Week 2 HW: DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
I made a free account on benchling and then imported the Lambda DNA (see below).
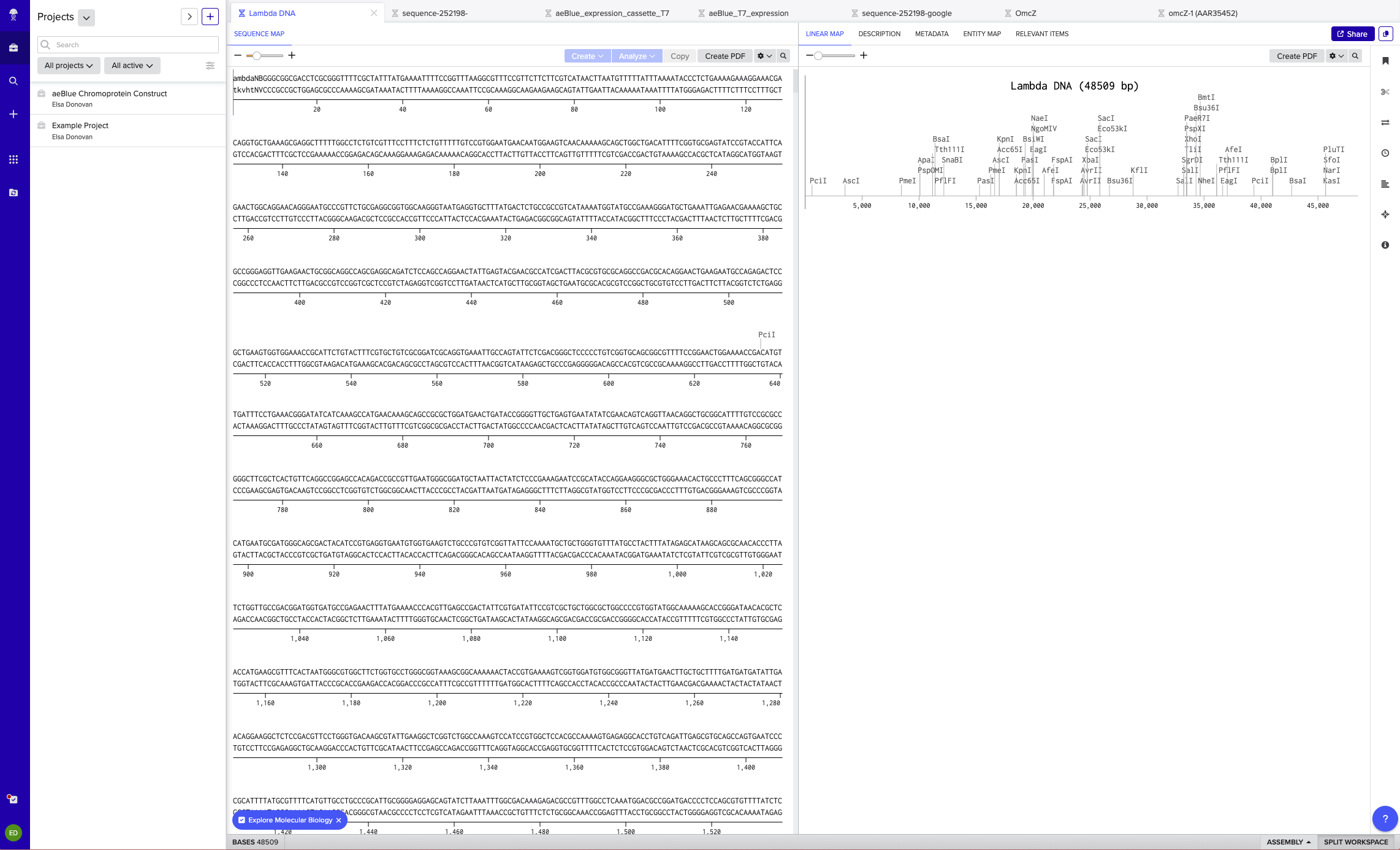
I simulated the restriction enzyme digestion with the following enzymes: EcoRI, EcoRV, SacI, SacII, KpnI, BamHI, HindIII.

I made an attempt at a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
It was supposed to be a semi circle of sorts, I’ve decided it’s ski goggles now. I suppose it’s a bit of a Rorschach Test and you can see what you want to see.
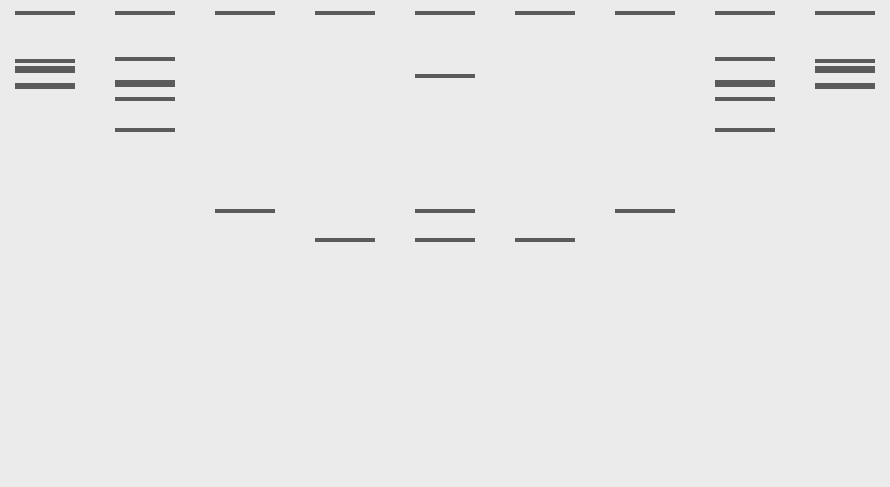
Here are the enzymes and enzyme combinations used for the ski goggles:

Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
Not applicable – No Lab Access
Part 3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why?
I am choosing the PilA protein which is derived from the bacteria Geobacter Sulfurreducens. PilA is the protein that is the main building block in the conductive nanowires (called e-pili) on Geobacter bacteria. Interestingly in a lab setting it is easier to work with the water-friendly shortened version of the protein, called PilA19, which can then used in bacteria such as E.coli. I will be finding my protein based on what is studied in the “Bottom-Up Fabrication of Protein Nanowires via Controlled Self-Assembly of Recombinant Geobacter Pilins” research paper.
References:
Cosert KM, Castro-Forero A, Steidl RJ, Worden RM, Reguera G. Bottom-Up Fabrication of Protein Nanowires via Controlled Self-Assembly of Recombinant Geobacter Pilins. mBio. 2019 Dec 10;10(6):e02721-19. doi: 10.1128/mBio.02721-19. PMID: 31822587; PMCID: PMC6904877.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
Using the reverse translate tool from bioinformatics.org I was able to get the most likely codons from my protein sequence.
3.3. Codon optimization.
I have used VectorBuilder’s online codon optimization tool to optimize my above protein sequence to be used in E.coli. I chose E.coli because it is one of the most commonly used chassis when it comes to genetic engineering and therefore it will make experimenting with the PilA protein easier. The codon optimization is important because different organisms have preferences for different codons. For example some organisms have more available tRNA’s for specific codons. By optimizing codon usage, you can take advantage of what is more available in the organism, which speeds up the protein-making process.
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
To produce the PilA protein from its DNA sequence, I could use the cell-dependent method. So the PilA19 (the adapted version of PilA) plasmid gene would be in the E.coli host. What the Central Dogma tells us is that the PilA DNA will be next to a promoter, which wil allow RNA polymersae to read the DNA sequence and make an mRNA copy. This copy can be scanned by a ribosomes in sets of 3 (codons) Each codon represents an amino acid (which is fetched by tRNA) and compiled by the ribosome to create a chain of amino acids which folds and becomes the PilA19 protein.
I would to use cell-dependent production with E.coli because E.coli can give higher yields for the protein as opposed to cell-free methods. It is also less expensive. And it is the method used in the reserach paper I referenced above.
Part 4: Prepare a Twist DNA Synthesis Order
I am creating a DNA sequence that would use GFP with PilA to create glowing green conductive nanowires that in theory could be visually detected and tested for conductivity via Scanning Tunneling Microscopes (STM).
I will be basing myself on several papers that have worked with these proteins before, which I will reference below.
I will be using a basic T7 promoter, found one here: https://parts.igem.org/Part:BBa_I719005.
I will be using the native RBS (Shine-Delgarno): https://parts.igem.org/Help:Ribosome_Binding_Sites/Mechanism.
I will be using this linker for my fusion protein (GFP-tag): https://parts.igem.org/Part:BBa_K5283021.
I will be linking it to this already e.coli optimized version of GFP (sfGFP): https://www.ncbi.nlm.nih.gov/nuccore/HQ873313.1?report=fasta.
That will be followed by the 7x His-Tag used in the homework example as well as the Terminator used in the example: https://parts.igem.org/Part:BBa_B0015.
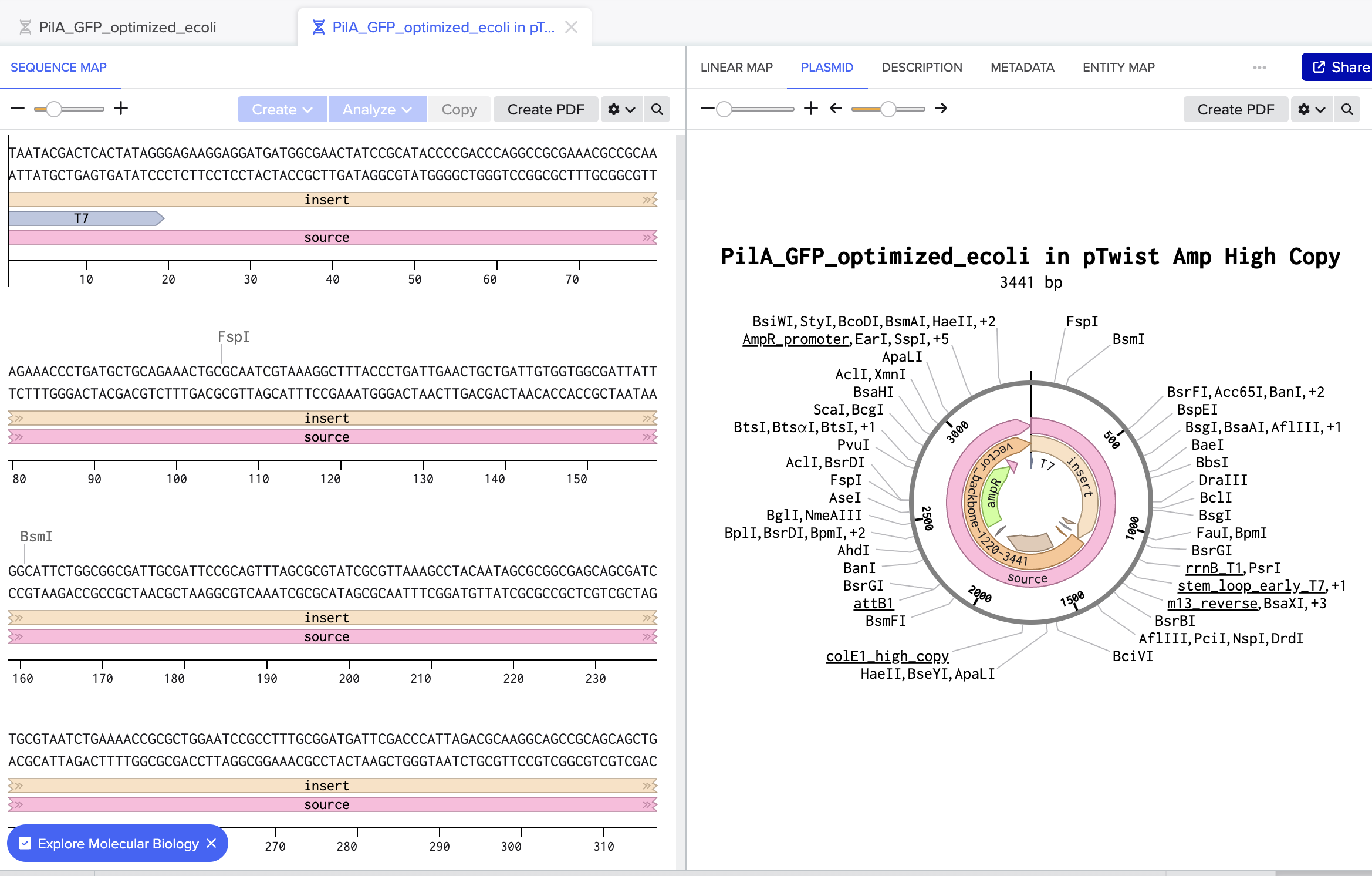
Link for Twist Order (using the homework example + pilA): https://benchling.com/s/seq-HNOuv8nh3BZpdko0cxxR?m=slm-Ei5fR7bWZIBnKBNornvb.
Link for Twist Order (PilA with sfGFP tag): https://benchling.com/s/seq-T1jlrWcLtLrD49qmLQDv?m=slm-ck0HqucQWeYd28sFDf3T.
There were some redundancies that Twist Bioscience optimized for with my PilA with sfGFP tag DNA sequence. The GenBank file, which I’ve added to my assets folder is the optimized version.

References:
Cosert, K. M., Castro-Forero, A., Steidl, R. J., Worden, R. M., & Reguera, G. (2019). Bottom-Up Fabrication of Protein Nanowires via Controlled Self-Assembly of Recombinant Geobacter Pilins. mBio, 10(6), 10.1128/mbio.02721-19. https://doi.org/10.1128/mbio.02721-19.
Ueki, T., Walker, D. J. F., Woodard, T. L., Nevin, K. P., Nonnenmann, S. S., & Lovley, D. R. (2020). An Escherichia coli Chassis for Production of Electrically Conductive Protein Nanowires. ACS Synthetic Biology, 9(3), 647–654. https://doi.org/10.1021/acssynbio.9b00506.
Wahlfors, J., Loimas, S., Pasanen, T., & Hakkarainen, T. (2001). Green fluorescent protein (GFP) fusion constructs in gene therapy research. Histochemistry and Cell Biology, 115(1), 59–65. https://doi.org/10.1007/s004180000219.
Part 5: DNA Read/Write/Edit
5.1 DNA Read
What DNA would you want to sequence (e.g., read) and why?


I have two projects that inspire me: one by Carolina Reyes about fungal batteries, another one by Tanguy Chotel about ressurecting ancestral proteins that were better suited at carbon capture and using them to do a “reboot” of the traditional Calvin Cycle. He uses Chlamydomonas reinhardtii as his chassis which is a microalgae.
References: Reyes, C., Fivaz, E., Sajó, Z., Schneider, A., Siqueira, G., Ribera, J., Poulin, A., Schwarze, F. W. M. R., & Nyström, G. (2024). 3D Printed Cellulose-Based Fungal Battery. ACS Sustainable Chemistry & Engineering, 12(43), 16001–16011. https://doi.org/10.1021/acssuschemeng.4c05494
Inckemann, R., Chotel, T., Brinkmann, C. K., Burgis, M., Andreas, L., Baumann, J., Sharma, P., Klose, M., Barrett, J., Ries, F., Paczia, N., Glatter, T., Willmund, F., Mackinder, L. C. M., & Erb, T. J. (2024). Advancing chloroplast synthetic biology through high-throughput plastome engineering of Chlamydomonas reinhardtii (p. 2024.05.08.593163). bioRxiv. https://doi.org/10.1101/2024.05.08.593163
(i) Building off the aforementioned fungal battery, I would be interestied in sequencing the white-rot fungus Trametes pubescens – which in the fungal battery serves as a cathode. There are laccase enzymes in the white-rot fugus that capture the electrons and close the circuit. I’d be curious to sequence other fungi and compare with the Trametes pubescens to see if there are other laccase enzymes in other species that could prove even more adept for bio-batteries. I could sequence specifically the laccase enzymes (lap1/2). Laccase enzymes have copper atoms that capture the electrons. Those electrons then are used to turn airborne oxygen to harmless water (no issue of hydrogen peroxide).
ii) I think I would use NGS (Next Generation Sequencing) to read the DNA from Trametes pubescens because it is one of the more cost-effective solutions and it also handles multiple samples well (for if I want to compare laccase enzymes from different fungi.) However if the genome is very long I should use Oxford Nanopore Technologies which can handle long reads.
- The NGS method is second generation (massively parallel), which means that as opposed to Sanger sequencing (first generation) this method uses multiplexing (not just one tube per reaction).
References Jiang, S., Chen, Y., Han, S., Lv, L., & Li, L. (2022). Next-Generation Sequencing Applications for the Study of Fungal Pathogens. Microorganisms, 10(10), 1882. https://doi.org/10.3390/microorganisms10101882
5.2 DNA Write
(i) An example of a DNA I would want to synthesize is what I wrote above in the Twist order – a GFP-tag version of the pilA protein in order to understand visually how and where the nanowires are constructed.
The link for the Twist order of this synthesized DNA can be found here: https://benchling.com/s/seq-T1jlrWcLtLrD49qmLQDv?m=slm-ck0HqucQWeYd28sFDf3T
To take this project a step further (and align with the project I investigated in my first homework) I could make a genetic circuit that would take a promoter from another bacteria which is activated by high levels of toxic metals. One such example is the czcCBA promoter that comes from Pseudomonas putida bacteria. This promoter is activated by zinc, cadmium, or lead. I could place this promoter in front of the pilA-GFP protein so that these proteins are expressed only when heavy metals are detected. The GFP would be useful for debugging the circuit, the conductivity could be hooked up to electrodes or other electric components to create a bio-hybrid senor.
(ii) To perform this particular DNA synthesis I would use Twist Bioscience’s web platform. It would be a similar process to what I created in the Twist Example order above. There are other options, like using Golden Gate cloning to assemble DNA. But with Twist I can order the fully built plasmid. They verify the sequences and there’s a guarantee that the sequence delivered is 99.5% exact. Practically speaking this is the best option for me, even if it is more expensive.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In a similar theme to my earlier responses I would edit DNA in order to be able to boost the conductive capabilities of Geobacter Sulfurreducens.
Based on a research paper I found there is a gene called ftsZ which limits the length of the host body. If this gene is repressed (according to the paper) the cell body becomes longer and more filamentous (ressembling a conductive thread). This creates a higher density of the nanowires which improves conductivity.
(ii) I’d use CRISPRi (CRISPR interference) witha dead or deactivated Cas9. CRISPRi is the perfect tool for this because it can pause certain genes without having to cut out parts of the DNA. This makes it perfect for testing.It functions similarly to CRISPR in that there is a “guide RNA” attached to the Cas9 that helps to find the target DNA. However instead of cutting out the DNA it blocks the transcription of that part of the DNA. In this way we can do gene silencing at the transcription level. Once I’ve tested with CRISPRi I can then move to CRISPR to do true gene editing. CRISPR is the same mechanism except is uses activated Cas9 protein which actually cuts out the DNA it is configured to cut out.
In order to do this (for either CRISPR or CRISPRi) I would need to: a. Find the sequence for the ftsZ gene. This will be the guide RNA for Cas9. b. Order the Cas9 proteins c. Grow host chassis (Geobacter or E.coli)
I would have to clone the guide RNA into the Cas9 plasmids. Then I would have to deliver that plasmid into the host cell, something I could do with electroporation.
CRISPR has some limitations, particularly that it does not always target the write cut. This is not only inefficient but can also cause bigger issues like cell death due to inaccurate cuts.
References:
Bird, L. J., Kundu, B. B., Tschirhart, T., Corts, A. D., Su, L., Gralnick, J. A., Ajo-Franklin, C. M., & Glaven, S. M. (2021). Engineering wired life: Synthetic biology for electroactive bacteria. ACS Synthetic Biology, 10(11), 2808–2823. https://doi.org/10.1021/acssynbio.1c00335