Week 4 HW: Protein Design
Part A
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Since meat is not entirely made of proteins, lets assume 20% of the whole meat mass = around 100 g. An amino acid is ~100 Da (=~100g/mol). 100 g/ (100 g/mol) = 1 mol = 6.022* 10^23 AA.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Proteins in processed meat are getting denatured in our stomach by HCl and the enzyme pepsin, cutting long polypeptides. Proteases continue cutting these peptides into smaller peptides and intestinal enzymes complete the digestion into amino acids.
Shortly, our bodies do not absorb animal proteins whole, but use different enzymes to break them down to get basic amino acids
Why are there only 20 natural amino acids?
20 amino acids are representing an ideal balance for biological efficiency and chemical necessity to build all known life on Earth.
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids were synthesized abiotically through high-energy interactions between gases.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
L-amino acids are all right-handed due to steric hindrance between side chains. Since the D-enantiomer is a mirror image of an L-enantiomer, we would expect left handed helix
Can you discover additional helices in proteins?
The new helices are being discovered every day using tools like Alpha Fold.
Why are most molecular helices right-handed?
Because of the dominance of L-aminoacids in life and their chirality, most of the helices are right-handed to be sterically and energetically favourable.
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
Beta sheets are characterised by their open structure, where the carbonyl and amide groups are exposed at the edges. This exposure promotes hydrogen bonding with neighbouring strands, that is forming a stack of “sheets”.
Why do many amyloid diseases form β-sheets?
Because of the stacking nature of beta-sheets, amyloid diseases occur when proteins misfold into flat, “sticky” layers that act as templates, forcing other healthy proteins to aggregate into insoluble, thread-like fibrils. The chain of reaction recruits new proteins that are resistant to clearing mechanisms
Can you use amyloid β-sheets as materials?**
This mechanism, though, can be quite beneficial for the biomaterials. Beta-sheets represent extreme stability and high tensile strength for such biomaterials as vascular grafts in medicine, which need to have resistance function inside the body
Part B
Briefly describe the protein you selected and why you selected it.
For this part, I have selected Clathrin Heavy Chain (CHC). This protein is widely known in biology as a self-assembly protein consisting of three light chains that join into a triskeleion. Triskelions them assemble inot a geometric closed shape that creates vesicles.
Identify the amino acid sequence of your protein.
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids. Protein: Clathrin Heavy Chain 2 (Human). Length: 1,645 amino acids. Most Frequent Amino Acid: Leucine (L) - appears 196 times.

Blast search revealed homology with other clathrins, confirming its belonging to the Clathrin heavy chain family
Identify the structure page of your protein in RCSB
PDB ID: 1XI4
The resolution is 2.30 Å, which is better (smaller) than the 2.70 Å
It was published in 2004 and apart from the protein, the structure contains water molecules and glycerol
Classification: It belongs to the 7-bladed beta-propeller family.
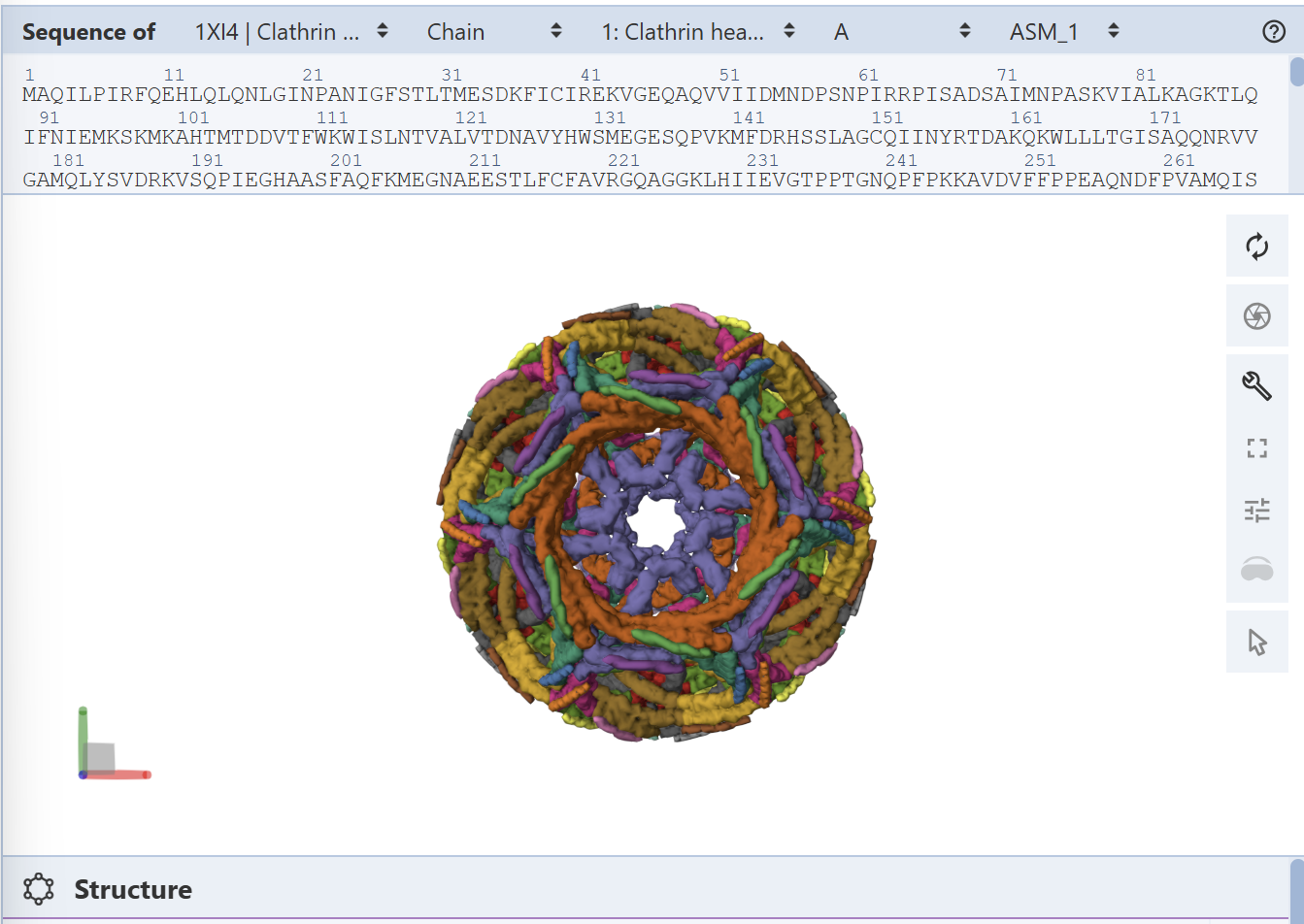
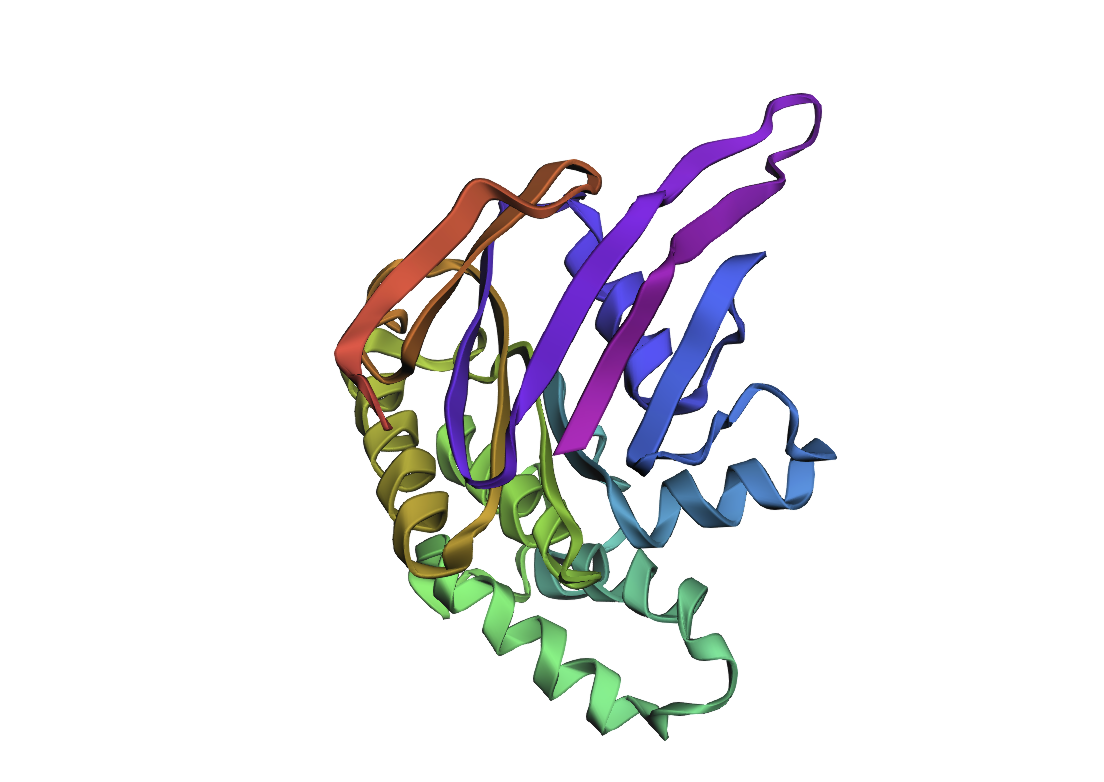
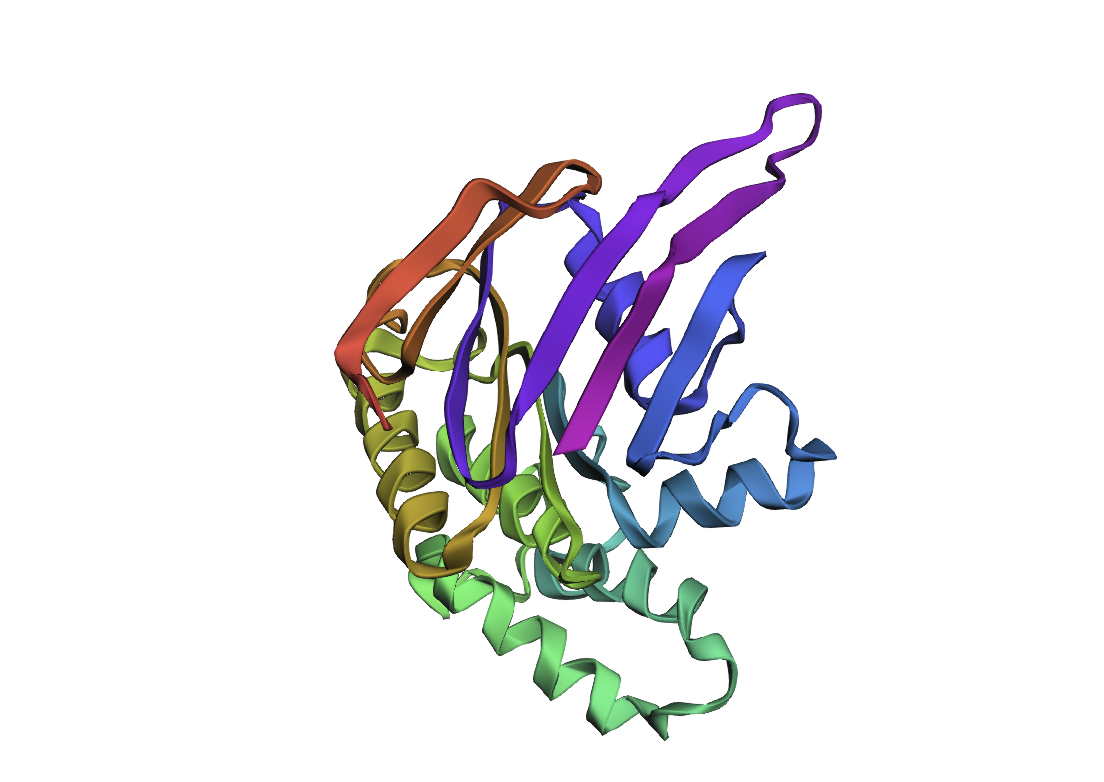
I analyzed clathrin D6 coat (PDB 1XI4), focusing on one heavy‑chain leg (chain A).

I viewed it as cartoon/ribbon, which shows a long curved backbone made almost entirely of α‑helices with only short loops.

I also added ball‑and‑stick on top of the ribbon to see individual atoms and side chains.
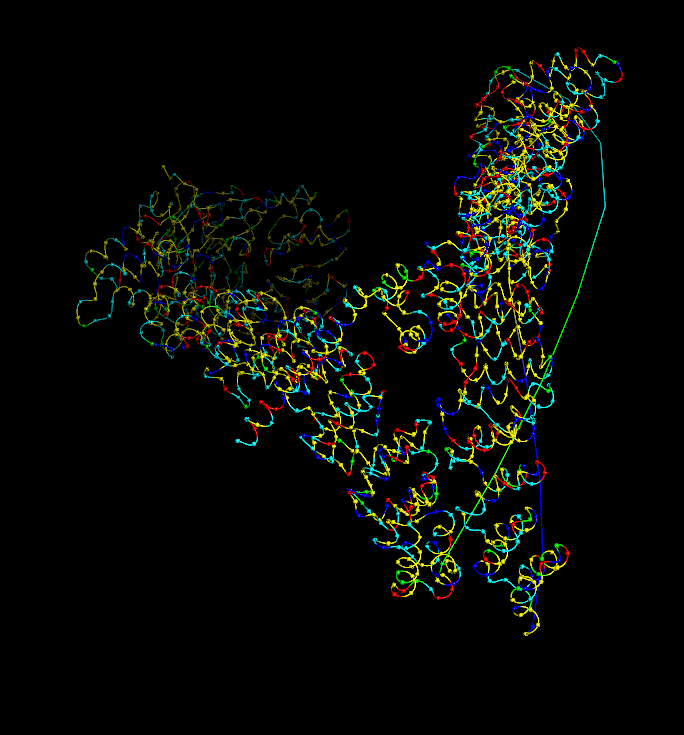
 Coloring by secondary structure (helices red, sheets yellow, loops green) showed that chain A is strongly helix‑rich, with almost no β‑sheets.
Coloring by secondary structure (helices red, sheets yellow, loops green) showed that chain A is strongly helix‑rich, with almost no β‑sheets.

Coloring by residue type (hydrophobic yellow; acidic red; basic blue; polar cyan) revealed that hydrophobic residues are mostly buried in the helical core, while charged/polar residues are on the surface.
When I displayed the surface, I saw grooves and shallow cavities between helices rather than one deep pocket, suggesting multiple shallow binding/interaction sites along the leg.
 Unfortunately, I wasn’t able to figure out how to color the structure by residues ;(
Unfortunately, I wasn’t able to figure out how to color the structure by residues ;(
Part C - KLC1 protein
C1. Protein Language Modeling
Deep Mutational Scans
Here is the protein that I used:
sp|P14679|TYRO_HUMAN Tyrosinase OS=Homo sapiens OX=9606 GN=TYR PE=1 SV=3 MLLAVLYCLLWSFQTSAGHFPRACVSSKNLMEKECCPPWSGDRSPCGQLSGRGSCQNILL SNAPLGPQFPFTGVDDRESWPSVFYNRTCQCSGNFMGFNCGNCKFGFWGPNCTERRLLVR RNIFDLSAPEKDKFFAYLTLAKHTISSDYVIPIGTYGQMKNGSTPMFNDINIYDLFVWMH YYVSMDALLGGSEIWRDIDFAHEAPAFLPWHRLFLLRWEQEIQKLTGDENFTIPYWDWRD AEKCDICTDEYMGGQHPTNPNLLSPASFFSSWQIVCSRLEEYNSHQSLCNGTPEGPLRRN PGNHDKSRTPRLPSSADVEFCLSLTQYESGSMDKAANFSFRNTLEGFASPLTGIADASQS SMHNALHIYMNGTMSQVQGSANDPIFLLHHAFVDSIFEQWLRRHRPLQEVYPEANAPIGH NRESYMVPFIPLYRNGDFFISSKDLGYDYSYLQDSDPDSFQDYIKSYLEQASRIWSWLLG AAMVGAVLTALLAGLVSLLCRHKRKQLPEEKQPLLMEKEDYHSLYQSHL
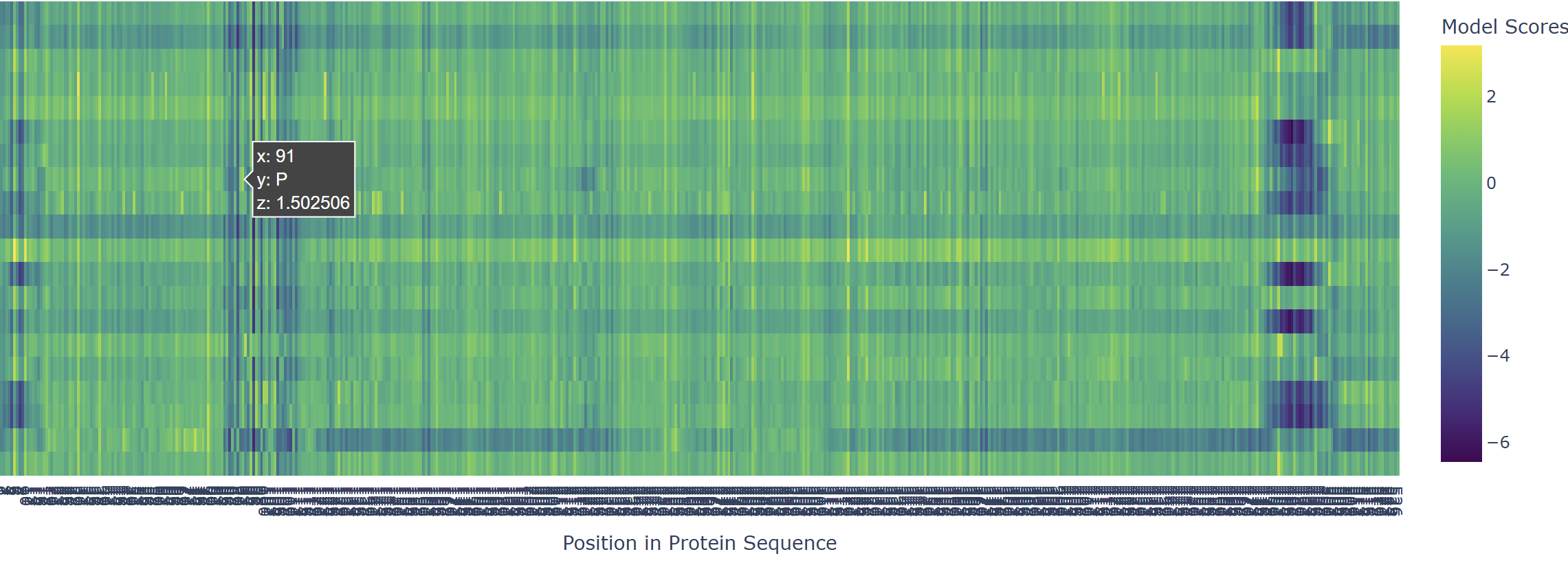
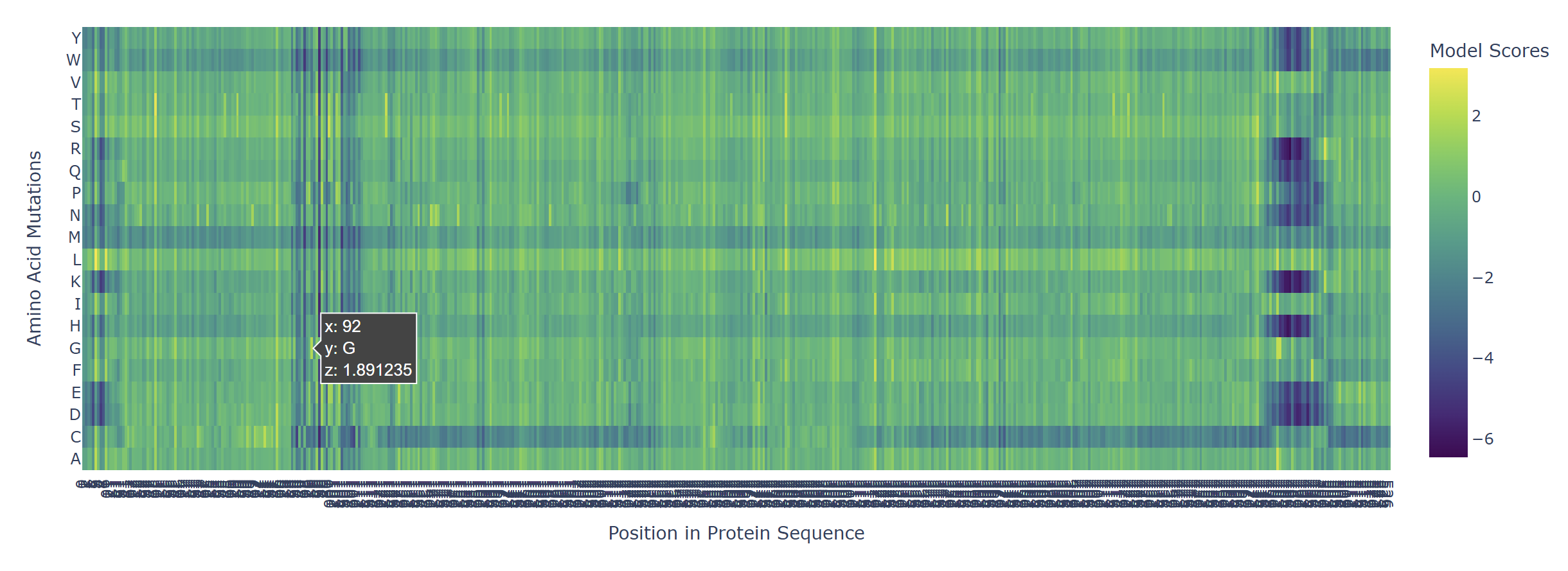
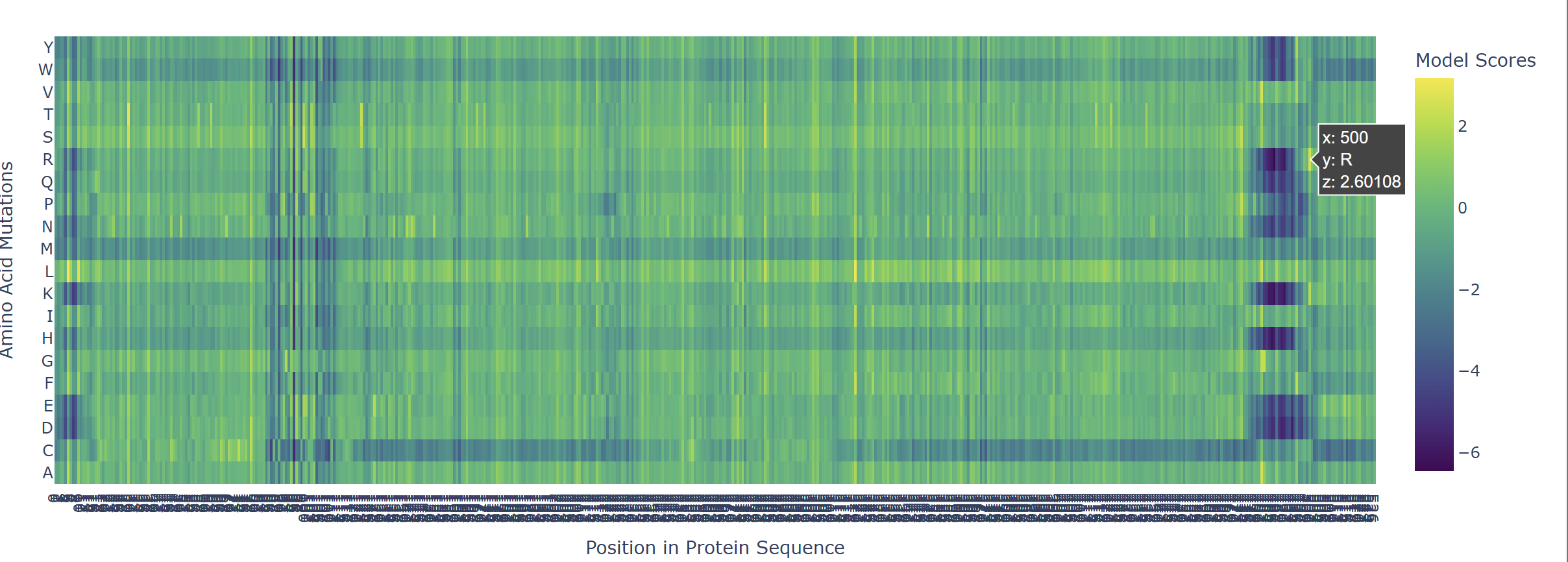
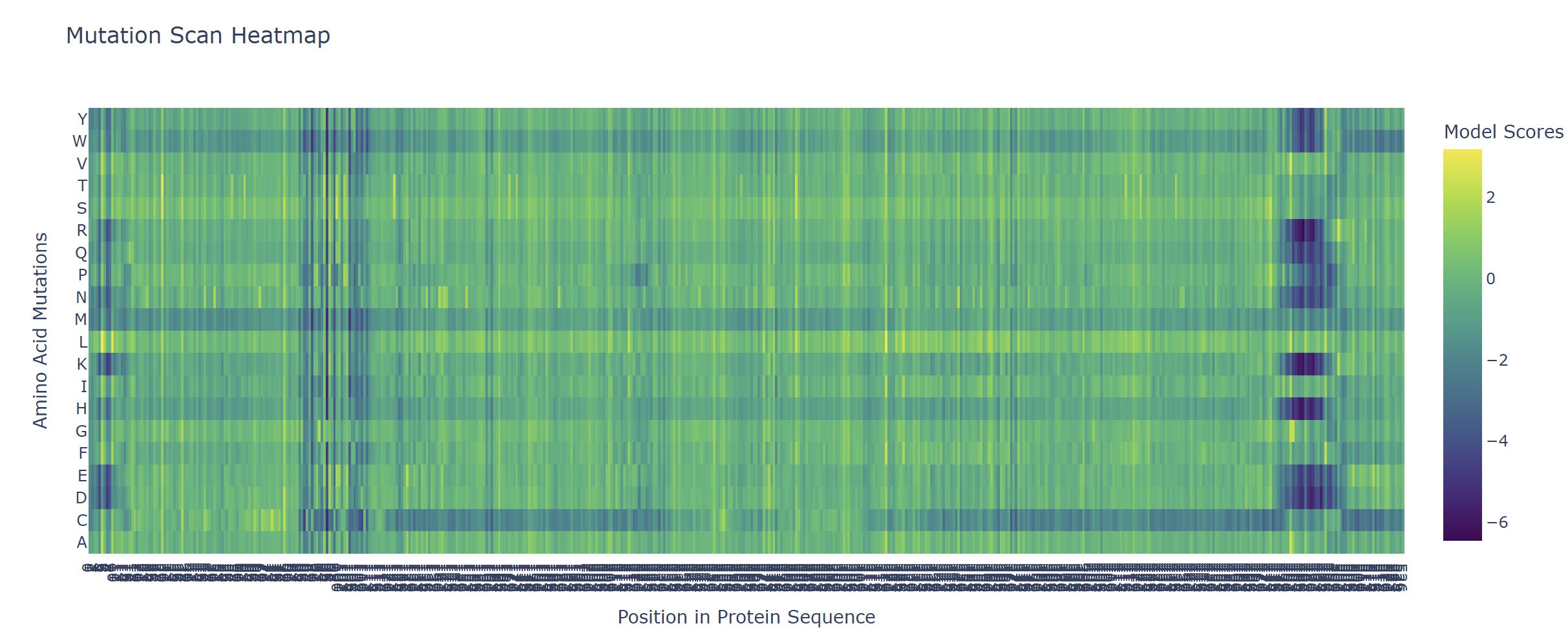 Based on the heatmap I got for my protein, I navigated to the locations where a sharp contrast was noticeable between highly sensitive sites (dark blue) and tolerant mutations (yellow). I identified three random locations (residues) that stood out by being next to dark blue. These yellow spots (see photo below) represent permessive mutations: specific amino acid substitutions that the language model predicted will preserve the protein structural and functional integrity despite being highly conserved regions.
Based on the heatmap I got for my protein, I navigated to the locations where a sharp contrast was noticeable between highly sensitive sites (dark blue) and tolerant mutations (yellow). I identified three random locations (residues) that stood out by being next to dark blue. These yellow spots (see photo below) represent permessive mutations: specific amino acid substitutions that the language model predicted will preserve the protein structural and functional integrity despite being highly conserved regions.
| 
| 
|
Latent Space Analysis
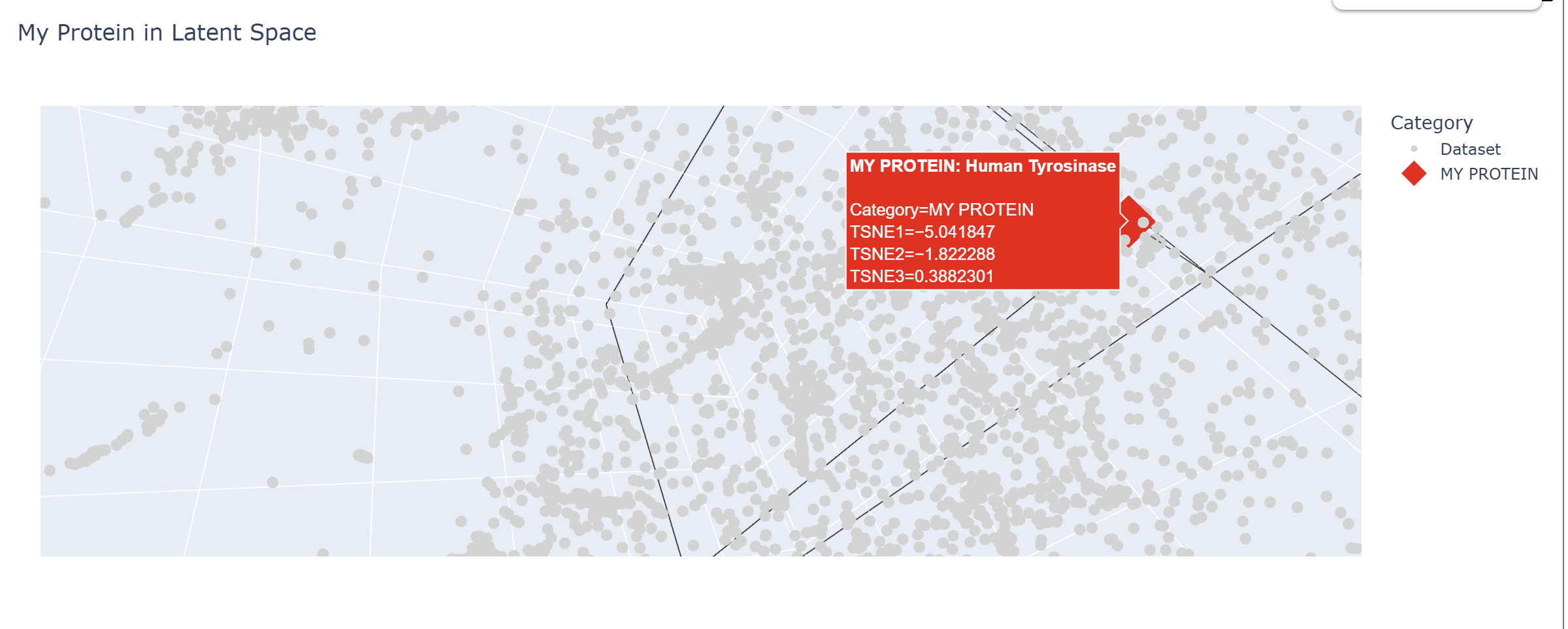
In my Latent Space Analysis, my protein (Human Tyrosinase) appeared within the class of All-Alpha protein neighborhood, which makes biological sense because both proteins share a conserved di-copper binding fold. This shows that the ESM2 model can accurately group proteins by their 3D shape and evolutionary ’language,’ even if they come from completely different species.
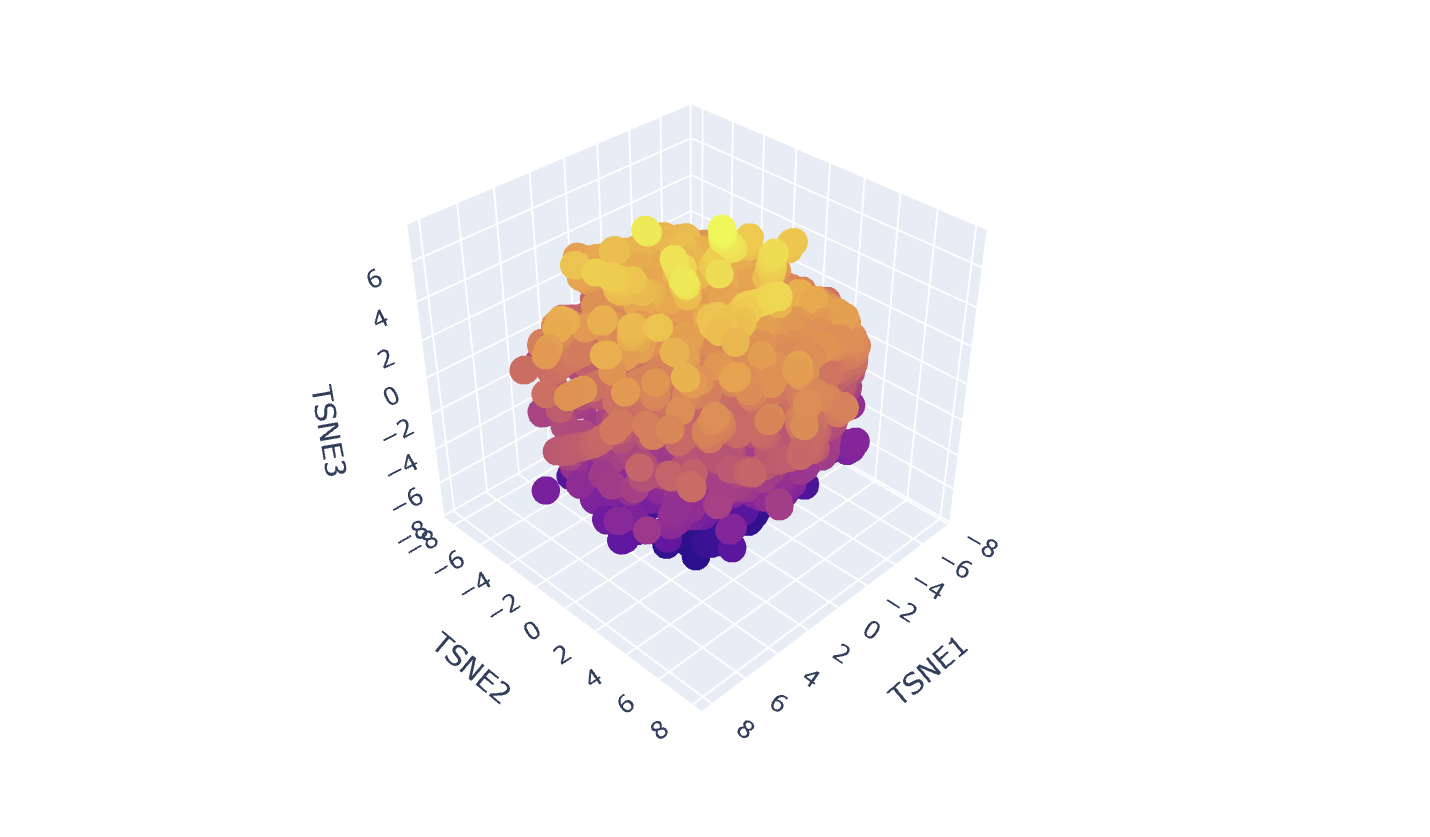
I changed the code a bit so my protein would be visible wihtin thousands of dots.
C2. Protein Folding
I chose a random protein I found on the ESM Metagenomic Atlas fro my Protein folding task. Amino Acid sequence:
MSIPTINAEGLNKSFGHRQVLNDISFRVAKGEMVALIGPSGSGKSTLLRHLVGLTCGNRHQGGRVSLMGREVQASGHLRRAARIERCRTGYIFQQFNLVGRLSVLTNVLVGQLGSMSRLRALFGRFTEQERQRARACLARVGLEELIDQRANTLSGGQMQRVAIARVLMQDAELILADEPIASLDPRSAREVMEILSRIHAEDGRTVVVTLHQVDVARRYCHRAVALKDGRLYFDGPINELTDERLQALYENADLDELRASEASNGEALSSDRRDRTPHTVVTPVLG
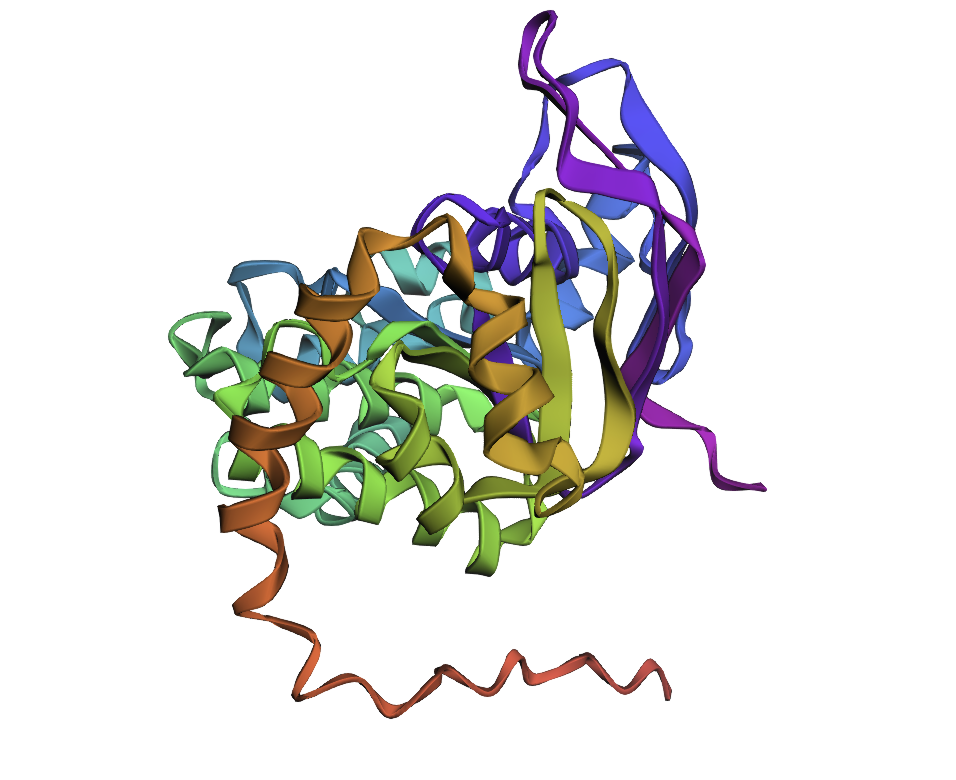
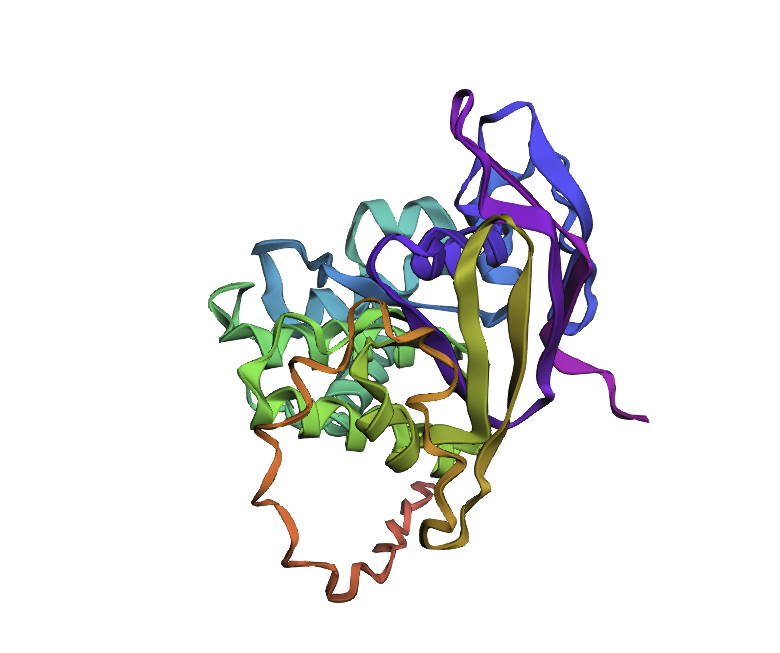
Few mutations haven’t changed the 3D structure at all, so I performed a ‘stress test’ on my protein by changing a large sequence of amino acids from position 170 to 200. The 3D model showed minor conformational change:
C3. Protein Generation
After performing Inverse Folding with ProteinPMNN, I’ve received the next sequence:
MLELENISYKVNLGDKIVTRLDNVNLSVPKGERVVILGEPGSGKSTLMDILACLAKPTSGKVLVDGEDVNDLSEEERERVRRTKIGLIDQEPGLDPDLTALENVMVPLRELYPGELTDEELEARARECLLLAQLPAELFDKRPAELTPLEQQRVQLARALAPEPPILLADEPTAALDPEDGAKLMDLLVYLADVLGKTVVIFTHNPEVARYGDRIIHLKNGKIASEEVLRPL
The resulted sequence appeared to 232 AA long, compared to the original 287 AA.
After inputting this sequence into ESMFold, next 3d structure formed:
By the comparison we can see it differs from the original structure but has some similarities.
| Original | New |
|---|---|
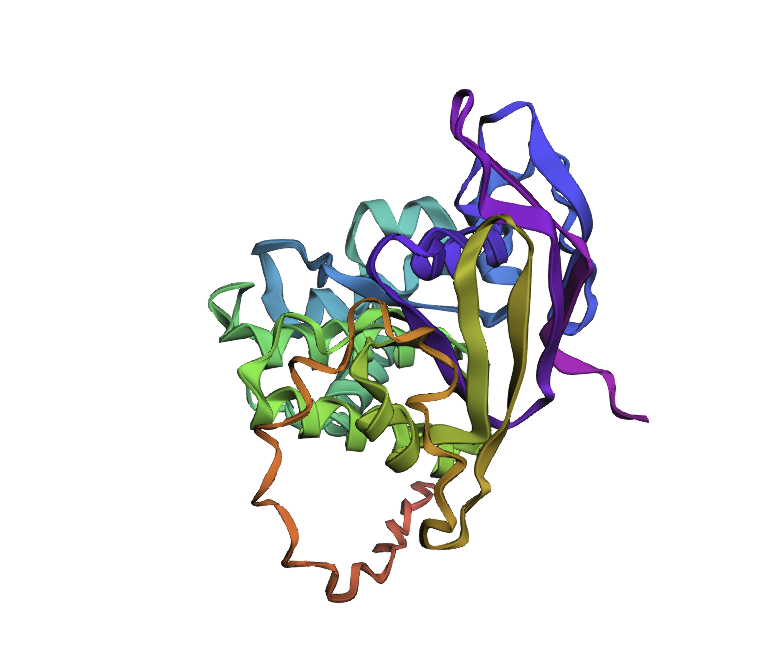
| 
|
Part D
Project Proposal
Chosen Goals
Increase toxicity (lytic efficiency) of the MS2 L protein by tuning its interaction with E. coli DnaJ and its putative target.
Improve thermal and conformational stability of L so that toxic variants remain well folded and functional across experimental conditions.
Computational approach
Protein language models (ESM-2 / ProGen) - to design
Run in silico mutagenesis on the MS2 L sequence to score single and small combinatorial substitutions for evolutionary “fitness” and tolerated diversity.
Use these scores to (i) preserve positions that are highly conserved or known to be essential for lysis and DnaJ dependency, and (ii) explore mutations at more flexible residues that may enhance toxicity or stability.
Structure prediction (AlphaFold-Multimer or AlphaFold3)
- Model the complex between full-length MS2 L and E. coli DnaJ, using the experimentally defined minimal lytic domain and the N‑terminal basic regulatory domain as guides.
- Map the predicted binding interface around residues implicated in DnaJ dependence and inactivating missense mutations (for example, the conserved Leu48–Ser49 motif and neighboring central-domain residues).
- Use these models to prioritize mutations predicted to strengthen productive L–DnaJ contacts or relieve autoinhibition of L while maintaining membrane association.
Sequence redesign for stability (ProteinMPNN, Foldseek/NGL/PyMOL)
For promising L variants from the pLM and AlphaFold stages, use ProteinMPNN on fixed backbones to propose alternative side chains that lower the estimated folding free energy (ΔG) without disrupting the DnaJ-contact surface.
Visualize candidate designs in NGL Viewer or PyMOL to check for clashes, loss of transmembrane character, or obvious disruption of the domain architecture suggested by mutational analysis.
Potential pitfalls and limitations
Mutations that increase toxicity may destabilize the protein or alter its membrane topology, leading to misfolding or loss of function DnaJ Conformational Flexibility: Chaperones like DnaJ are inherently flexible. A static AlphaFold model might not capture the dynamics for lysis and false positive
Pipeline Schematic
Input: Wild-type MS2 L Protein Sequence. Step 1 (Optimization): ESM-2 mutation scoring for fitness.
Step 2 (Binding): AlphaFold-Multimer modeling of L-Protein + DnaJ complex.
Step 3 (Refinement): ProteinMPNN sequence redesign for thermal stability.
Output: Top 5 candidate sequences for in vitro synthesis and plaque assay testing.