Hi, I’m Eleonora, a junior in Bio‑Convergence at Yonsei University.
This is my first time diving deeply into synthetic biology and testing my skills, so through this course, I hope to learn new tools, see different applications of bioengineering, build a trustworthy community, explore how creative I can be in this field, and how design and biology can work together. 🌱
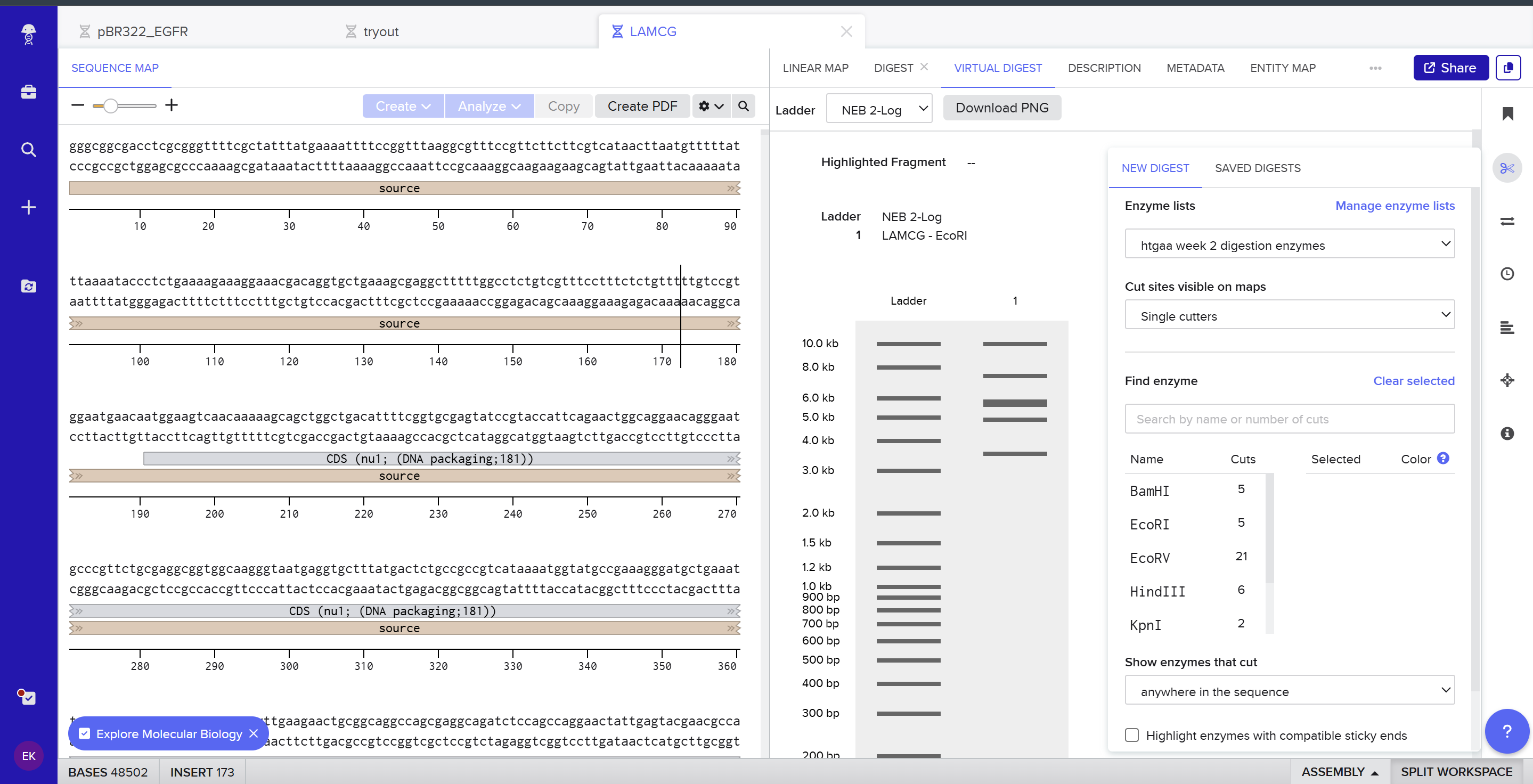
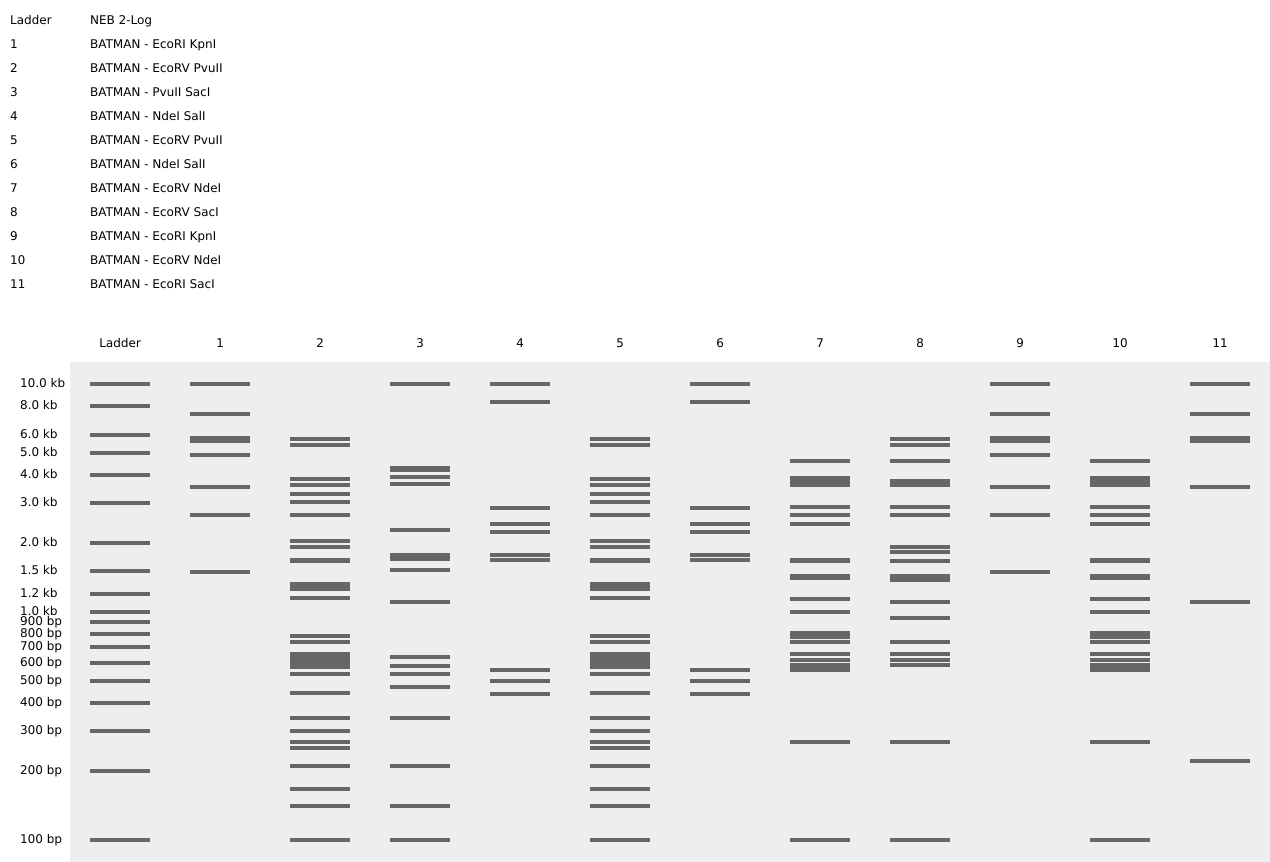
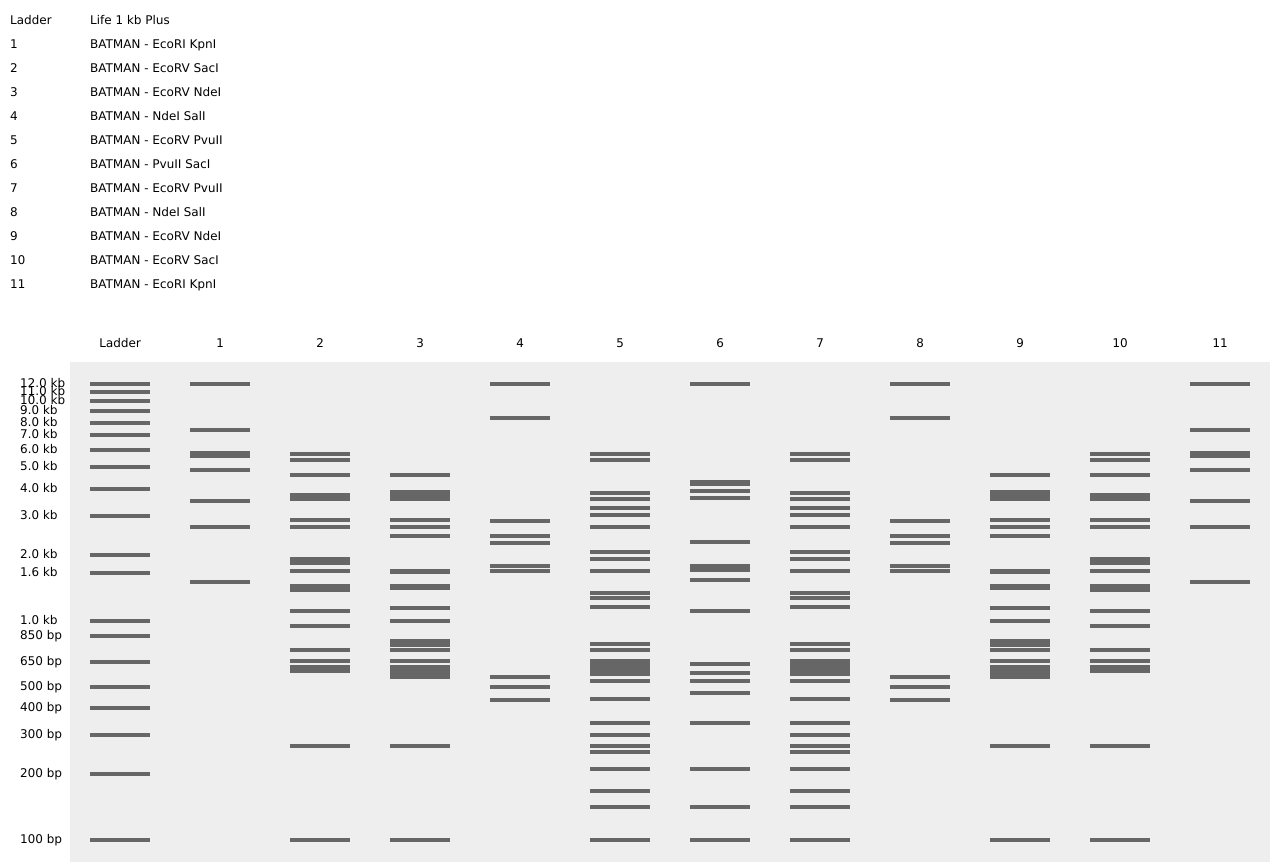
Part 1 – Benchling & In-silico Gel Art I used Benchling to design an in‑silico restriction digest of Lambda DNA. In Benchling, I created a customized restriction enzyme list for smoother later operations that included all the enzymes provided in the Week 2 HTGAA homework
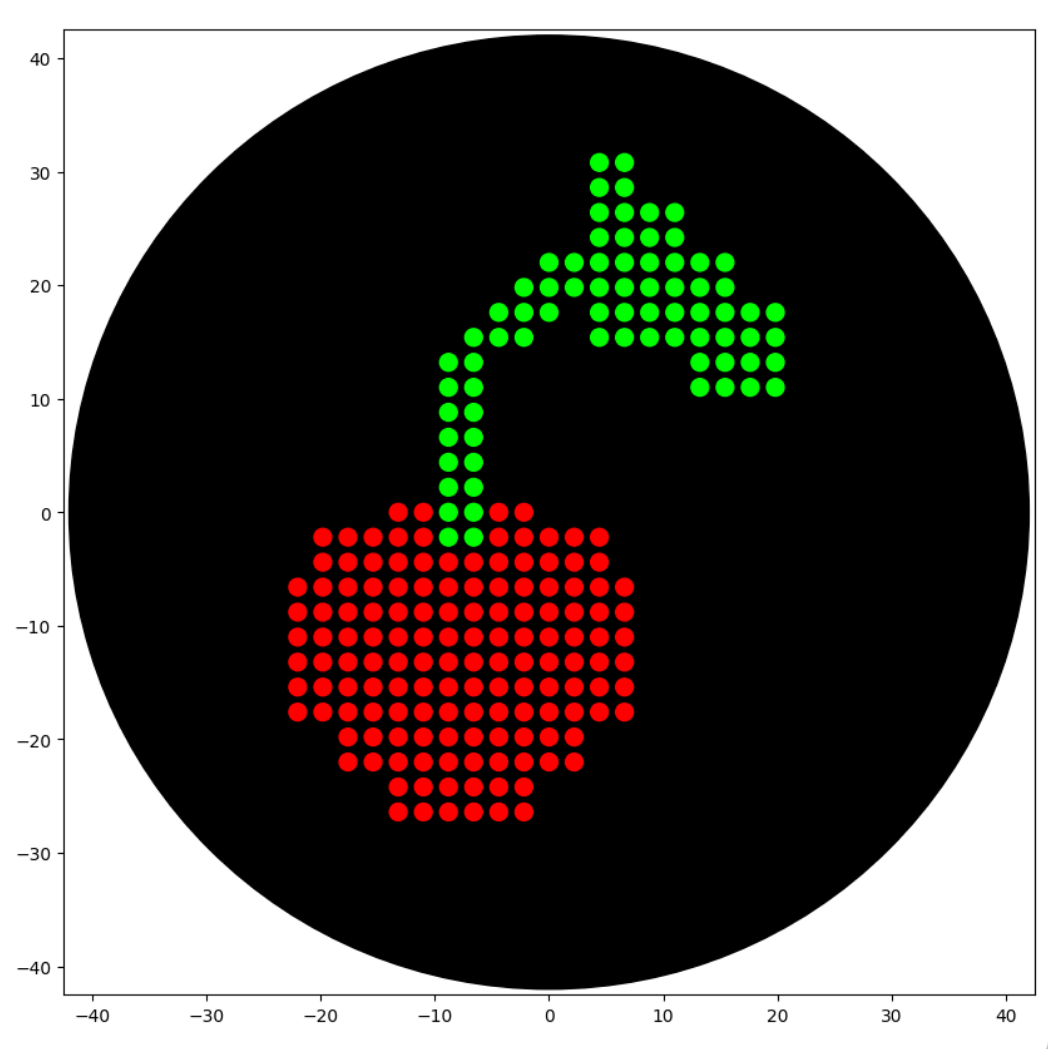
Assignment 1: Python Script for Opentrons Artwork This week we are creating a Python file to run on an Opentrons OT-2 liquid handling robot to create flourescent designs. Using provided website I created a small “Cherry” pattern. I have little experience in coding on such platofrms, so Google Gemini was a big help to assist while writing a code: https://colab.research.google.com/drive/1kZZStiHlPdG17vqHZPM2IhAQ3vTWkMRb#scrollTo=pczDLwsq64mk&line=76&uniqifier=1
Part A Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Since meat is not entirely made of proteins, lets assume 20% of the whole meat mass = around 100 g. An amino acid is ~100 Da (=~100g/mol). 100 g/ (100 g/mol) = 1 mol = 6.022* 10^23 AA.
PART A: Computational Peptide Design — SOD1 A4V Binder Generation Background
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS) — a severe neurodegenerative disorder characterized by adult-onset loss of upper and lower motor neurons, progressive paresis, skeletal muscle atrophy, quadriplegia, and fatal respiratory failure. The A4V mutation (Alanine → Valine at residue 4) is one of the most aggressive ALS-associated variants. It subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation. The task is to design short peptides that bind mutant SOD1 and evaluate which are worth advancing toward therapy.
DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Phusion DNA Polymerase
Chimeric enzyme that catalyzes the synthesis of ew DNA strand in the 5 -> 3 direction with high-fidelity dNTPs
four chemical building blocks ($dATP, dTTP, dCTP, dGTP$) used to construct the DNA. They provide both the physical material and the energy required for the polymerase to grow the new strand Reaction Buffer
Part 1 What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Traditional genetic circuits implement Boolean logic gates (AND, OR, NOT, NAND, etc.), hence their input/output relationships are discrete - a gene is either ON or OFF. This allows only binary decision-making and makes it difficult to represent graded, continuous, or context-dependent responses. IANNs provide continuos computation where inputs and outputs exist on a continuum, allowing cells to integrate multiple signals simultaneously.
General homework questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis is more flexible than in vivo expression because we can directly control the reaction conditions, such as DNA concentration, salts, cofactors, temperature, and additives. The in vivo model limits out experimet by time since we have atcually grow cells and wait for results, in cell free systems the speed of these procedures is much faster. It is more beneficial to use cell free systems for toxic proteins, membrane proteins, and rapid prototyping or diagnostics, because we do not need to keep a living cell alive while producing the protein.
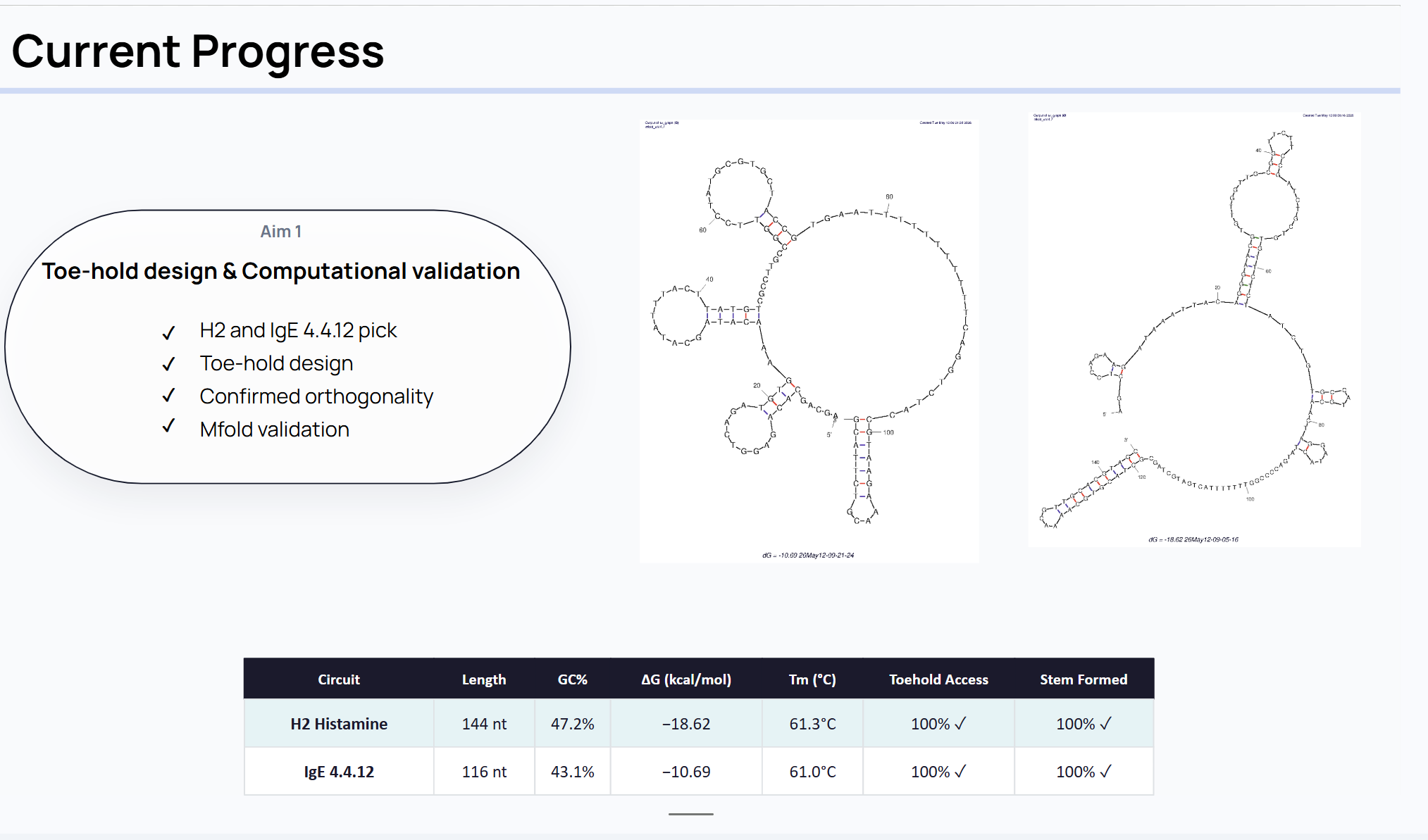
For final project In this project, I will measure several aspects of the DNA sensing system, including sequence correctness, predicted folding behavior, target response, orthogonality, and signal output. The most important biological measurements are whether the histamine and IgE circuits are correctly designed and whether they respond only to their intended targets. I will also measure the strength of the output signal after target binding, since the goal is to convert molecular recognition into a detectable readout. In addition, I will look at background activity and nonspecific activation to estimate how cleanly the system distinguishes true signal from noise. These measurements will help determine whether the platform is suitable for future wearable use.
Part A Unfortunately, I did not have the opportunity to contribute to the project before the deadline ended. However, for next semester, I think it would be a good idea to create several variations of the same artwork using different color palettes or design concepts. I noticed that many people were unsure about what exact pattern or style they were supposed to contribute, while others had their own creative ideas that did not fully match the overall design. Because everyone has different artistic preferences and interpretations, it could be helpful to divide the project into multiple themed sections or versions. This would make the collaboration process more flexible, reduce confusion, and allow more students to express their creativity in their own way.
Subsections of Homework
Week 1 HW: Principles and Practices
Question 1 – Application & why
1. First, describe a biological engineering application or tool you want to develop and why.
Introduction
My proposition for a biological engineering application is a synthetic cell circuit for neuroprotection in neurodegenerative diseases that is non-invasively controlled by a physical sound/ultrasound signal to help modulate inflammation and support brain health.
Motivation
During my junior year, I started learning about neurodegenerative diseases and current therapies. I came across lots of reading explaining non-pharmacological tools, such as music therapy, that are used as a complementary support rather than precise, controlled interventions. My interets was going beyond background music therapy and instead treating acoustic stimulation to its full potential as one possible non-invasive control channel for an engineered neuro-immune circuit.
Synthetic biology has already shown that mammalian cells can be engineered with mechanogenetic and sonogenetic switches to trigger therapeutic gene expression via receptor or responsive promoters. Music and music-like acoustical interventions could be engineered to play the role of an external controller that does not require being injected or physically contact witha patient
Design
A simple example would be an acoustic‑controlled promoter driving anti‑inflammatory cytokines such as IL‑10 or TGF‑β, neurotrophic factors like BDNF or GDNF, or enzymes that enhance clearance of toxic proteins such as Aβ.
The core logic gate would be an AND gate that requires both an acoustic input and a local inflammatory signal (for example, NF‑κB activation) before turning on the therapeutic gene, so that the circuit activates only when the brain is inflamed and the specific sound signal is applied.
Question 2 – Governance goals
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
Goal 1: Long-term biological safety of use
Ensure that sound-controllable synthetic immune circuits are designed and used in a way that is biologically safe and technically trustworthy.
Sub goal 1.1. Manage biological and technical risks
Identification and termination of key risks. Targeted circuit development design.
Sub goal 1.2. Robust testing and monitoring
Ensure there is detailed preclinical testing and long-term clinical monitoring before device deployment
Goal 2: Protection and respectful use in memory-impaired patients
Protect the rights and autonomy of neurodegenerative patients who receive this treatment and avoid health inequalities
Sub goal 2.1. Control and consent
Develop a consent and specialised process that would not violate rights of memory-impaired individuals patients
Sub goal 2.2. Ability to withdraw
Ensure patients can decline the intervention or request deactivation/removal of the circuit
Sub goal 2.2. Promote equity in access
Allow public health systems and diverse patient groups to benefit from this technology
Question 3 – Governance actions
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions …
Option 1: Establishing Regulation Rules and Technical Standards
Purpose: Outline clear guidelines for such circuits to create standardized safety requirements before any medical implementation and fabrication.
Design: The regulators for such action would include national FDA-like agencies, neurology societies, and expert committees. A specific category and preclinical studies would be defined to mitigate potential risks of off-target activation, long-term expression, response to repeated acoustic exposure, and biological safety.
The “safety checklist” could be developed for synthetic switches and minimum acoustic parameter requirements.
Assumptions: This assumes developers would agree to additional testing and expert review for approval.
Risks: In case of standards being considered too weak for fabrication without consideration of unknown long-term risks. On the contrary, overly complicated standards might make the whole project too expensive and unachievable.
Option 2: Setting Advance Directives
Purpose: Build a system that lets patients with neurodegenerative disease state their wishes in advance and appoint a trusted person to help control when and how the acoustic stimulation is used if their memory or decision‑making declines.
Design: Use advance directive forms specific to this intervention, completed while the patient still has capacity, where they can (a) record preferences about starting, pausing, or stopping stimulation, and (b) designate a person/guardian who is allowed to initiate, schedule, or terminate acoustic stimulation.
Assumptions: Assumes patients receive a diagnosis early enough, and with enough support, to complete advance directives; that legal systems recognize such documents and surrogate decision‑makers for neuromodulation or implantable synbio interventions; and that clinicians have time and training to revisit consent and preferences over time.
Risks: Some patients may never complete directives, leaving families and clinicians uncertain; designated guardians might have conflicts of interest or interpret wishes differently from what the patient would want. Strict reliance on old directives could also override a patient’s current expressions if they still have partial capacity or have changed their mind, which could undermine respect for present‑time autonomy.
Option 3: Set a transparency and public access
Purpose: Ensure the proven safety and effectiveness to the public with an understanding of all risks, benefits, and intervention procedures.
Design: Build a public interest campaign/communication platform with an explanation of the technology and treatment procedures, including uncertainty and possible side effects. Require recruiting diverse groups in clinical trials. Not limit the research to private research hospitals only.
Assumptions: Health systems are willing to invest in high-quality communication and marketing to reach diverse communities.
Risks: With too succesfull communication campaign, the public may overestimate benefits or underestimate uncertainty and risks. Policies to ensure inclusive trials and access may increase costs and administrative complexity for hospitals.
Question 4 – Scoring the options
Next, score (from 1–3 with 1 as the best, or n/a) each of your governance actions against your rubric of policy goals.
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
1
2
3
• By preventing incidents
1
2
3
• By helping respond
1
2
3
Foster Lab Safety
1
2
3
• By preventing incident
1
1
3
• By helping respond
1
2
3
Protect the environment
n/a
n/a
n/a
• By preventing incidents
n/a
n/a
n/a
• By helping respond
n/a
n/a
n/a
Other considerations
2
2
n/a
• Minimizing costs and burdens to stakeholders
3
2
2
• Feasibility?
2
1
2
• Not impede research
3
1
1
• Promote constructive applications
2
2
2
Question 5 – Recommendation & reflection
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why …
According to the scoring table, I prioritize both Option 1 and 2, which balances the hospital ethics and regulatory rules approved by national regulatory actors. This combination ensures that the biological tool is governed by both human-centric ethics and rigorous technical safety.
The target for this choice would be the FDA and NIS communities, with international groups working in neurology and the clinical trial approval committee.
Option 2 scores well (1) on feasibility, low costs, and patient autonomy—it uses existing hospital systems for quick consent processes and monitoring. Option 1 scores best (1) on biosecurity and lab safety prevention, adding uniform rules like safety checklists for acoustic frequencies. Together, they cover biological safety (Goal 1), patient rights (Goal 2), and fair access through trials (Goal 2) without major delays to research.
Considered Trade-Offs & Assumptions
This combination may have risks in uneven standards across hospitals, since each hospital may have its own patient consent, as well as higher costs and longer approval times.
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose … then propose any governance actions you think might be appropriate to address those issues.
From the first week’s lesson and recitation, the topic that caught my attention was genetic engineering and pathogen research/studying viruses in bats or building synthetic genetic circuits in these organisms. Even simple work, such as modulating pathogens or implementing circuits in cells, carries big biosecurity risks. If not handled carefully, a dangerous pathogen could escape the lab, spread to people, or be misused. This led to long thought for me on how this issue is being regulated now and how these experiments are conducted safely without stopping important science.
Governance solutions
Mandatory additional training: Require specialized training for all lab workers on incident reporting, strict entry/exit protocols, and emergency response. This builds skills to prevent accidents, like pathogen leaks during bat virus studies.
Screening panels with oversight: Create independent review panels of scientists and safety experts to screen high-risk experiments (e.g., pathogen modulation or synthetic circuits). These panels would approve protocols, monitor ongoing work, and ensure regular audits—similar to dual-use research reviews.
Another frequently mentioned topic from class was “core libraries” in synthetic biology. Biobanks, genetic databases, and DNA sequence archives are presented like reusable IP blocks. In many cases, patient data or cells are taken without permission and used for science or profit.
Governance solutions
Broader consent involvement with time-limited withdrawal rights. When patients enter treatment, get broad consent for future unknown uses. Allow donors or families to withdraw from data access within a clear time period (e.g., 6-12 months). This protects privacy early on while preventing disruptions after data is already shared and in open research use.
Rules for sharing and minor benefits to track the contribution by group.
Pre-lecture Questions
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
DNA polymerases have an error rate of about 10*-2 errors per base.
The human genome is ~3.2 × 10*9 bp in lenght, so this creates a significant disperancy which results in thousands of errors percopy.
Biology fixes this with proofreading by polymerase and post‑replication mismatch repair (MutS/MutL/MutH etc.), which together reduce the error rate.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
An average human protein is ~330–350 amino acids, giving the possibility of a massive number of DNA sequences (around 10*150), because of the portein redundancy of the genetic code.
Many possible codes “don’t work” because sseries of resons: secondary structure of mRNA; poor codon usage/tRNA availability; splicing or binding sites.
Homework Questions from Dr. LeProust:
What’s the most commonly used method for oligo synthesis currently?
The standard, most widely used method is solid‑phase phosphoramidite chemistry.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
It is difficult to make long oligos via direct synthesis due to comulative yiel loss. By ~200 bases there are many truncated and error‑containing products and it is hard to purify the correct full‑length oligo.
Why can’t you make a 2000bp gene via direct oligo synthesis?
A 2 000‑step phosphoramidite synthesis would give zero yield.
Instead, synthesizing many shorter oligos, then assembling them enzymatically (PCR assembly, Gibson, etc.) into longer gene fragments is used.
Essential for humans/animals: histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine, and arginine.
Animals already depend on the diet for multiple essential amino acids, including lysine, so making organisms “lysine‑dependent” is not a safe way to contain a synthetic organism. Though for movie purposes it is a fun scientific explanation.
Week 2 HW: DNA Read, Write, & Edit
Part 1 – Benchling & In-silico Gel Art
I used Benchling to design an in‑silico restriction digest of Lambda DNA. In Benchling, I created a customized restriction enzyme list for smoother later operations that included all the enzymes provided in the Week 2 HTGAA homework
Using Ronan’s website, I tried to create a “Bat signal” 🦇 pattern on the gel (hopefully you can see my vision too!)
This was my first attempt, where the lanes did not appear in the order I expected, so the pattern looked wrong…
To fix this, I renamed each “Digest” tab with numbers, because every new digest was appearing in a random order.
After running all the digests and then ordering the numbered lanes correctly, I finally obtained my intended DNA gel “Batman” pattern.
Part 3 - DNA Design Challenge
Protein – TRPV1 (heat and “spicy” pain sensation)
cation channel expressed in nociceptive sensory neurons, where it detects noxious heat, low pH, and capsaicin (main compound in chili peppers) 🌶️. I chose TRPV1 because it directly links physical stimuli at the skin (heat or spicy chemicals) to electrical activity in pain pathways, making it a clear molecular mediator of sensory perception. Engineering the DNA sequence that encodes TRPV1 could tune its expression or gating properties, which is relevant for altering thermal pain sensitivity or designing cells that report damaging levels of heat.
Codon Optimization
For codon optimization, I planned to take my reverse‑translated TRPV1 coding sequence and run it through an online codon optimization tool to adapt codon usage to E. coli, replacing rare codons, adjusting GC content, and removing unwanted motifs while keeping the amino‑acid sequence unchanged. However, the TwistBioscience optimization tool was unavailable and other available web tools repeatedly failed on my long TRPV1 sequence, so for this homework I kept the reverse‑translated sequence from Part 3.2 as my working TRPV1 coding sequence and discussed codon optimization conceptually instead of providing a fully optimized sequence.
3.4: What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into a protein. You may describe either cell-dependent or cell-free methods, or both.
Once I have a coding DNA sequence for TRPV1, I can synthesize it and clone it into an expression plasmid with a suitable promoter, ribosome binding site, and terminator. After transforming this plasmid into host cells such as E. coli or mammalian cells, RNA polymerase transcribes the TRPV1 gene into mRNA, and ribosomes translate the mRNA into the TRPV1 channel, which is inserted into the plasma membrane and opens in response to heat or capsaicin to generate pain signals. The same DNA sequence could also be used in a cell‑free transcription–translation mix to produce TRPV1 in vitro, still following the central dogma from DNA to RNA to protein
Part 4
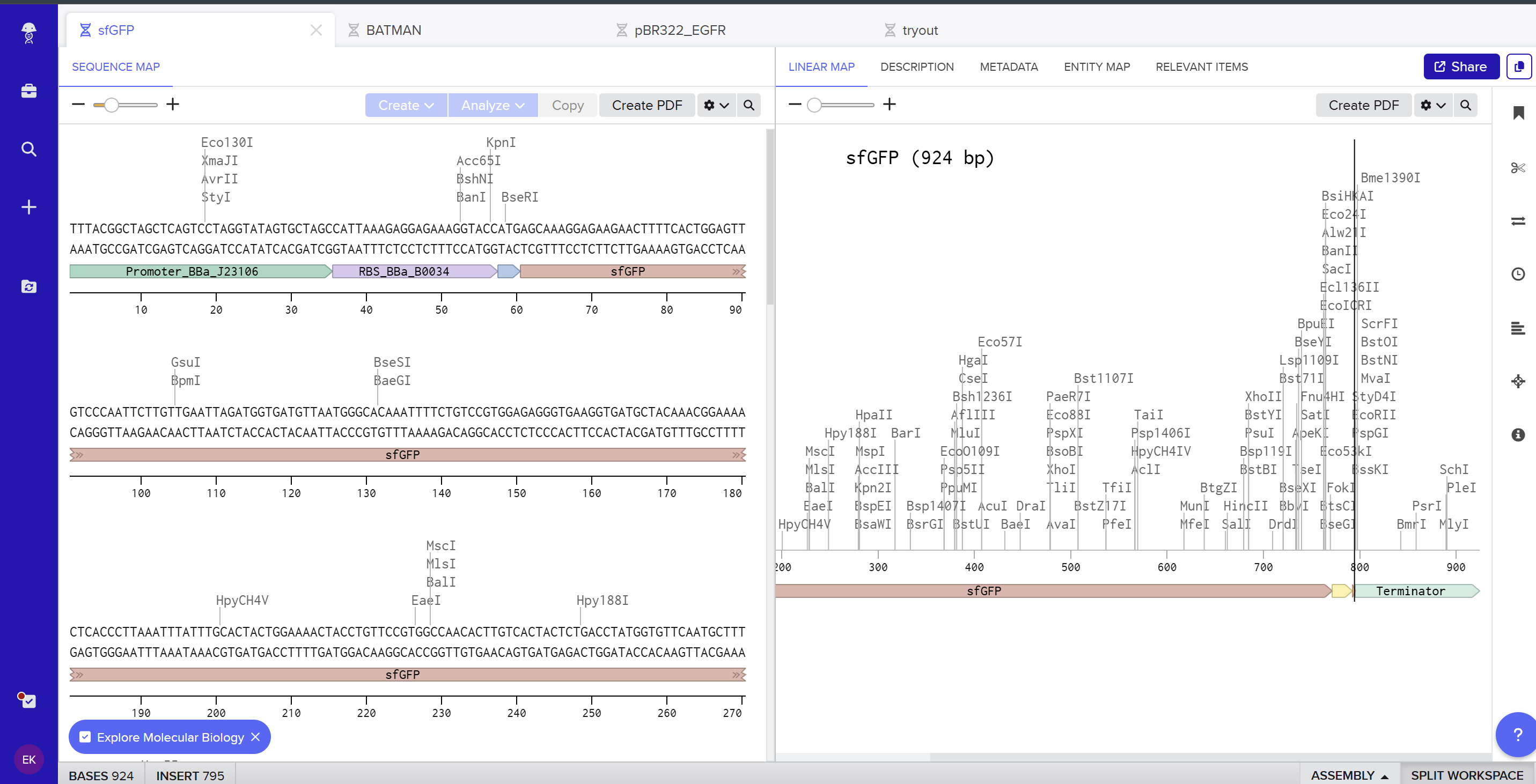
I created a new linear DNA sequence in Benchling named sfGFP, set the nucleotide type to DNA, and topology to Linear. In the sequence editor I pasted, in order, the example promoter BBa_J23106, RBS BBa_B0034 with spacer, start codon (ATG), the provided codon‑optimized sfGFP coding sequence, a 7×His tag at the C‑terminus, a stop codon (TAA), and the BBa_B0015 terminator, and added annotations for each feature (Promoter, RBS, sfGFP CDS, 7×His tag, Stop, Terminator).
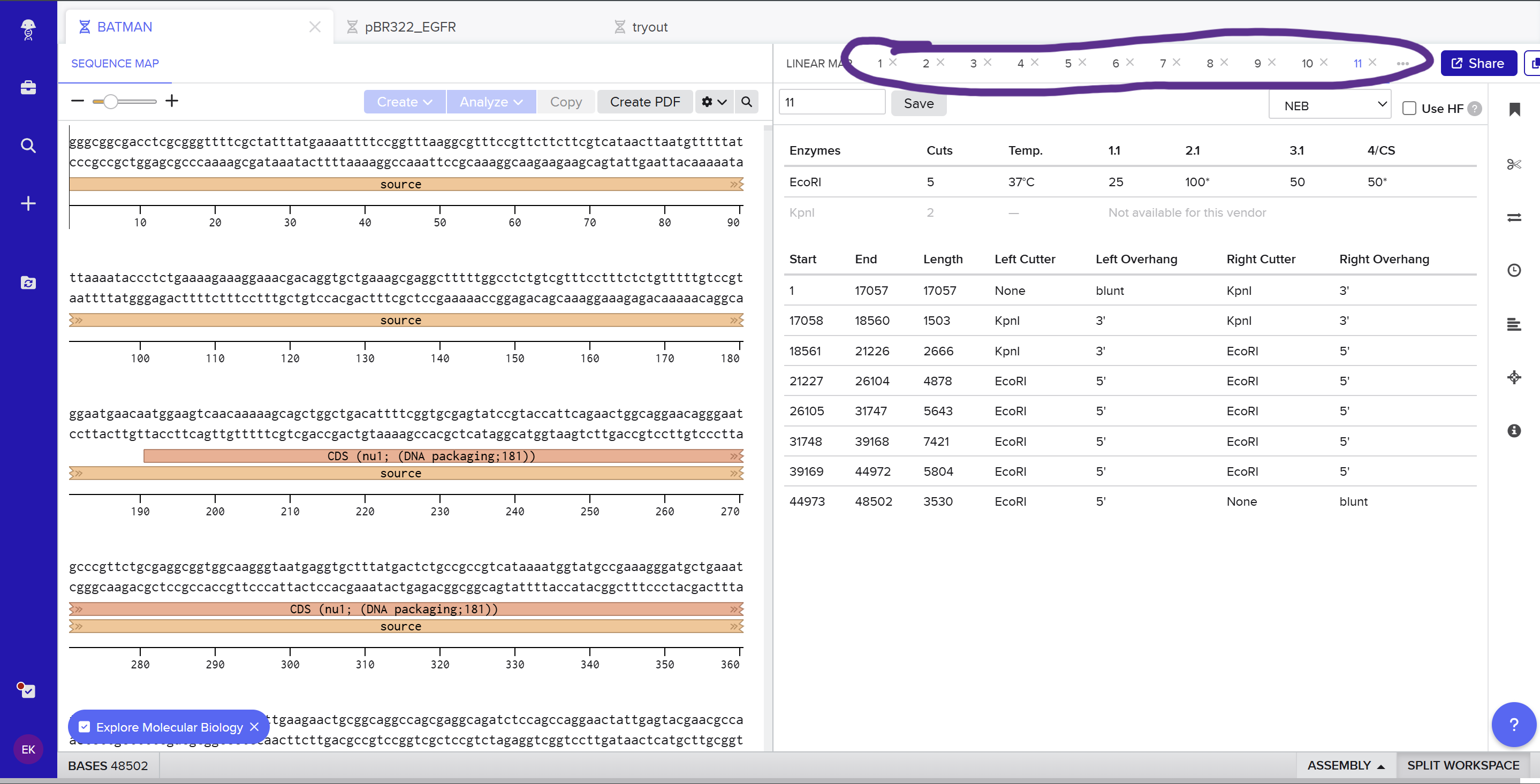
Here you can see the screenshot from Benchling showing the sequence map: (https://benchling.com/s/seq-KNkSG9FjYrEgCrgZE0Id?m=slm-aiflv0AFXb7Fro539sLk)
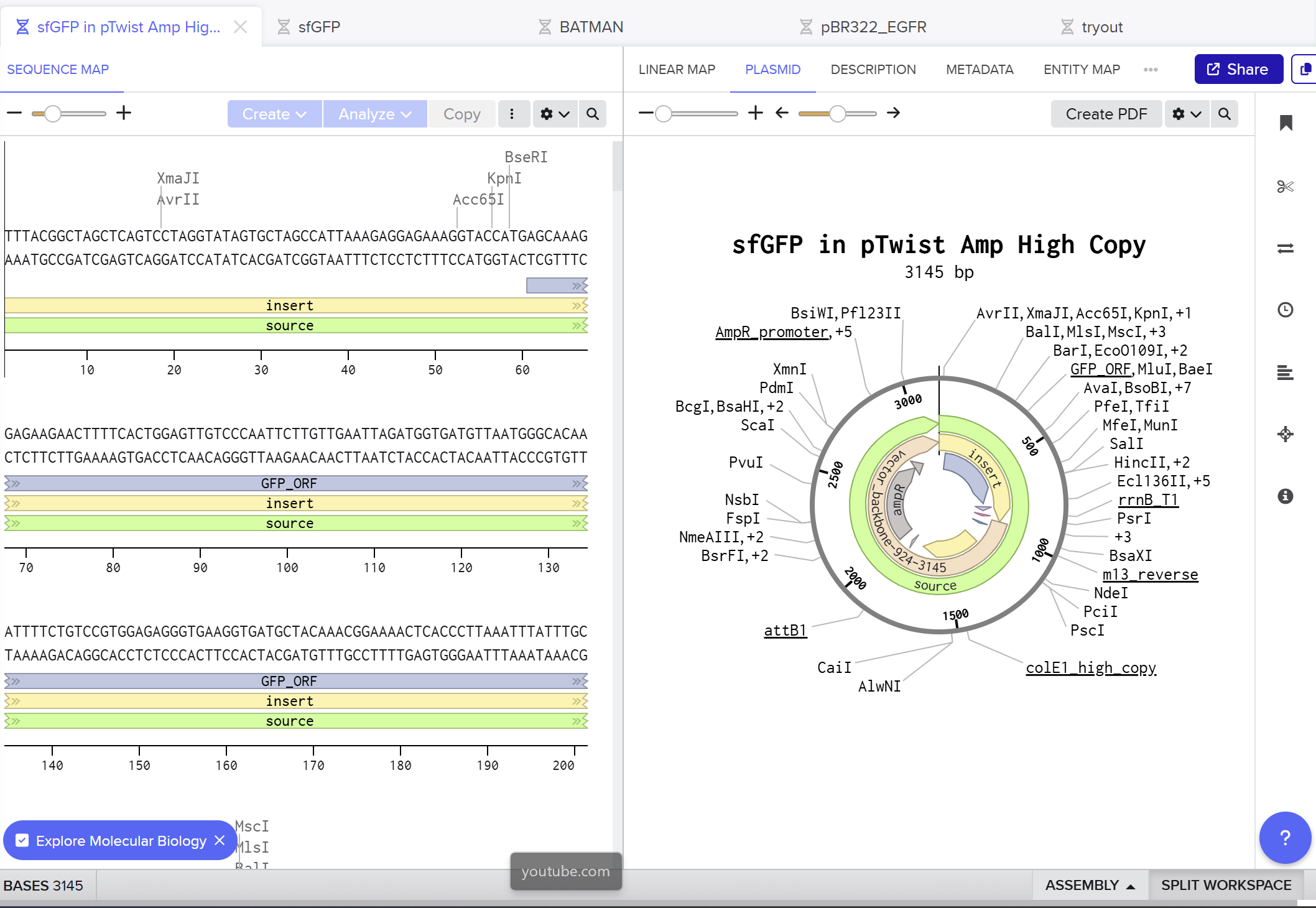
On the Twist portal I selected the “Genes” product and chose the “Clonal Genes” option, since this provides my insert in a circular plasmid that can be transformed directly into E. coli. I imported the FASTA file of my sfGFP expression cassette as a nucleotide sequence, then chose a Twist cloning vector (pTwist Amp High Copy) as the backbone so that the final construct includes an origin of replication and ampicillin resistance. After Twist generated the plasmid design, I downloaded the GenBank file and re‑imported it into Benchling to view the full plasmid map with my annotated sfGFP expression cassette inserted:
Part 5
DNA Read
What DNA would you want to sequence and why?
I would like to sequence DNA from banana (Musa species) to explore how similar or different it is from the human genome, especially because of the known fun fact stating that humans “share around half their genes” with banana.
By sequencing banana DNA, I would wanna compare it to human gene sets and get the idea where these similarities come from and what they lead to. 🍌
What technology would you use and why?
I would use Illumina sequencing‑by‑synthesis (second‑generation NGS), possibly complemented by nanopore (third‑generation) for long reads.
Input and prep: extract banana genomic DNA, fragment it, repair ends, ligate Illumina adapters, PCR‑amplify, then load on a flow cell
How it reads bases: clusters are formed on the flow cell. In each cycle, fluorescently labeled nucleotides are added, one base at a time, and the machine takes a picture. The color of each spot in each cycle tells you which base (A, T, C, or G) was added there.
Output: millions of short reads in FASTQ format, which can be assembled and compared to human genes
DNA Write ✍🏽
What DNA would you want to synthesize (e.g., write) and why?
I would like to synthesize a genetic circuit for a “self‑adjusting” biomaterial, where cells inside a hydrogel can sense mechanical stress and then change the stiffness of the material. The idea is to have a material that becomes stiffer when it needs more support and softer when stress is too high, using gene expression instead of external tools. This could be useful for tissue engineering and mechanobiology, because many studies show that cell fate and behavior depend not only on stiffness, but also on how stiffness changes over time
What technology would you use to perform this DNA synthesis and why?
To build this circuit, I would use chip‑based DNA oligo synthesis plus clonal gene synthesis, and then assemble the parts into an expression cassette. Chip‑based synthesis is good for designing and producing many regulatory variants (different mechanosensitive promoters, crosslinker genes, degradation domains) in parallel, which is important when tuning a dynamic material
Essential steps
Design the circuit in silico: pick mechanosensitive promoter elements, choose coding sequences for matrix‑building proteins and matrix‑remodeling enzymes, then add RBSs and terminators
Order synthetic DNA fragments or full clonal genes from a synthesis provider, using chip‑based oligo synthesis to keep costs down for complex designs.
Assemble the fragments into plasmids, transform them into the chosen cell chassis, and verify by sequencing
Limitations
Complex construction can have a high error rate
Synthesis and clonign might take several days to weeks
Mechanosensitive elements characterized in 2D cultures may behave differently in 3D hydrogels
DNA Edit 🖆
What DNA would you want to edit and why?
I would like to edit DNA in cartilage‑related cells for athletes. The example would be figure skaters who often perform repeated high jumps and landings that produce a very high impact on the knee and ankle. Most figure skaters frequently develop overuse injuries and early degenerative changes in the ankle/knee joints. This leads to the early retirement of athletes in their early teens and extensive health problems. Editing joint cartilage cells to be more regenerative, so that damaged cartilage can be repaired more effectively over time.
The target gene would be SOX9 and TGF-Beta pathway genes, since they are known to be the main pro-generative genes in cartilage.
The reason why I wouldn’t want to explicitly target genes related to the defensive functions of cartilage to prevent injuries is that it would raise some ethical concerns.
What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-based gene activation in joint-derived stem cells to upregulate SOX9 and TGF-Beta pathways genes. This technology would guide RNAs targeting promoters to boost cells’ own existing genes without cutting DNA. This would explicitly focus on existing injuries.
Essential steps
Confirm that SOX9 and key TGF genes are pro-generative in articular cartilage and design guide RNAs that bind promoter regions of SOX9 adn TGFB-pathways genes in human joint cells
Build dCas9-activator plasmids for designed gRNAs
Deliver dCas9-activator and gRNA to the cell
Culture and differentiate edited cells towards cartilage
Preparation and inputs
Extensive research and selection of targeted genes and regulatory regions in human joint cartilage
design of guide RNA
selection of dCas9-activator
Inputs: DNA templates, plasmids, viral vectors encoding dCas9-activator, plasmids for gRNAs, patient derived MSCs cells
Limitation
Since dCas9 does not cut DNA, there is a possibility of upregulation of unintended genes, because of the off-target binding
There should be controlled upregulation, since over-activation of these genes can lead to fibrosis or abnormal tissue growth
Description This paper addresses the difficulty of manual hydrogel fabrication, which is often prone to human error and low reproducibility due to the viscosity of the materials. The authors utilized an Opentrons OT-2 to automate the mixing and deposition of various hydrogel precursors (including methacrylated gelatin and others) into 96-well plates.
Relevance
The Opentrons OT-2 will be essential for the chemical formulation of the Bio-Blocks. Because the effectiveness of dissolution depends on the precise concentration of hexametaphosphate and citrate, the robot will be used to: Generate Concentration Gradients of alginate, HMPs and citarte & Ensure Consistency by automating the inoculation of cross linking agents
3D-Printed Holders & Custom Hardware would be developed for molding structural blocks
Creation of bylayer hydorgels can be achieved using robot to deposit a “structral layer” wiht high cross-linking density
Week 4 HW: Protein Design
Part A
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Since meat is not entirely made of proteins, lets assume 20% of the whole meat mass = around 100 g. An amino acid is ~100 Da (=~100g/mol).
100 g/ (100 g/mol) = 1 mol = 6.022* 10^23 AA.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Proteins in processed meat are getting denatured in our stomach by HCl and the enzyme pepsin, cutting long polypeptides. Proteases continue cutting these peptides into smaller peptides and intestinal enzymes complete the digestion into amino acids.
Shortly, our bodies do not absorb animal proteins whole, but use different enzymes to break them down to get basic amino acids
Why are there only 20 natural amino acids?
20 amino acids are representing an ideal balance for biological efficiency and chemical necessity to build all known life on Earth.
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids were synthesized abiotically through high-energy interactions between gases.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
L-amino acids are all right-handed due to steric hindrance between side chains. Since the D-enantiomer is a mirror image of an L-enantiomer, we would expect left handed helix
Can you discover additional helices in proteins?
The new helices are being discovered every day using tools like Alpha Fold.
Why are most molecular helices right-handed?
Because of the dominance of L-aminoacids in life and their chirality, most of the helices are right-handed to be sterically and energetically favourable.
Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation?
Beta sheets are characterised by their open structure, where the carbonyl and amide groups are exposed at the edges. This exposure promotes hydrogen bonding with neighbouring strands, that is forming a stack of “sheets”.
Why do many amyloid diseases form β-sheets?
Because of the stacking nature of beta-sheets, amyloid diseases occur when proteins misfold into flat, “sticky” layers that act as templates, forcing other healthy proteins to aggregate into insoluble, thread-like fibrils. The chain of reaction recruits new proteins that are resistant to clearing mechanisms
Can you use amyloid β-sheets as materials?**
This mechanism, though, can be quite beneficial for the biomaterials. Beta-sheets represent extreme stability and high tensile strength for such biomaterials as vascular grafts in medicine, which need to have resistance function inside the body
Part B
Briefly describe the protein you selected and why you selected it.
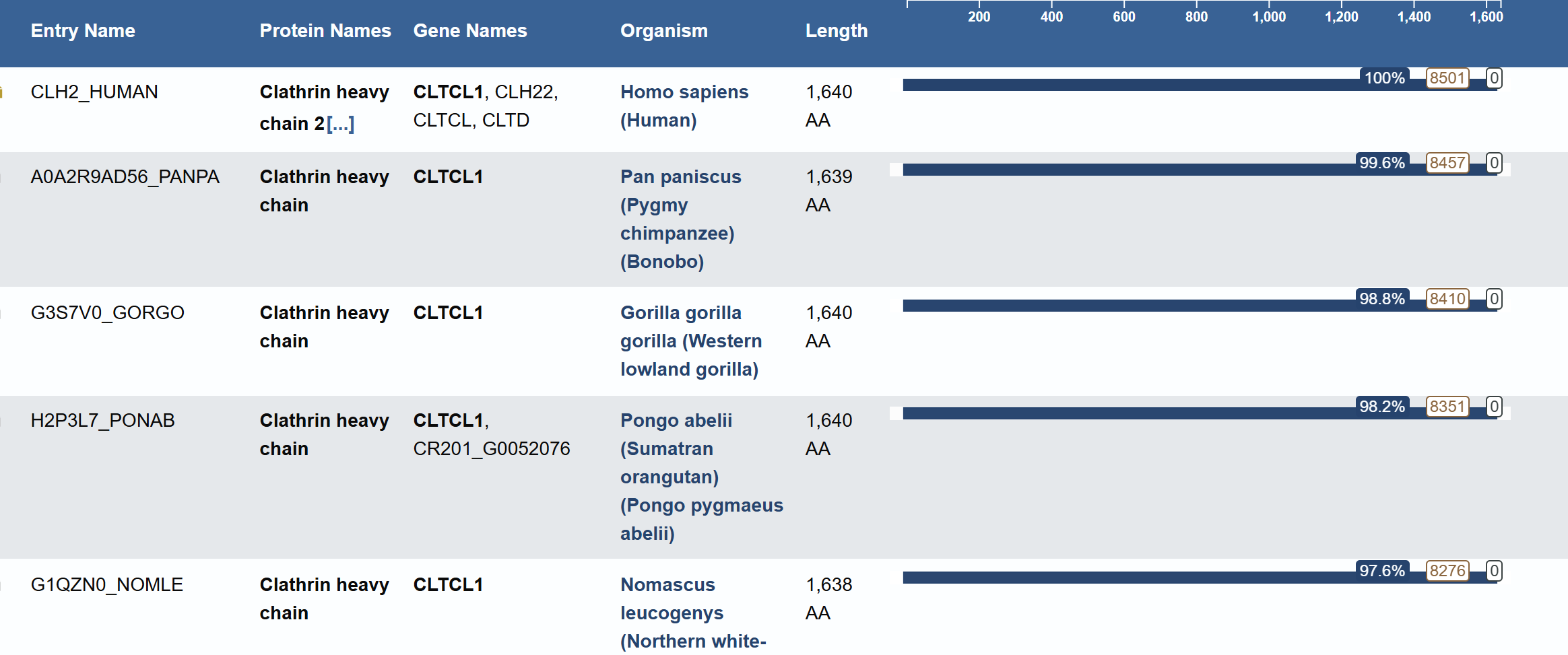
For this part, I have selected Clathrin Heavy Chain (CHC).
This protein is widely known in biology as a self-assembly protein consisting of three light chains that join into a triskeleion. Triskelions them assemble inot a geometric closed shape that creates vesicles.
Identify the amino acid sequence of your protein.
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
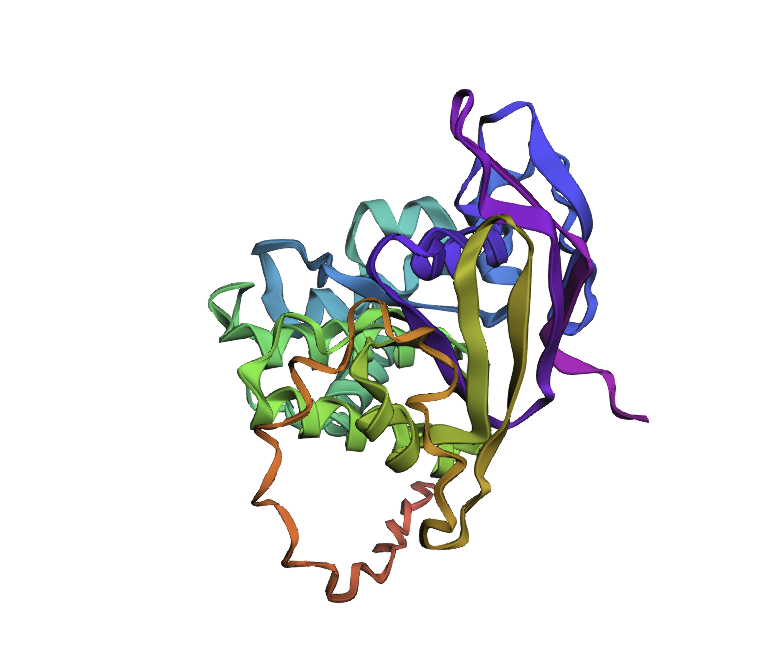
Protein: Clathrin Heavy Chain 2 (Human).
Length: 1,645 amino acids.
Most Frequent Amino Acid: Leucine (L) - appears 196 times.
Blast search revealed homology with other clathrins, confirming its belonging to the Clathrin heavy chain family
Identify the structure page of your protein in RCSB
PDB ID: 1XI4
The resolution is 2.30 Å, which is better (smaller) than the 2.70 Å
It was published in 2004 and apart from the protein, the structure contains water molecules and glycerol
Classification: It belongs to the 7-bladed beta-propeller family.
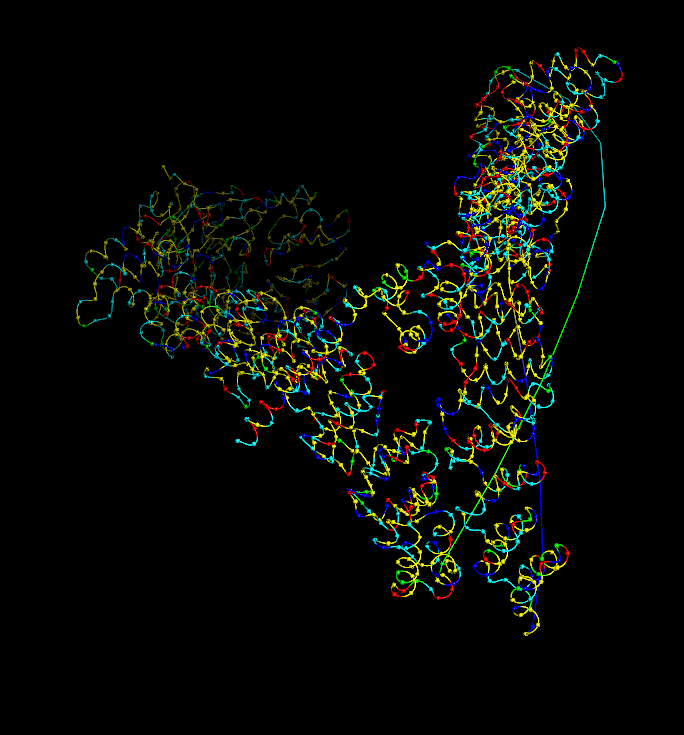
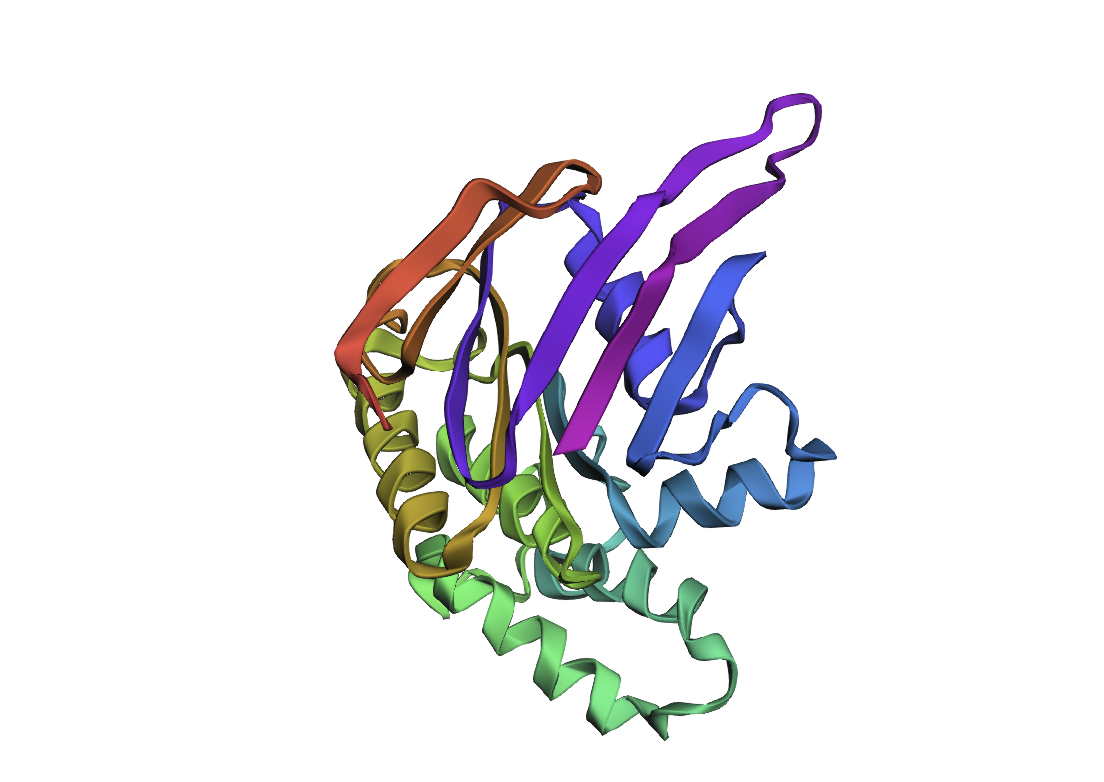
I analyzed clathrin D6 coat (PDB 1XI4), focusing on one heavy‑chain leg (chain A).
I viewed it as cartoon/ribbon, which shows a long curved backbone made almost entirely of α‑helices with only short loops.
I also added ball‑and‑stick on top of the ribbon to see individual atoms and side chains.
Coloring by secondary structure (helices red, sheets yellow, loops green) showed that chain A is strongly helix‑rich, with almost no β‑sheets.
Coloring by residue type (hydrophobic yellow; acidic red; basic blue; polar cyan) revealed that hydrophobic residues are mostly buried in the helical core, while charged/polar residues are on the surface.
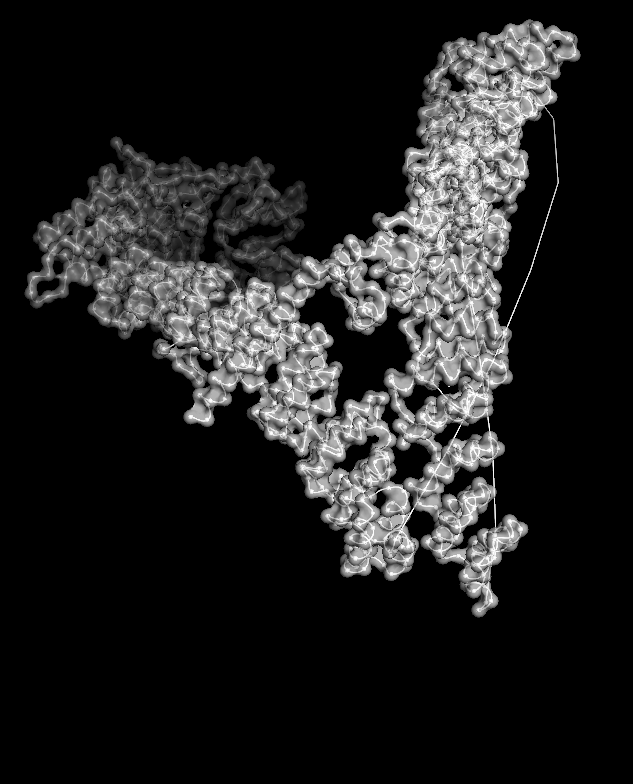
When I displayed the surface, I saw grooves and shallow cavities between helices rather than one deep pocket, suggesting multiple shallow binding/interaction sites along the leg.
Unfortunately, I wasn’t able to figure out how to color the structure by residues ;(
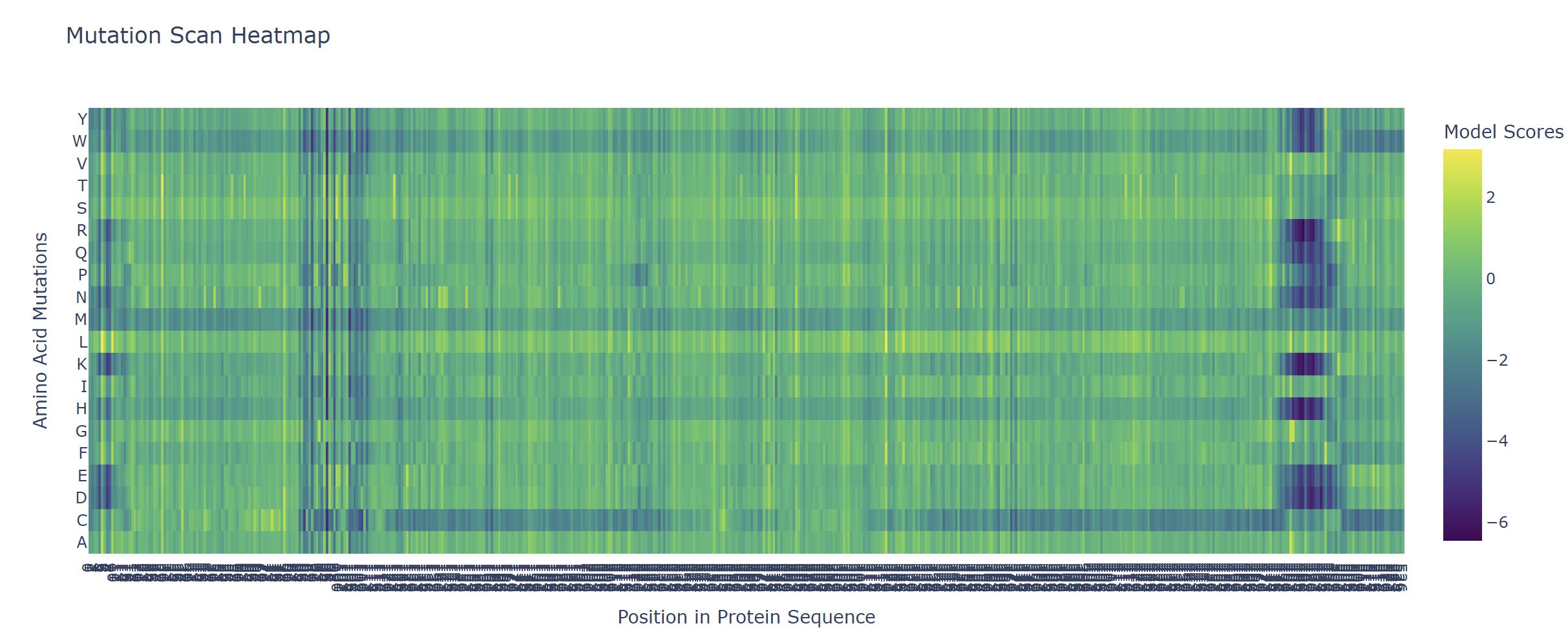
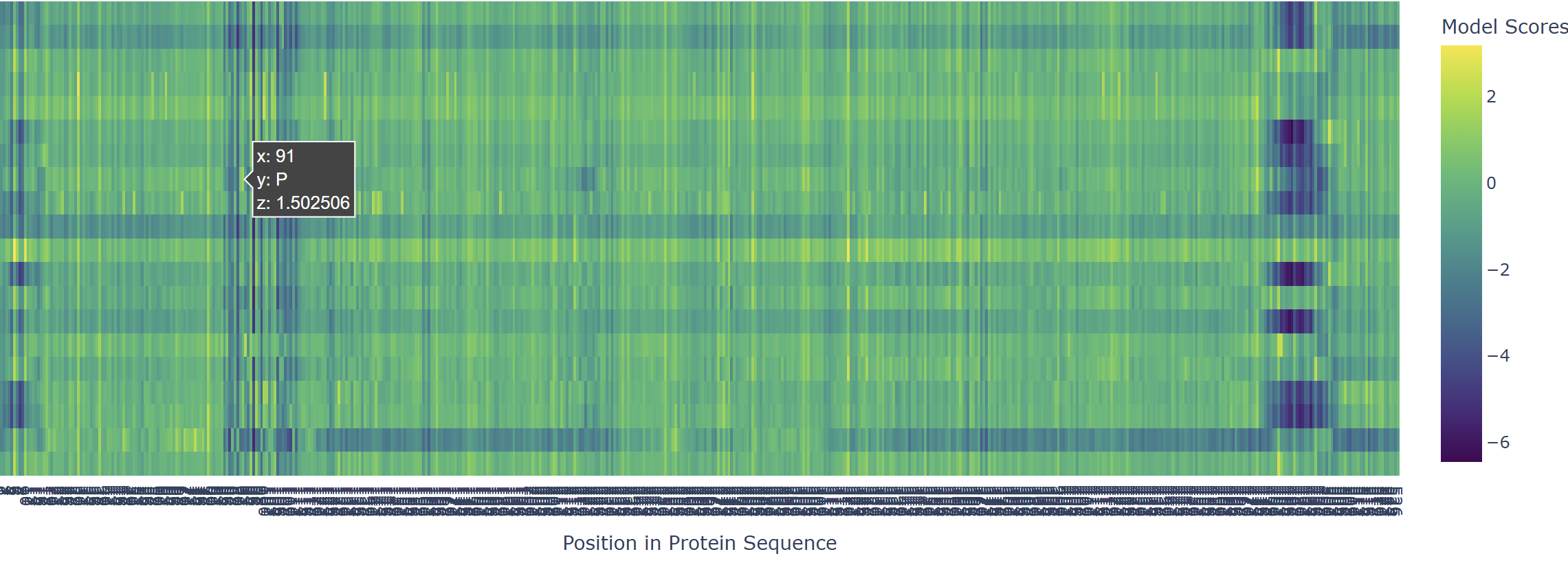
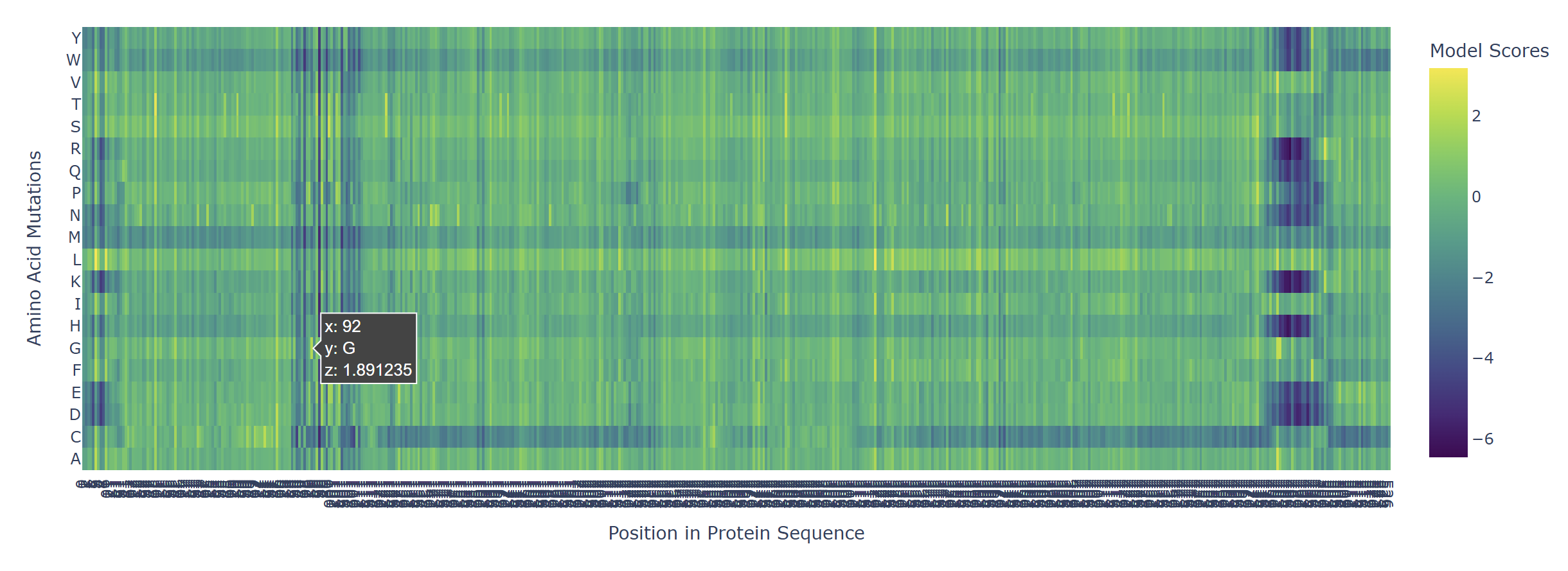
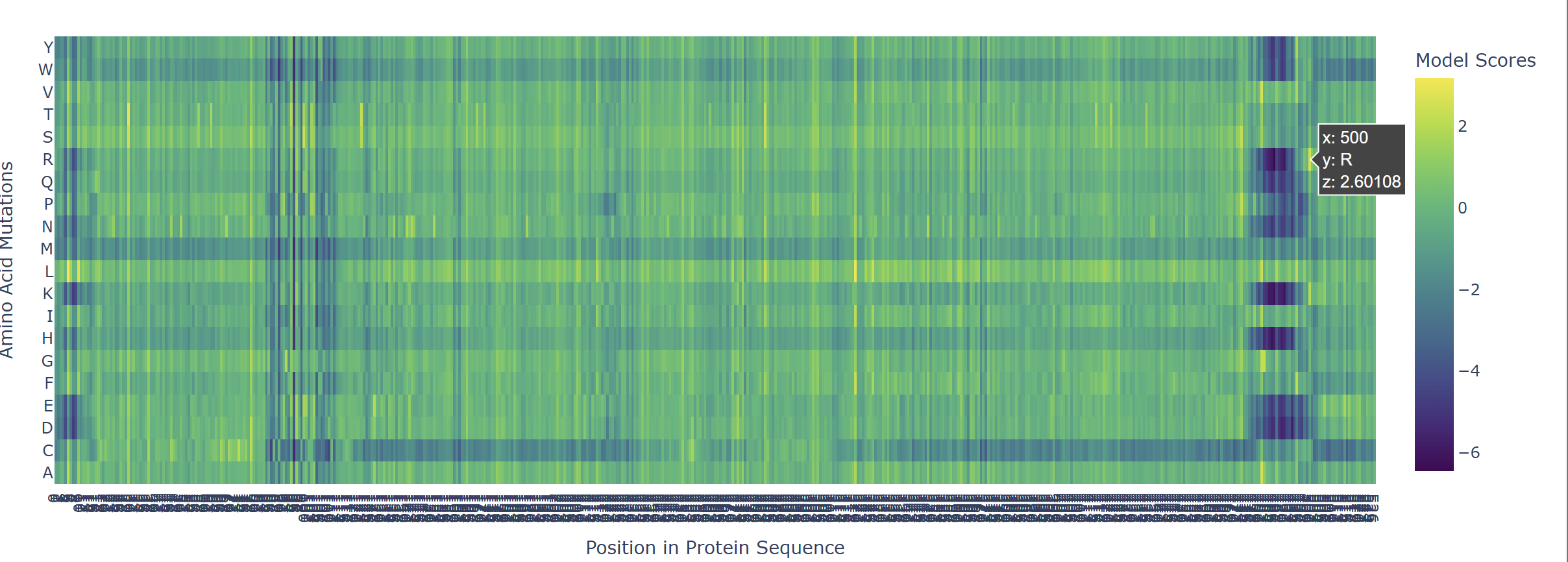
Based on the heatmap I got for my protein, I navigated to the locations where a sharp contrast was noticeable between highly sensitive sites (dark blue) and tolerant mutations (yellow). I identified three random locations (residues) that stood out by being next to dark blue. These yellow spots (see photo below) represent permessive mutations: specific amino acid substitutions that the language model predicted will preserve the protein structural and functional integrity despite being highly conserved regions.
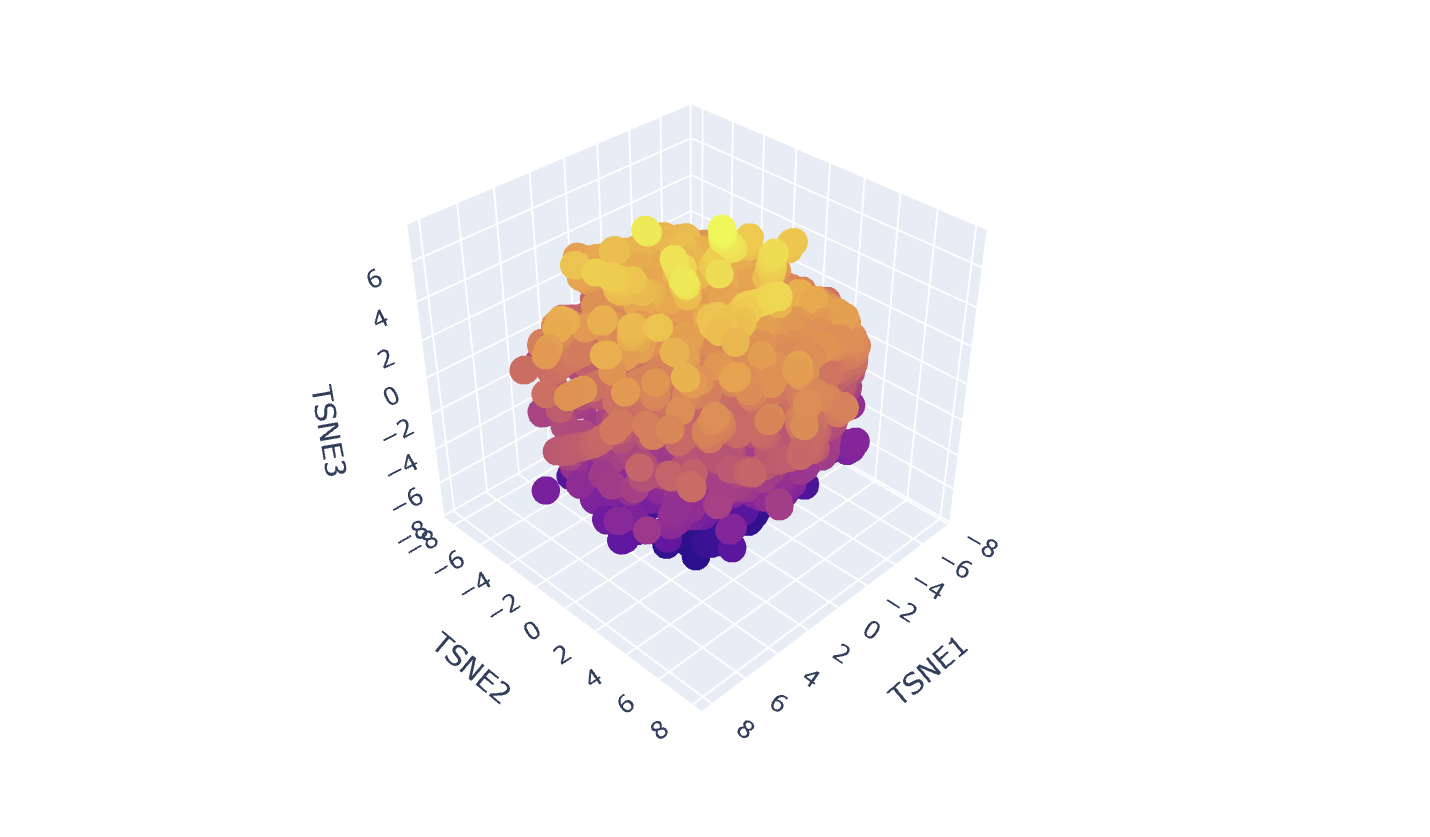
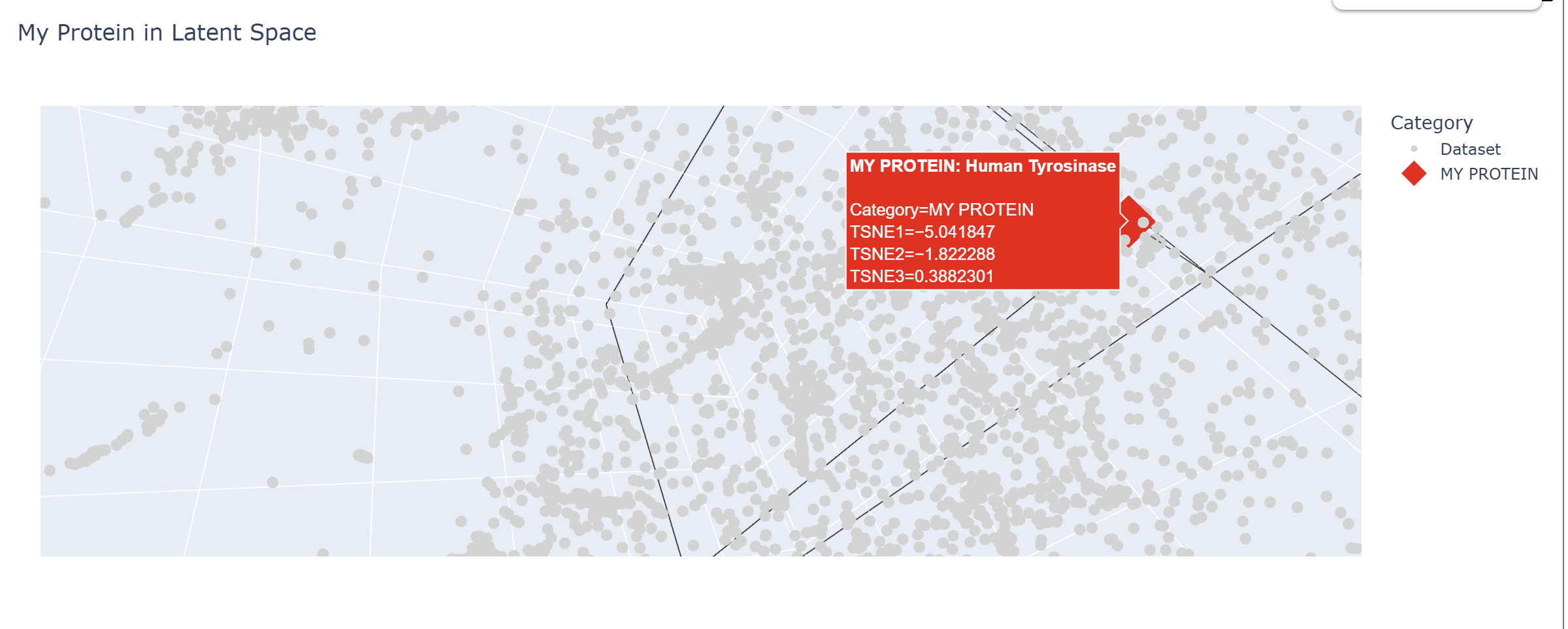
Latent Space Analysis
In my Latent Space Analysis, my protein (Human Tyrosinase) appeared within the class of All-Alpha protein neighborhood, which makes biological sense because both proteins share a conserved di-copper binding fold. This shows that the ESM2 model can accurately group proteins by their 3D shape and evolutionary ’language,’ even if they come from completely different species.
I changed the code a bit so my protein would be visible wihtin thousands of dots.
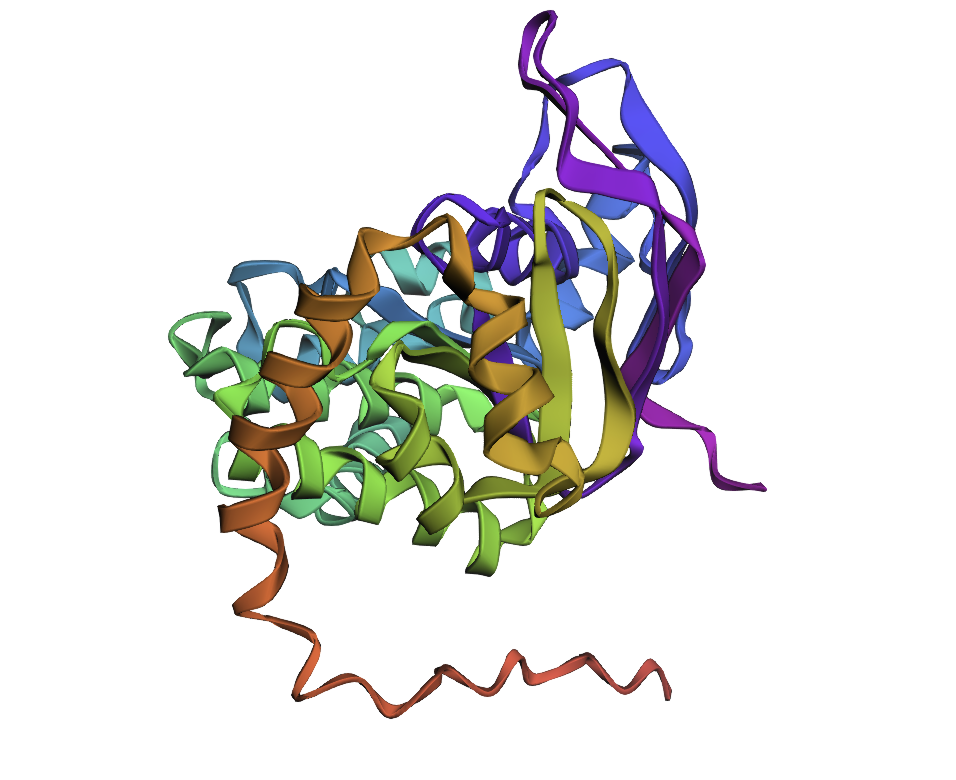
C2. Protein Folding
I chose a random protein I found on the ESM Metagenomic Atlas fro my Protein folding task. Amino Acid sequence:
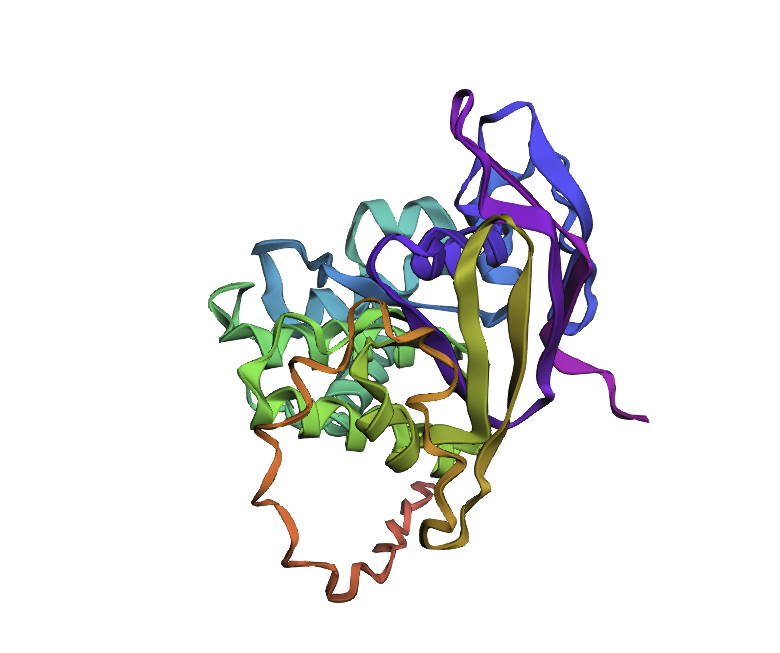
Few mutations haven’t changed the 3D structure at all, so I performed a ‘stress test’ on my protein by changing a large sequence of amino acids from position 170 to 200. The 3D model showed minor conformational change:
C3. Protein Generation
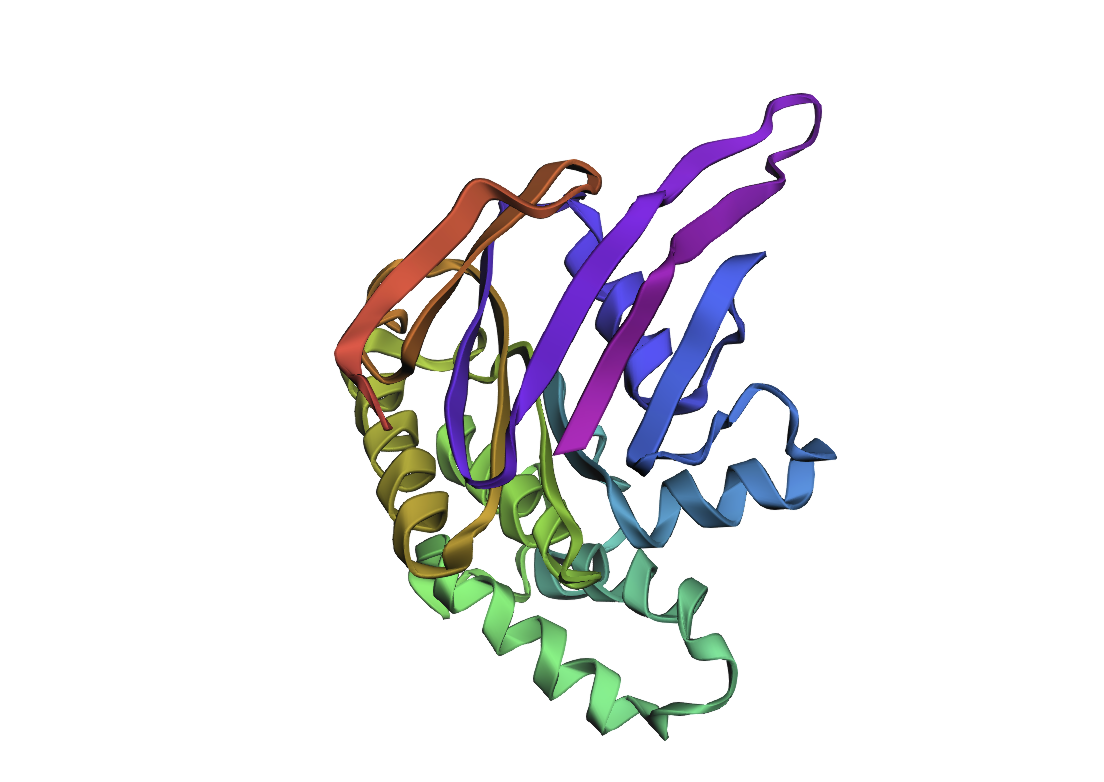
After performing Inverse Folding with ProteinPMNN, I’ve received the next sequence:
The resulted sequence appeared to 232 AA long, compared to the original 287 AA.
After inputting this sequence into ESMFold, next 3d structure formed:
By the comparison we can see it differs from the original structure but has some similarities.
Original
New
Part D
Project Proposal
Chosen Goals
Increase toxicity (lytic efficiency) of the MS2 L protein by tuning its interaction with E. coli DnaJ and its putative target.
Improve thermal and conformational stability of L so that toxic variants remain well folded and functional across experimental conditions.
Computational approach
Protein language models (ESM-2 / ProGen) - to design
Run in silico mutagenesis on the MS2 L sequence to score single and small combinatorial substitutions for evolutionary “fitness” and tolerated diversity.
Use these scores to (i) preserve positions that are highly conserved or known to be essential for lysis and DnaJ dependency, and (ii) explore mutations at more flexible residues that may enhance toxicity or stability.
Structure prediction (AlphaFold-Multimer or AlphaFold3)
Model the complex between full-length MS2 L and E. coli DnaJ, using the experimentally defined minimal lytic domain and the N‑terminal basic regulatory domain as guides.
Map the predicted binding interface around residues implicated in DnaJ dependence and inactivating missense mutations (for example, the conserved Leu48–Ser49 motif and neighboring central-domain residues).
Use these models to prioritize mutations predicted to strengthen productive L–DnaJ contacts or relieve autoinhibition of L while maintaining membrane association.
Sequence redesign for stability (ProteinMPNN, Foldseek/NGL/PyMOL)
For promising L variants from the pLM and AlphaFold stages, use ProteinMPNN on fixed backbones to propose alternative side chains that lower the estimated folding free energy (ΔG) without disrupting the DnaJ-contact surface.
Visualize candidate designs in NGL Viewer or PyMOL to check for clashes, loss of transmembrane character, or obvious disruption of the domain architecture suggested by mutational analysis.
Potential pitfalls and limitations
Mutations that increase toxicity may destabilize the protein or alter its membrane topology, leading to misfolding or loss of function
DnaJ Conformational Flexibility: Chaperones like DnaJ are inherently flexible. A static AlphaFold model might not capture the dynamics for lysis and false positive
Pipeline Schematic
Input: Wild-type MS2 L Protein Sequence.
Step 1 (Optimization): ESM-2 mutation scoring for fitness.
Step 2 (Binding): AlphaFold-Multimer modeling of L-Protein + DnaJ complex.
Step 3 (Refinement): ProteinMPNN sequence redesign for thermal stability.
Output: Top 5 candidate sequences for in vitro synthesis and plaque assay testing.
Week 5 HW: Protein Design. Part 2
PART A: Computational Peptide Design — SOD1 A4V Binder Generation
Background
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS) — a severe neurodegenerative disorder characterized by adult-onset loss of upper and lower motor neurons, progressive paresis, skeletal muscle atrophy, quadriplegia, and fatal respiratory failure.
The A4V mutation (Alanine → Valine at residue 4) is one of the most aggressive ALS-associated variants. It subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation. The task is to design short peptides that bind mutant SOD1 and evaluate which are worth advancing toward therapy.
Part 1: Peptide Generation with PepMLM
The human SOD1 sequence was retrieved from UniProt (P00441) and the A4V mutation was introduced manually (position 4: Ala → Val):
Using the PepMLM-650M model (ChatterjeeLab, HuggingFace) conditioned on the A4V mutant sequence, 4 peptides of length 12 amino acids were generated. The known SOD1-binding peptide FLYRWLPSRRGG was added as a control. Lower pseudo-perplexity = higher model confidence in binding potential.
Index
Peptide Sequence
Pseudo-Perplexity
Notes
1
WRSYATAAEHKE
10.552
Best candidate — lowest perplexity, highest model confidence
2
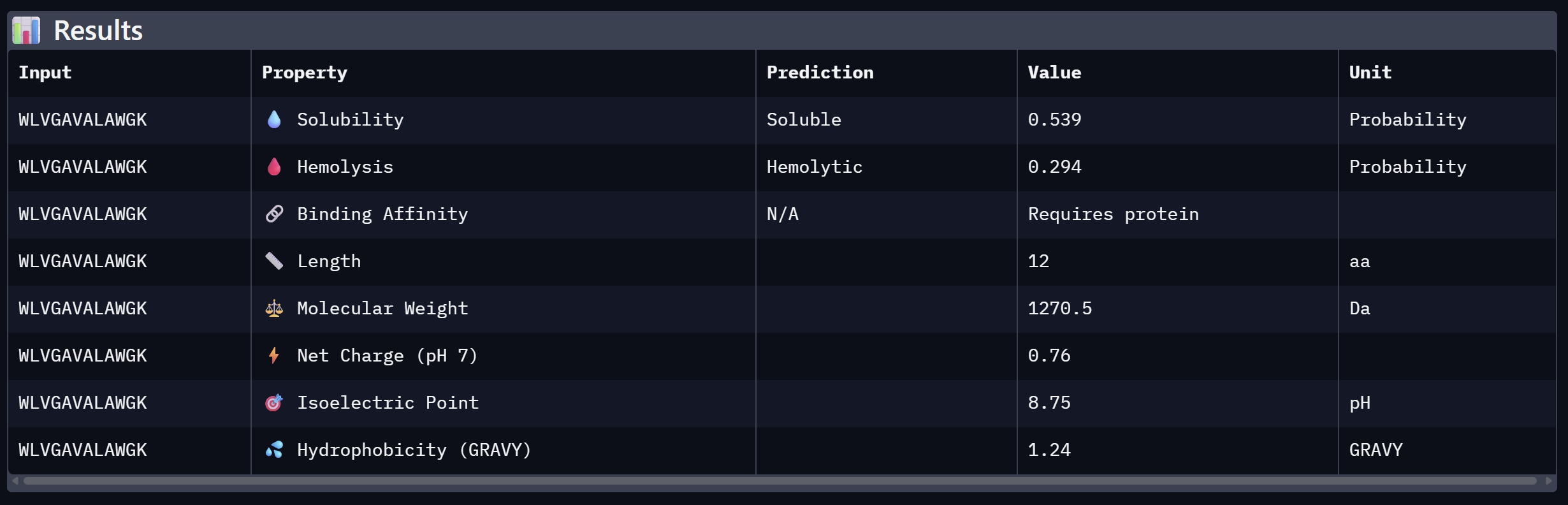
WLVGAVALAWGK
10.608
Close second; hydrophobic core with aromatic flanking
3
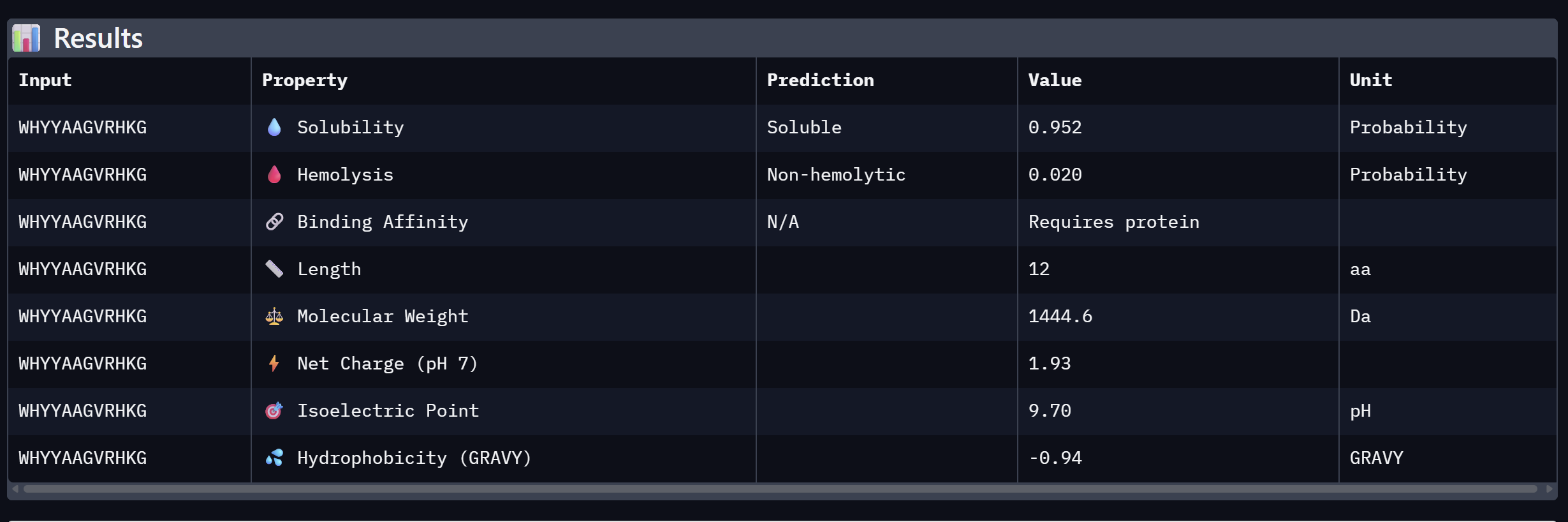
WHYYAAGVRHKG
16.820
Moderate confidence; aromatic-rich N-terminus
4
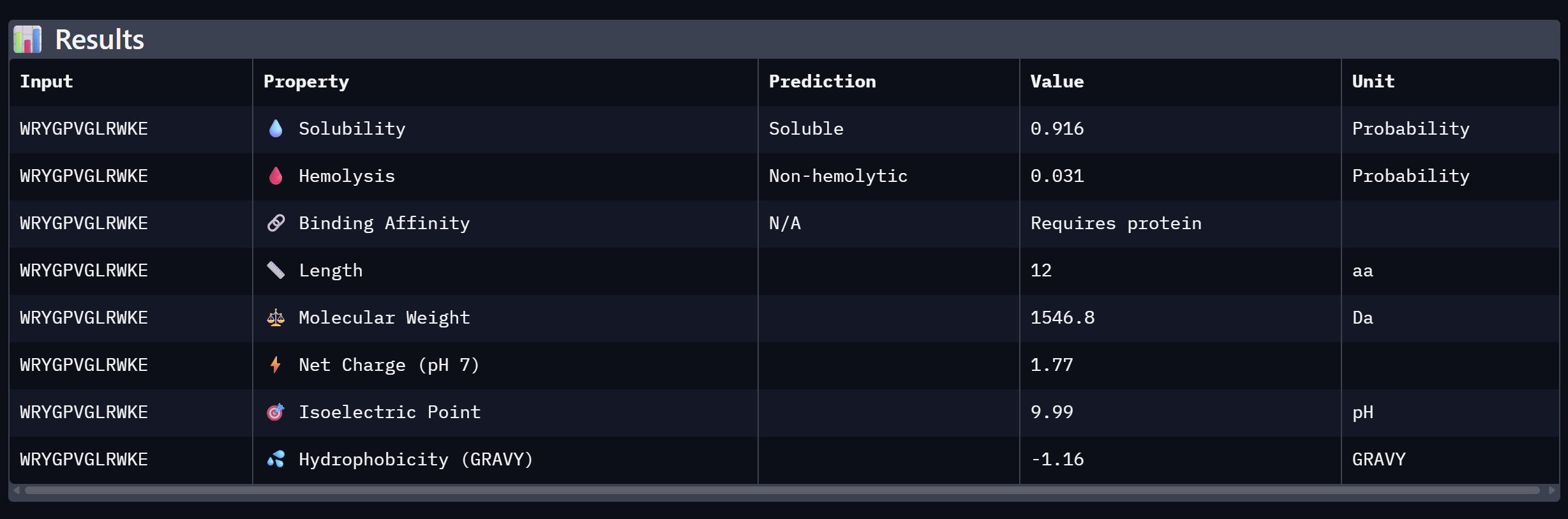
WRYGPVGLRWKE
19.620
Lowest confidence; highest perplexity
Control
FLYRWLPSRRGG
—
Known SOD1-binding peptide; reference benchmark
The best candidate is WRSYATAAEHKE (pseudo-perplexity = 10.552).
Part 2: Evaluate Binders with AlphaFold3
I submitted the mutant SOD1 sequence followed by the peptide sequence into Alphafold to model the protein-peptide complex and evulate their binding efficiecy.
Binder 1:
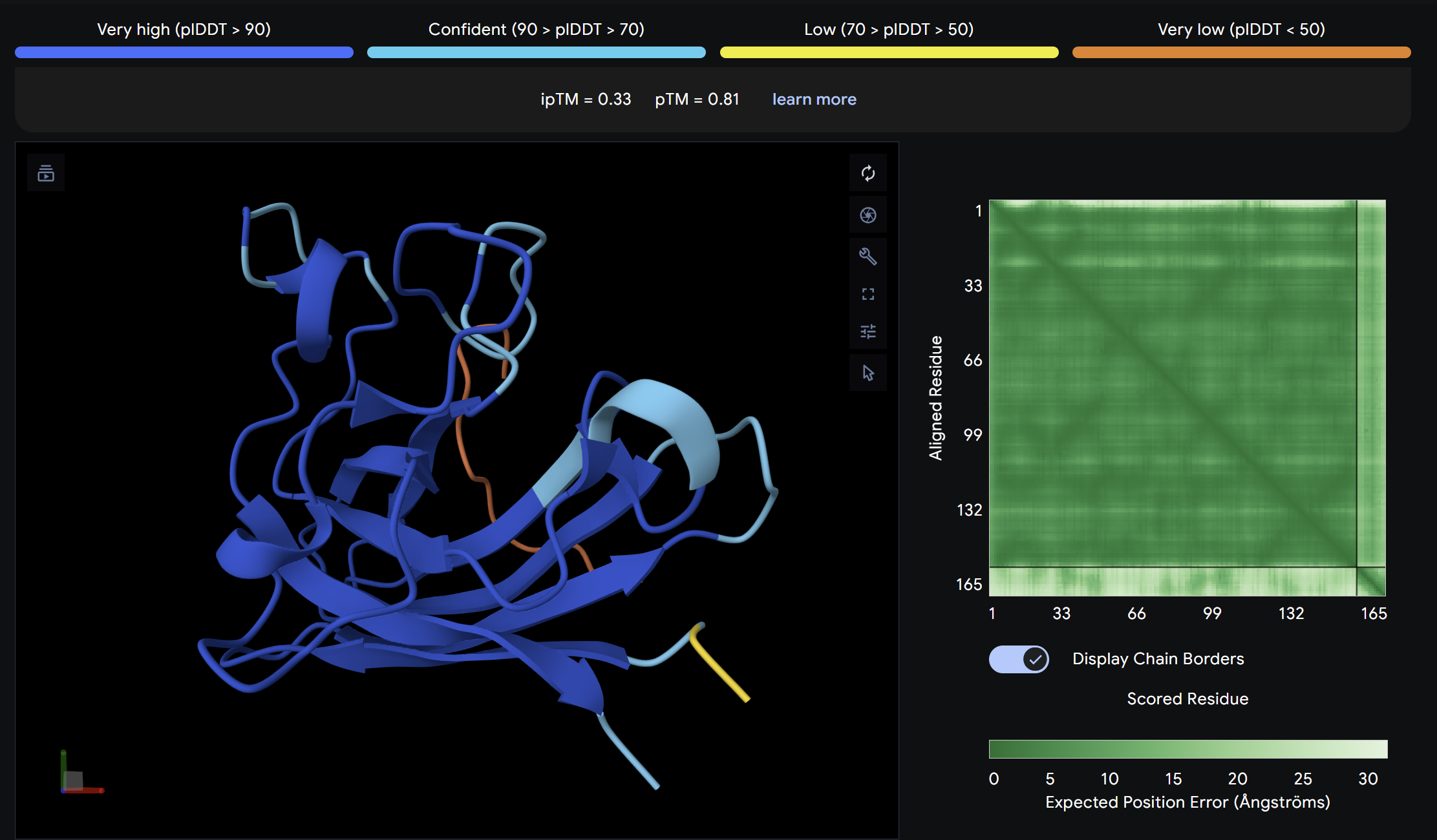
Alpha Fold ImageipTM = 0.33
ptm = 0.81
This peptide showed moderate predicted binding confidence, comparable to the control. In the AlphaFold3 structure, it appears to localize peripherally on the SOD1 surface, away from the N-terminal A4V mutation site. The peptide appears largely surface-bound with no significant burial into the protein core, suggesting a weak or non-specific interaction.
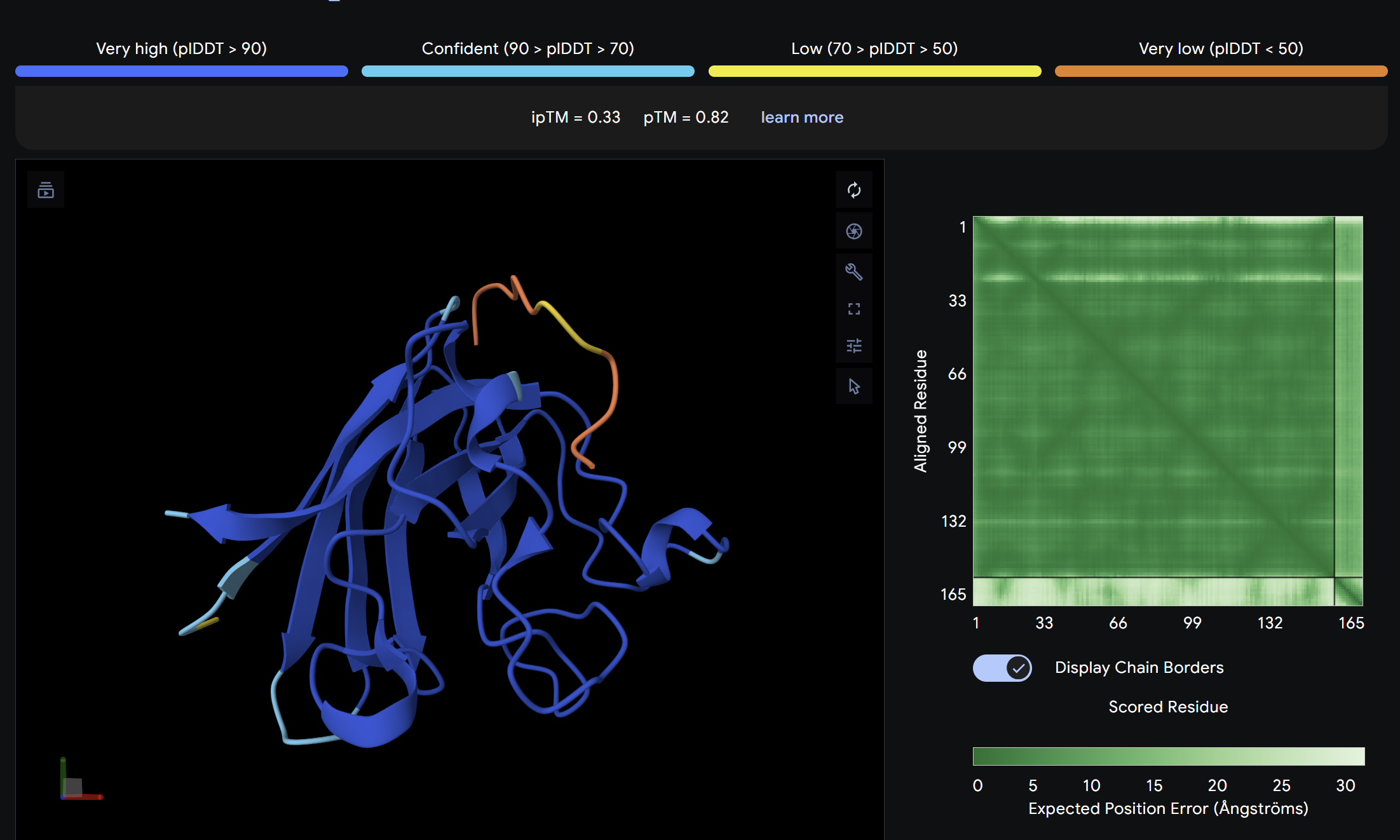
Binder 2:
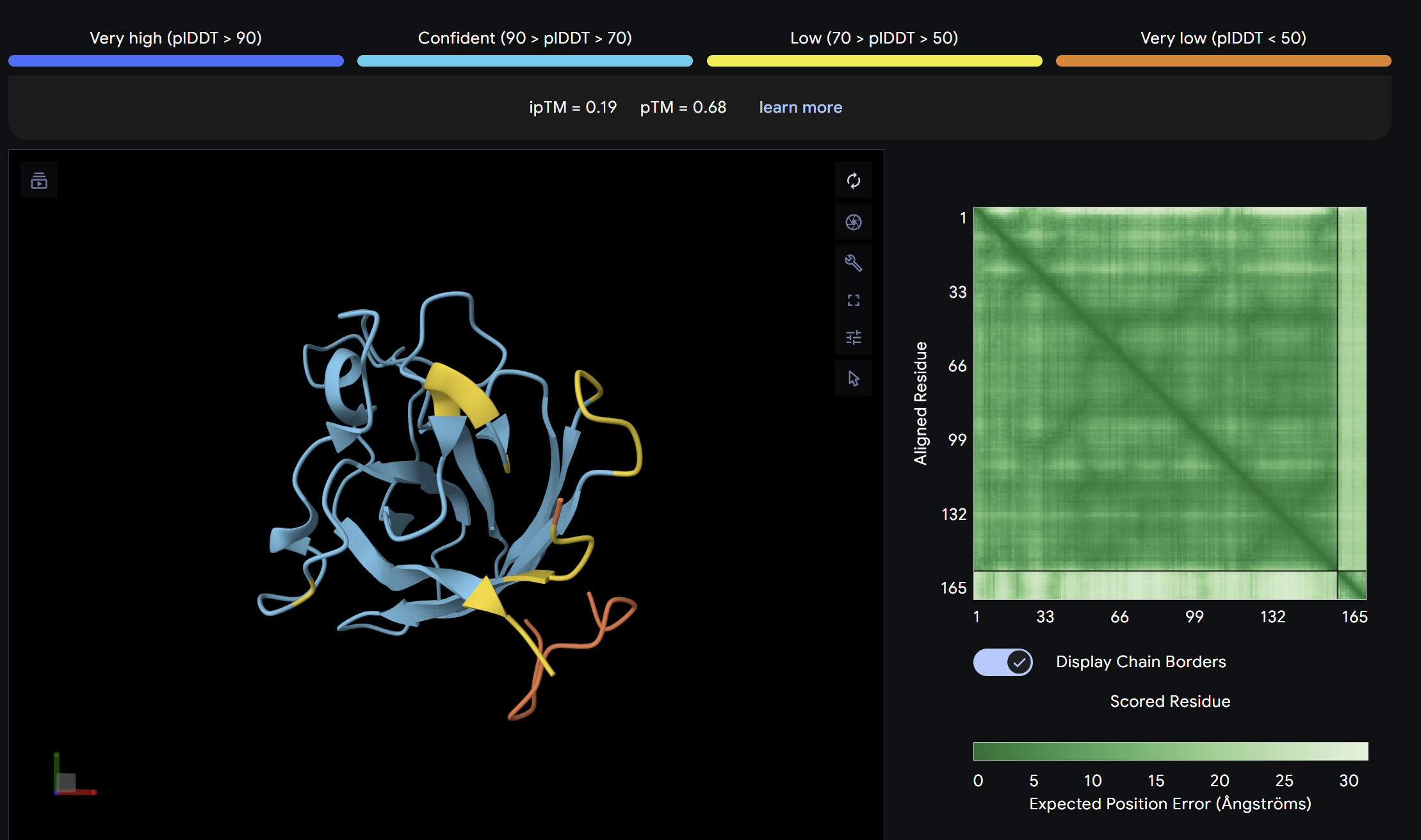
Alpha Fold ImageipTM = 0.19
ptm = 0.68
The lowest ipTM score of all tested peptides. The predicted complex shows a loosely associated peptide with high positional uncertainty across the PAE matrix. It does not appear to engage the N-terminus, β-barrel, or dimer interface in a meaningful way. This peptide is unlikely to be a functional binder despite its low perplexity score.
Binder 3:
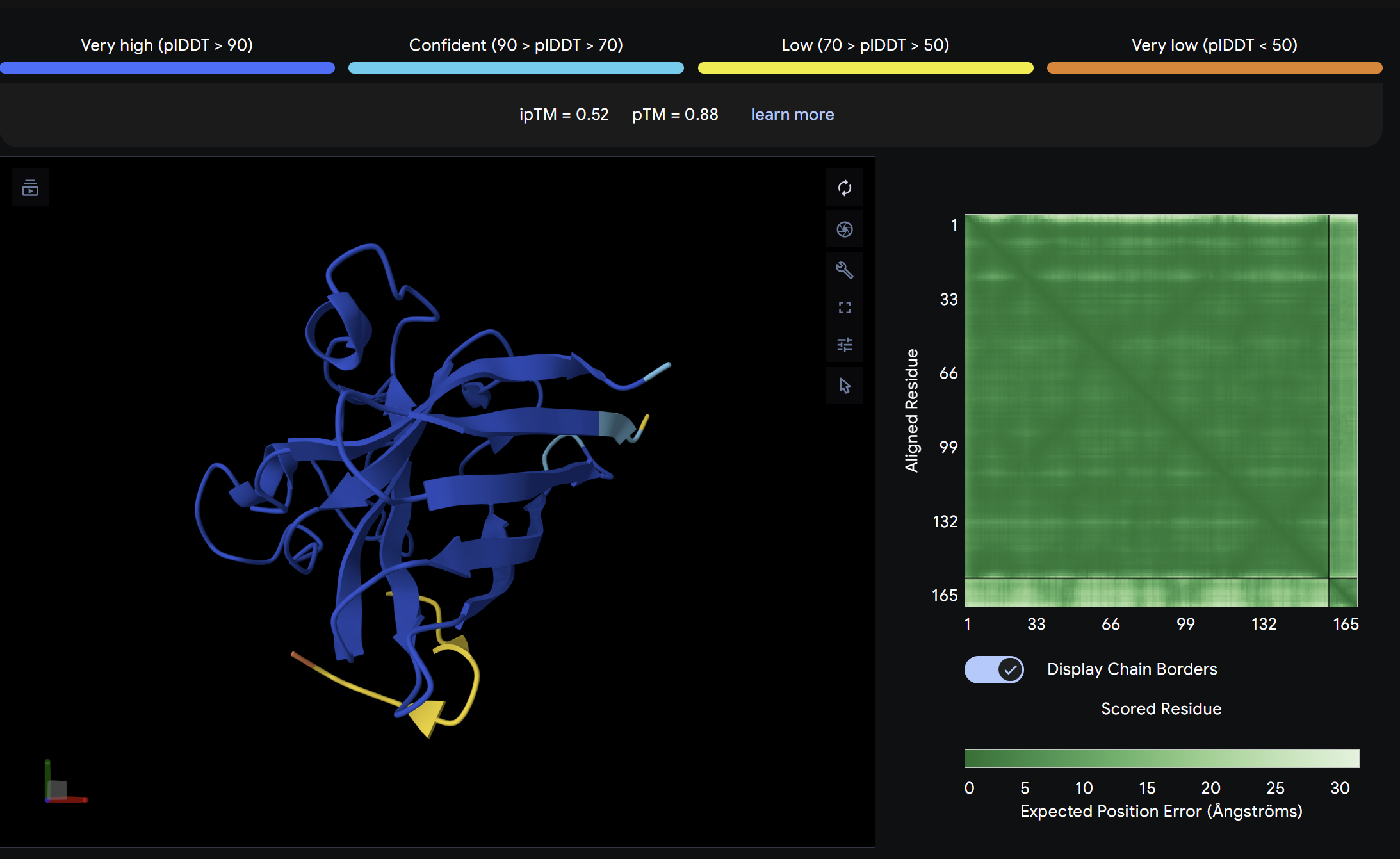
Alpha Fold ImageipTM = 0.52
ptm = 0.88
The strongest predicted binder of the set, exceeding the control. The structure shows the peptide engaging a region near the β-barrel domain with partial contact toward the N-terminal region where A4V sits. The peptide appears partially buried rather than fully surface-exposed, suggesting a more specific interaction interface. This is the most promising PepMLM-generated candidate.
Binder 4:
Alpha Fold ImageipTM = 0.55
ptm = 0.87
Slightly above the control in ipTM score. The peptide localizes near a peripheral interface region of SOD1 but does not clearly engage the A4V mutation site or dimer interface. It appears surface-bound with moderate positional confidence.
Binder 5:
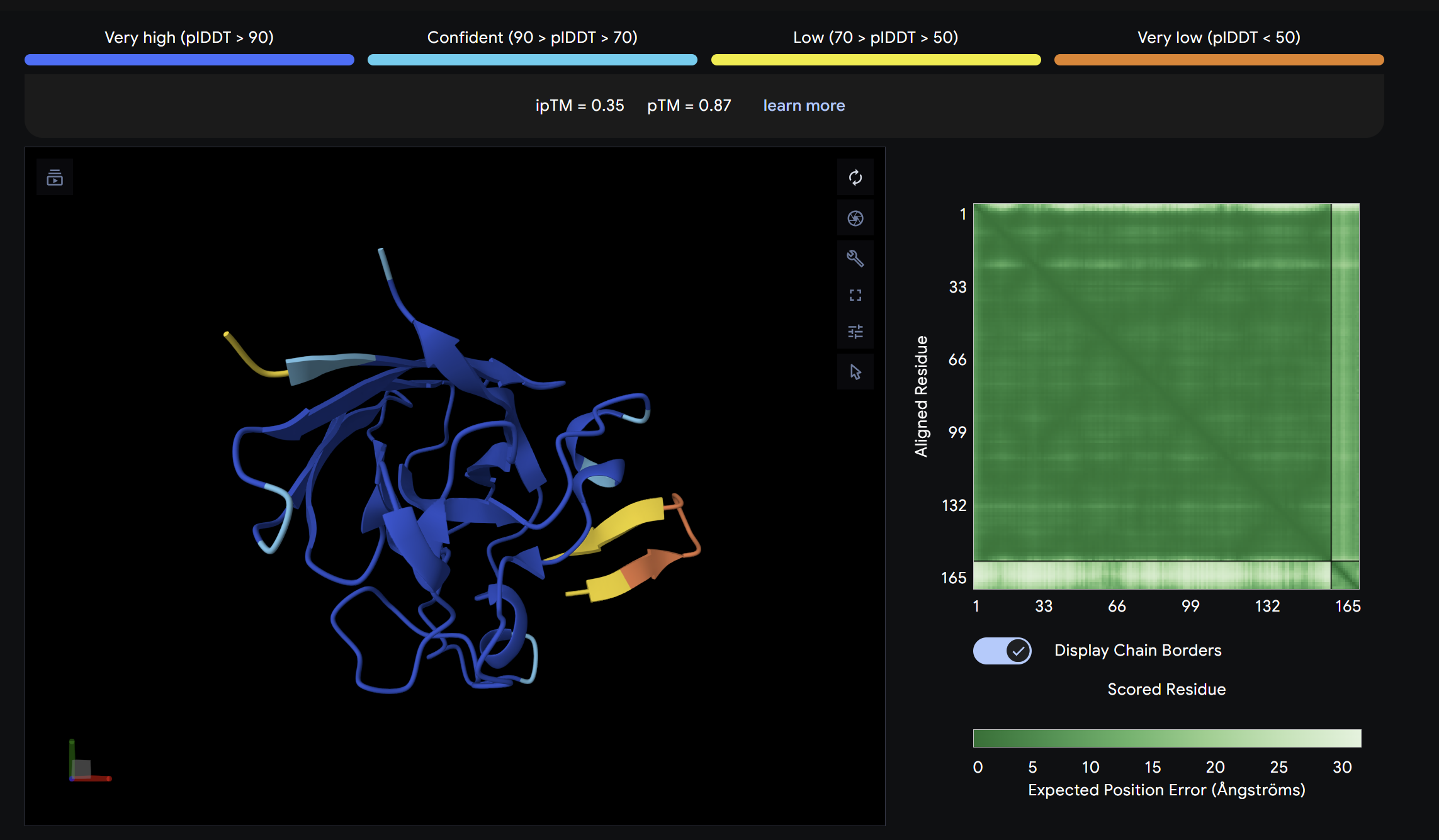
Alpha Fold ImageipTM = 0.33
ptm = 0.82
The known SOD1-binding peptide serves as the baseline reference. Its ipTM of 0.33 reflects moderate predicted binding, surface-associated without deep burial. Notably, Binder 3 (WHYYAAGVRHKK) exceeds the control with an ipTM of 0.52, suggesting PepMLM successfully generated at least one peptide with stronger predicted binding than the established reference. Overall, ipTM values across all peptides are in the low-to-moderate range (0.19–0.52), consistent with short peptide binders where full complex confidence is inherently limited.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Binder 1:
Binder 2:
Binder 3:
Binder 4:
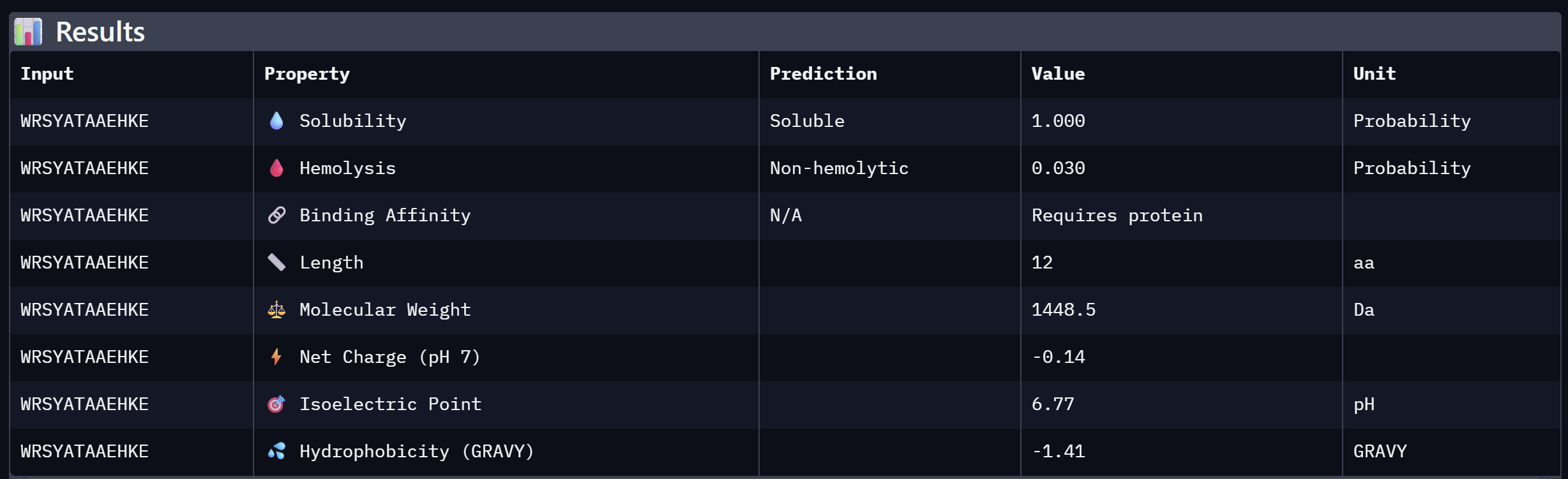
An analysis of PeptiVerse predictions alongside AlphaFold3 structural data highlights a significant alignment between structural binding and therapeutic potential. Binder 3 (WHYYAAGVRHKG) emerged as the most promising candidate, achieving the highest ipTM score of 0.52 and an optimal therapeutic profile. Its characteristics include superior solubility (0.952), a minimal hemolysis probability (0.020), and a +1.93 positive charge that likely facilitates electrostatic interactions with the negatively charged surface of SOD1. This correlation suggests that high predicted structural binding serves as a reliable indicator of therapeutic viability.
At the same time, Binder 2 (WLVGAVALAWGK) is the least viable candidate due to both structural and physicochemical deficiencies. It recorded the lowest ipTM score (0.19) and is categorized as hemolytic (0.294) with marginal solubility (0.539). Its elevated hydrophobicity (GRAVY = +1.24) is the probable cause for its poor solubility and associated hemolysis risk. While Binders 1 and 4 demonstrated solubility and were non-hemolytic, their moderate ipTM scores reduced their overall therapeutic appeal. Since PeptiVerse does not support direct protein target input for affinity calculations, AlphaFold3 ipTM scores were utilized as a proxy for binding affinity.
My chosen peptide for later advancement is WHYYAAGVRHKG (Binder 3). It is the only candidate that at the same time: exceeds the control binder in predicted structural bidning (ipTM = 0.52 vs. 0.33), hihgly soluble, non hemolytic and carreis a favorable charge for SOD1 interaciton. Despite possessing a higher perplexity score than Binders 1 and 2, its integrated therapeutic and structural profile establishes it as the most stongest candidate overall.
Part C: L-Protein Mutant Design — MS2 Phage Lysis Engineering
Background
Bacteriophage MS2 relies on its L-protein (lysis protein) to form pores in the E. coli cell membrane, ultimately lysing the host. A common bacterial resistance mechanism involves a point mutation in the chaperone DnaJ, which prevents proper L-protein processing and abolishes lysis. The objective is to engineer L-protein variants that either (1) fold independently of DnaJ, or (2) lyse bacteria faster, reducing the window for resistance to develop.
The wild-type L-protein sequence (UniProtKB P03609) is:
Soluble N-terminal domain (residues 1–40): responsible for DnaJ interaction
Transmembrane domain (residues 41–75): responsible for membrane insertion and lysis activity
Option 1: Mutagenesis Scoring via ESM2 Language Model
Step 1 — Computational scoring (ESM2 heatmap)
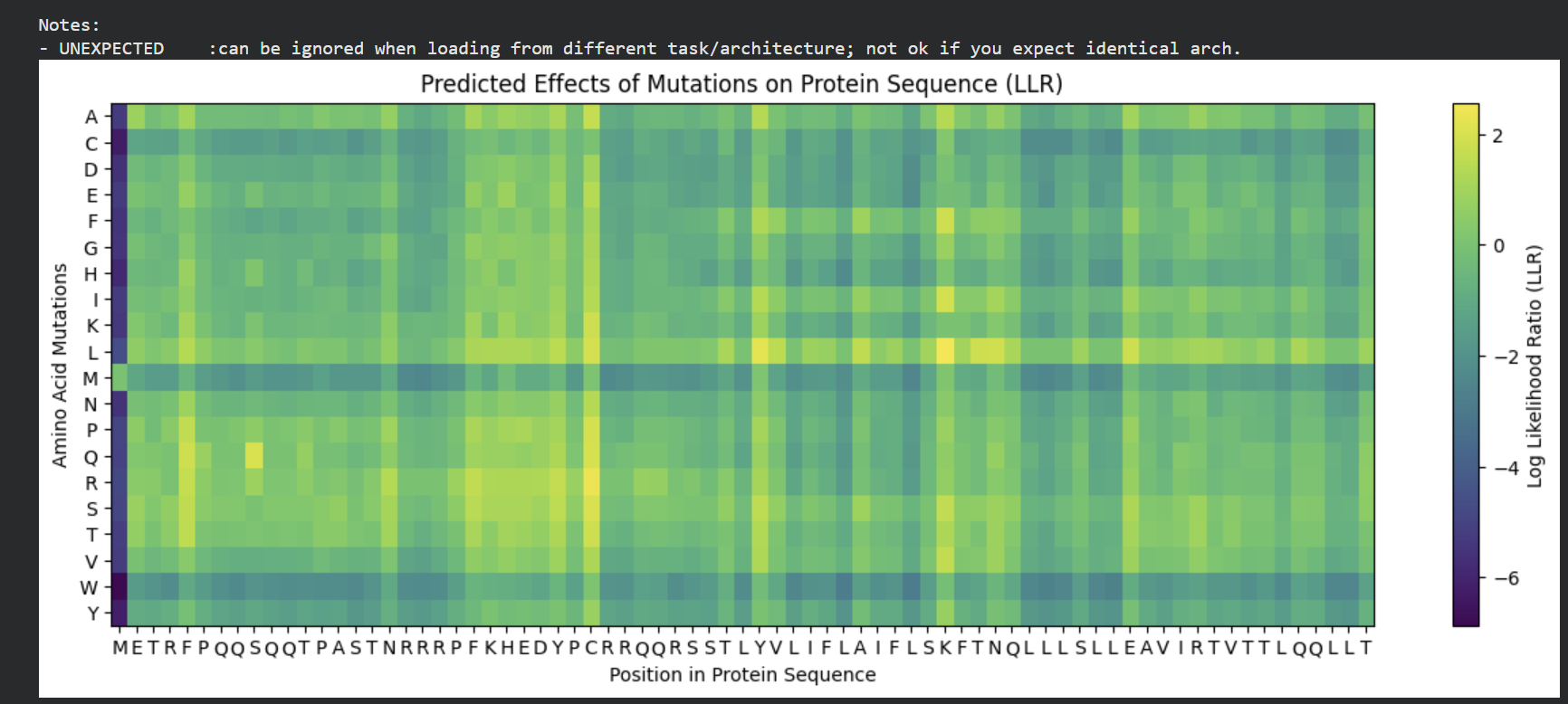
The mutagenesis scoring notebook was run using the ESM2 language model (facebook/esm2_t6_8M_UR50D). For each position in the L-protein sequence, a Log Likelihood Ratio (LLR) score was calculated for every possible amino acid substitution.
Positive LLR scores: indicates the model predicts the mutation is tolerated or beneficial
Negative LLR scores: indicates it would likely be harmful.
The resulting heatmap shows LLR values across all 75 positions and 20 amino acids. Bright yellow regions indicate high positive LLR (favorable mutations); dark blue/purple regions indicate strongly negative LLR (deleterious mutations). Notably, positions in the transmembrane region (right half of the heatmap) show more variability, reflecting the model’s sensitivity to hydrophobicity changes in membrane-spanning segments.
Step 2 — Cross-validation with experimental data
The experimental L-Protein Mutants dataset was compared against the ESM2 LLR scores. Key observations:
Mutations with Lysis = 0 (non-functional) generally corresponded to negative or near-zero LLR scores, confirming the model captures some biological signal.
Mutations with Lysis = 1 (functional) at positions 18, 25, 30, 31, 44, 45, and 46 were associated with positive LLR scores, suggesting reasonable agreement between computational and experimental data.
The model has limitations: some experimentally functional mutations (Lysis = 1) had modest LLR scores, and the model does not account for membrane topology or DnaJ interaction directly.
A selection of key entries from the experimental dataset is shown below (Lysis: 1 = functional, 0 = non-functional, N.D. = not determined):
AA Position
AA Change
Lysis
Protein Levels
1
M→I
0
0
1
M→T
0
0
13
P→L
1
1
15
S→A
1
1
18
R→G
1
1
18
R→I
1
1
18
R→Stop
0
N.D.
19
R→S
1
0
23
K→E
1
0
25
E→G
1
0
29
C→R
—
—
29
C→Stop
0
N.D.
30
R→Q
1
1
30
R→L
1
1
31
R→I
1
1
39
Y→H
0
0
39
Y→Stop
0
N.D.
44
L→P
1
1
45
A→P
1
1
46
I→F
1
1
50
K→N
0
1
Step 3 — Selected mutations
Five mutations were selected by prioritizing positions with (a) high positive LLR scores from the ESM2 notebook and (b) experimental lysis data showing Lysis = 1 where available. Conserved positions (no variation in BLAST alignments) were avoided.
#
Position
Domain
Wild-type AA
Mutant AA
LLR Score
Experimental Lysis
Rationale
1
18
Soluble
R
G
positive
Lysis = 1
Experimentally confirmed functional; removing the positively charged arginine may reduce DnaJ dependency by altering soluble domain surface charge
2
29
Soluble
C
R
2.395 (high)
Not tested
Highest LLR in soluble region; cysteine at position 29 may form inappropriate disulfide bonds — replacing with arginine adds a stabilizing charge interaction
3
39
Soluble/TM boundary
Y
L
2.242 (high)
Not tested
High LLR score; tyrosine at the domain boundary may be substituted with leucine to improve hydrophobic continuity into the TM domain and aid autonomous folding
4
45
Transmembrane
A
L
1.539 (positive)
A→P Lysis = 1 (nearby)
Positive LLR; increasing hydrophobicity at this TM position may improve membrane insertion efficiency; nearby A45P is experimentally confirmed functional
5
50
Transmembrane
K
L
2.561 (highest)
Not tested
Highest LLR score across the entire sequence; K50 is a charged residue embedded in the hydrophobic TM core — replacing it with leucine removes the charge mismatch and is predicted to strongly stabilize membrane insertion
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion DNA Polymerase
Chimeric enzyme that catalyzes the synthesis of ew DNA strand in the 5 -> 3 direction with high-fidelity
dNTPs
four chemical building blocks ($dATP, dTTP, dCTP, dGTP$) used to construct the DNA. They provide both the physical material and the energy required for the polymerase to grow the new strand
Reaction Buffer
Maintains the optimal pH and ionic environment for the reaction. It ensures the enzyme remains stable and functional throughout the high-temperature cycles of PCR.
Magnesium Chloride
Co-factor for the polymerase enzyme. Without magnesium ions, the enzyme cannot catalyze the chemical reaction needed to link the DNA building blocks together
Additives & Stabilizers
Chemicals like glycerol or detergents protect the enzyme from degradation. Their purpose is to keep the master mix stable during storage and prevent the proteins from sticking to the plastic tube walls.
What are some factors that determine primer annealing temperature during PCR?
The primer annealing temperature ensures primers stick specifically to the target DNA. Main factors are:
Primer Length and Composition: ratio of G-C to A-T bases (G-C pairs have three hydrogen bonds)
Primer Concentración: higher concentration can increase binding
Salt concentration: cations like K+ and Mg2+ stabilize the DNA backbone, which increases the melting temperature
Base Mismatches
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Both PCR and Restriction Enzyme Digests are fundamental techniques for generating DNA fragments; they differ in the process of generating these fragments
PCR
requires a template of DNA
uses heat and a polymerase to synthesize new copies of a specific region
main components are primers, dNTPs, DNA polymerase, and thermal cycler
depend on the designed primer
Use when we have a low DNA sample volume/ we want to create a fragment of a very specific, non-common length
Restriction Enzyme Digest
requires a high concentration of purified DNA
uses molecular scissors to physically cut existing DNA
main components are restriction enzymes and a stable heat incubator
depend on the presence of specific recognition sequences (sites)
Use when we want to cut out a gene or insert from a circular plasmid to move to another vector/we want to check a piece of DNA (Diagnostic Digest)/when we know the restriction sites already exist
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure DNA fragments are ready for Gibson Assembly, we have to focus on the end of the sequences since Gibson uses overlapping DNA sequences, not the ‘sticky ends’from restriction enzymes.
Check for overlapping ends - each fragment must share an identical sequence with the fragment next to it.
Verify Clean Ends - since Gibson relies on Exonuclease, we must ensure there are no extra A overhangs or that enzymes have reached complete digestion.
Check the chemical environment - must remove polymerase, dNTPs, and salts from the PCR reaction
Sequence accuracy - verify the final assembled plasmid with the Sanger Sequencing kit at the junction points
How does the plasmid DNA enter the E. coli cells during transformation?
The process is called Transformation.
Steps:
Preparation - before DNA enters, the cells are soaked in a solution of calcium chloride, so that Ca+ would neutralize the negative charge of the DNA and the cell membrane, allowing them to get close to each other.
Entry point = SHOCK - once mixed, the cells with DNA are moved to a 42 C water bath for 30-60 seconds to create a temporary “pressure difference” and physical holes in the cell membrane. Plasmid DNA is sweeped into these pores
Recovery - put the cells back on ice to seal holes.
Describe another assembly method in detail (such as Golden Gate Assembly)
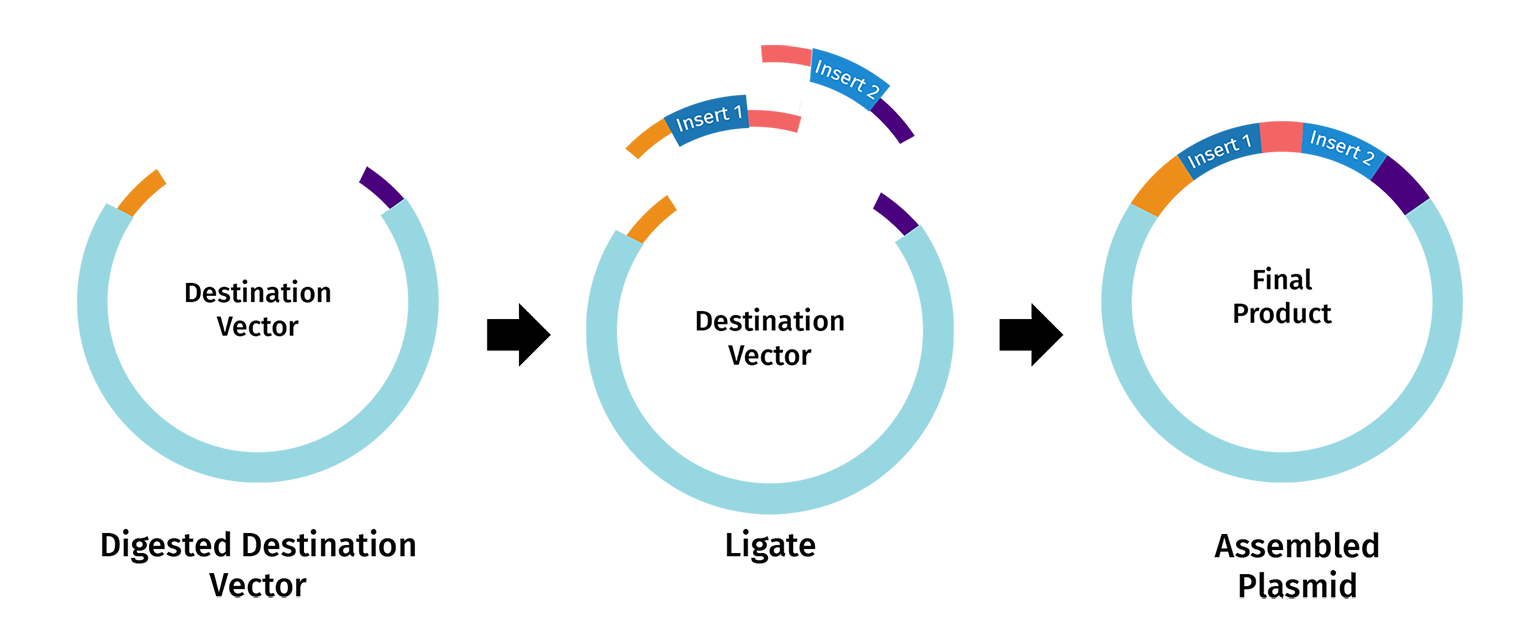
Golden Gate Assembly in essence is similar to the Gibbson assembly, but it allows for the simultaneous assembly of multiple DNA fragments using Type IIS restriction enzymes and T4 DNA ligase. Unlike standard enzymes, Type IIS enzymes (like BsaI) cut outside of their recognition sites, creating 4-base overhangs that can be customized to rule the assembly order. Because the recognition sites are placed at the very ends of the fragments and are “cut off” during the reaction, the final product is seamless and lacks the original restriction sites, preventing the enzyme from re-cutting the finished plasmid. This “scarless” assembly is highly efficient, often reaching nearly 100% accuracy even when joining ten or more fragments at once. The entire process occurs in a single tube through a series of temperature cycles that alternate between the optimal conditions for digestion and ligation.
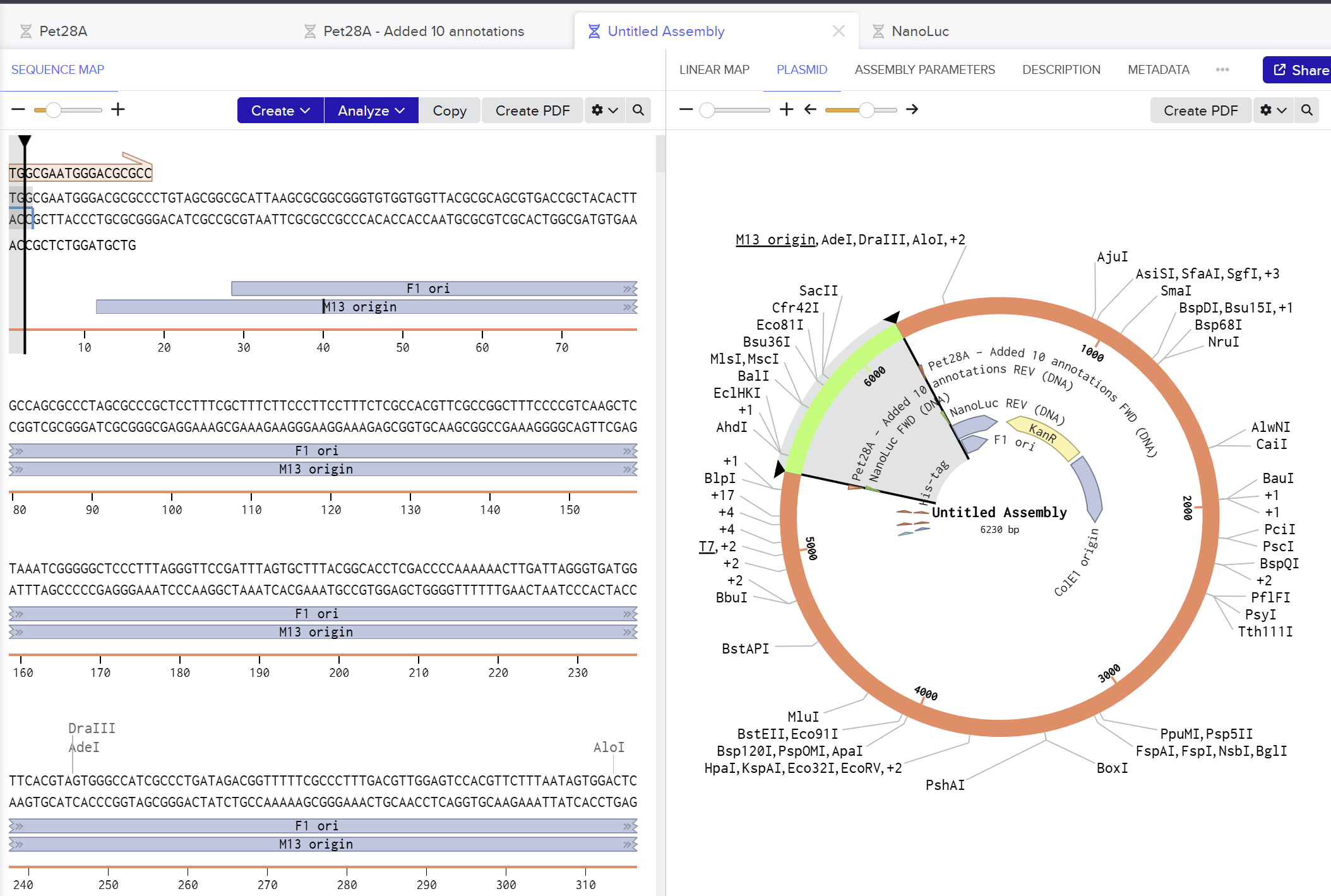
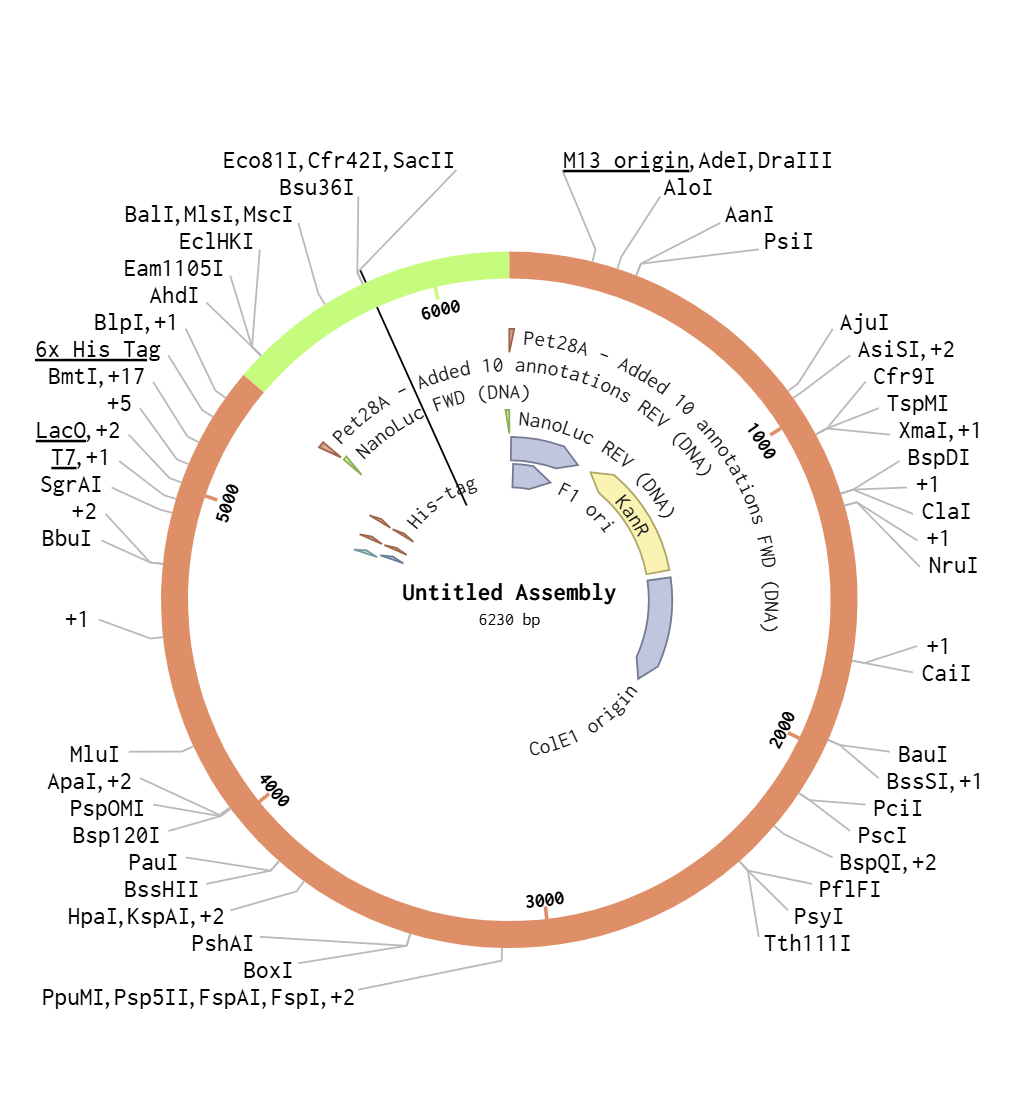
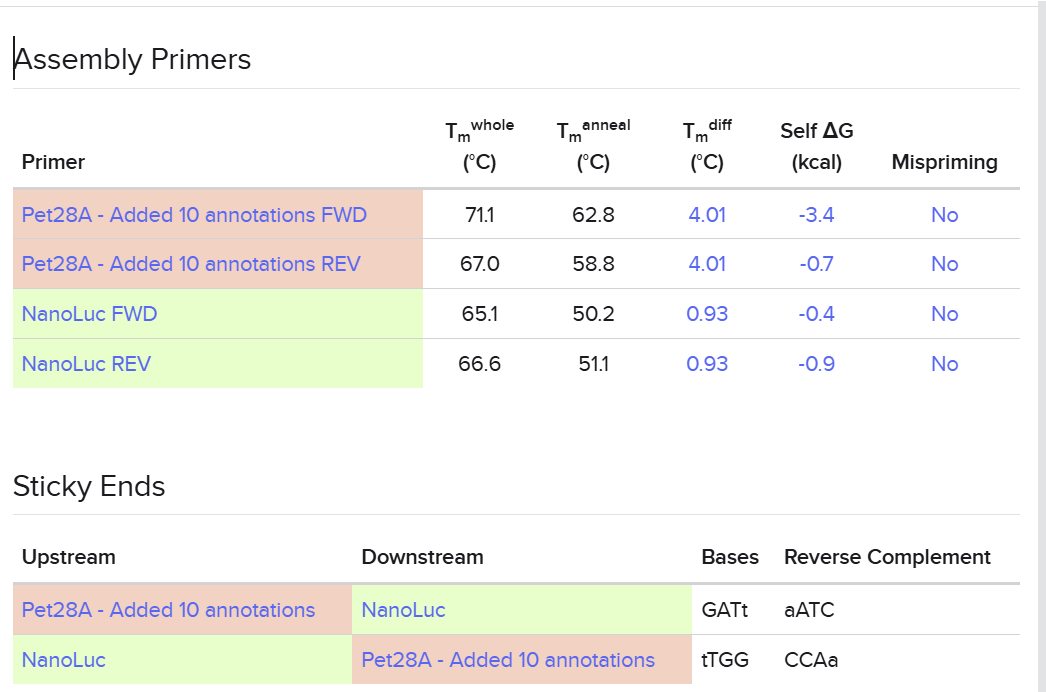
I used Benchling to simulate using Golden Gate Assembly to insert NanoLuc luciferase into a pET28a backbone.
I opened the pET28 plasmid in Benchling and copied it into my own workspace so I could edit it. Then I imported the NanoLuc DNA sequence as a separate linear DNA fragment.
After that, I used the Assembly Wizard to set up a Golden Gate assembly. I selected pET28 as the backbone and NanoLuc as the insert, then added the BsaI cut sites and matching overhangs needed for the assembly.
Finally, Benchling generated the assembled plasmid map for me. I checked the final circular construct to make sure the NanoLuc insert was placed in the correct position and orientation.
It was pretty easy and starighforward.
I have docuemnted all of my pr0gress in the COnstruct Log Notebook.
PART 1
PART 2
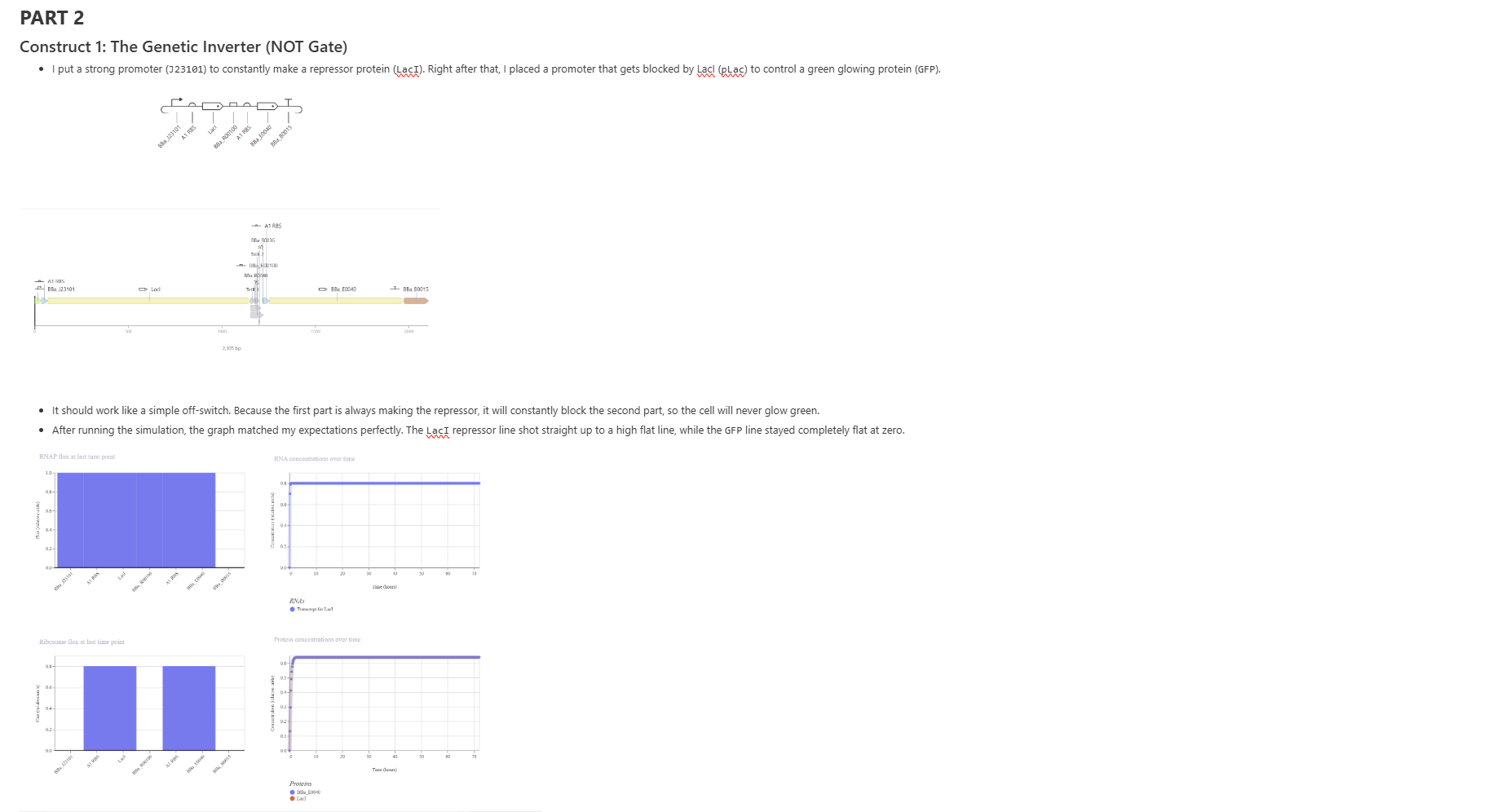
Construct 1: The Genetic Inverter (NOT Gate)
Construct 2: The Controlled T7 Cascade
Construct 3: Cell-to-Cell Signaling
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Part 1
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Traditional genetic circuits implement Boolean logic gates (AND, OR, NOT, NAND, etc.), hence their input/output relationships are discrete - a gene is either ON or OFF.
This allows only binary decision-making and makes it difficult to represent graded, continuous, or context-dependent responses.
IANNs provide continuos computation where inputs and outputs exist on a continuum, allowing cells to integrate multiple signals simultaneously.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
IANNS would be best implemented in the monitroing procedures in metabolic conditions.
In my particular exmaple, the IANNS would be good to monitor and asses PMOS (polyendocrine metabolic ovarian syndrome).
Since this disease is characterised by three co-occuring signals that can be read intracellularly: elevated androgens, insulin resistance and chronic low-grade inflammaiton.
The IANN’s strength is integrating all three continuously, which a Boolean circuit cannot do.
Some limitations:
Using multiple Csy4 variants risks cross-reactivity — they may cleave each other’s targets.
Cell-type specificity/Biological noise: A diagnostic device would need to specify the cellular context.
Baseline variability: Hormone levels fluctuate across the menstrual cycle even in healthy individuals, so the IANN would need calibration thresholds per individual rather than universal cutoffs.
Delivery: Getting the genetic construct into the relevant cells non-invasively remains an unsolved challenge for any in-vivo IANN.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Part 2
What are some examples of existing fungal materials and what are they used for?
Fungal materials utilize mycelium - the vegetative root network of fungi.
Currently, Alaska (AK) has emerged as a top user of fungal materials in daily life, pioneering their implementation to address specific environmental challenges.
One example is how fungal biocomposites are being used to manufacture insulated container materials designed to replace traditional plastic packaging, drastically helping with the persistent plastic pollution problem.
In Alaska, where the seafood export industry relies heavily on lightweight insulation, researchers have developed mycelium-based container materials combined with local wood pulp. These containers serve as biodegradable shipping boxes, directly replacing expanded polystyrene (Styrofoam) and preventing non-degradable plastic waste from accumulating in maritime ecosystems.
The second exmaple, would help with high heating costs and environmental degradation, by implementing fungus based insulation. These insulation panels are grown locally by feeding fungal strains on cellulose substrates harvested from beetle-killed spruce trees, transforming a major forest fire hazard into high-performance, sustainable housing insulation.
(tap on the pink text to see the supplementary links)
The core advantages are:
Drastic Reduction in Plastic Pollution: Unlike traditional plastics that persist in landfills and oceans for centuries, fungal materials are completely biodegradable and compostable, breaking down naturally after their operational lifespan.
Fire Retardancy: The natural presence of chitin in fungal cell walls gives mycelium-based insulation excellent fire-resistant properties, making it safer during combustion events compared to plastic foams, which release toxic volatile organic compounds (VOCs).
Moisture Breathability: Fungus-based insulation is vapor-permeable. In cold climates like AK, this allows trapped structural moisture to escape, preventing the structural rot and toxic mold growth often caused by vapor-impermeable plastic barriers.
Couple of disadvatages that I thought of were:
Production Time Constraints: Traditional plastics can be manufactured instantaneously via high-throughput chemical extrusion. Fungal materials, however, require a biological incubation period of several days to weeks to grow, making rapid mass production challenging.
Material variance and potential structural decay over time: The strength, density, and insulating properties of a mycelium block depend heavily on how evenly the fungus grew throughout its wood-pulp substrate. In highly humid environments or if a container gets scratched or punctured, dormant fungal spores or ambient microbes can re-activate.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
For me, it would be interesting to see how fungal materials can benefit human health by acting as living therapeutics.
If we use safe fungi such as yeast, they can be engineered to act as drug-delivery vehicles in the intestine. These engineered fungi could sense specific metabolites like bile acids or glucose and then release insulinotropic peptides, vitamins, or other compounds when needed.
Another promising application is using fungi as living wound dressings in hydrogel bandages. In this case, the fungi could secrete growth factors or antimicrobial peptides to help accelerate wound healing and prevent infection.
Fungi are also excpetionally beneficial for synthetic biology because they share more cellular features with human cells than bacteria, which can make them better at producing complex human proteins in a functional form. They also have rich metabolic capacity because they often have larger genomes and more enzymes. In addition, fungi coexist with the bacterial microbiota in the gut, so they could potentially provide beneficial functions without completely disrupting the existing microbial community.
Week 9 HW: Cell free systems
General homework questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis is more flexible than in vivo expression because we can directly control the reaction conditions, such as DNA concentration, salts, cofactors, temperature, and additives. The in vivo model limits out experimet by time since we have atcually grow cells and wait for results, in cell free systems the speed of these procedures is much faster. It is more beneficial to use cell free systems for toxic proteins, membrane proteins, and rapid prototyping or diagnostics, because we do not need to keep a living cell alive while producing the protein.
2. Describe the main components of a cell-free expression system and explain the role of each component.
There are several components of a cell-free expression systems:
Cell extract:
This is the liquid fraction from broken cells that supplies the machinery needed to make proteins.
DNA template:
This is the gene blueprint that tells the system which protein to produce.
Amino acids:
These are the raw materials used to build the protein chain.
NTPs:
These molecules are used to make RNA and also help power the reaction.
Energy source:
This keeps the system active by replacing the energy used during protein synthesis.
Cofactors:
These keep the reaction environment suitable so the enzymes can work efficiently.
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is critical because the cell free system does not provide naturally energy for wokring conditions. Transcription and translation consume ATP and GTP very quickly, so the reaction stops if energy runs out. A common method is to add an energy regeneration system such as phosphoenolpyruvate or creatine phosphate so ATP can be continuously recycled during the reaction.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic and eukaryotic cell-free systems are better for different kinds of proteins. A prokaryotic system, usually based on E. coli extract, is fast, inexpensive, and very efficient for making proteins that do not require complicated folding or post-translational modifications. Because of that, I would choose a simple bacterial protein, such as GFP or a bacterial enzyme, to produce in this system. These proteins are usually easier to express because they fold well in bacterial conditions and do not depend on glycosylation or other eukaryotic modifications.
A eukaryotic cell-free system, such as one based on wheat germ, insect, or mammalian extract, is better for proteins that are more complex and need additional folding help or cellular processing. I would choose a human membrane receptor or a secreted human protein for this system, because these proteins often need more than just translation to become functional. Eukaryotic systems are more suitable when the target protein needs correct folding, disulfide bond formation, or a more native-like environment to stay stable and active.
The main difference between the two systems is the source of the extract and also the type of protein they are best able to produce. Prokaryotic systems are usually preferred when speed, low cost, and high yield are most important. Eukaryotic systems are preferred when protein quality, folding, and biological realism are more important than maximum yield.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
To optimize the expression of a membrane protein in a cell-free system, I would choose a system that better supports membrane insertion and protein folding, such as a eukaryotic extract or an extract supplemented with membrane-mimicking components. Membrane proteins are difficult to express because their hydrophobic regions tend to aggregate in aqueous solution, so I would include liposomes, nanodiscs, or microsomal vesicles to provide a more natural lipid environment for folding and insertion.
I would also design the construct with a small fluorescent tag, such as GFP, so I can monitor whether the protein is being produced successfully and whether the expression level changes under different conditions. This would allow me to compare different reaction setups, such as varying temperature, magnesium concentration, extract type, and DNA template amount, to find the best conditions for expression. If the protein is especially sensitive, I would also test slower reaction temperatures because lower temperatures can sometimes improve folding and reduce aggregation.
Another important part of the design would be energy management. Since membrane protein synthesis can take a long time, I would use an energy regeneration system, such as creatine phosphate and creatine kinase, or a continuous exchange setup to keep ATP levels stable during the reaction. This would help extend the reaction and increase the chance of obtaining a properly folded product.
The main challenge is that membrane proteins are not only hard to synthesize, but also hard to keep soluble and functional after synthesis. To address this, I would compare several conditions side by side, including reactions with and without membrane mimics, and measure both yield and activity. In the end, the best setup would be the one that gives the highest amount of correctly folded membrane protein, not just the highest total protein amount.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Some possible low yield target protein cases:
Poor DNA template quality or low translation efficiency:
One possible reason for low protein yield is that the DNA template is not clean, damaged, or not optimized for expression in the cell-free system. This can be improved by purifying the plasmid or PCR product more carefully, and in some cases by using codon optimization or adding an RNA inhibitor to reduce template degradation and improve expression.
Insufficient energy supply:
Another reason is that the reaction may run out of ATP and GTP too quickly, so the protein synthesis machinery cannot keep working. To fix this, you can improve the ATP regeneration system by adding a stronger energy source or a better recycling strategy so the reaction stays active for longer.
Unfavorable reaction conditions:
A third reason is that the magnesium level, salt concentration, temperature, or extract quality may not be ideal for the protein you are trying to make. You can troubleshoot this by testing several reaction conditions one by one, such as different temperatures or ion concentrations, until you find the setup that gives the best yield.
Homework question from Kate Adamala
Pick a function and describe it. What would your synthetic cell do? What is the input and what is the output?
The synthetic minimal cell (SMC) I want to design would detect TDP-43 protein aggregates (the pathological hallmark of Frontotemporal Dementia (FTD)) and respond by producing and releasing a therapeutic anti-aggregation peptide. TDP-43 is an RNA-binding protein that under disease conditions mislocalizes from the nucleus to the cytoplasm, where it forms toxic aggregates that drive neurodegeneration.
Input: Extracellular TDP-43 aggregates in the cerebrospinal fluid or interstitial brain environment
Output: A designed TDP-43 aggregation-inhibiting peptide released into the local environment to disrupt fibril formation and reduce proteotoxic stress
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
No. Without encapsulation, the cell-free Tx/Tl system would constitutively produce the anti-aggregation peptide regardless of whether TDP-43 aggregates are present. Encapsulation is essential to couple detection (input sensing via an aptamer) to production (output peptide expression), creating a conditional, signal-responsive system rather than a constitutive one.
Could this function be realized by a genetically modified natural cell?
Yes, in theory. Natural cell could be engineered with a TDP-43-responsive promoter driving anti-aggregation peptide expression. However, introducing living genetically modified cells into the brain raises several biosafety, immune rejection, and ethical concerns. A synthetic minimal cell offers a safer, non-replicating, and fully controllable alternative that degrades naturally once its payload is delivered.
Describe the desired outcome of your synthetic cell operation.
In the presence of TDP-43 aggregates, the SMC detects them via a surface-anchored RNA aptamer, triggers internal gene expression of the anti-aggregation peptide, and releases it through a membrane pore into the surrounding tissue. The result is localized, on-demand therapeutic peptide delivery specifically where and when TDP-43 aggregation is occurring, slowing the progression of FTD neurodegeneration.
Design All Components
What would the membrane be made of?
Phospholipids (POPC + POPE) and cholesterol to mimic a stable bilayer. Some biotinylated lipids would be incorporated to anchor the TDP-43-sensing aptamer on the outer membrane surface via streptavidin linkage.
What would you encapsulate inside?
Bacterial cell-free Tx/Tl system (PURE system)
DNA construct: gene encoding the anti-aggregation peptide under a T7 promoter coupled to a TDP-43 aptamer-responsive riboswitch
Gene encoding α-hemolysin (aHL) pore-forming protein, also under aptamer control, to enable peptide release upon activation
Small molecule cofactors for Tx/Tl (ATP, amino acids, NTPs)
Which organism will your Tx/Tl system come from?
Bacterial (PURE system from E. coli) is sufficient here, as the riboswitch used for TDP-43 detection is compatible with bacterial transcription/translation machinery. A mammalian system is not required since no mammalian-specific promoters (e.g., Tet-ON) are needed.
How will your synthetic cell communicate with the environment?
The outer membrane surface displays a TDP-43 RNA aptamer. When TDP-43 aggregates bind this aptamer, a conformational signal is transduced intracellularly, activating the riboswitch and initiating Tx/Tl of both the anti-aggregation peptide and α-hemolysin. The expressed aHL inserts into the membrane and forms a pore through which the therapeutic peptide is released into the extracellular environment.
ahlA (α-hemolysin from Staphylococcus aureus) — membrane pore for peptide release
Synthetic gene encoding TDP-43 aggregation-inhibiting peptide (e.g., based on the YQ-rich domain inhibitor design) — under T7 promoter + TDP-43 aptamer riboswitch
Aptamer: Anti-TDP-43 aggregate RNA aptamer anchored to outer membrane via streptavidin-biotin linkage
How will you measure the function of your system?
Incubate SMCs with recombinant TDP-43 aggregates in vitro and measure peptide release via ELISA or fluorescently tagged peptide fluorescence
Use a ThT (Thioflavin T) aggregation assay to confirm that released peptide reduces TDP-43 fibril formation compared to controls without SMC
Confirm pore formation by aHL via dye leakage assay (encapsulate fluorescent dye, measure release upon TDP-43 addition)
Homework question from Peter Nguyen
One-sentence pitch: A living wall coating embedded with freeze-dried cell-free biosensors that detects black mold (Stachybotrys chartarum) VOC emissions and produces a visible color change before mold becomes visible to the naked eye.
How will the idea work?
Black mold (Stachybotrys chartarum) releases characteristic volatile organic compounds (VOCs) during early colonization — most notably 1-octen-3-ol — before any visible growth appears. The proposed system embeds freeze-dried cell-free Tx/Tl reactions into a breathable polymer wall coating (e.g., a porous silicone or hydrogel matrix). When ambient humidity reactivates the freeze-dried system and 1-octen-3-ol diffuses into the coating, it binds to an engineered transcription factor (based on a modified OBP — odorant binding protein) that activates a chromoprotein reporter gene. The wall visibly changes color (e.g., from clear to deep violet using the chromoprotein amilCP) in the region of mold colonization, providing a spatially precise early warning signal. No electronics, power, or human monitoring are required — the building itself becomes the sensor.
What societal challenge or market need does this address?
Black mold is a major public health hazard linked to respiratory illness, neurological symptoms, and immune disorders, particularly in children and immunocompromised individuals. Current detection relies on visible inspection or expensive air quality testing, by which point mold is already well-established and remediation is costly. An estimated 50% of buildings in developed countries have some form of problematic moisture/mold. An early, passive, low-cost detection system embedded directly into building materials would allow intervention before health impacts occur, reducing both healthcare costs and remediation expenses.
How do you envision addressing the limitations of cell-free reactions?
Activation with water: The freeze-dried system is formulated in a hygroscopic hydrogel matrix that reactivates only when local humidity exceeds the threshold typical of mold-favorable conditions (>70% RH), creating a built-in environmental trigger
Stability: Freeze-drying with trehalose as a cryoprotectant extends shelf life to 1–2 years at room temperature; the wall coating can be replaced as a panel every 2 years as part of standard building maintenance
One-time use: This is reframed as a feature — once the color change occurs, it serves as a permanent record of mold detection in that location, and the panel is replaced. Multiple overlapping panels can be layered to provide repeated sensing capacity over the building lifetime
Homework question from Ally Huang
Background (≤100 words)
Long-duration spaceflight profoundly suppresses astronaut immune function. A well-documented consequence is the reactivation of latent herpesviruses — including Epstein-Barr virus (EBV) and Varicella-Zoster virus (VZV) — which remain dormant in healthy individuals but reactivate under the immune dysregulation caused by microgravity, radiation, and psychological stress. Herpesvirus reactivation has been detected in over 50% of astronauts on ISS missions and poses risks ranging from mild illness to serious neurological complications on long-duration missions to the Moon or Mars, where return to Earth is not possible.
Molecular/Genetic Target (≤30 words)
EBV immediate-early gene BZLF1 (also called Zta/ZEBRA) — its expression is the molecular switch that triggers EBV reactivation from latency and is detectable in saliva.
How does the target relate to the challenge? (≤100 words)
BZLF1 mRNA expression is the earliest detectable signal of EBV reactivation — preceding viral shedding and any clinical symptoms by days. Detecting BZLF1 transcripts in astronaut saliva samples using a cell-free toehold switch biosensor would provide real-time, equipment-minimal immune status monitoring. Since EBV reactivation is directly driven by the cortisol-mediated immune suppression characteristic of spaceflight stress, BZLF1 acts as a functional readout of overall immune dysregulation, not just viral status — making it a highly informative single-target proxy for astronaut immune health.
Hypothesis/Research Goal (≤150 words)
Hypothesis: BZLF1 mRNA will be detectable in astronaut saliva samples collected during ISS missions using a freeze-dried BioBits® toehold switch biosensor, and its expression will correlate with mission duration and radiation exposure levels, providing a quantitative real-time index of immune suppression severity.
Rationale: Toehold switches are synthetic RNA regulators that activate cell-free reporter gene expression only when a specific target RNA sequence is present. By designing a toehold switch complementary to the BZLF1 mRNA sequence and freeze-drying it with a cell-free GFP reporter into BioBits® pellets, we can create a single-use, room-temperature-stable diagnostic that requires only saliva addition and the P51 fluorescence viewer to produce a quantitative readout. This requires no specialized laboratory equipment, making it fully compatible with ISS constraints and scalable to future deep space missions.
Experimental Plan (≤100 words)
Samples: Astronaut saliva collected at mission days 0, 30, 60, 90, and 180. Controls: BZLF1-positive saliva (EBV-reactivating donor, ground), BZLF1-negative saliva (healthy seronegative donor, ground), and a no-template BioBits® pellet.
Procedure: Add 2 µL saliva to rehydrated BioBits® toehold switch pellet. Incubate 37°C for 2 hours. Read GFP fluorescence using the P51 Molecular Fluorescence Viewer.
Data collected: GFP intensity (proxy for BZLF1 mRNA concentration), correlated with mission day, cumulative radiation dose (from personal dosimeters), and cortisol levels (parallel measurement). Statistical correlation analysis performed on ground post-mission.
Week 10 HW: Imagining and Measurement
For final project
In this project, I will measure several aspects of the DNA sensing system, including sequence correctness, predicted folding behavior, target response, orthogonality, and signal output. The most important biological measurements are whether the histamine and IgE circuits are correctly designed and whether they respond only to their intended targets. I will also measure the strength of the output signal after target binding, since the goal is to convert molecular recognition into a detectable readout. In addition, I will look at background activity and nonspecific activation to estimate how cleanly the system distinguishes true signal from noise. These measurements will help determine whether the platform is suitable for future wearable use.
I will begin by comparing the designed DNA sequences to the intended circuit architecture to make sure the correct aptamer, toehold, and trigger regions are present. Next, I will use secondary-structure prediction to check whether each circuit forms the expected hairpin and whether the toehold remains accessible for switching. To experimentally test conformational change, I would use native PAGE gel electrophoresis, which can reveal mobility shifts when the DNA switches structure or binds its trigger. If the circuit is extended to a functional reporter stage, I would also use a cell-free assay to measure whether target binding leads to a detectable output. Finally, I would compare matched, mismatched, and no-target controls to quantify specificity and background signal.
The main technologies in this project are computational DNA design tools, secondary-structure prediction software, native PAGE gel electrophoresis, and electrochemical sensing methods. Benchling or a similar platform will be used to organize and annotate the DNA circuits, while NUPACK or Mfold will help predict folding and accessibility. Native PAGE will be used to observe structural changes in the DNA constructs, and electrochemical readouts such as impedance or current measurements will be used for the wearable sensing concept. If needed, a cell-free expression system can provide an intermediate functional readout before moving to the wearable electrode platform. Together, these technologies allow both design validation and functional testing of the sensing system.
Waters Part I — Molecular Weight
For the following calculations, I will be using the provided eGFP sequence:
Using the online ExPASy Compute pI/Mw tool, the baseline expected molecular weight of this unmodified amino acid sequence was found to be
28006.60 g/mol
(Theoretical pI/Mw: 5.90 / 28006.60). Note that the His-tag (HHHHHH) and LE linker are included in this sequence and therefore in the calculated weight.
To calculate the experimental molecular weight of eGFP using the intact LC-MS data from Figure 1, I selected an adjacent pair of consecutive charge state peaks:
The accuracy of 0.0809% demonstrates that the difference between the experimental mass and theoretical mass is extremely small, indicating high accuracy in peak selection.
The charge state can also be observed directly from the zoomed-in peak structure. When zooming tightly into the mass spectrum for intact eGFP, individual isotopic lines become visible inside the peak envelope. This occurs because the protein carries multiple protonation states, producing a charge-state distribution where isotopic fine structure can be resolved.
Waters Part II — Secondary/Tertiary Structure
Q1: Native vs. Denatured Protein Conformations
The native state of a protein is its thermodynamically stable, biologically active three-dimensional conformation, maintained by non-covalent interactions including hydrogen bonding, hydrophobic interactions, and electrostatic forces.
When a protein unfolds (denatures), environmental stressors such as organic solvents (e.g., acetonitrile) or acidic conditions (e.g., formic acid) disrupt these interactions. The ordered secondary and tertiary structure collapses into a disordered polypeptide chain, while the primary amino acid sequence remains intact.
A mass spectrometer detects this through changes in the charge state distribution (CSD). In the native state, most protonatable residues (Lys, Arg, His) are buried inside the folded protein, so only a few are available for protonation. This produces a narrow distribution of low charge states at high m/z values (typically above 2500 m/z), as seen in the bottom spectrum of Figure 2.
In the denatured state, unfolding exposes all protonatable sites to solvent, generating a broad distribution of high charge states shifted toward lower m/z values (500–1500 m/z), as seen in the top spectrum of Figure 2.
Q2: Charge State of the ~2800 m/z Peak (Figure 3)
Yes, the charge state can be determined from the zoomed-in inset in Figure 3. Isotopic peaks within a single charge envelope are separated by 1 Da / z, so measuring the spacing between adjacent isotopic lines gives z directly:
z = 1 / Δ(m/z)
From the inset, the isotopic spacing is approximately 0.09 m/z, giving:
z = 1 / 0.09 ≈ 11
The peak at ~2800 m/z therefore carries a charge state of +11, consistent with the compact, tightly folded native conformation producing low charge states at high m/z.
Waters Part III — Peptide Mapping
1. Residue Quantification and Sequence Highlight
Trypsin cleaves peptide bonds at the C-terminal side of Lysine (K) and Arginine (R) residues unless followed by Proline (P).
After counting through the full 246-amino-acid eGFP sequence, there are:
Using the ExPASy PeptideMass tool with the parameters shown in Figure 4 (Trypsin, monoisotopic, [M+H]+, 0 missed cleavages, mass range 500–unlimited Da), a total of 19 theoretical peptides were predicted. Although there are 26 cleavage sites, some adjacent cleavage sites produce very small peptides that fall below the 500 Da mass filter, and the His-tag C-terminal segment does not end in K or R, reducing the reported count to 19.
3. LC-MS Chromatographic Peak Count
From the TIC chromatogram in Figure 5a, the tallest peak is at 4.87 minutes with approximately 1.2 × 107 counts. Counting all peaks above the 10% relative abundance threshold between 0.5 and 6.0 minutes gives 21 chromatographic peaks.
4. Comparison: Predicted vs. Observed Peaks
Predicted peptides (theory): 19
Observed chromatographic peaks (experiment): 21
There are more observed peaks than predicted. This can be explained by several factors:
Missed Cleavages
Trypsin does not always cleave at every K/R site with 100% efficiency. Peptides with one or more missed cleavages will appear as additional, larger peaks in the chromatogram.
Non-specific Cleavage
Trypsin may occasionally cleave at non-K/R sites, generating unexpected peptide fragments not predicted by the in silico digest.
Protein Modifications
Post-translational or chemical modifications (e.g., oxidized methionines) can shift peptide masses and cause modified and unmodified forms of the same peptide to appear as separate peaks.
5. Charge State and Mass of the Peak at 2.78 min (Figure 5b)
A. Charge State Determination
From the zoomed inset in Figure 5b, the two most abundant isotopic peaks are:
m/z1 = 525.76712
m/z2 = 526.25918
Isotopic spacing:
Δ(m/z) = 526.25918 − 525.76712 = 0.49206
Charge state:
z = 1 / 0.49206 = 2.032 ≈ 2
The dominant peptide ion carries a charge state of +2.
B. [M+H]+ Mass
Using the relationship [M+H]+ = (m/z × z) − H, with z = 2 and H = 1.00727 Da:
Matching the experimental [M+H]+ of 1050.527 Da to the PeptideMass theoretical output for eGFP, the closest peptide is FEGDTLVNR, with a theoretical monoisotopic mass of 1050.5214 Da.
This is excellent mass accuracy, consistent with the high-resolution Waters BioAccord QToF instrument.
7. Sequence Coverage
As shown in Figure 6, the amino acid coverage map reports 88% sequence coverage of eGFP based on the peptides positively identified by their calculated mass and fragmentation pattern.
8. Peptide Sequence from Fragmentation Spectrum (Bonus)
The peptide eluting at 2.78 minutes has a [M+H]+ of 1050.527 Da, which matches FEGDTLVNR (theoretical 1050.5214 Da) from the PeptideMass output. To confirm, the fragmentation spectrum in Figure 5c was compared to the predicted b/y ion series for FEGDTLVNR using the FragIon tool. The observed fragment masses are consistent with the expected y-ion series for this peptide sequence.
9. Does the Peptide Map Data Confirm eGFP? (Bonus)
Yes, the data strongly supports that the sample is eGFP. The peptide map shows 88% amino acid sequence coverage (Figure 6), meaning the large majority of the protein's primary structure was directly confirmed by mass and fragmentation matching. The observed peptide masses align with the theoretical tryptic digest of eGFP within 5.30 ppm accuracy, and the fragmentation spectrum of the 2.78-minute peak matches the expected ions for the eGFP peptide FEGDTLVNR. Together, these results provide high confidence that the protein standard is authentic eGFP.
Waters Part IV — Oligomers
Theoretical Mass Calculations
To identify each oligomeric assembly of KLH on the CDMS spectrum, the expected total mass was calculated by multiplying the subunit mass by the number of subunits in each complex:
Comparing the calculated masses to the labeled peaks in the CDMS spectrum:
Species
Theoretical Mass
Observed Peak (MDa)
Notes
7FU Decamer
3.40 MDa
3.4 MDa
Exact match
8FU Didecamer
8.00 MDa
8.33 MDa
Most abundant peak; highest intensity in spectrum
8FU 3-Decamer
12.00 MDa
12.67 MDa
Clearly resolved peak
8FU 4-Decamer
16.00 MDa
~16.0 MDa
Low intensity; visible as small broad peak in blue trace
All four oligomeric species are detectable in the spectrum. The 8FU Didecamer at 8.33 MDa is the dominant species. The observed masses for the 8FU assemblies are slightly higher than theoretical (~4–5% offset), which is expected for CDMS measurements of large MDa-scale complexes where instrument calibration and space-charge effects introduce mass shifts.
Waters Part V — Did I Make GFP?
The table below summarizes the theoretical and observed molecular weights for intact eGFP, using the data from the provided figure screenshots (lab work at Waters was not performed in person).
Theoretical
Observed / Measured on the Intact LC-MS
PPM Mass Error
Molecular weight (kDa)
28,006.60 Da (28.007 kDa)
27,983.93 Da (27.984 kDa)
809.45 ppm
The theoretical MW (28,006.60 Da) was obtained from ExPASy Compute pI/Mw using the full eGFP sequence including the His-tag. The observed MW (27,983.93 Da) was deconvoluted from the intact LC-MS spectrum in Figure 1 using the adjacent charge state method (z = 31, m/z = 903.7148). The mass error of 809.45 ppm reflects the difference between the sequence-predicted mass and the experimentally measured denatured protein mass, which is consistent with the resolution of the LC-MS system used.
Week 11 HW: Bioproduction & Cloud Labs
Part A
Unfortunately, I did not have the opportunity to contribute to the project before the deadline ended. However, for next semester, I think it would be a good idea to create several variations of the same artwork using different color palettes or design concepts. I noticed that many people were unsure about what exact pattern or style they were supposed to contribute, while others had their own creative ideas that did not fully match the overall design. Because everyone has different artistic preferences and interpretations, it could be helpful to divide the project into multiple themed sections or versions. This would make the collaboration process more flexible, reduce confusion, and allow more students to express their creativity in their own way.
E. coli Lysate / BL21 (DE3) Star Lysate: Provides the essential molecular machinery, such as ribosomes and translation factors, required to synthesize proteins from an RNA template. The inclusion of T7 RNA Polymerase specifically enables high-level, targeted transcription of genes cloned under a T7 promoter.
Potassium Glutamate: Serves as the primary source of potassium ions to maintain correct intracellular ionic strength and supports optimal ribosome stability during translation. The glutamate anion acts as a compatible solute that mimics the physiological conditions of living bacterial cells.
HEPES-KOH pH 7.5: Functions as a chemical buffer to maintain a stable, optimal pH environment for enzymatic activity throughout the course of the prolonged incubation. It resists pH fluctuations that can occur as metabolic byproducts accumulate in the reaction.
Magnesium Glutamate: Supplies essential magnesium ions (Mg2+) which act as mandatory cofactors for the structural stability of ribosomes and the proper catalytic function of polymerases. Precise concentration management is critical, as magnesium directly influences translation accuracy and efficiency.
Potassium phosphate monobasic / dibasic: Forms a secondary buffering system that stabilizes pH while simultaneously providing inorganic phosphate ions (Pi). This phosphate source is crucial for driving the enzymatic recycling and phosphorylation of nucleotides into energy-rich forms.
Ribose: Functions as a stable carbohydrate precursor that is enzymatically processed within the reaction to synthesize the sugar backbones of nucleosides. This enables the sustainable, long-term generation of nucleotides over extended incubation periods.
Glucose: Serves as a primary metabolic energy source that undergoes catabolism to generate adenosine triphosphate (ATP) through glycolysis-like pathways. This continuous energy generation sustains the metabolic demands of transcription and translation over many hours.
AMP / CMP / GMP / UMP: Represent the nucleoside monophosphates (NMPs) that serve as basic building blocks for the reaction. They are dynamically phosphorylated into nucleoside triphosphates (NTPs) to power both transcription and ongoing energy-consuming translation steps.
Guanine: Acts as a purine base precursor that can be salvaged by the bacterial enzymes in the lysate to supplement the nucleotide pool. This ensures that guanosine-based energy intermediates (GTP) remain sufficient for the protein synthesis elongation steps.
17 Amino Acid Mix: Supplies the fundamental monomeric building blocks necessary for assembling the primary peptide chains during protein translation. This core mix lacks certain low-solubility or sensitive amino acids that must be prepared and adjusted independently.
Tyrosine: An aromatic amino acid added separately due to its poor solubility at neutral pH, which requires precise preparation (often at pH 12) to ensure adequate concentration in the final master mix. It is essential for incorporating tyrosine residues into the nascent protein chain.
Cysteine: A sulfur-containing amino acid added independently because it is highly prone to oxidation and degradation when stored in complex mixtures. It is vital for the formation of disulfide bonds and maintaining proper tertiary protein structures.
Nicotinamide: Acts as a stabilizing additive and precursor for pyridine nucleotides like NAD+, supporting the active metabolic pathways within the lysate. It helps maintain the redox balance required for sustained, long-term enzymatic energy regeneration.
Nuclease Free Water: Used to backfill the reaction to its final volume, ensuring that all chemical reagents are precisely diluted to their intended target concentrations. It is strictly purified to remove any degrading nucleases that could destroy the DNA template or RNA transcripts.
Key Differences: 1-Hour vs. 20-Hour Master Mixes
The 1-hour optimized master mix relies on pre-supplied nucleoside triphosphates (NTPs) and phosphoenolpyruvate (PEP) to deliver immediate, high-burst energy for rapid transcription and translation. Conversely, the 20-hour system utilizes low-cost, stable precursors—specifically nucleoside monophosphates (NMPs), ribose, and glucose—which are gradually converted into functional energy molecules by the lysate’s internal metabolic enzymes. This structural shift prevents early energy exhaustion and byproduct inhibition, allowing for a highly sustainable, cost-effective, and prolonged protein production window.
E. coli Lysate / BL21 (DE3) Star Lysate: Provides the essential molecular machinery, such as ribosomes and translation factors, required to synthesize proteins from an RNA template. The inclusion of T7 RNA Polymerase specifically enables high-level, targeted transcription of genes cloned under a T7 promoter.
Potassium Glutamate: Serves as the primary source of potassium ions to maintain correct intracellular ionic strength and supports optimal ribosome stability during translation. The glutamate anion acts as a compatible solute that mimics the physiological conditions of living bacterial cells.
HEPES-KOH pH 7.5: Functions as a chemical buffer to maintain a stable, optimal pH environment for enzymatic activity throughout the course of the prolonged incubation. It resists pH fluctuations that can occur as metabolic byproducts accumulate in the reaction.
Magnesium Glutamate: Supplies essential magnesium ions (Mg2+) which act as mandatory cofactors for the structural stability of ribosomes and the proper catalytic function of polymerases. Precise concentration management is critical, as magnesium directly influences translation accuracy and efficiency.
Potassium phosphate monobasic / dibasic: Forms a secondary buffering system that stabilizes pH while simultaneously providing inorganic phosphate ions (Pi). This phosphate source is crucial for driving the enzymatic recycling and phosphorylation of nucleotides into energy-rich forms.
Ribose: Functions as a stable carbohydrate precursor that is enzymatically processed within the reaction to synthesize the sugar backbones of nucleosides. This enables the sustainable, long-term generation of nucleotides over extended incubation periods.
Glucose: Serves as a primary metabolic energy source that undergoes catabolism to generate adenosine triphosphate (ATP) through glycolysis-like pathways. This continuous energy generation sustains the metabolic demands of transcription and translation over many hours.
AMP / CMP / GMP / UMP: Represent the nucleoside monophosphates (NMPs) that serve as basic building blocks for the reaction. They are dynamically phosphorylated into nucleoside triphosphates (NTPs) to power both transcription and ongoing energy-consuming translation steps.
Guanine: Acts as a purine base precursor that can be salvaged by the bacterial enzymes in the lysate to supplement the nucleotide pool. This ensures that guanosine-based energy intermediates (GTP) remain sufficient for the protein synthesis elongation steps.
17 Amino Acid Mix: Supplies the fundamental monomeric building blocks necessary for assembling the primary peptide chains during protein translation. This core mix lacks certain low-solubility or sensitive amino acids that must be prepared and adjusted independently.
Tyrosine: An aromatic amino acid added separately due to its poor solubility at neutral pH, which requires precise preparation (often at pH 12) to ensure adequate concentration in the final master mix. It is essential for incorporating tyrosine residues into the nascent protein chain.
Cysteine: A sulfur-containing amino acid added independently because it is highly prone to oxidation and degradation when stored in complex mixtures. It is vital for the formation of disulfide bonds and maintaining proper tertiary protein structures.
Nicotinamide: Acts as a stabilizing additive and precursor for pyridine nucleotides like NAD+, supporting the active metabolic pathways within the lysate. It helps maintain the redox balance required for sustained, long-term enzymatic energy regeneration.
Nuclease Free Water: Used to backfill the reaction to its final volume, ensuring that all chemical reagents are precisely diluted to their intended target concentrations. It is strictly purified to remove any degrading nucleases that could destroy the DNA template or RNA transcripts.
Key Differences: 1-Hour vs. 20-Hour Master Mixes
The 1-hour optimized master mix relies on pre-supplied nucleoside triphosphates (NTPs) and phosphoenolpyruvate (PEP) to deliver immediate, high-burst energy for rapid transcription and translation. Conversely, the 20-hour system utilizes low-cost, stable precursors—specifically nucleoside monophosphates (NMPs), ribose, and glucose—which are gradually converted into functional energy molecules by the lysate’s internal metabolic enzymes. This structural shift prevents early energy exhaustion and byproduct inhibition, allowing for a highly sustainable, cost-effective, and prolonged protein production window.
Part C
Fluorescent Protein Properties in Cell-Free Systems
sfGFP (Superfolder GFP): sfGFP is engineered to fold very efficiently, even when the environment is a bit stressful or crowded. This means most of the protein that is translated in a cell‑free lysate becomes properly folded and fluorescent very quickly, so signal builds up fast.
mRFP1: mRFP1 has slower and less efficient maturation than newer red FPs, so freshly made protein often sits in non‑ or weakly fluorescent intermediate states for a while. As a result, early time‑points in a cell‑free experiment can show relatively low red signal even when translation itself is strong
mKO2: mKO2 depends on oxygen to complete its chromophore maturation, so its fluorescence strongly reflects how much O₂ is available during the reaction. In sealed or poorly mixed plates, oxygen can become limiting over time, and the orange signal will level off or stay low even if more protein continues to be produced.
mTurquoise2: mTurquoise2 is a very bright cyan protein with high quantum yield, so each correctly folded molecule gives a strong signal. However, its folding is somewhat demanding, so if the lysate has limited chaperone activity or suboptimal conditions, a noticeable fraction of the translated protein may misfold and never become fluorescent.
mScarlet_I: mScarlet_I combines high brightness with relatively fast maturation for a red FP, which makes it good for time‑course measurements in cell‑free systems. Its fluorescence, however, drops when the reaction becomes more acidic, so pH drift during long incubations can reduce the apparent signal even without changes in expression
Electra2: Electra2 matures very quickly, so blue fluorescence appears early and can track expression dynamics on short timescales. Over long experiments with repeated plate‑reader scans, its moderate photostability means the signal can fade due to photobleaching, making late‑time measurements underestimate the amount of protein present
Optimization Hypothesis for a 36-Hour Incubation
Proteins: mScarlet_I & mKO2
Reagents to Adjust: HEPES-KOH (Buffer) and oxygen
Hypothesis: Increasing the concentration of HEPES-KOH while supplementing the system with an active oxygenation will maximize the long-term fluorescence readout of mScarlet_I and mKO2 over a 36-hour incubation.
Expected Effect: A stronger buffer should reduce pH drops caused by metabolism in the lysate, and because mScarlet_I is sensitive to acidic conditions, this should help maintain a brighter and more consistent signal over time.
Wearable Electrochemical Immunosensor for Dual Allergy Detection Abstract Allergic diseases affect over 500 million people worldwide and represent a critical public health challenge. Current allergy diagnostics rely on laboratory-based immunoassays or skin-prick tests that require physical visits to the clinic and specialized equipment, without existence of real-time data during acute exposure events. This project addresses the need by creating a minimally invasive wearable device that simultanesly monitros both histamine, the trigger, and immunoglobin (IgE), antibody for allergic sensation, in real time. The plan is to create two independent, computaitonally designed DNA toehold switch circuits where one switch is triggered by histamine-bound aptamer output and one by IgE-bound aptamer output. The central hypothesis is that two switched can undergoe confromaitonal changes upon target engagement and produce a deetctable signal in a cell-free system.
Subsections of Projects
Group Final Project
Individual Final Project
Wearable Electrochemical Immunosensor for Dual Allergy Detection
Abstract
Allergic diseases affect over 500 million people worldwide and represent a critical public health challenge. Current allergy diagnostics rely on laboratory-based immunoassays or skin-prick tests that require physical visits to the clinic and specialized equipment, without existence of real-time data during acute exposure events.
This project addresses the need by creating a minimally invasive wearable device that simultanesly monitros both histamine, the trigger, and immunoglobin (IgE), antibody for allergic sensation, in real time.
The plan is to create two independent, computaitonally designed DNA toehold switch circuits where one switch is triggered by histamine-bound aptamer output and one by IgE-bound aptamer output. The central hypothesis is that two switched can undergoe confromaitonal changes upon target engagement and produce a deetctable signal in a cell-free system.
To test this hypothesis, this project will pursue the next three aims:
Computational aptamer construct design and scoring
Cell-free electrochemical validation
Integration of the validated sensing elements into a flexible wearable platform with multiplexed readout capability
Project Aims
Aim 1: Computational Design
The first aim of my project is to computationally design and score DNA aptamer toehold switch circuits - one for histamine detection and one for IgE detection. Each circuit concsisit of: validated seqeunce, toehold switch hairpin and a trigger strand.
Aim 2: cell-free electrochemical validation of aptamer-target binding
Aim 2 would be ordering Tiwst designs and validating each circuit independently using native PAGE gel shift assays and, if possible, a cell-free transcription/translation (TX-TL) reporter system. Each aptamer-toehold circuit will be incubated with its target (histamine or recombinant IgE), and band shifts will confirm conformational switching. The two circuits will then be tested together to confirm orthogonality — that each responds only to its own target.
Aim 3: integration of the validated sensing elements into a flexible wearable platform with multiplexed readout capability
The long-term vision of this aim is to develop a multiplexed wearable patch that embeds both aptamer-toehold circuits within an electrochemical transduction layer and samples interstitial fluid through microneedles. Each circuit would produce a measurable impedance or current change upon target binding, enabling continuous, simultaneous monitoring of both allergy markers. In the broader context, this modular framework could be adapted to other biomarker pairs with known aptamers, including cytokines, infectious disease markers, or performance-related biomarkers.
Current microneedle-based sensing platforms show that minimally invasive access to interstitial fluid is feasible, but most systems remain limited to single-analyte detection or fixed recognition chemistries. By contrast, this project would expand the concept to establish a modular multiplexed wearable strategy for real-time monitoring of multiple allergy-related biomarkers, offering a more dynamic view of immune activation than traditional clinic-based tests.
A major barrier is biological noise, including biofouling, nonspecific adsorption, and interference from complex interstitial fluid components. These factors can reduce specificity, mask low-abundance targets, and undermine long-term sensing performance in real-world conditions.
To address this challenge, I would integrate the validated aptamer-toehold sensing elements into a flexible microneedle-electrode patch and test whether each module can generate a distinct electrochemical signal without cross-talk. If successful, this approach could shift allergy testing from infrequent clinical measurements toward continuous, personalized, at-home monitoring during natural exposure events.
Background
I was refering to multiple supproting studies and would like two highlight next two peer-reviewed researches that are relevant to my project:
Aptamer biosensor for label-free detection of human IgE
This paper shows that a DNA aptamer can be used as a label-free recognition element for human IgE, allowing allergy-related detection without antibody labeling. The study demonstrates real-time IgE sensing with good sensitivity and selectivity, which makes it a strong precedent for your planned IgE module in a wearable allergy-monitoring system.
Microneedle Aptamer-Based Sensors for Continuous, Real-Time Therapeutic Monitoring
This paper demonstrates that aptamer-based electrochemical sensors can be integrated into microneedle arrays to monitor molecules continuously in interstitial fluid. It is especially relevant to your project because it supports the idea that aptamer recognition can be translated into a minimally invasive, wearable platform with real-time and multiplexed readout potential.
This project is novel in three persepctives.
First, it applies toehold switch logic to DNA aptamer-based detection of protein and small-molecule targets, expanding synthetic biology tools into a new biosensing context. Second, it combines two orthogonal aptamer-toehold circuits for simultaneous histamine and IgE detection, enabling a dual-marker allergy sensor that has not yet been demonstrated. Third, it introduces an electrochemical signal readout for aptamer-toehold switching, creating a more practical and wearable-compatible output than the fluorescence or reporter-based systems commonly used in cell-free sensing.
The project addresses a major real-world problem: allergic diseases affect hundreds of millions of people worldwide, and severe reactions such as anaphylaxis can develop within seconds and become life-threatening. Current diagnostic tools are typically clinic-based and do not provide real-time information during active exposure, which limits their usefulness for prevention and rapid response. By simultaneously tracking histamine, the immediate chemical trigger, and IgE, the immune marker associated with allergic sensitivity, this platform could provide a more complete picture of allergic state than single-analyte approaches. The cell-free design also improves stability, manufacturability, and accessibility, making the system more practical for low-resource or decentralized settings. If successful, this framework could shift allergy monitoring from intermittent testing toward continuous, personalized sensing, and its modular logic could later be extended to other biomarkers beyond allergy.
This project raises several ethical considerations related to beneficence, non-maleficence, and justice. From a beneficence perspective, earlier and more precise detection of allergic responses could improve patient safety and support better clinical decision-making. At the same time, non-maleficence is important because false-positive or inaccurate signals could cause unnecessary anxiety, inappropriate medication use, or delayed trust in the device. Justice is also relevant because one goal of the platform is to create a low-cost and scalable sensing technology that could be accessible beyond well-resourced clinical environments. In addition, continuous wearable monitoring raises concerns about privacy, data ownership, and possible misuse of sensitive immunological information.
To ensure the project is ethical, the research should be conducted with careful validation, transparent reporting, and open sharing of design logic where appropriate. Any future human-subject or clinical use would require informed consent, institutional review board approval, and clear policies for data security and access. One potential unintended consequence is that users may overinterpret imperfect sensor readings, so the system should be framed as a supportive monitoring tool rather than a standalone diagnostic replacement. There are also scientific uncertainties, including whether computationally predicted binding behavior will hold under real biological conditions and whether the two circuits will remain orthogonal in complex samples. If cross-reactivity or instability occurs, alternative strategies could include redesigning the toehold regions, introducing stronger antifouling interfaces, or using a different sensing architecture such as antibody-based or protein-based recognition elements.
SECTION 4: Experimental Design, Techniques, Tools and Technology
Step 1Literature review & Seqeunce Collection identify validated histamine-binding and IgE-binding aptamer sequences from NCBI/literature. Collect information on sequence length, reported affinity (Kd), secondary structure, and experimental conditions used for validation.
Timeline ~ Day 1-3
Step 2
Import the selected histamine aptamer into Benchling and design the first toehold switch hairpin. Add a 5’ toehold region of approximately 7–10 nucleotides that remains single-stranded and is complementary to the trigger strand. Annotate all functional domains, including the aptamer stem, toehold, and trigger-binding region.
Timeline ~ Day 4-5
Step 3
Run the full Circuit 1 sequence through NUPACK or Mfold at 37°C and physiological ionic conditions. Confirm that the aptamer domain forms the expected stem-loop structure and that the toehold region remains accessible and unpaired.
Timeline ~ 6
Step 4
Design the Circuit 1 trigger strand to be complementary to the toehold and adjacent stem region, with a target length of approximately 20–25 nucleotides. Model strand displacement in NUPACK or Mfold by simulating the switch and trigger as interacting strands. Confirm that the hairpin opens upon trigger binding.
Timeline ~ 7
Step 5
Repeat the design workflow for the IgE-binding aptamer to construct Circuit 2. Use a different toehold sequence from Circuit 1 to reduce cross-reactivity and improve orthogonality. Annotate all sequence domains in Benchling.
Timeline ~ 8-9
Step 6
Analyze the full Circuit 2 sequence in NUPACK or Mfold under the same conditions used for Circuit 1. Verify correct folding of the aptamer stem-loop and accessibility of the toehold region.
Timeline ~ 10
Step 7
Design the Circuit 2 trigger strand and simulate strand displacement. Then test orthogonality computationally by pairing Circuit 1 with the Circuit 2 trigger and vice versa. Confirm that each switch responds preferentially to its own trigger and shows minimal cross-binding.
Timeline ~ 11
Step 8
Generate a script that reads NUPACK output files from both circuits and generates a ranked CSV table. The output should include sequence name, minimum free energy ΔG, GC content, toehold accessibility score, and orthogonality flag. This analysis will provide a standardized way to compare candidates across design variants.
Timeline ~ 12-13
Step 9
Generate three variants of each circuit by varying toehold length, such as 7, 9, and 11 nucleotides. Run the ranking script on each version and select the best-performing variant based on high toehold accessibility and the most favorable ΔG.
Timeline ~ 14-15
Step 10
Submit the final designs for synthesis through a commercial DNA synthesis provider such as Twist or a comparable vendor. Order the Circuit 1 switch, Circuit 1 trigger, Circuit 2 switch, Circuit 2 trigger, and scrambled control oligos for each circuit.
Timeline ~ 16
Step 11
Prepare a native PAGE workflow to test each switch strand alone and in combination with its trigger and target aptamer. Incubate the samples in binding buffer, then run them on a 12% native gel. A shift in band mobility is expected when the switch adopts an opened conformation after target or trigger engagement.
Timeline ~ 17
Step 12
Test each circuit with matched and mismatched triggers, scrambled oligos, and no-target controls. Compare band patterns to determine whether each switch is selective for its intended input.
Timeline ~ 18-19
Step 13
If the gel shifts are successful, move to a cell-free transcription/translation assay using a fluorescence or colorimetric reporter downstream of the switch output. Measure reporter activation in the presence of histamine or IgE and compare signal intensity across controls.
Timeline ~ 20-21
Step 14
Integrate the validated sensing elements into a simple electrochemical setup and measure impedance or current changes after target binding. Compare signal before and after activation to determine whether the switch can support wearable-compatible transduction.
Timeline ~ 22-23
Step 15
Compile all computational and experimental results into a final comparison of performance across circuit variants. Evaluate stability, accessibility, specificity, and signal output to determine whether the central hypothesis is supported.
Timeline ~ 24-29
Techniques relevant to my project
• Bioethical Considerations ✓
• DNA Construct Design ✓
• Designing a Twist Order ✓
• Gel Electrophoresis ✓
• Databases ✓
• Designing a Twist Order ✓
• Models and Notebooks ✓
• Cell-Free Reactions ✓
1. DNA Construct Design (Benchling): Each aptamer-toehold circuit will be designed as a single linear DNA strand in Benchling, with distinct annotated features: aptamer binding domain, stem region, loop, and toehold overhang. Benchling’s sequence editor enables visualization of domain boundaries, automatic Tm calculation for the toehold region, and export in formats compatible with NUPACK and Twist. Two separate constructs (Circuit 1: histamine; Circuit 2: IgE) will be designed in parallel, with the toehold sequences chosen to have <30% complementarity to each other to minimize cross-reactivity. The trigger strands will be designed as separate oligos in the same Benchling project and linked as a ‘part’ for version control.
2. Computaitonal Modeling: Mfold will be used to predict the thermodynamic ensemble of secondary structures for each switch strand at 37°C in physiological salt conditions (150 mM NaCl, 0 mM Mg2+), using both MFE analysis and partition function calculation. The partition function output provides per-base pairing probabilities, which will be parsed by the BioPython script to quantify toehold accessibility as the fraction of toehold nucleotides with pairing probability <0.1 (i.e., reliably single-stranded). Strand displacement will be modeled by inputting switch + trigger as a two-strand complex and verifying that the lowest free energy state corresponds to the open (trigger-bound) conformation. This automated pipeline will screen multiple toehold length variants and output a ranked CSV for construct selection.
SECTION 5: Results & Quantative Expectations
I chose to validate the DNA design logic for Aim 1, specifically the computational design of the histamine- and IgE-responsive aptamer-toehold switch circuits. This validation focused on whether the designed sequences formed the expected secondary structures and whether the toehold regions remained accessible for strand displacement. Because this project is still in the design stage, validating the computational construct design was the most feasible first step.
Detailed protocol
I collected validated histamine-binding and IgE-binding aptamer sequences from the literature and selected candidate sequences with reported target specificity.
I designed two independent DNA circuits in Benchling, each containing an aptamer-responsive switch region and a unique trigger-binding toehold sequence.
I exported the full sequences and analyzed them using NUPACK and/or Mfold under physiologically relevant conditions.
For each construct, I checked whether the predicted secondary structure formed the intended hairpin and whether the toehold region remained unpaired.
I then compared multiple design variants by adjusting toehold length and sequence composition to identify the most stable and accessible version.
Finally, I ranked the constructs based on predicted minimum free energy, toehold accessibility, and orthogonality between the two circuits.
The main synthetic biology techniques used in this validation were DNA circuit design, aptamer-based biosensor engineering, and computational secondary structure modeling. I also used sequence annotation in Benchling to organize functional domains such as the aptamer stem, toehold region, and trigger-binding sequence. In addition, I applied strand-displacement design principles, which are central to synthetic gene regulation and nucleic acid circuit behavior. This validation also involved rational design optimization, since I compared multiple sequence variants before selecting the best candidate. Together, these techniques allowed me to test whether the proposed sensing architecture was structurally plausible before experimental implementation.
I generated a comparison table of candidate designs using sequence features such as minimum free energy, GC content, and toehold accessibility. The data showed which constructs were predicted to be the most stable while still keeping the trigger-binding region open and available for strand displacement. I also compared the two circuits to assess whether they were orthogonal and unlikely to cross-react. This analysis helped identify the best design candidates for future synthesis and experimental validation.
One challenge was that computational predictions do not always reflect behavior in real biological conditions. For example, a design that appears stable in NUPACK may still misfold or behave differently in the presence of salts, competing nucleic acids, or target molecules. Another limitation is that orthogonality in silico does not guarantee orthogonality in solution, especially when sequences are short or partially similar. To address this, I would test multiple design variants with different toehold lengths and sequence compositions, and then prioritize the constructs with the strongest predicted accessibility and weakest cross-reactivity. If the designs still showed instability, an alternative strategy would be to simplify the circuit architecture or switch to a different recognition-output coupling strategy.
DNA oligonucleotides: histamine switch, IgE switch, trigger strands, scrambled control strands. Estimated budget: $150–$400 depending on length and purification.
High-purity oligo synthesis: HPLC-purified final switch strands for better experimental quality. Estimated budget: $100–$250.
Computational tools: Benchling, NUPACK, Mfold, Python. Budget: $0 if using student/free access.
Gel imaging access: shared lab imaging system or institutional equipment. Estimated budget: $0–$50.
Cell-free TX-TL reagents: if you test reporter output, use a commercial cell-free kit, reporter plasmid, and buffer components. Estimated budget: $200–$500.