DOMENICA LILIA VIZCAINO ANDRADE — HTGAA Spring 2026
About me
I’m a computer engineering student interested in the intersection of biology, technology, and medicine. I enjoy exploring how emerging tools can be designed and used to benefit both people and the environment.
Biological engineering application / tool One biological engineering tool I would like to develop is a programmable biological computing platform based on synthetic genetic circuits that can sense biological inputs, perform simple computational operations, and produce interpretable outputs such as a visible color change or measurable signal. This idea comes from my interest in using synthetic biology as a medium for computation that could complement or eventually reduce reliance on energy-intensive electronic systems.
Part 0: Basics of Gel Electrophoresis Completed
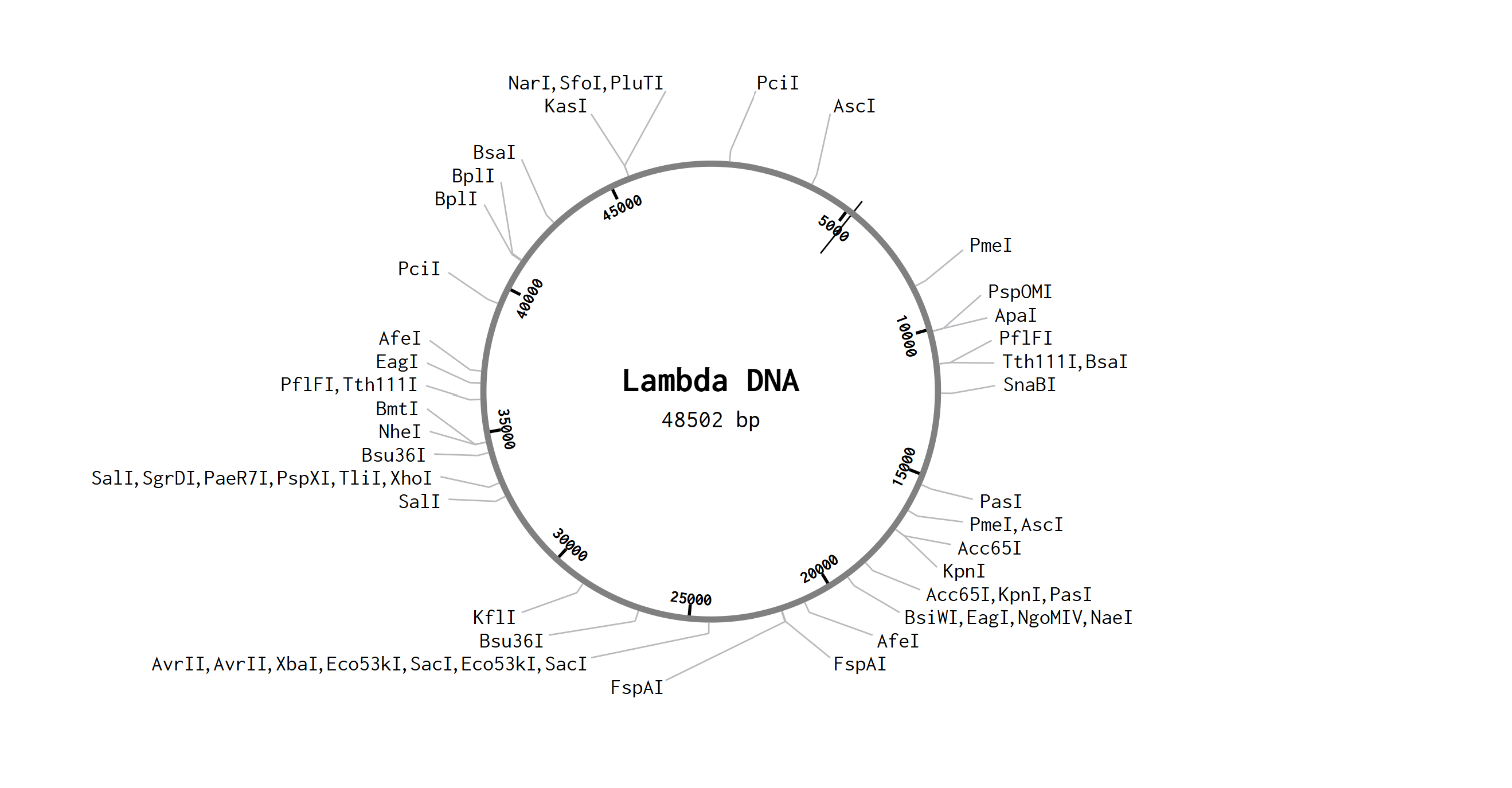
Part 1: Benchling & In-silico Gel Art After opening my Benchling account and joining the HTGAA group in Benchling, I imported the Lambda DNA. A restriction enzyme digestion was simulated in Benchling with the following enzymes:
Python Script for Opentrons Artwork For this lab, I generated an artistic design using the GUI at opentrons-art.rcdonovan.com. I decided to design some undersea animals because I really like being underwater and observing their nature. Given that in my node’s lab there are only fluorescent green and red, I decided to draw a red crab, a green turtle, and a red and green fish.
Part A. Conceptual Questions 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Amino acids have an average mass of about 100 Daltons, which is approximately 100 g per mole. If we assume that most of the mass of meat comes from proteins, we can estimate the number of amino acids in 500 g of meat to be 3*10^24 amino acid molecules.
Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? There is currently a need for non-binary biological computing, where an input for example is analog, or we want an output to be graded, this is where Intracellular Artificial Neural Networks are useful. This means that outputs can be low, medium, high. We can manupulate the inputs/outputs as if they were signals (using low-pass, high-pass, band-pass, etc filters) and with mathematical functions, rather than strictly Boolean ON/OFF outputs. IANNs combine many inputs simultaneously, and each input has a different “weight”. They can also detect patterns that are nonlinear and create thresholds. IANNs also help with scalability, as we can include many layers (multi-layers) while dealing with boolean circuits, and gates can get very messy.
General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. The main advantage of cell-free protein synthesis is the complete control we can get over experimental conditions. Unlike in vivo systems, where the cell regulates gene expression, metabolism, and resource allocation, in cell-free systems, the researcher directly defines all components and conditions.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Unfortunately, I was not able to contribute to the canvas that week as I had no signal. The idea of people from all over the world collaborating on a single piece, everyone bringing their own little piece of creativity into something unified through synthetic biology, is such a cool concept.
Subsections of Homework
Week 1 HW: Principles and Practices
1) Biological engineering application / tool
One biological engineering tool I would like to develop is a programmable biological computing platform based on synthetic genetic circuits that can sense biological inputs, perform simple computational operations, and produce interpretable outputs such as a visible color change or measurable signal. This idea comes from my interest in using synthetic biology as a medium for computation that could complement or eventually reduce reliance on energy-intensive electronic systems.
Rather than using biological systems only as sensors, I am interested in engineering circuits that can process multiple inputs and perform basic forms of computation within living or cell-free systems. For example, a biological circuit could detect a combination of biological markers and compute a response based on their relationships, producing an output that reflects the processed information rather than a single input. These types of systems could act as early models for more advanced biological computation and could eventually contribute to sustainable technologies that merge biology and information processing.
I am also interested in how neural network concepts and quantum computing approaches might inform the design of such biological circuits. Neural networks provide a framework for pattern recognition and adaptive systems, while quantum and quantum-inspired optimization methods may help solve complex design problems involved in assembling reliable biological circuits. Although these approaches are still emerging, integrating them with synthetic biology could help advance a future in which biological systems play a role in computation, diagnostics, and personalized medicine.
In the long term, platforms like this could support more personalized and environmentally sustainable medical technologies by allowing biological systems to process information locally and respond early to complex biological states. However, because tools that combine sensing, computation, and biological data processing could be misused or misinterpreted, it is important to consider governance and ethical safeguards alongside technical development.
2. Governance and policy goals for an ethical future
Main goal
Enable beneficial innovation in biological computing while preventing unsafe deployment, privacy violations, and misleading medical use.
Sub-goal 1: Reduce dual-use and misuse risk (non-malfeasance)
Because programmable biocircuits and design workflows can lower barriers to engineering biology, governance should reduce the chance they’re repurposed for harm (intentional misuse) and reduce accidental risks (lab accidents, unintended releases). This is the core concern in synthetic genomics governance options and WHO dual-use governance.
Sub-goal 2: “Trustworthy outputs” for health contexts (avoid harm from errors)
If biocircuits are ever used to inform health decisions (even as screening tools), governance should prevent harm from false positives/negatives, unclear limitations, and overconfident marketing. The Bioethics Commission stresses ongoing risk analysis, transparency, and public trust; this fits that logic.
Sub-goal 3: Protect autonomy, privacy, and consent
Biological systems that sense/compute on biological states could enable non-consensual testing or sensitive inference. Governance should promote consent norms, privacy-preserving design, and clear boundaries for acceptable uses—consistent with ethics frameworks emphasizing transparency, stewardship, and justice/fairness.
Sub-goal 4: Promote equitable access and responsible innovation
Ensure benefits (diagnostics, sustainability, safer computing) are broadly shared and not limited to wealthy settings; align incentives so safety and equity are not afterthoughts. This aligns with “societal goals” framing in the White House biotech R&D goals report.
3. Governance actions
Action 1 — “Architecture/code” + industry norm: sequence screening + provenance for risky designs
Actors: DNA synthesis & gene/genome services, protein design platforms, funders, and journals.
Purpose: Today, many safeguards rely on institutional biosafety review and some DNA-order screening, but AI/protein design and synthesis are moving fast. A proposed change is to strengthen screening and add provenance/traceability for high-risk designed sequences so dangerous designs are harder to order and easier to flag. This mirrors the biosecurity direction argued in the Baker & Church Science editorial and fits WHO-style biorisk mitigation.
Design:
Require/expand sequence screening for orders (DNA and other relevant synthesis inputs) using updated threat databases and better detection of modified/obfuscated sequences.
Store minimal audit logs (provenance) for flagged/high-risk categories with clear governance on who can access logs and under what conditions.
Tie compliance to funding/journal policies (e.g., “must use screened providers”) and to procurement requirements. (This is similar in spirit to governance options discussed for providers/users in synthetic genomics governance thinking.)
Assumptions:
Assumes screening algorithms keep up with rapid design methods.
Assumes there’s agreement on what counts as “high-risk” and how to avoid overblocking legitimate research.
Assumes audit logging can be done without creating new privacy/security problems.
Risks of failure & “success”:
Failure: adversaries route around regulated providers; screening misses novel threats; smaller labs get locked out.
Success: more surveillance infrastructure than necessary; chilling effect on open science; inequity if only wealthy groups can access compliant supply chains.
Action 2 — “Norms + law” in research & community biology: tiered biosafety/biosecurity readiness + external support
Actor: universities, community labs (DIYbio spaces), iGEM-like education programs, local regulators, biosafety professionals.
Purpose: Right now, safety practices vary widely across institutions and community spaces. The change is a tiered, “right-sized” readiness system: clear requirements for training, risk assessment, and project review that scale with risk—especially for projects involving novel circuits, pathogen-adjacent work, or anything that could be misused. This aligns with the Bioethics Commission’s call for ethics education and governance that anticipates DIY contexts, and with WHO’s emphasis on governance tools/mechanisms for biorisk and dual-use.
Design:
Adopt a tiered checklist + review model (low-risk fast path; higher-risk requires review).
Require documented training aligned with recognized biosafety references and iGEM-style responsibility practices (risk forms, safety planning, escalation paths).
Build an “ask-an-expert” escalation channel for community labs (modeled on DIYbio’s biosafety expert portal concept).
Encourage periodic external audits for higher tiers (WHO mentions external audit as a mechanism).
Assumptions:
Assumes community labs will opt in if requirements aren’t too burdensome.
Assumes there are enough trained biosafety professionals to support audits/advice.
Assumes tiers map well to risk in fast-changing tech areas.
Success risk: could reduce accessibility for low-resource groups unless paired with funding/support; may centralize power in institutions and exclude community innovation.
Purpose: Today, the jump from “cool demo” to “health claim” is where harm happens. For biocircuits that might be used in personalized medicine pathways, require that products can’t be marketed as diagnostic/therapeutic guidance without evidence standards and clear labeling of limitations. This supports the Bioethics Commission’s emphasis on transparency, ongoing risk analysis, and public trust.
Design:
A staged pathway: research-use-only → screening/decision-support → clinical diagnostic, with increasing evidence requirements.
Clear performance metrics (sensitivity/specificity, failure modes, bias across populations) and user-facing communication.
Post-market monitoring for real-world failures and misuse (aligns with “ongoing” oversight logic rather than one-time approval).
Align incentives: procurement and reimbursement favor validated tools (a “market lever”), consistent with “biotech to further societal goals” framing.
Assumptions:
Assumes regulators can classify and evaluate novel “bio-computing” tools cleanly.
Assumes companies won’t avoid regulation by making vague “wellness” claims.
Assumes validation datasets represent diverse populations.
Risks of failure & “success”:
Failure: regulatory gaps lead to misleading products; over-regulation slows beneficial innovation.
Success risk: big companies with money for trials dominate; smaller innovators struggle unless there are grants/partnership pathways.
4. Scoring governance actions
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
1
2
3
• By helping respond
2
1
3
Foster Lab Safety
• By preventing incident
2
1
3
• By helping respond
2
1
3
Protect the environment
• By preventing incidents
1
2
3
• By helping respond
2
1
3
Other considerations
• Minimizing costs and burdens to stakeholders
2
2
3
• Feasibility?
2
1
2
• Not impede research
2
1
3
• Promote constructive applications
2
1
1
5. Reflection on trade-offs
Based on the scoring, I would prioritize a combination of Option 2 and Option 1, while treating Option 3 as a later-stage priority once the technology begins to move toward health-facing applications. Option 2, which proposes a tiered biosafety and responsibility framework for academic and community laboratories, stands out as the most feasible and immediately impactful starting point. It scored highest for fostering lab safety, improving response capacity, and not impeding research, because similar training and review systems already exist in many research environments and can be expanded without excessive cost. Strengthening education, risk assessment, and oversight in a tiered way allows governance to scale with the level of risk rather than applying overly strict requirements to all projects. This aligns with governance approaches that emphasize responsible stewardship, ethics training, and adaptive oversight while still supporting innovation. Option 1, which involves sequence screening and provenance tracking for higher-risk designs, complements this by addressing upstream biosecurity risks that cannot be fully mitigated through local lab training alone. Screening and traceability can help prevent misuse and enable investigation if something goes wrong, making it an important second priority that strengthens prevention at the supply-chain level.
Option 3, focused on pre-market validation and clear rules for health-related claims, becomes essential once biological computing tools begin to be used in medical or personalized medicine contexts. Although it scored lower on feasibility and cost because regulatory validation can be burdensome and slow, it is critical for preventing harm from inaccurate or misleading outputs and for maintaining public trust. For that reason, I would treat Option 3 as a second-phase priority: not the first governance action for early-stage research, but something that should be planned early so that clear evidence standards exist before technologies are deployed in real health settings.
Several trade-offs influenced this prioritization. Strong screening and regulatory measures can improve safety and security but may also increase costs, slow research, or concentrate power in larger institutions that can more easily comply with requirements. Conversely, focusing only on education and local oversight could leave gaps in system-wide protection if risky materials can still be obtained without screening. The combined approach of Option 2 and Option 1 balances these concerns by strengthening safety culture and responsibility at the research level while also adding upstream safeguards to reduce misuse. However, this approach assumes that risk categories can be defined clearly, that screening systems will keep pace with advances in design technologies, and that enough biosafety expertise exists to support tiered review processes. There is also uncertainty about how regulators will classify and evaluate emerging biological computing tools that blur boundaries between research, diagnostics, and wellness technologies.
My recommendation is directed primarily toward university leadership, biosafety offices, research funders, and community biology networks, because these actors are well positioned to implement tiered safety frameworks and require the use of screened synthesis providers. A secondary audience includes biotechnology companies and synthesis provider consortia, whose cooperation would make screening and traceability more consistent across the field. Finally, as the technology matures toward personalized medicine applications, health regulators and clinical partners would become key audiences for implementing validation and marketing standards. Overall, prioritizing Option 2 and Option 1 provides a practical and balanced foundation for responsible development, while Option 3 ensures that future clinical uses are introduced in a safe and trustworthy way.
WEEK 2 LECTURE PREP
Homework Questions from Professor Jacobson
QUESTION 1
From the slide comparing chemical vs biological DNA synthesis, the error-correcting polymerase is shown with an error rate ~ 1 in 10⁶ (10⁻⁶) per base. The slides list the human genome length as ~3.2 Gbp. If you multiply that out: 3.2×10⁹ bases × 10⁻⁶ ≈ 3.2×10³, so ~3,200 errors per genome copy at that raw polymerase error rate. That’s a big discrepancy. Biology reduces discrepancy in layers, including:
Proofreading exonuclease activity (noted on the polymerase slide as proofreading/error-correcting exonuclease functions).
Post-replication mismatch repair, e.g., the slide explicitly mentions the MutS repair system as an “Error Correction” mechanism.
Plus broader cellular responses (conceptually): damage repair pathways, cell-cycle checkpoints, and removal of heavily damaged cells.
QUESTION 2
The slides give an average human protein coding length of around 1036 bp. Because the genetic code is redundant since most amino acids have multiple codons, the number of distinct DNA sequences that could encode the same protein is very large. In practice, many of these sequences don’t work well because the same protein does not equal the same expression.
Some reasons why not all codes work in practice are:
The tRNA abundance differs by organism which therefore affects translation speed and accuracy.
GC content and repeats which can make DNA hard to synthesize or clone. It can cause recombination instability.
mRNA secondary structure can block ribosome binding or slow translation.
Restriction sites or assembly constraints.
Homework Questions from Dr. LeProust
QUESTION 1
The standard workhorse method is solid-phase phosphoramidite chemical DNA synthesis, shown as the “Phosphoramidite DNA Synthesis Cycle” in the slides.
QUESTION 2
Because direct chemical synthesis is stepwise: each added base has <100% coupling efficiency, so the yield of full-length product drops exponentially with length. It also accumulates deletions or substitutions, and longer products are harder to purify away from the huge mixture of truncated failure products.
QUESTION 3
The compounded stepwise yield becomes extremely low, and the error burden becomes too high to get enough correct full-length molecules. Instead, many shorter oligos can be synthesized, then assembled into longer DNA using Gibson Assembly, and later the sequence gets verified and corrected if needed.
Homework Question from George Church
The 10 essential amino acids for animals are:
Arginine
Histidine
Isoleucine
Leucine
Lysine
Methionine
Phenylalanine
Threonine
Tryptophan
Valine
(Google search)
Lysine contingency is a scientific procedure created for Jurassic Park, which consists of using lysine dependence and availability as a safety control. The fact that lysine is essential for animals doesn’t automatically make it a strong containment strategy. Animals need lysine from diet, but lysine is still common in many real-world environments because it’s present in food, biomass, and is produced by microbes/plants. So lysine dependence alone could be a weak containment barrier outside carefully controlled settings. This makes me think lysine-based containment could work as one layer in a broader safety plan, but it likely shouldn’t be the only line of defense.
Week 2 HW: DNA Read, Write, & Edit
Part 0: Basics of Gel Electrophoresis
Completed
Part 1: Benchling & In-silico Gel Art
After opening my Benchling account and joining the HTGAA group in Benchling, I imported the Lambda DNA.
A restriction enzyme digestion was simulated in Benchling with the following enzymes:
The image I tried to recreate in the style of Paul Vanouse’s latent Figure Protocol was a bird flying as seen below.
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
I do not have lab access.
Part 3: DNA Design Challenge
3.1. Choose your protein
I chose bacteriorhodopsin because it is a light-activated membrane protein that can convert light into electrochemical signals. This makes it interesting for synthetic biology applications in bio-electronics and biological computing which is what I would like to do my final project on. In particular, bacteriorhodopsin has been used in optical memory systems and protein-based neural networks, where its light-induced conformational changes allow it to function similarly to a switch or synaptic element. I am interested in future applications where biological molecules are integrated with electronic or neuromorphic systems for sensing, computing, or personalized medical technologies, so bacteriorhodopsin is a useful model protein to study.
The following information was taken from UniProt:
UniProt entry: P02945
Organism: Halobacterium salinarum
Protein: bacteriorhodopsin
Organism taxonomy ID: 64091
Protein existence evidence: 1
Sequence version: 2
Although many different DNA sequences can encode the same protein, organisms prefer certain codons over others. This is called codon bias. If a gene uses codons that are rare in the host organism, the protein may be expressed poorly because the necessary tRNAs are less abundant. Codon optimization adjusts the DNA sequence so that it uses codons preferred by the host organism while still encoding the same amino-acid sequence. This improves translation efficiency, protein yield, and overall stability of expression.
Codon optimization can also remove problematic sequences such as restriction enzyme sites, repeats, or regions that form strong secondary structures, which can interfere with cloning or gene synthesis.
I optimized the bacteriorhodopsin coding sequence for Escherichia coli expression. E. coli is one of the most commonly used organisms in synthetic biology because it grows quickly, is easy to genetically engineer, and has well-characterized expression systems. Optimizing the gene for E. coli would make it easier to clone and express bacteriorhodopsin in a laboratory setting for research or synthetic biology applications.
The sequence was codon optimized using the IDT codon optimization tool, avoiding Type IIs enzyme recognition sites BsaI, BsmBI, and BbsI. These were avoided because restriction enzymes like BsaI, BsmBI, and BbsI are commonly used in synthetic biology cloning methods (especially Golden Gate assembly). These enzymes recognize very specific DNA sequences and cut at those sites. If the gene contains one of those recognition sequences internally, the enzyme will cut the gene in the middle when trying to clone it. That would break the construct and prevent proper assembly.
ATG TTG GAA CTG TTA CCG ACC GCG GTT GAA GGA GTC TCT CAG GCG CAG ATT ACG GGG CGT CCA GAA TGG ATC TGG TTG GCA CTG GGA ACA GCT TTA ATG GGC TTA GGA ACT CTG TAT TTT TTA GTC AAG GGA ATG GGC GTT TCC GAC CCT GAT GCT AAA AAA TTT TAT GCA ATT ACC ACA CTG GTG CCA GCT ATT GCC TTC ACC ATG TAC TTG TCT ATG CTT CTG GGG TAT GGA CTT ACA ATG GTT CCG TTT GGC GGT GAG CAA AAT CCG ATT TAT TGG GCA CGT TAC GCG GAC TGG CTT TTT ACG ACG CCG TTA CTG TTA TTG GAT CTT GCA CTT CTG GTG GAC GCG GAT CAA GGT ACC ATT CTT GCA TTG GTG GGG GCT GAT GGG ATA ATG ATA GGC ACC GGT TTA GTG GGT GCA CTG ACA AAA GTA TAT TCA TAT CGC TTC GTG TGG TGG GCC ATC TCA ACA GCC GCC ATG CTT TAC ATA TTG TAT GTA TTG TTT TTT GGC TTT ACC TCG AAA GCA GAG AGT ATG CGT CCG GAA GTA GCG TCT ACA TTC AAA GTG TTG CGC AAT GTT ACG GTG GTG TTA TGG TCA GCC TAC CCC GTA GTA TGG CTG ATT GGT AGC GAG GGA GCG GGT ATT GTA CCG CTT AAT ATC GAA ACC CTG CTG TTC ATG GTA CTG GAC GTG TCG GCC AAG GTG GGC TTC GGC CTG ATA CTG TTA CGT AGC AGA GCA ATT TTC GGA GAA GCT GAA GCT CCA GAG CCT AGT GCA GGT GAT GGT GCA GCC GCA ACG TCA GAT TAA
3.4. You have a sequence! Now what?
The bacteriorhodopsin protein can be produced by first introducing the codon-optimized DNA sequence into an expression system. This can be done using either cell-dependent (living cells) or cell-free protein expression technologies.
Cell-dependent
One common method is to insert the codon-optimized gene into a plasmid vector and transform it into a host organism such as Escherichia coli. Inside the cell, the DNA is first transcribed into messenger RNA (mRNA) by RNA polymerase. The mRNA is then translated by ribosomes, which read the codons in the mRNA and assemble the corresponding amino acids into the bacteriorhodopsin protein. Transfer RNAs (tRNAs) bring amino acids to the ribosome based on codon-anticodon pairing, allowing the polypeptide chain to form. After translation, the protein folds into its functional structure and can be integrated into membranes or purified for further use.
Cell-free
The gene can be expressed using a cell-free protein synthesis system, which contains ribosomes, enzymes, nucleotides, and tRNAs without living cells. In this system, the DNA template is added directly to the reaction mixture, where it is transcribed into mRNA and translated into protein in vitro. Cell-free systems allow rapid protein production and easier control of conditions, which can be useful for synthetic biology and protein engineering experiments.
Part 4: Prepare a Twist DNA Synthesis Order
After opening my Twist and Benchling accounts, I followed the steps mentioned and obtained the following plasmid.
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I would want to sequence DNA used in synthetic biological computing systems, particularly DNA strands that encode digital information or act as molecular logic circuits. DNA can be used to store data, perform computations, and respond to biological signals. Sequencing these DNA molecules would allow us to read stored information, verify computational outputs, and detect mutations or errors that occur over time.
In the long term, sequencing DNA-based computing systems could enable personalized medicine applications. For example, DNA circuits inside cells could record molecular events such as exposure to drugs, stress signals, or disease biomarkers. Sequencing these DNA records would allow clinicians to understand how a patient’s cells responded to treatment and adjust therapies accordingly. Accurate DNA reading is therefore essential for both bio-computing and medical diagnostics.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use a combination of Illumina sequencing (second-generation) and Oxford Nanopore sequencing (third-generation).
Illumina sequencing provides high accuracy, which is useful for verifying computational DNA outputs and detecting small errors. Nanopore sequencing allows long reads and real-time sequencing, which is useful for quickly decoding DNA circuits or recording information from living cells. Input would be synthetic DNA strands or DNA extracted from engineered cells.
Preparation steps:
DNA extraction
Fragmentation (if needed)
Adapter ligation
PCR amplification (for Illumina)
Loading onto sequencing platform
Illumina sequencing uses fluorescently labeled nucleotides. As each base is incorporated during synthesis, a fluorescent signal is detected and recorded to determine the sequence. Nanopore sequencing detects changes in electrical current as DNA passes through a nanopore, and software converts those signals into base calls.
The output is digital sequence data (FASTQ files) that can be decoded into stored information or analyzed to determine how the DNA circuit functioned.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would synthesize DNA sequences that function as biological computing circuits. These sequences could encode logic operations, data storage elements, or sensing modules that respond to cellular signals. For example, a DNA circuit could detect inflammation markers in a patient and record that information into DNA for later sequencing.
Such systems could eventually be used in personalized medicine, where engineered cells monitor patient health and record molecular events. The synthesized DNA could include logic gates, memory elements, and protein-coding sequences that allow cells to process information and respond to disease states.
(ii) What DNA synthesis technology would you use and why?
I would use phosphoramidite-based oligonucleotide synthesis followed by DNA assembly methods such as Gibson Assembly or Golden Gate assembly.
Essential steps
Chemical synthesis of short DNA oligos
Purification
Assembly into longer constructs
Cloning into plasmids
Sequence verification
Limitations
Errors increase with longer DNA
Cost of long constructs
Some sequences are difficult to synthesize
Assembly required for long circuits
Despite these limitations, modern DNA synthesis allows rapid construction of custom genetic circuits for research and therapeutic applications.
DNA Edit
(i) What DNA would you want to edit and why?
I would want to edit DNA in engineered cells used for biological computing and personalized medicine. For example, cells could be edited to contain DNA circuits that sense disease markers, process information, and respond by producing therapeutic molecules. Editing could also be used to improve stability and accuracy of DNA-based memory systems. In medicine, editing patient-derived cells could allow personalized treatments tailored to an individual’s genetic and molecular profile.
(ii) What editing technology would you use and why?
I would use CRISPR-Cas9 and base editing technologies to make precise modifications to DNA. A guide RNA directs the Cas9 enzyme to a specific DNA sequence. Cas9 creates a cut at that location, and the cell’s repair machinery introduces the desired edit using a provided template or repair process.
Week 3 HW: Lab automation
Python Script for Opentrons Artwork
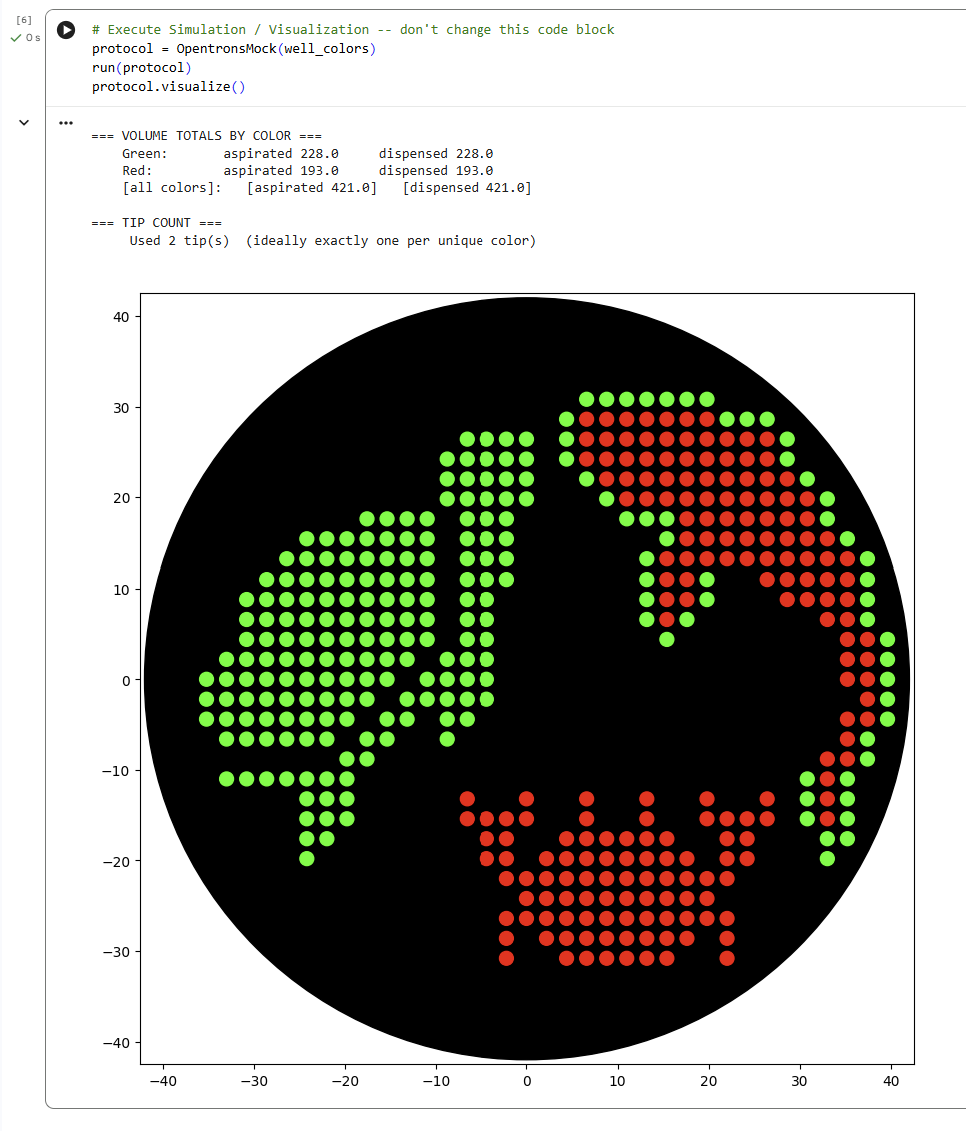
For this lab, I generated an artistic design using the GUI at opentrons-art.rcdonovan.com. I decided to design some undersea animals because I really like being underwater and observing their nature. Given that in my node’s lab there are only fluorescent green and red, I decided to draw a red crab, a green turtle, and a red and green fish.
Later, I simulated the design in the Opentrons Colab. This protocol programs the Opentrons robot to create an artistic pattern on an agar plate using two fluorescent proteins (mRFP1 in red and sfGFP in green).
First, the robot loads the necessary labware: a 20 µL tip rack, a temperature module holding the color plate (agar plate with dyes), and the agar plate where the design will be drawn. The pipette is initialized and set to start from a specific tip.
The script defines helper functions:
location_of_color() finds the well containing a specific color.
dispense_and_detach() carefully deposits a droplet onto the agar surface by approaching from above, dispensing, and lifting back up to avoid smearing.
Two lists of (x, y) coordinates define the artwork. These coordinates represent positions relative to the center of the agar plate.
The paper I found describes how researchers used automated liquid handling systems to process biological samples in a standardized way. Instead of manually pipetting every reaction, they used robotic tools to precisely mix reagents, prepare samples, and run assays with much less human intervention. This allowed them to reduce variability, increase reproducibility, and process many samples at the same time. Automation was especially important because small pipetting differences can affect biological results. By using robotics, the researchers were able to make their workflow more scalable and reliable, which is critical in biomedical research where consistency matters.
For my final project, I would like to use an Opentrons robot to automate the testing of DNA-based logic circuits using cell-free protein synthesis. The robot would transfer DNA constructs into a well plate, add the expression mix, dispense cofactors, and incubate the reactions before measuring fluorescence output. This would allow me to test many different circuit designs at once and compare their outputs automatically. I could also design a small 3D-printed holder to support custom plates or patterned substrates. Overall, automation would help me prototype biological computation systems faster and more accurately, while reducing human error and making the experiments more scalable.
Final Project Ideas
In slides for committed listeners.
Week 4 HW: Protein Design Part 1
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Amino acids have an average mass of about 100 Daltons, which is approximately 100 g per mole. If we assume that most of the mass of meat comes from proteins, we can estimate the number of amino acids in 500 g of meat to be 3*10^24 amino acid molecules.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When we eat food, our digestive system breaks proteins down into individual amino acids using enzymes. These amino acids are absorbed into the bloodstream and used by our cells to build human proteins, not cow or fish proteins.
The instructions for building proteins come from our DNA, which determines the structure of the proteins our cells produce. Therefore, even though the amino acids come from other organisms, they are reassembled according to human genetic instructions, so we remain human.
3. Why are there only 20 natural amino acids?
There are 20 standard amino acids used in proteins because evolution selected a set that provides enough chemical diversity to build stable and functional proteins. These amino acids include different properties such as hydrophobic, polar, charged, aromatic.
Together, they allow proteins to fold into many complex structures and perform many functions. While other amino acids exist, the 20 canonical ones became part of the genetic code early in evolution, and this system remained conserved because it works efficiently for life.
5. Where did amino acids come from before enzymes that make them, and before life started?
Before life existed, amino acids were likely formed through prebiotic chemical reactions on early Earth. Experiments like the Miller-Urey experiment showed that amino acids can form when simple molecules such as methane, ammonia, water, and hydrogen are exposed to energy sources like lightning.
Amino acids may also have come from meteorites, which have been found to contain organic molecules. These sources suggest that amino acids could form naturally in the environment before enzymes or living organisms existed.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Proteins made from L-amino acids, which are the natural form in biology, usually form right-handed α-helices.
If the helix were made from D-amino acids, the geometry would be mirrored, so the helix would form a left-handed α-helix.
7. Can you discover additional helices in proteins?
Yes, it is possible to discover additional helices. The α-helix is the most common helix found in proteins, but other helical structures exist, such as 3₁₀ helices and π-helices.
Using computational protein design, structural biology, and protein engineering, scientists can also design new helices with different properties. Advances in synthetic biology and protein modeling tools like AlphaFold or Rosetta make it easier to identify or design new helical structures.
8. Why are most molecular helices right-handed?
Most helices in biological proteins are right-handed because proteins are built from L-amino acids. The stereochemistry of L-amino acids favors a right-handed helix because it allows better bond angles and fewer steric clashes between atoms.
This configuration is therefore energetically more stable, which is why evolution selected it.
9. Why do β-sheets tend to aggregate? - What is the driving force for β-sheet aggregation?##
β-sheets tend to aggregate because their structure allows strong hydrogen bonding between neighbouring strands. When β-sheets from different proteins align, they can form extended networks of hydrogen bonds.
The main driving forces are hydrogen bonding between peptide backbones, hydrophobic interactions, and stacking of β-strands. These interactions stabilize large sheet-like structures, which can lead to aggregation.
9. Why do many amyloid diseases form β-sheets? - Can you use amyloid β-sheets as materials?
Many amyloid diseases form β-sheets because misfolded proteins can rearrange into stable β-sheet-rich structures. These sheets stack together into amyloid fibrils, which are very stable and difficult for cells to break down. This aggregation can disrupt normal cellular function and lead to diseases such as Alzheimer’s or Parkinson’s disease.
However, amyloid β-sheets also have useful properties. Because they are extremely strong, stable, and self-assembling, scientists are studying them as materials for nanofibers, biomaterials, scaffolds for tissue engineering, and nanotechnology applications. So although amyloids can cause disease, their structural properties could also be used in synthetic biology and materials science.
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
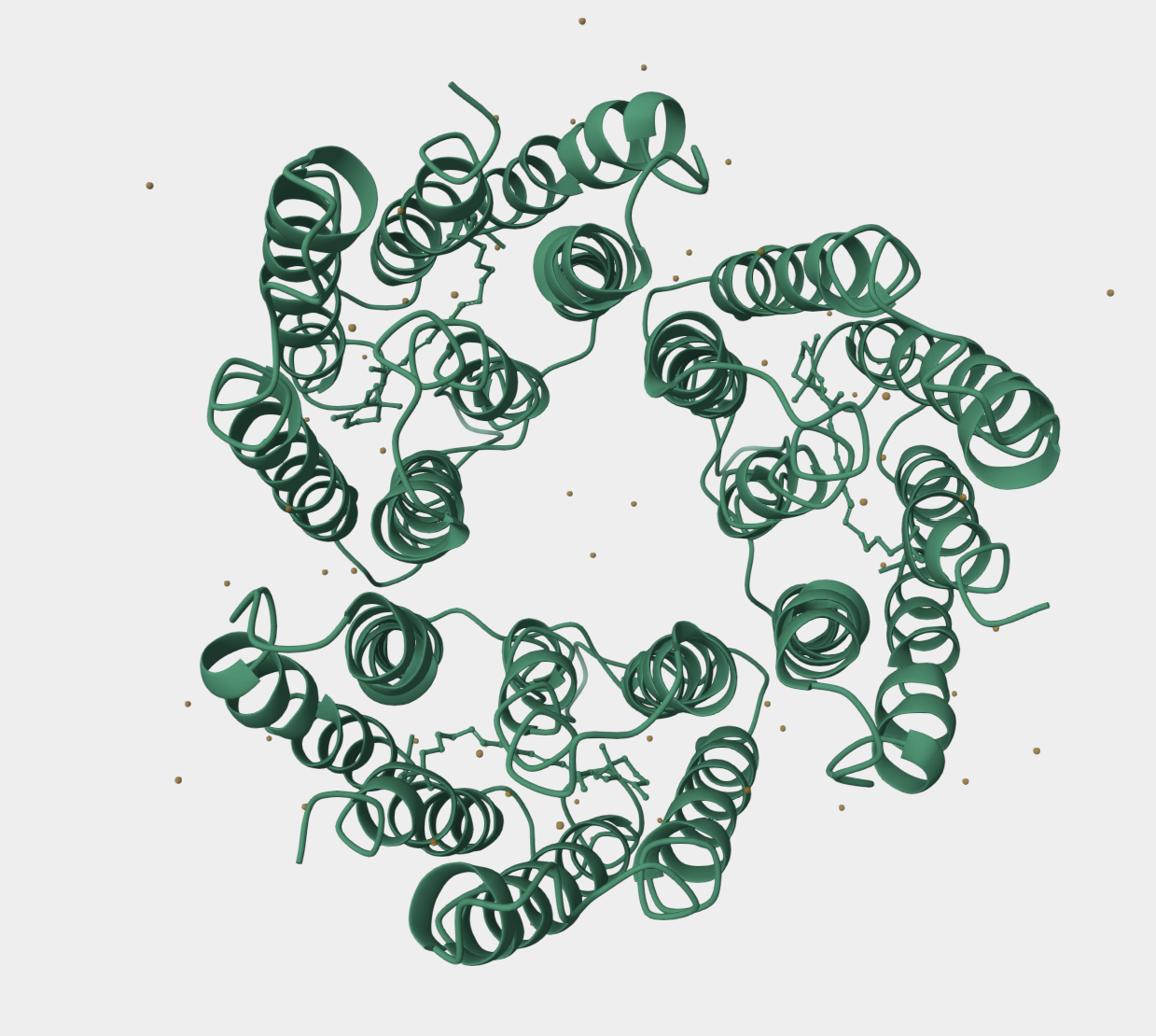
The protein I selected is NaChBac (Bacterial Voltage-Gated Sodium Channel). NaChBac is a voltage-gated sodium ion channel originally discovered in bacteria. It allows sodium ions to pass through the cell membrane when the membrane voltage changes. This flow of ions can create electrical signals similar to action potentials in neurons. I selected NaChBac because sodium channels are essential for neuronal signaling and spike generation, which are key mechanisms in biological neural networks. NaChBac is simpler than human sodium channels and easier to study or engineer, which makes it useful for synthetic biology and bio-inspired computing systems. Using proteins like NaChBac could help design biological circuits that behave similarly to neurons, which could eventually contribute to new approaches to neuromorphic or bio-based AI systems which is what I would like to work in for my final project.
After searching for its sequence in PDB, there were multiple structures for this protein. I selected PDB structure 6VWX, which represents the NaChBac sodium channel in a lipid nanodisc at 3.1 Å resolution determined by cryo-electron microscopy. This structure provides the highest resolution available among the listed options and represents the channel in a membrane-like environment, which is important because NaChBac is a transmembrane ion channel. Using a lipid nanodisc preserves the native conformation of membrane proteins better than detergent conditions. Therefore, 6VWX provides the most biologically relevant structure for analyzing the architecture and function of the NaChBac channel.
How many protein sequence homologs are there for your protein?
2: A0A4Q0VN24 and A0ABS6JN79
Does your protein belong to any protein family?
Yes. NaChBac belongs to the voltage-gated sodium channel (Nav) protein family.
Identify the structure page of your protein in RCSB
When was the structure solved?
The structure was solved in 2020, deposited on February 20, 2020 and released to PDB on June 24, 2020.
Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better.
The resolution is 3.10 Å, determined using cryo-electron microscopy. This number indicates that the overall protein structure and most side chains can be reliably modeled.
Are there any other molecules in the solved structure apart from protein?
Yes, besides the protein, the structure also contains sodium ions (Na⁺) and lipid molecules (POV). These molecules help stabilize the sodium channel and mimic the membrane environment in which the protein normally functions.
Does your protein belong to any structure classification family?
Yes. NaChBac belongs to the voltage-gated sodium channel structural family, which is part of the larger voltage-gated ion channel superfamily.
Structure of your protein in the 3D molecule visualization software PyMol
Cartoon visualization
Ribbon visualization
Ball and stick visualization
Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
C2. Protein Folding
C3. Protein Generation
Part D. Group Brainstorm on Bacteriophage Engineering
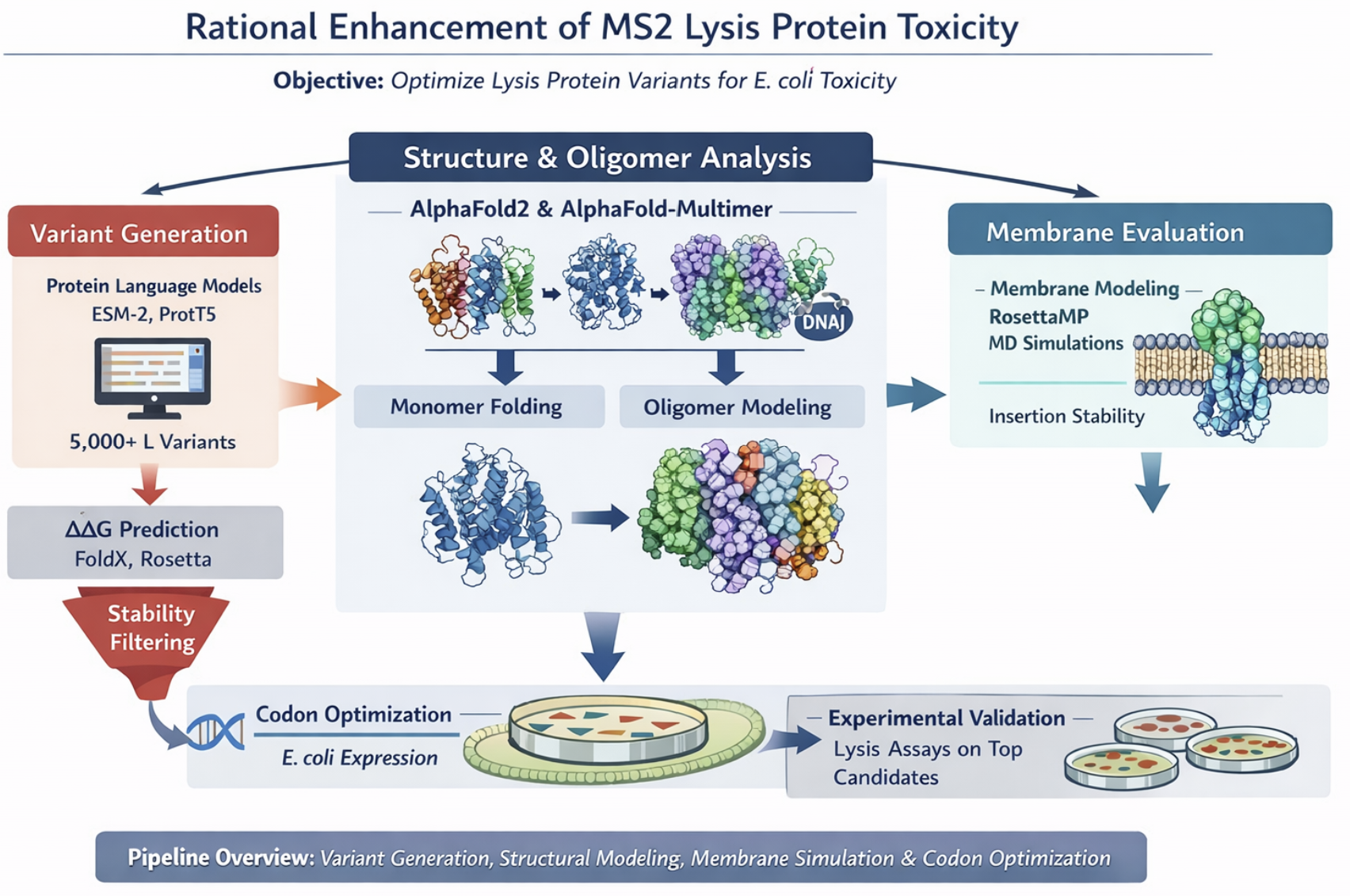
Computational Engineering of the MS2 Lysis Protein to Improve Stability, Titers, and Toxicity
Group members: Emmanuel Pereyra, Sergio Cuiza, Domenica Vizcaino, Diana Grimaldos
Selected Goals
After reviewing the provided literature on the MS2 lysis protein (L) and discussing the project aims, our group has decided to focus on three interconnected goals:
Goal 1: Increase the stability of the L protein.
Rationale: As the “easiest” goal, it is the most computationally tractable. A stabilized protein is less prone to degradation and misfolding, which could directly lead to higher functional titers and serve as a robust starting point for any subsequent engineering.
Goal 2: Increase bacteriophage titers through improved lysis efficiency.
Phage therapy relies on high phage titers for effective bacterial killing and scalable manufacturing, but phage production can be limited by inefficient lysis or poor coordination between phage replication and host destruction. Improving the efficiency and timing of host cell lysis can therefore directly increase the number of phage particles released per infected cell.
The MS2 L protein is a small 75–amino acid membrane protein that triggers bacterial lysis and is essential for the release of new phage particles. In the paper Mutational analysis of the MS2 lysis protein L, it is described how MS2 L functions as a single-gene lysis protein that disrupts bacterial cell envelope integrity without classical enzymatic activity. Additionally, L interacts with the host chaperone DnaJ, which modulates its activity and timing of lysis. In MS2 Lysis of Escherichia coli Depends on Host Chaperone DnaJ it is shown that lysis timing strongly affects the number of virions produced before the host cell bursts, meaning that engineering improved L variants may increase overall phage titers.
Goal 3: Increase the toxicity of the lysis protein.
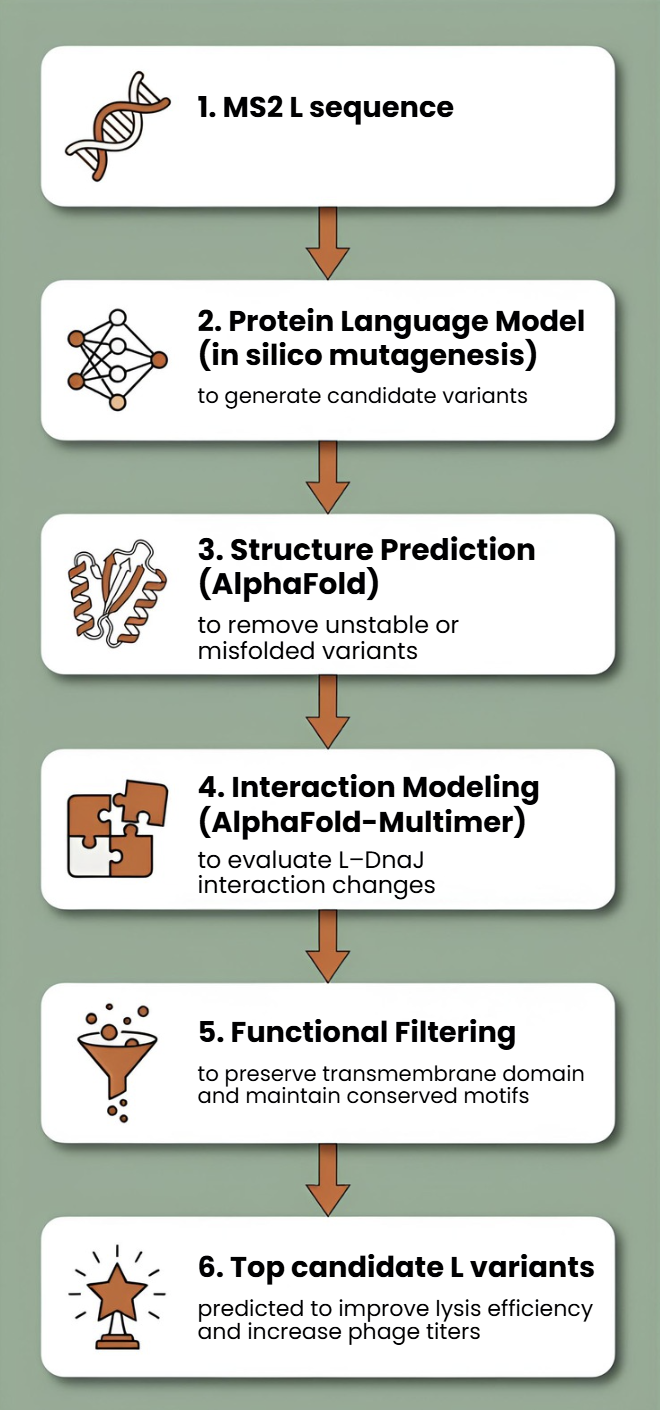
This proposal addresses the subproblem of increasing the toxicity of the L lysis protein from Bacteriophage MS2. Instead of random mutagenesis, toxicity will be approached as a multi-factor optimization problem involving structural stability, membrane insertion, oligomerization efficiency, and expression kinetics in Escherichia coli. The objective is to design L variants that enhance membrane disruption while maintaining proper folding and stability.
Additionally, we will explore disrupting the interaction between the L protein and the E. coli chaperone DnaJ.
Rationale: The reading “Identification MS2 lysis protein dependency on DnaJ” establishes this interaction as critical for function. By computationally predicting and then disrupting this interface, we can test its necessity and potentially create a DnaJ-independent lysis mechanism, offering a new avenue for controlling lysis timing.
Together, these three goals form a coherent strategy: stabilizing the L protein may improve its folding and expression, which can increase functional titers, while further engineering of membrane disruption and host interactions may increase toxicity and lysis efficiency.
Proposed Computational Tools and Approaches
Proposed Tools and Approaches We will build a computational pipeline using the tools introduced in recitation and the provided resources. The key steps and tools are:
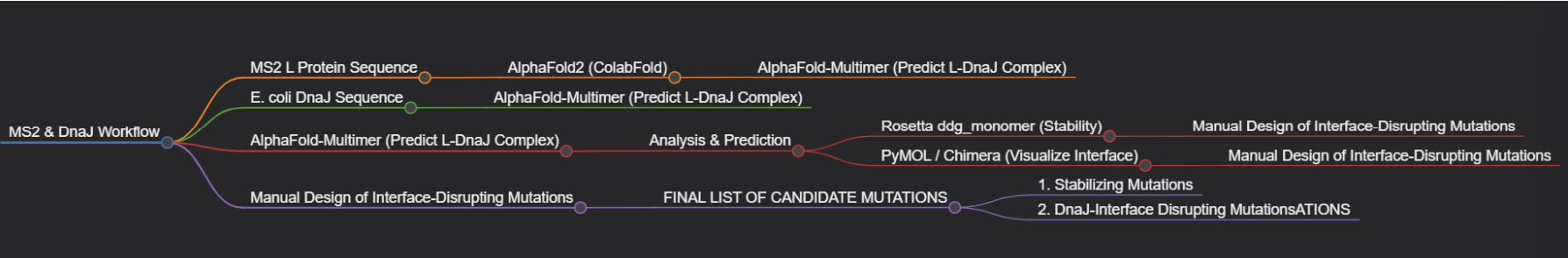
Step 1: Structural Modeling of the L Protein
Tool: AlphaFold2 (via ColabFold for ease of use).
Why: No high-resolution experimental structure of the full-length MS2 L protein exists. A reliable 3D model is the absolute foundation for all downstream analysis, allowing us to visualize which parts are structured vs. disordered.
Step 2: Modeling the L-DnaJ Complex
Tool: AlphaFold-Multimer.
Why: To disrupt the interaction, we first need to know where it occurs. AlphaFold-Multimer is the current state-of-the-art for predicting protein-protein complexes and will generate a testable model of the L protein bound to E. coli DnaJ.
Step 3: In Silico Mutagenesis for Stability
Tool: Rosetta (or FoldX). Specifically, the ddg_monomer application for predicting changes in folding free energy (ΔΔG).
Why: These tools are parameterized using vast amounts of experimental data on protein stability. They can systematically mutate each residue in our L protein model and predict whether the change (e.g., A->V) makes the protein more stable (negative ΔΔG) or less stable (positive ΔΔG).
Step 4: Visualizing and Selecting Interface Mutations
Tool: PyMOL and the HTGAA Protein Engineering Tools spreadsheet.
Why: We will use PyMOL to visually inspect the predicted L-DnaJ complex from Step 2 and select residues at the interface. We will then use the spreadsheet to check the conservation of those residues and manually design mutations (e.g., swapping a large hydrophobic residue for a charged one) predicted to break the interaction.
Protein Language Models (PLMs)
Protein language models such as ESM or ProtBERT will be used to perform in silico mutagenesis on the MS2 L protein sequence. These models can suggest mutations that preserve structural and functional constraints learned from large protein datasets.
This approach allows us to generate multiple candidate mutations across the L protein, avoid mutations likely to disrupt folding, and explore sequence space beyond naturally occurring variants.
AlphaFold Structure Prediction
Each candidate L variant will be analyzed using AlphaFold to predict protein structure and membrane topology. Since the C-terminal transmembrane region is essential for lytic activity, structural prediction will help identify mutations that preserve this functional domain.
Structural predictions will also help identify:
misfolded variants
mutations that destabilize the transmembrane region
variants that may alter oligomerization or membrane insertion
Interaction Modeling with Host Proteins
Because MS2 L interacts with the DnaJ chaperone, which affects lysis timing, candidate variants can be evaluated using AlphaFold-Multimer to predict changes in the L–DnaJ interaction.
This could help identify variants that:
maintain necessary folding assistance
reduce excessive dependency on host chaperones
improve robustness of lysis across physiological conditions
Proposed Computational Strategy
First, protein language models (e.g., ESM-2, ProtT5) will be used to perform directed in silico mutagenesis. These models capture evolutionary constraints and residue interactions, enabling the generation of structurally plausible variants while identifying mutation-tolerant and functionally critical positions. This step efficiently reduces the combinatorial search space.
Second, predicted variants will be structurally evaluated using AlphaFold2 for monomer folding and AlphaFold - Multimer to assess oligomerization and interaction with host factors such as DnaJ.
Third, membrane compatibility will be analyzed using membrane-aware modeling (RosettaMP) and selected molecular dynamics simulations.
Fourth, ΔΔG prediction tools (e.g., FoldX, Rosetta energy functions) will filter out destabilizing mutations.
In parallel, codon optimization algorithms will redesign selected variants for improved expression in E. coli, as toxicity depends on both structure and intracellular concentration.
Why These Tools Will Help
Why These Tools Will Help This pipeline is powerful because it moves from the general to the specific.
AlphaFold2/3 provides the necessary atomic-resolution context, transforming a sequence into a tangible structure we can analyze.
Rosetta leverages that structural context to make quantitative, physics-based predictions about stability.
AlphaFold-Multimer extends this to the biological mechanism, allowing us to generate a hypothesis about the DnaJ interaction that is currently unknown.
PyMOL enables the crucial final step of human intuition, allowing us to filter computational predictions through biological reasoning.
Rationale
Toxicity emerges from the combination of folding stability, cooperative oligomerization, membrane insertion, and sufficient expression levels.
These computational tools allow us to screen large numbers of protein variants without performing wet-lab experiments first.
Previous studies show that mutations in specific regions of L can abolish lysis function, indicating that the protein’s structure and interactions are highly sensitive to sequence changes.
Additionally, new AI-based methods are increasingly being used to design bacteriophages and improve phage performance.
Potential Pitfalls
Pitfall 1: Dynamic Regions and Model Quality
The L protein is small and likely has flexible/disordered regions, especially in its N-terminal domain.
Pitfall 2: Stability vs. Function Trade-off
A mutation that makes the protein more stable in its monomeric state might prevent it from undergoing the necessary conformational changes to oligomerize and form a pore in the membrane.
Pitfall 3: Lack of Membrane Context
Our stability predictions (Rosetta) are performed in a virtual “aqueous” environment and do not account for the energetic complexity of the lipid bilayer.
Limited biological data: There is still limited structural and mechanistic knowledge about MS2 L.
Cellular context not captured computationally Protein modeling tools may not fully capture membrane environment.
One limitation is the scarcity of quantitative datasets linking specific mutations to measured lysis kinetics.
Pipeline
We have developed three different pipelines to address each goal more specifically. The images were generated with AI:
Goal 1.
Group’s Short Plan for Engineering a Bacteriophage: Our group will computationally engineer the MS2 lysis protein to enhance its utility. First, we will use AlphaFold to model the protein and its complex with the host factor DnaJ. We will then employ Rosetta to perform in silico saturation mutagenesis, identifying point mutations that increase the protein’s predicted stability. Concurrently, using the AlphaFold-Multimer model, we will design mutations at the L-DnaJ interface intended to disrupt this key interaction.
Goal 2.
Goal 3.
Generate ~5,000 variants with protein LLMs, filter by ΔΔG stability, predict structure and oligomers with AlphaFold, evaluate membrane behavior, optimize codons, select top candidates for experimental lysis assays.
Week 5 HW: Protein Design Part 2
Week 7 HW: Genetic Circuits Part 2
Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
There is currently a need for non-binary biological computing, where an input for example is analog, or we want an output to be graded, this is where Intracellular Artificial Neural Networks are useful. This means that outputs can be low, medium, high. We can manupulate the inputs/outputs as if they were signals (using low-pass, high-pass, band-pass, etc filters) and with mathematical functions, rather than strictly Boolean ON/OFF outputs. IANNs combine many inputs simultaneously, and each input has a different “weight”. They can also detect patterns that are nonlinear and create thresholds. IANNs also help with scalability, as we can include many layers (multi-layers) while dealing with boolean circuits, and gates can get very messy.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
A useful application of an intracellular artificial neural network (IANN) is in early cancer detection and targeted therapy. In this system, multiple intracellular biomarkers such as oncogenic mRNA levels, microRNA expression, and hypoxia signals serve as inputs. Each input produces regulatory molecules, such as endoribonucleases, which act on engineered mRNA transcripts with specific cleavage sites. These interactions allow the system to integrate signals in a weighted and nonlinear manner, producing a graded response.
The output can be either a diagnostic signal, such as fluorescence, or a therapeutic response, such as expression of a pro-apoptotic protein. The system is designed so that only a strong and specific combination of cancer-related signals produces a high output, enabling selective targeting of cancer cells while minimizing effects on healthy cells.
However, several limitations must be considered. Biological noise in gene expression can reduce reliability, while precise tuning of regulatory weights is difficult to achieve. Off-target effects of endoribonucleases may disrupt normal cellular processes, and delivering the system efficiently into target cells remains a major challenge. Additionally, the metabolic burden of maintaining complex circuits and the risk of mutations affecting stability over time further complicate implementation. Despite these challenges, IANNs offer a powerful framework for implementing complex decision-making in living cells.
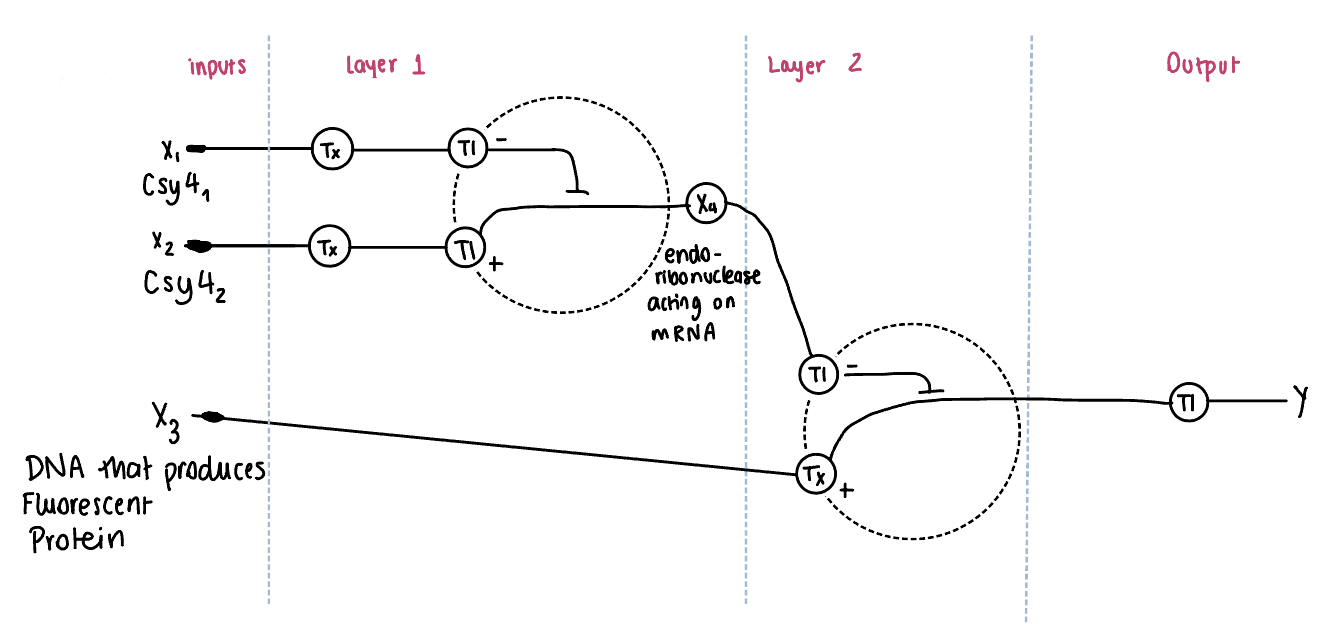
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
In this diagram, an intracellular multilayer perceptron composed of two regulatory layers is represented. In Layer 1, inputs X1 and X2 encode for endoribonucleases (Csy4 variants), which are produced through transcription (Tx) and translation (Tl). These endoribonucleases act on mRNA and generate an intermediate regulatory signal X4, representing the hidden layer. We need both inputs in layer one to downregulate and control the system from the difference or the sum of the inputs. This would be regulated right after the promoter and before the CDS.
In Layer 2, the intermediate regulator X4, controls the expression of a downstream gene X3, which encodes a fluorescent protein. The regulation occurs at the mRNA level. Finally, the fluorescent protein is produced through translation, resulting in the output Y.
Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
An example of existing fungal materials are Mycelium-based materials used for packaging, leather-like materials or other textiles for fashion, and bricks for construction.
Some advantages are that it is biodegradable, sustainable, customizable, and it has a low carbon footprint. In some cases, it is fire-resistant. However, some disadvantages are that they often have lower mechanical strength, are sensitive to moisture, and can be difficult to scale consistently.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
One potential application of synthetic biology in fungi is the development of self-healing, environmentally responsive building materials. In this system, fungal mycelium would be engineered to form structural materials such as bricks or insulation panels, while also incorporating genetic circuits that enable sensing and response to environmental conditions.
Inputs to the system could include the presence of environmental toxins (such as heavy metals or pollutants) or physical damage to the material. These signals would activate engineered biosensing pathways that regulate gene expression. The outputs could include the production of fluorescent proteins to signal contamination or damage, as well as the activation of growth pathways that promote self-repair of the material.
This system would allow the material to both detect and respond to changes in its environment, making it more adaptive and functional than traditional materials. However, limitations include challenges in controlling fungal growth, maintaining long-term stability, and ensuring safety when deploying genetically engineered organisms in real-world environments. Additionally, precise tuning of gene expression and scalability remain significant challenges.
Fungi are particularly well-suited for this application because they naturally form interconnected three-dimensional networks and can grow into large structures, unlike bacteria. This makes them ideal for material-based applications where both structure and function are required.
Week 9 HW: Cell Free Systems
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
The main advantage of cell-free protein synthesis is the complete control we can get over experimental conditions. Unlike in vivo systems, where the cell regulates gene expression, metabolism, and resource allocation, in cell-free systems, the researcher directly defines all components and conditions.
This means that there is flexibility on what you want or not, such as specific DNA, enzymes, cofactors, salts, or inhibitors. There are no cellular constraints, such as toxicity issues. There is no need for cloning, transformation, or cell growth, which makes experimentation faster. There is overall full control over experimental variables like transcription and translation rates, the environment and energy.
Examples where cell-free protein synthesis is more beneficial:
1.- Proteins that would kill or stress a cell can be easily and safely produced.
2.- When there is a need to do a quick prototype to test a genetic circuit without actually needing to grow a cell
3.- In case you want to incorporate non-natural amino acids, it is easier to do it in a cell-free system.
Describe the main components of a cell-free expression system and explain the role of each component.
DNA
Encodes the target protein
RNA polymerase
Transcribes DNA into mRNA
Ribosomes
Translates mRNA into protein
tRNAs and amino acids
Subparts to synthesize protein
Energy system
Powers transcription and translation. Some examples are ATP, GTP.
Enzymes and cofactors
Provides the necessary biological machinery
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Protein synthesis consumes large amounts of ATP and GTP. Without regeneration, the reaction stops quickly, and the protein yield is very low. A method to ensure continuous ATP supply would be the Phosphoenolpyruvate (PEP) system, which consists of adding PEP to ADP to obtain ATP.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic
Eukaryotic
Fast and cheap
Slower and more expensive
High yield
Support proper folding
Limited post-translational modifications
Support post-translational modifications
I would produce GFP in the prokaryotic system because its simple, we can make a lot for cheap, and it does not need complex folding or modification.
I would produce antibodies in the eukaryotic system because it requires complex foldings and modifications.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Membrane proteins are difficult to express because they are hydrophobic, require a lipid environment, and tend to misfold or aggregate in aqueous systems.
To design a successful cell-free experiment, it is important to mimic the natural membrane environment. This can be done by adding lipid-based structures such as liposomes or nanodiscs, or by using mild detergents to stabilize the protein during synthesis.
Additionally, experimental conditions should be optimized to improve folding and stability. This includes adjusting temperature (often lower temperatures improve folding), controlling the expression rate to avoid aggregation, and adding molecular chaperones to assist in proper protein folding.
By taking advantage of the flexibility of cell-free systems, the environment can be engineered to support correct membrane protein insertion and function.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
One possible reason for low protein yield is energy depletion. Protein synthesis requires large amounts of ATP and GTP, and if these are not regenerated, the reaction will stop early. This can be addressed by including an energy regeneration system such as phosphoenolpyruvate or a glucose-based system.
A second possible cause is non-optimal reaction conditions, such as incorrect magnesium concentration, pH, or salt levels. These factors strongly affect enzyme activity and ribosome function. The solution is to systematically optimize buffer conditions for the specific system being used.
A third reason could be protein misfolding or degradation. Some proteins require specific folding conditions or assistance from chaperones. This issue can be mitigated by lowering the reaction temperature, adding chaperone proteins, or including stabilizing agents in the reaction mixture.
Homework question from Kate Adamala
Example design: synthetic minimal cell that detects doxycycline and activates bacteria
This system is not directly measuring bacterial killing, but rather detecting the presence of an antibiotic and converting it into a readable biological signal.
Function
A synthetic minimal cell that acts as a chemical translator between the environment and bacteria
It detects doxycycline and converts that signal into IPTG release
IPTG then activates GFP expression in nearby engineered bacteria
Input and output
Input: doxycycline
Output of synthetic cell: IPTG
Final system output: GFP fluorescence in bacteria
Could this be done with cell-free Tx/Tl alone?
No, not properly
Without encapsulation:
IPTG would diffuse freely
No controlled release
No real “cell-like” behavior
Encapsulation is necessary to:
store the output molecule
release it only after activation
Could this be done with a genetically modified natural cell?
Yes, it could be engineered directly in bacteria
However, synthetic minimal cells offer:
modular design
easier swapping of sensing modules
no need to maintain living cell viability
Synthetic and natural systems can work together
Desired outcome
Without doxycycline:
no IPTG release
bacteria remain non-fluorescent
With doxycycline:
synthetic cell activates
IPTG is released
bacteria express GFP
Components of the synthetic minimal cell
Lipid vesicle membrane
Cell-free transcription/translation system
DNA encoding sensing and response
Encapsulated IPTG
Energy regeneration system
Enzymes, ribosomes, tRNAs, cofactors
Buffer and salts
Membrane composition
POPC
Cholesterol
Maybe phospholipids to tune membrane fluidity
Encapsulated contents
Bacterial cell-free Tx/Tl extract
Amino acids
Nucleotides (ATP, GTP, CTP, UTP)
Energy regeneration substrate
Salts and buffer
Plasmid DNA
IPTG
Tx/Tl system source
E. coli (bacterial system)
Reasons:
Simpler
Faster
Cheaper
Does not require complex post-translational modifications
Communication with environment
Input (doxycycline):
diffuses through membrane
Output (IPTG):
released through membrane pore
Mechanism:
doxycycline enters vesicle
activates gene expression
pore protein is produced
IPTG exits vesicle
bacteria detect IPTG
Experimental details
Lipids
POPC
Cholesterol
Optional: DOPC
Genes (synthetic cell)
tetR
hla (alpha-hemolysin pore protein)
Reporter bacteria
E. coli with:
gfp
lac promoter system
Measurement
Measure GFP fluorescence in bacteria using:
flow cytometry
plate reader
fluorescence microscopy
Controls:
no doxycycline
no pore gene
no IPTG
bacteria without reporter
Expected result:
GFP only when doxycycline is present and system is functional
Homework question from Peter Nguyen
Application: Smart antimicrobial and contamination-detecting fabric
A wearable fabric embedded with freeze-dried cell-free systems that detects harmful bacteria or toxins and responds by producing a visible signal and releasing antimicrobial compounds.
How the idea works
The fabric contains microcapsules with freeze-dried cell-free transcription/translation systems embedded into the fibers
These systems are activated when exposed to moisture (for example, sweat, rain, or environmental humidity)
Once activated, the system detects specific bacterial molecules using designed genetic circuits
In response, the cell-free system produces:
a colorimetric signal, so basically a colour change in fabric
antimicrobial peptides that help reduce bacterial growth on the surface
Because cell-free systems allow full control over components, the sensing and response modules can be easily redesigned for different pathogens or environments
sportswear and everyday clothing exposed to bacteria
Current fabrics are passive and cannot detect or respond to contamination
This system provides:
real-time detection of harmful microbes
active response to reduce contamination
improved safety and hygiene without requiring complex electronics
It could be especially useful in:
hospitals (infection prevention)
military or field work
travel and high-density environments
Addressing limitations of cell-free systems
Activation with water:
The system is designed to activate only in the presence of moisture, making activation natural and controlled
Stability:
Freeze-drying increases long-term stability
Encapsulation in protective polymers or hydrogels within the fabric helps preserve function over time
One-time use:
The system can be designed as replaceable or layered patches within the fabric
Alternatively, multiple microcapsules can be distributed throughout the material so activation occurs gradually over time
Control and reliability:
Because all components are specified manually, the system can be tuned for sensitivity, response time, and output intensity
Homework question from Ally Huang
Mock Genes in Space proposal
Background information (max 100 words)
Long-duration spaceflight increases radiation exposure and physiological stress, which can damage DNA and threaten astronaut health. A lightweight, freeze-dried cell-free platform is attractive in space because it is easy to transport, activated on demand, and does not require maintaining live cells. The Genes in Space toolkit specifically supports BioBits® as a “protein factory in a tube,” with miniPCR for DNA amplification and the P51 viewer for simple fluorescence readout. This makes space-based DNA damage detection both practical and scientifically valuable for astronaut monitoring, mission safety, and future deep-space habitation.
Molecular or genetic target (max 30 words)
A double-stranded DNA sequence containing a T7 promoter, GFP coding region, and defined UV/radiation-damage sites used to test whether damage reduces BioBits® expression output.
Relation of target to the challenge (max 100 words)
DNA damage can block transcription and translation, so a damaged template should produce less GFP in a cell-free reaction than an intact template. By comparing fluorescence from intact versus damaged DNA, we can estimate how strongly template integrity affects biological function in a space-compatible system. This is scientifically interesting because it connects a major space biology problem, radiation-induced molecular damage, to a direct functional readout. It also follows the logic from the recording that cell-free systems are powerful because we manually specify the parts and can rebuild and test pathways step by step.
Hypothesis / research goal (max 150 words)
My hypothesis is that DNA templates exposed to damaging conditions will produce measurably lower GFP fluorescence in the BioBits® cell-free system than matched undamaged templates. The reasoning is that radiation-related lesions can interfere with transcription by RNA polymerase and reduce the amount of functional protein produced. A cell-free system is ideal for this because it isolates the molecular effect of template damage without the added complexity of living cells, matching the recording’s emphasis on full manual control over system components. The research goal is to develop a simple, portable assay for functional DNA damage detection in space using only the Genes in Space toolkit. In the future, this approach could help evaluate DNA stability in spacecraft environments or screen protective strategies for long-duration missions.
Experimental plan (max 100 words)
I would test two DNA samples: an intact GFP template and a deliberately damaged GFP template, plus a no-DNA negative control. miniPCR would amplify the template if needed, then equal amounts of DNA would be added to separate BioBits® reactions. After hydration, reactions would incubate for protein expression, and GFP fluorescence would be measured with the P51 Molecular Fluorescence Viewer. The main data would be fluorescence intensity for each condition. Lower fluorescence in the damaged-template reaction compared with the intact-template reaction would support the hypothesis that DNA damage reduces functional gene expression in space-compatible cell-free systems.
Individual Final Project
The slide has been posted in the slide deck and I am still working on filling the form.
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Unfortunately, I was not able to contribute to the canvas that week as I had no signal. The idea of people from all over the world collaborating on a single piece, everyone bringing their own little piece of creativity into something unified through synthetic biology, is such a cool concept.
What I really liked was how the project showcased everyone’s creativity in such a tangible way. I also loved reading the comments where people were playfully “fighting” over pixels and trying not to get their work erased. That kind of interaction made it feel like a real community experience rather than just an assignment.
For next year, it would be great if the editing window opened earlier or lasted longer, maybe two full weeks instead of just three days. I really wanted to participate. It would also be cool if there was some kind of live view or timelapse so you could watch the artwork evolve in real time, seeing how the whole thing came together pixel by pixel would be a good addition to the experience.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
1. Component Roles in the NMP-Ribose Cell-Free Reaction
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): The lysate provides the core cellular machinery required for transcription and translation, including ribosomes, tRNAs, translation factors, and metabolic enzymes. The inclusion of T7 RNA Polymerase enables efficient transcription of genes placed under a T7 promoter.
Salts/Buffer
Potassium Glutamate: Serves as the primary monovalent salt in the reaction, maintaining ionic strength and stabilizing ribosome activity and protein folding.
HEPES-KOH pH 7.5: Acts as a buffering agent to maintain a stable physiological pH throughout the reaction, which is critical for enzymatic activity.
Magnesium Glutamate: Provides magnesium ions essential for ribosome assembly, stabilization of nucleic acid structures, and as a cofactor for many enzymatic reactions.
Potassium Phosphate Monobasic & Dibasic (1.6:1 ratio): Together these contribute to phosphate buffering capacity and provide inorganic phosphate that supports metabolic reactions within the system.
Energy / Nucleotide System
Ribose: Serves as a simple sugar precursor that, together with cellular enzymes from the lysate, can be used to regenerate nucleotides via the pentose phosphate pathway, providing a sustainable energy and nucleotide source.
Glucose: Functions as an additional carbon and energy source, feeding into central metabolic pathways to support ATP regeneration and overall reaction sustainability.
AMP, CMP, UMP: These nucleoside monophosphates serve as precursors that are phosphorylated by endogenous kinases into their triphosphate forms (ATP, CTP, UTP), which are then directly used as energy currency and building blocks for RNA synthesis.
GMP: Similarly serves as a precursor to GTP, supporting both transcription (as a nucleotide building block) and energy-dependent translation processes — though notably listed at 0.00 µM in this formulation (see Bonus).
Guanine: Provides a nucleobase that can be salvaged by the cell’s purine salvage pathway enzymes present in the lysate to synthesize GMP and ultimately GTP, compensating for the absence of exogenously added GMP.
Translation Mix (Amino Acids)
17 Amino Acid Mix: Supplies the standard amino acids required as substrates for ribosomal translation of the target protein, excluding those provided separately.
Tyrosine: Provided separately due to its low solubility at neutral pH, requiring preparation at pH 12 before addition; it is an essential amino acid for protein synthesis.
Cysteine: Also supplied separately as it is particularly sensitive to oxidation and must be handled carefully; it is essential for proteins requiring disulfide bonds or cysteine residues.
Additives
Nicotinamide: Serves as a precursor to NAD⁺, supporting redox reactions and metabolic pathways within the lysate that are necessary for sustained energy regeneration.
Backfill
Nuclease-Free Water: Used to bring the reaction to the final desired volume without introducing RNases or DNases that would degrade the nucleic acid components of the reaction.
2. Main Differences Between the 1-Hour PEP-NTP and 20-Hour NMP-Ribose-Glucose Master Mixes
The most fundamental difference lies in how each system supplies energy and nucleotides. The 1-hour PEP-NTP mix provides energy directly through phosphoenolpyruvate (PEP) as a high-energy phosphate donor and supplies pre-formed NTPs (ATP, GTP, CTP, UTP), enabling rapid transcription and translation but depleting quickly. In contrast, the 20-hour NMP-Ribose-Glucose mix uses a more metabolically regenerative strategy, supplying nucleoside monophosphates and simple sugars (ribose and glucose) that are processed by enzymes in the lysate to continuously regenerate NTPs, thereby sustaining the reaction over a much longer period. Additionally, the 20-hour formulation includes fewer additives (e.g., no spermidine, DMSO, cAMP, NAD, or folinic acid) but adds nicotinamide to support NAD⁺ regeneration, reflecting an overall shift toward a simpler, more sustainable, and cost-effective design.
3. Bonus: How Can Transcription Occur if GMP Is Listed at 0.00 µM?
Transcription can still occur because guanine is included in the reaction and can be converted to GMP, then to GTP, through the purine salvage pathway. Enzymes present in the E. coli lysate, such as hypoxanthine-guanine phosphoribosyltransferase (HGPRT), catalyze the transfer of a ribose-5-phosphate group from phosphoribosyl pyrophosphate (PRPP) onto guanine to generate GMP directly. This GMP is then phosphorylated to GDP and GTP by nucleoside monophosphate and diphosphate kinases also present in the lysate, making exogenous GMP unnecessary as long as the salvage pathway remains active.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
1. Biophysical/Functional Properties of Each Fluorescent Protein
i. sfGFP (Superfolder GFP)
sfGFP was specifically engineered to fold more robustly and faster than standard reporter GFP, and its maturation process involves folding of the β-barrel, torsional rearrangements, cyclization, and oxidation of the chromophore. This exceptionally fast and robust folding makes sfGFP one of the most reliable reporters in cell-free systems, where molecular chaperones are limited and proteins must fold without cellular assistance. sfGFP demonstrated a 3.5-fold faster initial folding rate compared to its predecessor and tolerated challenging fusion partners, making it highly suitable for cell-free expression contexts.
ii. mRFP1
mRFP1 is reported to be a somewhat slowly-maturing monomer, and although it has a lower extinction coefficient, quantum yield, and photostability than DsRed, it matures more than 10 times faster than its tetrameric predecessor. In a cell-free context, the slow maturation relative to sfGFP is a relevant limitation, as the oxidation step required for chromophore formation depends on available dissolved oxygen in the reaction, meaning fluorescence signal readout may be delayed and underrepresent total protein yield, particularly in longer incubation formats.
iii. mKO2 (monomeric Kusabira Orange 2)
mKO2 shows a strong dependence on oxygen tension during the maturation process, and its fluorescence recovery kinetics after reoxygenation are notably slower than those of green fluorescent proteins. This heightened oxygen dependence is particularly relevant in cell-free systems, where oxygen availability can be limited in sealed or anaerobic reaction conditions, meaning mKO2 may yield a significantly reduced fluorescence signal even if the protein is being actively translated.
iv. mTurquoise2
mTurquoise2 is reported to be a rapidly-maturing monomer with very low acid sensitivity. Its exceptionally high quantum yield, improved photostability, and faster maturation relative to its predecessor mTurquoise make it well-suited for cell-free systems, where the pH of the reaction mixture can drift over long incubation periods — the low acid sensitivity ensures that fluorescence readout remains stable and reliable throughout the 20- or 36-hour incubation windows.
v. mScarlet-I
The single amino acid substitution T74I found in mScarlet-I results in a marked maturation acceleration in cells, but at the cost of a moderate decrease in fluorescence quantum yield (0.54) and fluorescence lifetime (3.1 ns), although both values are still higher than those of all previously engineered bright monomeric RFPs. In a cell-free context, this trade-off is generally favorable, faster maturation means a greater fraction of translated protein becomes fluorescent within the incubation window, making mScarlet-I a better real-time reporter than the brighter but slower-maturing mScarlet.
vi. Electra2
Electra2 is a monomeric blue fluorescent protein engineered with optimized intracellular brightness, developed through hierarchical screening in both bacterial and mammalian cells. A relevant functional property for cell-free systems is that, like all blue fluorescent proteins, Electra2 requires molecular oxygen for chromophore maturation and is derived from a GFP-like scaffold — blue FPs as a class are characterized by lower brightness compared to green, yellow, and red counterparts, which means that Electra2’s signal-to-noise ratio in a cell-free readout will be inherently lower and may require longer incubation times or higher DNA template concentrations to produce a detectable fluorescent signal.
2. Hypothesis for Mastermix Optimization
Protein: mKO2
As described above, mKO2 exhibits a strong dependence on oxygen availability during the final oxidation step of chromophore maturation. In a sealed cell-free reaction run over 36 hours, dissolved oxygen in the mastermix is progressively consumed by metabolic activity in the lysate, which is expected to severely limit mKO2 maturation over time, even if transcription and translation remain productive.
Hypothesis: Supplementing the 36-hour cell-free mastermix with a controlled oxygen-releasing compound, such as a dilute concentration of hydrogen peroxide (H₂O₂) or a solid oxygen-releasing material, or alternatively conducting the reaction in a gas-permeable vessel with atmospheric oxygen exchange, would increase the available dissolved oxygen concentration throughout the incubation period. This is expected to accelerate and sustain chromophore oxidation in mKO2, resulting in a greater fraction of translated protein reaching the fully mature, fluorescent state and therefore producing a higher cumulative fluorescence signal at the 36-hour endpoint compared to a standard sealed reaction.
3. Composition
I have not received the e-mail.
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
\section{Project Overview}
My project explores how synthetic biology can be used to build a neuron-like system for basic computation. Instead of relying on traditional electronics, this idea is inspired by the way real neurons use ionic signals to communicate. Recent work has shown that microscale ionic devices can generate controlled ionic currents and modulate neuronal activity, suggesting that ion-based systems could support biological information processing. :contentReference[oaicite:6]{index=6} :contentReference[oaicite:7]{index=7}
The goal of this project is to design a synthetic biological unit that receives an ionic input, processes it through a genetic circuit, and produces an output such as fluorescence. In the future, this system could be expanded with memory and network behavior to support neuromorphic computing applications.
My project explores how synthetic biology can be used to build a neuron-like system for basic computation. Instead of relying on traditional electronics, this idea is inspired by the way real neurons use ionic signals to communicate. Recent work has shown that microscale ionic devices can generate controlled ionic currents and modulate neuronal activity, suggesting that ion-based systems could support biological information processing. :contentReference[oaicite:6]{index=6} :contentReference[oaicite:7]{index=7}
The goal of this project is to design a synthetic biological unit that receives an ionic input, processes it through a genetic circuit, and produces an output such as fluorescence. In the future, this system could be expanded with memory and network behavior to support neuromorphic computing applications.
\section{Main Aims}
\begin{itemize}
\item Build a basic synthetic neuron-like system with ionic input and fluorescent output
\item Add threshold and memory behavior for simple computation
\item Explore how multiple units could form a biological neuromorphic network
\end{itemize}