Week 5 HW: Protein Design. Part 2
PART A: Computational Peptide Design — SOD1 A4V Binder Generation
Background
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS) — a severe neurodegenerative disorder characterized by adult-onset loss of upper and lower motor neurons, progressive paresis, skeletal muscle atrophy, quadriplegia, and fatal respiratory failure.
The A4V mutation (Alanine → Valine at residue 4) is one of the most aggressive ALS-associated variants. It subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation. The task is to design short peptides that bind mutant SOD1 and evaluate which are worth advancing toward therapy.
Part 1: Peptide Generation with PepMLM
The human SOD1 sequence was retrieved from UniProt (P00441) and the A4V mutation was introduced manually (position 4: Ala → Val):
Wild-type: MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V mutant: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM-650M model (ChatterjeeLab, HuggingFace) conditioned on the A4V mutant sequence, 4 peptides of length 12 amino acids were generated. The known SOD1-binding peptide FLYRWLPSRRGG was added as a control. Lower pseudo-perplexity = higher model confidence in binding potential.
| Index | Peptide Sequence | Pseudo-Perplexity | Notes |
|---|---|---|---|
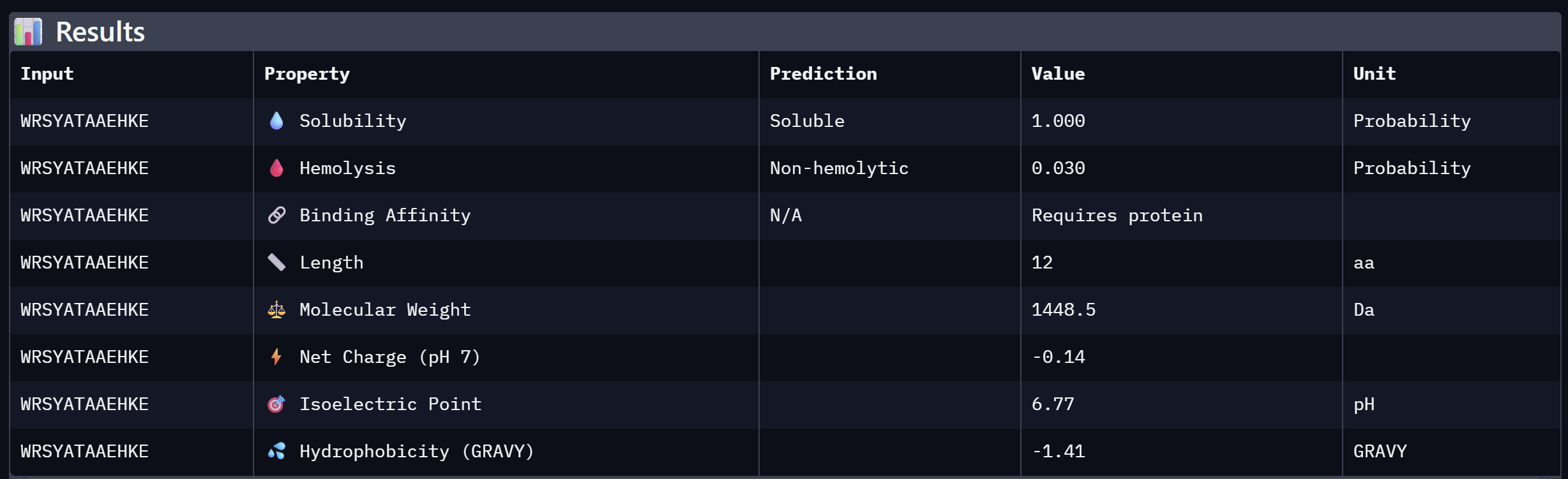
| 1 | WRSYATAAEHKE | 10.552 | Best candidate — lowest perplexity, highest model confidence |
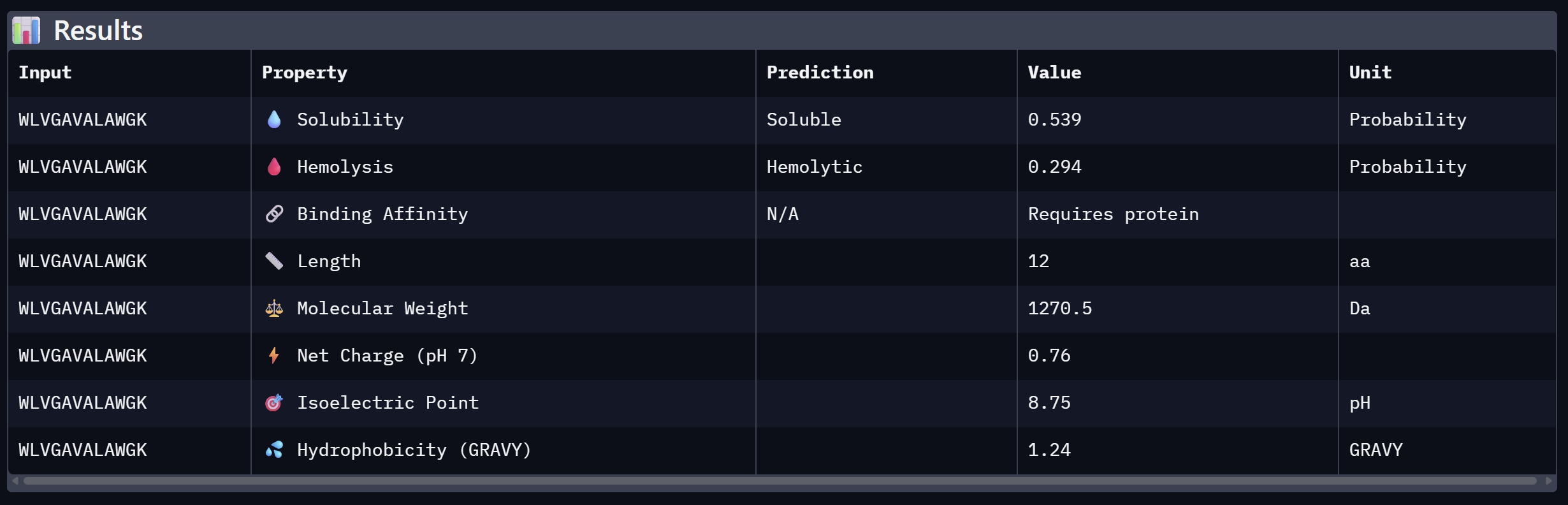
| 2 | WLVGAVALAWGK | 10.608 | Close second; hydrophobic core with aromatic flanking |
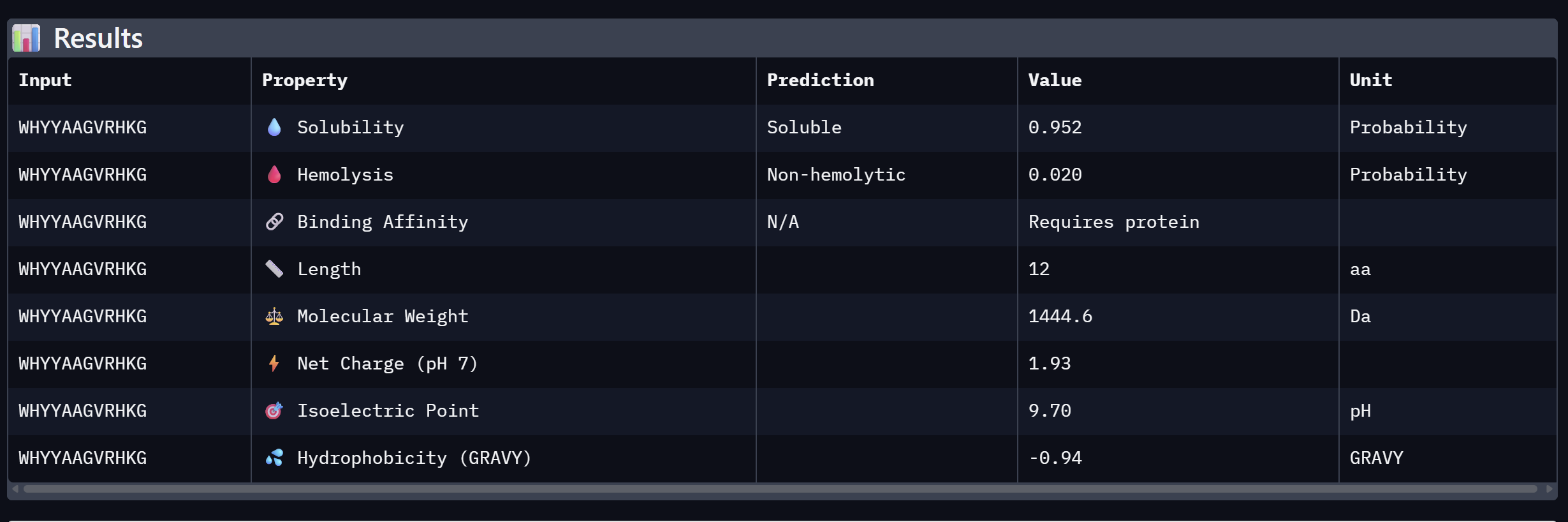
| 3 | WHYYAAGVRHKG | 16.820 | Moderate confidence; aromatic-rich N-terminus |
| 4 | WRYGPVGLRWKE | 19.620 | Lowest confidence; highest perplexity |
| Control | FLYRWLPSRRGG | — | Known SOD1-binding peptide; reference benchmark |
The best candidate is WRSYATAAEHKE (pseudo-perplexity = 10.552).
Part 2: Evaluate Binders with AlphaFold3
I submitted the mutant SOD1 sequence followed by the peptide sequence into Alphafold to model the protein-peptide complex and evulate their binding efficiecy.
Binder 1:
Alpha Fold Image
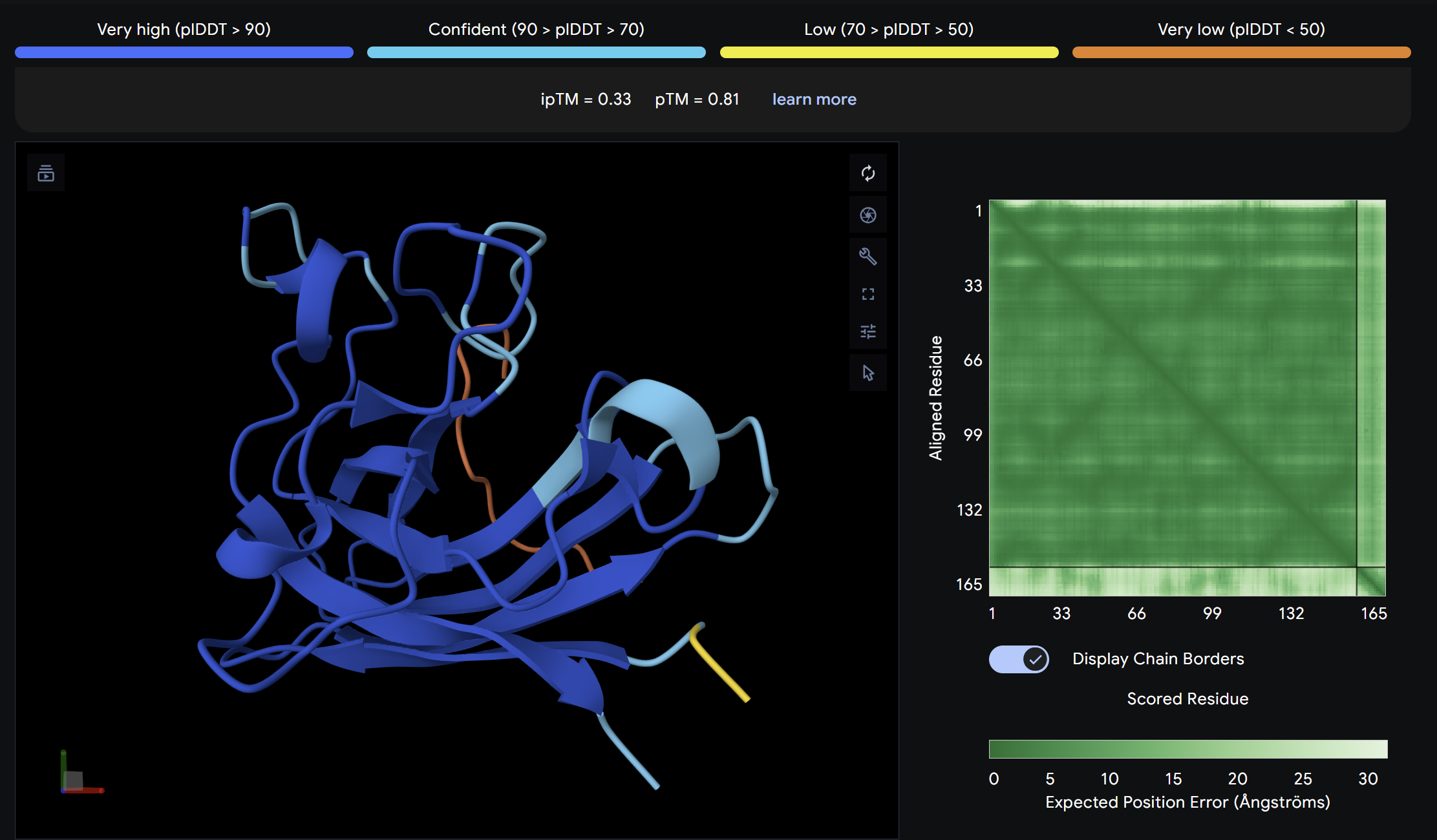 ipTM = 0.33
ptm = 0.81
This peptide showed moderate predicted binding confidence, comparable to the control. In the AlphaFold3 structure, it appears to localize peripherally on the SOD1 surface, away from the N-terminal A4V mutation site. The peptide appears largely surface-bound with no significant burial into the protein core, suggesting a weak or non-specific interaction.
ipTM = 0.33
ptm = 0.81
This peptide showed moderate predicted binding confidence, comparable to the control. In the AlphaFold3 structure, it appears to localize peripherally on the SOD1 surface, away from the N-terminal A4V mutation site. The peptide appears largely surface-bound with no significant burial into the protein core, suggesting a weak or non-specific interaction.
Binder 2:
Alpha Fold Image
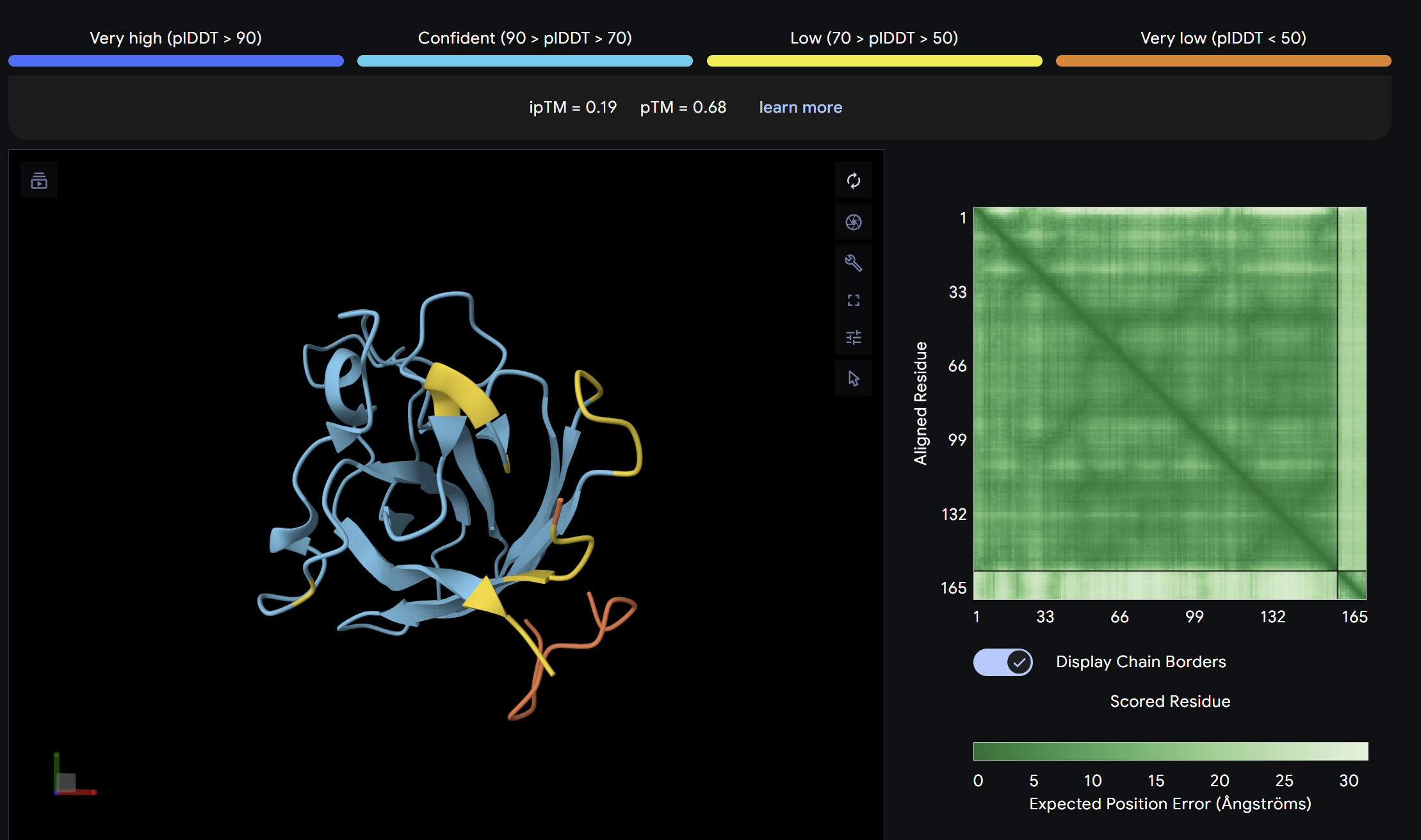 ipTM = 0.19
ptm = 0.68
The lowest ipTM score of all tested peptides. The predicted complex shows a loosely associated peptide with high positional uncertainty across the PAE matrix. It does not appear to engage the N-terminus, β-barrel, or dimer interface in a meaningful way. This peptide is unlikely to be a functional binder despite its low perplexity score.
ipTM = 0.19
ptm = 0.68
The lowest ipTM score of all tested peptides. The predicted complex shows a loosely associated peptide with high positional uncertainty across the PAE matrix. It does not appear to engage the N-terminus, β-barrel, or dimer interface in a meaningful way. This peptide is unlikely to be a functional binder despite its low perplexity score.
Binder 3:
Alpha Fold Image
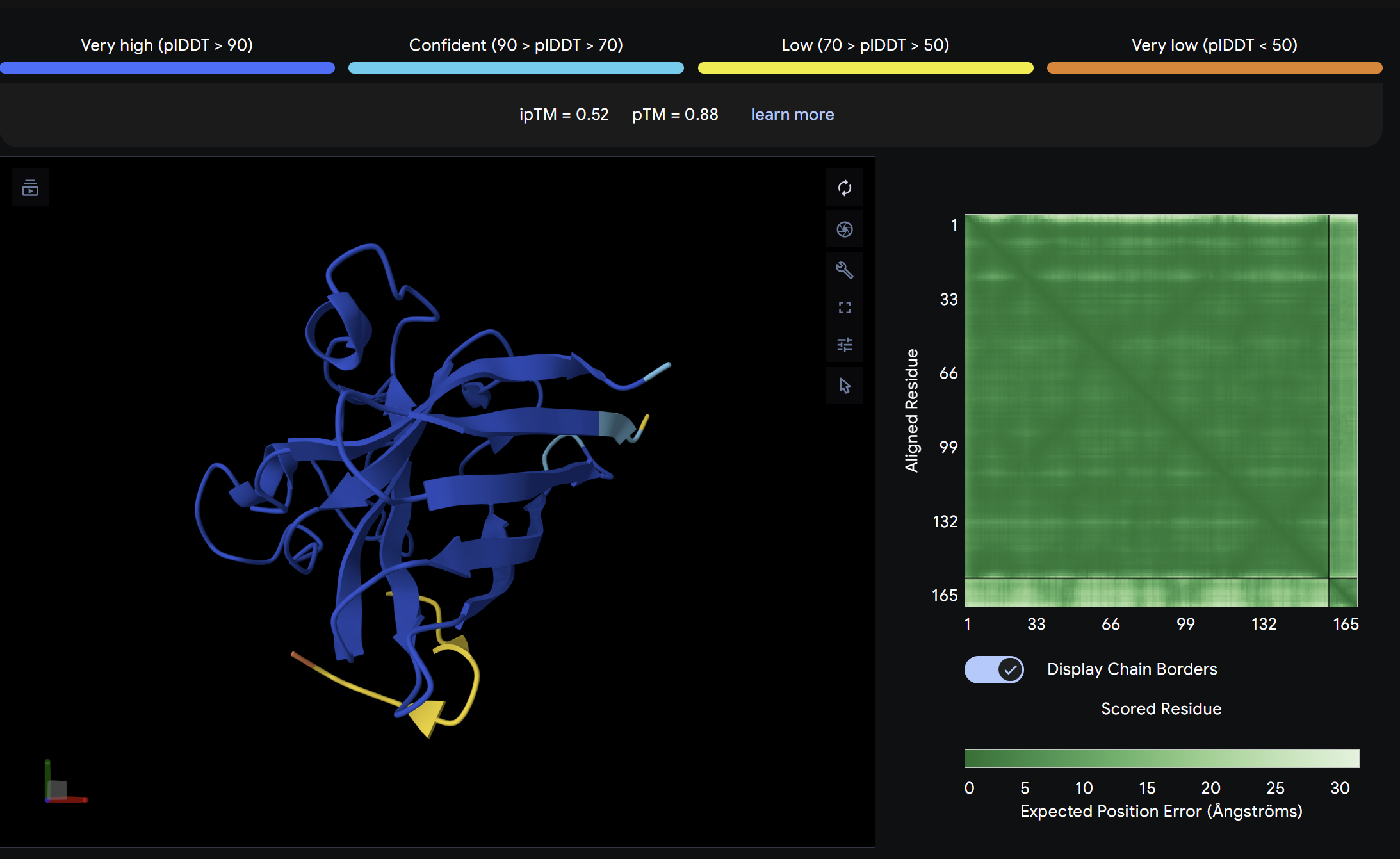 ipTM = 0.52
ptm = 0.88
The strongest predicted binder of the set, exceeding the control. The structure shows the peptide engaging a region near the β-barrel domain with partial contact toward the N-terminal region where A4V sits. The peptide appears partially buried rather than fully surface-exposed, suggesting a more specific interaction interface. This is the most promising PepMLM-generated candidate.
ipTM = 0.52
ptm = 0.88
The strongest predicted binder of the set, exceeding the control. The structure shows the peptide engaging a region near the β-barrel domain with partial contact toward the N-terminal region where A4V sits. The peptide appears partially buried rather than fully surface-exposed, suggesting a more specific interaction interface. This is the most promising PepMLM-generated candidate.
Binder 4:
Alpha Fold Image
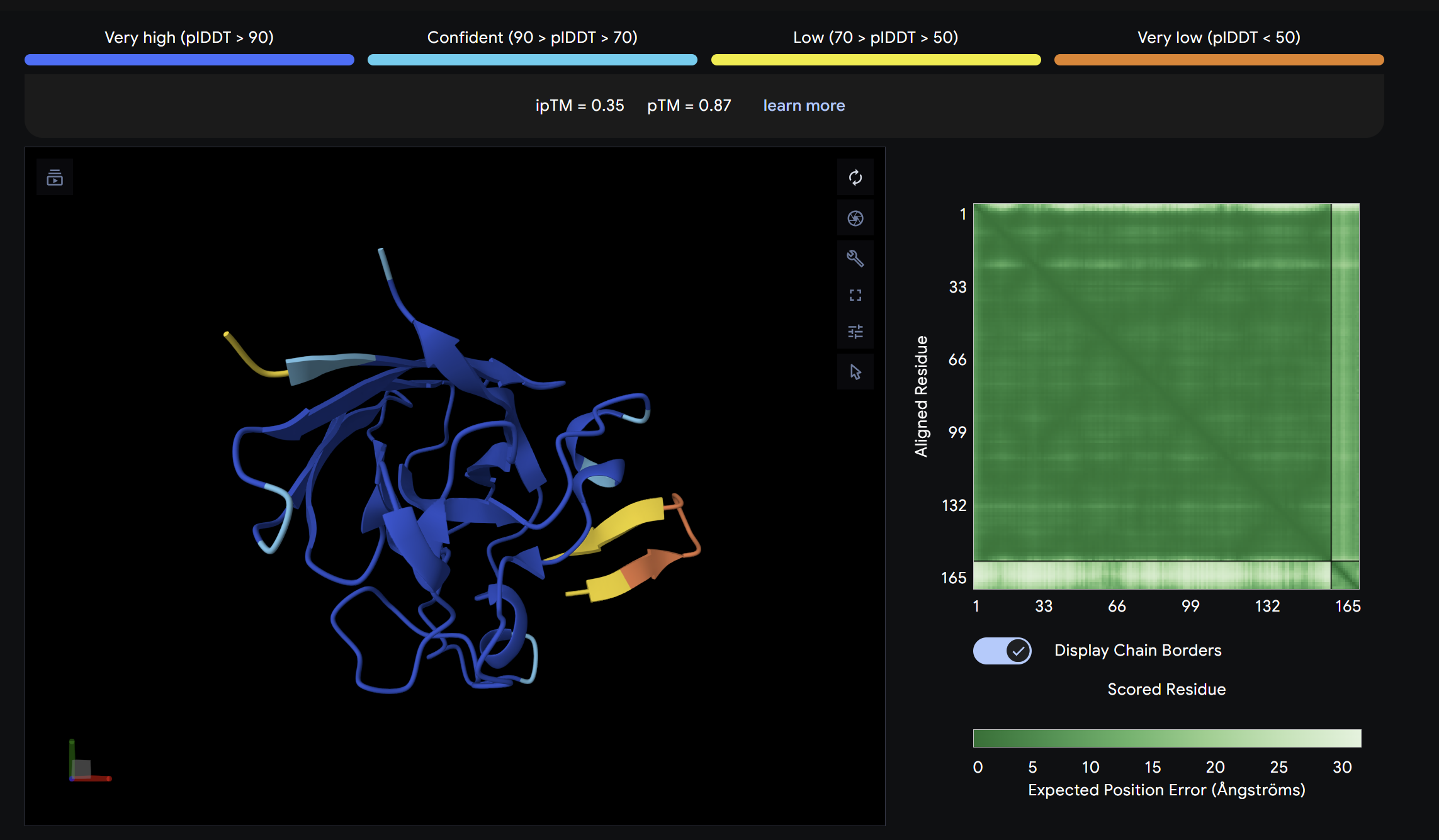 ipTM = 0.55
ptm = 0.87
Slightly above the control in ipTM score. The peptide localizes near a peripheral interface region of SOD1 but does not clearly engage the A4V mutation site or dimer interface. It appears surface-bound with moderate positional confidence.
ipTM = 0.55
ptm = 0.87
Slightly above the control in ipTM score. The peptide localizes near a peripheral interface region of SOD1 but does not clearly engage the A4V mutation site or dimer interface. It appears surface-bound with moderate positional confidence.
Binder 5:
Alpha Fold Image
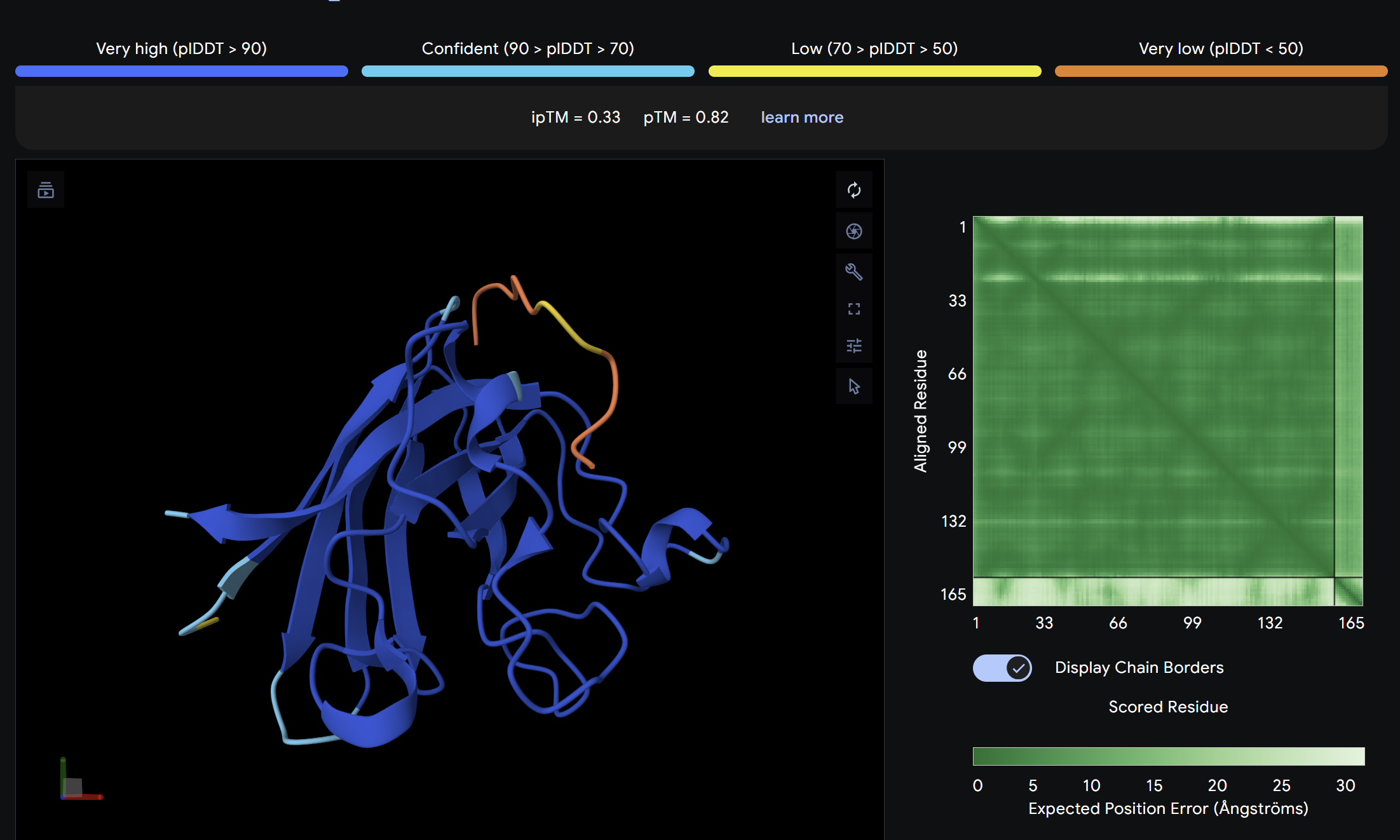 ipTM = 0.33
ptm = 0.82
The known SOD1-binding peptide serves as the baseline reference. Its ipTM of 0.33 reflects moderate predicted binding, surface-associated without deep burial. Notably, Binder 3 (WHYYAAGVRHKK) exceeds the control with an ipTM of 0.52, suggesting PepMLM successfully generated at least one peptide with stronger predicted binding than the established reference. Overall, ipTM values across all peptides are in the low-to-moderate range (0.19–0.52), consistent with short peptide binders where full complex confidence is inherently limited.
ipTM = 0.33
ptm = 0.82
The known SOD1-binding peptide serves as the baseline reference. Its ipTM of 0.33 reflects moderate predicted binding, surface-associated without deep burial. Notably, Binder 3 (WHYYAAGVRHKK) exceeds the control with an ipTM of 0.52, suggesting PepMLM successfully generated at least one peptide with stronger predicted binding than the established reference. Overall, ipTM values across all peptides are in the low-to-moderate range (0.19–0.52), consistent with short peptide binders where full complex confidence is inherently limited.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Binder 1:
Binder 2:
Binder 3:
Binder 4:
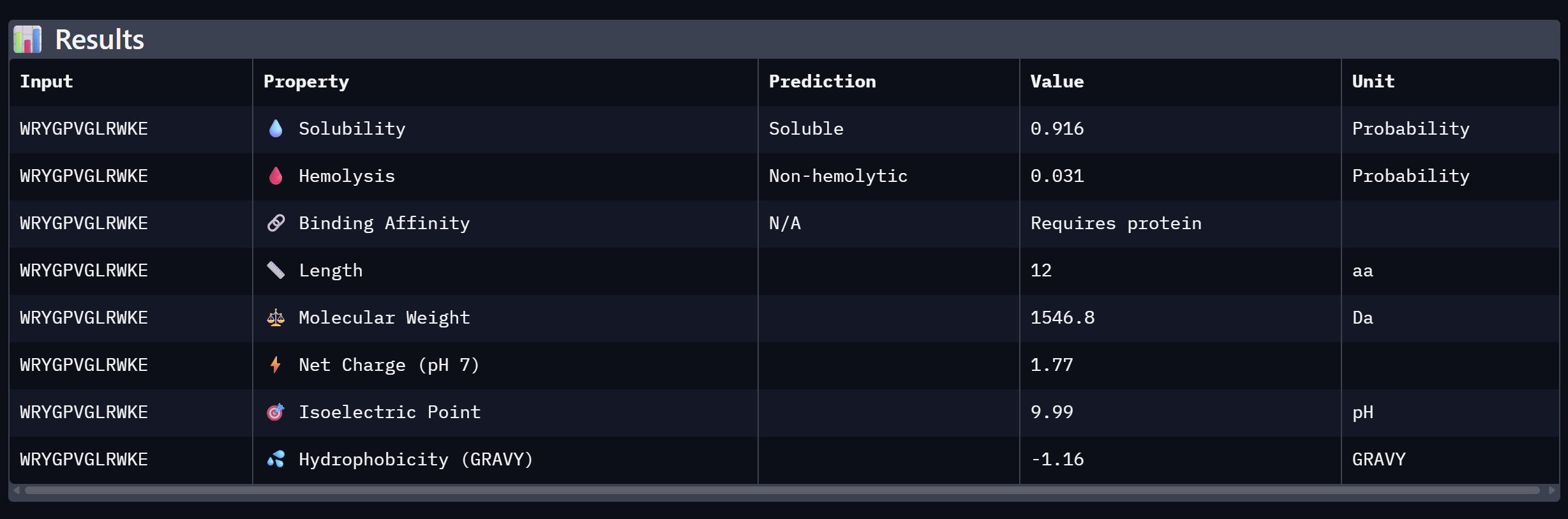
An analysis of PeptiVerse predictions alongside AlphaFold3 structural data highlights a significant alignment between structural binding and therapeutic potential. Binder 3 (WHYYAAGVRHKG) emerged as the most promising candidate, achieving the highest ipTM score of 0.52 and an optimal therapeutic profile. Its characteristics include superior solubility (0.952), a minimal hemolysis probability (0.020), and a +1.93 positive charge that likely facilitates electrostatic interactions with the negatively charged surface of SOD1. This correlation suggests that high predicted structural binding serves as a reliable indicator of therapeutic viability. At the same time, Binder 2 (WLVGAVALAWGK) is the least viable candidate due to both structural and physicochemical deficiencies. It recorded the lowest ipTM score (0.19) and is categorized as hemolytic (0.294) with marginal solubility (0.539). Its elevated hydrophobicity (GRAVY = +1.24) is the probable cause for its poor solubility and associated hemolysis risk. While Binders 1 and 4 demonstrated solubility and were non-hemolytic, their moderate ipTM scores reduced their overall therapeutic appeal. Since PeptiVerse does not support direct protein target input for affinity calculations, AlphaFold3 ipTM scores were utilized as a proxy for binding affinity. My chosen peptide for later advancement is WHYYAAGVRHKG (Binder 3). It is the only candidate that at the same time: exceeds the control binder in predicted structural bidning (ipTM = 0.52 vs. 0.33), hihgly soluble, non hemolytic and carreis a favorable charge for SOD1 interaciton. Despite possessing a higher perplexity score than Binders 1 and 2, its integrated therapeutic and structural profile establishes it as the most stongest candidate overall.
Part C: L-Protein Mutant Design — MS2 Phage Lysis Engineering
Background
Bacteriophage MS2 relies on its L-protein (lysis protein) to form pores in the E. coli cell membrane, ultimately lysing the host. A common bacterial resistance mechanism involves a point mutation in the chaperone DnaJ, which prevents proper L-protein processing and abolishes lysis. The objective is to engineer L-protein variants that either (1) fold independently of DnaJ, or (2) lyse bacteria faster, reducing the window for resistance to develop.
The wild-type L-protein sequence (UniProtKB P03609) is:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
The protein has two functional domains:
- Soluble N-terminal domain (residues 1–40): responsible for DnaJ interaction
- Transmembrane domain (residues 41–75): responsible for membrane insertion and lysis activity
Option 1: Mutagenesis Scoring via ESM2 Language Model
Step 1 — Computational scoring (ESM2 heatmap)
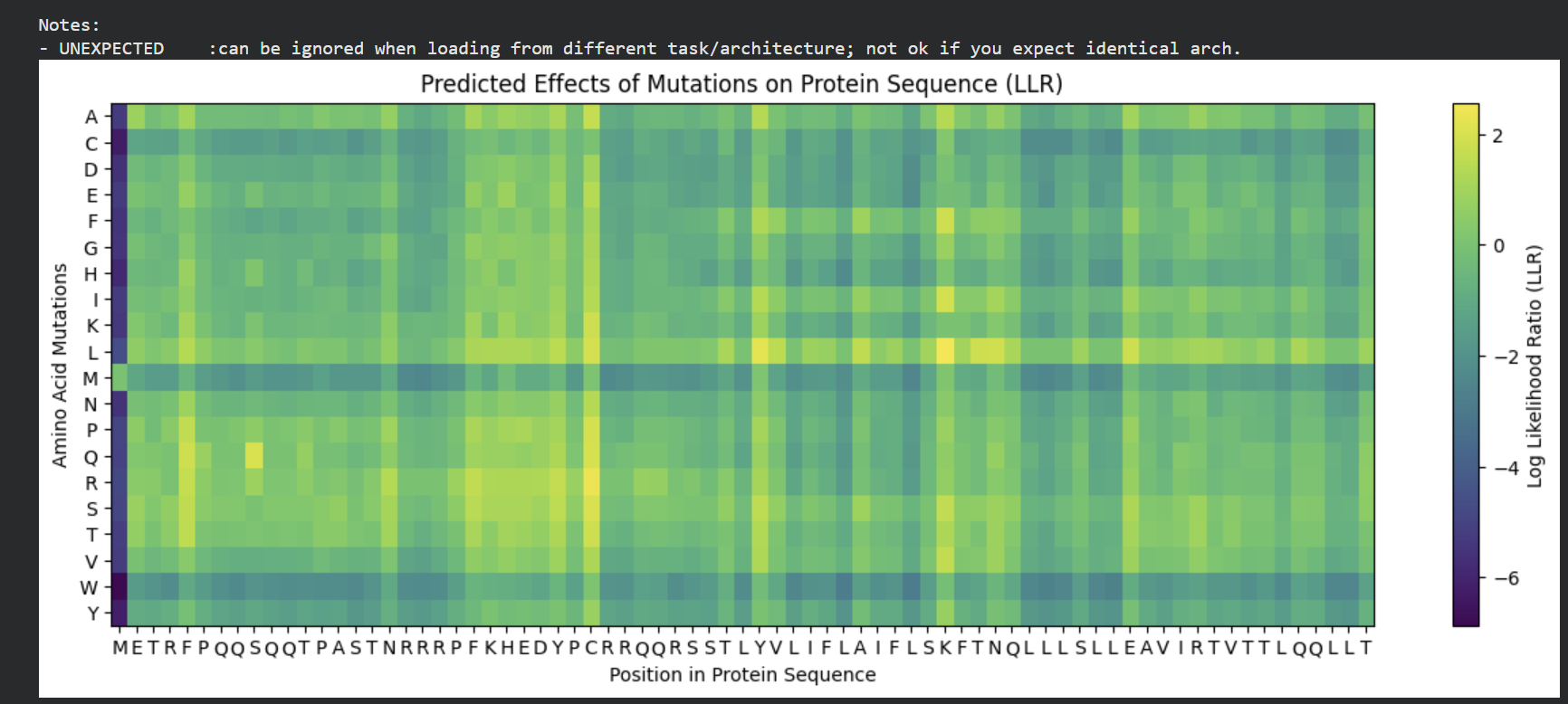
The mutagenesis scoring notebook was run using the ESM2 language model (facebook/esm2_t6_8M_UR50D). For each position in the L-protein sequence, a Log Likelihood Ratio (LLR) score was calculated for every possible amino acid substitution.
Positive LLR scores: indicates the model predicts the mutation is tolerated or beneficial
Negative LLR scores: indicates it would likely be harmful.
The resulting heatmap shows LLR values across all 75 positions and 20 amino acids. Bright yellow regions indicate high positive LLR (favorable mutations); dark blue/purple regions indicate strongly negative LLR (deleterious mutations). Notably, positions in the transmembrane region (right half of the heatmap) show more variability, reflecting the model’s sensitivity to hydrophobicity changes in membrane-spanning segments.
Step 2 — Cross-validation with experimental data
The experimental L-Protein Mutants dataset was compared against the ESM2 LLR scores. Key observations:
- Mutations with Lysis = 0 (non-functional) generally corresponded to negative or near-zero LLR scores, confirming the model captures some biological signal.
- Mutations with Lysis = 1 (functional) at positions 18, 25, 30, 31, 44, 45, and 46 were associated with positive LLR scores, suggesting reasonable agreement between computational and experimental data.
- The model has limitations: some experimentally functional mutations (Lysis = 1) had modest LLR scores, and the model does not account for membrane topology or DnaJ interaction directly.
A selection of key entries from the experimental dataset is shown below (Lysis: 1 = functional, 0 = non-functional, N.D. = not determined):
| AA Position | AA Change | Lysis | Protein Levels |
|---|---|---|---|
| 1 | M→I | 0 | 0 |
| 1 | M→T | 0 | 0 |
| 13 | P→L | 1 | 1 |
| 15 | S→A | 1 | 1 |
| 18 | R→G | 1 | 1 |
| 18 | R→I | 1 | 1 |
| 18 | R→Stop | 0 | N.D. |
| 19 | R→S | 1 | 0 |
| 23 | K→E | 1 | 0 |
| 25 | E→G | 1 | 0 |
| 29 | C→R | — | — |
| 29 | C→Stop | 0 | N.D. |
| 30 | R→Q | 1 | 1 |
| 30 | R→L | 1 | 1 |
| 31 | R→I | 1 | 1 |
| 39 | Y→H | 0 | 0 |
| 39 | Y→Stop | 0 | N.D. |
| 44 | L→P | 1 | 1 |
| 45 | A→P | 1 | 1 |
| 46 | I→F | 1 | 1 |
| 50 | K→N | 0 | 1 |
Step 3 — Selected mutations
Five mutations were selected by prioritizing positions with (a) high positive LLR scores from the ESM2 notebook and (b) experimental lysis data showing Lysis = 1 where available. Conserved positions (no variation in BLAST alignments) were avoided.
| # | Position | Domain | Wild-type AA | Mutant AA | LLR Score | Experimental Lysis | Rationale |
|---|---|---|---|---|---|---|---|
| 1 | 18 | Soluble | R | G | positive | Lysis = 1 | Experimentally confirmed functional; removing the positively charged arginine may reduce DnaJ dependency by altering soluble domain surface charge |
| 2 | 29 | Soluble | C | R | 2.395 (high) | Not tested | Highest LLR in soluble region; cysteine at position 29 may form inappropriate disulfide bonds — replacing with arginine adds a stabilizing charge interaction |
| 3 | 39 | Soluble/TM boundary | Y | L | 2.242 (high) | Not tested | High LLR score; tyrosine at the domain boundary may be substituted with leucine to improve hydrophobic continuity into the TM domain and aid autonomous folding |
| 4 | 45 | Transmembrane | A | L | 1.539 (positive) | A→P Lysis = 1 (nearby) | Positive LLR; increasing hydrophobicity at this TM position may improve membrane insertion efficiency; nearby A45P is experimentally confirmed functional |
| 5 | 50 | Transmembrane | K | L | 2.561 (highest) | Not tested | Highest LLR score across the entire sequence; K50 is a charged residue embedded in the hydrophobic TM core — replacing it with leucine removes the charge mismatch and is predicted to strongly stabilize membrane insertion |