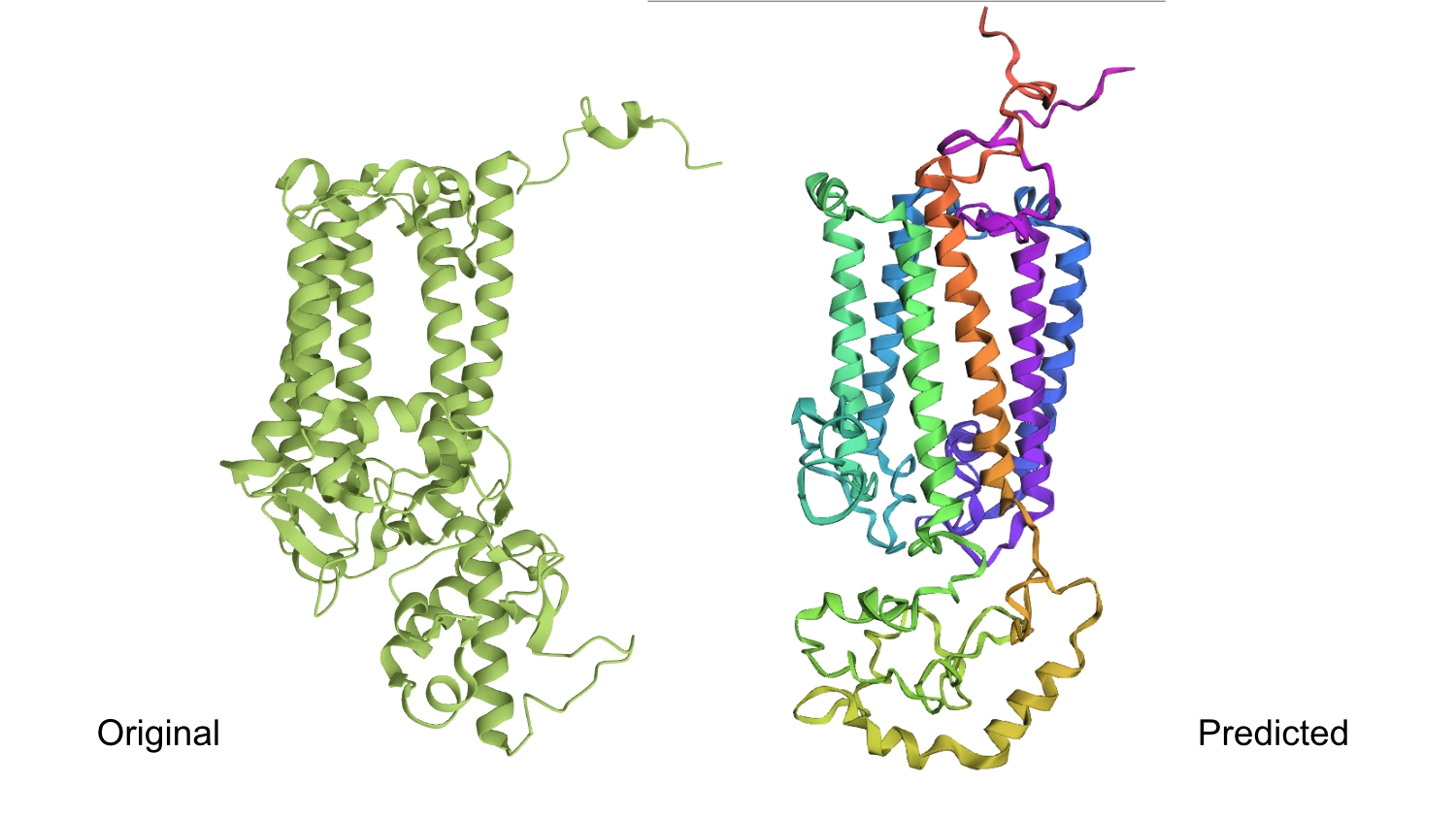
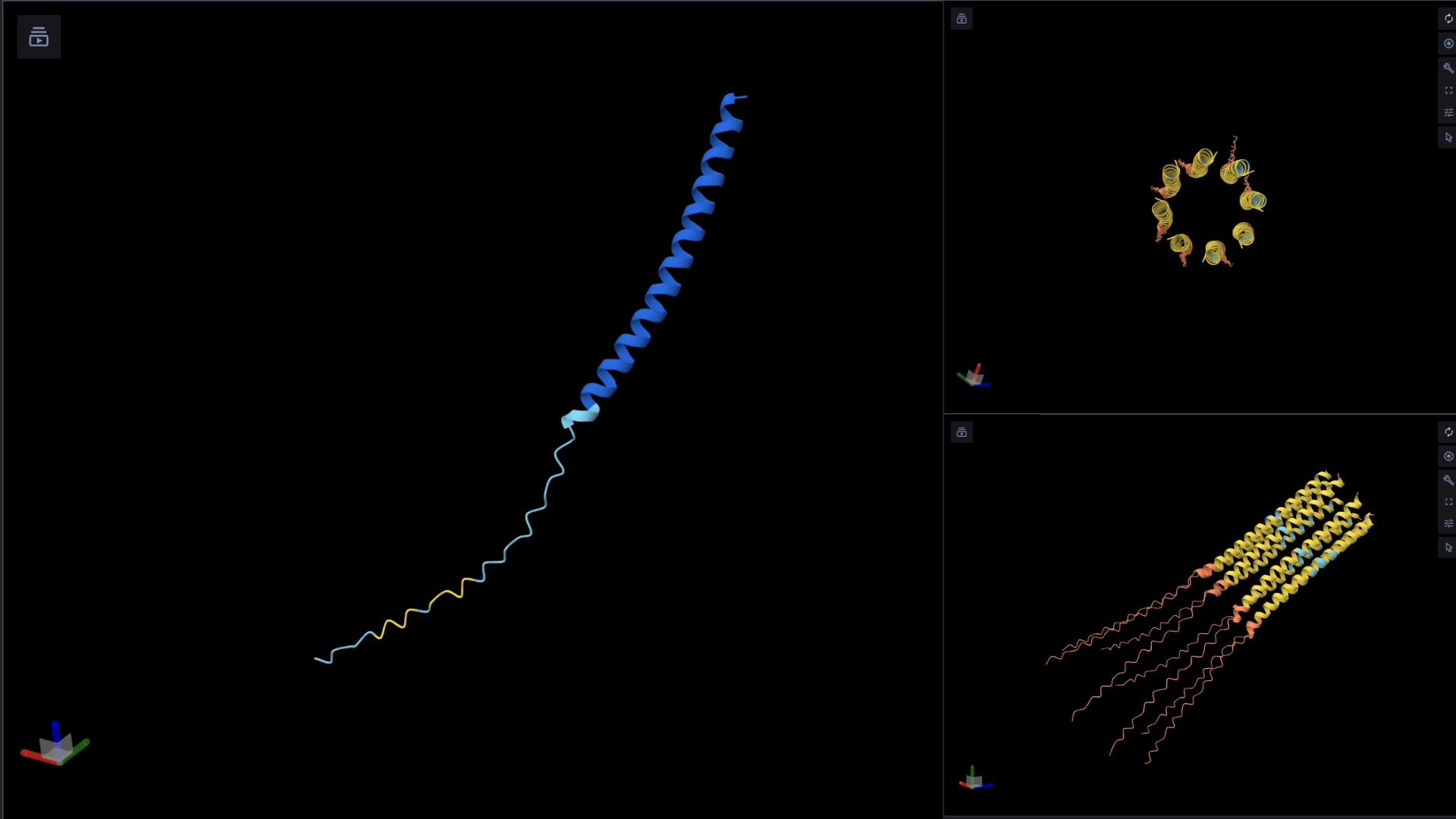
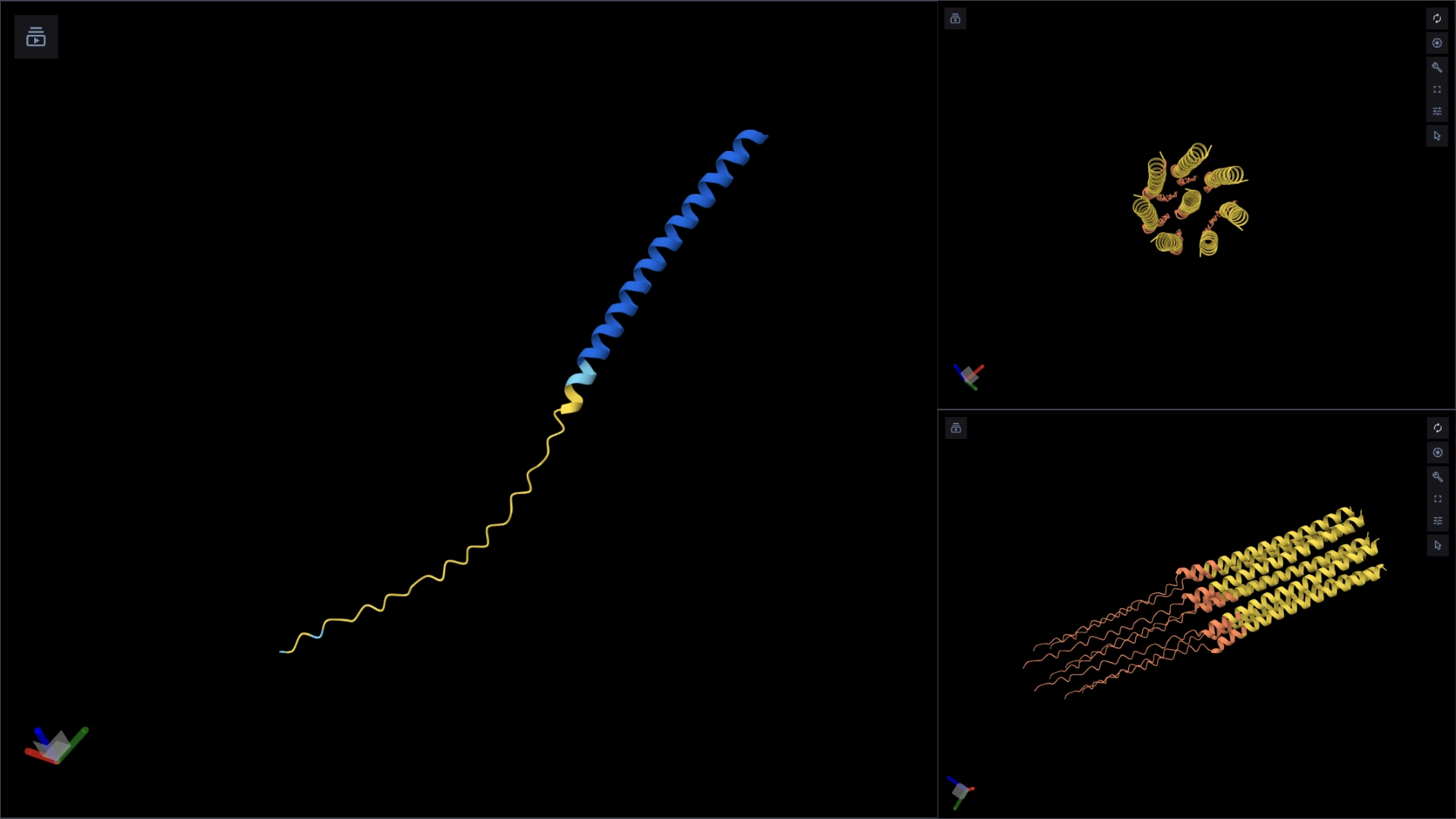
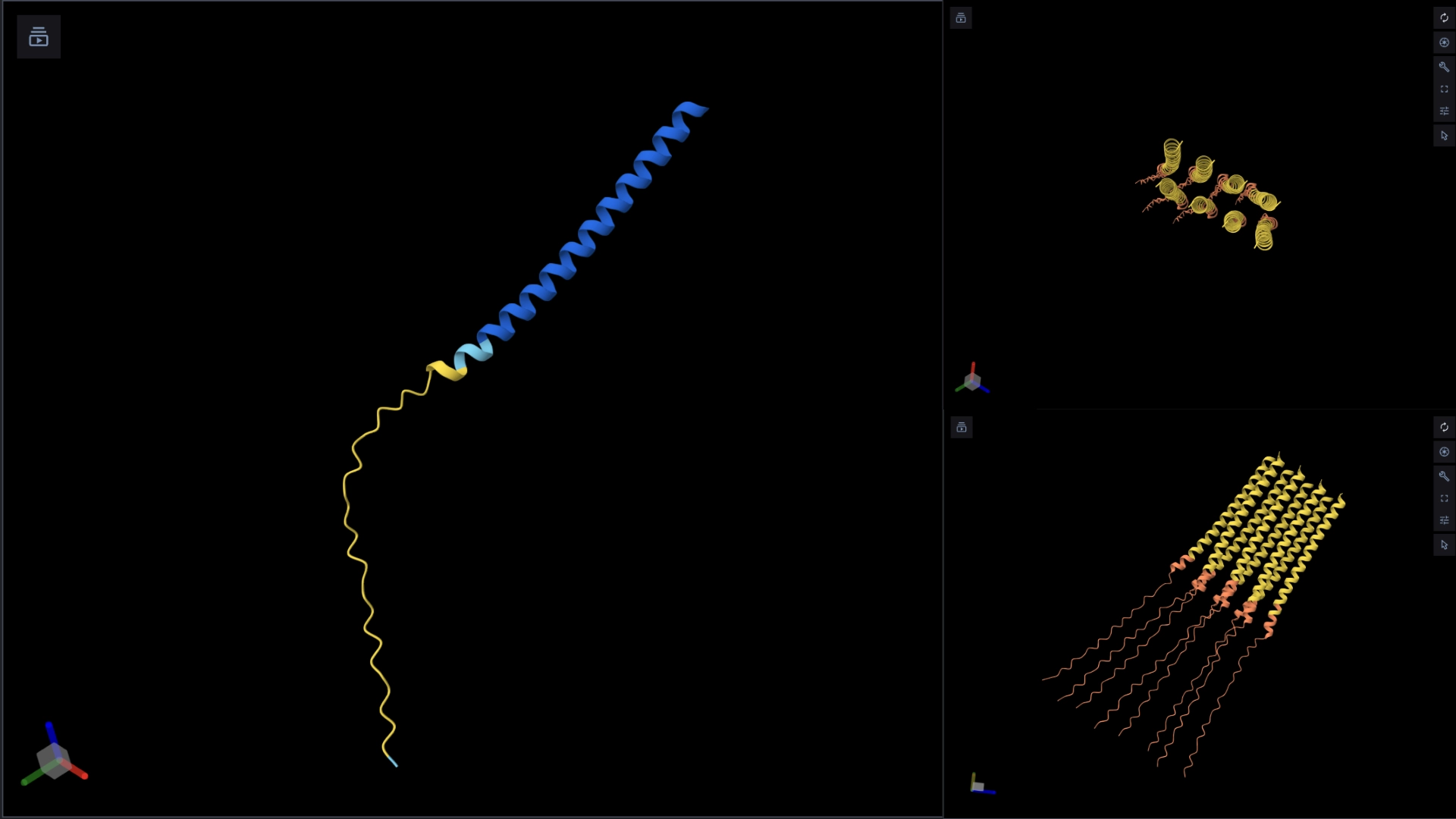
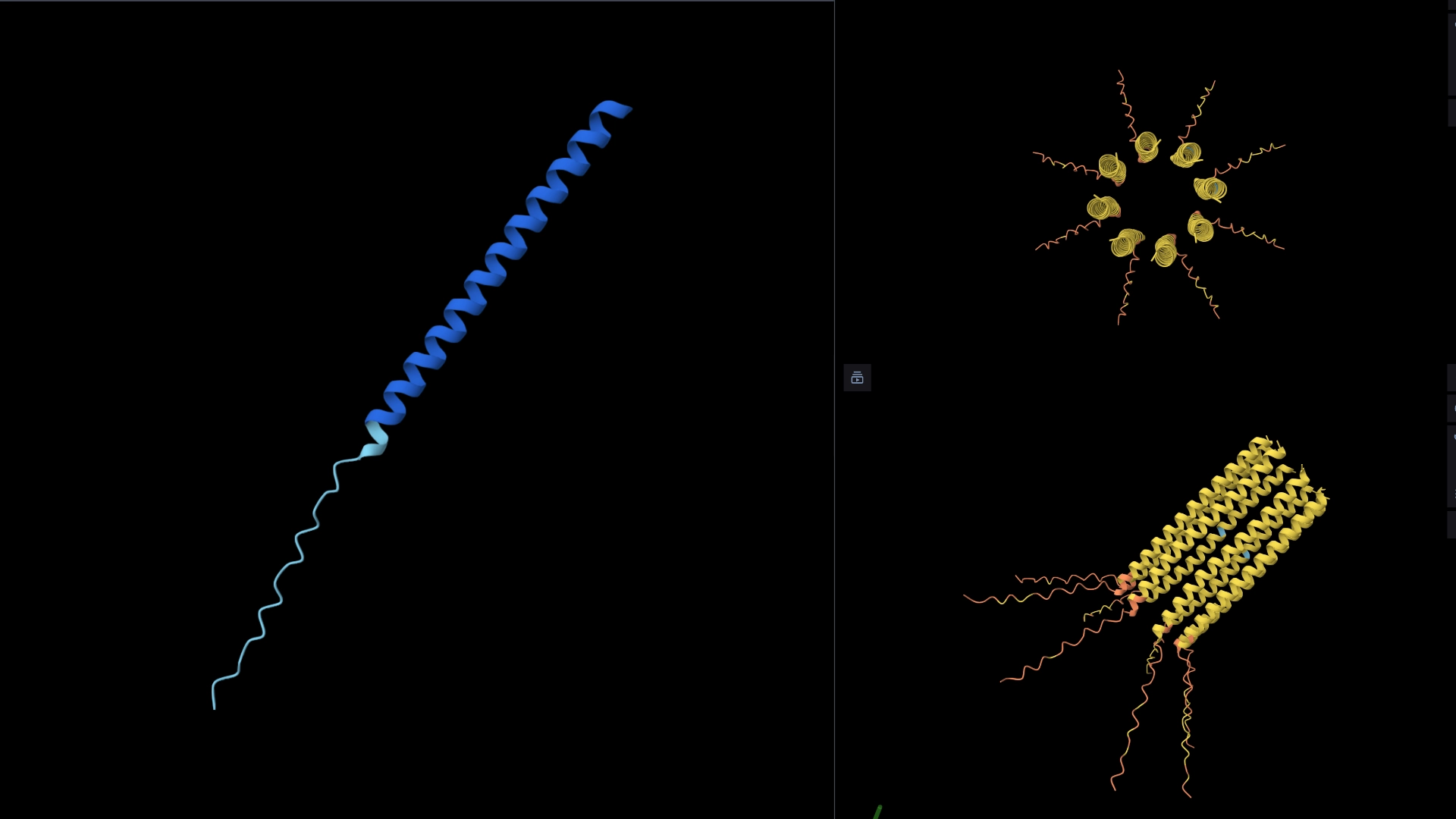
The protein I selected is CP43 (encoded by the psbC gene) which is an essential chlorophyll-binding core antenna protein in Photosystem II (PSII). I chose this protein since I’m doing research into light harvesting proteins in cyanobacteria. I had already done some research into Pcb proteins (chlorophyll binding proteins specific to Prochlorococcus and Prochloron), so now I thought it would be interesting to do some research into a more widely used protein.
2. Identify the amino acid sequence of your protein.
First solved in 2020 with 2.58 resolution, version of 2021 is the one with best resolution 1.93
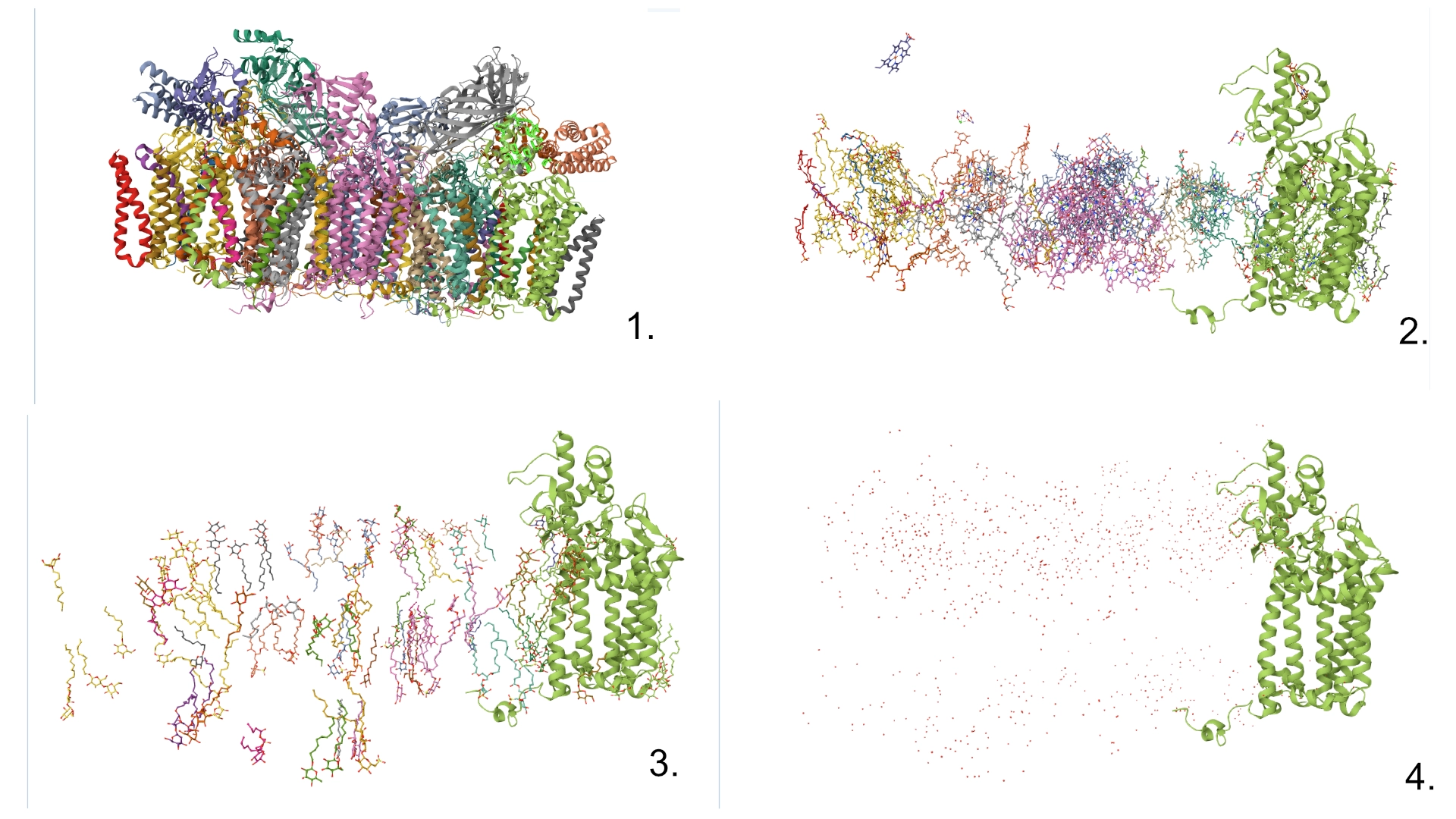
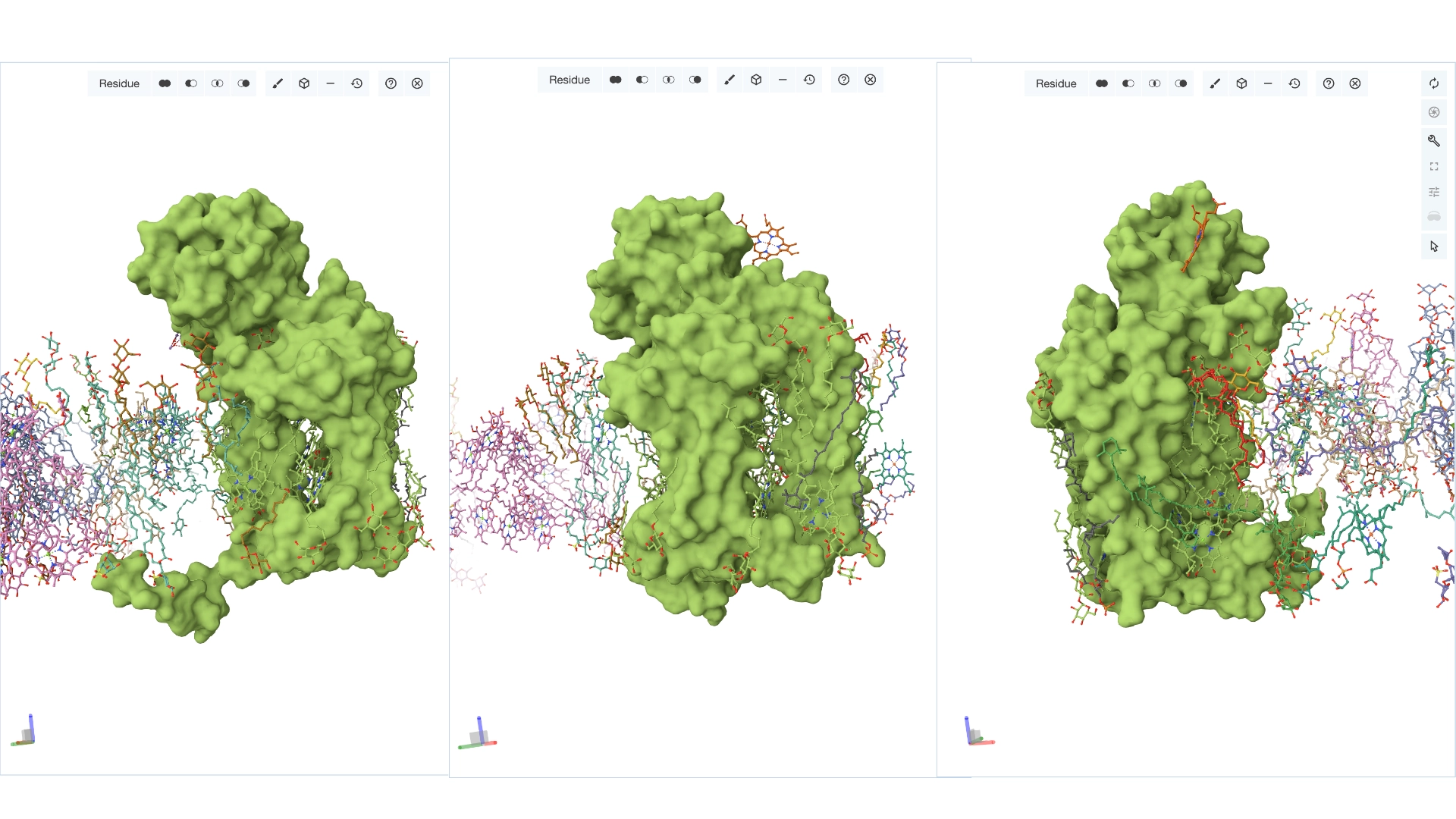
Yes, there are present the other proteins(1.) that constitute Photosystem II as well as ligands(2.), sugars(3.), ions and water molecules(4.)
4. Open the structure of your protein in any 3D molecule visualization software
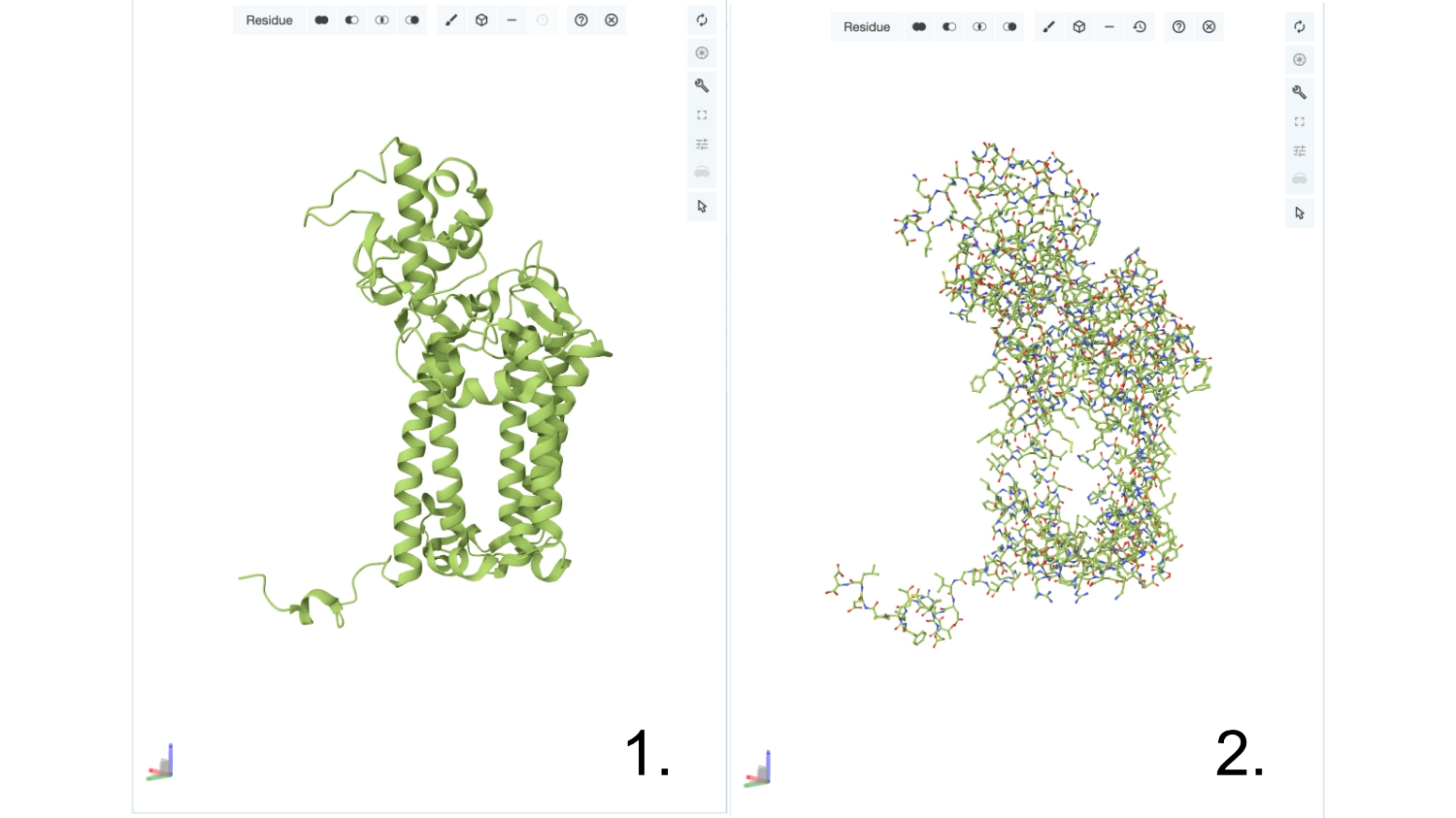
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
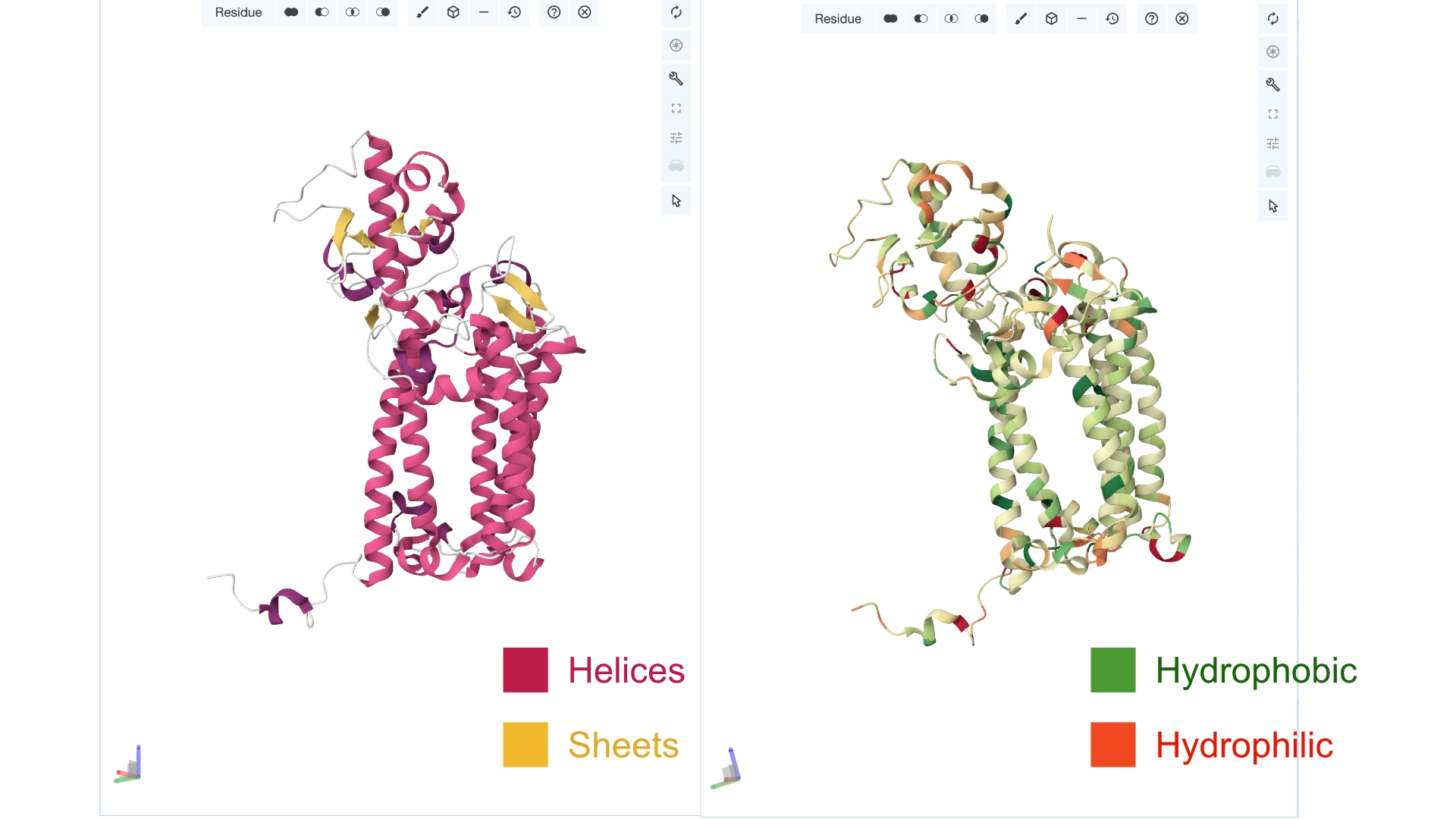
Color the protein by secondary structure. Does it have more helices or sheets? Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
More helices, Hydrophobic
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Yes it has several binding pockets. I turned on the ligands and sugars visualization to try to understand the binding pockets better
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Documentation
When I first ran the code there were way too many proteins in the visualization, then I understood that the fasta file being loaded had around 15 thousand sequences which were being rendered. So I asked Chat GPT (in order to not break the whole code by experimenting) where I could limit the amout of pronteins that were being embeded to a more manageable array.
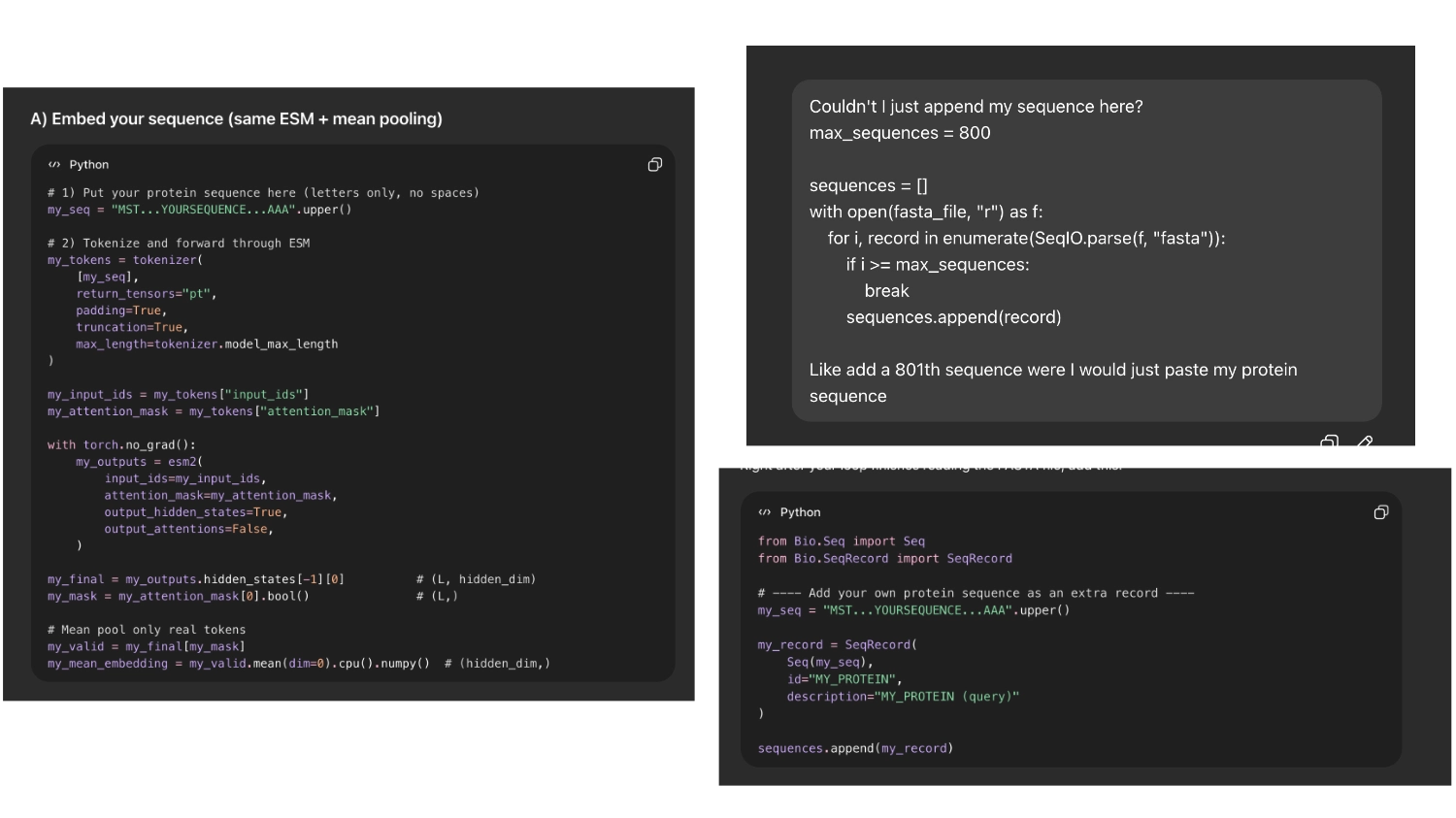
Then I asked chat gpt how I could insert my protein and it first tried to code it into a token. But it seemed easier and less risky If I could append my sequence to the array of sequences imported so I asked it to do that
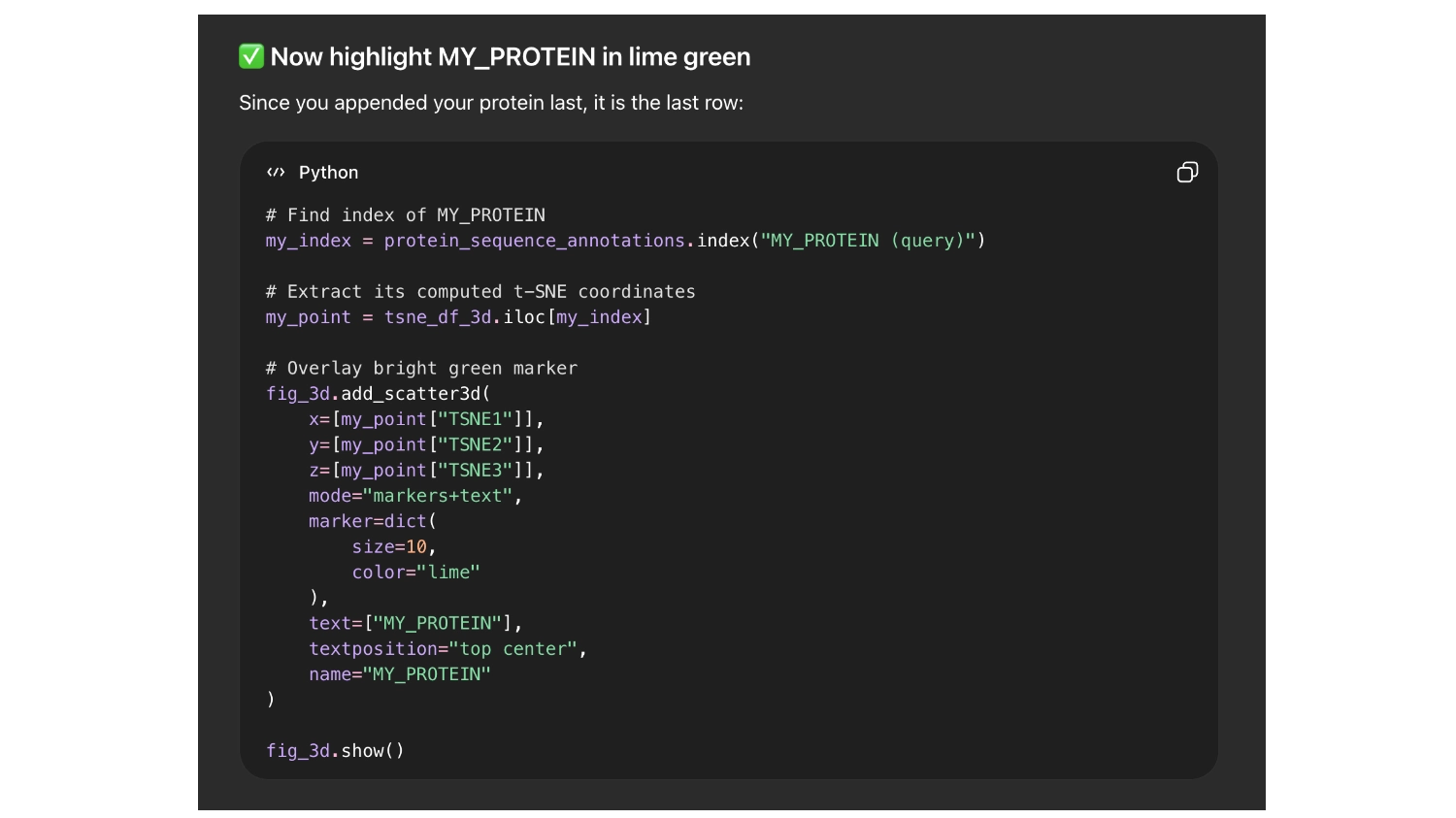
So it would be easier to find my protein I asked chat GPT to label it with the name ““My protein”” and color it bright green
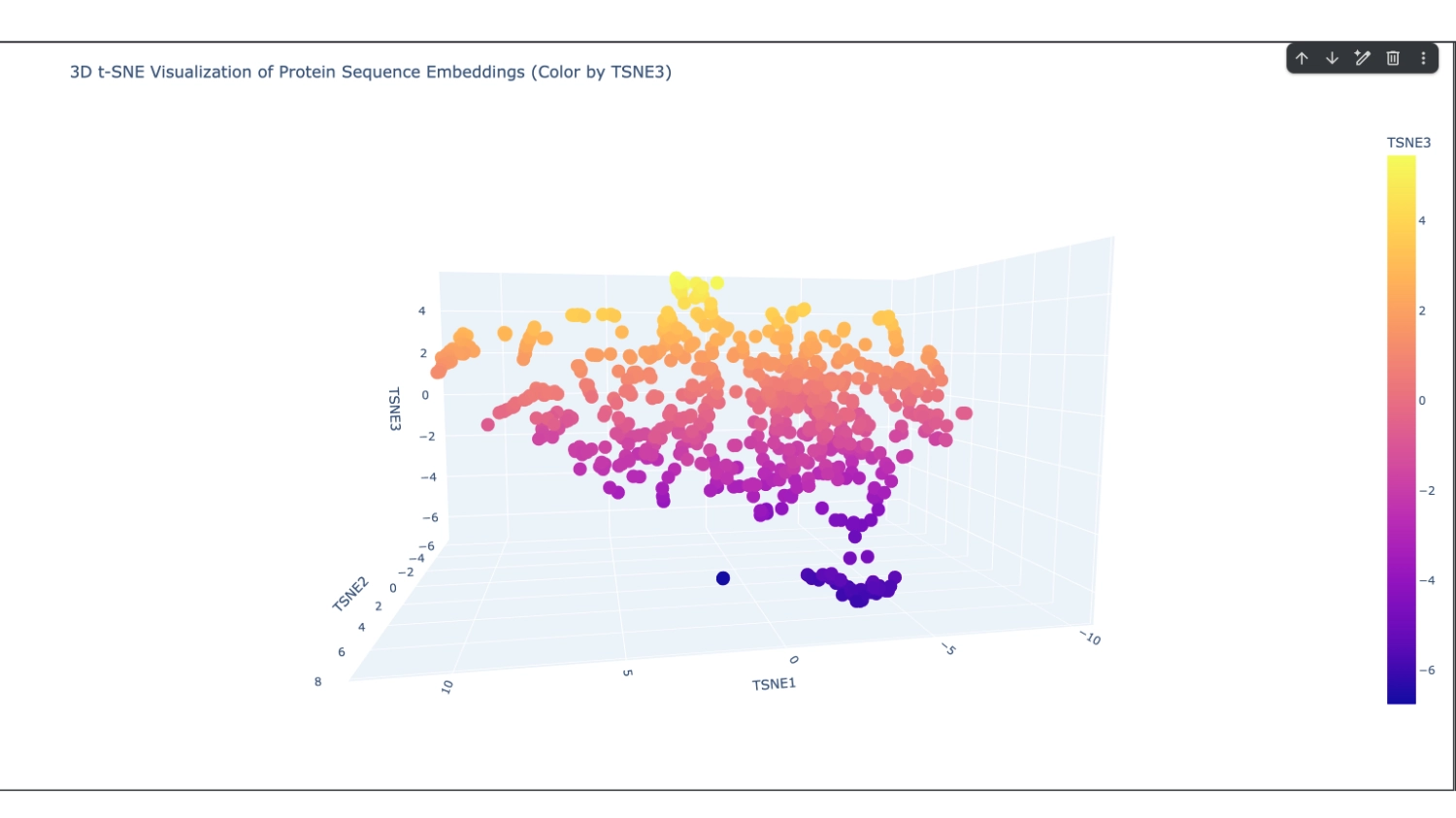
I set it to 800 which seemed to have a good amount of variation for me to understand what was happening. After that, I increased the array to 8000 because I didn’t have too many direct neighbours to my protein
1. Deep Mutational Scans
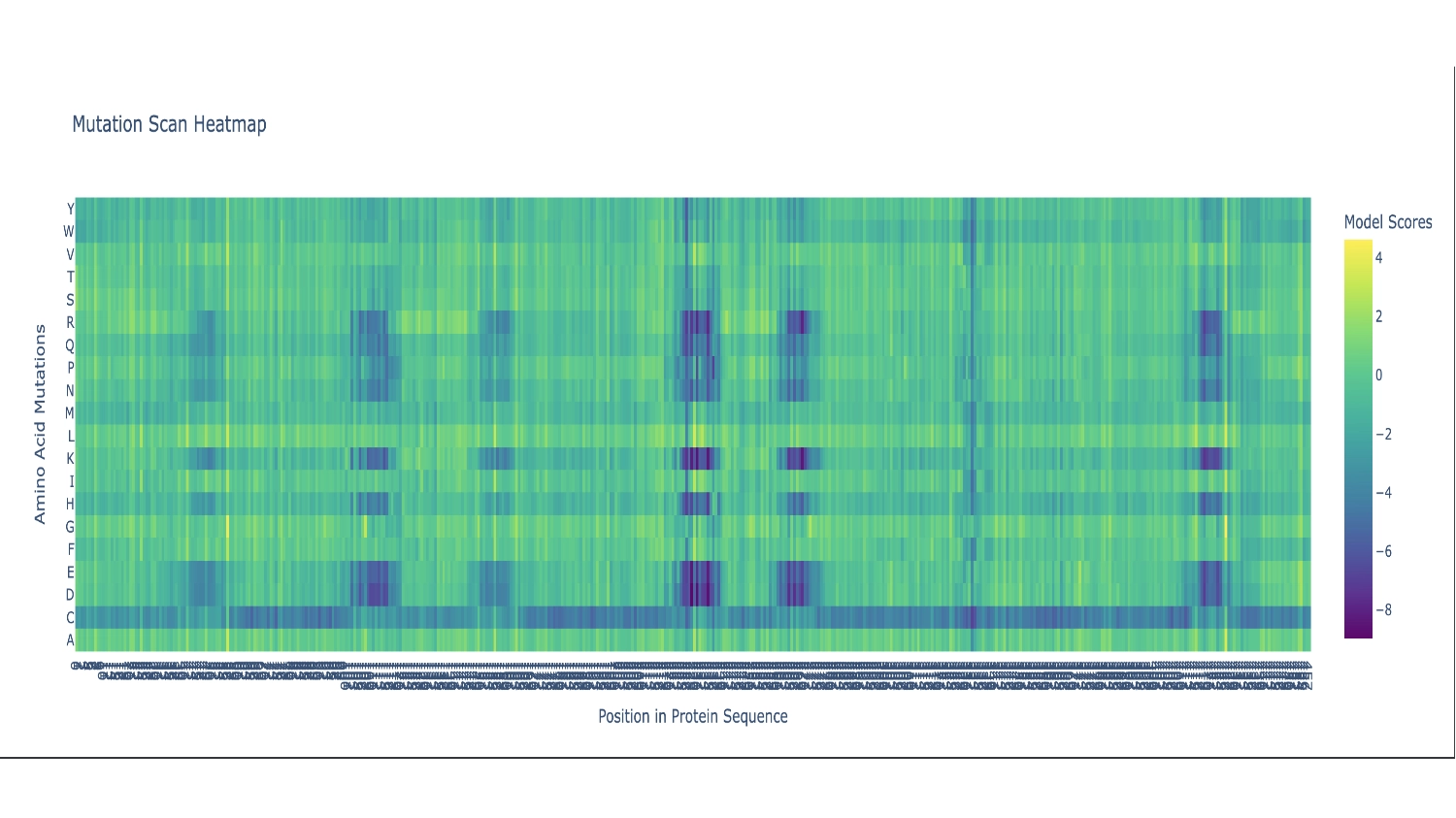
There are two primarily noticable patterns. The first one is the Cystine residue which mostly seems like a bad fit throughout the whole sequence, persumably because the chemistry of this amino acid would disrupt the function of this protein in most places— the actual protein sequence only contains 2 Cystines. The second pattern is the vertical blue columns throughout the sequence that indicate parts of the protein sequence which are propably very functionaly critic zones, thus toleratig very little exchange of residues. In these columns there are some interuptions for certain amino acids which seem to be fairly well accepted along the whole sequence which include: G (the most comon AA), and L, F, V and I which are all hidrophobic, since the protein as a whole is mainly hidrophobic this makes sence.
2. Latent Space Analysis
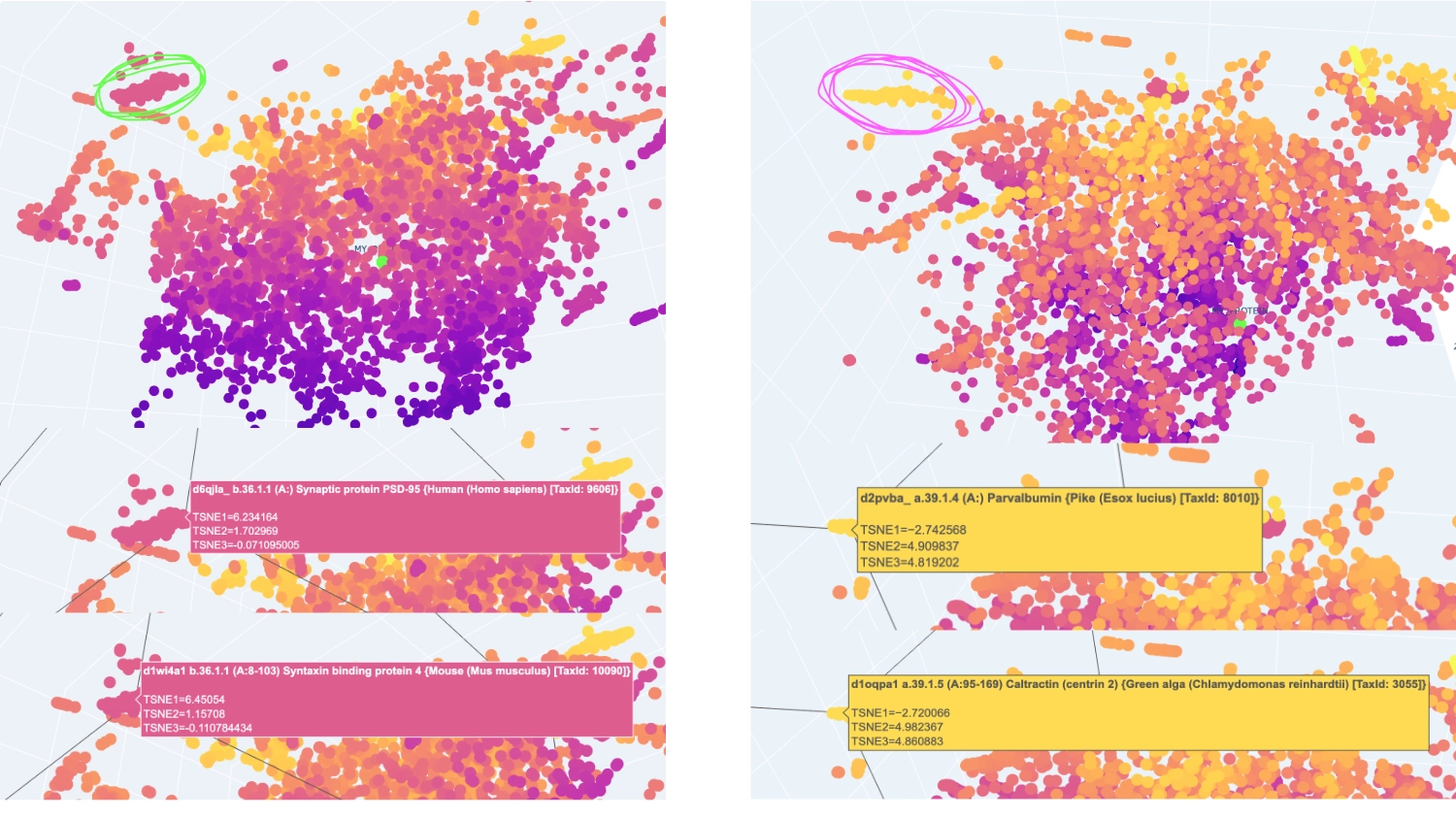
I analysed 2 well identifiable distinct neighborhoods. The one selected in green has several proteins related to neural function, while the one in pink has several calcium-binding proteins that serve different functions.
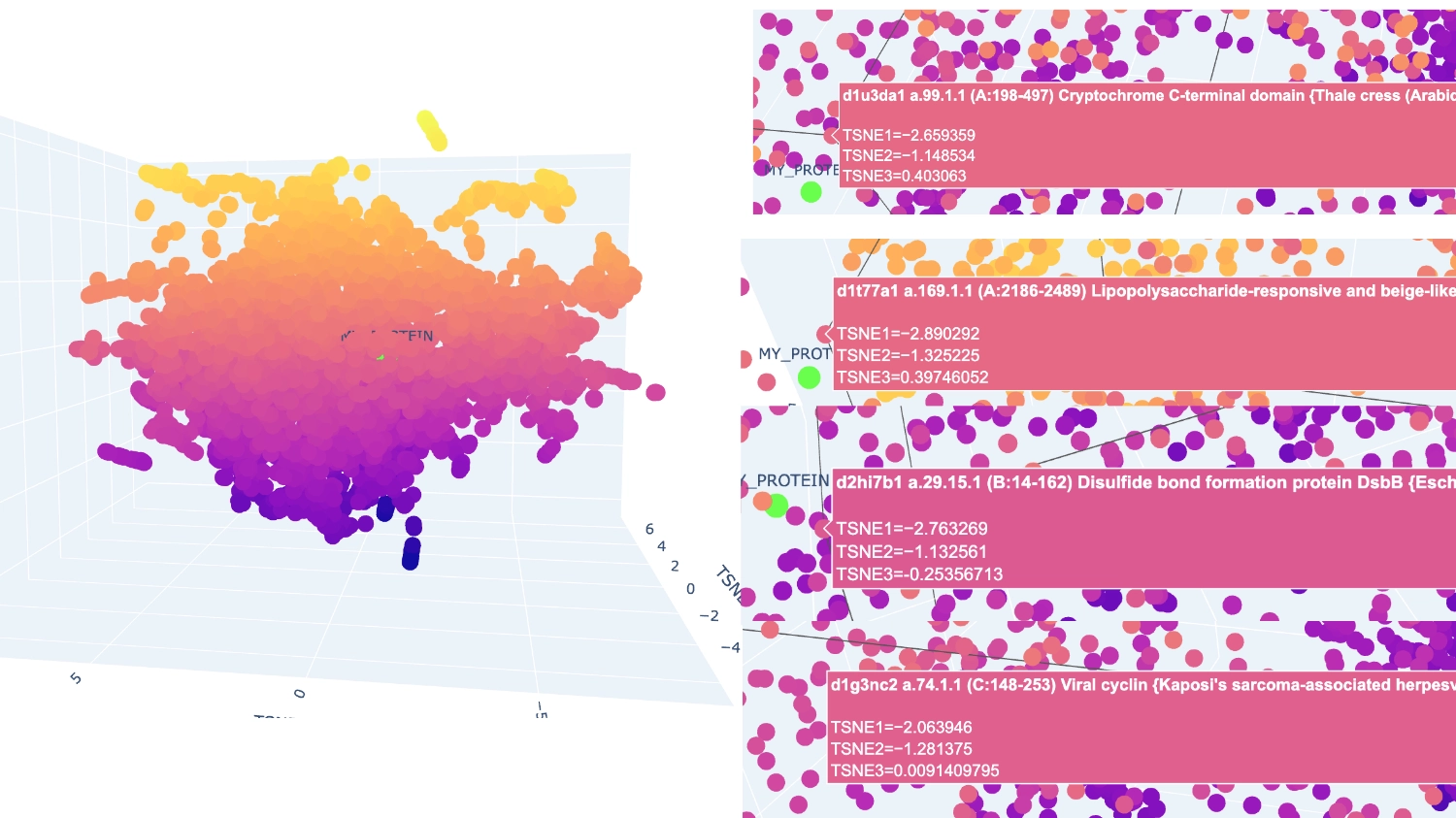
The closest and most interesting neighbour that I could identify as aproximate in function was a Cryptochrome C-terminal domain which is a flavoprotein blue light-sensing photoreceptors found in plants, animals, and microorganisms that regulate circadian rhythms and developmental processes
Most other neighbours seemed to be related by being membrane-related proteins or also being rich in helices like Disulfide bond formation protein, E. coli (membrane protein with transmembrane helices), Cytochrome c peroxidase (predominantly alpha-helical) and Viral nucleoproteins (helix-rich folds).
C2. Protein Folding
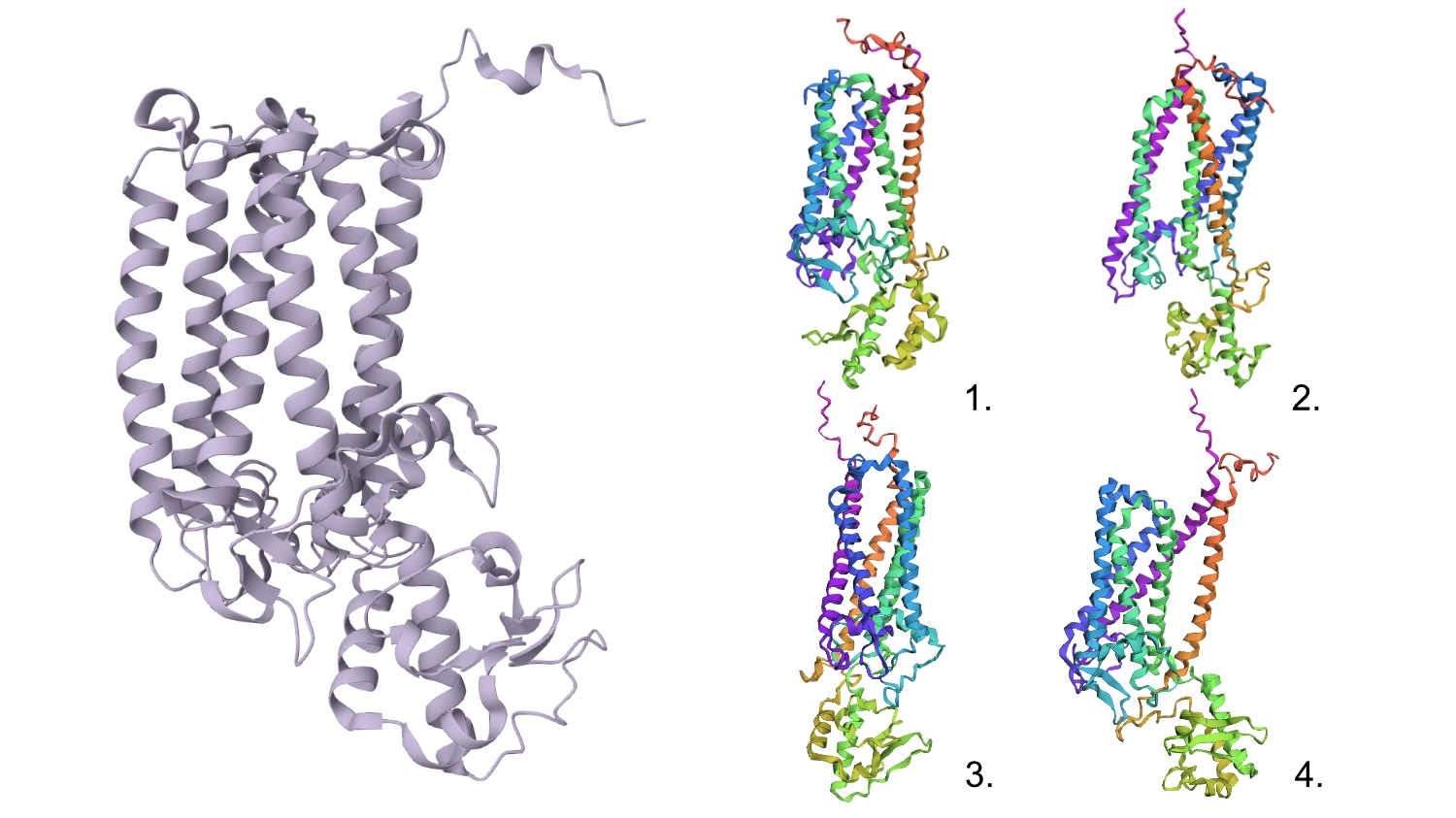
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
The predicted structure seems slightly different regarding the helices on the bottom part than the actual protein.
2. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
1.First I changed a random base for a G since it is one of the most well accepted residues and the protein seemed to maintain its shape.
2.After that, I changed 4 residues to Cs (the least accepted residue) and nothing seemed to change too much in the conformation.
3.Changed 4 sets of 6 to Cs
4.Changed 1 random set of 20 to Cs
5.Deleted 4 random residues — one of the six helixes partially uncoiled and smaller horizontal helices formed
6.Deleted 10 random residues — seems to have aggravated the latter step but still resembles the original protein
All these changes were cumulative so this protein seems relatively resilient to mutations
C3. Protein Generation
Documentation
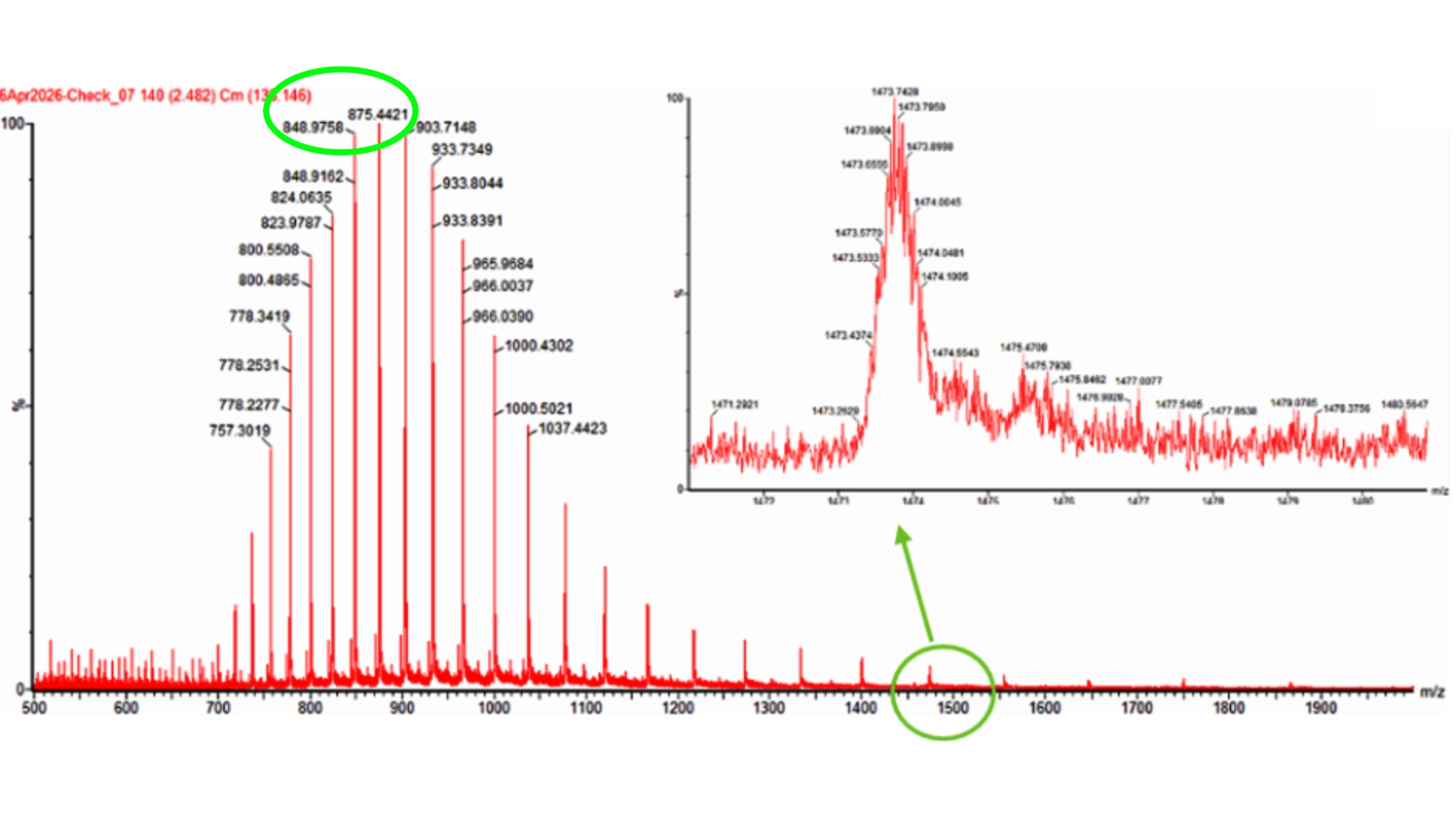
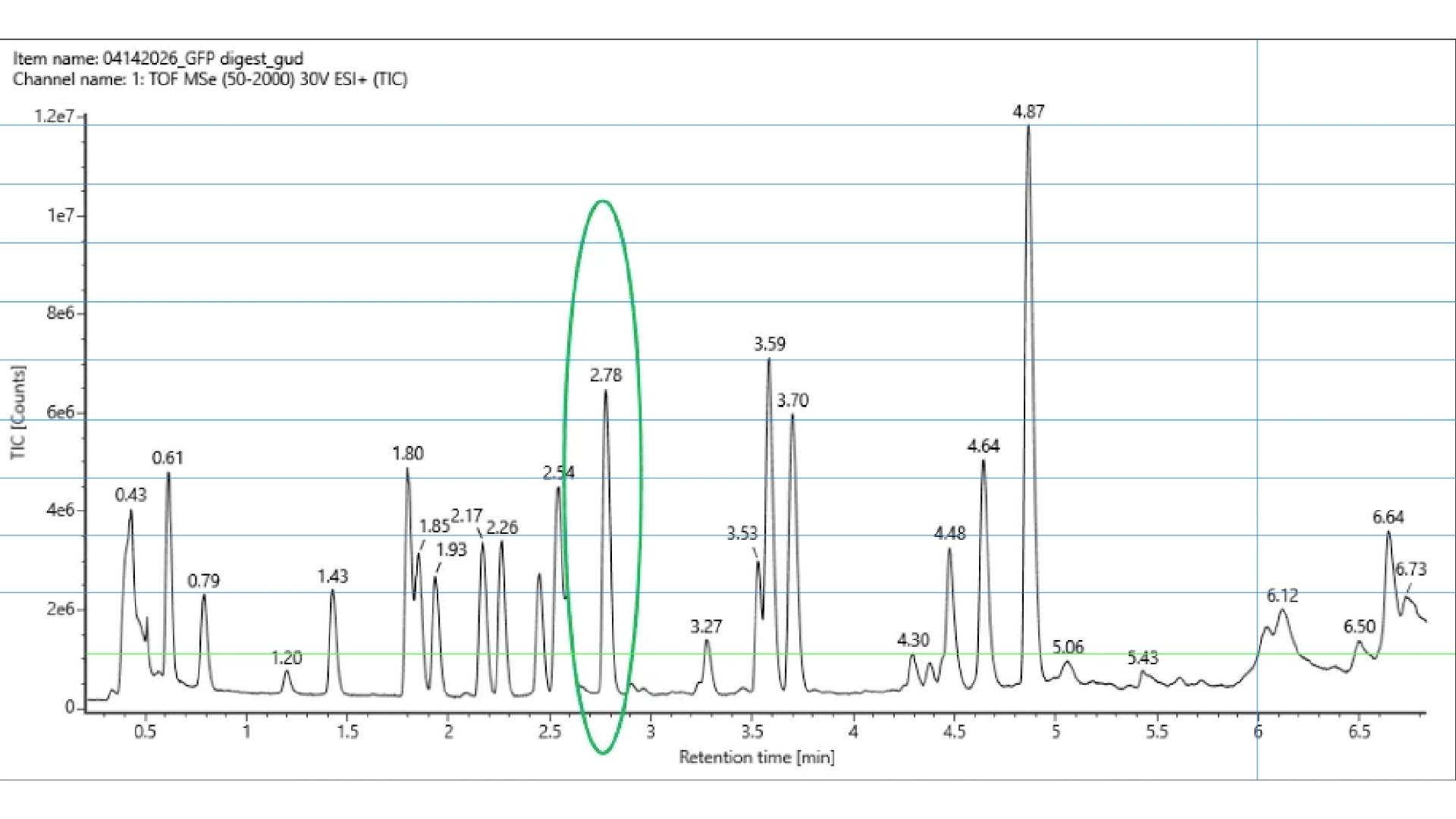
The first problem I ran into was that I couldn’t download the file for the 3D structure of CP43 protein alone, as it is part of photosystem II, all the proteins involved were also included in the in the files. I tried several ways through the RCBS website, but with no success. And so, I resorted to Chat GPT that spat out a python script to run on my computer terminal in order to download the correct file, and it worked.
Once I had the right file (checked it by opening it on the RCBS site), with both the backbone and all ligands to that specific protein. I tried to run the inverse folding code on the Collab notebook but with no success and couldn’t find a way to import hat was missing so I tried to use an online tool for inverse folding from neurosnap.ai and it seemed to work just fine!
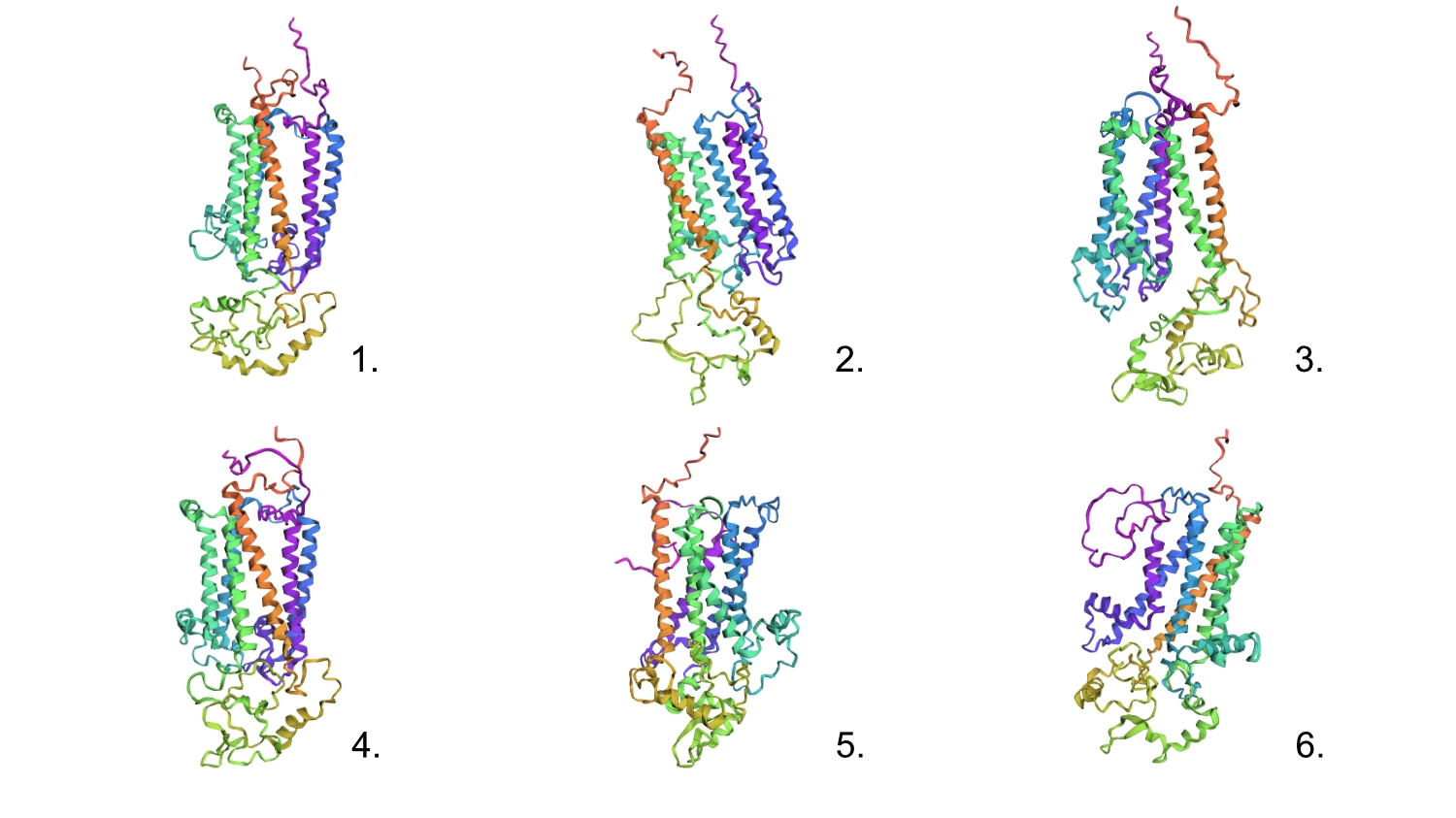
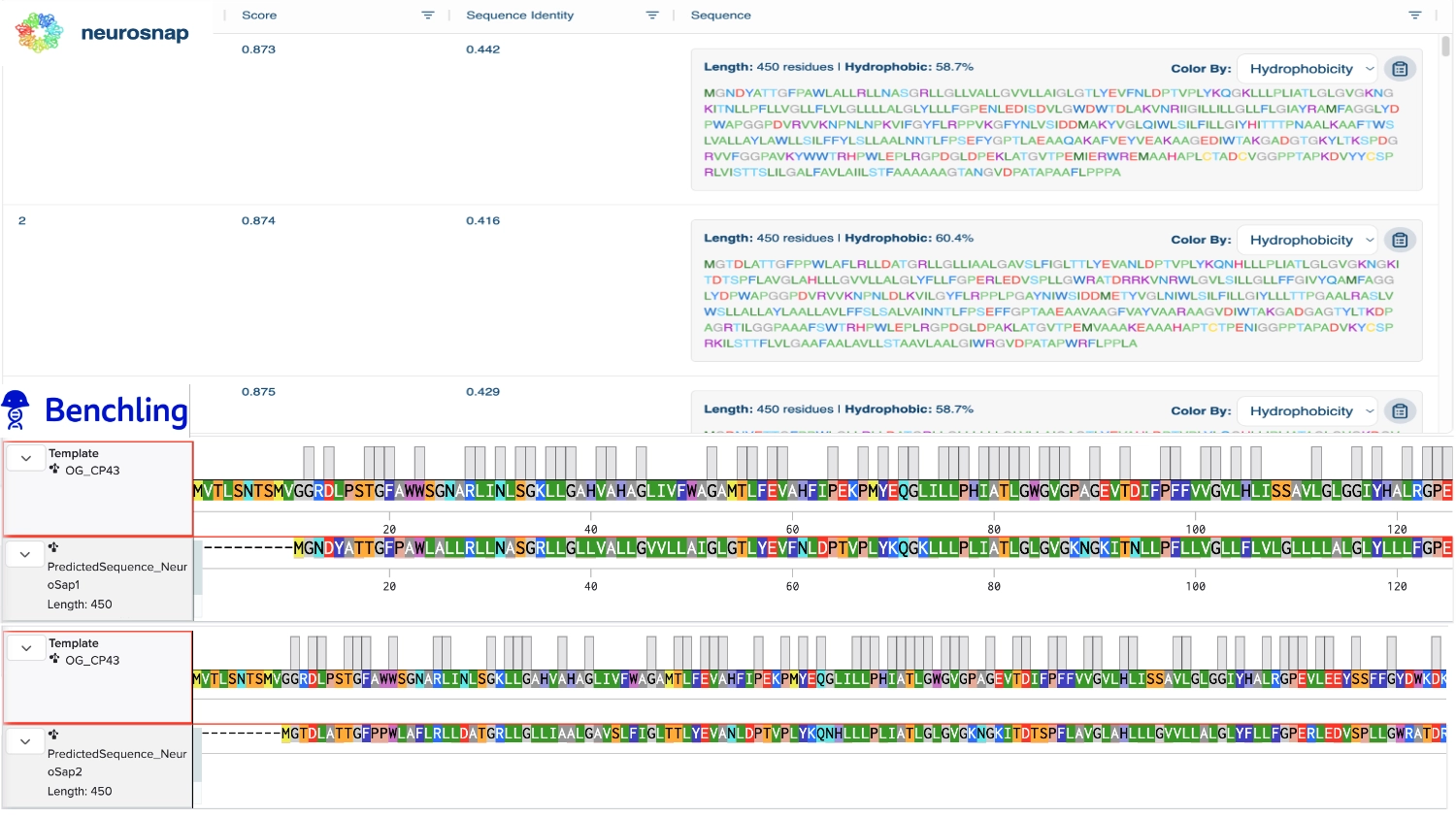
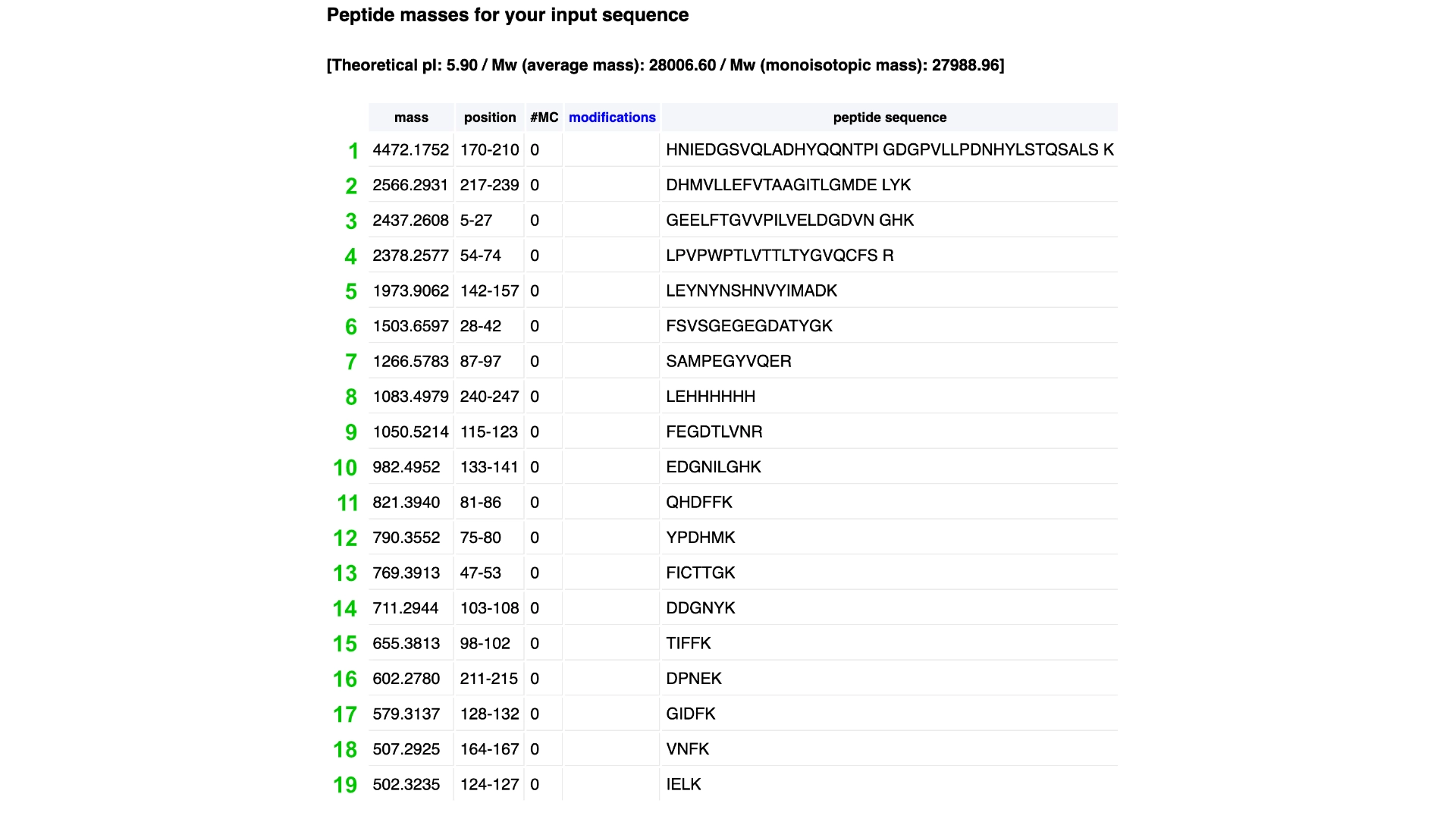
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Then compared some of the predicted sequences to the original one by performing alignments on Benchling to see what residues were used on the predictions and how similar they were to the original. As observed on the heatmap from earlier the C residues were kept at a minimum and the most used residues fluctuated between the best accepted ones like L and G. It was also interesting to observe that between predicted sequences there were similar areas that have the same residues in common as the original, which must be the most crutial for that type of folding. Also, the predicted sequences have -10 residues at the very beggining, you can see it from the benchling screenshot, and which I interpret as a N-terminus or C-terminus that the inverse folding model didn’t replicate since they probably weren’t solved in the 3D structure.
2. Input this sequence into ESMFold and compare the predicted structure to your original.
Regarding the 3D structure, the new predicted proteins were similar to the original, however, the main differences rested on the width of the 6 helices barrel which in wider on the original CP43 and the same helices are not as well organized, straight and vertical as the original.
Part D. Group Brainstorm on Bacteriophage Engineering
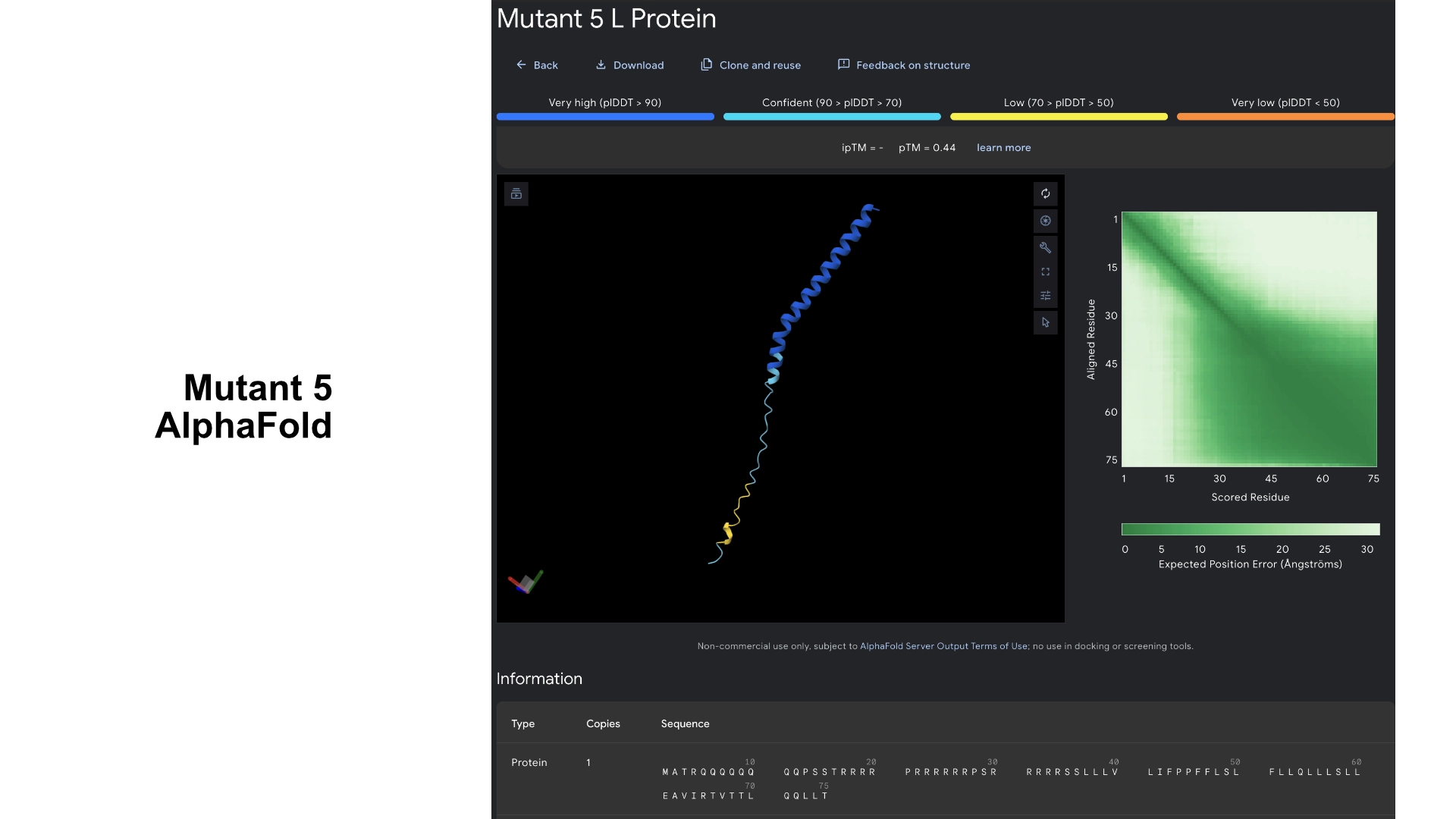
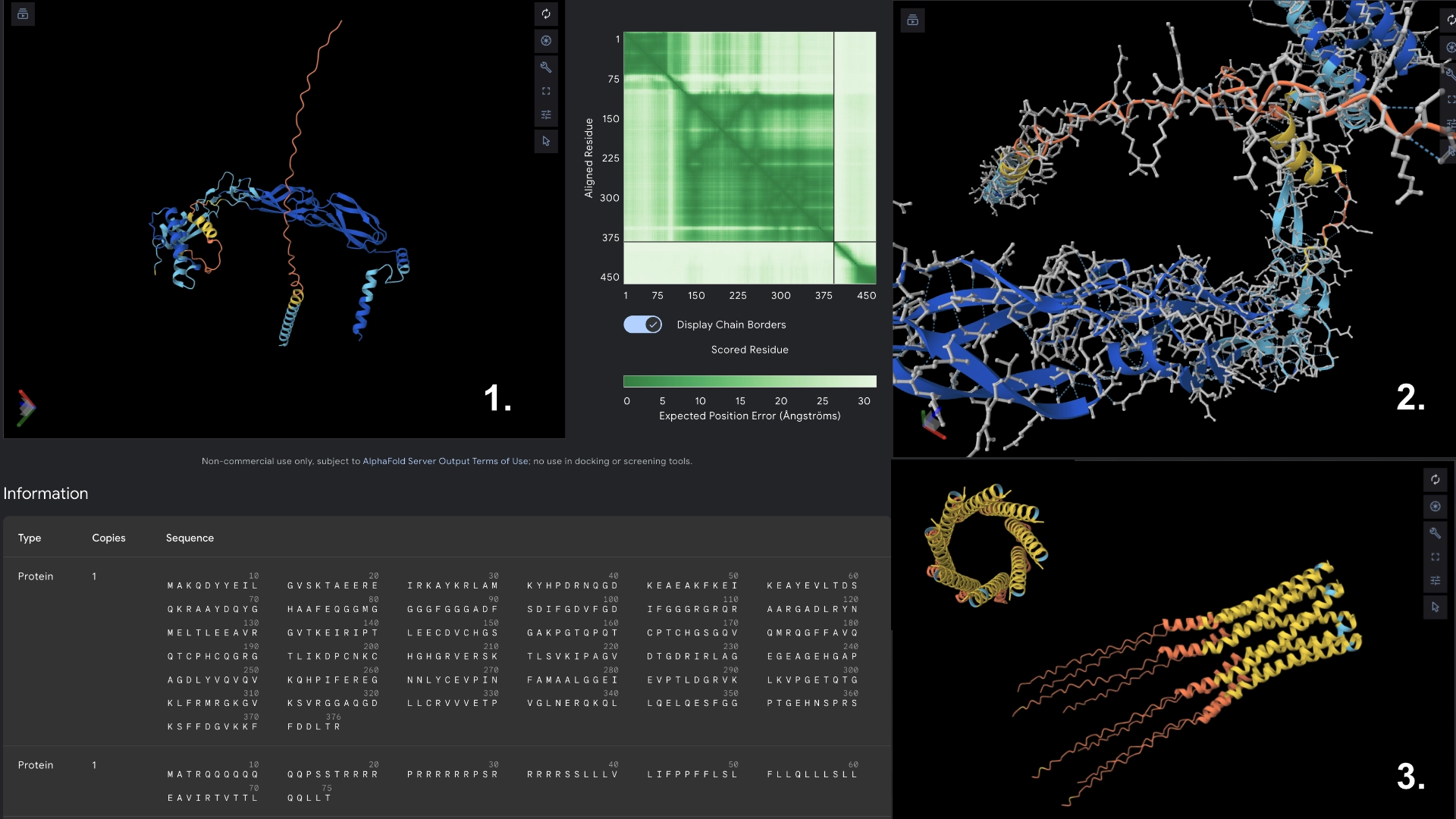
This was the result of my initial research for the group phage project. My group has setup a shared docs and we are working in the goal of stabilizing the L protein.
After generating the 4 sequences I resorted to Chat GPT to generate code to help me input the known sequence into the model so that PepMLM would grade its confidence in the known binder as well.
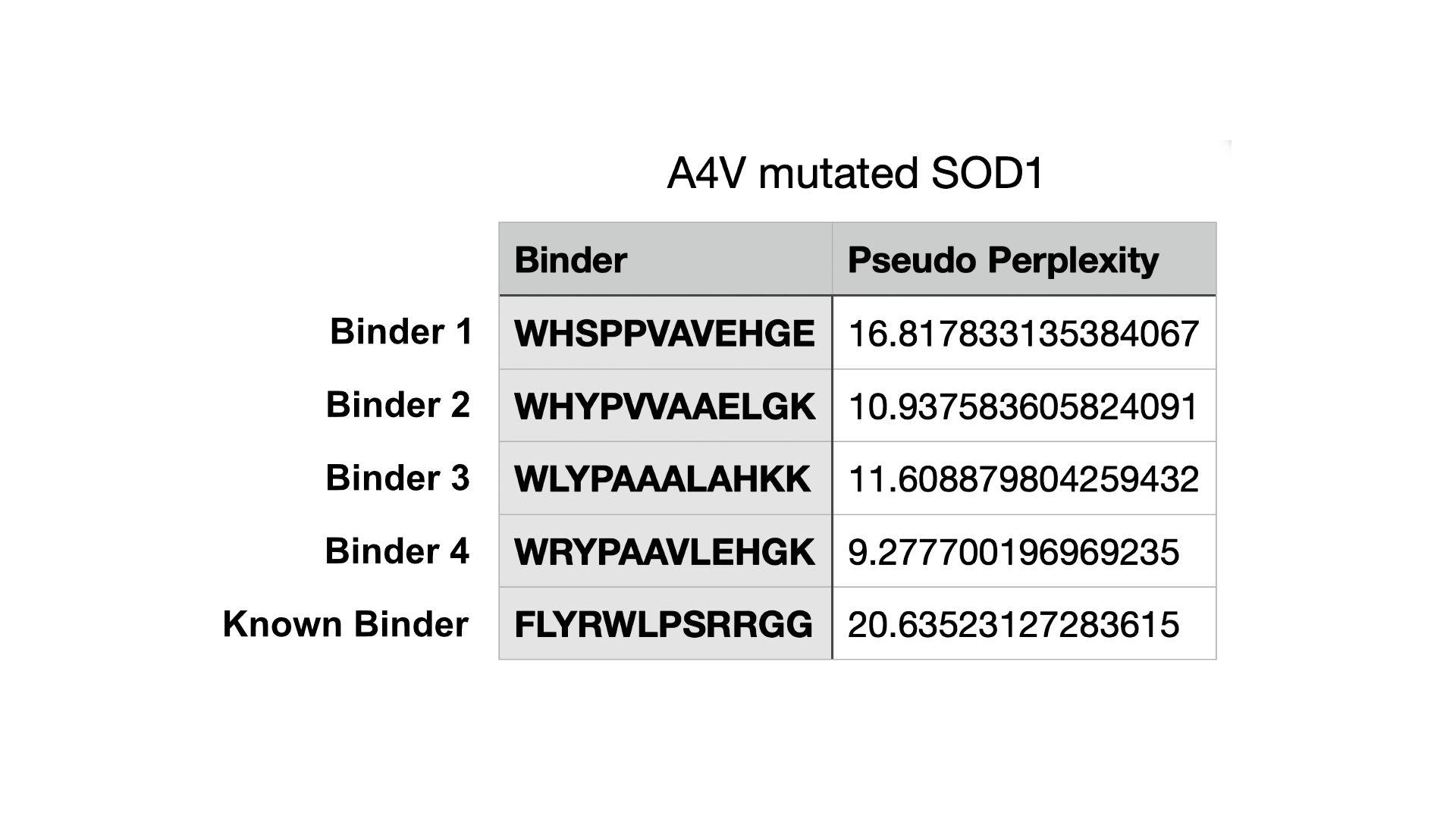
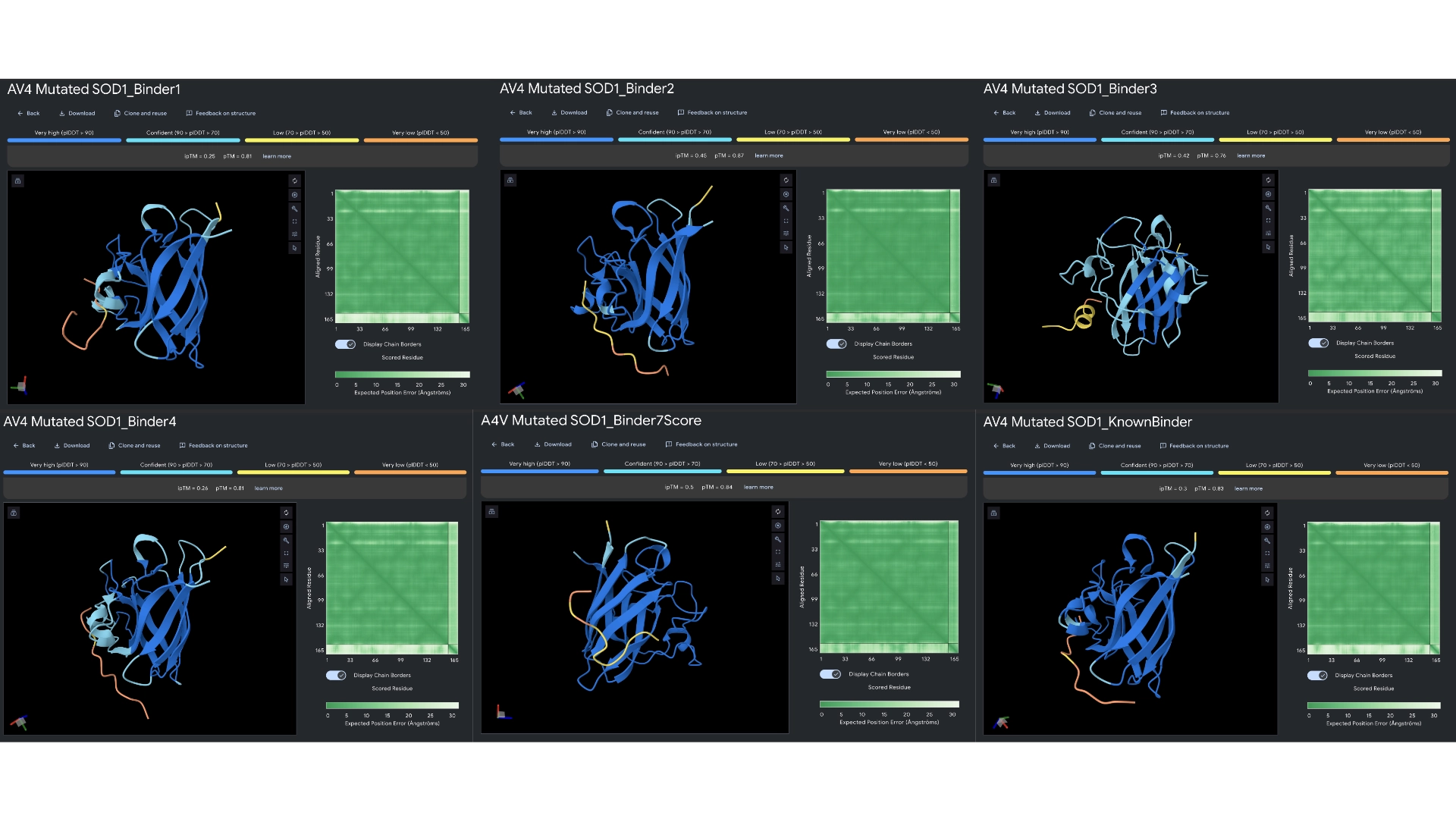
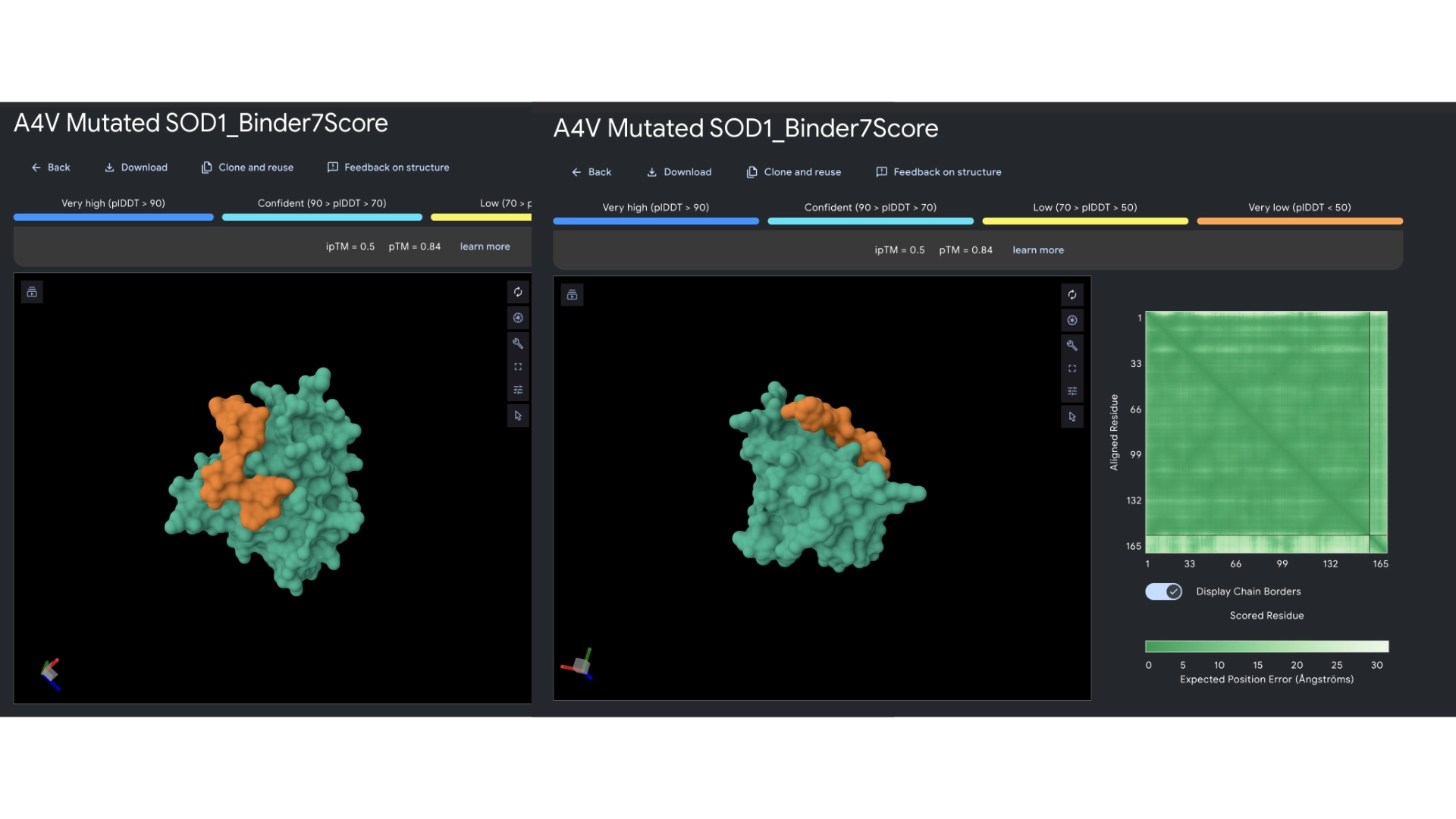
3. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
All the pepties I visualized bonded to the barrel region. The one I was able to generate which had the best perplexity score (of 7) and the best ipTM (of 5) bonded slightly closer to the N-Terminal, but still on the barrel region. I included a molecular surface visualization for that one so it could be better analized. It seems to bind at the surface, doesn’t seem to be partially burried.
The known binder got a perplexity score of 20 and a ipTM score of 0.3, so I’m not sure how indicative these scores are being of the actual binding abilities of these peptides
4. In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
All ipTM values I got were bellow 0.6 and so it would suggest the predictions failed in terms of relative positions between enzyme and binder peptide. But these might be due to the small nature of this structure and chain where the TM score is very strict. However, binders 2, 3 and the extra one with preplexity score of 7 all got better ipTM scores than the known binder, so that would supposedly indicate peptides that exceed the known binder.
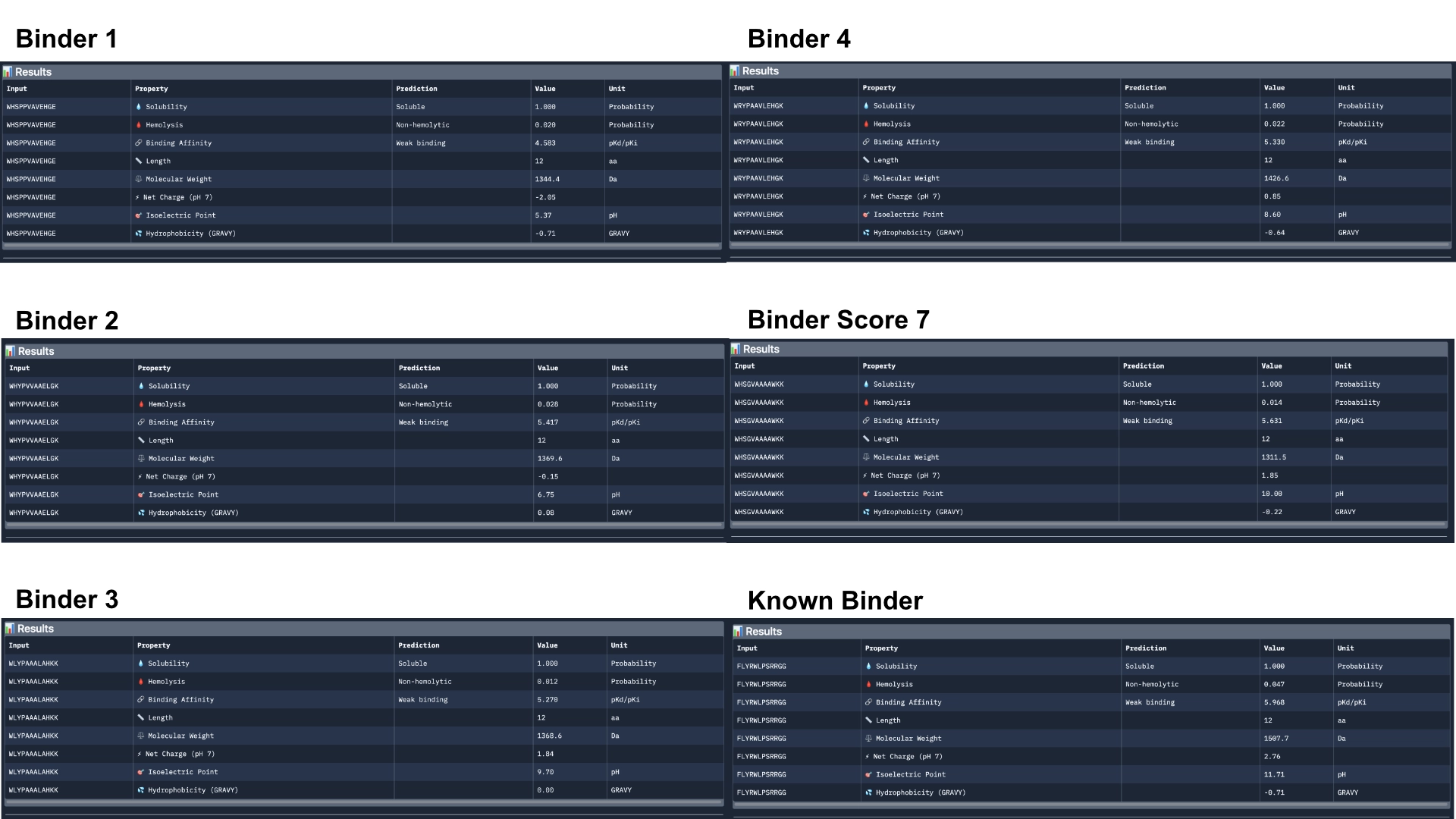
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
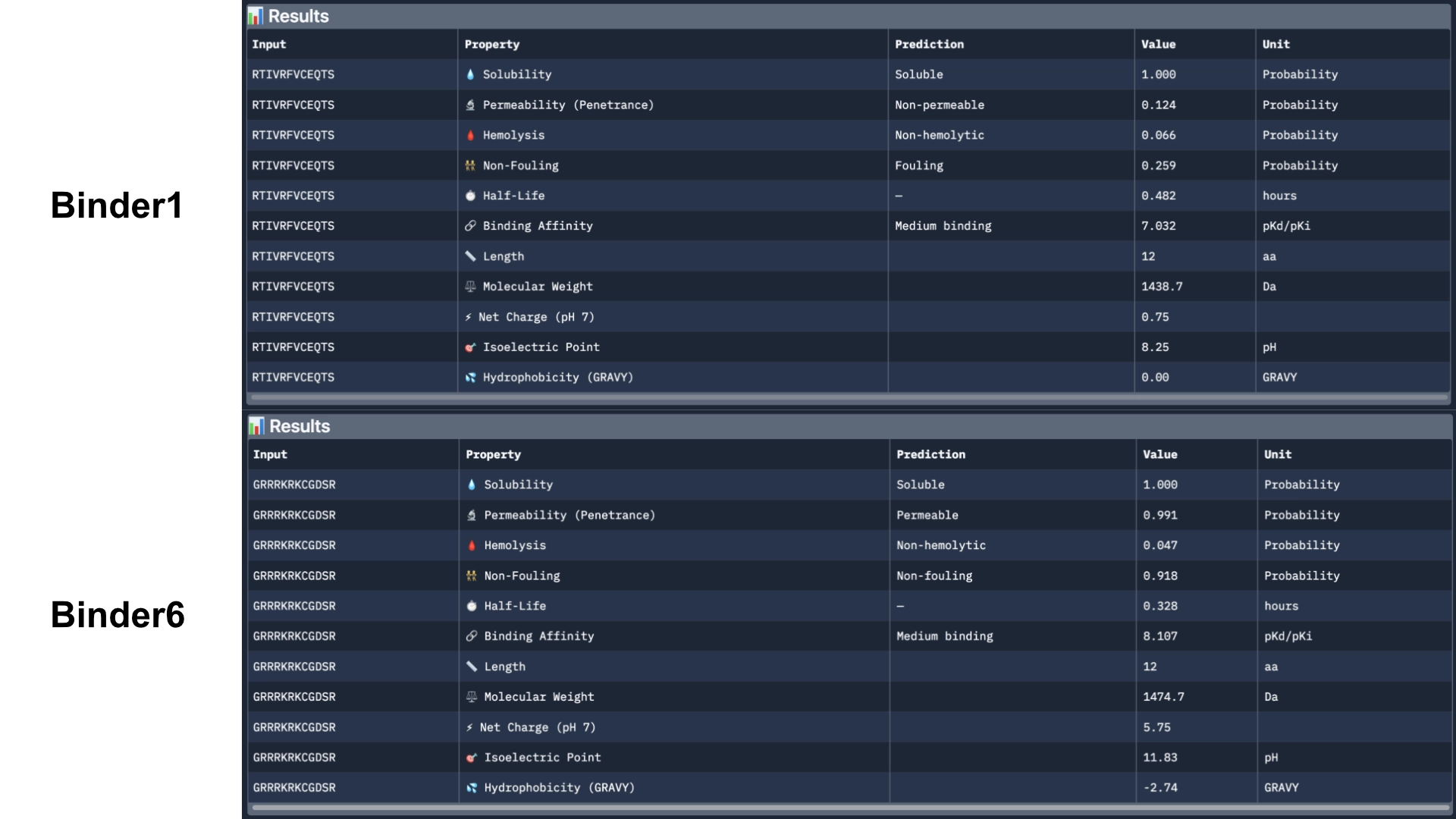
Overall, Peptiverse considers all generated binder and known binder as weak binders, the known binder having the highest score of 5.968, followed by the binder which had the highest ipTM score, with a binding affinity 5.631. However, binders 2 and 3, which surpassed the known binder in ipTM score, were not attributed better binding affinitty, having the scores 5.417 and 5.270 respectively.
All predicted binders had good therapeutic properties scores and none were non-soluble nor hemolytic.
The best overall in balancing all aspects was the known binder with 5.968 of predicted binding and 0.047 of hemolytic probability.
The best predicted binder, althought, it has a slightly lower hemolytic probability of 0.014, that might not balance out the 0.337 difference in binding affinity (5.631). However, this would probably be the best to advance with, even though it might not exceed the qualities of the already known binder.
Part 4: Generate Optimized Peptides with moPPIt
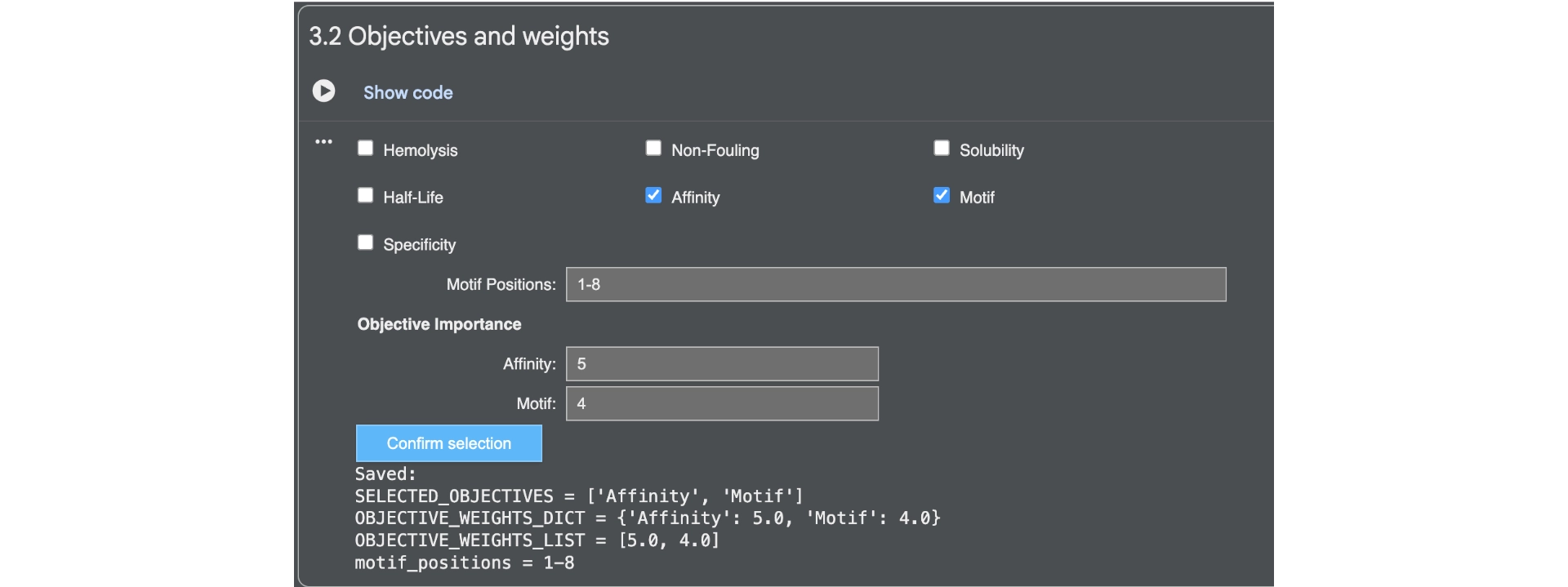
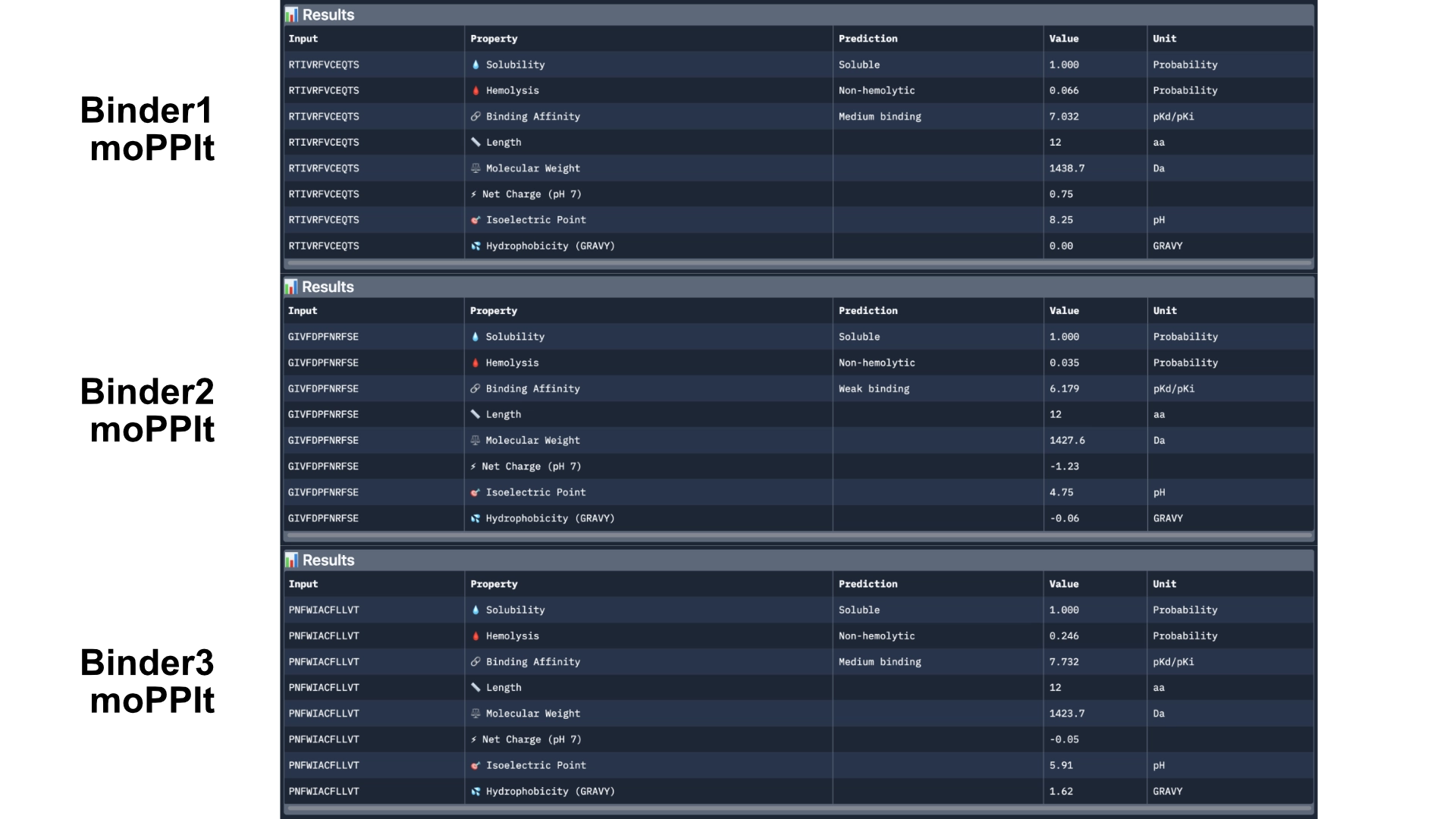
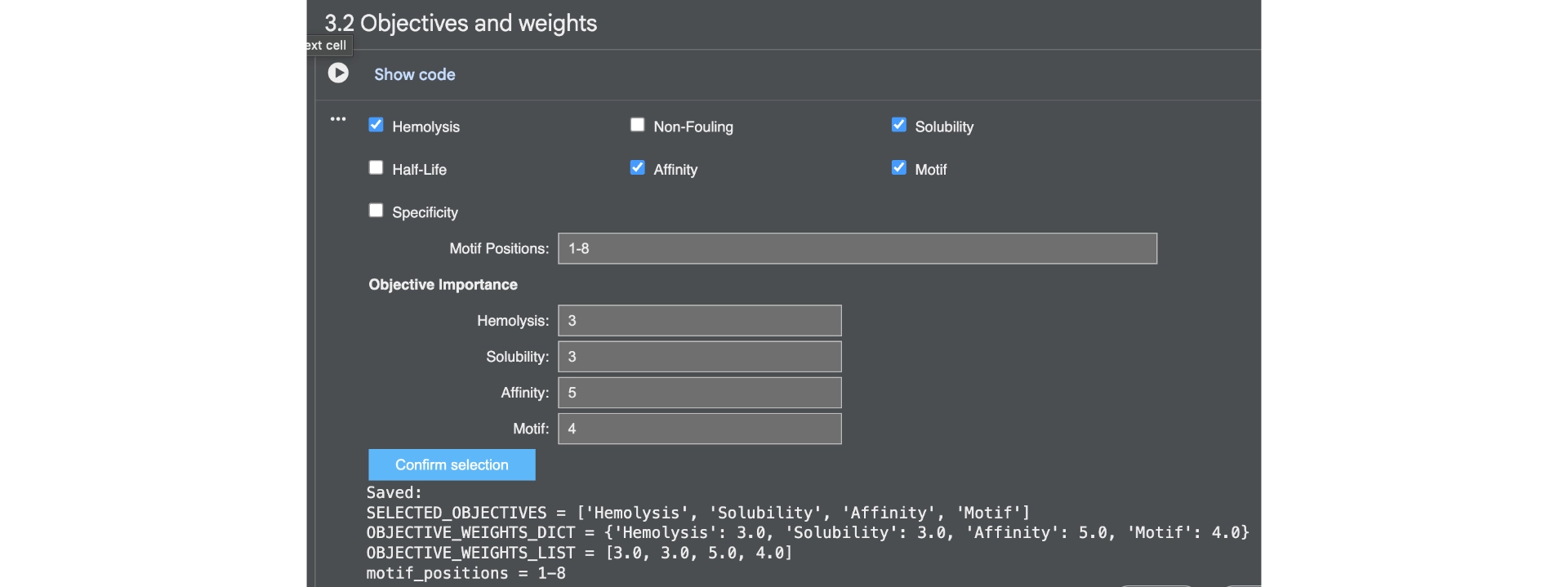
First of all I generated 3 peptides for the A4V mutated SOD1 with the only parameters being affinity and motif whith the following weights:
Then analysed the resulting peptides in PeptiVerse.
Comparing these to the PepMLM ones, there already was a good improvement as they were all soluble and non-hemolytic and had better affinities with two of them reaching medium binding instead of the previous weak binding levels.
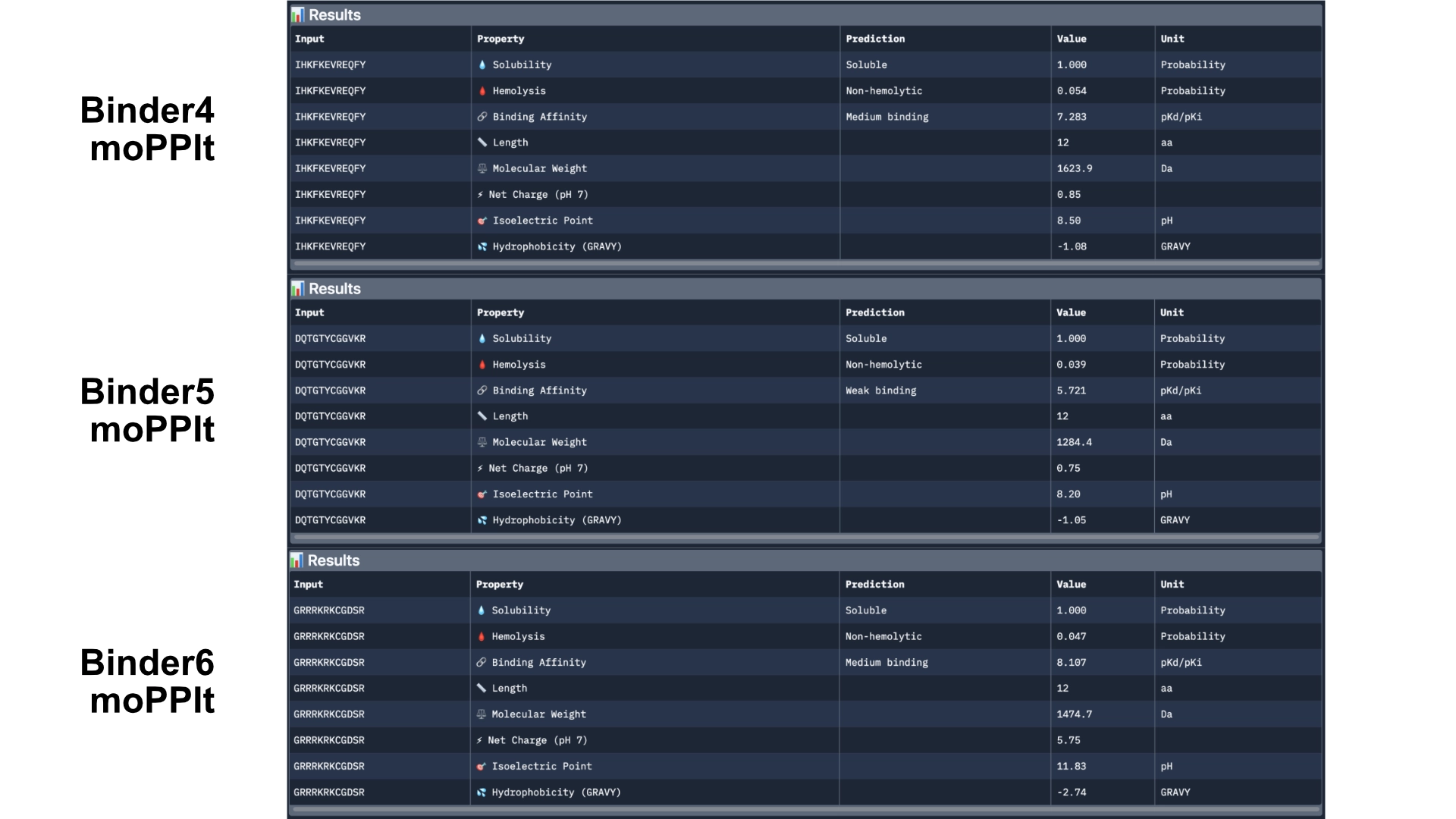
Then generated other 3 peptides with non-hemolytic and solubility objectives to watch how they would differ from the previous ones.
Then analysed with PeptiVerse again.
The results came back with low homlytic probabilities, soluble and with good affinity
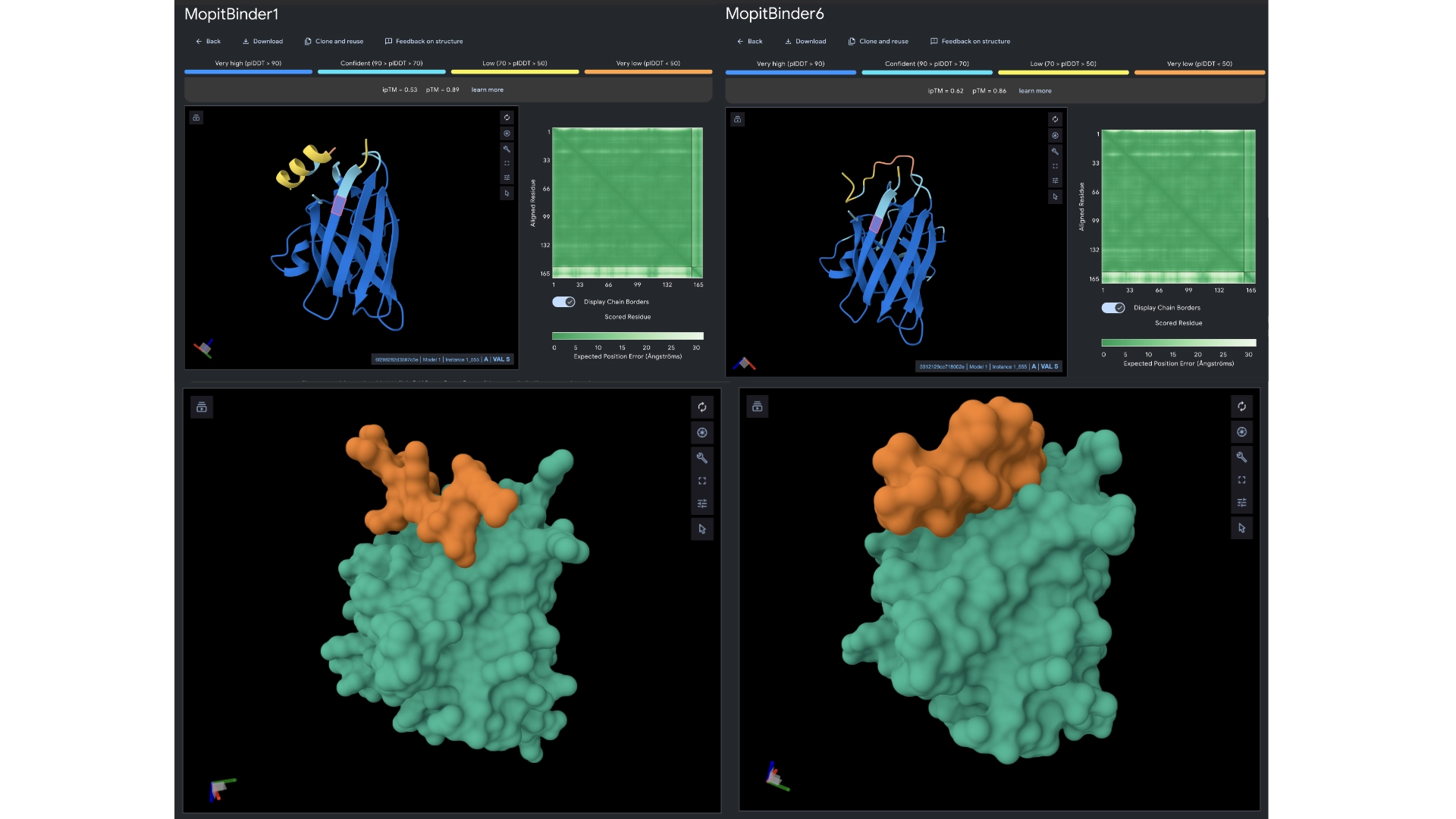
In orther to reach some more conclusions I fed the best 2 peptides— Binders 1 and 6— to Alphafold to see if they would bind closer to the N-terminus which was the motif I had input and see if the ipTM score had gotten any better.
The ipTM scores came back the highest yet, with binder 6 reaching 0.62. They did bind closer to the N-terminus and not to the barrel region. Binder 1 did form a helix in the visualization and this might be interesting since a helical structure is more stable and could be further improved to have really high binding affinity.
Then ran them against more therapeutic parameters— non-fouling, halflife and permeability— to see how these would hold up even not having been optimized for those purposes.
Binder 1 did come back as fouling and non-permeable. However, Binder 6 came back as permeable, non-fouling and with a half-life of 0.328 (which might be on the lower side). I would further optimize these 2 best peptides for the different theurapeutic qualities where they are weakest— fouling and permeability and half-life for binder 1 and half-life for binder 6— using a MOG-DMF model and run them against those parimeters using PeptiVerse.
Part C: Group Project: L-Protein Mutants
Documentation
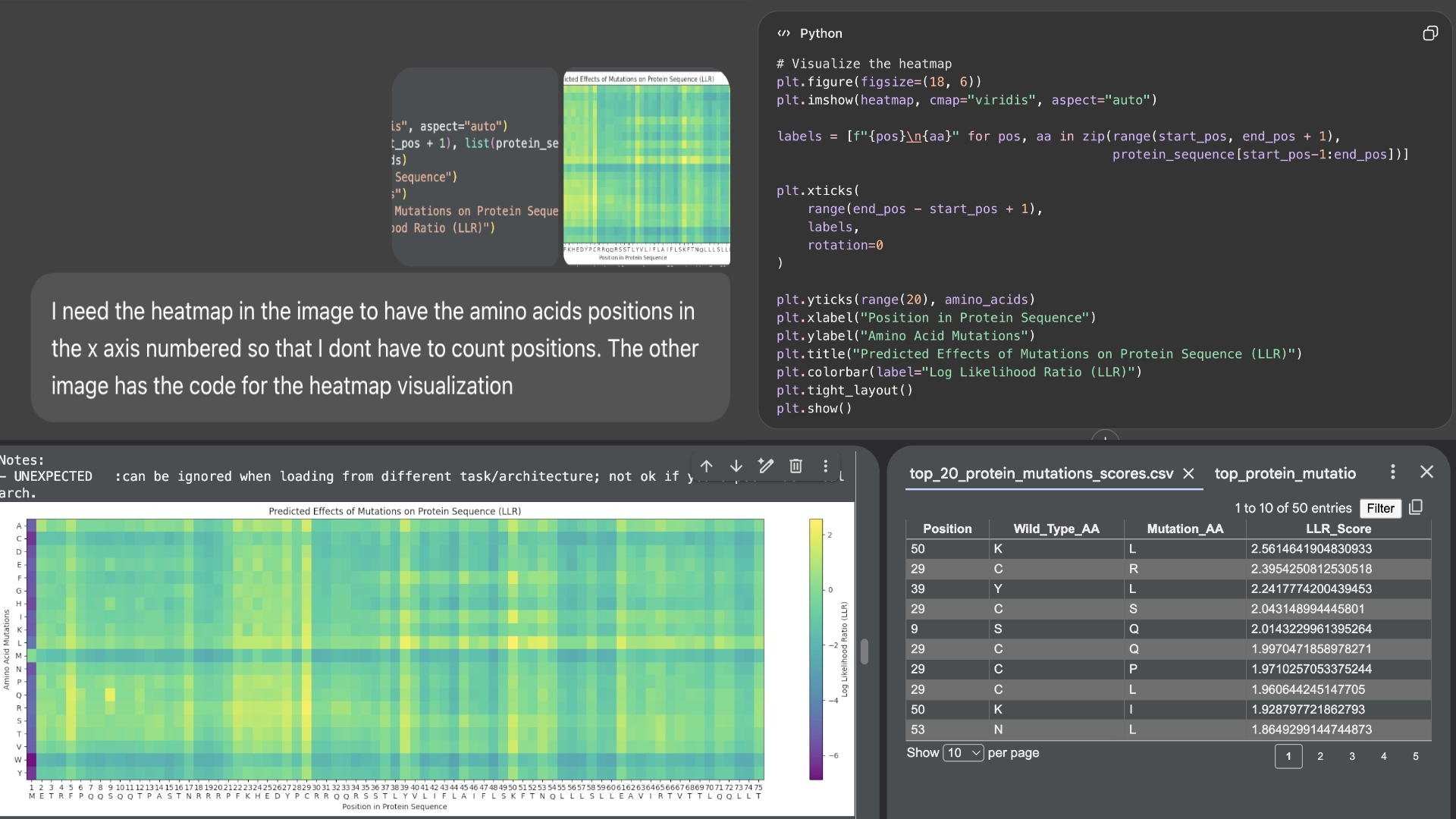
First of all, I used Chat GPT to number the residue sequence in the heatmap for me to compare the residues more easily
I started by making 2 mutants with several mutations to the hydrophilic domain, would be interesting to make it more stable and not dependent on DnaJ, by making changes in the positions best scored by the LLR and confirming them with the experimental data.
Mutant 1
6 best LLR scored residues in hydrophilic zone (no changes to transmembrane zone)
29 C->S (C->R was the best according to LLR score but was negative for lysis on the experimental data)
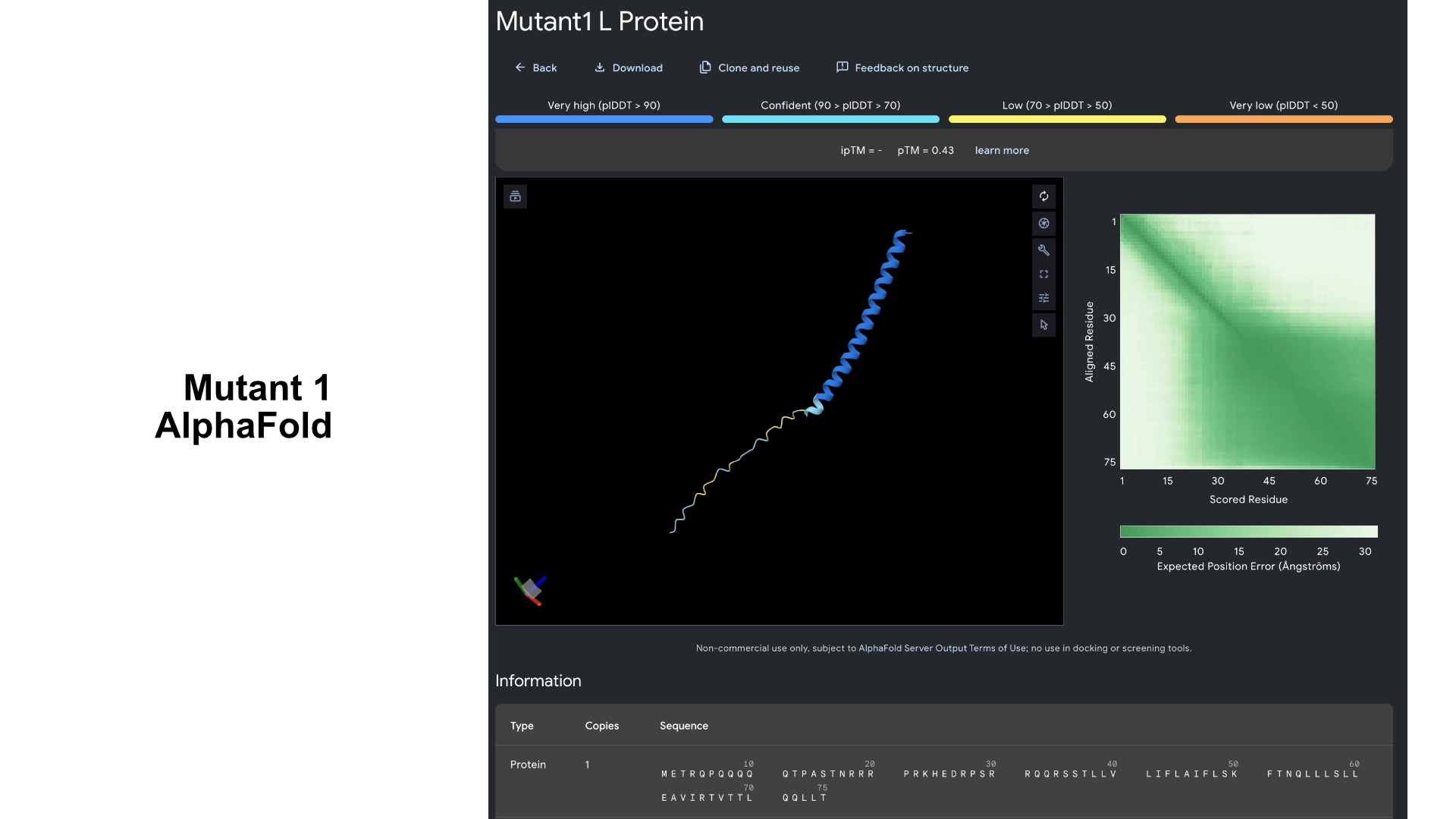
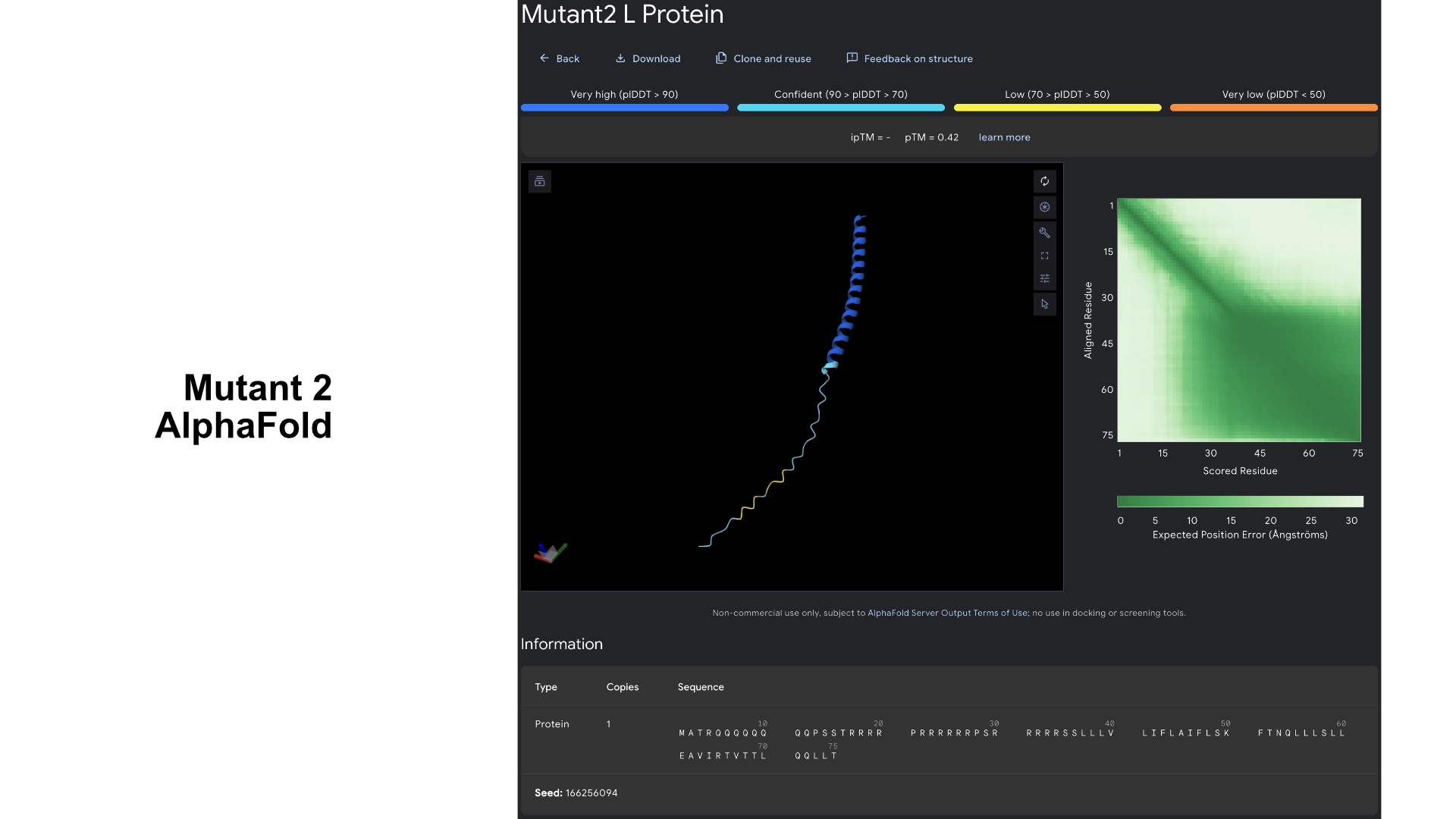
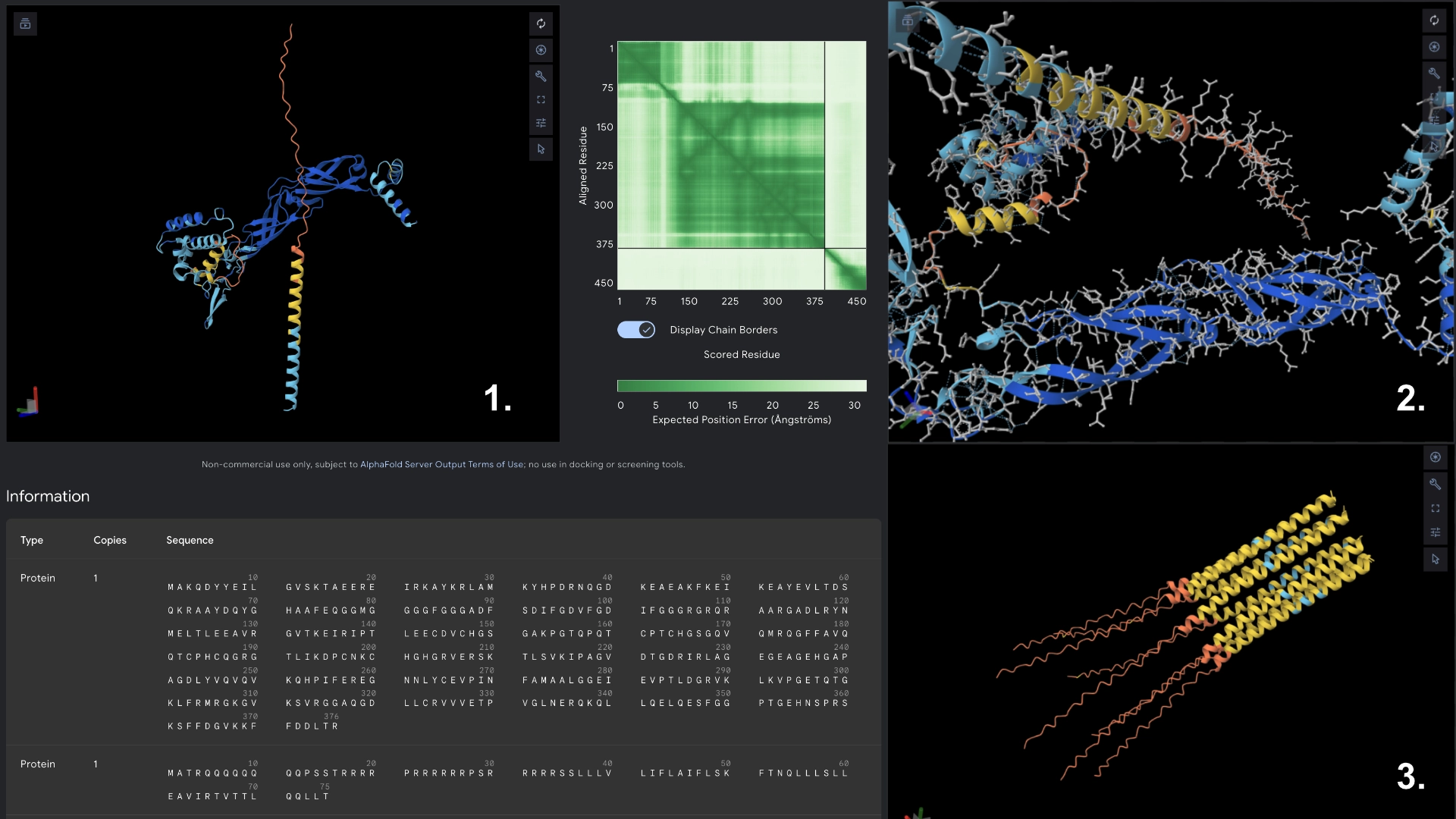
Then I visualized it in Alpha fold to see what had changed. While the hydrophilic region in the original L protein had low accuracy in the visualization and wasn’t uniform, this Mutant 1 had better values of accuracy and more uniformity in the hydrophilic tail.
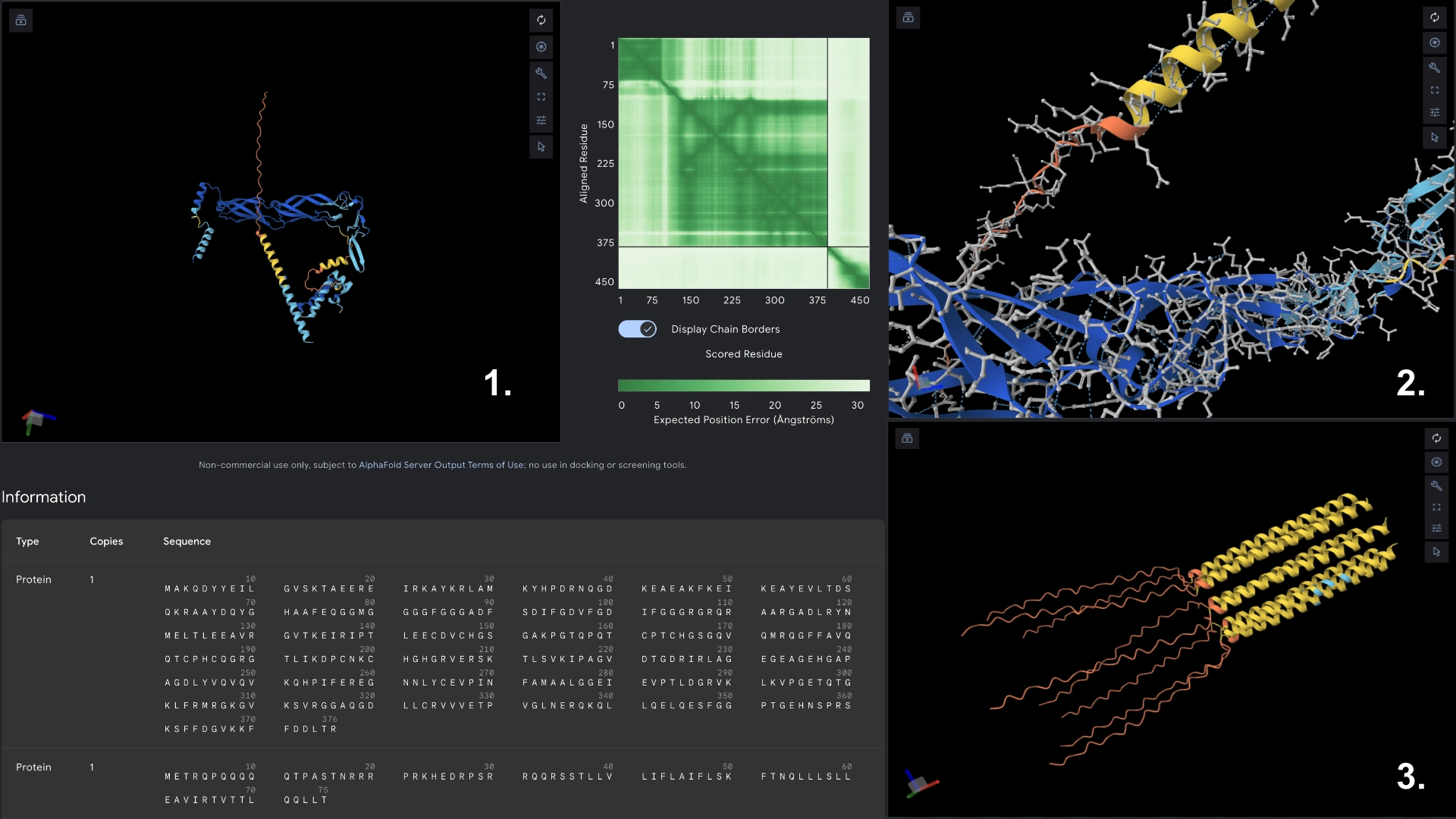
Redered interaction with DnaJ
Detail where we can see no bonds between DnaJ and L protein Mutant
Multimer made up of 8 Mutant L proteins. The aggregation seems close to what would be expected of wild type L Protein
Mutant 2
17 best residues in hydrophilic zone (no changes to transmembrane zone)
Added to the previous best LLR scored residues the best mutant for other 11 residues always double checking with experimental data sheet
This version had slightly better scoring in regards to prediction confidence and was slightly more uniform, but no significant changes in visualization
Redered interaction with DnaJ
Detail where we can see no bonds between DnaJ and L protein Mutant
Multimer made up of 8 Mutant L proteins. The aggregation seems close to what would be expected of wild type L Protein
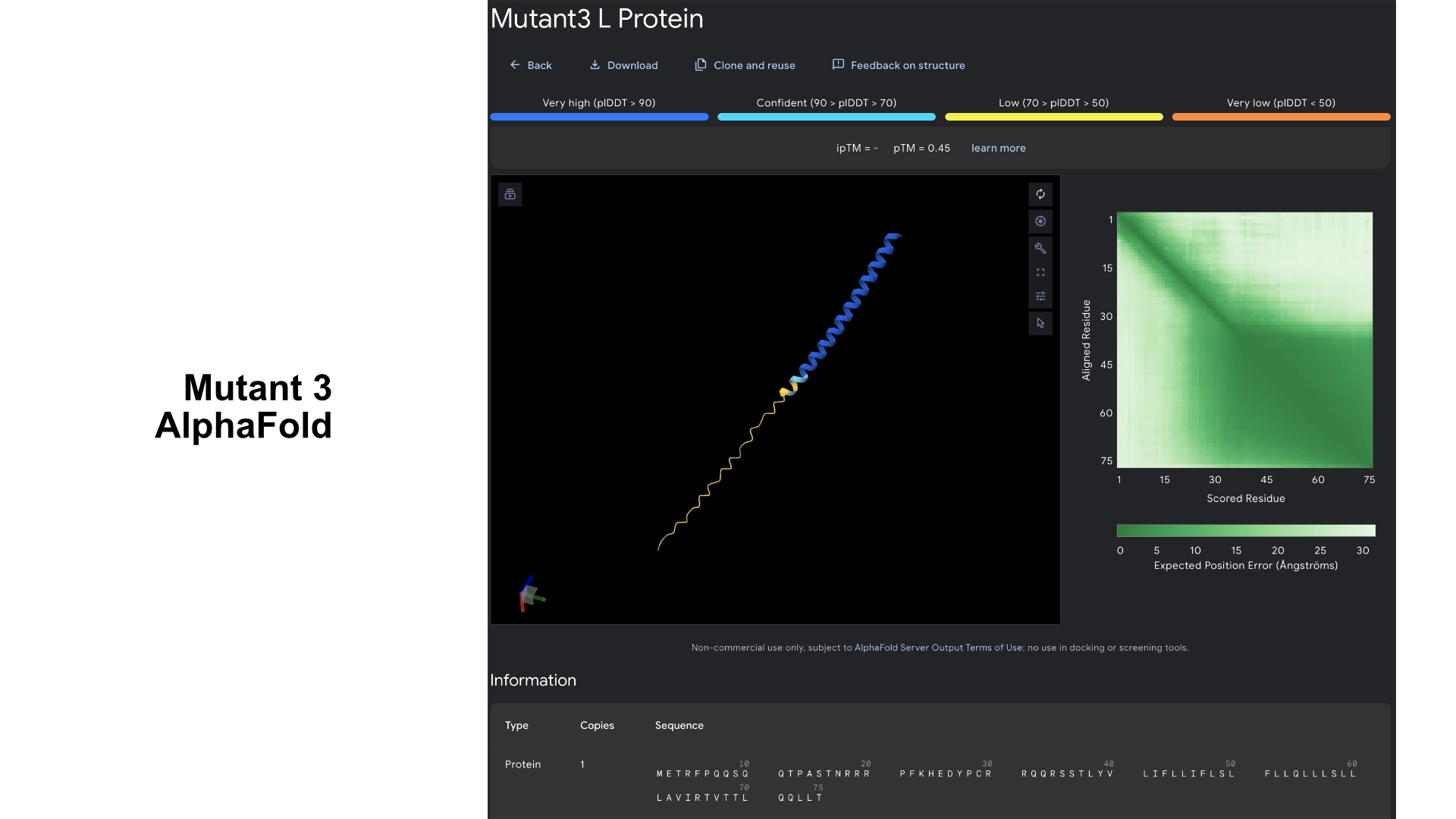
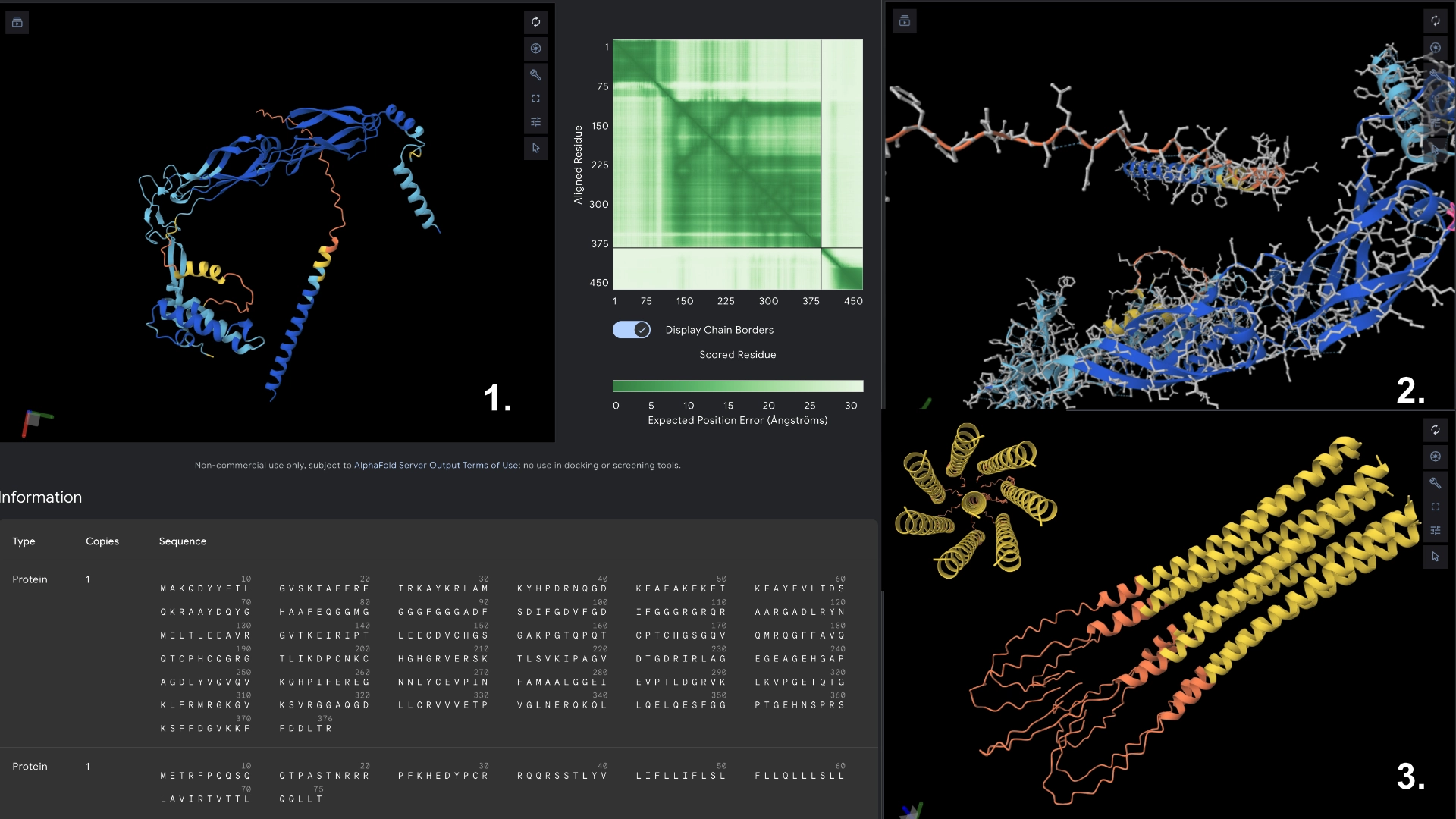
Mutant 3
5 best LLR scored residues in transmembrane region always double checking with experimental data sheet (no changes to hydrophylic zone)
These residues correspond to the areas in the heatmap where mutation seems to be more accepted. As the transmembrane region already has a well defined structure and good prediction confidence in the original protein, no significant changes were noticeable in the visualization.
Redered interaction with DnaJ
Detail where we can see no bonds between DnaJ and L protein Mutant
Multimer made up of 8 Mutant L proteins. The aggregation here changed a lot, having a circular conformation, but with the 8th monomer at the center, and the hydrophylic tails folded inwards at the end
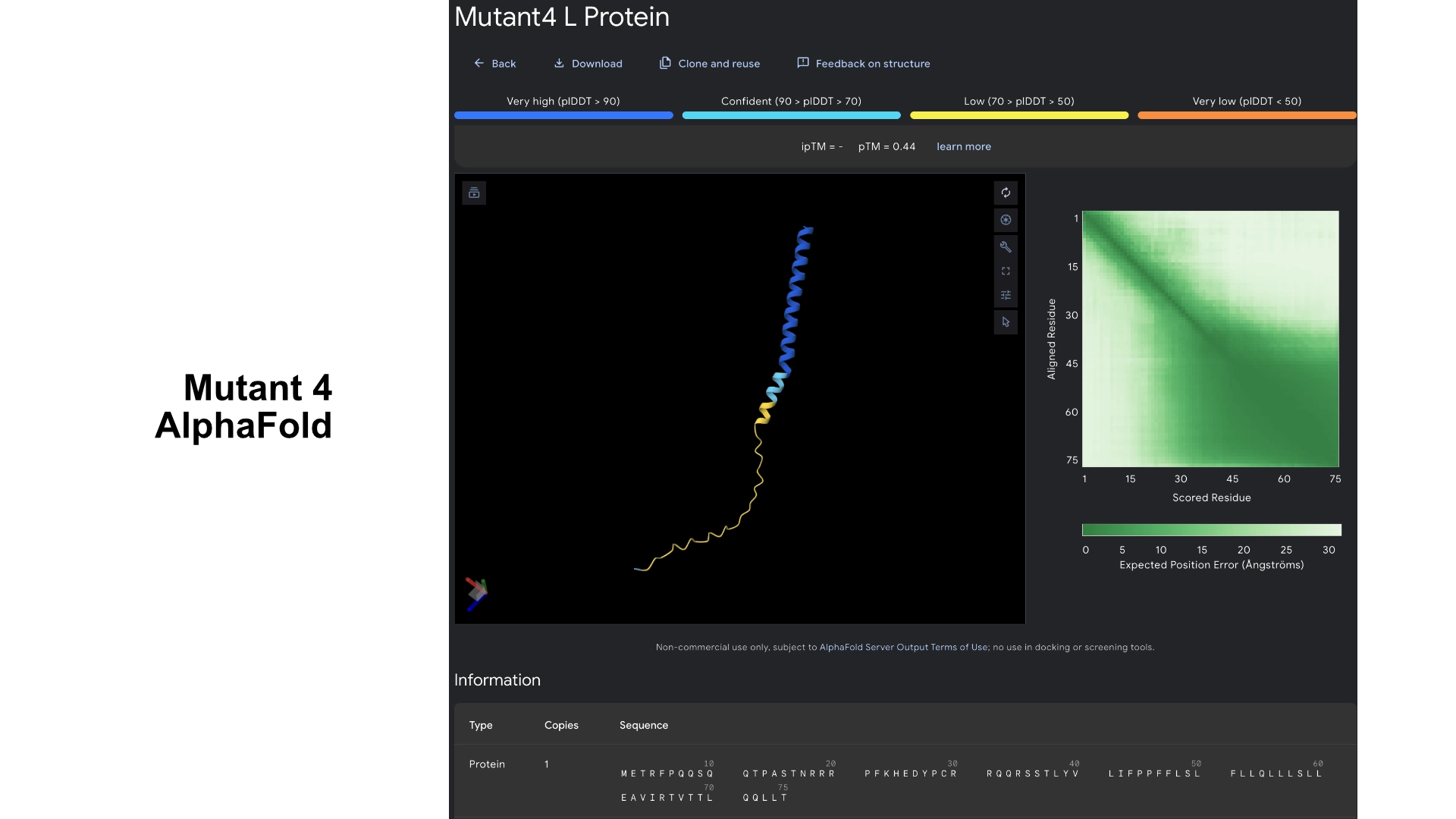
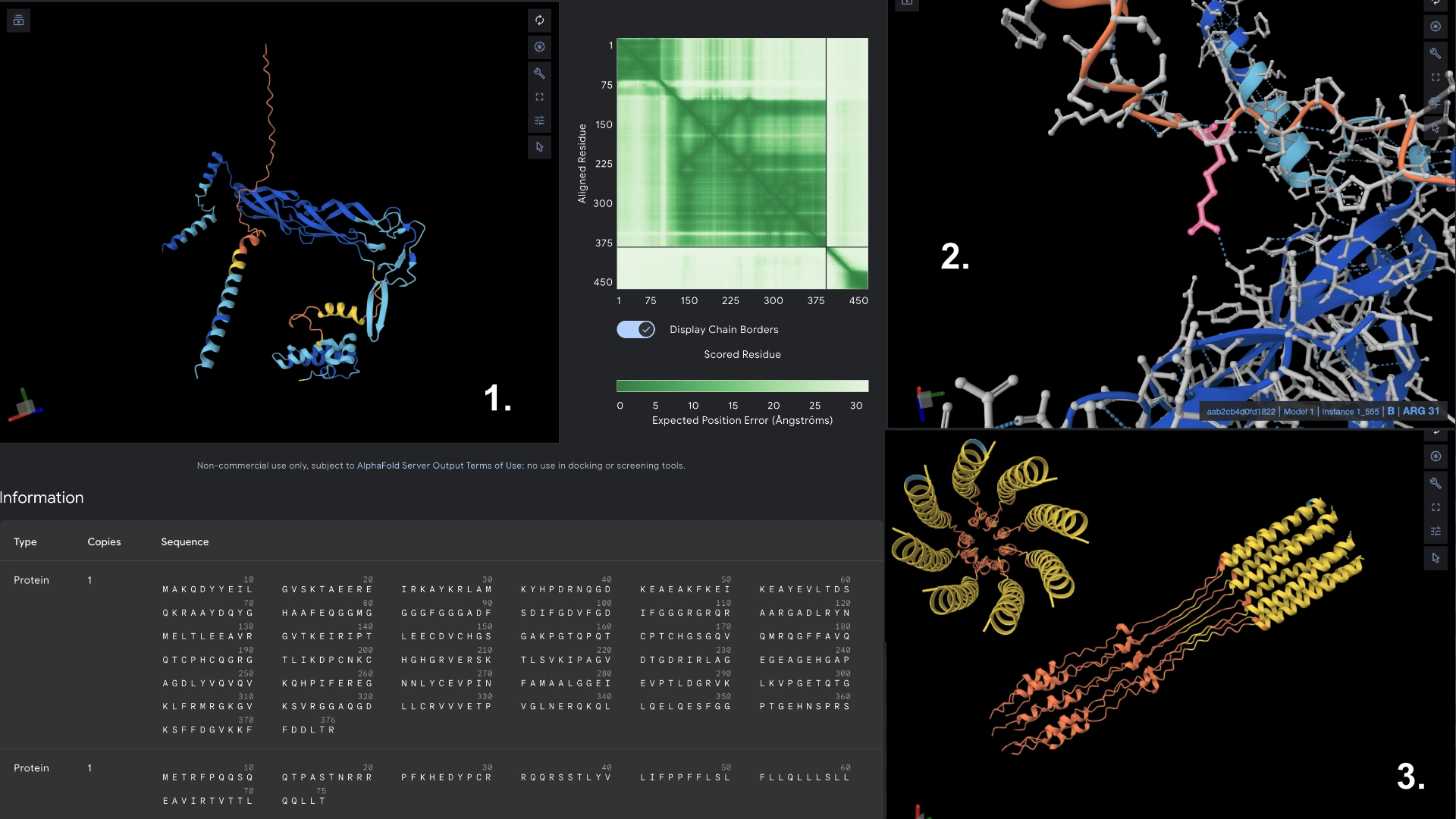
Mutant 4
Added the known positive mutations for lysis from experimental data to mutant 3 (no changes to hydrophylic zone)
For this version I thought that maybe if the transmembrane region has a mix of better residues for Lysis (from experimental sheet) combined with better structure from best scored residues in the less conserved areas it could have an overall benefit for Lysis and anchoring to the membrane.
It was interesting to see that most changes to residues in the transmembrane region in the experimental data resulted in negative impact on lysis, and that the residues that came back positive for lysis in experimental data don’t necessarily correspond to the structural scoring of the LLR
Redered interaction with DnaJ
Detail where we can see 1 bond to DnaJ on the residue 31 of Mutant 4
Multimer made up of 8 Mutant L proteins. The aggregation here changed even more, uncoilig some of the transmembrane portions. It was interesting to understand how mutating a protein might not affect it’s individual folding but can affect a lot its interactions with other proteins, and the low tolerance of the transmembrane zone to mutations.
It was interesting to observe that in the visualization this version formed a small helix segment in the hydrophilic region
Redered interaction with DnaJ
Detail where we can see no bonds between DnaJ and L protein Mutant
Multimer made up of 8 Mutant L proteins. The aggregation seems close to what would be expected of wild type L Protein, although the monomers bend slightly outwards
Week 6 HW: Genetic Circuits Part1
Assignment: DNA Assembly
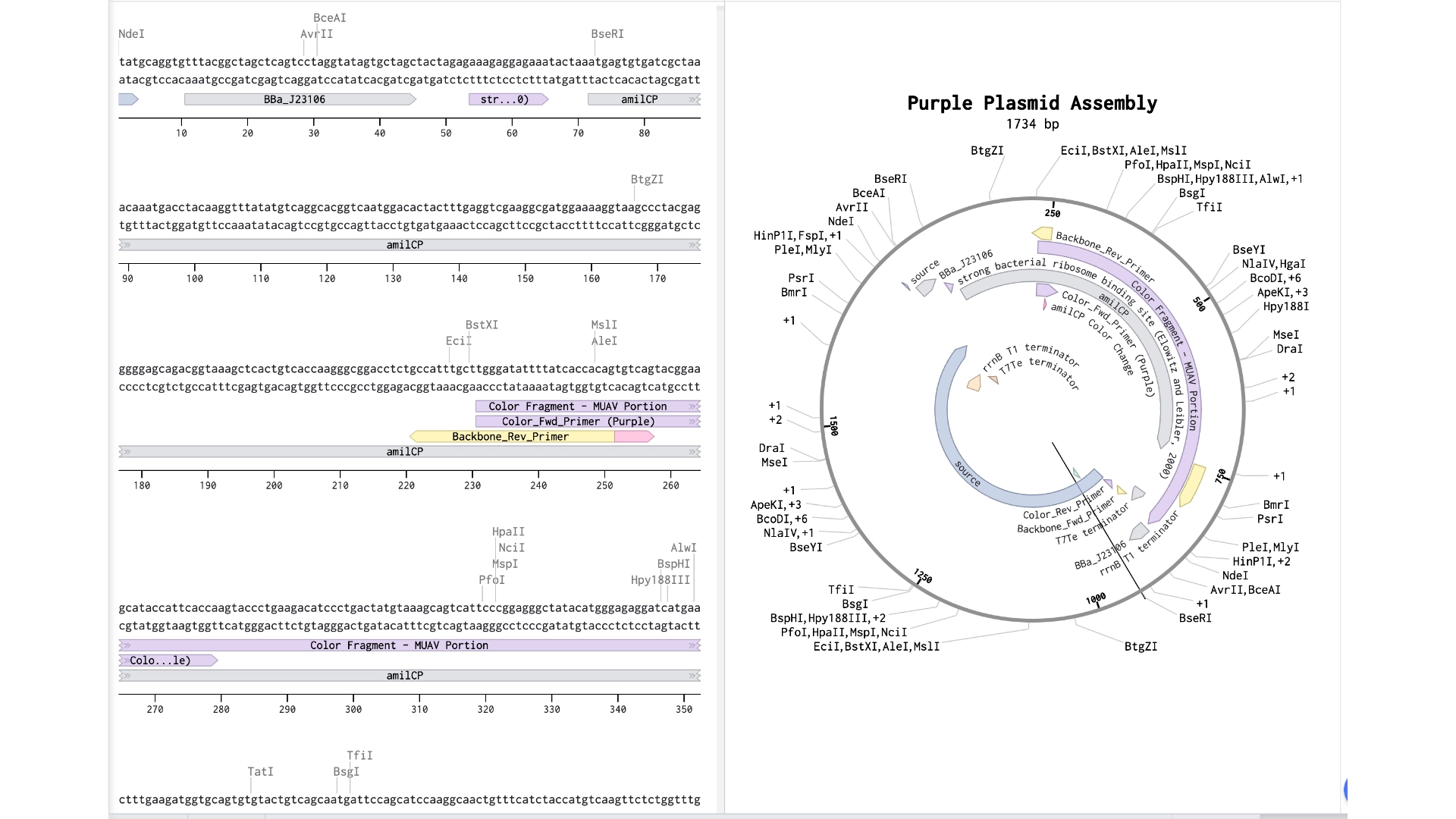
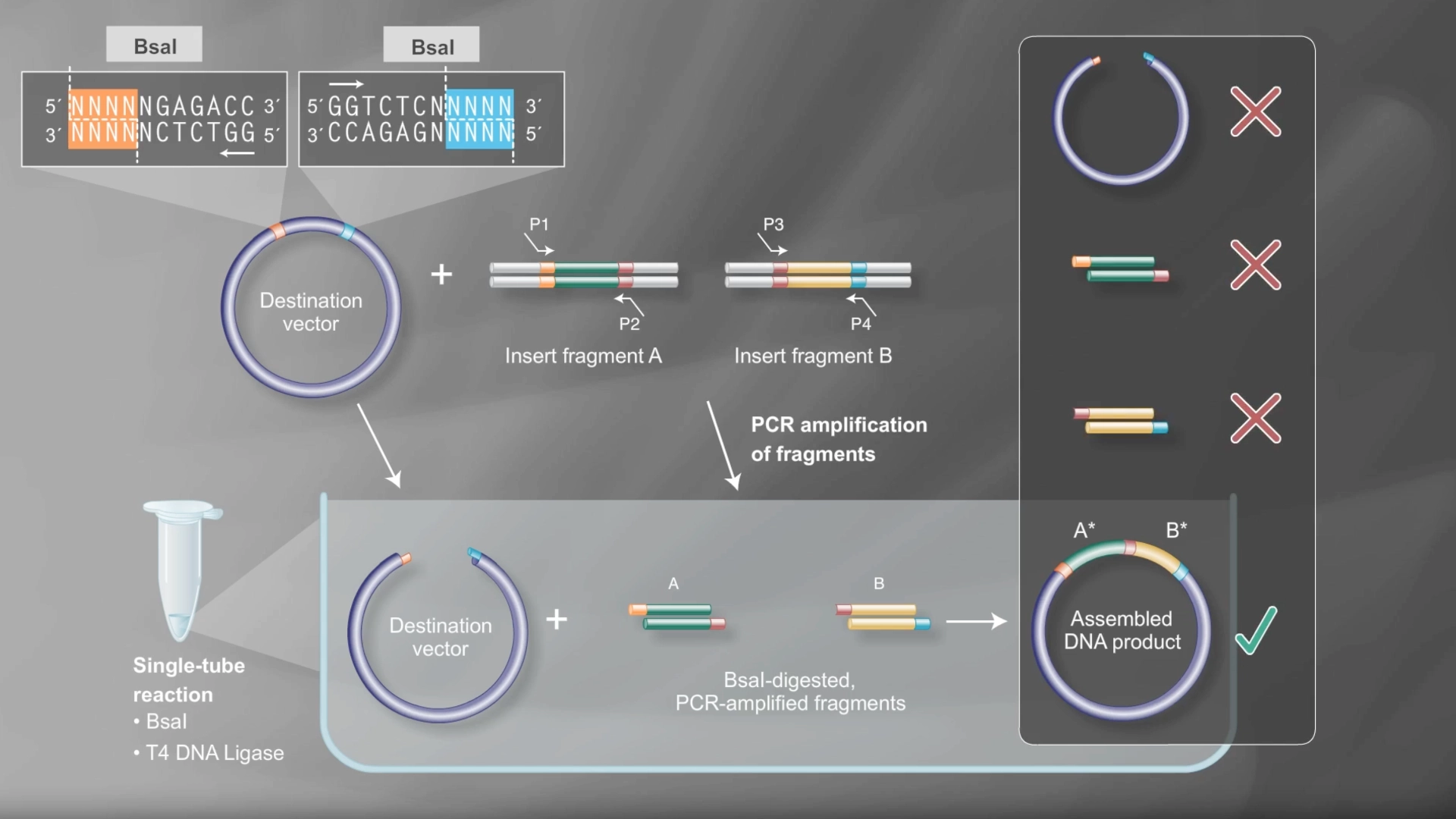
Documentation
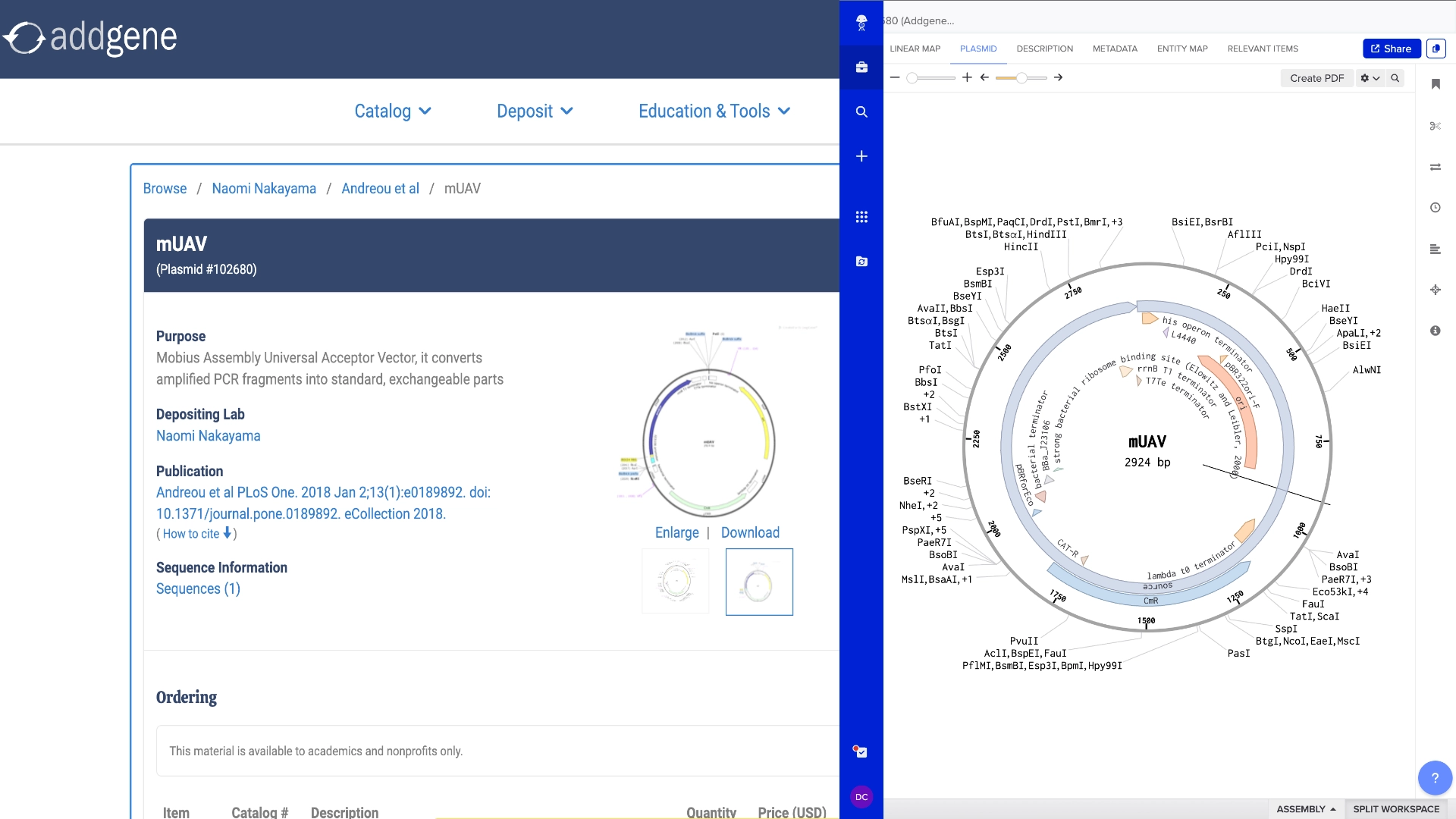
In order to model a Golden Gate assembly on Benchling I first got a mUAV plasmid backbone from Addgene
Then created a new linear DNA sequence by copying the amilCP gene from the HTGAA Benchling
Both fragments already had a Type IIS enzyme cut site in the right zones for assembly, using BsaI
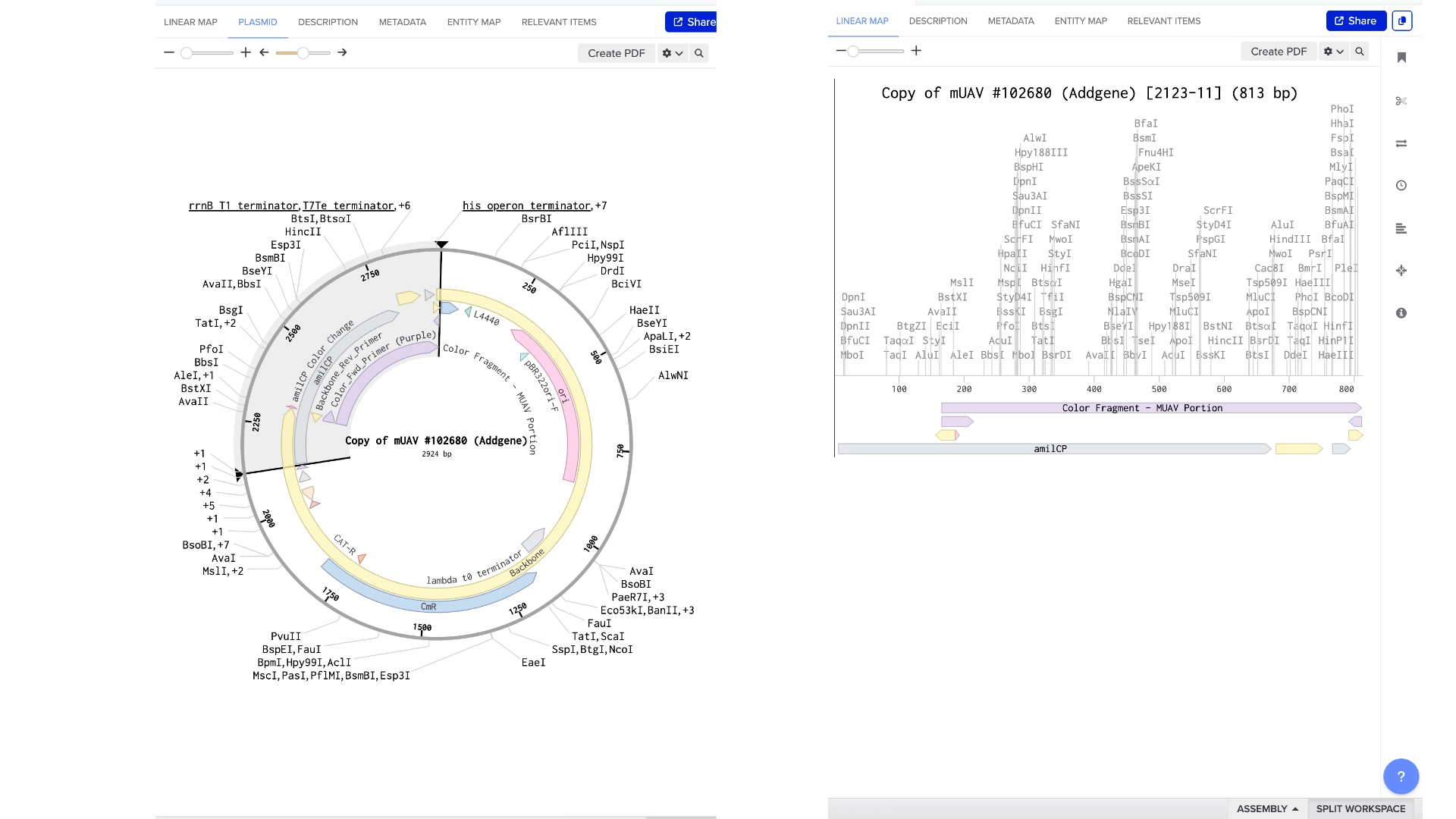
Created a Golden Gate assembly, using the mUAV as backbone and the amilCP (Purple) as insert
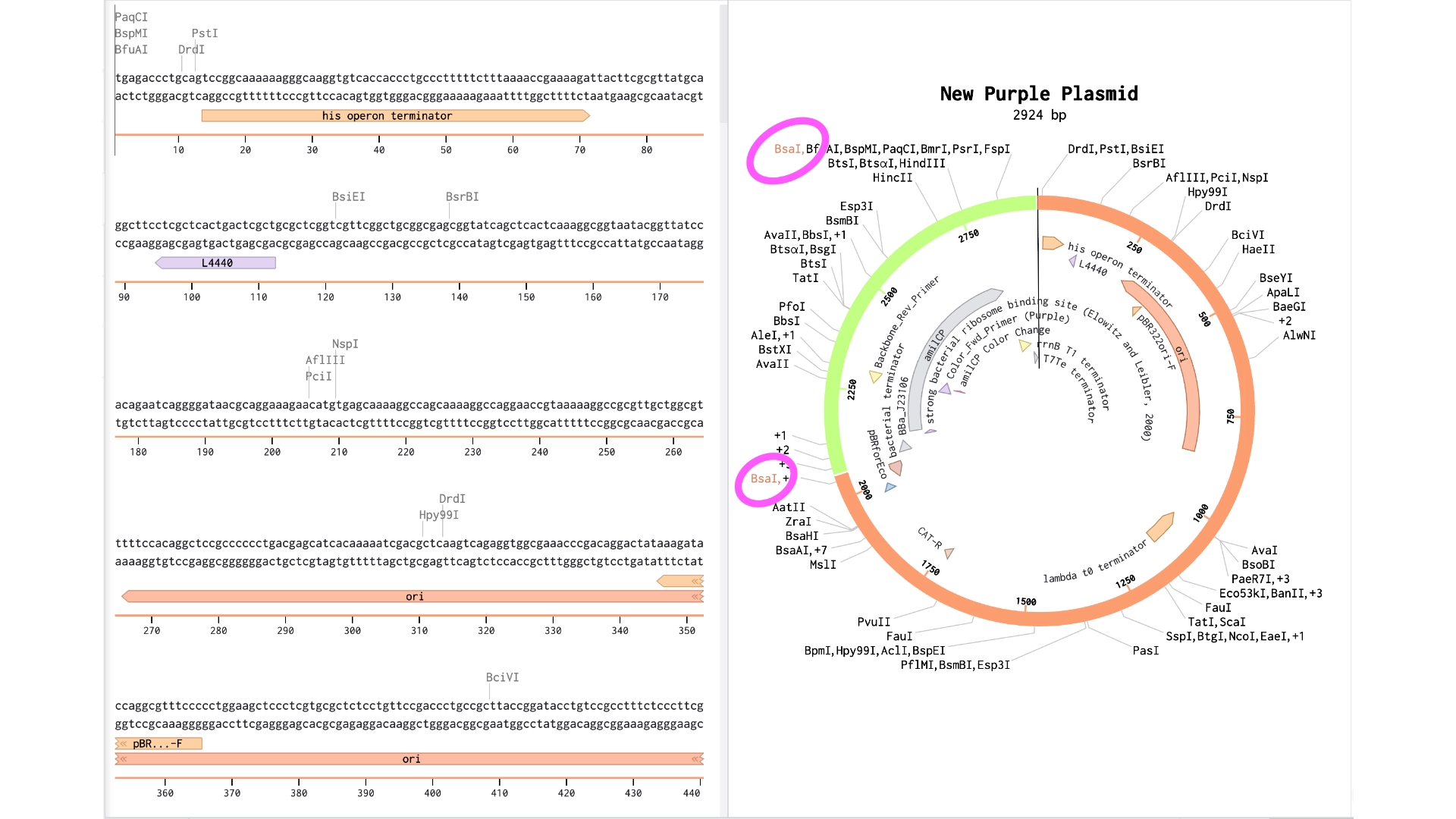
When the assembly was put together, I realized that it had been put together the wrong way— because of the orientation of the BsaI enzymes, the backbone being used was the 867 bp long instead of 2057 bp long
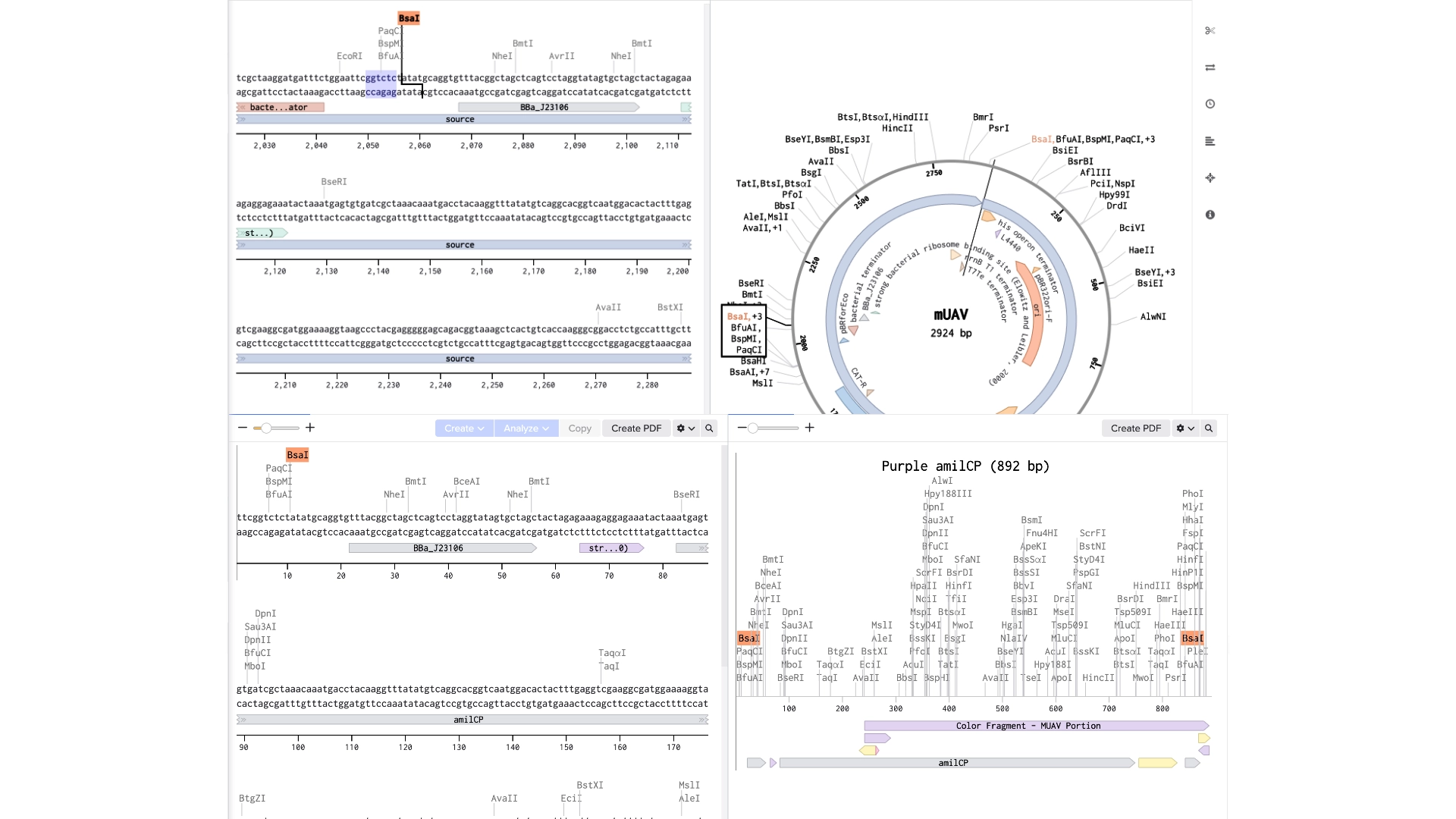
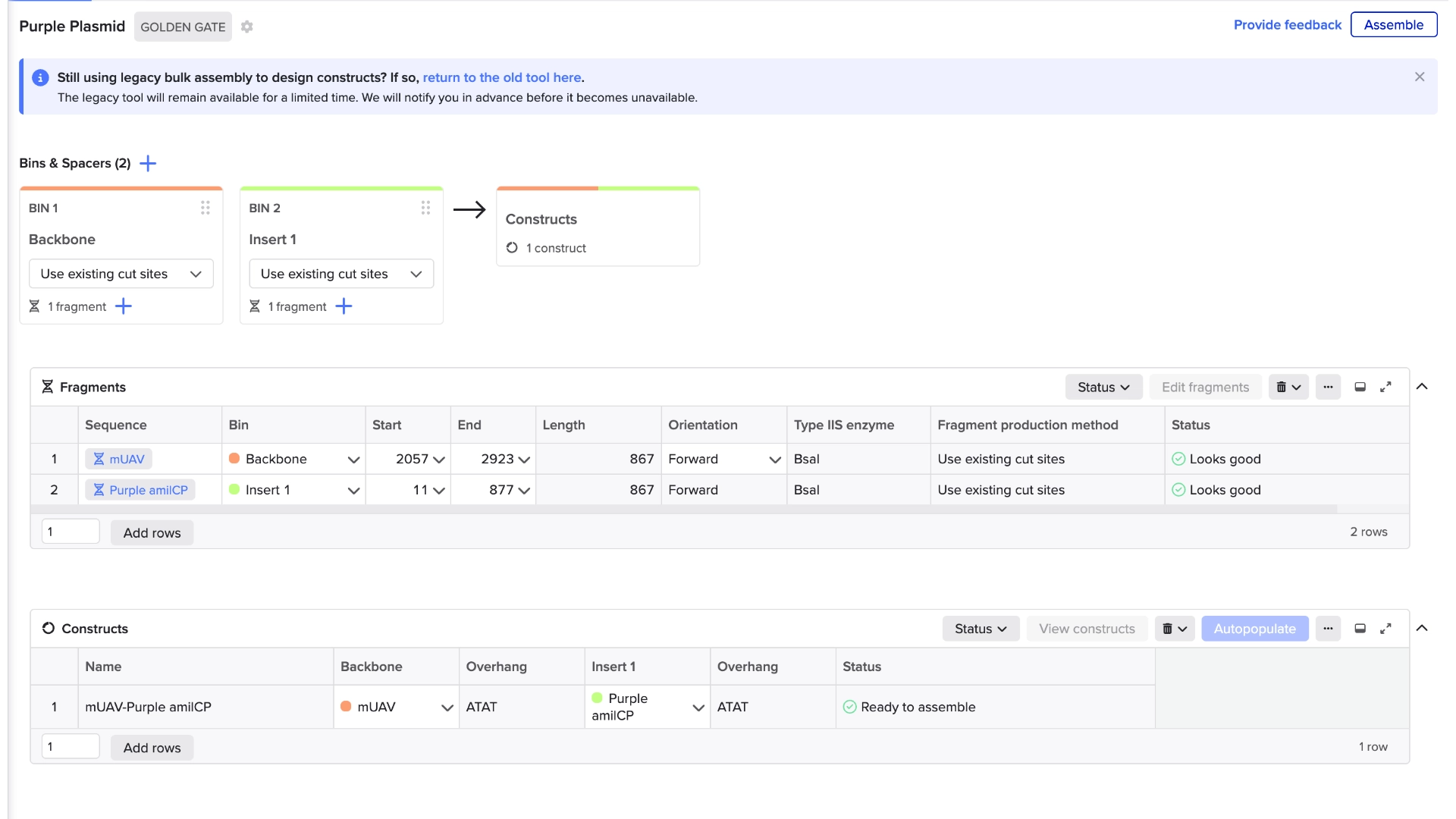
Created a new Golden Gate assembly and was able to output the plasmid in the correct order, although the recognition sites of BsaI enzymes of the backbone stayed inside the final plasmid which wasn’t supposed and would make a real golden gate assembly not work properly
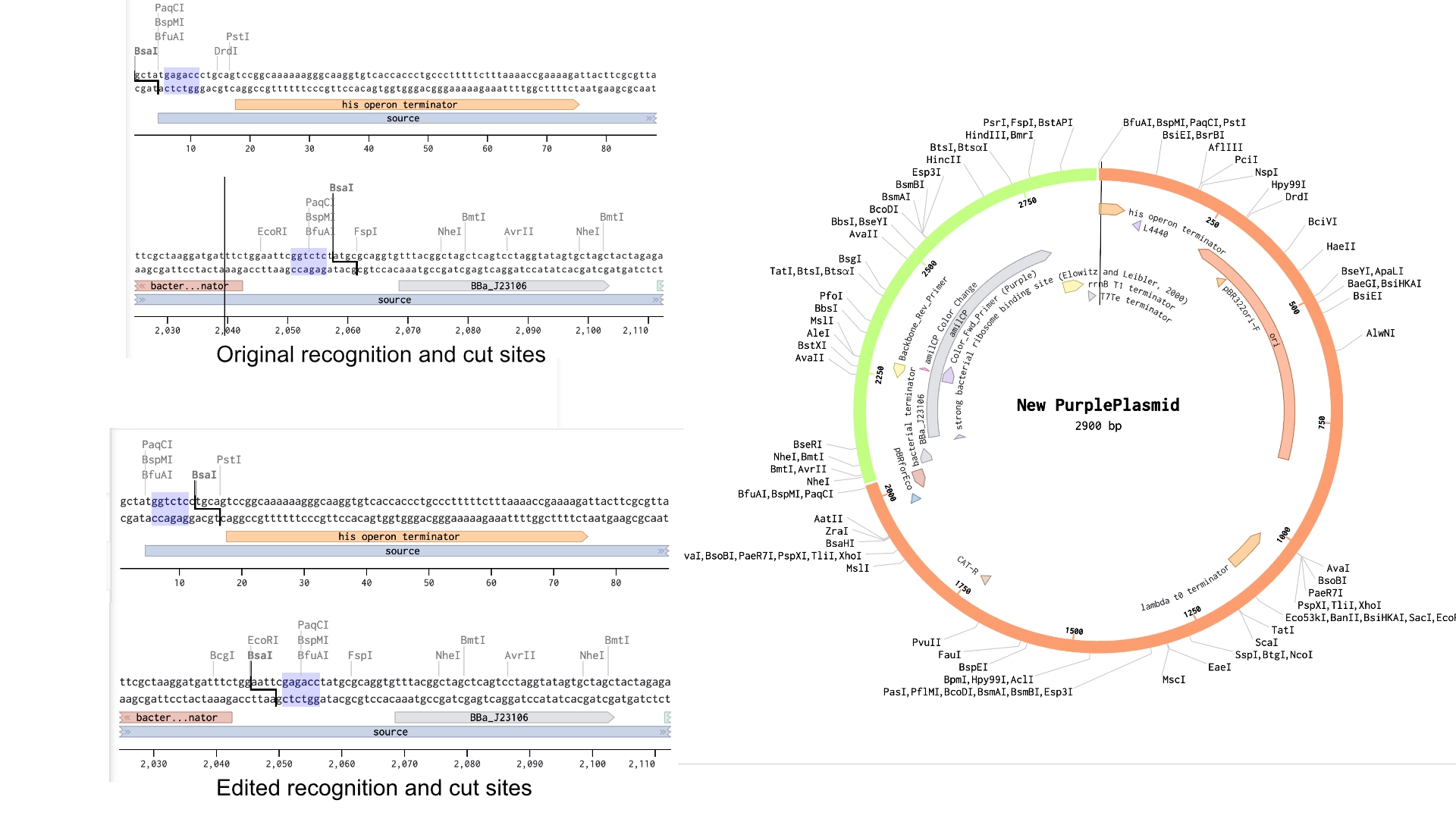
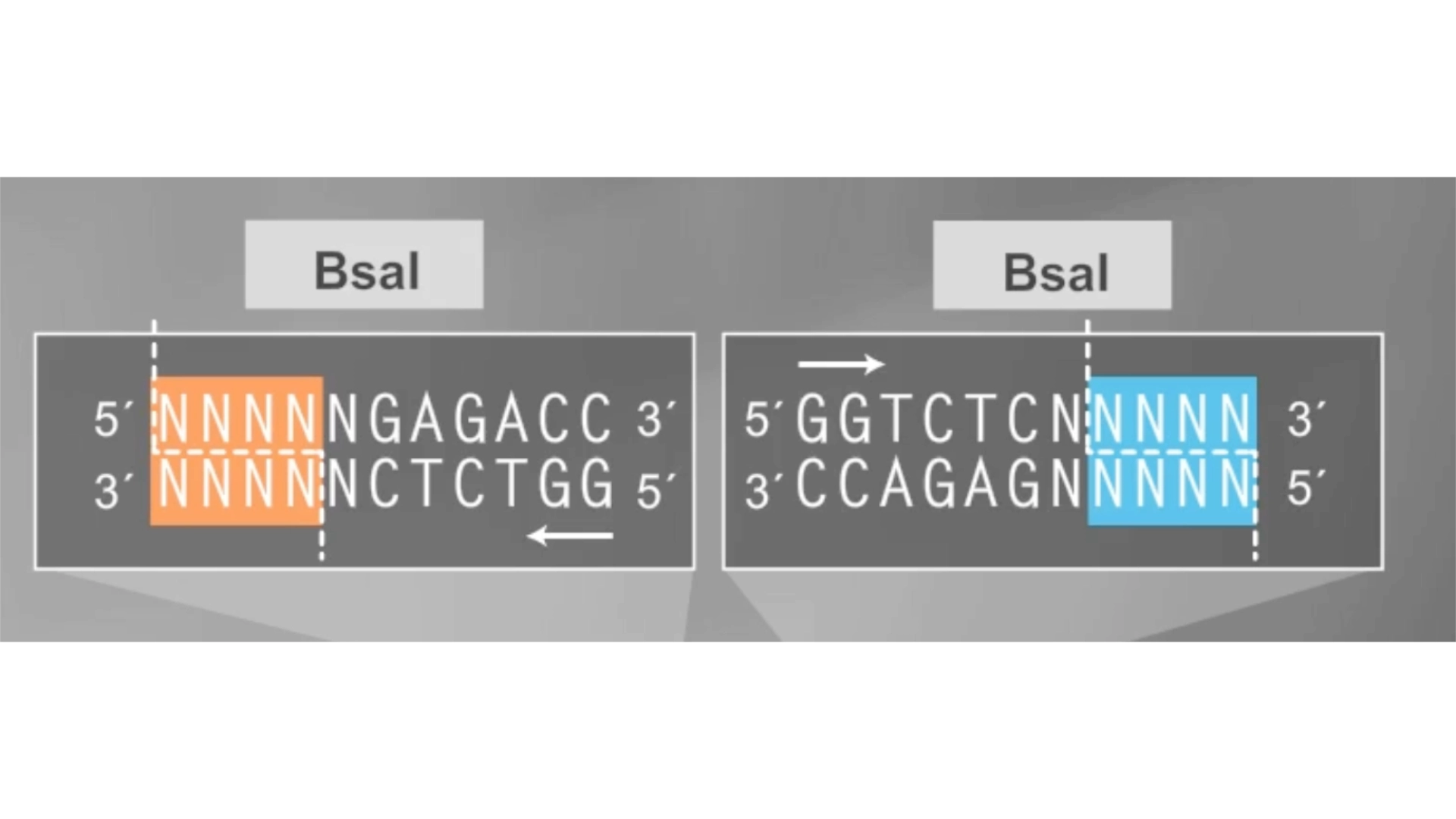
To solve this the solution would be to create primers for the BsaI enzymes to be sure they pointed in the right direction and that their recognition sites would be eliminated with the process
In the context of this exercise I manually edited the recognition sites of the BsaI enzymes in order to invert their cut site direction and was finally able to get a correct assembly
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Some of the key components of the Master Mix are: Phusion High-Fidelity DNA Polymerase (1 unit/50 µl) which are thermostable DNA polymerase with proofreading activity to ensure minimal error rate; dNTPs (200 µM of each) — deoxynucleotide building blocks that allow the synthesizing of DNA strands and HF Reaction Buffer — an optimized buffer which ensures good polymerase activity, including MgCl₂ (magnesium ions, 1.5 mM) – essential cofactor for DNA polymerase and other salts and ions to maintain proper enzyme activity and fidelity. Optionally, if the desired segments have high CG content, DMSO (dimethyl sulfoxide) can be used — is often provided separately and can be added to improve amplification of GC-rich templates
2. What are some factors that determine primer annealing temperature during PCR?
The primary factor that influences the annealing temp. is the melting temperature of the primers — temperature at which 50% of the primer-template duplex dissociates— and which, if met correctly, allows for optimal interaction between DNA and primers resulting in correct binding. This Tm (melting temperature) is heavily influenced by the CG to AT content— high CG content, which bind more strongly to each other result in higher TMs—, also, the longer the primers are the higher Tm is.
In addition to these, the concentration of salts in the master mix also influences the temperature to which the reaction can be heated up.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests.
My mental depiction of these two processes is that PCR is used to identify one target sequence and multiplicate it through synthesizing new segments that are used to synthesize even more, with this process you end up with millions or billions of copies of the original target. While restriction enzyme digests are more of an identify and cut out process, through which you extract the desired segment from the remaining DNA, you don’t end up with more copies of the target segment.
In terms of protocol:
PCR amplifies a specific DNA region using primers and a DNA polymerase, through temperature cycling between the following steps
Denaturation (~95°C)
Double-stranded DNA separates.
Annealing (~50–65°C)
Primers bind to complementary regions flanking the target DNA.
Extension (~72°C)
DNA polymerase synthesizes new DNA strands.
These steps are repeated 25–35 cycles, producing millions of copies of the target fragment.
Restriction Enzyme Digests
Restriction enzymes are endonucleases that cut DNA at specific recognition sequences (usually palindromic).
By using DNA, the specific restriction enzyme, an appropriate buffer and incubating (usually 37°C), DNA is cut into fragments based on enzyme’s recognition sites. This results in fragments dependent on restriction sites of the enzymes used (might produce unintended fragments if restriction sites repeat). Generates sticky ends or blunt ends useful for cloning.
In terms of use, PCR is more useful when you need to isolate and amplify a certain gene from a limited DNA source, ensuring that you have enough quantity and purity of the desired segment, small edits can also be added by creating primers that induce a small “error” to the copying. Restriction Enzyme Digests are preferable when the fragment already exists and you want clean, mutation-free cuts with compatible ends for cloning or to analyze DNA fragments (for gel electrophoresis)
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
You must ensure the ends of the different sequences you want to assemble through Gibson cloning are homologous matches, by using primers that overlap— the primers for the different segments must have an overlapping site of around 20-40 bp that allow for the formation of complementary sticky ends during assembly. Or, in case of restriction digests, the fragments that are cut must already have zones that are homologous, which is harder because in this method you don’t have a way to induce these sites like in PCR.
5. How does the plasmid DNA enter the E. coli cells during transformation?
During transformation the objective is to be able to disrupt the E.coli’s cell wall just enough for the plasmid to be able to enter and then allow them to return to their natural form. This can be done through heat shock or electroporation that induces formation of reversible aqueous pores through the creating of an external electric field causes a transmembrane potential difference.
6. Describe another assembly method in detail (such as Golden Gate Assembly)
The Golden Gate Assembly relies on two Type IIS Restriction Enzymes that cut DNA outside their recognition sites, allowing for the use of unique and custom 4bp long sticky overhangs, and therefore, a really precise assembly of multiple small sequences at the same time. Having the constraint of ensure the used sequences are “domesticated”— free of internal Type IIS recognition sites.
The process goes as follows:
Creation of PCR primers that contain the Type IIS enzyme’s recognition sites and the unique 4bp overhangs in the right order for each of the segments.
(Screenshot from Golden Gate Assembly video by New England Biolabs)
Restriction digestion, where the Type IIS enzyme cuts all fragments and the vector, producing the designed overhangs. Because the cut site is outside the recognition sequence, the recognition sequence itself is removed during assembly.
DNA ligase joins fragments that have matching overhangs. Since the restriction sites are removed after ligation, the assembled DNA cannot be cut again, making the reaction efficient.
Cycling digestion and ligation is performed through alternating temperatures for restriction digestion and ligation. This increases assembly efficiency because incorrectly assembled fragments can be cut again until the correct construct forms.
(Screenshot from Golden Gate Assembly video by New England Biolabs)
6.2 Model this assembly method with Benchling or Asimov Kernel!
Final result of assembling a mUAV backbone with an insert of purple amilCP
Simulation
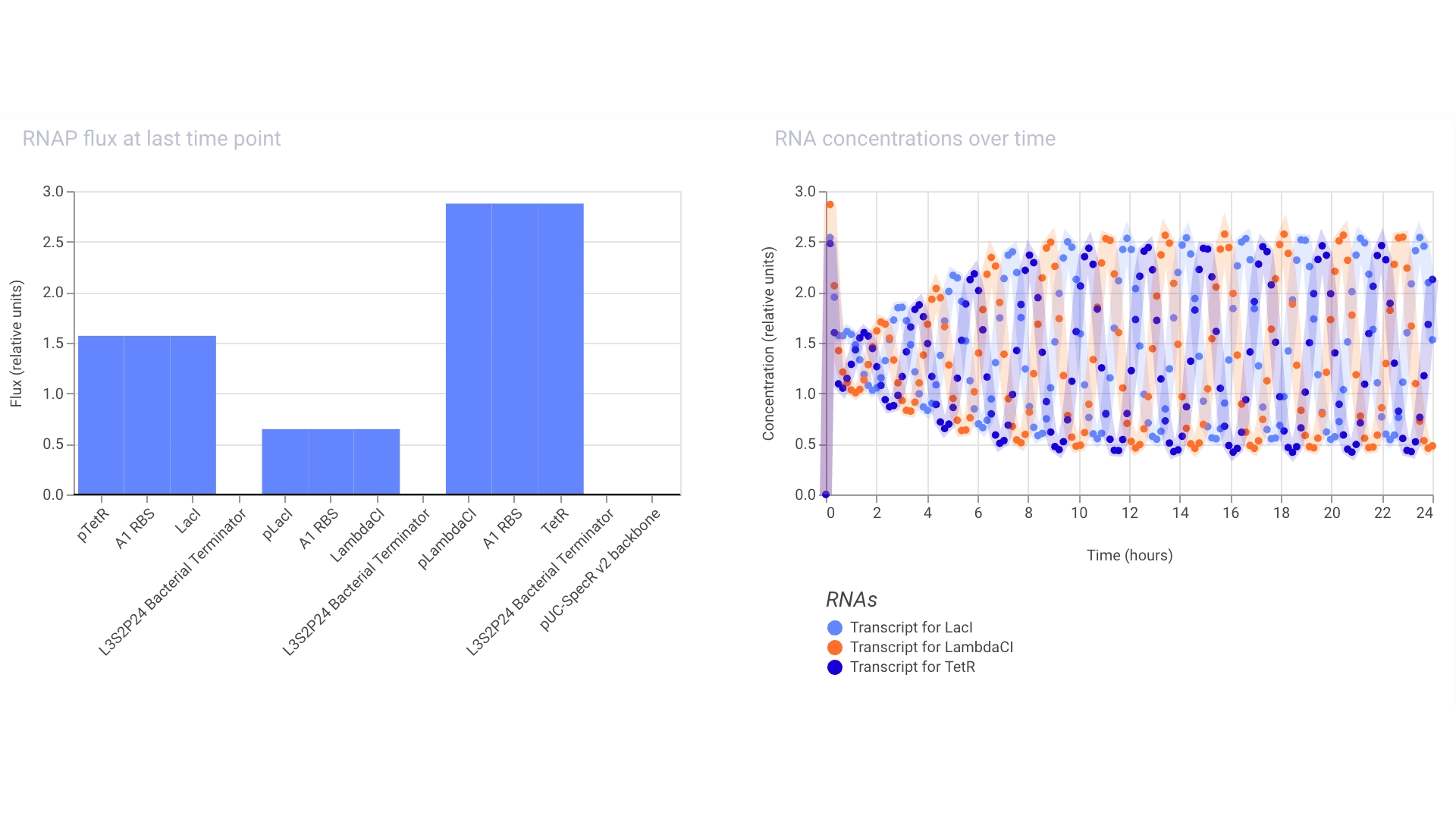
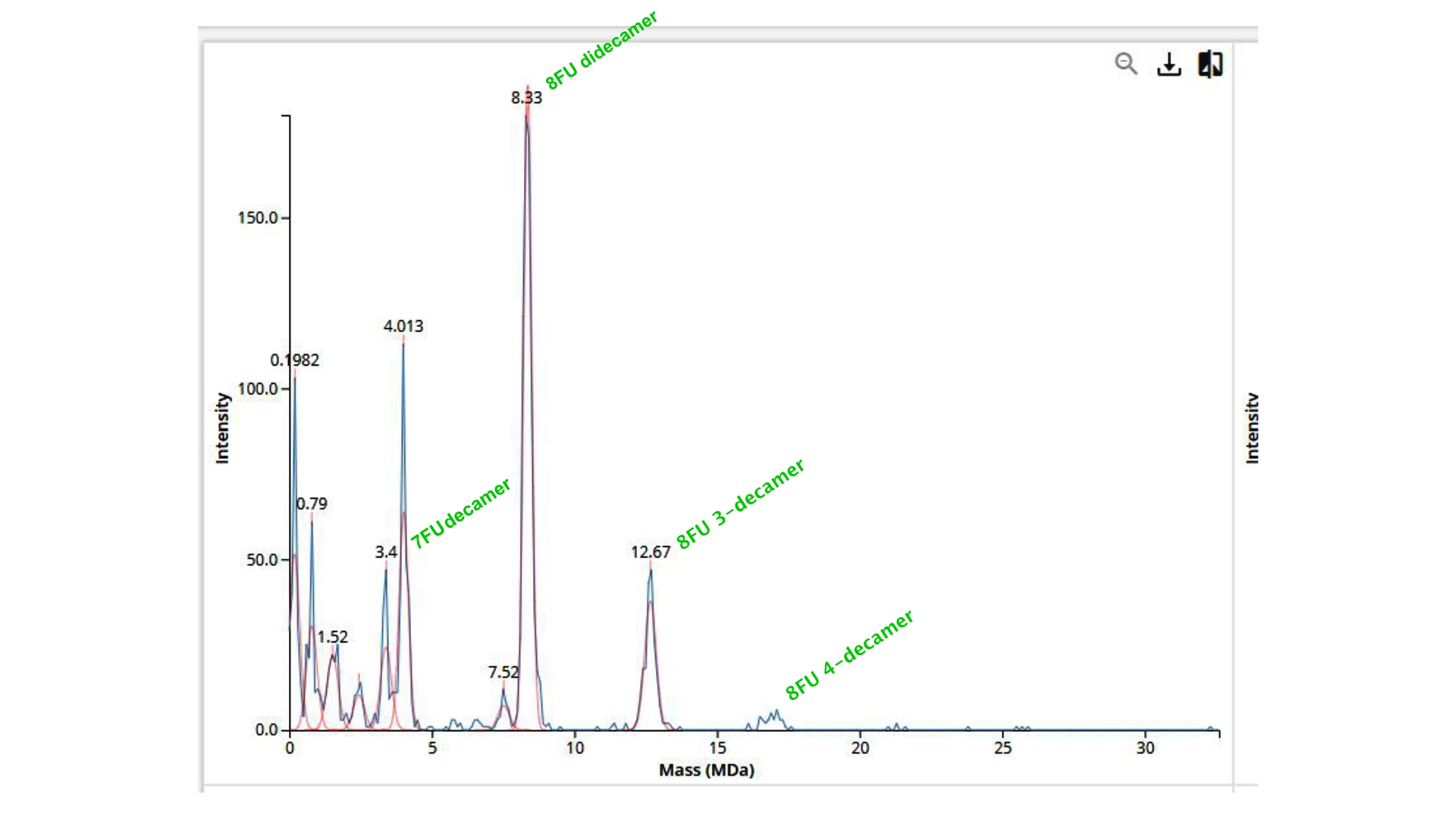
Proteins expressed by the Repressilator seem to alternate in the expected way!
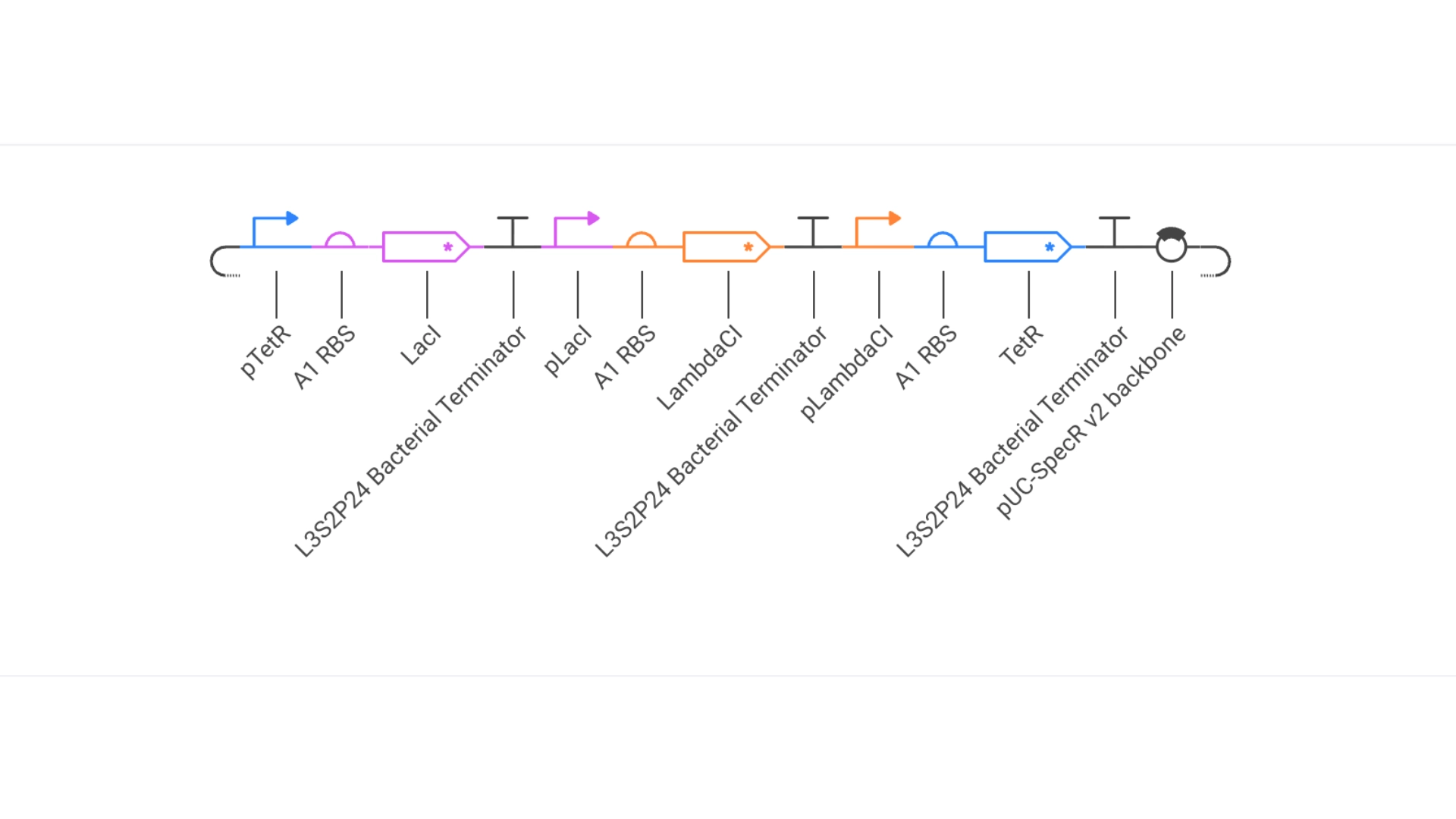
Construct #1:
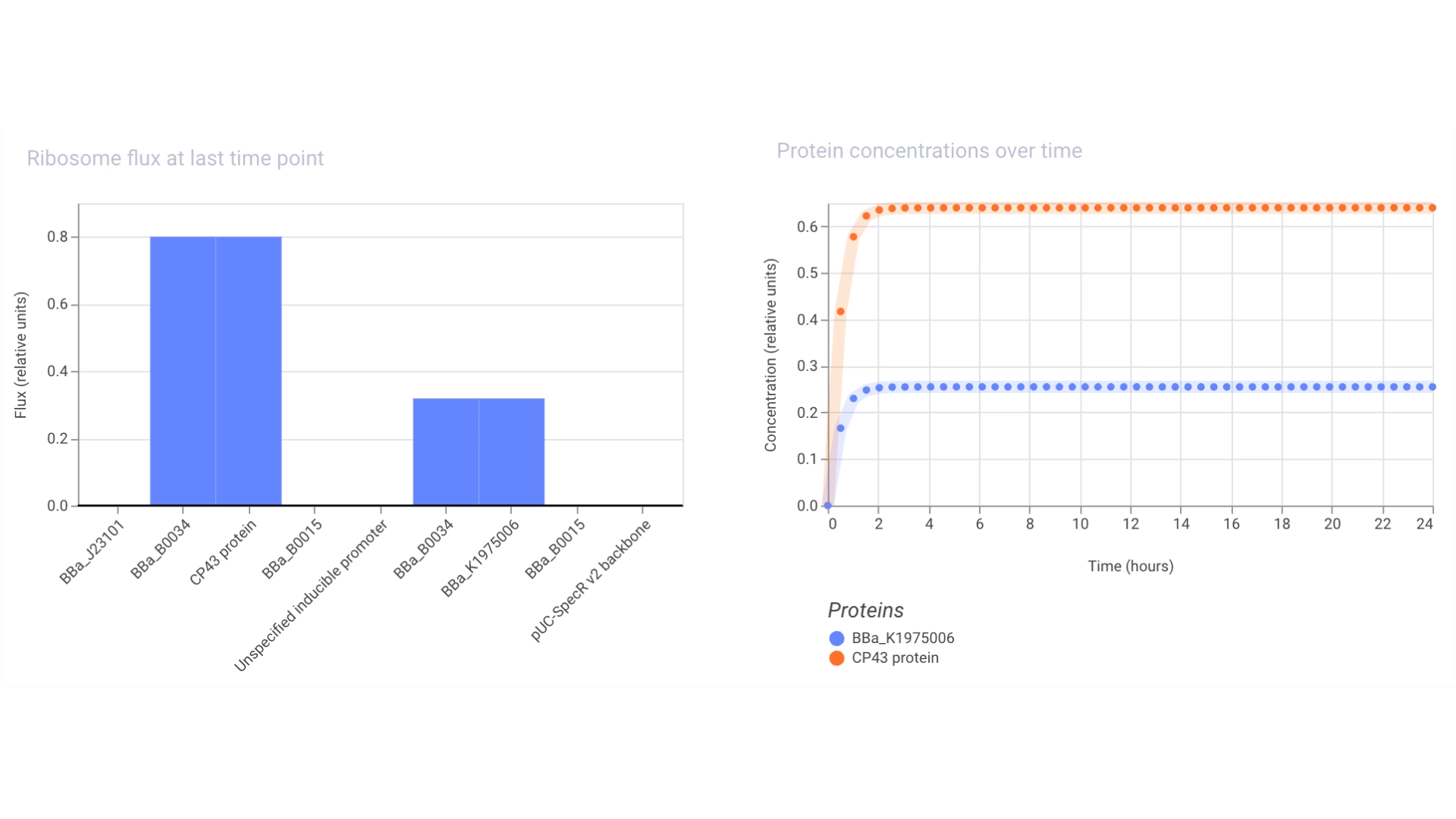
Objective: Create a simple construct that expresses the chlorophyll binding protein CP43
For this construct I created a new part — a CDS part coding for the CP43 protein
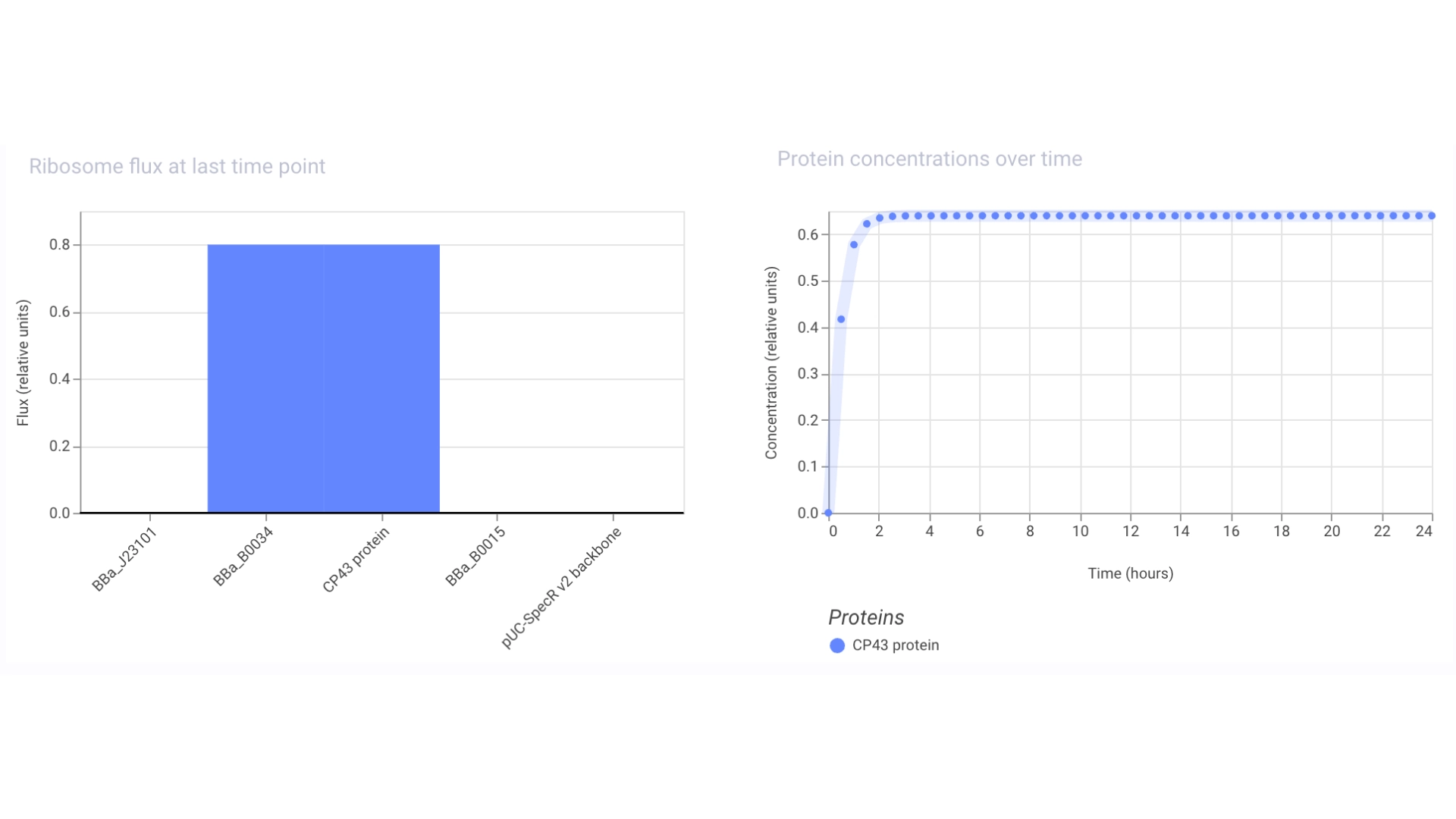
Then assembled the circuit using a strong promoter, RBS, terminator and pUC-SpecR v2 backbone
At first only the RNA for the protein was being expressed but not translated into the actual protein, then I found out that the construct had been created for Chinese hamster instead of E. coli
Objective: Create a simple construct that expresses a Red Fluorescent Protein
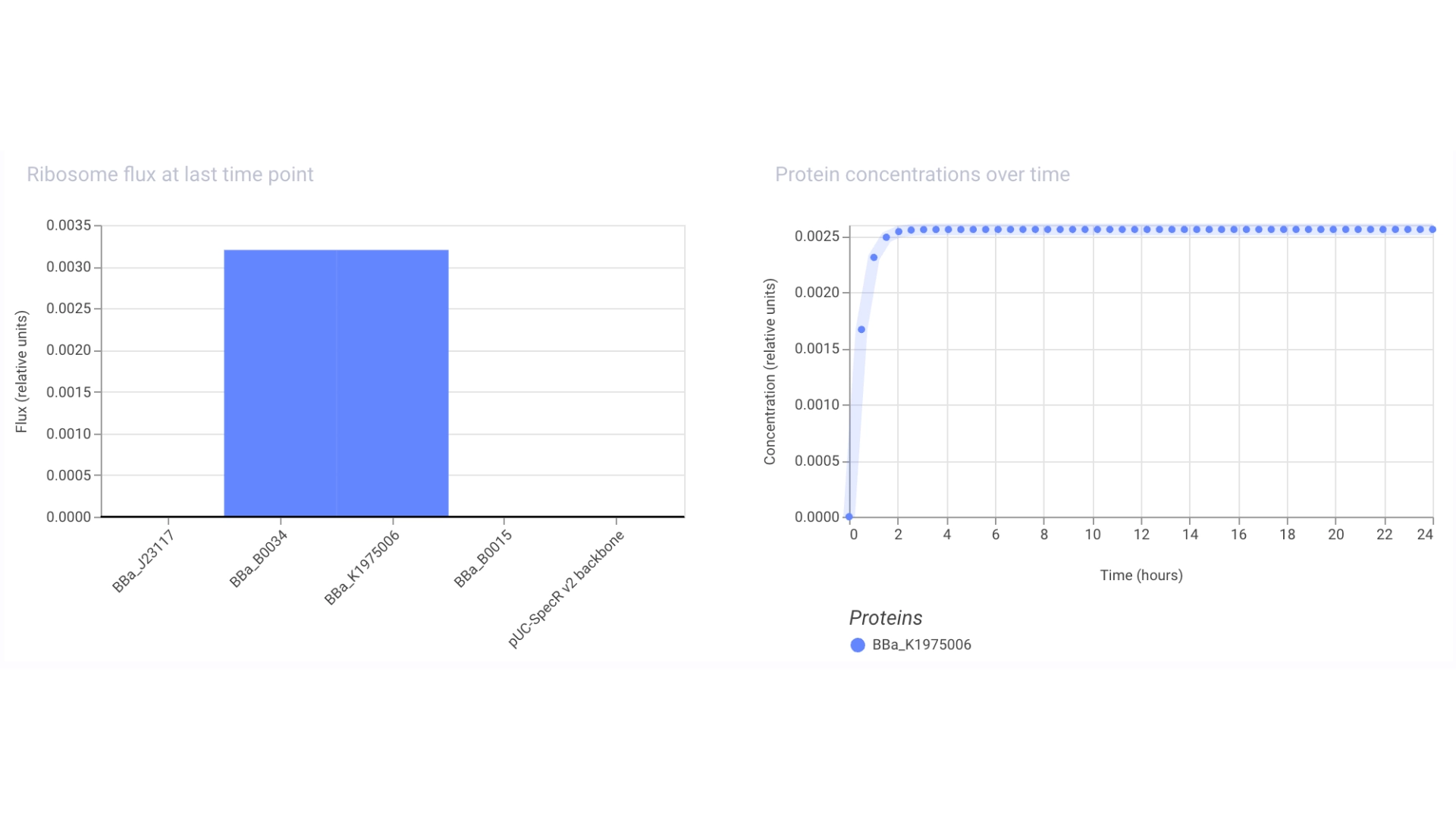
For this I had to change the RBS part I was using for the CP43 protein, presumably because the CP43 is a CDS (-Start,+Stop) and the RFP is CDS (+Start,+Stop)
The objective of this construct would be to create a circuit that only expressed a fluorescent protein if the CP43 folded correctly— as it is a lipoprotein it needs an environment with chlorophyll which acts as folding co-factor— and to achieve this an inducible promoter could be designed to be induced by the folded CP43 protein. This way we would have a construct that both expresses the needed protein and gives feedback on the success of the reaction.
For this exercise I just used an Unspecified Inducible Promoter part
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Traditional genetic circuits based on digital logic are primarily limited by low complexity of operations and needing an expressive amount of metabolic effort from cells. IANNs have the advantage of using analog logic. By leveraging non-linear functions and using continuous input ranges, instead of Boolean circuits that require discrete thresholds, neuromorphic systems allow for more nuanced decision-making, while reducing the metabolic burden since fewer components are needed.
Intracellular Neural Networks take advantage of the type of chaotic organization that already happens inside a cell, instead of trying to impose a translation of logic into more readable inputs and outputs like digital logic does, providing access to more complexity, scalability and adaptability to different environments. The main drawback of neuromorphic systems is noise, like any biologic system, but this can be mitigated through the aggregation of information across a population of cells which diminishes the intrinsic noise of cellular environments.
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
The most interesting application for IANNs that I see is as preventive therapy that can have an intravenous delivery and regulate the immune system in response to really early and small changes in cells, only activating the immunotherapy in the right tissue when inputs meet certain specific criteria. In this type of approach different circuits with different targets can be introduced into the body and each of them have a small numbers of inputs, lowering the complexity needed for each different circuit but creating a system that can analyze and potentially respond to a wide variety of targets. So, with each circuit having two or three biomarker inputs that only trigger a immunotherapy response if specific non-linear patterns are met, a full “body scan” can be performed with really high precision.
3. Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
The diagram I drew represents an XOR function where the output is high when the inputs have “opposite” values. The output is only high when X1 is high and x2 is low or when x2 is high and x1 is low. If x1 and x2 are both high or low the output is low
Assignment Part 2: Fungal Materials
1. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials range from textiles like leather — which fungi are really good at mimicking since the formation of mycelium creates really strong and flexible networks— to more structural elements like building block for walls— mycelium has the ability to grow in many types of substrates like grain or straw, and while growing it aggregates these fibers or grains into solid chunks that can be later dried. This creates a strong material which can support some considerable charge and isn’t brittle, also having really good insolation qualities both for sound and heat.
2. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Besides the possibilities for materials derived from fungi, another really interesting scope through which I’m interested in fungi is the possibility of therapeutic solutions by engeneering human microbiomes with adaptogenic fungi that could regulate our imune systems through the amazing methabolic pathways they posess, which bacteria don’t.
Week 9 HW: Cell Free Systems
General homework questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production cell production.
Cell-free synthesis excells in enabeling open system manipulation by allowing the direct addition of additives; rapid experimentation and high-throughput since the transformation and cultivation steps can be skiped, moving directly from PCR to protein production; and tolerance to unnatural aminoacids via custom tRNA.
In the case of my final project, cell-free synthesis is more benificial since the first exeriments I would like to perform are based in the synthesis and analisys of chlorophyll-binding light-harvesting proteins, which depend on the presence of chlorophyll to fold properly. If cell-free synthesis is used, chlorophyll can be added as an additive to enable correct folding in a single reaction, otherwise, using cultivation of E.coli, the apoprotein form of the LHP would have to be purified and only after react with chlorophyll.
Another case that I see cell-free synthesis being really usefull is in polymer design, where the possibilities are much more vast and interesting if using de novo amino acids. Cell-free synthesis allows to bipass the in vivo systems that possess established translational machinery adapted for natural occuring amino acids and tRNA.
2. Describe the main components of a cell-free expression system and explain the role of each component.
The main components include:
Genetic template either in linear segments or plasmid form
Cell Extract (Lysate) which contains necessary transcriptional/translational machinery: ribosomes, aminoacyl-tRNA synthetases, translation factors, and enzymes. This can be either whole cell extract, which also contain metabolic enzymes and other cellular components typically from E.coli or can be a PURE system which only contains the needed purified machinery.
Buffer which maintains optimal PH for enzymatic activity and reaction stability
ATP that provides energy; GTP, CTP, and UTP contribute to RNA synthesis.
Nucleotides for transcription and Amino acids for translation
Co-folding factors if needed to ensure proper folding of proteins
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is essential in cell-free systems because ATP is rapidly consumed during transcription and translation, and without replenishment the reaction quickly stops. One way to maintain ATP levels is to add creatine kinase and phosphocreatine, which regenerate ATP from ADP and provide a continuous energy supply during the experiment.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free systems (e.g., E. coli) are fast, cost-effective, and high-yielding, and allow direct control over reaction conditions, but they lack post-translational modifications. Eukaryotic systems are more complex and lower-yielding, but enable proper folding, disulfide bond formation, and post-translational modifications such as glycosylation.
A prokaryotic system can be used to express, e.g., chlorophyll-binding proteins such as CP43 or PcbA, since chlorophyll can be added directly to the reaction as a folding cofactor, promoting correct assembly of these hydrophobic proteins.
In contrast, a eukaryotic system is suitable for expressing, e.g., fungal lectins (glycoproteins), as their biological activity depends on correct folding, disulfide bond formation, and glycosylation. These modifications are essential for carbohydrate-binding function and are only supported in eukaryotic cell-free systems.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Using chlorophyll-binding membrane proteins such as CP43 or PcbA as example. To optimize their expression in a cell-free system, I would screen different reaction conditions while supplying a membrane-mimicking environment, such as detergent micelles, liposomes, or nanodiscs, so the hydrophobic transmembrane regions can insert properly instead of aggregating. Because these proteins also depend on chlorophyll as a folding cofactor, I would add chlorophyll directly into the reaction to promote correct folding and assembly.
The main challenges are poor solubility, aggregation, misfolding, and low functional yield, since membrane proteins are highly hydrophobic. In the case of CP43 or PcbA, another challenge is that without chlorophyll the protein may not fold correctly or remain stable. To address this, I would optimize variables such as temperature, magnesium concentration, reaction time, membrane mimetics, and chlorophyll concentration, and then evaluate expression by SDS-PAGE/Western blot and functionality by measuring pigment binding or spectroscopic properties. This makes cell-free expression especially useful because the folding environment and cofactors can be controlled directly in one reaction.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Low yield can arise from degraded or low-concentration DNA, as well as from poorly designed constructs (e.g., weak promoter, inefficient RBS, incorrect spacing, or unfavorable codon usage). These issues reduce transcription and translation efficiency. This can be addressed by using high-quality DNA and optimizing the construct with a stronger promoter, improved RBS, and codon optimization.
Energy depletion or suboptimal reaction conditions, given that cell-free systems rapidly consume ATP and cofactors, which can limit protein synthesis. This can be improved by optimizing the energy regeneration system and adjusting parameters such as Mg concentration, temperature, and reaction time.
Protein misfolding or aggregation, especially for membrane or complex proteins, improper folding can reduce yield. This can be addressed by lowering the reaction temperature, adding chaperones, or including detergents, liposomes, or nanodiscs.
These questions where actually really helpful for my project because while searching for cell-free expression of LHPs and the difficulties of expressing membrane proteins in cell free systems I found WSCPs which are water soluble chlorophyll-binding proteins which provide a promising platform for organizing and stabilizing chlorophyll within aqueous materials, enabling experiments of how protein-mediated pigment structuring affects light-induced degradation processes
Homework question from Kate Adamala
1. Pick a function and describe it: Design a light-responsive synthetic minimal cell that uses chlorophyll to sense light and converts light exposure into a measurable biological signal. This system would function as a biohybrid light sensor or smart material for monitoring sunlight exposure.
What would your synthetic cell do? What is the input and what is the output?: Input: light; Output: production of a detectable reporter signal (e.g., fluorescence or color change) proportional to light exposure
Chlorophyll inside the synthetic cell absorbs light and generates a photochemical/redox signal (e.g., reactive oxygen species). This activates a redox-sensitive transcription factor, e.g. OxyR, which triggers Tx/Tl of a reporter protein such as GFP or an enzyme that produces a colored product.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?: Only partially. In a bulk reaction, the signal would diffuse and lack spatial organization. Encapsulation is important because it creates discrete microreactors, protects the components, and allows integration into materials such as coatings or hydrogels.
Could this function be realized by genetically modified natural cell?: Yes, but synthetic cells are preferable because they are non-living, easier to control, and more compatible with material applications. Natural cells introduce metabolism, growth, and variability that can interfere with stable sensing.
Describe the desired outcome of your synthetic cell operation: Upon illumination, synthetic cells produce a signal proportional to light exposure. When embedded in a material, they form a light-responsive layer that can map or record light intensity.
2. Design all components that would need to be part of your synthetic cell
What would the membrane be made of?: Phospholipids + cholesterol
What would you encapsulate inside?: E. coli cell-free Tx/Tl system; DNA encoding: OxyR (redox-sensitive transcription factor); reporter protein (e.g., GFP or luciferase); chlorophyll; amino acids, NTPs, ATP regeneration system
Which organism your Tx/Tl system will come from?: A bacterial (E. coli) cell-free system is sufficient, as no complex post-translational modifications are needed.
How will your synthetic cell communicate with the environment?: Light freely crosses the membrane and activates chlorophyll. Small molecules such as oxygen can diffuse through the membrane. If enhanced exchange is required, a membrane pore such as α-hemolysin (aHL) can be included.
3. Design all components that would need to be part of your synthetic cell
List all lipids and genes: Lipids— POPC; cholesterol. Genes— oxyR (E. coli); gfp or luciferase gene
How will you measure the function of your system?: Measure fluorescence (GFP) or luminescence (luciferase); Compare signal across different light intensities; Quantify output as a relation of exposure time
Homework question from Peter Nguyen
Develop a system that allows the transformation of almost any surface into a photographic support through the microencapsulation of a freeze-dried cell-free system embedded within a polymer matrix. This system would remain inert in the dry state and be activated upon exposure to moisture, initiating a series of biochemical and photochemical processes that generate light-sensitive chlorophyll domains.
Rather than synthesizing chlorophyll de novo, the system would incorporate pre-encapsulated chlorophyll or chlorophyll–protein complexes, stabilized within the material. Upon hydration, the cell-free system would become active and could produce supporting components (such as stabilizing proteins or enzymes) that help organize or maintain these pigment domains.
Once activated, the surface would gradually develop a more visible green coloration, indicating the presence of functional chlorophyll domains. When exposed to light, chlorophyll would undergo photodegradation into derivatives capable of chelating iron ions present in the surrounding matrix. These reactions would lead to the formation of a permanent image, effectively allowing the surface to “self-develop” without the need for external chemical processing.
The system could be tuned to respond to specific wavelengths of light, enabling controlled image formation while minimizing unwanted degradation. Visually, one could imagine walls or textiles that, once hydrated, slowly become photosensitive, capture light patterns over time, and then darken as the iron-based reaction fixes the image.
The idea behind this system— chlorophyll-based analog photography— has the main objective to substitute the toxic silver-based chemistry in current photographic emulsions. This application could be a way of demonstrating this technology, since photographic technology is usually a Blackbox. Chlorophyll Photographic Surfaces could be an amazingly visual way to observe the fascinating chemistry of chlorophyll as it captures a moment in time.
The limitations of cell-free systems are treated as design features. Activation by water allows the material to remain stable in its dry state and only become functional upon hydration, preventing premature chlorophyll degradation. Stability is ensured through freeze-drying and microencapsulation within a polymer matrix, which protects the components during storage. The one-time-use nature of the system aligns with its role as a photographic process, where a single, irreversible transformation is required to record and fix an image.
Homework question from Ally Huang
Background:
Spaceflight exposes astronauts to increased radiation, which leads to the production of reactive oxygen species (ROS) and oxidative stress. This can damage DNA, proteins, and cellular function, posing risks to astronaut health during long-duration missions. Understanding how oxidative stress affects biological systems is therefore critical for developing protective strategies. Cell-free systems provide a simplified platform to study these effects without the complexity of living cells. This project proposes using a cell-free protein expression system to model how oxidative stress influences gene expression, providing insight into biological damage mechanisms relevant to space environments.
Molecular / genetic target:
OxyR transcription factor and an OxyR-responsive promoter controlling GFP expression.
Relation to space biology:
Radiation in space generates reactive oxygen species (ROS), which induce oxidative stress in biological systems. The OxyR transcription factor in bacteria is activated by oxidative conditions and regulates gene expression in response to ROS. By coupling OxyR activation to GFP expression in a cell-free system, this experiment models how space-induced oxidative stress affects transcriptional responses. This provides a simplified and controllable way to study how oxidative conditions influence gene regulation and molecular damage in space.
Hypothesis / research goal:
The hypothesis is that exposure to space-induced oxidative stress will activate OxyR, resulting in increased GFP expression up to a threshold, beyond which excessive oxidative damage will reduce protein production. This is based on the known mechanism of OxyR, which is activated by oxidation, enabling it to promote transcription of target genes. However, high levels of oxidative stress can damage transcriptional and translational machinery, reducing overall protein synthesis. The goal of this experiment is to characterize how oxidative stress in space affects gene expression in a cell-free system and to identify conditions where biological systems remain functional versus when damage becomes inhibitory.
Experimental plan:
BioBits cell-free reactions containing OxyR and GFP under an OxyR-responsive promoter will be prepared and flown to the ISS. Samples will be exposed to the space environment, where radiation is expected to generate oxidative stress. Parallel ground controls will be maintained on Earth. Additional onboard controls will include reactions lacking OxyR or containing constitutive GFP expression. Fluorescence will be measured using the P51 Molecular Fluorescence Viewer to quantify GFP production. The miniPCR may be used to amplify DNA templates if needed. Differences between flight and ground samples will reveal how space-induced oxidative stress affects gene expression.
My project will measure several aspects related to the behavior of pure chlorophyll versus when bound to water-soluble chlorophyll-binding proteins (WSCPs). The main goal is to determine whether WSCP binding changes light sensitivity, photodegradation behavior, and its subsequent reaction with iron. For this end the following measurements will be needed:
1. Correct expression of the WSCPs
First of all, to check for the expression of WSCPs, a gel electrophoresis analisys would be conducted, this would be done after purification of the protein using a platform like Revvity LabChip (as used at Ginkgo Bioworks).
The preparation steps for the electrophoresis input go as follows:
Prepare Denaturing Solution: Prepare a mixture of Protein Express Sample Buffer and a reducing agent (if needed, such as BME, DTT, or TCEP).
Mix Sample with Buffer: Add 2 µL of protein sample to 7 µL of the denaturing solution in a 96-well PCR plate or 0.6 mL centrifuge tube.
Heat Denature: Seal the plate or tube and heat to 100°C for 5 minutes to denature the proteins.
Dilute and Mix: After cooling, add 35 µL of molecular biology grade water (or 32 µL for High Sensitivity) to each sample and mix by pipetting up and down.
Remove Bubbles: Centrifuge the sample plate at 3000 rpm for 5 minutes to eliminate bubbles, which can cause erratic results.
The expected output would be 20-22 kDa per subunit. This would allow for a first assessment of correct protein expression.
2. Absorbance spectrum of chlorophyll vs chlorophyll-bound WSCPs
The next measurement I would take would be a uv–vis absorbance spectrum analisys, comparing the pure chlorophyll vs the chlorophyll-bond WSCPs. In this case I believe absorbance would be enough to determine if there was a correct folding of WSCPs and consequent binding to chlorophylls since the main absorption bands of chlorophyll, including the Soret and Qy bands, will be compared in terms of peak position, intensity, and shape. Binding to WSCP is expected to induce shifts in absorption maxima and produce sharper, more defined peaks due to the organized protein environment.
To prepare the samples a chlorophyll stock solution would be prepared in a suitable solvent and diluted into buffer to generate the free chlorophyll sample. The purified WSCP–chlorophyll complex would be prepared in the same buffer conditions and adjusted to comparable chlorophyll concentrations.
Important to understand how WSCPs influence the absorbance spectrum of chlorophyll and check if WSCPs are correctly folded and bond to chlorophyll
3. Reaction with iron of exposed chlorophyll vs exposed chlorophyll-bound WSCPs
Third, I would measure the reaction with iron after light exposure in free chlorophyll compared with chlorophyll-bound WSCPs. This is central to the photographic concept, because chlorophyll is expected to degrade under illumination into derivatives capable of chelating iron and forming a darker final product. To test this, I would expose both samples to controlled light conditions for defined times, then add iron salts under standardized conditions and monitor the reaction. This could be achieved in solution and measured by UV–Vis spectroscopy to detect changes in absorbance associated with iron complex formation, or by embedding the two different samples in polymer and performing a visual test on the change of color after reaction with iron.
This will indicate how the degradation of chlorophyll is carried out in pure chlorophyll vs chlorophyll-bound WSCPs
It will also be important to understand the differences of light sensitivity in the two samples
Homework: Waters Part I — Molecular Weight
1. Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight?
According to Expasy Compute pI/Mw tool the molecular weight of eGFP with added linker and His-tag is 28006.60 Da
2. Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1)
I chose to work with the two peaks circled in green
$$
\begin{aligned}
z &= \frac{m/z_{n+1}}{(m/z_n - m/z_{n+1})} \
z &= \frac{848.9758}{875.4421 - 848.9758} \
z &= \frac{848.9758}{26.4663} = 32.08
\end{aligned}
$$
3. Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
No, the charge state cannot be determined for the zoomed-in peak from this figure alone. Determining the charge state requires at least two adjacent peaks, so their spacing can be used to calculate z. In the zoomed region, only a single isolated peak is shown and no neighboring charge-state peak is visible. Therefore, there is not enough information to assign its charge state.
Homework: Waters Part III — Peptide Mapping - primary structure
1. How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above.
There are 20 Lysines (K) and 6 Arginines (R) highlighted bellow:
2. How many peptides will be generated from tryptic digestion of eGFP?
According to the Expasy PeptideMass tool, 19 peptides will be generated
3. Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
Divided the highest peak in 10
There seem to be 19, which would match the prediction
4. Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
If all peaks are taken into account then there would be 22 which is more than the prediction
5. Identify the mass-to-charge of the peptide shown in Figure 5b. What is the charge of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H])+ based on its m/z and z.
In mass spectrometry, the instrument measures the mass-to-charge ratio (m/z) of a peptide that carries multiple protons, so to recover the true peptide mass (as the singly charged [M+H]+), we multiply the measured m/z by the charge z to remove the charge scaling and then subtract the mass of the extra proton that was added during ionization.
The most abundant peak is:
$$
m/z = 525.76712
$$
Adjacent isotope peaks:
(526.25918 - 525.76712 = 0.49206)
(526.76845 - 526.25918 = 0.50927)
Average spacing ≈ 0.50
$$
\Delta(m/z) \approx \frac{1}{z} ;\Rightarrow; z \approx \frac{1}{0.5} = 2
$$
Charge state: ( z = 2 )
$$
[M + H]^+ = z(m/z) - (z - 1)\times 1.0073
$$
$$
[M + H]^+ = 2(525.76712) - 1.0073
$$
$$
[M + H]^+ = 1051.53424 - 1.0073
$$
$$
[M + H]^+ \approx 1050.53
$$
6. Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
$$
MW_{\text{experiment}} = 1050.52438
$$
The closest predicted peptide mass from PeptideMass is:
$$
MW_{\text{theory}} = 1050.5214
$$
From peptide FEGDTLVNR
A 857ppm error is quite large, so I also tried to predict the molecular weight without the linker and His-tag in case that wasn’t used in the experimental version, but the results would then be even worse (Theoretical (kDa) — 26941.48) with an extremely large 38,600 ppm error, so I assume that can’t be the case.
Given the 857 ppm error and that Proteins are considered confidently identified if the mass accuracy is < 50 ppm, I assume something might be off regarding the theoretical mass probably
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Colaborated in the stage where a single picture was being formed throughout the 4 plates
It was a fun premise to explore could lab automation collaboratively
Next year there could be the “constraint” of creating a single image/pattern throughout all the plates to really engage in the collective activity of trying to figure out how to expand on what other people started like a Cadavre Exquis logic
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
1. Referencing the cell-free protein synthesis reaction composition, provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
In a cell-free master mix, the lysate, in this case E. coli, has all the machinery needed to process DNA to RNA to protein. In order for this to work, there is a mix of salts such as Potassium Glutamate, Magnesium Glutamate and Potassium phosphate, which maintain proper ionic concentrations to allow for correct DNA/RNA folding, enzymes to bind, and protein synthesis to occur efficiently, and buffers (HEPES + Potassium phosphate monobasic/dibasic) which maintain the optimal ph conditions for enzymatic activity.
2. Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix.
The main differences rest on how they supply energy and nucleotides, as well as in their intended performance (for fast expressions or for expressions that might need more time)
The 1-hour PEP–NTP system, uses pre-supplied NTPs (ATP, GTP, CTP, UTP) and the energy comes from PEP (phosphoenolpyruvate)— which is a high-energy molecule that can donate a phosphate group to regenerate ATP—, designed for rapid expression, by including high-energy, ready-to-use components.
While the 20-hour NMP–Ribose–Glucose system uses NMPs (AMP, CMP, UMP) instead of full NTPs. Nucleotides and energy are generated in situ from Ribose and Glucose, by relying on native enzymatic pathways in the extract. It is designed for longer expressions, having fewer synthetic additives— includes nicotinamide— which supports metabolic pathways that convert glucose into usable energy— instead of multiple cofactors. Therefore, being more metabolically integrated and sustainable.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
1. Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems.
sfGFP — Engineered for robust folding, even under non-optimal conditions, making it highly reliable in cell-free systems
mRFP1 — Has a relatively slow maturation time, meaning fluorescence appears later, which can delay readout despite successful expression.
mKO2 — Exhibits pH sensitivity, so fluorescence intensity can decrease in more acidic environments typical of some cell-free reactions over time
mTurquoise2 — Known for high quantum yield and brightness, but requires proper folding, making its performance sensitive to reaction conditions
mScarlet_I — Designed for fast maturation and high brightness, allowing rapid and strong fluorescence readout in cell-free systems.
Electra2 — Displays oxygen-dependent chromophore formation, so fluorescence requires sufficient oxygen availability, which can be limiting in dense or sealed reactions.
2. Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation.
mKO2 — Exhibits pH sensitivity, so fluorescence intensity can decrease in more acidic environments typical of some cell-free reactions over time
As mKO2 exhibits sensitivity to ph shifts which decrease fluorescence over-time as the reaction gets more acidic, one solution could be making a stronger buffer concentration, both with HEPES and the potassium phosphate (mono/dibasic)
In order to validate and optimize this hypothesis the following experimental set could be performed:
Sample 1 — Control
HEPES-KOH pH 7.5: 45 mM
K-phosphate dibasic: 5.63 mM
K-phosphate monobasic: 5.63 mM
Sample 2 — Moderate buffer increase
HEPES-KOH pH 7.5: 60 mM
K-phosphate dibasic: 5.63 mM
K-phosphate monobasic: 5.63 mM
Sample 3 — Stronger buffer increase
HEPES-KOH pH 7.5: 75 mM
K-phosphate dibasic: 5.63 mM
K-phosphate monobasic: 5.63 mM
Sample 4 — HEPES + phosphate support
HEPES-KOH pH 7.5: 60 mM
K-phosphate dibasic: 7.5 mM
K-phosphate monobasic: 7.5 mM
mTurquoise2 — Exhibits high brightness and quantum yield but depends on efficient translation and proper protein folding, making its fluorescence sensitive to ionic conditions in cell-free reactions.
As mTurquoise2 fluorescence depends on correct folding and efficient translation, one approach to improve its performance is to optimize magnesium concentration, since Mg²⁺ plays a key role in ribosome function and protein folding in cell-free systems.
In order to validate and optimize this hypothesis, the following experimental set could be performed:
Sample 1 — Control
Magnesium Glutamate: 6.975 mM
Condition 2 — Moderate Mg increase
Magnesium Glutamate: 8.000 mM
Condition 3 — Strong Mg increase
Magnesium Glutamate: 10.000 mM
Condition 4 — Very strong Mg increase
Magnesium Glutamate: 12.000 mM
3. The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment.
According to the hypothesis presented in the previous step, I created these 8 wells, 4 for the mKO2 and 4 for the mTurquoise2.
Nature’s Latent Image is a project which explores the possibility of using chlorophyll and light-harvesting proteins a novel photographic system to substitute toxic silver in current analog photography media.
Engineer a shorter version of L protein that permits an independent folding and conserves essential parts of the sequence such as the transmembrane domain needed for the insertion of the L protein into the membrane and the C-terminal domain needed for the clustering of the L proteins that leads to the lysis of the bacteria.
Subsections of Projects
Individual Final Project — Nature's Latent Image
SECTION 1: ABSTRACT
Analog photography has been experiencing a growing revival and with it a growing ecological concern, especially regarding the impacts of its “magical” component — silver halides. Much of the movement of trying to address the environmental impact of analogue film has fallen on individual artists and researchers, by trying to mitigate the consequences of silver. However, despite the efforts of exploring plant-based developers, and darkroom procedures to prevent damaging disposal of silver contaminated solutions, (extremely toxic for the environment affecting primarily microbial life) we are still left with the need to use this toxic metal in lack of any other option for analog camera photography.
Nature’s Latent Image is a project which explores the possibility of using chlorophyll and light-harvesting proteins a novel photographic system to substitute toxic silver in current analog photography media, with the ultimate objective of developing working photographic emulsions which can be applied to film rolls, paper, etc.
This hypothesis is based on personal experiments that show that chlorophyll, when exposed to light, rapidly degrades into derivatives— pheophytin/pheophorbide that have a porphyrin type ring which (without the central magnesium ion) has the capacity of chelating iron ions. Therefore, chlorophyll can be used to create positive latent images (acting as light-sensitive agent) which can be developed with iron that forms the final negative image (acting as density builder).
Chlorophyll is highly prone to degradation, particularly through photooxidation, where light excitation leads to the formation of reactive oxygen species (ROS) such as singlet oxygen. These ROS can oxidize chlorophyll and nearby molecules, resulting in localized propagation of degradation. In addition, chlorophyll is also sensitive to air oxidation and environmental conditions, especially when not stabilized within a protein complex. And so, the main focus of this research project is understanding the influence of chlorophyll-binding proteins in order to maximize this pigment’s properties and stabilize it so it doesn’t degrade under unwanted situations.
Chlorophyll-binding proteins are here explored as a way to aggregate chlorophyll molecules and amplify their potential as a novel photographic substance, by creating matrixes of organized chlorophyll light-sensitive domains which should improve light absorption and allow for a controlled degradation in order to obtain photographic exposure speeds, stable latent images with good contrast and resolution, which can be later developed.
In order to achieve this project’s objectives, water soluble chlorophyll-binding proteins (WSCPs), CauWSCPs from Brassica oleracea, will be expressed to understand their impact on chlorophyll’s stability, light-sensitive capacity, degradation rate and ability to chelate iron. Another challenge rests on finding the right polymer in which to embed the protein-chlorophyll emulsion as to be able to achieve good experimental results. In further experiments, optimized chlorophyll-binding proteins could be design, if natural occurring ones do not meet photographic needs. Thinking in terms of scalability it would be interesting to synthesize these chlorophyll light-sensitive domains with cyanobacteria with the objective of having a renewable source to produce these emulsions. Finally, the ultimate step would be to develop a fully working photographic emulsion which could be applied to film rolls, paper, etc.
The first experimental approaches (as to achieve Aim 1 of this project) will start with the extraction and purification of chlorophyll from a plant source Urtica dioica (Nettles), as they are abundant and have high concentrations of Chlorophyll.
E.coli plasmids for the expression of WSCPs will be design and expressed using an E.coli cell-free system where the purified chlorophyll will be used as co-folding factor. The purified chlorophyll-protein will be embedded into a polymer either chitosan based or algae based like agar to allow for visual tests of the formation of latent images and reaction with iron.
SECTION 2: PROJECT AIMS
Aim 1 — Experimental Aim:
Aim 1 is based on further understanding chlorophyll-binding proteins and their effect on chlorophyll from a photographic point of view. These proteins seem to be the biological way to organize and make the most use of this photosensitive pigment.
Some challenges that are comprised in this aim are: understanding to what extent the organization of the chlorophyll through WSCPs is favorable for photographic purposes; understand if iron will still react with chlorophyll if it is bonded to WSCPs; and stopping chlorophyll degradation by air oxidation which might be aided by being bond to proteins and by being embedded into the right polymer;
To achieve these goals
Use Twist clonal genes tool
Create a plasmid ready for cell free expression and order it to be delivered at Ginkgo
Ginkgo Cell Free Protein Expression Validation automation workflow — Gel imaging of protein expression
Due to the unknown exact behavior of chlorophyll-protein complexes in terms of degradation speed, at least 2 different samples of WSCP assembly should be prepared:
Sample1 with purified whole chlorophylls
Sample2 with saponified chlorophyll— converted into chlorophyllin through treatment with an alkali, which removes the pythol tail
This will allow for a range of photostability and controlled degradation, since the presence of the pythol tail in WSCP chlorophyll-protein complexes drastically increases the photostability of chlorophyll according to Agostini et al., 2017
Analyse absorprion spectrum of proteins + chlorophyll vs pure chlorophyll
Check for correct protein-pigment complex formation
Embed both proteins + chlorophyll and pure chlorophyll in polymer to and test one against another in light sensitivity and reaction with iron
This allows for an easily identifiable visual test to acess latent image formation and ability to chelate iron
Aim 2 — Development Aim:
Designing an optimized version of WSCPs so it better suits photographic objectives, if needed. These adjustments are yet to be determined and rest on the experimental data from Aim 1.
After having understood the mechanisms of chlorophyll and WSCPs, develop a system where cyanobacteria, which already have both the mechanisms to produce chlorophyll and the proteins in question, could be used, instead of always having to resort to cell-free synthesis, as they would be a renewable source for scalability — the most appropriate chasis for this step would probably be Synechocystis sp. PCC 6803, due to its well-characterized genomes and natural competency.
Aim 3 — Visionary Aim:
The visionary aim of this project would be to develop fully working emulsions that could be applied to the fabrication of film rolls and paper, as well as creating Open-source low-tech protocols to produce chlorophyll-based emulsions so that the experimental analog photography community could expand on the findings of this research.
In the diagram above I drew a conceptual cycle in which this new media could participate in. It would open the possibility of a photographic practice completely compatible with the environment and new ways of producing images could be born, for example, through the partial decomposition of the photographic materials themselves, allowing for a direct intervention of other-than-human beings like bacteria, fungi and plants on the photographic medium.
This would enable an ecologically involved photographic practice that doesn’t rest on an active degradation of the ecosystems from the extraction of silver and its processing to the use of film rolls and other photographic materials and the darkroom waste they produce and their hazardous disposal.
1. Briefly summarize two peer-reviewed research citations relevant to your research
Experimental photography has increasingly foregrounded the material and ecological implications of its processes. In Blue Mud: Entangled Geologies and Lives of Photographic Silver, Alice Cazenave reframes photography as an extractive practice, entangled with mining, toxicity, and planetary infrastructures. Similarly, The Ecology of Grain exposes the environmental and animal-derived dependencies of gelatin-based film, while Andrés Pardo’s Back to Basics Vol. 1 and 2 explore alternative and low-toxicity processes. However, while these works critically examine photographic materials and propose process-based alternatives, they do not replace the reliance on silver as the core photosensitive element.
In parallel, research in biotechnology offers insight into chlorophyll as a light-sensitive molecule. Studies such as An unusual role for the phytyl chains in the photoprotection of the chlorophylls bound to Water-Soluble Chlorophyll-binding Proteins demonstrate that chlorophyll-binding proteins (WSCPs) can stabilize chlorophyll and protect it from degradation. Additionally, Fine tuning of chlorophyll spectra by protein-induced ring deformation shows that protein environments can modulate chlorophyll’s optical properties, suggesting that its behavior as a photosensitive material is not fixed but can be engineered.
The gap this project addresses lies at the intersection of these fields. While experimental photography has critically engaged with the ecological cost of its materials, it has yet to fully develop a non-toxic, non-silver-based photosensitive system. Conversely, scientific research on chlorophyll and WSCPs has not explored their potential within image-forming processes. This project proposes to bridge this gap by investigating chlorophyll as an alternative photosensitive agent, combined with plant-based polymers as an emulsion matrix, aiming to develop a biologically derived photographic process that replaces both silver and animal-based materials.
2. Explain how your project is novel or innovative. (Minimum 3 sentences.)
Analog photography has always stood in a paradox with ecology, while being the media many use through artistic practices to document and explore the problem, it actively contributes to it through its materiality based on toxic silver and also gelatine components. This project could bring about a change in the way analog photography relates itself with the natural environment, and allow for practices that participate in ecological cycles
3. Explain why your project matters and what impact it could have
The significance of this work lies in the absence of viable, non-toxic alternatives to silver-based camera photography that retain both functional and artistic potential. By exploring biologically derived materials as active components in image formation, the project seeks to expand the possibilities of experimental photography while reducing its environmental impact.
Beyond its technical aims, the project contributes to a broader rethinking of how artistic practices can engage with ecological systems—not as subjects to be represented, but as processes to work within. It also opens a critical dialogue within the experimental ecological community on whether synthetic biology should be considered a viable tool to address environmental challenges, even within artistic contexts.
If successful, this research could shift experimental photography toward more sustainable, biologically integrated methods, contributing for the artistic practice as a site for both ecological awareness and material innovation.
4. Describe the ethical implications associated with your project and identify relevant ethical principles (e.g., non-maleficence, beneficence, justice, or responsibility). (Minimum 2 paragraphs.)
This project raises ethical considerations primarily related to the use of biotechnology, material sustainability, and the environmental impact of artistic practices. By proposing chlorophyll and plant-based polymers as alternatives to toxic silver-based photographic materials, the project aligns with the principle of non-maleficence, aiming to reduce harm to ecosystems and human health. At the same time, it engages with responsibility in the use of synthetic biology, particularly in the potential development of genetically modified cyanobacteria as a production system. While the final photographic materials would not contain viable genetically modified organisms, the use of such systems during production raises questions about containment, ecological risk, and the broader normalization of biotechnology within artistic contexts. The project also seeks to contribute to more sustainable material practices, and opens a critical discussion on whether synthetic biology can be ethically integrated into ecological and artistic frameworks.
To ensure the project remains ethical, several measures should be implemented. The initial use of cell-free expression systems minimizes risk by avoiding living genetically modified organisms, while any later use of genetically modified cyanobacteria would require strict laboratory containment, controlled handling, and responsible disposal protocols. A key action is to ensure that no viable GM organisms are present in the final materials, clearly separating production from outcome.
Potential unintended consequences include the accidental release of modified organisms, or the broader risk of legitimizing biotechnological interventions without sufficient critical reflection.
Additionally, one possible incorrect assumption in this project is that chlorophyll is the most suitable photosensitive pigment for these purposes; although it has shown strong conceptual and experimental promise so far, alternative pigments or systems may prove more effective or sustainable. To address this uncertainty, the project remains open to alternative approaches and iterative testing.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Experimental Design
Written with use of HTGAA AI Tutor
Step 1 — DNA Design Using Twist Clonal Genes (Day 1–2)
Purpose: Generate a plasmid optimized for cell-free WSCP1 expression.
Method: Use the Twist Clonal Genes tool to design a codon-optimized WSCP1 construct for E. coli cell-free expression. Include:
17 best residues in hydrophilic zone (no changes to transmembrane zone) Added to the previous best LLR scored residues the best mutant for other 11 residues always double checking with experimental data sheet
Selection Rationale: Hydrophobic substitution in the transmembrane segment. Ala→Leu increases hydrophobicity and may stabilize membrane helix packing/insertion and oligomer stability.
Selection Rationale: Polar→hydrophobic change in the TM region. Thr→Leu may increase membrane compatibility and reduce local insertion/misfolding penalties.
PROPOSAL from Flo’s research: Shorten the L protein.
Rationale: MS2 bacteriophages kill E. coli bacteria via the protein L. L proteins insert themselves into the membrane and cause the lysis of the bacteria. A weakness of the L protein is that its folding and/or stabilization depends on a bacterial chaperone (DnaJ), making it vulnerable to adaptive mutations of the latter. A mutational study demonstrated that shorter versions of the L protein do not depend on the chaperone anymore [1], making it more resistant to bacterial mutations.
Strategy: Engineer a shorter version of L protein that permits an independent folding and conserves essential parts of the sequence such as the transmembrane domain needed for the insertion of the L protein into the membrane [2] and the C-terminal domain needed for the clustering of the L proteins that leads to the lysis of the bacteria [3].
Possible pitfalls:
We lack knowledge about the precise interactions between the L protein and DnaJ and thus, can’t exclude other purposes than the folding.
The absence of folding in the shortened versions of the protein might make them more susceptible to aggregation or/and degradation by endoribonucleases/proteases
We might underestimate the role of the “non-essential” regions
The bacteria can still keep adapting to survive (e.g. membrane composition, upregulation of proteases)
Rationale: After our individual efforts on reaching a more stable mutant, we decided to make a shorter version out of the more apparently stable mutant— which seemed to fold the best according to AlphaFold.



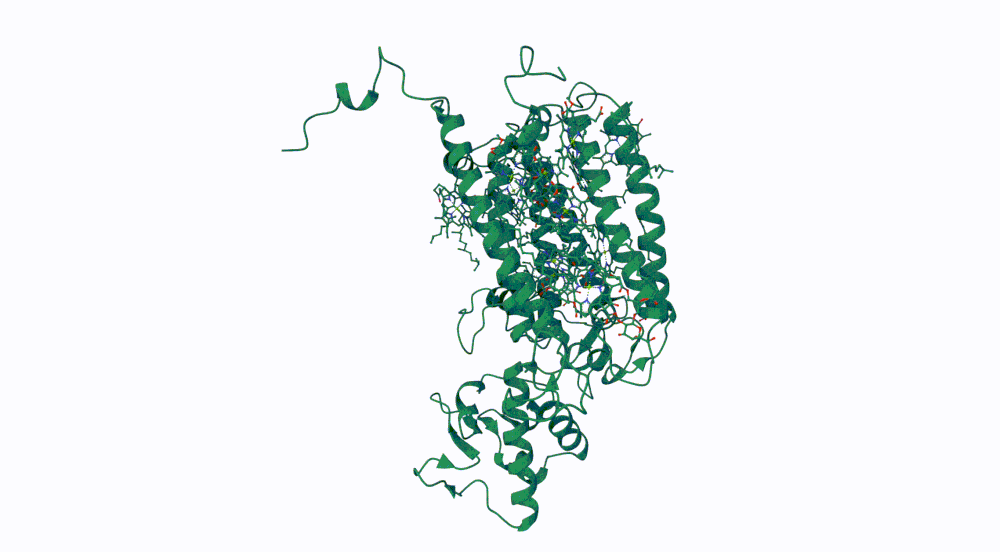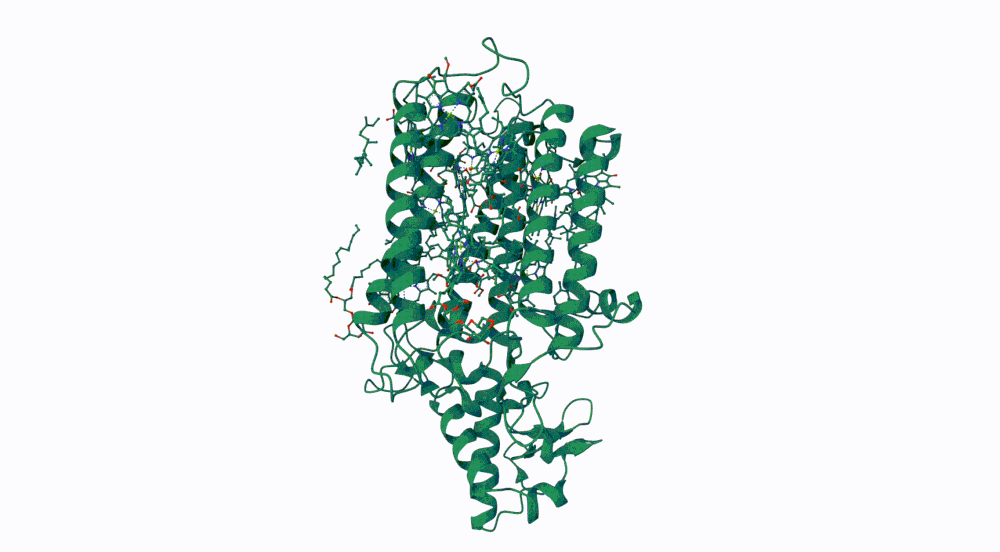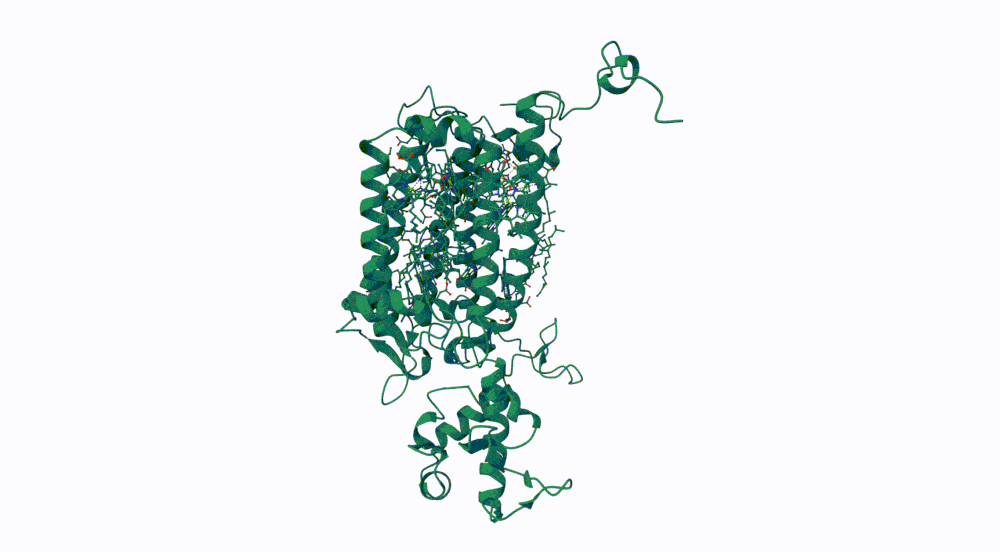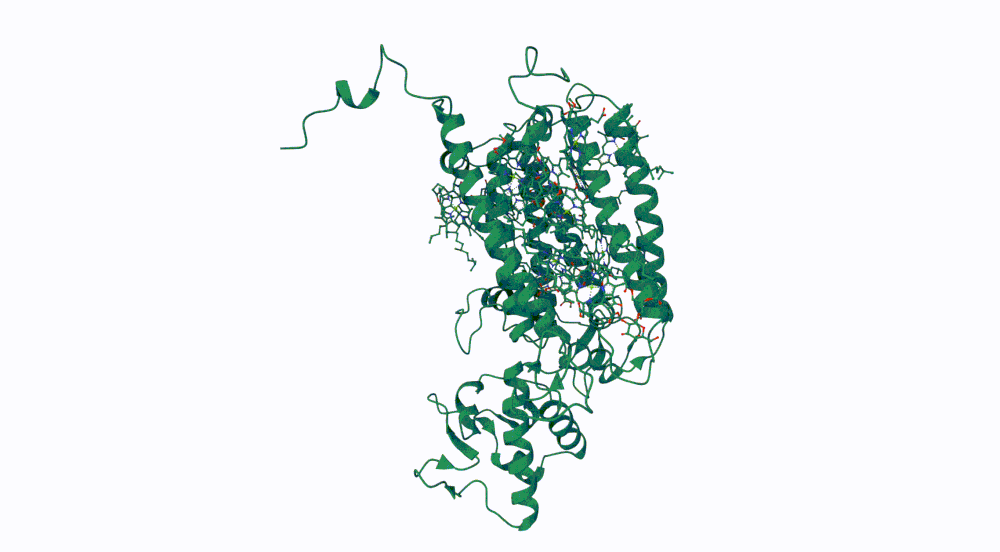
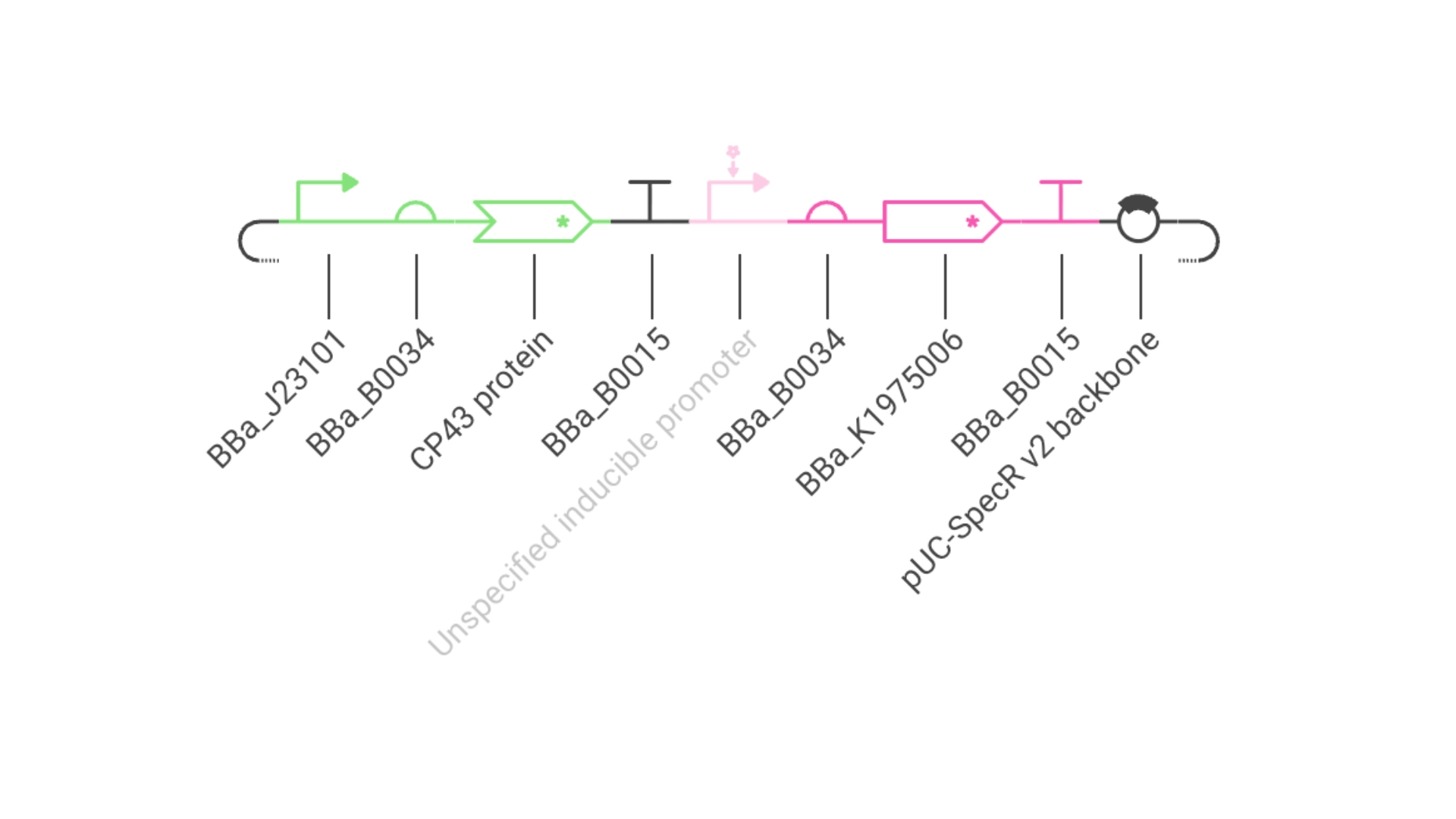
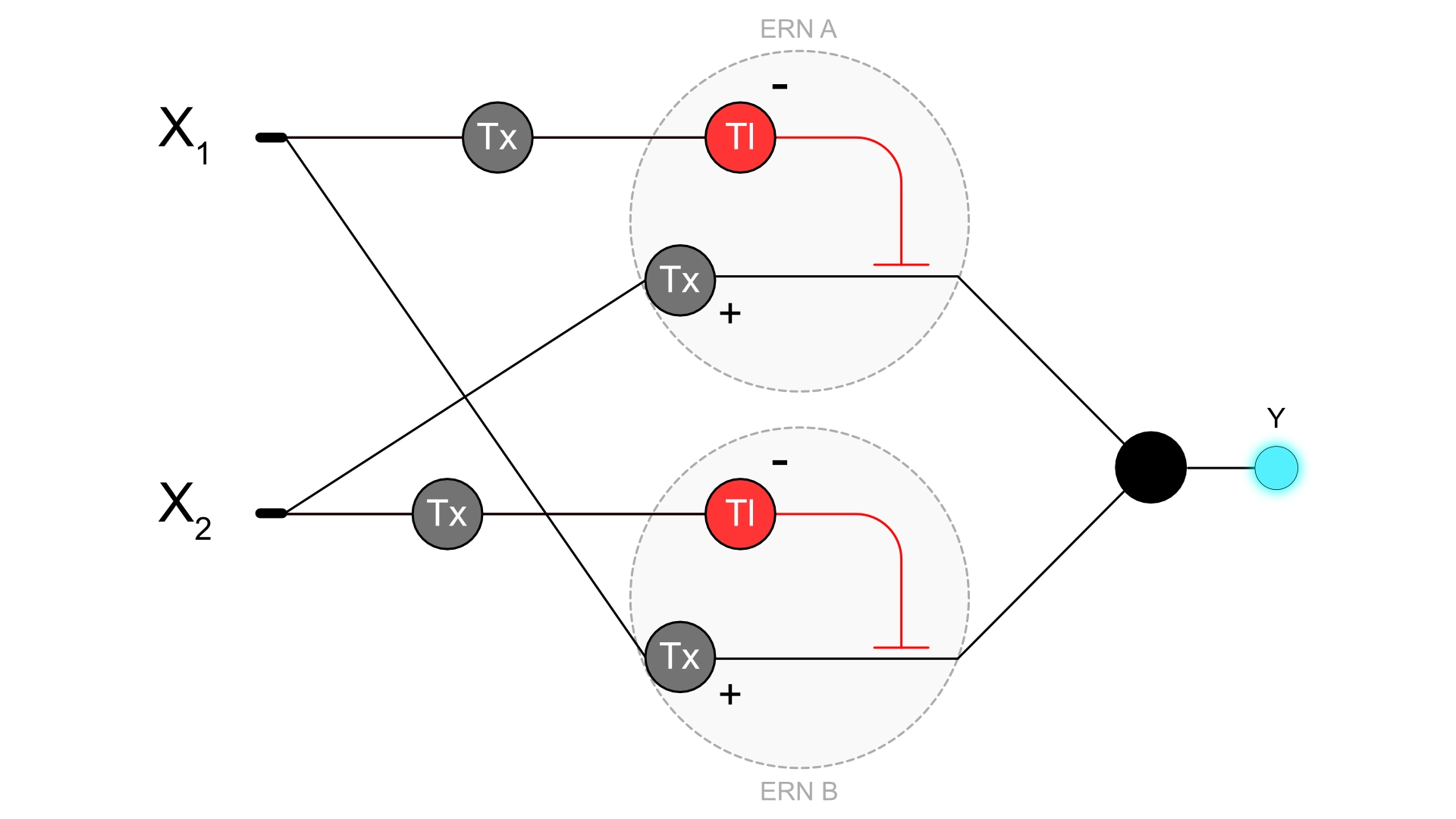


.webp)

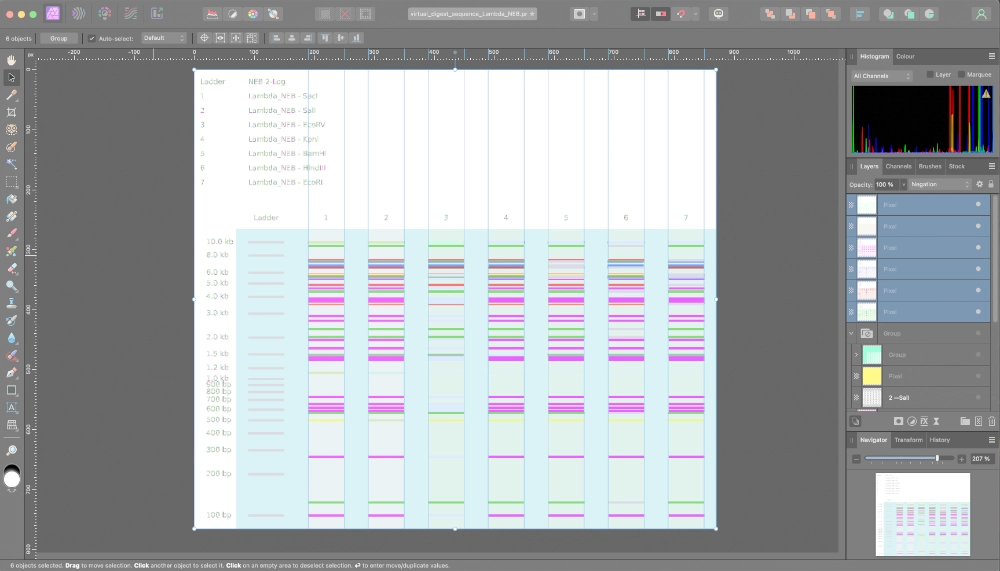
.png)