A happy, curious soul who loves working with livestock 🐄, though if you dangle schistosomiasis, I might just cave 😆.I’m proudly biased toward animal health, deeply passionate about One Health.
On a typical day, you’ll find me doing science communication, policy advocacy for biotechnology, and figuring out how to better support One Health governance 😊 all while innovating, experimenting, and pushing the needle forward to make the world a little better ⭐ ⭐
I am a HTGAA Committed Listener, my responsibilities are:
• Watching class lectures and recitations
• Participating in node reviews
• Developing and documenting my homework
• Actively communicating with other students and TAs on the forum
• Allowing HTGAA and BioClub to share my work (with attribution)
• Honestly reporting on my work, and appropriately attributing and citing the work of others (both human and non-human)
• Following locally applicable health and safety guidance
• Promoting a respectful environment free of harassment and discrimination
• Signed by committing this file to my documentation page/repository,
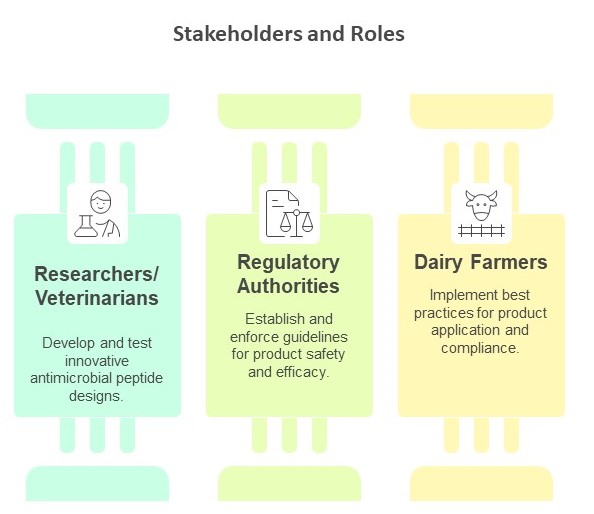
(The Challenge) Mastitis is an inflammation of the mammary gland in dairy cows, often caused by bacterial pathogens. It is a common and costly issue in dairy farms, leading to significant economic losses and affecting overall milk quality(Damasceno et al., 2025). The condition can arise from various factors, including poor hygiene, stress, and injuries to the udder. Common bacterial pathogens responsible for mastitis include Staphylococcus aureus, Escherichia coli, and Streptococcus uberis, which can enter the udder through damaged skin or during milking. Mastitis presents in either clinical or subclinical form. Coliforms from Escherichia coli, Klebsiella spp., and Enterobacter spp. account for 40% of clinical mastitis cases.
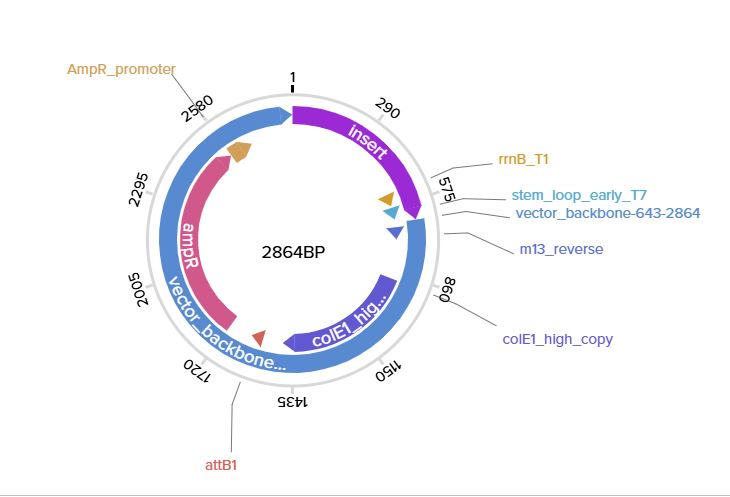
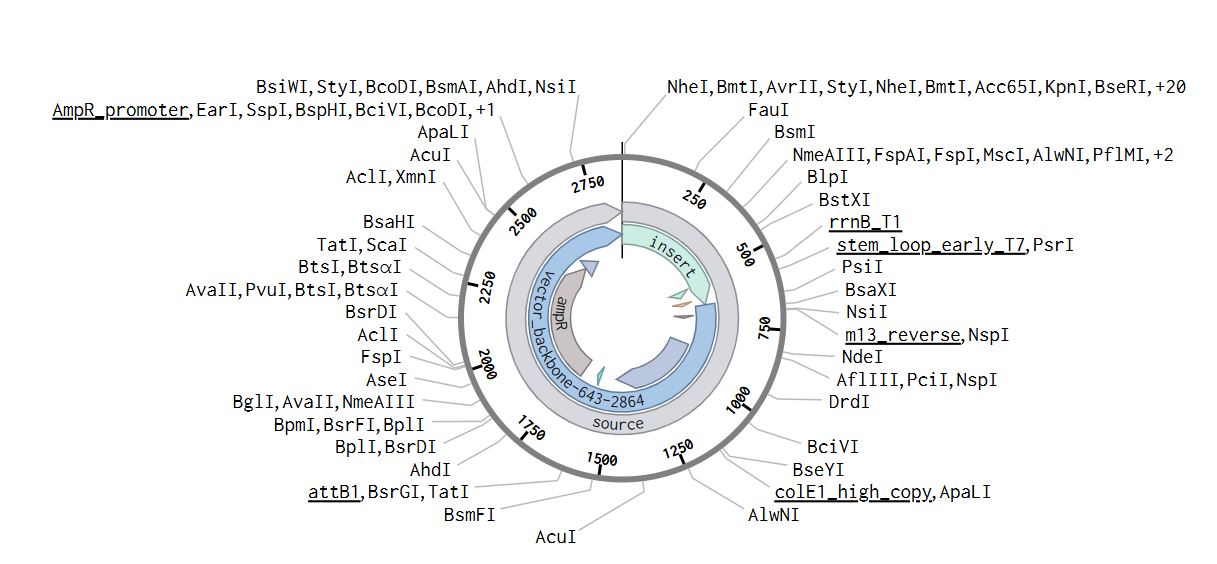
Part 1: Benchling & In-silico Gel Art I began by signing into my Benchling account and creating a new folder called HTGAA-read, write, and edit I then added a new sequence that is the Lambda_NEB fasta sequence On the right-hand side, I used the icon that looks like scissors to enter the different restriction enzymes and then viewed the different digestion sites on the virtual digest tab. Below is the image from the virtual digest for the different enzymes.
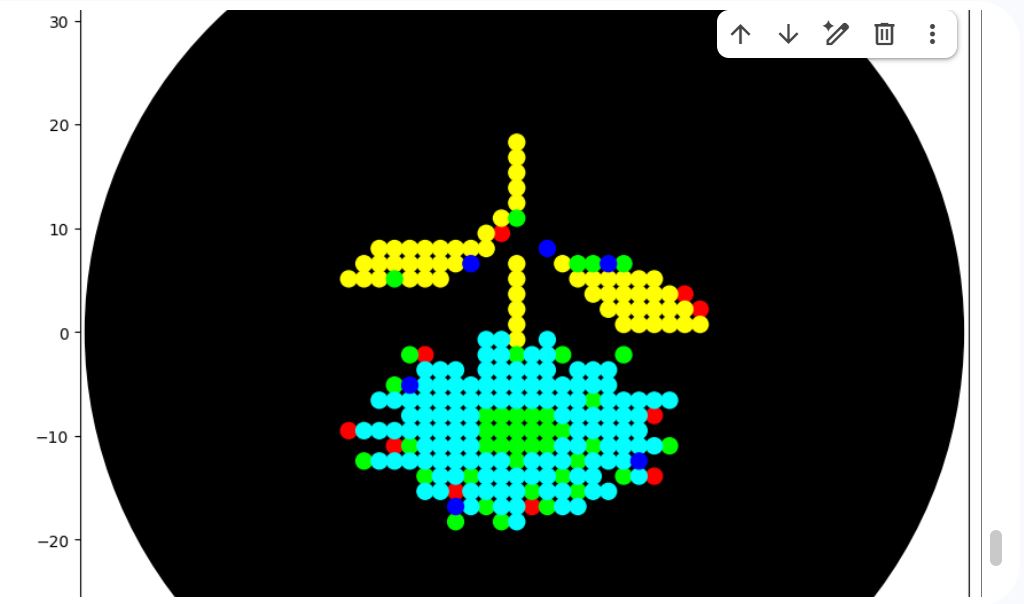
Python Script for Opentrons Artwork I first designed the sunflower using the Opentrons-Art Website. For the design, I went with a sunflower design. https://opentrons-art.rcdonovan.com/?id=e3z1i8r73863y1k
I then downloaded the Excel file, and on HTGAA26 Opentrons Colab, I leveraged Gemini assistance to be able to write the Python script. I uploaded the Excel sheet and promted gemini to assist in developing a script that would give me the sunflower design. Link to the Colab:https://colab.research.google.com/drive/1arsozAVNQhs-4Ol0LMIRKZ4QGVld0Kgf?usp=sharing
Part A. Conceptual Questions How many molecules of amino acids in 500g of meat? A quick search shows that, for example, beef contains ~20–22g of protein per 100g of meat. Also, it would be good to mention that Raw meat contains more water, so the total weight of amino acids is lower per 100g compared to cooked meat, where water loss concentrates the nutrients (often increasing protein to 28–36g per 100g).
Part 1: Generate binders with PepMLM Step 1 — Get the mutant SOD1 sequence Original SOD1 sequence from Uniprot
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V mutant SOD1 sequence
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Step 2 — Run PepMLM Used google colab notebook for the step.
DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? What you add yourself (not in the master mix) Forward primer + Reverse primer — define what region to amplify Template DNA — the sequence to be copied Nuclease-free water — to bring the reaction to final volume
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Integrated Artificial Neural Networks (IANNs) offer superior advantages over traditional Boolean genetic circuits by enabling analog processing, high-dimensional pattern recognition, and robust adaptability. Unlike Boolean circuits (on/off), IANNs handle continuous input levels, allowing complex decision-making, such as classifying disease states based on weighted molecular signatures rather than simple threshold switches.
Homework Part A: General and Lecturer-Specific Questions General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. In traditional in vivo (inside the cell) methods, the cell’s primary goal is its own survival. In CFPS, the goal is purely production.
Homework: Final Project Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements. What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Make a note on your HTGAA webpages including:
what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”) I made 3 dots on the lower right quadrant. In green.
what you liked about the project It was something new and also interesting to see how creative people can be
Subsections of Homework
Week 1 HW: Principles and Practices
(The Challenge)
Mastitis is an inflammation of the mammary gland in dairy cows, often caused by bacterial pathogens. It is a common and costly issue in dairy farms, leading to significant economic losses and affecting overall milk quality(Damasceno et al., 2025). The condition can arise from various factors, including poor hygiene, stress, and injuries to the udder. Common bacterial pathogens responsible for mastitis include Staphylococcus aureus, Escherichia coli, and Streptococcus uberis, which can enter the udder through damaged skin or during milking. Mastitis presents in either clinical or subclinical form. Coliforms from Escherichia coli, Klebsiella spp., and Enterobacter spp. account for 40% of clinical mastitis cases.
While antibiotics remain the primary treatment for clinical mastitis, improper use contributes to antimicrobial resistance (AMR), making infections harder to control. The overuse and misuse of antibiotics in farms contribute to the emergence of drug-resistant mastitis-causing pathogens, further complicating disease management. Mastitis is difficult to eradicate, but its prevalence can be significantly reduced through proper farm management and preventive measures to ensure profitable production in dairy farms. One of such methods is the use of teat dipping disinfectants before and after milking. Current post-milking disinfectants are designed to reduce bacterial exposure and control infections. However, their effectiveness can vary, and there are limitations to their use.
Commercial teat dips typically contain active ingredients such as iodine, chlorhexidine, and lactic acid, each chosen for their bactericidal properties. Iodine-based products have long been favored for their broad-spectrum efficacy; however, concerns about iodine residues in milk are prompting farmers to consider alternatives. Chlorhexidine is another common disinfectant known for its effectiveness against various pathogens, but its performance can vary significantly depending on the formulation. Lactic acid is gaining popularity as well, particularly when combined with hydrogen peroxide, which has shown promising results in reducing bacterial loads. Nevertheless, these traditional disinfectants are not without limitations. For example, their effectiveness might diminish in the presence of organic matter, which can inhibit their action on bacteria (Fitzpatrick et al., 2021).
Reliance on these products may lead to issues such as the development of resistant bacterial strains, raising questions about their long-term efficacy(Damasceno et al., 2025). Given these challenges, the dairy industry is increasingly looking for innovative solutions to enhance mastitis control. This exploration includes synthetic antimicrobial peptides, which could provide a more effective and safer alternative to conventional disinfectants, addressing both efficacy and the concerns associated with traditional products (Ózsvári & Ivanyos, 2022)
(The Intervention)
Synthetic antimicrobial peptides (AMPs) is a promising substitute for traditional post-teat dips in dairy farming. The peptides can be designed to control a broad range of bacteria, making them effective against the pathogens responsible for mastitis. The mechanism of action of AMPs can involve disrupting the bacterial cell membranes, which leads to cell death. This method of attack is different from that of many conventional disinfectants that often rely on chemical reactions.
(The Promise)
A safer, more effective preventative solution to control mastitis.
‘Week 1 HW: Governance Policy Goals’
‘Week 1 HW: ‘Current Status & Potential Governance Actions’
In Kenya, currently, there are no defined regulations for products developed through synthetic biology. Medicines and veterinary products are regulated mainly through the Pharmacy and Poisons Board and the Directorate of Veterinary Services. For products that may have foreign DNA they would be considered as GMOs and therefore would be regulated by the National Biosafety Authority.
There is a need to explore the development of a regulatory pathway within existing Kenyan institutions for synthetic biology-based products. This will ensure that the products meet all safety requirements and that there is public confidence in their safety. However, there is concern that the introduction of these regulations may over-extend approval timelines and regulatory processes, potentially delaying research progress and the deployment of synthetic biology–derived products. The following governance actions are meant to help balance building public trust and ensuring that the products from synthetic biology reach the end users.
Development of a regulatory pathway for the regulation of Syn-Bio products
Picking lessons from the development of gene editing regulatory products, the same aspect can be used to decide on how syn-bio products are regulated. It can be 2-phased, where level 1 is those that do not incorporate foreign DNA, and level 2 is those that involve incorporated foreign DNA. In terms of regulation, a clear pathway can be developed for the two levels, whereby the different government agencies can work together to develop factors such as setting the maximum residue limit in Milk & Milk products, as in the case of antimicrobial peptides.
For this to be achieved, there is a need for buy-in from the National Government, researchers, and regulatory bodies such as NBA-Biotech regulation, NEMA- environment regulation, and directorate of Veterinary Service, and the Public Health Institutes.
One of the main assumptions is that the various organizations have the technical capacity and know-how to regulate synthetic biology products. There is also an assumption that there is an existing risk assessment structure that can easily be adapted for antimicrobial peptides and even those for ranking syn-bio products as high, moderate, and low risk.
Develop and Implement a stewardship plan
Drawing from best practices from stewardship plans from biotech products, a detailed stewardship plan can be developed by regulators and researchers to ensure that farmers use the products as stipulated and that there is a way of ensuring that they are not abused. A quick example would be guidelines on how to ensure traceability and compliance, and use AMP-based dips as part of integrated mastitis management plans and not as the magic bullet that would solve all the problems.
The assumptions made are that the products can be easily traceable and that the farmers would be willing to keep proper records without tokenism involved. The expectation is that the farmers will follow the farm management and record-keeping plan provided, and that they will be honest and not falsify the records.
‘Week 1 HW: ‘Week 2 Lecture Prep’
Homework Questions from Professor Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Error Rate: 1:106
Biology deals with this through proofreading and replacing the mismatched base pairs.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Homework Questions from Dr. LeProust
What’s the most commonly used method for oligo synthesis currently? Phosphoramidite synthesis
Why is it difficult to make oligos longer than 200nt via direct synthesis? There would be compounding inefficiencies, since the addition process is not perfect
Why can’t you make a 2000bp gene via direct oligo synthesis? There would be higher error rates
Homework Question from George Church
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Lysine is an essential amino acid, meaning that we have to eat certain foods to obtain it. Based on the story from Jurassic Park, the scientist inserted a gene that created a single faulty enzyme in protein metabolism. The animals could therefore not manufacture the amino acid lysine. This technically does not matter because with or without the engineered enzyme, the dinosaurs would still be dependent on the food they consume to get the nutrients.
In the event they escaped, they would still eat other lysine-rich foods in nature and would get lysine and therefore survive without being in captivity.
Week 2 HW: Read, write and Edit
Part 1: Benchling & In-silico Gel Art
I began by signing into my Benchling account and creating a new folder called HTGAA-read, write, and edit
I then added a new sequence that is the Lambda_NEB fasta sequence
On the right-hand side, I used the icon that looks like scissors to enter the different restriction enzymes and then viewed the different digestion sites on the virtual digest tab. Below is the image from the virtual digest for the different enzymes.
Part 3: DNA Design Challenge
3.1. Choose your protein
Through a literature search using pubmed I searched for papers that have explored naturally occurring peptides that have antimicrobial activity towards both Gram-positive and Gram-negative bacteria. Staphylococcus aureus (Gram-positive), Escherichia coli (Gram-negative), and Streptococcus uberis (Gram-positive) are the most common mastitis-causing bacteria in dairy farming.
Pituitary adenylate cyclase-activating polypeptide (PACAP) is a naturally occurring cationic peptide known for its strong immunosuppressive and cell-protective effects. It belongs to the secretin, growth hormone-releasing hormone (GHRH), and vasoactive intestinal peptide (VIP) family, and exhibits significant anti-inflammatory and cytoprotective capabilities. While PACAP is most concentrated in the brain, it is also present in notable amounts in other tissues, such as the thymus, spleen, lymph nodes, and duodenal mucosa (Vaudry et al., 2009). Despite its therapeutic potential, the use of PACAP as a pharmaceutical agent is hindered by its extremely short half-life in the bloodstream after systemic delivery, largely due to rapid degradation—particularly by the enzyme DPP IV, which targets the peptide’s amino terminus through exopeptidase activity (Green et al., 2006). PACAP occurs in two bioactive forms, both amidated, consisting of either 38 or 27 amino acids.
Producing PACAP synthetically is crucial for scalability, uniformity, and ethical reasons(Starr et al., 2018). Since the peptide is primarily found in sensitive tissues like the brain and immune organs, extracting it from natural sources is neither feasible nor appropriate for widespread agricultural applications. Instead, recombinant methods using engineered microorganisms enable large-scale, cost-efficient production through fermentation, ensuring consistent quality and purity.
Green, B. D., Irwin, N., & Flatt, P. R. (2006). Pituitary adenylate cyclase-activating peptide (PACAP): Assessment of dipeptidyl peptidase IV degradation, insulin-releasing activity and antidiabetic potential. Peptides, 27(6), 1349–1358. https://doi.org/10.1016/j.peptides.2005.11.010
Starr, C. G., Maderdrut, J. L., He, J., Coy, D. H., & Wimley, W. C. (2018). Pituitary adenylate cyclase-activating polypeptide is a potent broad-spectrum antimicrobial peptide: Structure-activity relationships. Peptides, 104, 35–40. https://doi.org/10.1016/j.peptides.2018.04.006
Vaudry, D., Falluel-Morel, A., Bourgault, S., Basille, M., Burel, D., Wurtz, O., Fournier, A., Chow, B. K. C., Hashimoto, H., Galas, L., & Vaudry, H. (2009). Pituitary Adenylate Cyclase-Activating Polypeptide and Its Receptors: 20 Years after the Discovery. Pharmacological Reviews, 61(3), 283–357. https://doi.org/10.1124/pr.109.001370
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence
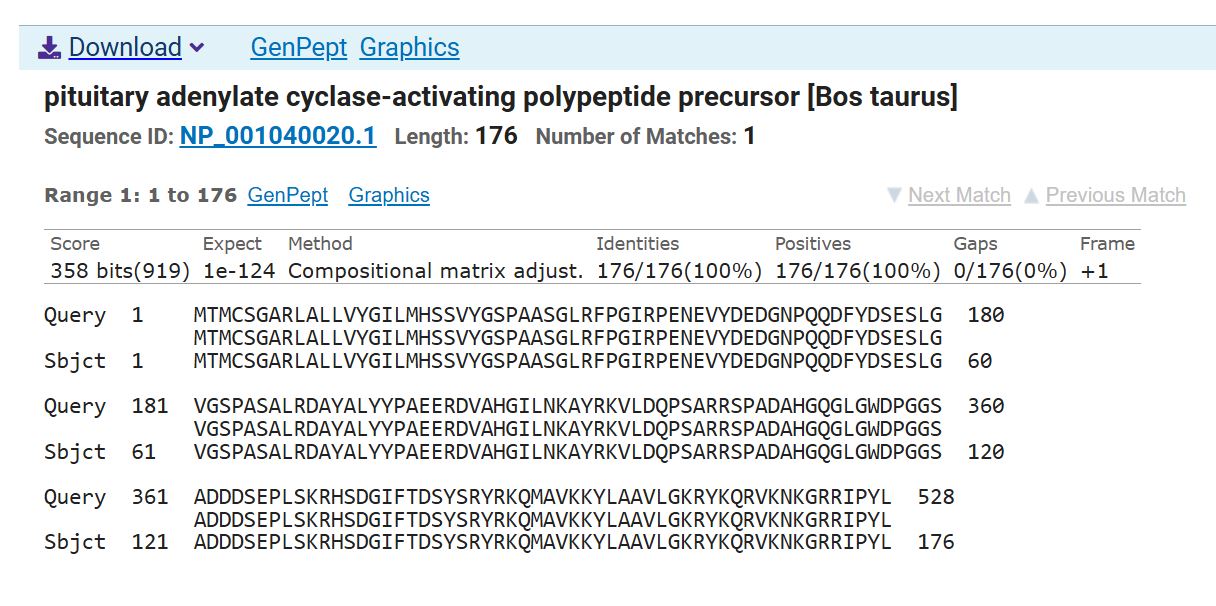
I used the “Reverse Translate” tool of the Sequence Manipulation Suite to get the nucleotide sequence. And confirmed that the sequence was accurate using the NCBI BlastX to confirm that it is accurate.
Screenshot of the BlastX results confirming accurate reverse translation
3.3. Codon optimization
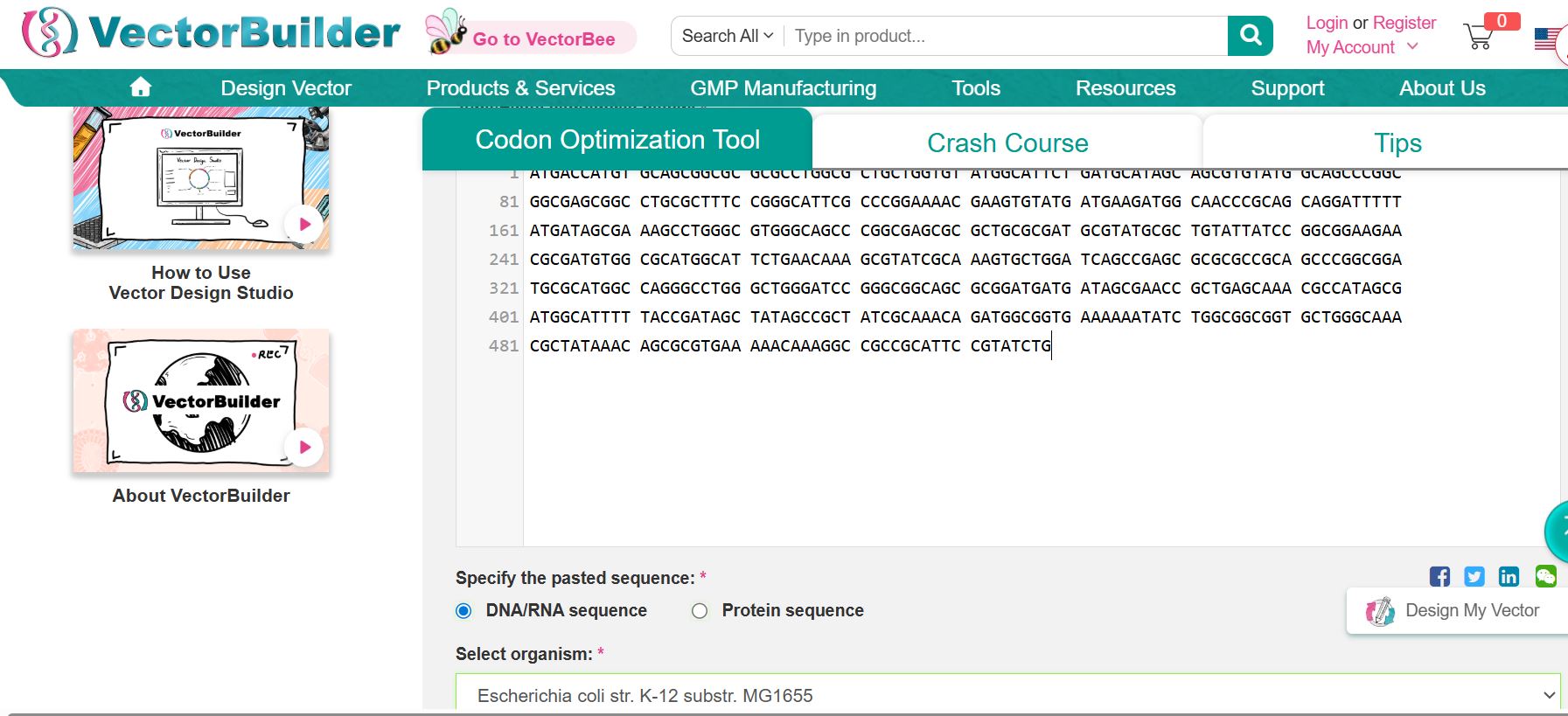
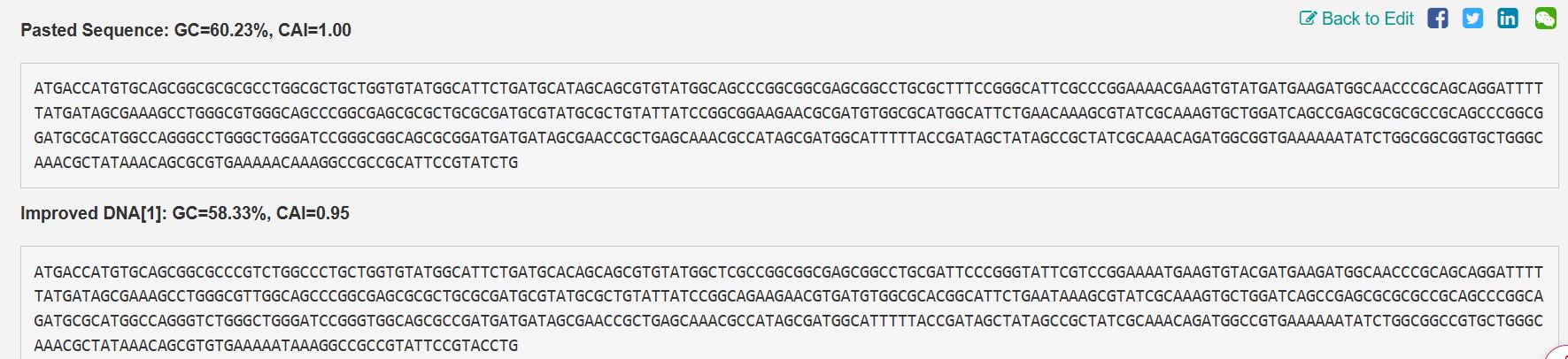
Codon optimization is essential because, while several DNA codons can code for the same amino acid, different species show distinct preferences in codon usage. Expressing the bovine PACAP sequence directly in a microbial host without adjustment could lead to poor translation efficiency and, consequently, low protein output. To support economical, large-scale production, the gene sequence was adapted for use in Escherichia coli, a widely adopted system for recombinant protein expression. By aligning codon usage with the host’s natural preferences, translation becomes more efficient, protein yields improve, and overall production costs decrease, enabling viable industrial synthesis of the antimicrobial peptide.
Codon optimization using VectorBuilder
Improved sequences from VectorBuilder
The following is the improved DNA sequence post-codon optimization
ATGACCATGTGCAGCGGCGCCCGTCTGGCCCTGCTGGTGTATGGCATTCTGATGCACAGCAGCGTGTATGGCTCGCCGGCGGCGAGCGGCCTGCGATTCCCGGGTATTCGTCCGGAAAATGAAGTGTACGATGAAGATGGCAACCCGCAGCAGGATTTTTATGATAGCGAAAGCCTGGGCGTTGGCAGCCCGGCGAGCGCGCTGCGCGATGCGTATGCGCTGTATTATCCGGCAGAAGAACGTGATGTGGCGCACGGCATTCTGAATAAAGCGTATCGCAAAGTGCTGGATCAGCCGAGCGCGCGCCGCAGCCCGGCAGATGCGCATGGCCAGGGTCTGGGCTGGGATCCGGGTGGCAGCGCCGATGATGATAGCGAACCGCTGAGCAAACGCCATAGCGATGGCATTTTTACCGATAGCTATAGCCGCTATC
3.4. You have a sequence! Now what?
Once the codon-optimized DNA sequence for PACAP is available, it needs to be integrated into a biological system capable of transcribing the DNA into mRNA and subsequently translating that mRNA into the PACAP protein. Given the objective of producing PACAP as an affordable antimicrobial peptide for managing mastitis, a cell-based production system using E. coli fermentation is the most suitable choice. This approach offers high protein output, low manufacturing costs, scalability for industrial use, and practical potential for commercial application.
The PACAP gene would first be incorporated into an expression vector, typically a plasmid, which is then delivered into Escherichia coli cells. Within the bacterial host, RNA polymerase recognizes the promoter region and initiates transcription of the PACAP gene into mRNA. Ribosomes then attach to the mRNA and begin translation, synthesizing the PACAP peptide with the help of tRNAs that correspond to the optimized codons. As the bacteria grow and divide, they generate substantial quantities of the peptide, which can later be harvested and purified.
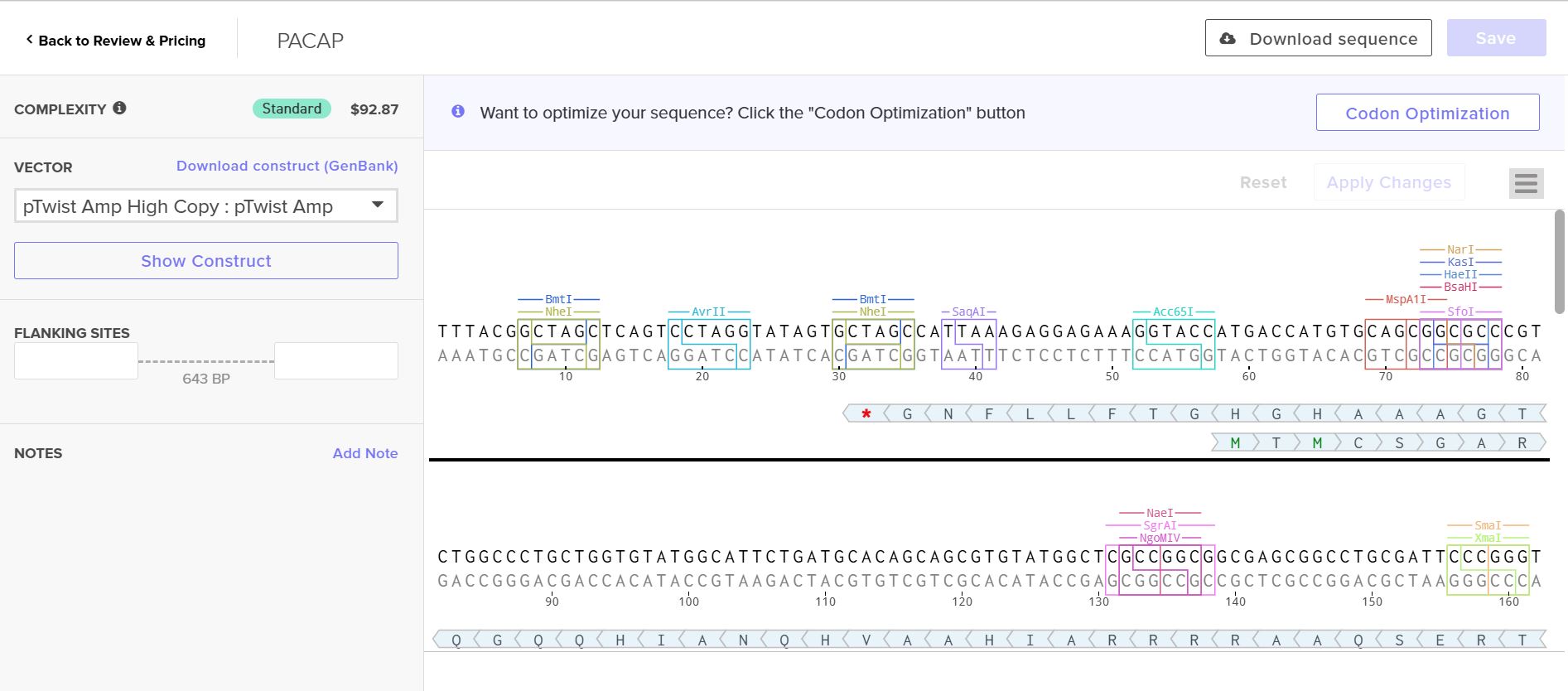
Part 4: Prepare a Twist DNA Synthesis Order
4.2. Build Your DNA Insert Sequence
Following the instructions from the homework, I manually inserted the following sequences sequentially into the bases section
Promoter (e.g. BBa_J23106): TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC
RBS (e.g. BBa_B0034 with spacers for optimal expression): CATTAAAGAGGAGAAAGGTACC
Coding Sequence (The codon optimized sequence already had a start codon so I did not put two start codons)
7x His Tag (Let’s add a 7×His tag at the C-terminus of the protein to enable protein purification from E. coli): CATCACCATCACCATCATCAC
Stop Codon: TAA
Terminator (e.g. BBa_B0015): CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
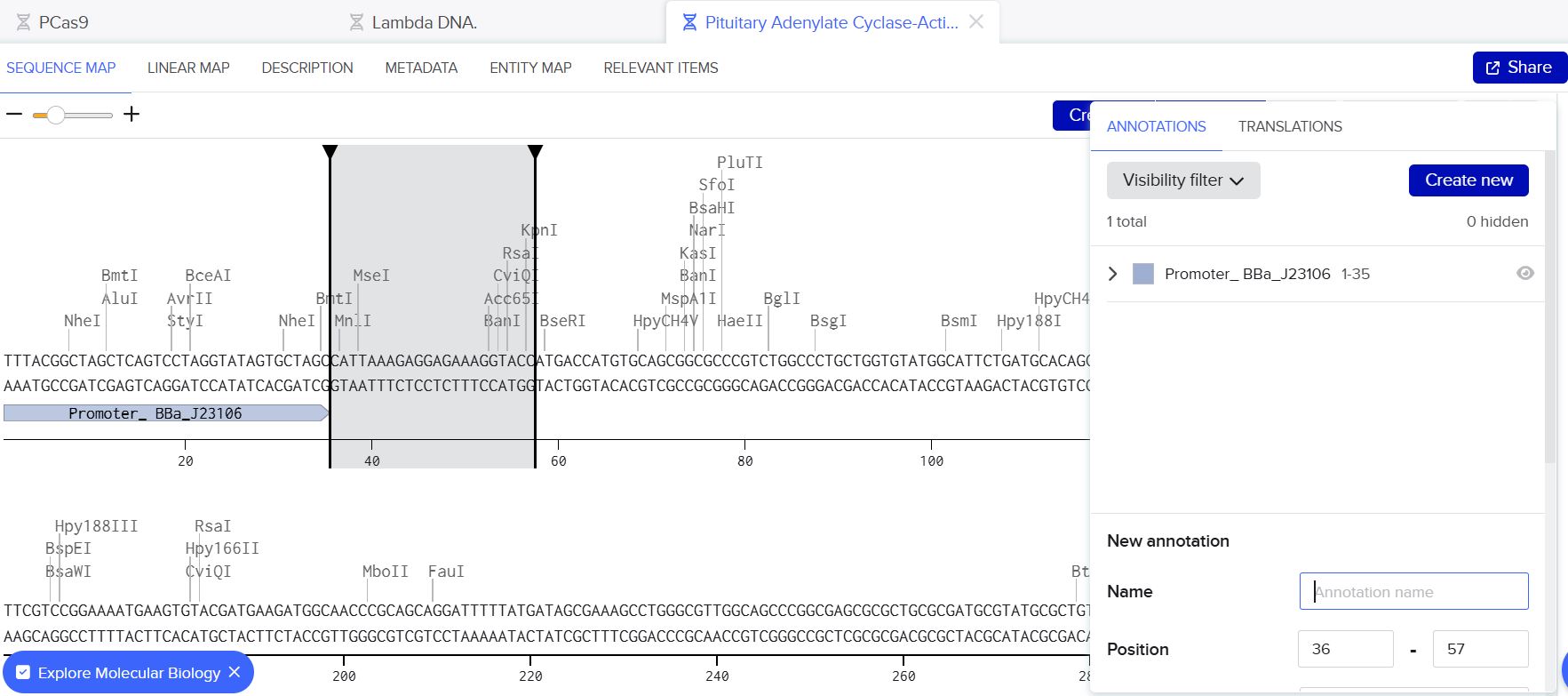
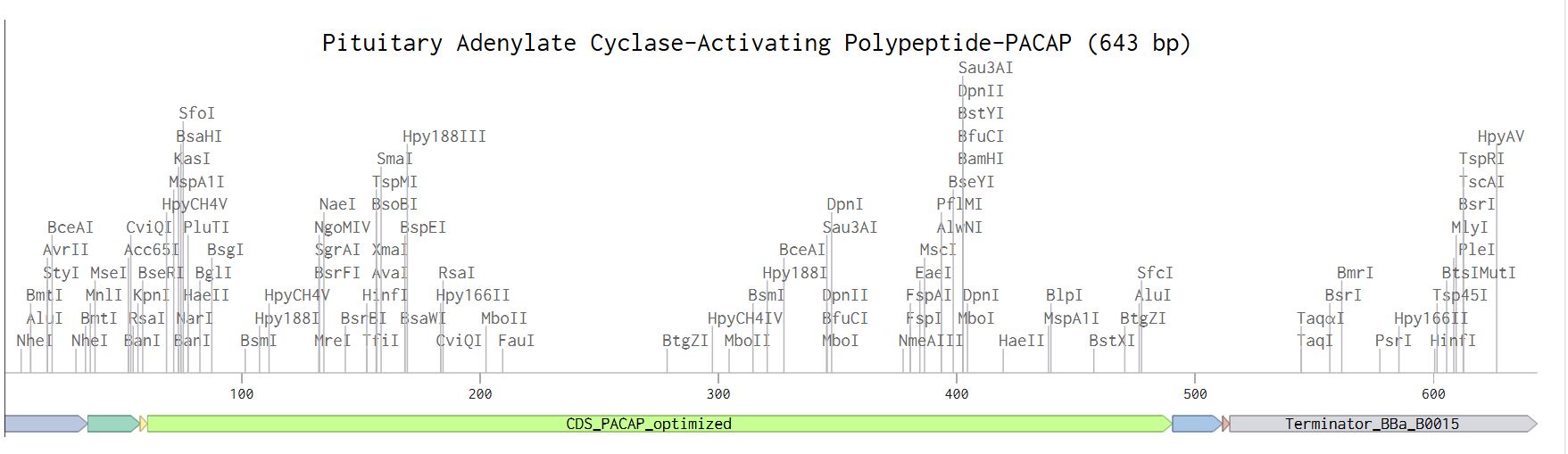
Below are the screenshots from the various steps during the annotation process.
Annotation window
Linear map of the annotated sequence.
Twist Workflow
Construct- Twist
Plasmid with expression cassette - Benchling
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
In the context of mastitis control and epidemiological surveillance, I would sequence metagenomic DNA extracted from milk, teat swabs, and environmental samples collected from dairy farms with and without biosecurity measures. Sequencing this DNA would allow identification of the dominant mastitis-causing bacteria circulating within these farms and enable detection of antibiotic resistance genes present in the microbial populations. This information would provide real-world insight into pathogen prevalence and resistance patterns, which would directly inform the rational design and optimisation of my PACAP-based antimicrobial peptide to ensure it is effective against the most relevant and resistant strains. In addition, I would sequence my final PACAP expression construct to confirm sequence accuracy and ensure that no mutations were introduced during cloning or synthesis.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
For metagenomic analysis of dairy farm samples, I would use shotgun next-generation sequencing (NGS), specifically a short-read platform such as Illumina sequencing. This method is classified as a second-generation sequencing technology.
The input for this method would be total DNA extracted from milk, teat swabs, or environmental samples collected from dairy farms. After extraction, the DNA must be prepared into a sequencing library. (Usually, there is a step by step protocal shared on how to prepare the libraries, including pulling and how to send them for sequencing)
The output of this sequencing technology is a large dataset of short DNA reads in FASTQ format, which includes both the nucleotide sequence and associated quality scores for each base. These reads can then be assembled or mapped against reference databases to identify bacterial species present in the samples and detect antibiotic resistance genes.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would synthesise a codon-optimised version of the PACAP-27 gene designed for efficient expression in Escherichia coli. The sequence would be optimised to match the preferred codon usage of the host organism in order to maximise protein yield. Additionally, based on insights gained from metagenomic surveillance data, I may incorporate rational modifications to improve antimicrobial activity, stability, or resistance to proteolytic degradation. Synthesising the gene rather than amplifying it from a natural source allows full control over sequence design and ensures the construct is tailored specifically to the pathogens identified in dairy farm environments.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use commercial chemical DNA synthesis technology, which relies on phosphoramidite chemistry to generate short oligonucleotides that are enzymatically assembled into full-length genes. This method allows precise sequence customisation, incorporation of optimised codons, and removal of unwanted restriction sites. It is reliable, accurate, and particularly suitable for small genes such as PACAP, making it an efficient approach for generating a ready-to-clone expression construct.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would edit the PACAP gene sequence to enhance its antimicrobial properties while maintaining safety and specificity. Edits may include amino acid substitutions that increase cationic charge, improve membrane disruption, or enhance activity against Gram-negative bacteria and biofilm-forming strains identified in the metagenomic analysis. These modifications would be guided by epidemiological data to ensure the peptide is effective against the most prevalent mastitis pathogens. I may also edit regulatory elements within the expression vector to optimise protein yield or secretion efficiency.
(ii) What technology or technologies would you use to perform these DNA edits and why?
To introduce precise amino acid substitutions into the PACAP gene, I would use site-directed mutagenesis, as it enables targeted and controlled sequence modifications without altering the rest of the construct. Site-directed mutagenesis would be sufficient to generate improved peptide variants informed by metagenomic surveillance data.
I then downloaded the Excel file, and on HTGAA26 Opentrons Colab, I leveraged Gemini assistance to be able to write the Python script.
I uploaded the Excel sheet and promted gemini to assist in developing a script that would give me the sunflower design.
Link to the Colab:https://colab.research.google.com/drive/1arsozAVNQhs-4Ol0LMIRKZ4QGVld0Kgf?usp=sharing
Publication Title: Automation of biochemical assays using an open-sourced, inexpensive robotic liquid handler
While going through the paper, I analyzed two things.
How the Opentrons OT-2 was used.
How was it making it easier?
The Opentrons OT-2 was being used to automate biochemical assays for protein and DNA measurement. Specifically, the researchers developed two assays: the Bradford assay for protein quantification and the PicoGreen assay for double-stranded DNA measurement. These assays are highly relevant to vaccine development, as they provide information on efficacy, potency, safety, and quality control.
Based on this article, the OT-2 simplifies laboratory automation in several ways:
Cost-effective: Unlike traditional commercial liquid handlers that cost over $250,000, this system costs under $10,000, making automation accessible to smaller labs.
User-friendly programming: It uses Python programming language with open-source flexibility, reducing training requirements.
Accurate and precise: The study found the pipettes exhibited excellent accuracy and precision, with relative inaccuracy of 1.30% for the P20 pipette and 0.53% for the P300 pipette.
Time-saving: The Bradford assay completed in 75 minutes and the PicoGreen assay in 41 minutes, reducing manual labor.
How would I leverage this for my work
Based on the work I want to do that is aimed at prpoducing Antimicrobial Peptide at industrial scaled there are several ways I can leverage it to my advantage.
The OT-2 could be adapted for this application in the following ways:
Quality control testing: The automated protein and DNA quantification assays demonstrated here could monitor protein expression levels and detect residual DNA contamination during antimicrobial peptide synthesis and purification.
Assay automation: The system’s ability to run customized Python protocols means I could develop automated workflows for peptide synthesis optimization, mixing reagents, and conducting potency assays.
High-throughput screening: The medium-throughput capability would allow testing multiple peptide variants or production conditions simultaneously to identify the most effective formulations.
Week 4 HW: protein-design-part-i
Part A. Conceptual Questions
How many molecules of amino acids in 500g of meat?
A quick search shows that, for example, beef contains ~20–22g of protein per 100g of meat.
Also, it would be good to mention that Raw meat contains more water, so the total weight of amino acids is lower per 100g compared to cooked meat, where water loss concentrates the nutrients (often increasing protein to 28–36g per 100g).
If:
100g = 20g of protein
500g = ?
(500*20)/100= 100g of protein
An average amino acid molecular weight of ~100 Da (100 g/mol):
Moles of amino acids = 100g ÷ 100 g/mol = 1 mol
Number of molecules = 1 mol × 6.022 × 10²³
≈ 6 × 10²³ amino acid molecules
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The short answer is because of digestion.
Proteases in the gut break all dietary proteins down to their constituent free amino acids, meaning the informational content (sequence) of the cow’s proteins is destroyed. Ribosomes then reassemble amino acids in the order dictated by ones own mRNA, encoding human proteins.
Interesting fact I discovered Any intact foreign protein that did slip through is attacked by ones immune system as a non-self antigen which is the basis of food allergies (fragments sometimes escape digestion).
Why are there only 20 natural amino acids?
One of the main theories as to why there are only 20 amino acids in the world is attributed to evolution.
The “frozen accident” / historical contingency
The genetic code was fixed early in evolution (~3.5–4 billion years ago) and became locked in. Changing it would be catastrophic (every codon reassignment would corrupt thousands of proteins simultaneously).
Can you make other non-natural amino acids? Design some new amino acids.
Yes, numerous non-natural (or non-canonical) amino acids (ncAAs) can be designed and synthesized using advanced chemical methods, such as palladium-catalyzed C-H bond functionalization. By incorporating unnatural amino acids, scientists can extend the genetic code, creating proteins with unique structural and functional properties.
Where did amino acids come from before enzymes that make them, and before life started?
Some of the theories of the origin of amino acids include:
a) The Miller-Urey experiment (1953)
b) Meteorite delivery
c) Hydrothermal vents
d) HCN and formaldehyde chemistry
e) The “RNA world” scenario
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
It would be LEFT-HANDED.
Proteins built from D-amino acids form left-handed α-helices, which are mirror images of the right-handed helices formed by L-amino acids.
Can you discover additional helices in proteins?
Proteins can adopt several types of helical structures, each with distinct geometric and hydrogen-bonding characteristics. The α-helix is the most common helical structure found in proteins. Beyond the classic α-helix, several other helical structures exist.
There is:
3₁₀-helix is a tighter helix with 3.0 residues per turn. Its hydrogen bonds form between residue i and i + 3, resulting in a rise of about 2.0 Å per residue. This helix often appears at the termini of α-helices rather than as long, independent structures.
π-helix is a rare and wider helix, containing 4.4 residues per turn. Its hydrogen bonding occurs between residue i and i + 5, with a rise of approximately 1.1 Å per residue. π-helices are uncommon but are frequently found at functional sites in proteins.
Polyproline II (PPII) helix has 3.0 residues per turn and lacks intramolecular hydrogen bonds. Instead, it adopts an extended left-handed conformation with a rise of about 3.1 Å per residue. This structure plays an important role in signaling, particularly in binding interactions such as those involving SH3 domains.
Polyproline I (PPI) helix contains 3.3 residues per turn and also lacks intramolecular hydrogen bonds. It is right-handed and more compact, with a rise of about 1.9 Å per residue. This form is typically observed in organic solvents.
Collagen helix consists of approximately 3.3 residues per turn and is stabilized by interchain hydrogen bonds rather than intramolecular ones. Each chain has a rise of about 2.9 Å per residue, and three chains wind together to form a characteristic triple helix. This structure is distinguished by its repeating Gly–X–Y sequence pattern.
Why are most molecular helices right-handed?
Natural proteins are composed mainly of L-amino acids. The stereochemistry of the peptide backbone energetically favors right-handed α-helices.
Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation?
β-sheets hydrogen bond through their edge strands, which have free NH and C=O groups pointing outward, ready to hydrogen bond with another β-sheet edge. Unlike α-helices (where all H-bond donors/acceptors are internally satisfied), β-sheet edges are “sticky.”
Part B: Protein Analysis and Visualization
One of the final individual projects I want to do is the development of a biosensor for the diagnosis of Vibrio Cholerae. To help me learn more about V. Cholerae I will use this assignment to study one of the main toxins that is associated with toxicity.
Cholera toxin (CT) is defined as the major virulence factor of the bacterium Vibrio cholerae, comprising a single A subunit and five identical B subunits in an A-B5 architecture, which causes severe diarrheal symptoms in infected individuals. Cholera toxin is a member of the AB toxin family and is composed of a catalytically active heterodimeric A-subunit linked with a homopentameric B-subunit.
Briefly describe the protein you selected and why you selected it.
The protein I have selected is the Cholera enterotoxin subunit A. Cholera toxin subunit A (CTA) is the catalytically active component of the AB₅ cholera enterotoxin secreted by Vibrio cholerae, and it is the central molecular component responsible for the devastating secretory diarrhea that defines cholera disease.
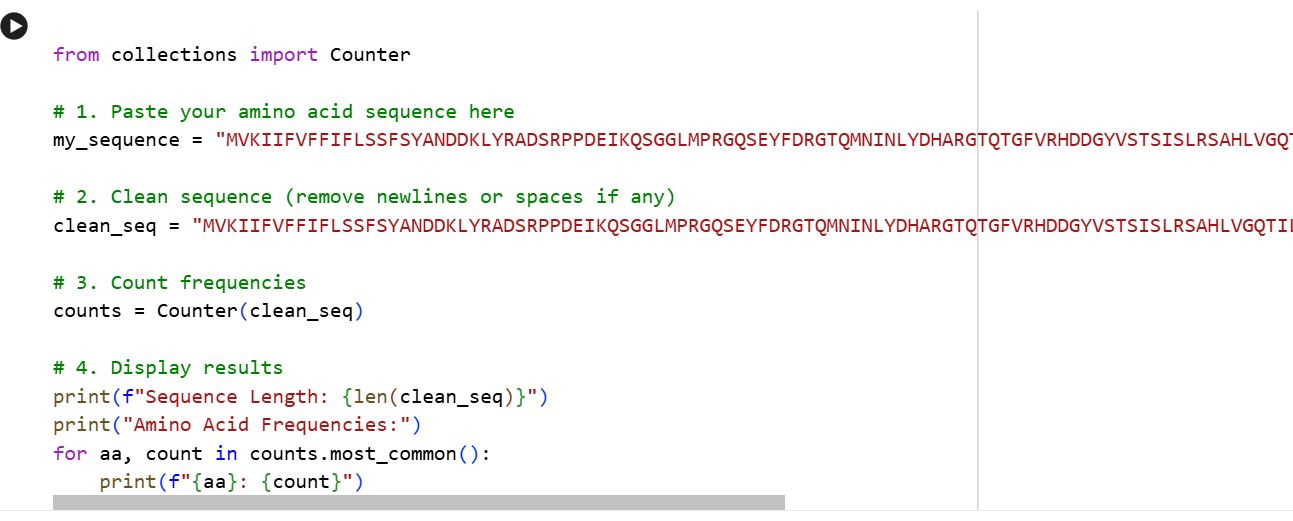
Identify the amino acid sequence of your protein. (For this part I search the Cholera toxin on UniProt, after which i selected the Cholera enterotoxin subunit A)
The amino acid sequence
MVKIIFVFFIFLSSFSYANDDKLYRADSRPPDEIKQSGGLMPRGQSEYFDRGTQMNINLYDHARGTQTGFVRHDDGYVSTSISLRSAHLVGQTILSGHSTYYIYVIATAPNMFNVNDVLGAYSPHPDEQEVSALGGIPYSQIYGWYRVHFGVLDEQLHRNRGYRDRYYSNLDIAPAADGYGLAGFPPEHRAWREEPWIHHAPPGCGNAPRSSMSNTCDEKTQSLGVKFLDEYQSKVKRQIFSGYQSDIDTHNRIKDEL
Sequence Lenght 258 Amino Acids
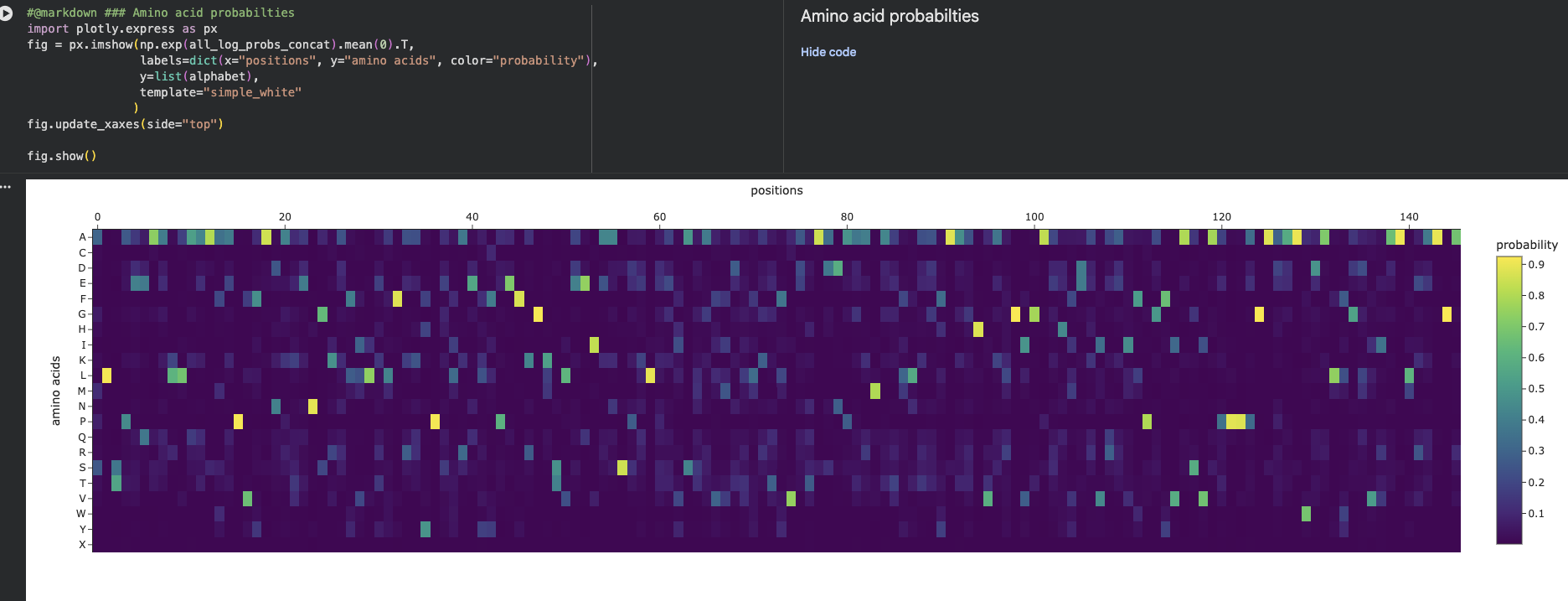
Amino Acid Frequency Using the provided colab notebook I was able to get the amino acid count.
The output is as follows: S: 23, G: 22, D: 19, Y: 18, R: 17, I: 16, L: 16, A: 15, P: 14, V: 13, F: 12, Q: 12, N: 11, E: 11, H: 11, T: 10, K: 8, M: 5, W: 3, C: 2
The letters represent each of the amino acids: S = Serine, G = Glycine, D = Aspartate, Y = Tyrosine, R = Arginine, I = Isoleucine, L = Leucine, A = Alanine, P = Proline, V = Valine, F = Phenylalanine, Q = Glutamine, N = Asparagine, E = Glutamate, H = Histidine, T = Threonine, K = Lysine, M = Methionine, W = Tryptophan, C = Cysteine.
The most abundant amino acid is Serine (S, n=23), closely followed by Glycine (G, n=22) and Aspartate (D, n=19) The overall composition hints at a hydrophilic, surface-exposed protein given the prevalence of polar and charged residues (S, D, R, Y, Q, N, E, H).
Protein Sequence Homologs
Protein homologs are proteins that share a common evolutionary origin. They come from the same ancestral gene, even if their sequences or functions have changed over time. There are two main types:
Orthologs – Proteins in different species that evolved from a common ancestral gene after a speciation event. They often retain similar functions.
Paralogs – Proteins within the same species that arose by gene duplication. They may evolve new or specialized functions over time.
Through BlastKB there were 250 hits
A comprehensive search for cholera toxin homologs across biological databases yielded 250 results distributed across three domains. The vast majority of results (207 sequences, 83%) came from fungi, particularly Ascomycota species, with notable representations from entomopathogenic fungi in the families Ophiocordycipitaceae, Clavicipitaceae, and Cordycipitaceae, as well as the plant pathogen Colletotrichum (84 results). Bacterial homologs comprised 42 sequences (17%), dominated by Pseudomonadota, including pathogenic species such as Escherichia coli, Vibrio cholerae, and Paraburkholderia, alongside environmental bacteria like Bartonella and Leptospira. A single viral sequence (1 result) was identified as Vibrio phage CTXphi, which is notably the well-characterized prophage known to carry the cholera toxin genes themselves, validating the search methodology. The predominance of fungal and bacterial sequences suggests that toxin-like proteins with structural or functional homology to cholera toxin are widespread across microbial organisms, though the biological significance of these fungal matches warrants further investigation given that toxin production is not a typical trait of this kingdom.
Protein Family
Based on the results, the protein belongs to the Cholera enterotoxin subunit A family.
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
It was solved on 2004-04-06. It has a resolution of 1.9 Å which was obtained through X-RAY DIFFRACTION. I would say it has good quality since the resolution is smaller than 2.70 Å.
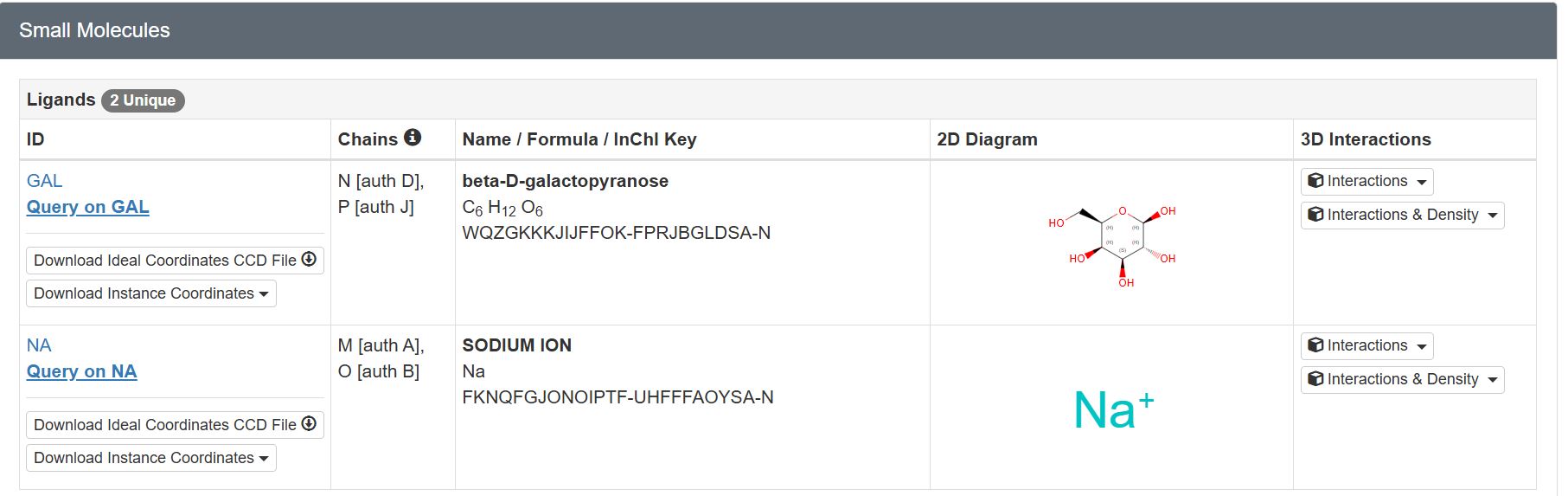
Are there any other molecules in the solved structure apart from protein?
There are two unique ligands present in the structure, specifically: beta-D-galactopyranose and Sodium Ion.
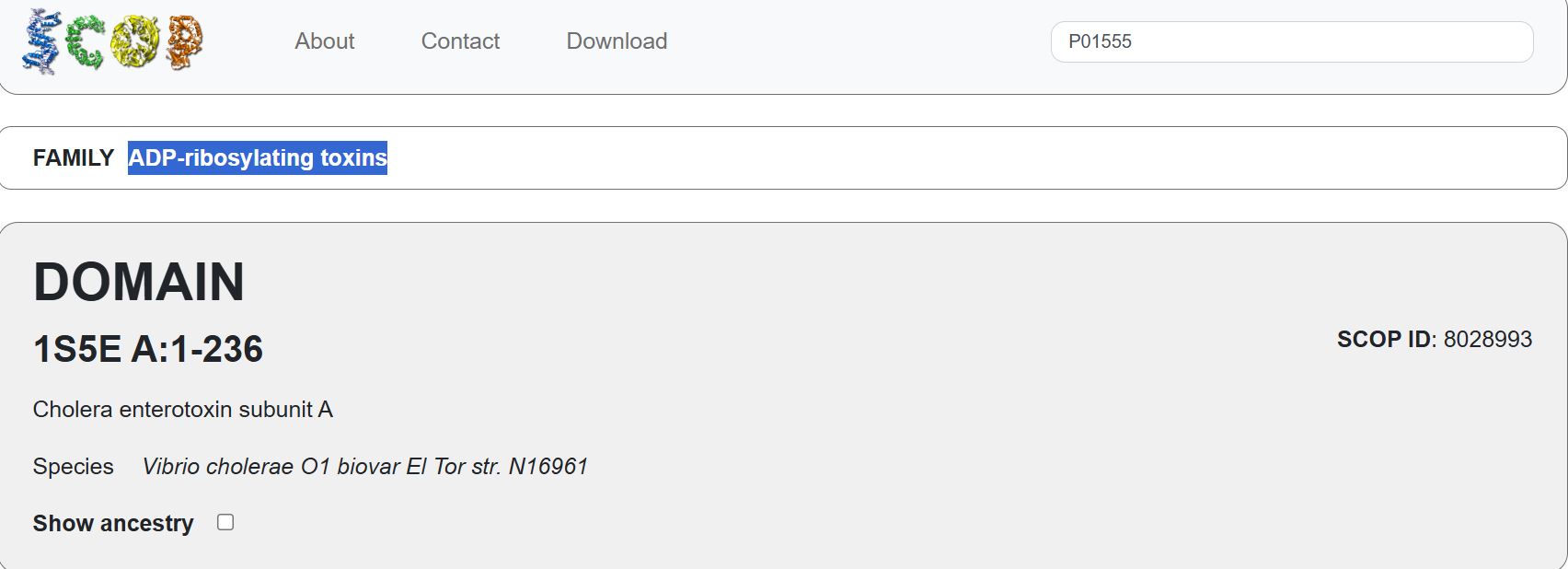
Does your protein belong to any structure classification family?
Yes. It belongs to the ADP-ribosylating toxins. (NOT SURE ABOUT THIS ANSWER)
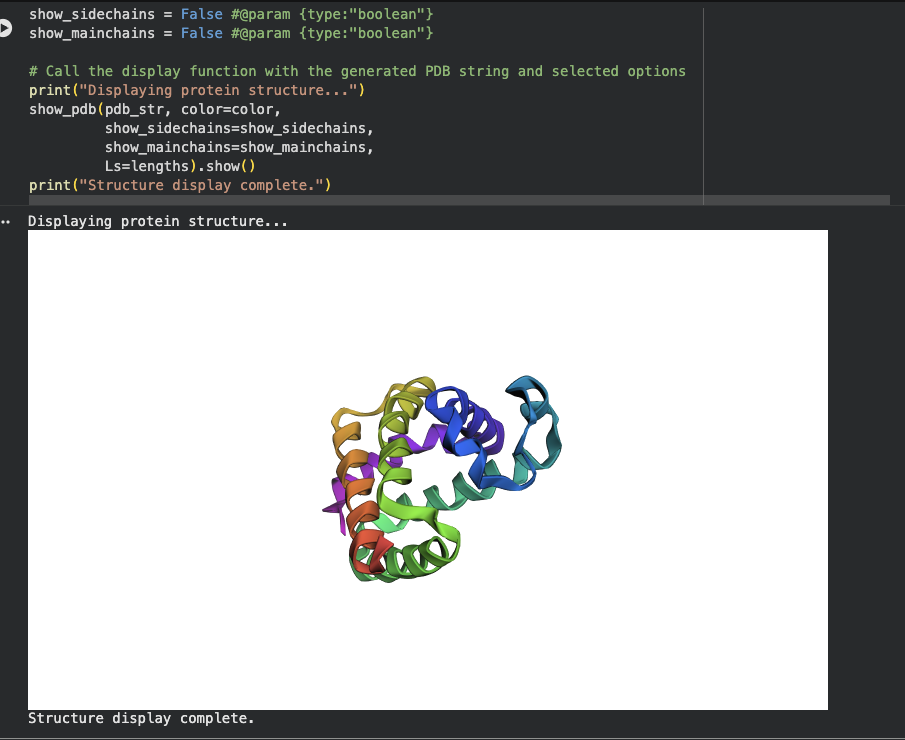
Open the structure of your protein in any 3D molecule visualization software:
Using PyMol I wanted to visualize my protein of interest.
Download the PDB file for the protein, then on PyMol, go to file then open, select the downloaded PDB file. (Chatgpt helped with the python code for the different visualizations)
Cartoon
I used the following commands
PyMOL>hide everything
PyMOL>show cartoon
PyMOL>color red
PyMOL>util.cbc (color by secondary structure)
Ribbon
I used the following commands
PyMOL>hide everything
PyMOL>show ribbon
Ball and Stick
I used the following command
yMOL>hide everything
PyMOL>show sticks
PyMOL>show spheres
To Improve structure
PyMOL>set sphere_scale, 0.25
PyMOL>set stick_radius, 0.15
To answer the following questions I used chapgpt to explain how to identify the different structures and also to understand the residues
Color the protein by secondary structure. Does it have more helices or sheets?
By default, PyMOL colors:
Red → α-helices
Yellow → β-sheets
Green → loops/coils
The protein has α-helices that appeared as a spiral / corkscrew shape, Long cylindrical coils and was red
It also has β-Sheets that appeared as Flat arrow-shaped strands, Multiple arrows next to each other and were yellowSpiral/corkscrew
It also had Loops that appeared as Thin connecting regions that were green
To tell whether I have more helices or sheets, I ran the following code
PyMOL>select helices, ss h
Selector: selection “helices” defined with 3854 atoms.
PyMOL>select sheets, ss s
Selector: selection “sheets” defined with 2744 atoms.
My protein has more Helices
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
“hydrophobic” defined with 4211 atoms.
“hydrophilic” defined with 3626 atoms.
There are more hydrophobic atoms than hydrophilic atoms.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
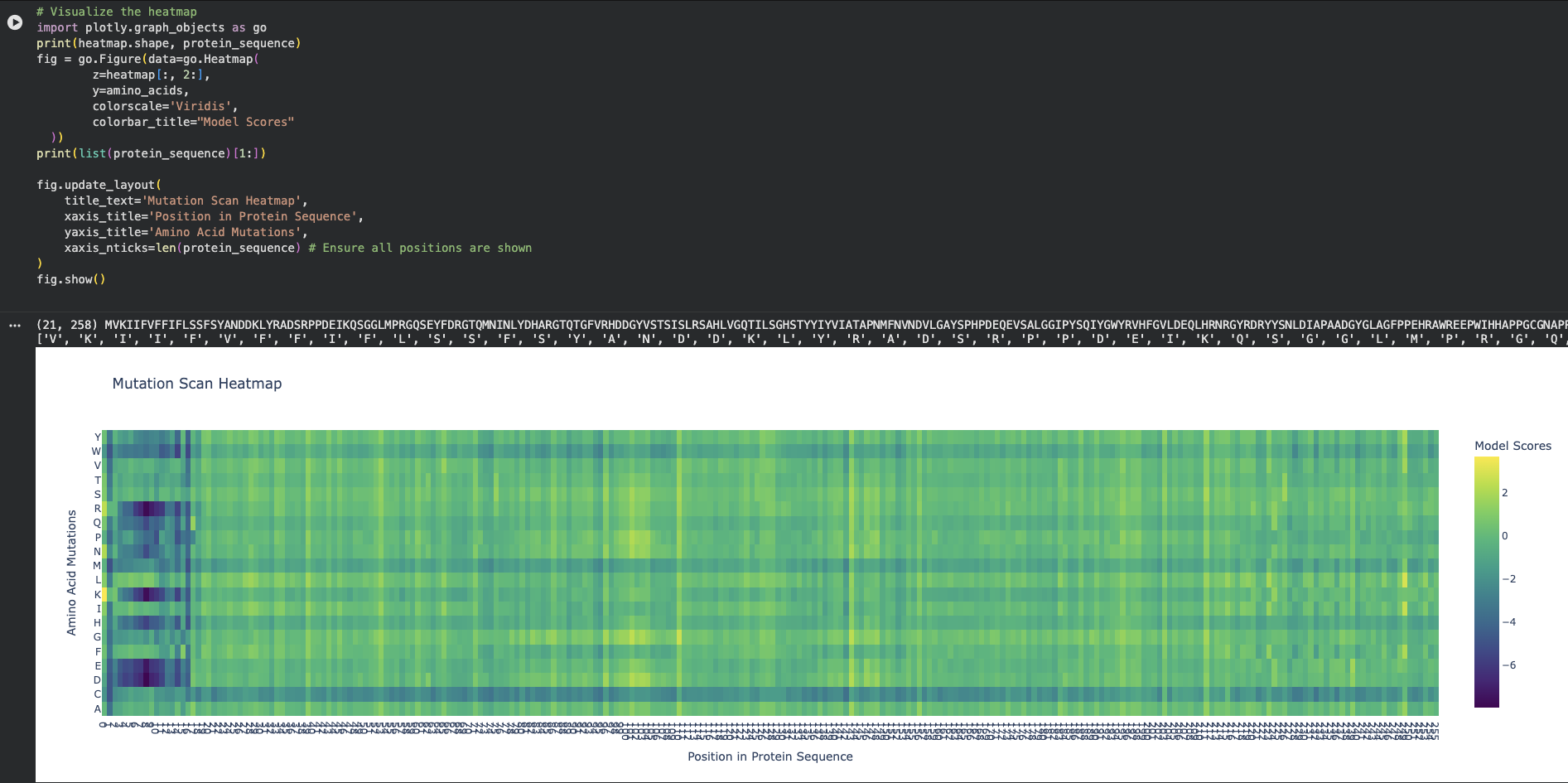
On the left side of the heatmap (approximately positions 5 to 15 in the protein sequence), there is a massive cluster of dark purple columns.
At at Position 6 (Phenylalanine - F) or Position 9 (Phenylalanine - F) there are clear mutations
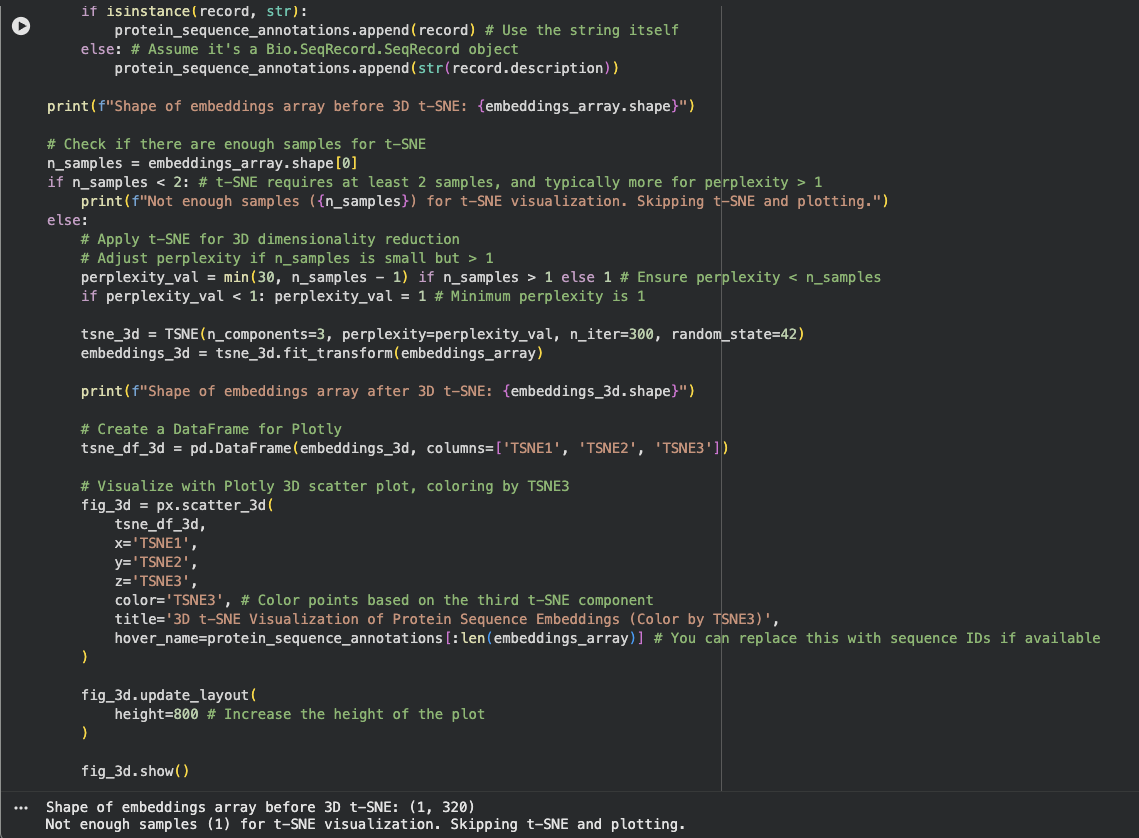
Latent Space Analysis
I am not sure what I did wrong here. The output gave me Shape of embeddings array before 3D t-SNE: (1, 320)
Not enough samples (1) for t-SNE visualization. Skipping t-SNE and plotting.
I need to review this later
C2. Protein Folding
There is a very distinct, long, prominent alpha-helix colored in a brown/orange gradient at the bottom right in both structures.
The protein structure from ESMFold looks less dense than the original structure.
C3. Protein Generation
Inverse-Folding a protein
Amino Acid possibilities heatmap
The New Sequence:
ALSAEQAALLAAAFAPVAADAEANGRAFILRLFEAYPELRELFPEFKGLSLEEIAASPKLGEIAGAHFALMAEFVATAADAAAMAALLADFAARHVALGIGAAHVEAIRAIHPGFVASVAAPPPGAAAAWDALFGMVIDALRAAGA
There is a significant difference between the original sequence and the new sequence as pictured when using ESMFold.
(lower perplexity = the model is more confident the peptide is a good binder)
Part 2: AlphaFold3 structure prediction
For each result, I looked at the below:
The ipTM score (higher is better, closer to 1.0 means confident binding prediction)
Binder
Pseudo Perplexity
ipTM score
WLSYPVVLEWGE
16.4334209
0.21
WHYYVVAVRWKE
31.112519
0.35
WHSYAAAAALWE
12.9540785
0.27
WLYYAVGLAWKX
14.2779037
Could not be generated because it had a letter X
FLYRWLPSRRGG
XXXX
0.3
The ipTM score (interface predicted TM-score) measures how confidently AlphaFold3 predicts the two chains interact. It ranges from 0 to 1.
Below 0.4 = poor
0.4–0.6 = moderate
Above 0.6 = good
Where the peptide appears to dock (look at the 3D viewer — is it near position 4 at the N-terminus? At the dimer interface? Surface-exposed?)
I have a question on the fact that my second binder has a high Pseudo Perplexity value of 31 therefore the assumption is that the binding confidence level is very low. However through alphafold it has the best ipTM score suggesting that it had the best confident binding prediction
Part 3: PeptiVerse therapeutic properties
Binder
Solubility
Hemolysis
Binding Affinity
Length
Molecular Weight
Net Charge (pH 7)
Isoelectric Point
Hydrophobicity (GRAVY)
WLSYPVVLEWGE
Soluble - 1.00
Non-hemolytic 0.112
Weak binding - 5.952
12
1477.7
-2.23
4.24
0.26
WHYYVVAVRWKE
Soluble - 1.00
Non-hemolytic 0.069
Weak binding - 6.345
12
1635.9
0.85
8.5
-0.42
WHSYAAAAALWE
Soluble - 1.00
Non-hemolytic 0.035
Weak binding - 5.863
12
1375.5
-1.15
5.47
0.18
WLYYAVGLAWKX
Soluble - 1.00
Non-hemolytic 0.115
Medium binding - 7.101
12
1351.8
0.76
8.5
0.56
the best one balances strong binding + not hemolytic + good solubility.
I initially started with trying to use the google colab shared and input what I thought was right. I continously kept on getting an error that is IndexError.
I used claude to help me work out what i can do, and this is what it provided me with the instructions below.
After that I did the following and this is a summary of my concluison from the work.
Steps taken
I then went to Gemini and copy pasted the above and let it run.
After completing the run the output it gave was as below
moPPIt uses discrete flow matching to steer generation toward specific binding sites AND simultaneously optimize multiple properties.
moPPIt peptides are expected to have higher binding affinity and better drug-like properties (solubility, safety) than PepMLM outputs
SOD1 Structure Guide: Targeting Residues for moPPIt
What Is SOD1?
Superoxide Dismutase 1 (SOD1)
Role: Antioxidant enzyme in cells; protects against free radical damage
Structure: Homodimer (~32 kDa monomer × 2) with zinc and copper cofactors
Disease Link: >180 mutations cause Familial ALS (fALS); A4V is one of the most severe
A4V Mutation Impact:
Position 4: Alanine → Valine
Makes protein unstable, prone to misfolding and aggregation
Leads to neuronal toxicity → motor neuron death → ALS
Position: 1 2 3 4 5 6 7 8 9 10
Sequence: M E T A→V K S Q V V Q
Index: 0 1 2 3 4 5 6 7 8 9
↑↑↑↑↑↑↑↑↑↑↑↑↑
TARGET INDEX: 3
Why target here?
✓ Direct site of pathogenic mutation
✓ Likely destabilized region
✓ Peptide here could stabilize or mark for degradation
✓ Highest specificity to A4V disease
Recommended for: Stabilization or selective targeting of mutant
2. Metal Cofactor Binding Sites (Stabilization)
Zinc Binding (Zn²⁺):
Position: 45 46 47 48 49
Sequence: R P D E D
Index: 44 45 46 47 48
↑ ↑↑↑↑↑↑↑↑↑↑↑↑↑
| Zn²⁺ binding motif
Loop
Why target here?
✓ Zinc stabilizes SOD1 structure
✓ A4V mutants have weaker Zn²⁺ binding
✓ Peptide here could enhance Zn²⁺ coordination
✓ Stabilizes fold → prevents aggregation
Recommended indices: [44, 45, 46, 47, 48]
Or 1-indexed positions: 45-49
Copper Binding (Cu²⁺):
Position: 70 71 72 73 74 75 76 77 78 79 80
Sequence: H S V Y V D Q W D W E
Index: 69 70 71 72 73 74 75 76 77 78 79
↑ ↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑
| Cu²⁺ binding cluster
Loop
Key residues:
- H71 (Histidine 71) - coordinates Cu²⁺
- D74 (Aspartate 74) - coordi
nates Cu²⁺
- W76 (Tryptophan 76) - structural support
Why target here?
✓ Critical for catalytic activity
✓ A4V mutants lose Cu²⁺ stability faster
✓ Peptide here could improve metal retention
✓ Even small improvements help
Recommended indices: [70, 71, 73, 74, 76, 78, 79]
Or 1-indexed positions: 71, 72, 74, 75, 77, 79, 80
3. Dimer Interface (Aggregation Prevention)
MONOMER A: MONOMER B:
┌──────────────────┐ ┌──────────────────┐
│ │ │ │
│ Position 50-60 │◄─────────►│ Position 50-60 │
│ (contacts B) │ Interface │ (contacts A) │
│ │ │ │
│ Position 85-100 │◄─────────►│ Position 85-100 │
│ (contacts B) │ Interface │ (contacts A) │
│ │ │ │
└──────────────────┘ └──────────────────┘
Dimer Interface A (Region 1):
Position: 50 51 52 53 54 55 56 57 58 59 60
Sequence: L G Q H D F S A G E G
Index: 49 50 51 52 53 54 55 56 57 58 59
↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑
Dimer Interface B (Region 2):
Position: 85 86 87 88 89 90 91 92 93 94 100
Sequence: G I E Q L P D G Q K ...
Index: 84 85 86 87 88 89 90 91 92 93 99
↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑↑
Why target here?
✓ A4V mutants hyperaggregate (misfolded dimers)
✓ Peptide here could disrupt "bad" dimers
✓ Or stabilize native dimers
✓ Critical for ALS pathogenesis
Recommended indices: [49, 51, 53, 55, 57, 84, 86, 88, 90]
Or 1-indexed positions: 50, 52, 54, 56, 58, 85, 87, 89, 91
4. General Surface Patches (High Accessibility)
SOD1 accessible surface:
- N-terminal region (1-20): Accessible, flexible
- Loop regions (40-50, 60-70): Flexible, exposed
- C-terminal region (140-153): Accessible, flexible
- Anywhere NOT in active site or dimer interface
Benefits:
✓ Easier for peptide to access
✓ Less steric clashes
✓ Multiple contact points
Recommended indices: [0-20] or [100-153] or scattered accessible residues
Goal: Keep SOD1 folded (prevent A4V misfolding)
Strategy: Reinforce metal binding sites
Biology positions: 45 46 47 48 49 70 71 72 73 74 75 76
Code indices: 44 45 46 47 48 69 70 71 72 73 74 75
For Colab input:
binding_residue_indices = [44, 45, 46, 47, 48, 69, 70, 71, 72, 73, 74, 75]
Explanation: "I'm targeting the Zn²⁺ and Cu²⁺ coordination sites. A4V
mutants are unstable, so a peptide that reinforces these metal-binding
regions could restore structural integrity."
Example 2: Anti-Aggregation Strategy
Goal: Prevent dimerization (block aggregation)
Strategy: Disrupt dimer interface
Biology positions: 50 52 54 56 58 85 87 89 91
Code indices: 49 51 53 55 57 84 86 88 90
For Colab input:
binding_residue_indices = [49, 51, 53, 55, 57, 84, 86, 88, 90]
Explanation: "I'm targeting the dimer interface. A4V causes pathogenic
aggregates, so a peptide at the dimer interface could selectively bind
and disaggregate or prevent formation of toxic SOD1 oligomers."
Example 3: Multi-Site Strategy (Recommended)
Goal: Multi-pronged approach
Strategy: Combine mutation site + metal binding + accessible surface
Targets:
- A4V site: position 4 → index 3
- Zn binding: positions 45-49 → indices 44-48
- Cu binding: positions 71, 74, 76 → indices 70, 73, 75
- Surface accessibility: position 100 → index 99
For Colab input:
binding_residue_indices = [3, 44, 45, 46, 47, 48, 70, 73, 75, 99]
Explanation: "I'm targeting multiple sites: the A4V mutation site (direct
targeting), the Zn²⁺ and Cu²⁺ binding regions (structural stabilization),
and a surface-accessible region (for cell recognition). This multi-target
approach should generate peptides with both high affinity and therapeutic
breadth."
INDEX CONVERSION QUICK TABLE
If you see position X in publications/UniProt:
Biology Position
Code Index
Region
4
3
A4V mutation
45-49
44-48
Zn²⁺ site
50-60
49-59
Dimer interface 1
71-80
70-79
Cu²⁺ site
85-100
84-99
Dimer interface 2
1-20
0-19
N-terminus
140-153
139-152
C-terminus
Formula: Code Index = Biology Position - 1
SAMPLE OUTPUT FROM moPPIt
Once you run generation, you’ll see output like this:
Affinity Score:
8.5-10.0 → Excellent (strong, specific binding)
7.0-8.4 → Good (decent binder)
6.0-6.9 → Okay (marginal)
<6.0 → Poor (weak or no binding)
Solubility:
0.8-1.0 → Excellent (soluble, won't aggregate)
0.6-0.79 → Good (mostly soluble)
0.4-0.59 → Fair (some precipitation risk)
<0.4 → Poor (will aggregate)
Hemolysis (non-toxicity):
0.8-1.0 → Excellent (safe to blood)
0.6-0.79 → Good (low toxicity)
0.4-0.59 → Fair (some toxicity risk)
<0.4 → Poor (likely toxic)
Overall = Average of normalized scores
Best candidates: All three scores >0.8
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
What you add yourself (not in the master mix)
Forward primer + Reverse primer — define what region to amplify
Template DNA — the sequence to be copied
Nuclease-free water — to bring the reaction to final volume
The star component is Phusion polymerase itself — it has a fused processivity domain that makes it faster than standard Taq, and crucially it has 3′→5′ proofreading (exonuclease) activity, giving it an error rate ~50× lower than Taq. This is why it is preferred when accuracy matters, such as before Gibson assembly or cloning.
What are some factors that determine primer annealing temperature during PCR?
The four main factors are GC content, primer length, the calculated melting temperature (Tm), and the buffer’s salt concentration. With Phusion specifically, NEB recommends using their online Tm calculator rather than generic formulas, because the enzyme’s processivity domain affects optimal conditions. A practical starting point is to set annealing temperature to the Tm of your lower-Tm primer minus 5°C, then optimise from there.
Consequences of wrong annealing temperature
Ta too LOW → primers bind non-specifically → multiple bands, wrong products
Ta too HIGH → primers cannot bind stably → no product or very low yield
Rule of thumb for Phusion: use the NEB Tm Calculator and set Ta = Tm of lower primer − 5°C
Gradient PCR can be used to empirically optimise Ta when uncertain
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Both methods produce linear DNA fragments, but through fundamentally different mechanisms. PCR synthesises new copies of a specific region exponentially from a template, using primers to define the exact endpoints — which means you can engineer the ends freely by adding sequences to your primers. A restriction enzyme digest cuts existing DNA at fixed recognition sequences, producing ends that are dictated entirely by where those sequences happen to sit in the molecule.
In terms of error risk, PCR introduces the possibility of polymerase errors (minimised by Phusion’s proofreading), while RE digests are faithful to the original sequence but can only cut where recognition sites exist. If those sites don’t exist in your insert, or if a cut site would fall in an undesirable location, PCR is the only option. Conversely, if you already have a plasmid carrying your insert flanked by known restriction sites, a digest is faster and simpler than designing and running PCR.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Gibson assembly works by joining fragments that share 15–40 bp of identical sequence at their junctions. The exonuclease in the Gibson mix chews back 5′ ends to create single-stranded overhangs, which then anneal and are ligated. For your fragments to be compatible, you must ensure overlapping homology at every junction.
For PCR fragments, the key step is primer design — your forward primer for each insert should carry a 5′ tail of 20–40 bp that matches the end of the upstream fragment (or linearised vector). Tools like Benchling let you design these overlaps visually and verify them in silico before ordering primers. For RE-digested fragments, you need to confirm that the sticky or blunt ends of adjacent fragments are complementary and that no sequence is lost or mismatched at the junction. After generating your fragments by either method, always run a gel to confirm band size, then column-purify or gel-extract to remove primers, enzymes, and salts that would interfere with the Gibson reaction.
Key things that can go wrong
Overlaps too short (<15 bp) → poor annealing, assembly fails
Overlaps contain repeats or secondary structure → mis-assembly or rearrangements
Impure fragments (primers still present) → compete with correct annealing
How does the plasmid DNA enter the E. coli cells during transformation?
The most common lab method is heat-shock transformation of chemically competent cells. Cells are treated with divalent cations (typically CaCl₂) during preparation, which partially neutralises the negative charge on both the cell membrane and the DNA, reducing electrostatic repulsion. When the cold cell-DNA mixture is briefly shifted to 42°C, transient pores open in the membrane through which DNA can enter. The cells are then recovered in rich medium at 37°C (called the recovery or outgrowth step) to allow antibiotic resistance genes on the plasmid to be expressed before plating on selective media.
What happens at the molecular level
CaCl₂ treatment: Ca²⁺ ions coat DNA and neutralise the membrane’s negative charge
Ice incubation: DNA-cell complexes form and associate with the outer membrane
Heat shock: membrane fluidity spike creates transient pores → DNA enters cytoplasm
Return to ice: pores reseal, DNA is trapped inside
Outgrowth: plasmid replicates, antibiotic resistance gene is expressed before selection
An alternative to chemical transformation is electroporation, where a brief electrical pulse physically creates pores in the membrane to allow DNA entry. Electroporation generally gives 10–100× higher efficiency and is preferred for large plasmids or when transformation efficiency is critical. However, it requires electrocompetent cells (washed to remove salts) and specialised equipment.
Describe another assembly method in detail (such as Golden Gate Assembly)
Golden Gate Assembly is an cloning method that uses Type IIS restriction enzymes — enzymes that cut outside their recognition sequence to generate custom 4-bp overhangs. Because the enzyme cuts away from its own recognition site, the recognition sequence is eliminated from the final product, leaving only the overhangs you designed. This enables scarless, seamless assembly.
Golden Gate uses a Type IIS enzyme (most commonly BsaI or BsmBI) whose recognition sequence and cut site are spatially separated. Each DNA part is flanked by Type IIS recognition sequences oriented so that cutting releases the recognition site and leaves behind a unique 4-bp overhang. Because every junction has a distinct 4-bp overhang, the parts can only assemble in one correct order — the overhangs provide directionality and specificity simultaneously. The restriction enzyme and T4 DNA ligase are added to the same tube and thermocycled together: digestion and ligation alternate in repeated cycles, so misligated or incorrect assemblies are continuously re-cut and corrected while the correct assembly accumulates. The final assembled product lacks all Type IIS sites, so it can never be re-cut by the enzyme in the reaction. This makes Golden Gate highly efficient even for assemblies of 5–10 fragments in a single reaction, which makes it faster and more scalable than sequential cloning strategies.
As a comparison to Gibson: Golden Gate is preferable when you are assembling many parts (5+) in a defined order and want precise scarless junctions, while Gibson is simpler to set up for 2–3 fragments and does not require specific restriction sites to be absent from your inserts. Golden Gate requires that no internal BsaI sites exist within any of your parts (you may need to silently mutate them out), whereas Gibson has no such constraint.
Two repressors that inhibit each other — the circuit “toggles” to one stable state.
Expected result: Simulator shows one protein high, the other low — a bistable switch
These two genes repress each other. Whichever starts higher will suppress the other, locking the circuit into one state.
The results are not what I expected. The simulator shows State B of the toggle switch, where LacI is highly expressed via pTetR, and this LacI represses pLacI, keeping TetR near zero.
This is one valid stable state of a bistable toggle. To confirm bistability, I would need to re-run with reversed initial protein concentrations to observe State A."
Construct 2: Simple Gene Expression
A gene that’s always “on” — no regulation, just constant expression.
Expected result: Flat, steady line in the simulator (constant protein level
A constitutive promoter drives constant transcription. No repressors or activators, so the protein level stays flat.
The constitutive construct using BBa_J23100 performed as expected.
The promoter drives constant RNAP flux through the RBS and EGFP coding sequence, with the terminator correctly stopping transcription.
Ribosome flux is slightly lower than RNAP flux, which is expected since translation efficiency is never 100%. There is no regulation here, the gene is simply always on, producing a steady level of EGFP protein.
Construct 3: Inducible Expression System
A gene that’s off by default but turns on when a repressor is absent.
Expected result: Protein rises quickly and plateaus
Without TetR repressor present, the pTetR promoter is unblocked and drives GFP expression continuously.
The inducible construct using pTetR drove strong EGFP expression (~3.75 RNAP flux) because no TetR repressor was present in the construct to block it.
This represents the induced/active state of the system. The terminator correctly stopped transcription.
Notably, pTetR produced ~3x higher flux than the constitutive BBa_J23100 promoter, suggesting it is a stronger promoter when unrepressed.
In a complete inducible system, a TetR-expressing cassette would silence this promoter unless aTc is added to relieve repression."
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Integrated Artificial Neural Networks (IANNs) offer superior advantages over traditional Boolean genetic circuits by enabling analog processing, high-dimensional pattern recognition, and robust adaptability. Unlike Boolean circuits (on/off), IANNs handle continuous input levels, allowing complex decision-making, such as classifying disease states based on weighted molecular signatures rather than simple threshold switches.
Advantages of IANNs
They can handle more complex signals (not just ON/OFF).
They can combine many inputs at once.
They give graded responses (not just yes/no).
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Use: Detect cancer cells inside the body
Inputs:
X1 = cancer marker 1
X2 = cancer marker 2
Output:
If both markers are high → produce drug (kill cell)
If not → do nothing
Limitations:
Hard to control inside real cells
Can be slow
May make mistakes (wrong output)
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Examples
Mycelium packaging
Used instead of plastic/foam
Mycelium leather
Used for clothes, bags
Building materials
Used for bricks/insulation
Advantages:
Biodegradable
Eco-friendly
Renewable
Disadvantages:
Not as strong as plastic/metal
Can be damaged by water
Slower to produce
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
What we could make fungi do:
Produce stronger materials
Glow (for lighting)
Clean pollution
Why fungi (vs bacteria):
Grow into solid shapes (good for materials)
Naturally make strong structures
Better for large-scale materials
Week 9 HW: Cell free systems
Homework Part A: General and Lecturer-Specific Questions
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
In traditional in vivo (inside the cell) methods, the cell’s primary goal is its own survival. In CFPS, the goal is purely production.
Experimental Control: You can directly manipulate the environment. You can add non-natural amino acids, adjust redox potential, or add chaperones at precise concentrations without the cell’s homeostatic mechanisms fighting back.
Flexibility: There is no “transformation” step. You can add linear DNA or mRNA directly to the mix, enabling rapid prototyping (Design-Build-Test cycles).
Two Cases where CFPS is superior:
Cytotoxic Proteins: Producing proteins that would kill a living host (e.g., antimicrobial peptides or certain toxins) is much easier in vitro because the system doesn’t need to stay “alive.”
Incorporation of Non-Canonical Amino Acids (ncAAs): CFPS allows for the site-specific insertion of synthetic amino acids to create proteins with new chemical properties, which is often difficult in cells due to transport issues or toxicity.
Describe the main components of a cell-free expression system and explain the role of each component.
Cell Extract (Crude Lysate): Provides the core machinery: Ribosomes, aminoacyl-tRNA synthetases, and initiation/elongation factors.
Cell-free protein synthesis template DNA or mRNA: Encodes the protein of interest.
Energy Source High-energy molecules (e.g., Phosphoenolpyruvate or Creatine Phosphate): to fuel the reaction.
Amino Acids: The raw building blocks for the protein chain.
Nucleotides (NTPs): Required for transcription (if starting from DNA) and energy transfer.
Salts and Buffers (e.g., Magnesium and Potassium): To maintain pH and stabilize the folding of the machinery.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Protein synthesis is energetically expensive. Every peptide bond requires the hydrolysis of multiple ATP and GTP molecules. If the energy supply is exhausted, translation stops, leading to low yields.
One common method is the Secondary Energy Solution using Creatine Phosphate and Creatine Kinase. The kinase enzyme transfers a phosphate group from creatine phosphate to ADP, constantly “recharging” the ATP pool within the tube.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic (e.g., E. coli): High yield, fast, and inexpensive. However, it lacks post-translational modification (PTM) capabilities.
Protein to produce: Insulin. While complex, it is a small protein that can be efficiently produced and folded in optimized E. coli systems.
Eukaryotic (e.g., CHO or Wheat Germ): Lower yield but capable of complex folding and PTMs like glycosylation.
Protein to produce: Monoclonal Antibodies. These require complex disulfide bond formation and glycosylation to be functional, which prokaryotic systems cannot easily handle.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Membrane proteins are notoriously difficult because they are hydrophobic and require a lipid environment to fold correctly; otherwise, they aggregate and become useless.
Challenges and Solutions:
Challenge: Lack of a lipid bilayer.
Solution: Supplement the CFPS reaction with nanodiscs or synthetic liposomes. These provide a “landing pad” for the protein to insert itself into as it is being synthesized.
Challenge: Detergent toxicity.
Solution: Use detergent-free CFPS or mild surfactants that stabilize the protein without denaturing the translation machinery.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Template Degradation: Check the purity of your DNA/mRNA. Use RNase inhibitors to prevent the degradation of the blueprint during the reaction.
Energy Depletion: Analyze the duration of the reaction. If it stops prematurely, increase the concentration of the secondary energy source or implement a dialysis system to remove byproduct phosphate.
Poor Protein Folding: If the protein is being made but is insoluble (forming pellets), try lowering the incubation temperature or adding molecular chaperones (like DnaK/J) to the extract.
Homework question from Kate Adamala
1. Pick a Function and Describe It
1a. What would your synthetic cell do? What is the input and what is the output?
I would like to make a cell free system for diagnosis of Vibrio cholerae.
Function: Detect Vibrio cholerae presence in environmental samples (water, food, clinical samples) through recognition of cholera toxin A, and produce a visible/measurable output signal.
Input: Cholera Toxin A protein (CTA) or V. cholerae bacterial lysate containing CTA
Intermediate Process: CTA enters the SMC through a permeable membrane or receptor-mediated endocytosis-like mechanism. Inside the SMC, CTA is recognized by a detection system.
Output of SMC: Production of ADP-ribosylated protein (marking detection), followed by activation of a reporter gene
Output of whole system: Fluorescent protein (GFP) or enzymatic output (luciferase) proportional to CTA concentration
1b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
No.
Without encapsulation, the CTA would immediately diffuse away and not accumulate
There would be no way to separate the detection reaction from background noise in the environment
The synthetic cell’s advantage is creating a compartmentalized detection zone where high local concentrations of detection machinery exist
The membrane creates a controlled microenvironment where we can detect low concentrations of CTA that would be undetectable in bulk solution
Encapsulation allows us to recycle detection components and amplify the signal within the confined space
1c. Could this function be realized by genetically modified natural cell?
Yes, but with significant limitations:
Theoretically, I can transform E. coli or V. cholerae with genes for CTA-binding proteins and reporter genes
However, this approach has major problems:
Toxicity: CTA’s ADP-ribosylation activity could damage the host cell’s essential proteins, killing the cell or triggering SOS response
Biocontainment: Releasing genetically modified bacteria for field diagnostics is problematic (regulatory, safety, environmental concerns)
Specificity: Natural cells have their own metabolic activities that interfere with clean detection
Standardization: Cell-free systems are more reproducible and easier to quality-control than living cells
Cost: A synthetic cell biosensor is cheaper to produce and deploy than maintaining living cell cultures
We avoid the toxicity problem because cell-free translation doesn’t depend on maintaining cellular homeostasis or viability.
1d. Describe the desired outcome of your synthetic cell operation.
Desired Outcome:
When a water sample (potentially contaminated with V. cholerae) is added to the SMC suspension:
Detection Phase (0-5 minutes): CTA molecules from the sample cross the SMC membrane and encounter our detection machinery inside
Signal Amplification (5-30 minutes): Positive detection triggers transcription/translation of GFP reporter gene
Output (30+ minutes): Fluorescence increases proportionally to CTA concentration
Readout: Using a simple fluorimeter (or even a smartphone with UV light), the presence of V. cholerae is confirmed - no bacterial culture needed, results in <1 hour
Clinical Application: A field-deployable diagnostic tool for cholera detection in resource-limited settings.
2. Design All Components of the Synthetic Cell
2a. What would be the membrane made of?
Membrane Composition:
POPC (1-palmitoyl-2-oleoyl-glycero-3-phosphocholine): 60-70%
→ Main phospholipid providing bilayer structure
→ Chosen because it’s fluid at room temperature and biologically relevant
Cholesterol: 20-25%
→ Increases membrane mechanical stability
→ Reduces permeability to small hydrophilic molecules we want to control
→ Improves long-term storage stability of vesicles
DSPE-PEG2000 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000]): 5-10%
→ Provides stealth properties (reduces non-specific protein absorption)
→ Improves biocompatibility if used in human samples
→ Prevents aggregation of SMC vesicles with each other
Optional: Sphingomyelin (5-10%)
→ Increases membrane rigidity and lifespan
→ Creates more ordered membrane domains
This formulation is based on successful demonstrations and mimics the complexity of biological membranes
The POPC:cholesterol ratio (60:25) provides optimal fluidity while maintaining stability
We keep it simple enough to be reproducible but complex enough to be functional
2b. What would you encapsulate inside? Enzymes, small molecules.
Encapsulated Components:
A. Cell-Free Translation/Transcription System (Tx/Tl):
Complete bacterial extract (PURE system or S30 extract from E. coli)
NTPs (ATP, GTP, CTP, UTP) - 2-4 mM each
Amino acids (all 20 standard ones) - 200-500 µM each
Energy regeneration system: PEP (phosphoenolpyruvate) and PEP synthetase
Ribosomes, tRNAs, various factors for transcription and translation
B. Detection Cassette (encoded in plasmid DNA):
CTA-Binding Domain: Nanobody (single-domain antibody) against cholera toxin A, expressed as a fusion protein
Nanobodies are smaller than full antibodies, easier to express in cell-free systems
Signal Transduction: Upon CTA binding to our detection protein, we trigger transcription of reporter gene
Use a constitutive T7 promoter (CTA presence alone triggers transcription)
C. Reporter Gene (in plasmid or added separately):
GFP (green fluorescent protein) gene under constitutive T7 promoter
Output: Green fluorescence at 509 nm
Easy to measure with standard equipment, visible signal
D. Supporting Small Molecules:
Magnesium chloride (MgCl₂): 5-10 mM (cofactor for ribosomes and polymerases)
Potassium glutamate: 100-150 mM (osmolyte, maintains ionic balance)
DTT (dithiothreitol): 1-2 mM (reduces oxidative stress, keeps proteins in reduced state)
EDTA: 0.1-0.5 mM (chelates metal contaminants)
E. DNA Encoding the Detection Circuit (Plasmid DNA):
2c. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system?
Bacterial (E. coli) is appropriate and optimal here. It costs less to use a bacterial system, more stable and robust, can incooperate the T7 promoter mechanism, and there is no need for post translation modification.
2d. How will your synthetic cell communicate with the environment?
Passive Diffusion
CTA is a ~25 kDa protein - too large to freely diffuse across lipid bilayer. However the following can be done to ensure that their is diffusion
Make the membrane slightly permeable to proteins
Use higher cholesterol content (to 30-35%) to increase membrane fluidity, this allows some leakage of proteins across the membrane
Proteins and enzymes: T7 RNA Polymerase, E. coli ribosomes
Gene Construct 1: CTA Detection & Signal Transduction
small molecules: ATP,GTP, CTP, UPT, All 20 amino acids
3b. How will you measure the function of your system?
Fluorescence-based detection
Homework question from Ally Huang
(Queried ChtaGpt, Gemini and Claude on this to see what the best approach would be for solving DNA damage, with a bias to rapid diagnostics as this is an area i am interested in)
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Long-duration space missions expose astronauts to cosmic radiation, causing DNA damage and muscle/bone loss. Preserving astronaut health is critical, but traditional medical diagnostic equipment is too heavy and resource-intensive for spacecraft. We need lightweight, on-demand tools to detect cellular stress early. Solving this is significant for humanity’s future as a multiplanetary species, relevant for safe Mars exploration, and scientifically interesting because it tests how synthetic biology can replace massive labs with paper-based, freeze-dried cellular sensors.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
The p53 tumor suppressor protein. It is known as the “guardian of the genome” and is rapidly produced by human cells in response to radiation-induced DNA damage.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
When astronauts are exposed to harmful space radiation, their cells experience DNA double-strand breaks. This damage activates the p53 pathway to either repair the cell or trigger cell death. By targeting the expression of the p53 gene, we can create a direct, biological warning system. If an astronaut’s blood or saliva sample triggers high p53 activity, it indicates dangerous levels of radiation exposure. Monitoring this target allows real-time health tracking before severe symptoms, like cancer or radiation sickness, physically manifest during a mission.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Freeze-dried BioBits® cell-free reactions can be successfully engineered to produce a visible green fluorescent protein (GFP) when activated by synthetic DNA plasmids mimicking astronaut p53 expression.
BioBits® contains all the cellular machinery (ribosomes, enzymes) needed to translate DNA into proteins without needing live cells. In space, live cell cultures easily die and are hard to maintain. Freeze-dried BioBits® are shelf-stable, lightweight, and activate simply by adding water. By linking a p53-responsive promoter sequence to a GFP gene, any p53 activation will trigger GFP production. This will cause the sample to glow brightly under the P51 Molecular Fluorescence Viewer, proving that cell-free systems can reliably diagnose radiation stress without requiring a traditional, heavy laboratory footprint.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Synthetic DNA templates representing astronaut samples under radiation stress will be tested.
Experimental Sample: BioBits® rehydrated with DNA containing the p53-activated GFP gene.
Positive Control: BioBits® with a standard, always-active GFP control DNA (to ensure the kit works).
Negative Control: BioBits® rehydrated with sterile water (no DNA) to check for background glow.
Samples will be incubated in the miniPCR® thermal cycler to mimic body temperature.
Data will be collected by placing the tubes into the P51 Fluorescence Viewer and measuring visual fluorescence intensity (glowing vs. dark).
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
A paper-based, low-cost diagnostic tool that utilizes freeze-dried cell-free systems and RNA toehold switches to provide a visible color-change signal in the presence of Vibrio cholerae within 60 minutes.
How will the idea work, in more detail? Write 3-4 sentences or more.
The device consists of a paper strip embedded with a freeze-dried cell-free transcription-translation (TX-TL) system and a synthetic “toehold switch” gene circuit. When a suspected water sample is dropped onto the paper, the water rehydrates the biological machinery, “waking it up.” If the specific RNA sequence of the Vibrio cholerae bacteria is present in the sample, it binds to the toehold switch, unfolding the RNA structure and allowing the ribosome to translate a reporter protein, such as LacZ or purple chromoprotein, which turns the paper from white to a distinct color.
What societal challenge or market need will this address?
Cholera remains a major global health threat in resource-limited settings where traditional lab-based testing (like bacterial culture or PCR) is too slow, expensive, and dependent on a “cold chain” for refrigerated transport. This kit addresses the need for decentralized, rapid outbreak detection, allowing local health workers to confirm cases in the field and immediately initiate life-saving interventions like water chlorination and rehydration therapy.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Activation: The limitation of needing water for activation is turned into a benefit here, as the liquid sample being tested serves as the rehydration agent for the freeze-dried reagents.
Stability: By incorporating lyoprotectants (such as trehalose) during the freeze-drying process, the enzymatic machinery is stabilized against high tropical temperatures, eliminating the need for refrigeration during shipping.
One-Time Use: To ensure the tool is economically viable despite being one-time use, the system is engineered onto cellulose paper, which is biodegradable and costs only cents per test, making it a scalable solution for mass surveillance during floods or humanitarian crises.
Week 10 HW: Imaging and Measurement
Homework: Final Project
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
I want to develop a synthetic biology-based point-of-care diagnostic platform that can rapidly detect cholera toxin and toxin co-regulated pilus antigens in stool and environmental samples within 15-30 minutes using engineered cell-free biosensors integrated into a paper-based microfluidic device.
The system employs single-domain antibodies recognition elements fused to split T7 RNA polymerase domains that reconstitute active enzyme only upon antigen binding, driving colorimetric reporter expression for visual result interpretation requiring no laboratory infrastructure or specialized training.
Target analytes: CT B-subunit and TCP major pilin proteins in stool and environmental water samples
Detection threshold: sensitivity down to ~1–10 ng/mL (clinically relevant concentration)
Specificity: cross-reactivity with other Vibrio species and common fecal flora
Biosensor Reconstitution & Activation
RNA polymerase (T7 RNAP) activity levels as a function of antigen binding
Kinetics of split-RNAP domain reconstitution upon antigen-induced proximity
Reporter Gene Expression Output
mRNA transcript abundance (real-time via fluorescent readout, or endpoint colorimetric)
Protein product accumulation (visual color development in paper device)
Time-to-positive result (assay kinetics within 15–30 min window)
Paper Device Integration & Performance
Antigen capture efficiency (how much target binds to sdAb immobilized on paper)
Cell-free extract stability in paper matrix (shelf-life validation)
Color intensity uniformity and visual interpretability (naked-eye readout without instrumentation)
To validate this system, I will employ LC-MS or MALDI-TOF mass spectrometry to confirm antigen identity and establish absolute quantification standards, real-time fluorescence assays to measure split-RNAP reconstitution kinetics and activation efficiency, endpoint colorimetric transcription-translation assays to verify reporter protein production within the 15–30 minute window, high-speed video imaging with ImageJ analysis to quantify color development uniformity and visual readability on the paper microfluidic device, SDS-PAGE and Western blotting to confirm correct fusion protein expression and reporter accumulation over time, fluorescence microscopy to validate antigen-capture specificity, qPCR targeting ctxA and tcpA genes as a gold-standard correlation, and thermal stability assays to assess shelf-life at multiple temperatures.
Together, these complementary approaches will establish your device’s sensitivity, specificity, positive/negative predictive values, and field-deployability while confirming that your split-RNAP biosensor engineering achieves rapid, naked-eye-readable detection without laboratory infrastructure.
This was one of the topics I really struggled to grasp, maybe beacuse it did not feel as practical and was harder to picture or implement in the project I want to do. I leveraged on Ai alot to explain what exactly I was doing and what the answers meant
Homework: Waters Part I — Molecular Weight
We were analyzing an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
1. Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight?
The chemical formula and average molecular weight.
Sequence Components:
Using an average isotopic mass calculator (like ExPASy Compute pI/Mw), the breakdown for this specific sequence is:
Calculated MW: 28,124.08 Da
2. Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
From Figure 1, two adjacent peaks are visible at approximately m/z = 1002.0 (charge n) and m/z = 1079.6 (charge n−1, so n+1 peaks to the left).
Using the formula:
Step 1 — Find z:
Using peaks at m/z₁ = 1079.6 and m/z₂ = 1002.0 (adjacent charge states differing by +1H):
z = m/z₂ ÷ (m/z₁ − m/z₂) = 1002.0 ÷ (1079.6 − 1002.0) = 1002.0 ÷ 77.6 ≈ 25
(So the lower m/z peak has charge z = 27, the higher has z = 25 — typical for a ~27 kDa protein)
Step 2 — Find MW:
MW = (m/z × z) − z × 1.0073 (mass of a proton)
Using peak at m/z = 1002.0 and z = 27:
MW = (1002.0 × 27) − (27 × 1.0073) = 27,054 − 27.2 ≈ 27,027 Da = ~27.0 kDa
3. Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
No, its is not clearly readable.
Homework: Waters Part II — Secondary/Tertiary structure
Skipping this as it is optional. Will revist incase I get some more time.
Waters Part III — Peptide Mapping - primary structure
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
How many peptides will be generated from tryptic digestion of eGFP?
After running the sequence through the ExPASy PeptideMass tool with the parameters shown in Figure 4 (trypsin, 1 missed cleavage, monoisotopic masses):there are approximately 26 peptides
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
From the Total Ion Chromatogram in Figure 5a, counting peaks above 10% relative abundance between 0.5–6 minutes: approximately 12–15 distinct peaks are visible
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
No. Fewer peaks are visible in the chromatogram than predicted peptides.
Identify the mass-to-charge
From Figure 5b:
Most abundant peak: m/z = 525.76
The zoomed inset shows isotope peaks spaced ~0.5 Da apart → charge state z = 2
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
From the PeptideMass results, the peptide with mass closest to ~1050.5 Da from eGFP tryptic digest is the peptide GIDFK or similar small peptide.
What is the percentage of the sequence that is confirmed by peptide mapping?
From Figure 6 (the amino acid coverage map), the highlighted/colored regions show confirmed residues. The coverage map shows approximately 85–95% sequence coverage,
Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c?
…….
Does the peptide map confirm it’s eGFP?
Yes. The peptide map confirms the protein is eGFP
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
Yes — the measured MW matches the theoretical MW of eGFP to within ~1 ppm, confirming successful production of the eGFP protein.
Week 11 HW: Building genomes
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Make a note on your HTGAA webpages including:
what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
I made 3 dots on the lower right quadrant. In green.
what you liked about the project
It was something new and also interesting to see how creative people can be
what about this collaborative art experiment could be made better for next year.
People discovered how to beat the system and be able to contribute more than once. So by the time I was joining alot of the spots had already been filled. Next year it would be nice for maybe the team to know what art work they would like done so that one can know how best to contribute. But all in all it was a new unique experience.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
1. Role of each component:
E. coli Lysate (BL21 DE3 Star + T7 RNA Polymerase)
Provides all the cellular machinery needed for transcription and translation — ribosomes, tRNA, elongation factors, and T7 RNAP to transcribe DNA into mRNA.
Salts/Buffer:
Potassium Glutamate — Mimics intracellular ionic conditions; supports ribosome function and enzyme activity.
HEPES-KOH pH 7.5 — Maintains a stable physiological pH throughout the reaction.
Magnesium Glutamate — Mg²⁺ is essential for ribosome assembly and stabilizing nucleic acids.
Potassium Phosphate Monobasic / Dibasic — Act as a pH buffer and provide phosphate for energy metabolism.
Energy / Nucleotide System:
Ribose & Glucose — Carbon/energy sources that feed metabolic pathways regenerating ATP and other nucleotides.
AMP, CMP, GMP, UMP — Nucleoside monophosphates used as building blocks for RNA synthesis (transcription).
Guanine — A nucleobase that can be salvaged and converted to GTP, supporting transcription without requiring pre-formed GMP to be included directly.
Translation Mix (Amino Acids):
17 Amino Acid Mix, Tyrosine, Cysteine — The complete set of 20 amino acids needed for protein synthesis. Tyrosine and Cysteine are kept separate due to their poor solubility or tendency to oxidize.
Nicotinamide — A precursor to NAD⁺; supports cellular redox reactions and energy regeneration.
Nuclease-Free Water — Brings the reaction to the correct final volume without introducing RNases that would degrade mRNA.
2. Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour PEP-NTP mix uses phosphoenolpyruvate (PEP) as a fast energy source and pre-formed nucleoside triphosphates (NTPs), enabling rapid but short-lived transcription/translation. The 20-hour NMP-Ribose-Glucose mix uses nucleoside monophosphates plus ribose and glucose as slower-releasing energy sources, allowing the reaction to sustain itself over a much longer incubation. The tradeoff is speed vs. duration — PEP-NTP is optimized for quick protein yield, while NMP-Ribose-Glucose is designed for extended, lower-intensity expression.
3. Bonus question: How can transcription occur if GMP is not included but Guanine is?
Guanine can be salvaged by the cell-free system’s enzymes and converted to GMP, then GDP, and finally GTP through phosphorylation steps driven by the energy system in the mix. So transcription can still proceed because GTP is synthesized in situ from the guanine base.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP — Superfolder GFP is engineered for extremely robust and rapid folding, making it highly tolerant to misfolding conditions; it matures quickly and fluoresces reliably even in cell-free environments.
mRFP1 — mRFP1 has a relatively slow chromophore maturation time and requires oxygen for oxidation of the chromophore, which can limit its fluorescence in oxygen-limited cell-free reactions.
mKO2 — mKO2 (monomeric Kusabira Orange 2) has a moderate maturation rate and is sensitive to acidic pH, which can reduce fluorescence if the reaction pH drifts below 7.
mTurquoise2 — mTurquoise2 has a high quantum yield and fast maturation, making it well-suited for cell-free systems; it is relatively photostable and performs well over long incubations.
mScarlet-I — mScarlet-I is a fast-maturing red fluorescent protein with high brightness; the “I” variant was specifically engineered for improved monomericity and fast maturation compared to the original mScarlet.
Electra2 — Electra2 is a recently engineered protein optimized for cell-free expression; it is designed for low oxygen dependence and fast maturation, making it particularly well-suited for extended cell-free reactions.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect
Hypothesis for improving fluorescence:
Increase Magnesium Glutamate concentration slightly above baseline.
Hypothesis: mRFP1 has slow maturation and relies heavily on correct folding of its β-barrel structure. Increasing Mg²⁺ availability can stabilize ribosome activity and improve translational fidelity, leading to more correctly folded mRFP1 and higher total fluorescence over a 36-hour incubation.
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
Project Description I want to develop a synthetic biology-based point-of-care diagnostic platform that can rapidly detect cholera toxin and toxin co-regulated pilus antigens in stool and environmental samples within 15-30 minutes using engineered cell-free biosensors integrated into a paper-based microfluidic device.
The system employs single-domain antibodies recognition elements fused to split T7 RNA polymerase domains that reconstitute active enzyme only upon antigen binding, driving colorimetric reporter expression for visual result interpretation requiring no laboratory infrastructure or specialized training.
I want to develop a synthetic biology-based point-of-care diagnostic platform that can rapidly detect cholera toxin and toxin co-regulated pilus antigens in stool and environmental samples within 15-30 minutes using engineered cell-free biosensors integrated into a paper-based microfluidic device.
The system employs single-domain antibodies recognition elements fused to split T7 RNA polymerase domains that reconstitute active enzyme only upon antigen binding, driving colorimetric reporter expression for visual result interpretation requiring no laboratory infrastructure or specialized training.
Aim 1: Design and validate a dual-channel cell-free biosensor that detects cholera toxin and toxin co-regulated pilus, demonstrating reliable, rapid results without living cells or laboratory infrastructure.
Aim 2: Embed the biosensor into a paper-based diagnostic strip and test it on real clinical and environmental samples, confirming accuracy, stability in tropical climates, and readiness for field deployment.
Aim 3: Create an adaptable platform for rapid infectious disease detection anywhere in the world from cholera to pandemic pathogens enabling real-time outbreak surveillance and early containment in resource-limited settings.
Unmet Global Need
2.9M
Annual Cases Worldwide
Despite being treatable, Cholera remains a major threat in resource-limited areas. Current gold-standard culture tests take 3 days and require intensive lab infrastructure.
Our Mission: To bridge the diagnostic gap with a $3 test that delivers results in under 30 minutes.
Mechanism: Split T7 Polymerase
The system utilizes Split T7 RNA Polymerase as a proximity-dependent biosensor.
Separated: T7N and T7C domains remain inactive when floating separately.
Detection: Specific nanobodies bind to the target protein (Cholera Toxin or TCP).
Signal: The active enzyme drives the production of a colorimetric output.
The dual-target detection strategy simultaneously targeting cholera toxin and TCP antigens provides unprecedented specificity for toxigenic Vibrio cholerae strains while distinguishing from non-toxigenic variants and other diarrheal pathogens. The integration of nanobody-based recognition elements with split T7 RNA polymerase systems creates a modular biosensor architecture that can be rapidly adapted to detect different pathogen targets by simply replacing the binding domains, representing a platform technology rather than a single-use diagnostic.
Engineered Fusion Proteins
Construct
Fusion Protein
Domain
Target Marker
Construct 1
CT-Nanobody A
T7N (1-179)
Cholera Toxin (Signal: BLUE)
Construct 2
CT-Nanobody B
T7C (180-883)
Cholera Toxin (Signal: BLUE)
Construct 3
TCP-Nanobody A
T7N (1-179)
TCP Protein (Signal: YELLOW)
Construct 4
TCP-Nanobody B
T7C (180-883)
TCP Protein (Signal: YELLOW)
Performance Targets
Parameter
Target Value
Public Health Significance
Sensitivity
≥ 95%
High true case detection rate
Specificity
≥ 98%
Elimination of false positives
Time-to-result
15-30 Minutes
Immediate clinical action
Cost Per Test
$3.00 USD
Affordable for global outbreaks
Storage Temp
2-30°C
No cold chain required
Reference
Core Synthetic Biology & Cell-Free Systems
Pardee, K., Green, A. A., Takahashi, M. K., Braff, D., Lambert, G., Lee, J. W., … & Collins, J. J. (2016). Rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell, 165(5), 1255-1266.
o Key for: Paper-based cell-free biosensors, freeze-dried systems, toehold switches
Split Protein Systems & Protein Engineering
Galarneau, A., Primeau, M., Trudeau, L. E., & Michnick, S. W. (2002). β-Lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein–protein interactions. Nature Biotechnology, 20(6), 619-622.
o Key for: Protein complementation assays, biosensor design
Shekhawat, S. S., & Ghosh, I. (2011). Split-protein systems: beyond binary protein–protein interactions. Current Opinion in Chemical Biology, 15(6), 789-797.
o Key for: Advanced split-protein applications, biosensor engineering
Nanobodies & Antibody Engineering
Muyldermans, S. (2013). Nanobodies: natural single-domain antibodies. Annual Review of Biochemistry, 82, 775-797.
o Key for: Nanobody fundamentals, single-domain antibodies
De Meyer, T., Muyldermans, S., & Depicker, A. (2014). Nanobody-based products as research and diagnostic tools. Trends in Biotechnology, 32(5), 263-270.
o Key for: Diagnostic applications of nanobodies
Jovčevska, I., & Muyldermans, S. (2020). The therapeutic potential of nanobodies. BioDrugs, 34(1), 11-26.
o Key for: Therapeutic and diagnostic nanobody applications