Week 4 HW: protein-design-part-i
Part A. Conceptual Questions
- How many molecules of amino acids in 500g of meat? A quick search shows that, for example, beef contains ~20–22g of protein per 100g of meat.
Also, it would be good to mention that Raw meat contains more water, so the total weight of amino acids is lower per 100g compared to cooked meat, where water loss concentrates the nutrients (often increasing protein to 28–36g per 100g).
If: 100g = 20g of protein 500g = ? (500*20)/100= 100g of protein An average amino acid molecular weight of ~100 Da (100 g/mol): Moles of amino acids = 100g ÷ 100 g/mol = 1 mol Number of molecules = 1 mol × 6.022 × 10²³ ≈ 6 × 10²³ amino acid molecules
- Why do humans eat beef but do not become a cow, eat fish but do not become fish? The short answer is because of digestion. Proteases in the gut break all dietary proteins down to their constituent free amino acids, meaning the informational content (sequence) of the cow’s proteins is destroyed. Ribosomes then reassemble amino acids in the order dictated by ones own mRNA, encoding human proteins.
Interesting fact I discovered Any intact foreign protein that did slip through is attacked by ones immune system as a non-self antigen which is the basis of food allergies (fragments sometimes escape digestion).
Why are there only 20 natural amino acids? One of the main theories as to why there are only 20 amino acids in the world is attributed to evolution. The “frozen accident” / historical contingency The genetic code was fixed early in evolution (~3.5–4 billion years ago) and became locked in. Changing it would be catastrophic (every codon reassignment would corrupt thousands of proteins simultaneously).
Can you make other non-natural amino acids? Design some new amino acids. Yes, numerous non-natural (or non-canonical) amino acids (ncAAs) can be designed and synthesized using advanced chemical methods, such as palladium-catalyzed C-H bond functionalization. By incorporating unnatural amino acids, scientists can extend the genetic code, creating proteins with unique structural and functional properties.
Where did amino acids come from before enzymes that make them, and before life started? Some of the theories of the origin of amino acids include: a) The Miller-Urey experiment (1953) b) Meteorite delivery c) Hydrothermal vents d) HCN and formaldehyde chemistry e) The “RNA world” scenario
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? It would be LEFT-HANDED. Proteins built from D-amino acids form left-handed α-helices, which are mirror images of the right-handed helices formed by L-amino acids.
Can you discover additional helices in proteins? Proteins can adopt several types of helical structures, each with distinct geometric and hydrogen-bonding characteristics. The α-helix is the most common helical structure found in proteins. Beyond the classic α-helix, several other helical structures exist. There is:
3₁₀-helix is a tighter helix with 3.0 residues per turn. Its hydrogen bonds form between residue i and i + 3, resulting in a rise of about 2.0 Å per residue. This helix often appears at the termini of α-helices rather than as long, independent structures.
π-helix is a rare and wider helix, containing 4.4 residues per turn. Its hydrogen bonding occurs between residue i and i + 5, with a rise of approximately 1.1 Å per residue. π-helices are uncommon but are frequently found at functional sites in proteins.
Polyproline II (PPII) helix has 3.0 residues per turn and lacks intramolecular hydrogen bonds. Instead, it adopts an extended left-handed conformation with a rise of about 3.1 Å per residue. This structure plays an important role in signaling, particularly in binding interactions such as those involving SH3 domains.
Polyproline I (PPI) helix contains 3.3 residues per turn and also lacks intramolecular hydrogen bonds. It is right-handed and more compact, with a rise of about 1.9 Å per residue. This form is typically observed in organic solvents.
Collagen helix consists of approximately 3.3 residues per turn and is stabilized by interchain hydrogen bonds rather than intramolecular ones. Each chain has a rise of about 2.9 Å per residue, and three chains wind together to form a characteristic triple helix. This structure is distinguished by its repeating Gly–X–Y sequence pattern.
Why are most molecular helices right-handed? Natural proteins are composed mainly of L-amino acids. The stereochemistry of the peptide backbone energetically favors right-handed α-helices.
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation? β-sheets hydrogen bond through their edge strands, which have free NH and C=O groups pointing outward, ready to hydrogen bond with another β-sheet edge. Unlike α-helices (where all H-bond donors/acceptors are internally satisfied), β-sheet edges are “sticky.”
Part B: Protein Analysis and Visualization
One of the final individual projects I want to do is the development of a biosensor for the diagnosis of Vibrio Cholerae. To help me learn more about V. Cholerae I will use this assignment to study one of the main toxins that is associated with toxicity.
Cholera toxin (CT) is defined as the major virulence factor of the bacterium Vibrio cholerae, comprising a single A subunit and five identical B subunits in an A-B5 architecture, which causes severe diarrheal symptoms in infected individuals. Cholera toxin is a member of the AB toxin family and is composed of a catalytically active heterodimeric A-subunit linked with a homopentameric B-subunit.
Briefly describe the protein you selected and why you selected it. The protein I have selected is the Cholera enterotoxin subunit A. Cholera toxin subunit A (CTA) is the catalytically active component of the AB₅ cholera enterotoxin secreted by Vibrio cholerae, and it is the central molecular component responsible for the devastating secretory diarrhea that defines cholera disease.
Identify the amino acid sequence of your protein. (For this part I search the Cholera toxin on UniProt, after which i selected the Cholera enterotoxin subunit A)
The amino acid sequence MVKIIFVFFIFLSSFSYANDDKLYRADSRPPDEIKQSGGLMPRGQSEYFDRGTQMNINLYDHARGTQTGFVRHDDGYVSTSISLRSAHLVGQTILSGHSTYYIYVIATAPNMFNVNDVLGAYSPHPDEQEVSALGGIPYSQIYGWYRVHFGVLDEQLHRNRGYRDRYYSNLDIAPAADGYGLAGFPPEHRAWREEPWIHHAPPGCGNAPRSSMSNTCDEKTQSLGVKFLDEYQSKVKRQIFSGYQSDIDTHNRIKDEL
Sequence Lenght 258 Amino Acids
Amino Acid Frequency Using the provided colab notebook I was able to get the amino acid count.
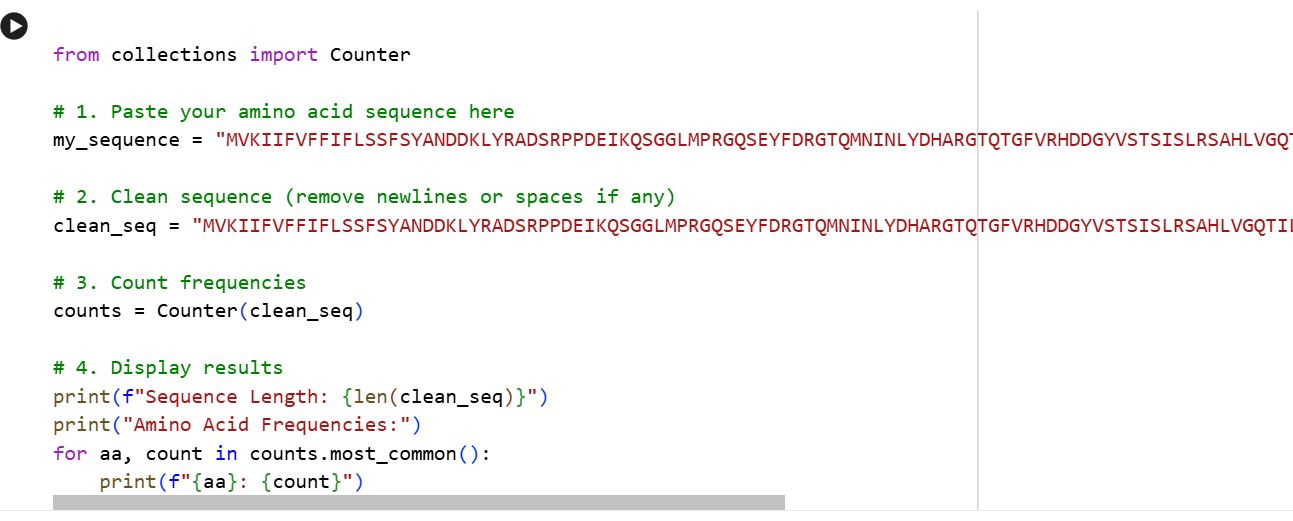 The output is as follows: S: 23, G: 22, D: 19, Y: 18, R: 17, I: 16, L: 16, A: 15, P: 14, V: 13, F: 12, Q: 12, N: 11, E: 11, H: 11, T: 10, K: 8, M: 5, W: 3, C: 2
The letters represent each of the amino acids: S = Serine, G = Glycine, D = Aspartate, Y = Tyrosine, R = Arginine, I = Isoleucine, L = Leucine, A = Alanine, P = Proline, V = Valine, F = Phenylalanine, Q = Glutamine, N = Asparagine, E = Glutamate, H = Histidine, T = Threonine, K = Lysine, M = Methionine, W = Tryptophan, C = Cysteine.
The output is as follows: S: 23, G: 22, D: 19, Y: 18, R: 17, I: 16, L: 16, A: 15, P: 14, V: 13, F: 12, Q: 12, N: 11, E: 11, H: 11, T: 10, K: 8, M: 5, W: 3, C: 2
The letters represent each of the amino acids: S = Serine, G = Glycine, D = Aspartate, Y = Tyrosine, R = Arginine, I = Isoleucine, L = Leucine, A = Alanine, P = Proline, V = Valine, F = Phenylalanine, Q = Glutamine, N = Asparagine, E = Glutamate, H = Histidine, T = Threonine, K = Lysine, M = Methionine, W = Tryptophan, C = Cysteine.
The most abundant amino acid is Serine (S, n=23), closely followed by Glycine (G, n=22) and Aspartate (D, n=19) The overall composition hints at a hydrophilic, surface-exposed protein given the prevalence of polar and charged residues (S, D, R, Y, Q, N, E, H).
Protein Sequence Homologs Protein homologs are proteins that share a common evolutionary origin. They come from the same ancestral gene, even if their sequences or functions have changed over time. There are two main types:
- Orthologs – Proteins in different species that evolved from a common ancestral gene after a speciation event. They often retain similar functions.
- Paralogs – Proteins within the same species that arose by gene duplication. They may evolve new or specialized functions over time.
Through BlastKB there were 250 hits
A comprehensive search for cholera toxin homologs across biological databases yielded 250 results distributed across three domains. The vast majority of results (207 sequences, 83%) came from fungi, particularly Ascomycota species, with notable representations from entomopathogenic fungi in the families Ophiocordycipitaceae, Clavicipitaceae, and Cordycipitaceae, as well as the plant pathogen Colletotrichum (84 results). Bacterial homologs comprised 42 sequences (17%), dominated by Pseudomonadota, including pathogenic species such as Escherichia coli, Vibrio cholerae, and Paraburkholderia, alongside environmental bacteria like Bartonella and Leptospira. A single viral sequence (1 result) was identified as Vibrio phage CTXphi, which is notably the well-characterized prophage known to carry the cholera toxin genes themselves, validating the search methodology. The predominance of fungal and bacterial sequences suggests that toxin-like proteins with structural or functional homology to cholera toxin are widespread across microbial organisms, though the biological significance of these fungal matches warrants further investigation given that toxin production is not a typical trait of this kingdom.

Protein Family Based on the results, the protein belongs to the Cholera enterotoxin subunit A family.
- Identify the structure page of your protein in RCSB When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
It was solved on 2004-04-06. It has a resolution of 1.9 Å which was obtained through X-RAY DIFFRACTION. I would say it has good quality since the resolution is smaller than 2.70 Å.

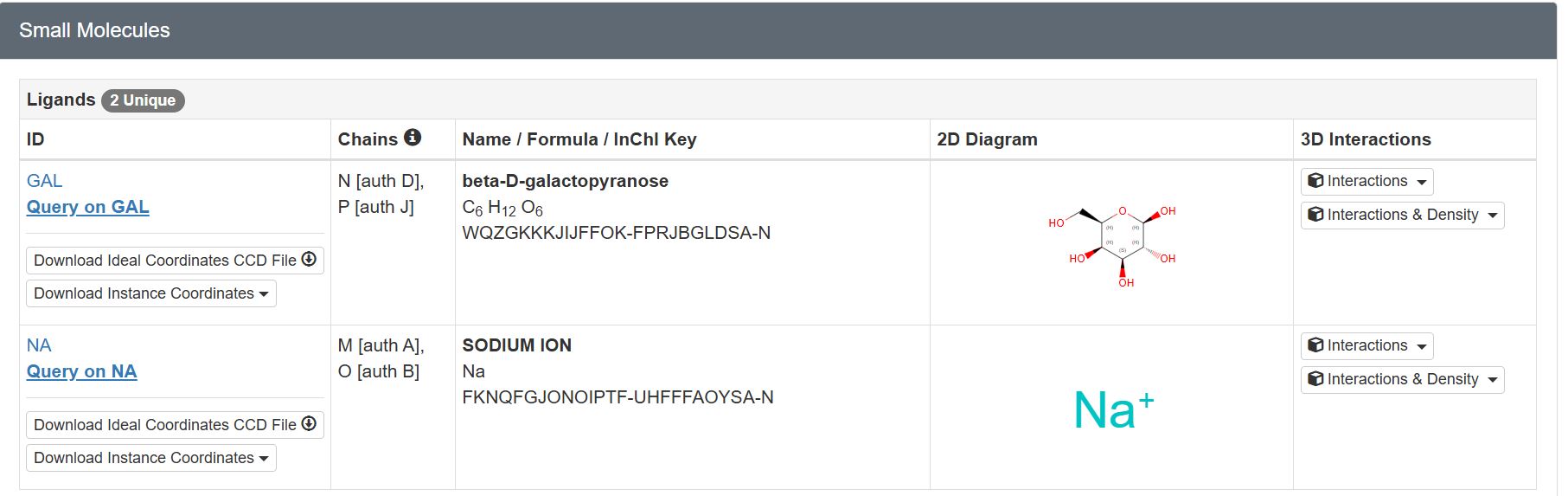
Are there any other molecules in the solved structure apart from protein?
There are two unique ligands present in the structure, specifically: beta-D-galactopyranose and Sodium Ion.
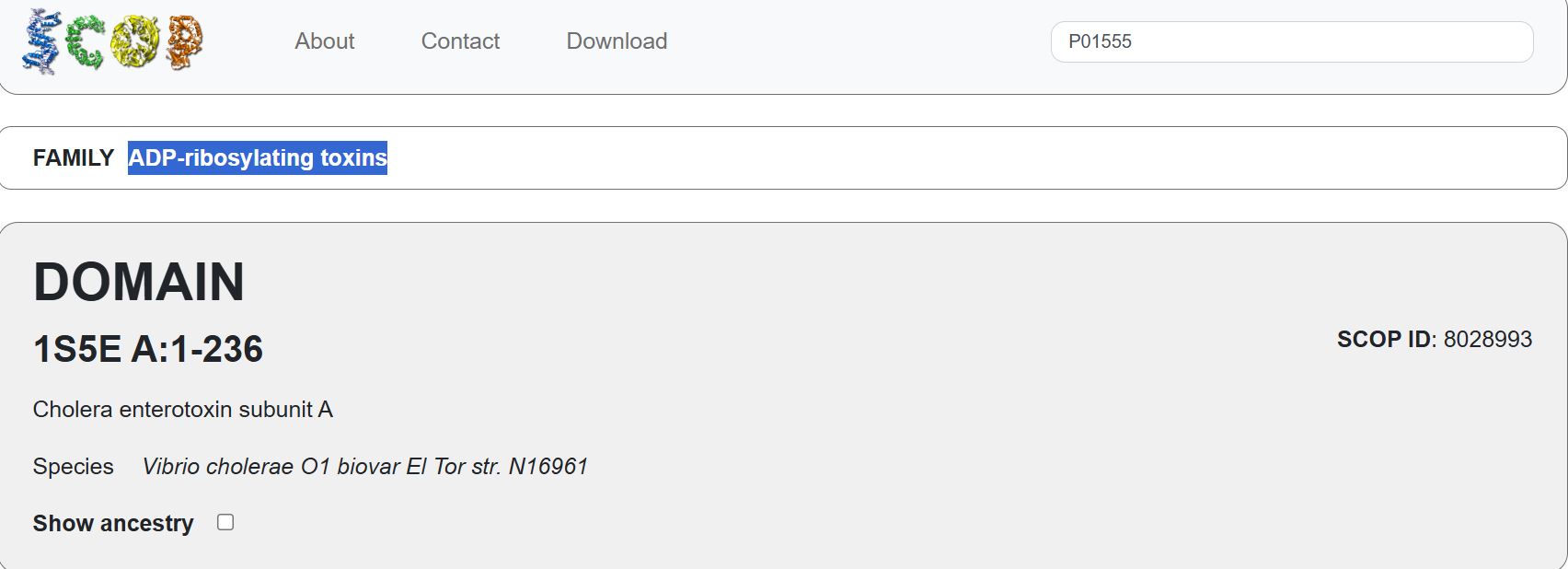
Does your protein belong to any structure classification family?
Yes. It belongs to the ADP-ribosylating toxins. (NOT SURE ABOUT THIS ANSWER)
- Open the structure of your protein in any 3D molecule visualization software: Using PyMol I wanted to visualize my protein of interest. Download the PDB file for the protein, then on PyMol, go to file then open, select the downloaded PDB file. (Chatgpt helped with the python code for the different visualizations)
- Cartoon
I used the following commands
PyMOL>hide everything
PyMOL>show cartoon
PyMOL>color red
PyMOL>util.cbc (color by secondary structure)
- Ribbon
I used the following commands
PyMOL>hide everything
PyMOL>show ribbon
Ball and Stick
I used the following command
yMOL>hide everything
PyMOL>show sticks
PyMOL>show spheres
To Improve structure
PyMOL>set sphere_scale, 0.25
PyMOL>set stick_radius, 0.15

To answer the following questions I used chapgpt to explain how to identify the different structures and also to understand the residues
- Color the protein by secondary structure. Does it have more helices or sheets? By default, PyMOL colors:
Red → α-helices
Yellow → β-sheets
Green → loops/coils
The protein has α-helices that appeared as a spiral / corkscrew shape, Long cylindrical coils and was red It also has β-Sheets that appeared as Flat arrow-shaped strands, Multiple arrows next to each other and were yellowSpiral/corkscrew It also had Loops that appeared as Thin connecting regions that were green
To tell whether I have more helices or sheets, I ran the following code PyMOL>select helices, ss h Selector: selection “helices” defined with 3854 atoms. PyMOL>select sheets, ss s Selector: selection “sheets” defined with 2744 atoms.
My protein has more Helices
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues? “hydrophobic” defined with 4211 atoms. “hydrophilic” defined with 3626 atoms.
There are more hydrophobic atoms than hydrophilic atoms.
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Part C. Using ML-Based Protein Design Tools
https://colab.research.google.com/drive/1MudK4WnxhWlNfMYfOA1UV0x40TB91vxE#scrollTo=f2a1f254
C1. Protein Language Modeling
- Deep Mutational Scans
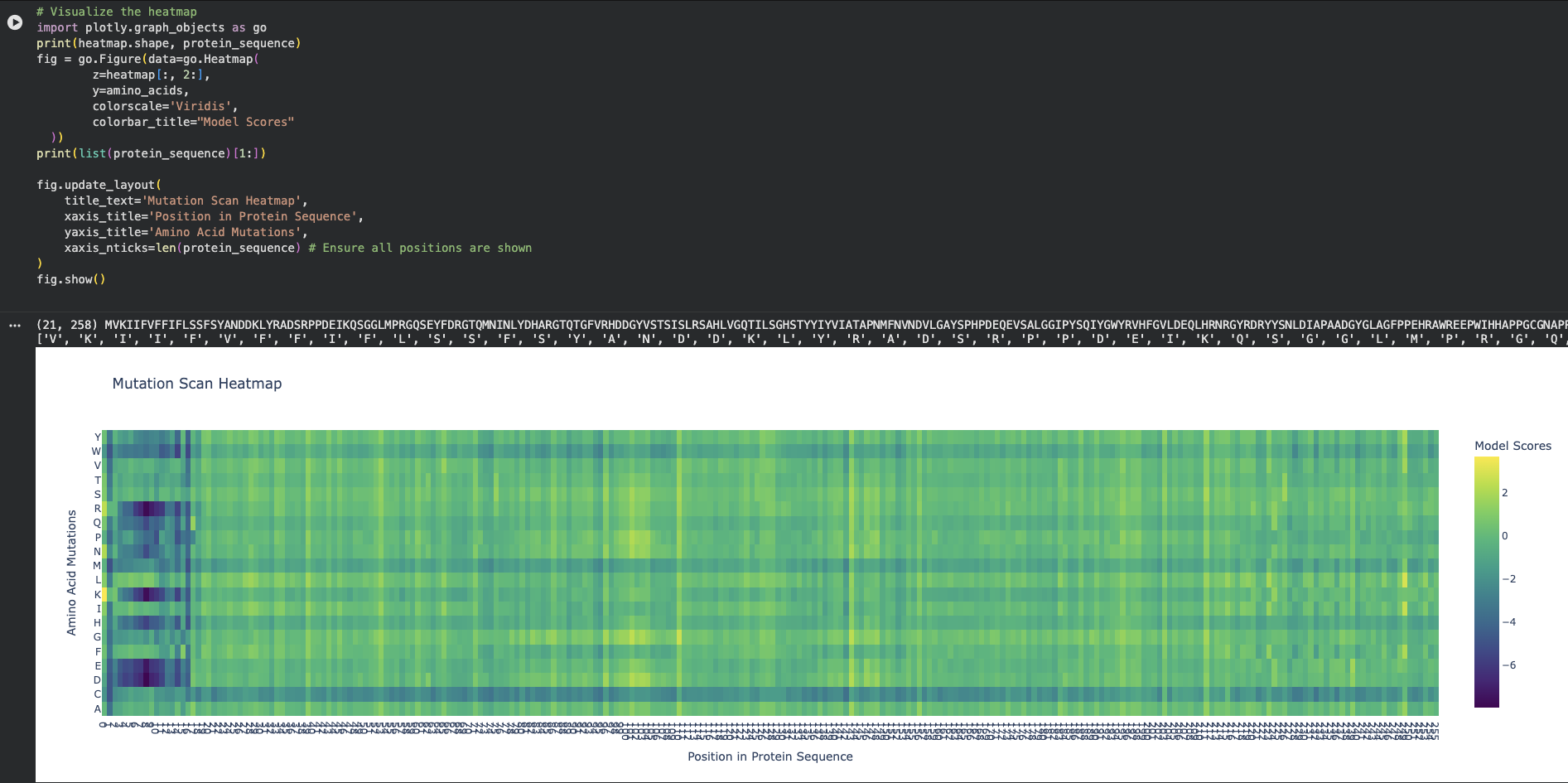
- On the left side of the heatmap (approximately positions 5 to 15 in the protein sequence), there is a massive cluster of dark purple columns.
- At at Position 6 (Phenylalanine - F) or Position 9 (Phenylalanine - F) there are clear mutations
- Latent Space Analysis
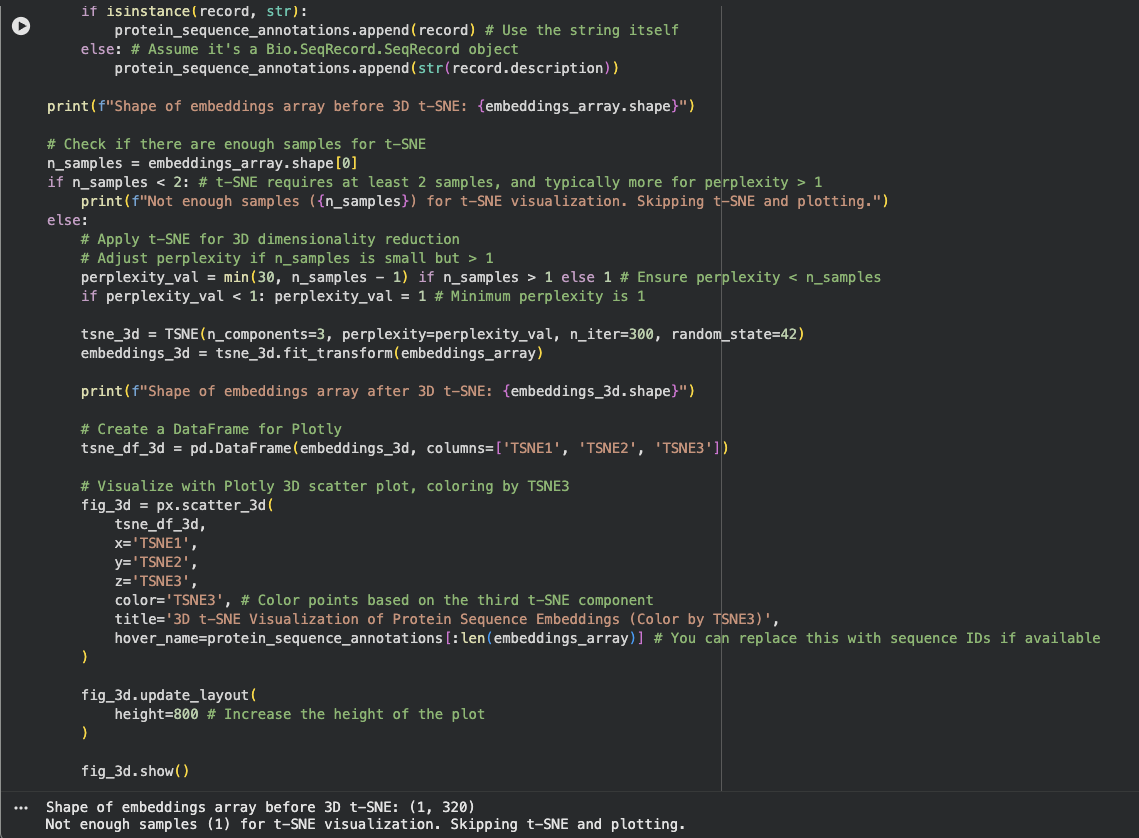
I am not sure what I did wrong here. The output gave me Shape of embeddings array before 3D t-SNE: (1, 320) Not enough samples (1) for t-SNE visualization. Skipping t-SNE and plotting.
I need to review this later
C2. Protein Folding
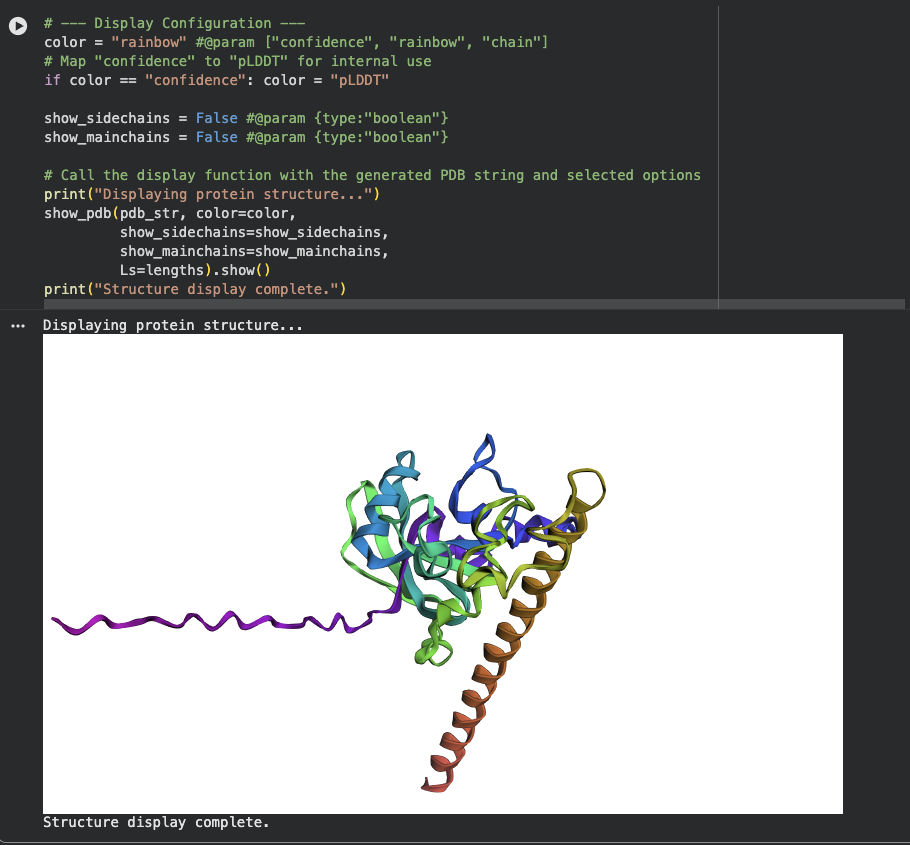
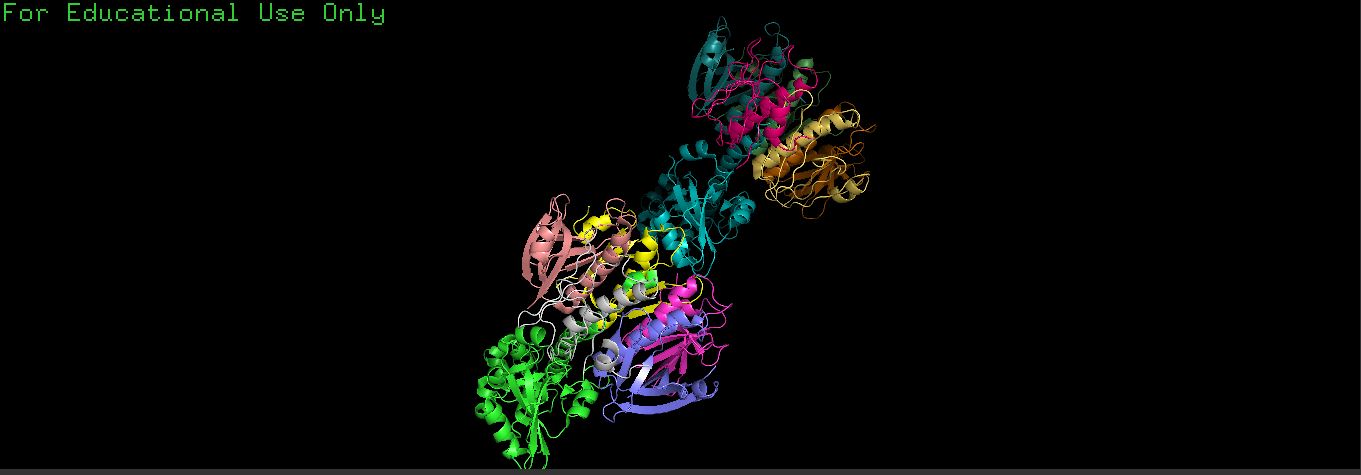
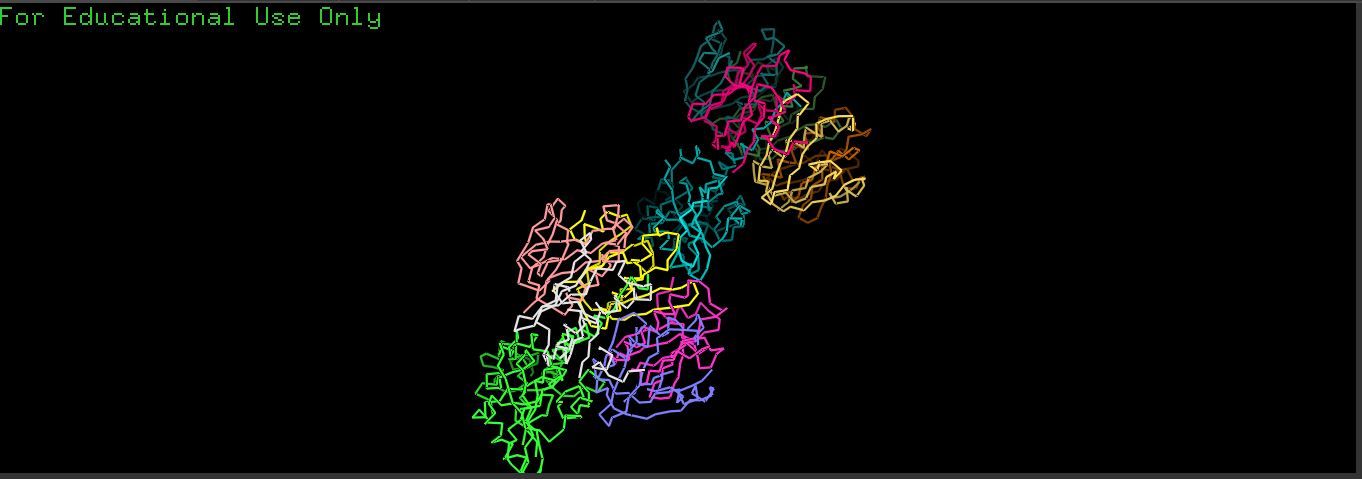
- There is a very distinct, long, prominent alpha-helix colored in a brown/orange gradient at the bottom right in both structures.
- The protein structure from ESMFold looks less dense than the original structure.
C3. Protein Generation
Inverse-Folding a protein
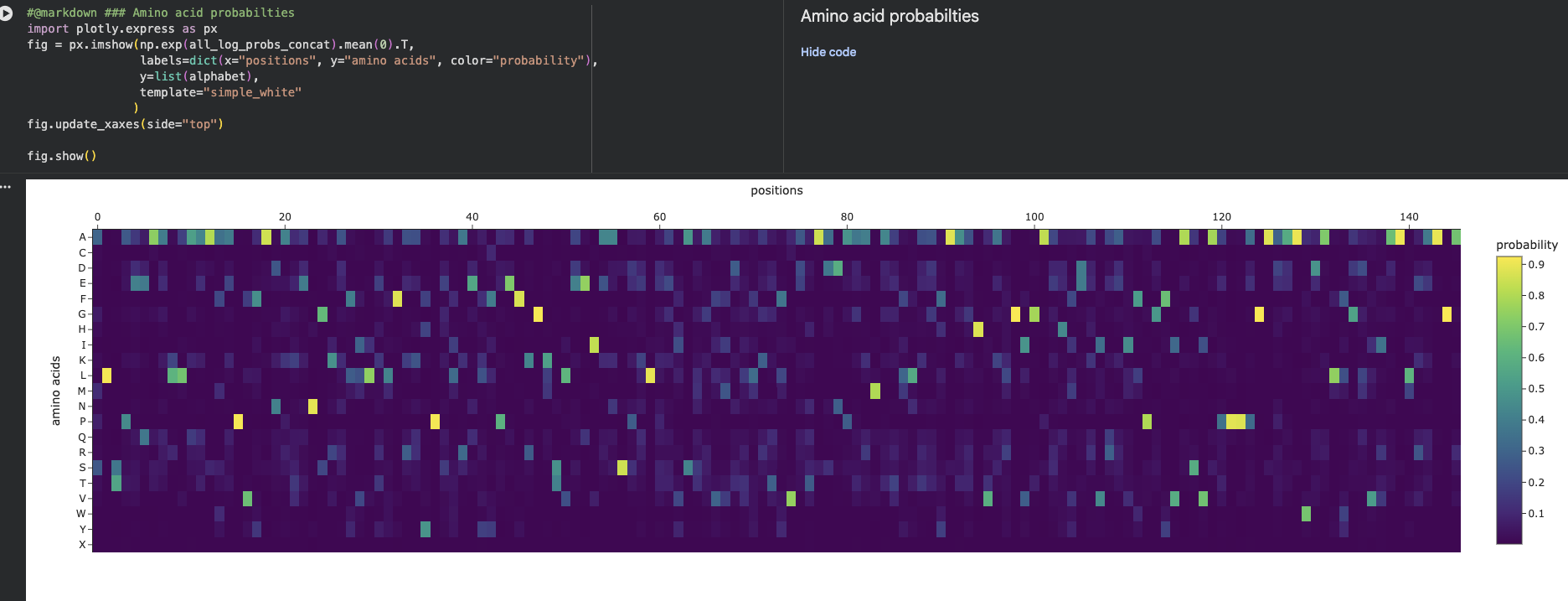
Amino Acid possibilities heatmap
The New Sequence: ALSAEQAALLAAAFAPVAADAEANGRAFILRLFEAYPELRELFPEFKGLSLEEIAASPKLGEIAGAHFALMAEFVATAADAAAMAALLADFAARHVALGIGAAHVEAIRAIHPGFVASVAAPPPGAAAAWDALFGMVIDALRAAGA
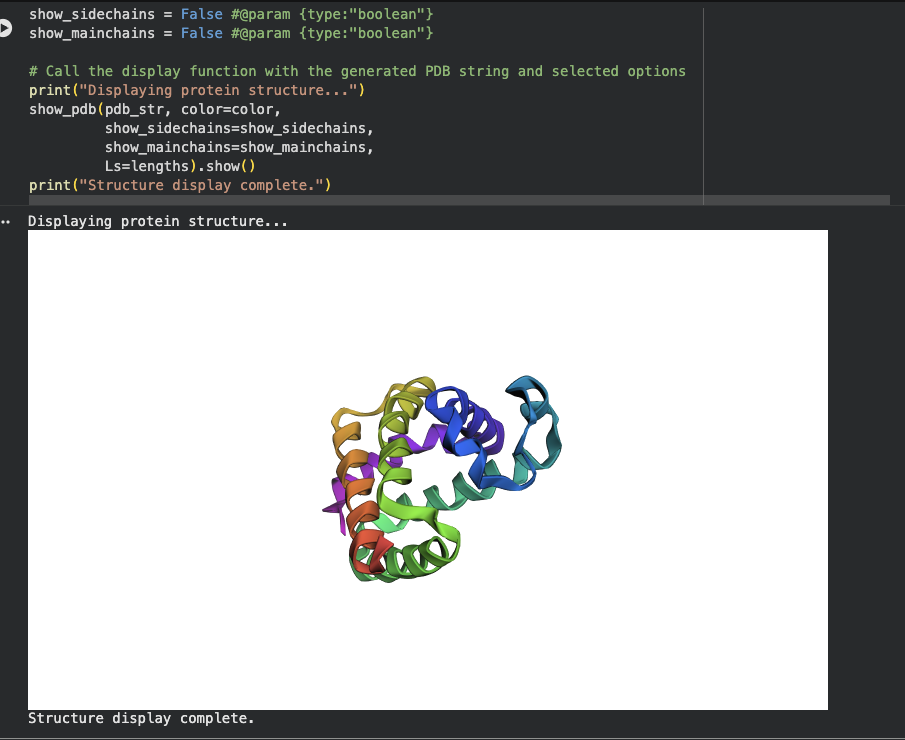
There is a significant difference between the original sequence and the new sequence as pictured when using ESMFold.