Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
- Human SOD1 sequence from Uniprot (no Mutation): (https://www.uniprot.org/uniprotkb/P00441/entry)
A4V Mutation refers to Alanine being changed to Valine at the 4th amino acid in the protein.
It’s called A4V in the homework but I’m pretty sure it’s now referred to as A5V based on the literature (or maybe actually the other way around - it used to be A5V and is now A4V). So I found this link for the A5V mutation on Uniprot here: https://web.expasy.org/variant_pages/VAR_007131.html
The entry with the mutation would now start with MATKVVC
- Using PepMLM colab: Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
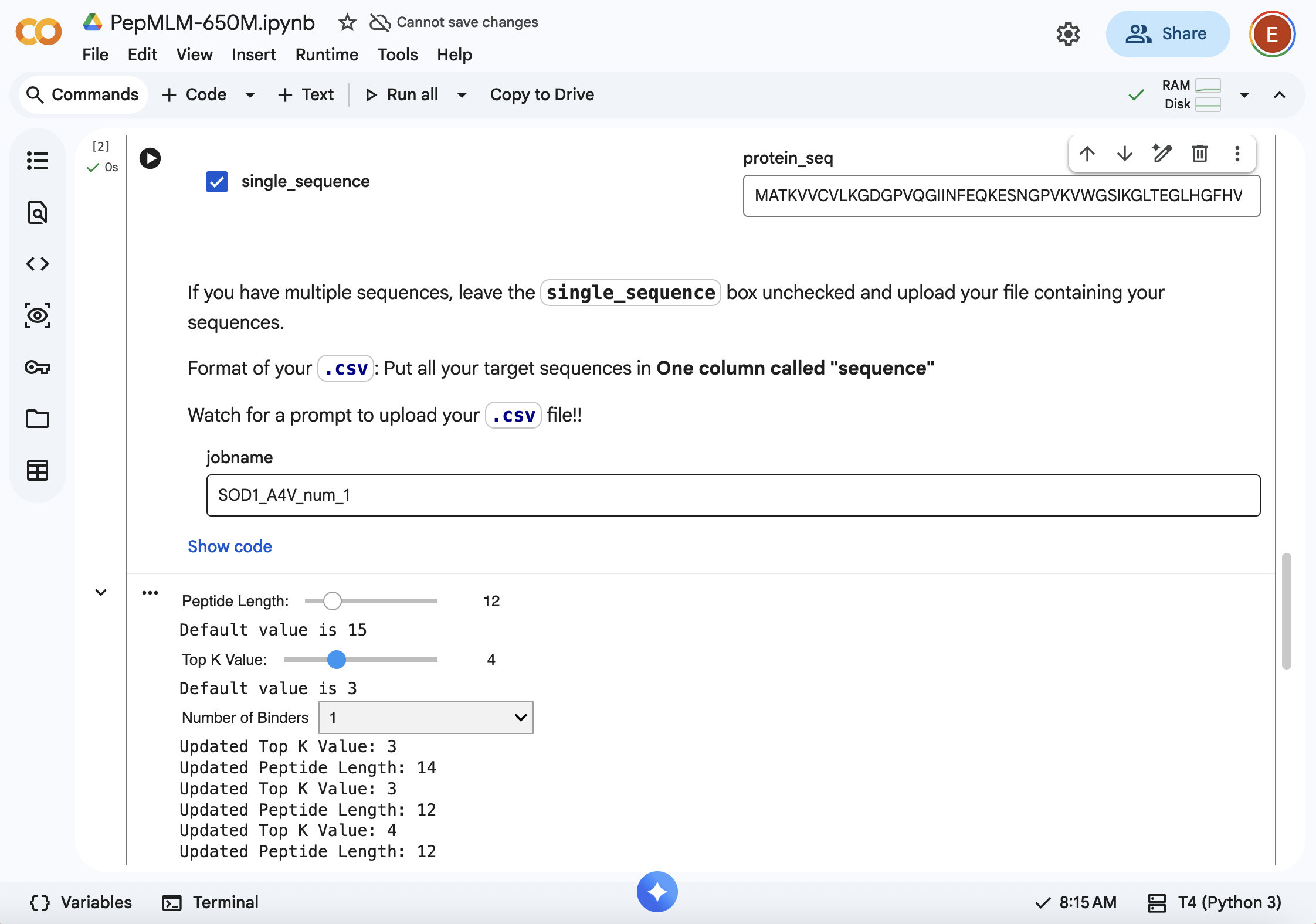
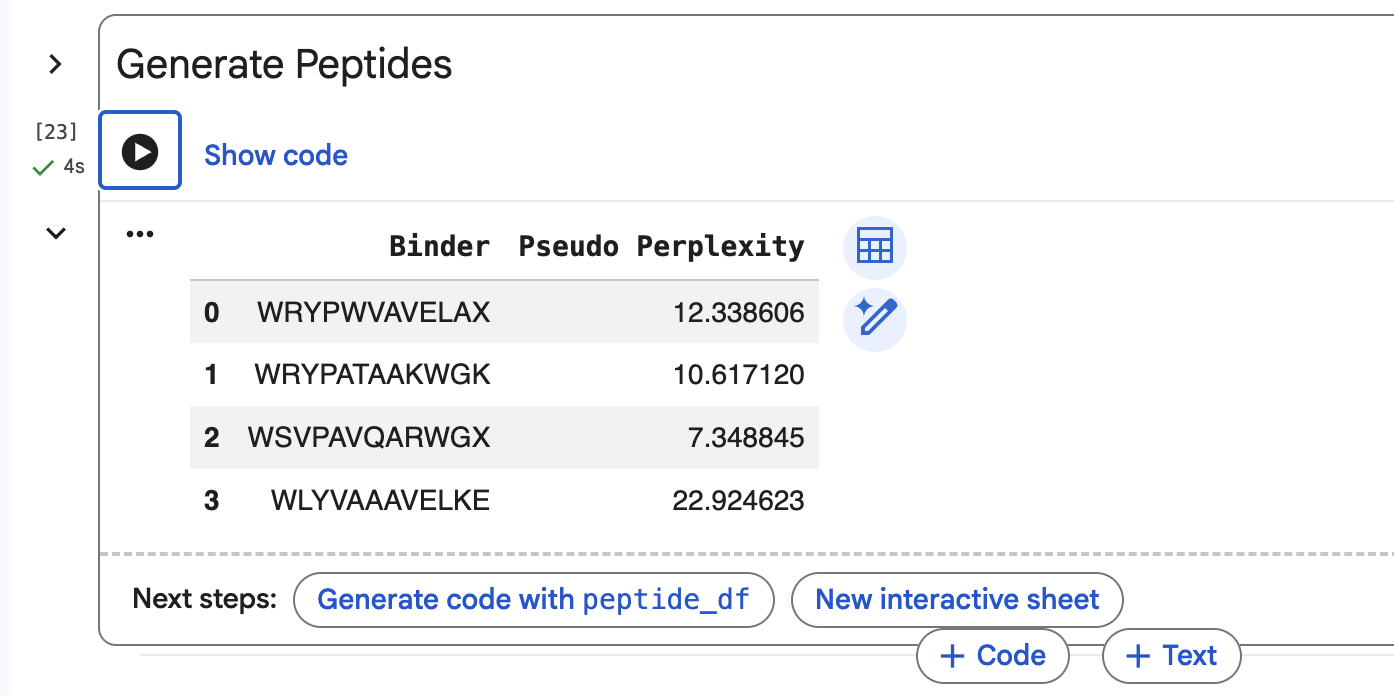
SOD1-binding peptide FLYRWLPSRRGG put through the PepMLM colab:
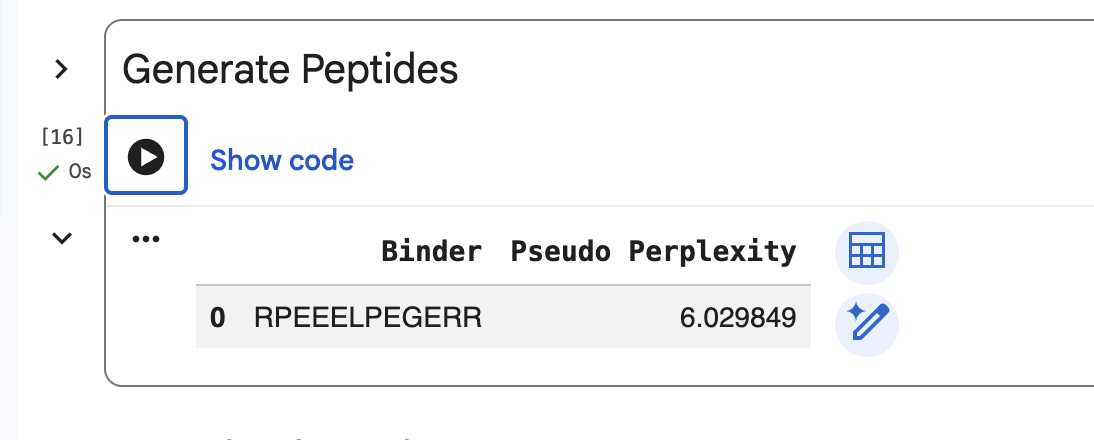
Part 2: Evaluate Binders with AlphaFold3
- Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
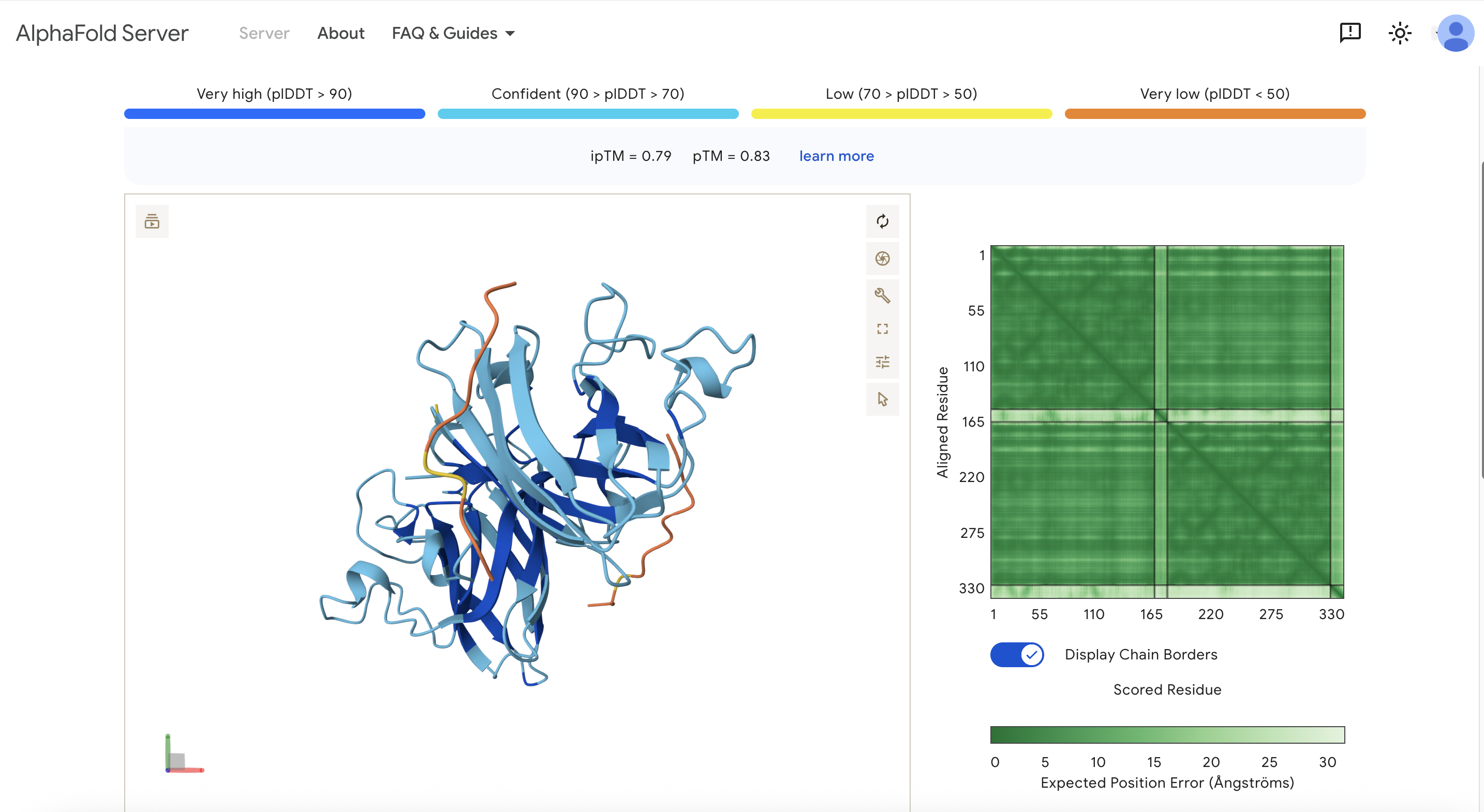
Note: Only 2 of my generated peptides were able to be used for the Alphafold model. It didn’t like the ones that ended with “X”.
These are the scores: ipTM = 0.79 pTM = 0.83
One of the yellow ends of the protein is the N-terminus. The peptides are near each of the ends of the protein but not directly on it. I would have expected it to be closer to the N-terminus since that’s where the mutation is. The peptides are surface-bound. The peptide doesn’t seem to bind to the β-barrel region, and it also doesn’t appear to bind at the dimer interface.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
For the peptide: WHSYPAQLRHKE (a new peptide I generated) The values for this peptide are: ipTM = 0.29 pTM = 0.84
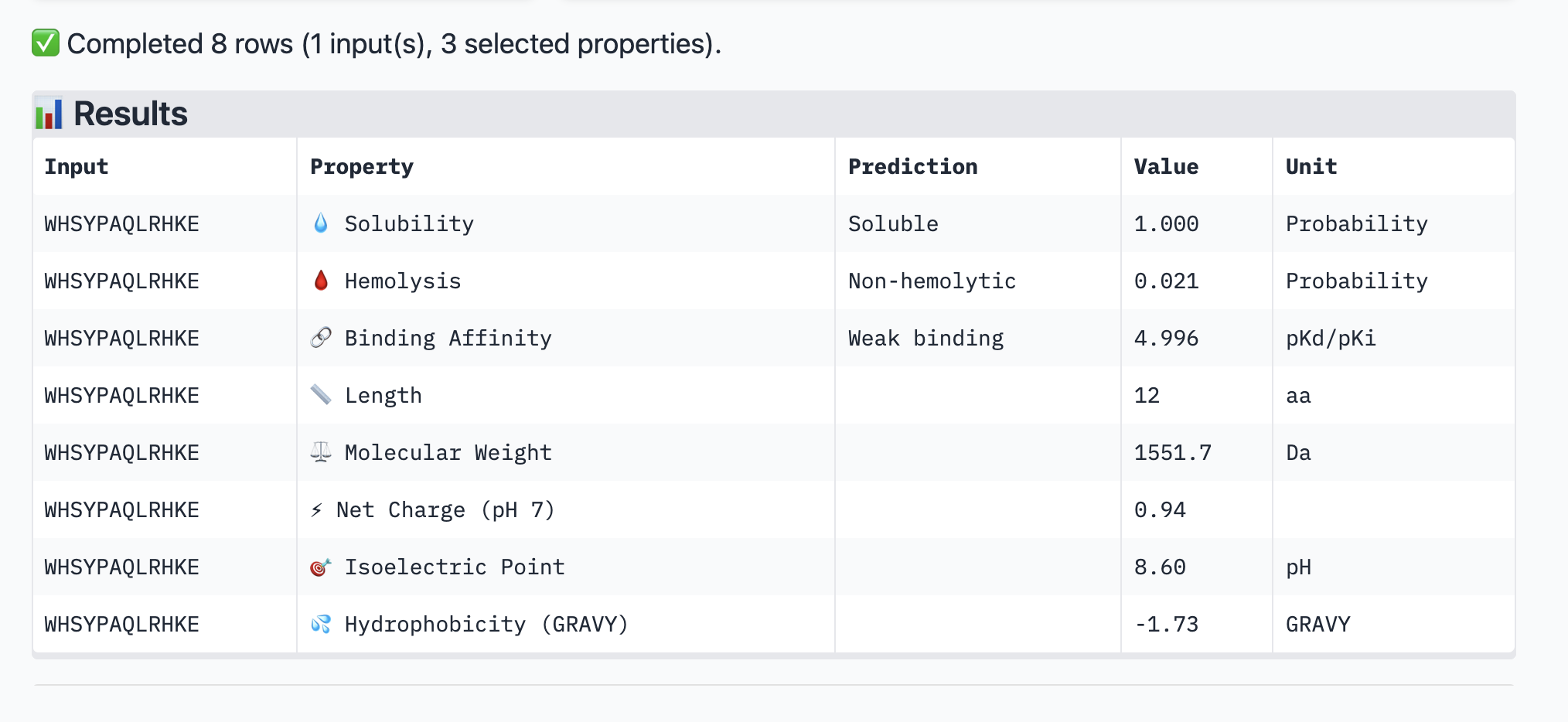
This particular peptide has a low ipTM. It also has a weak binding affinity. It is soluble but not hemolyctic. I assume that the weaker the binding, the more soluble it is due to the poor bonds.
If I want a protein that is not soluble I will choose one with a high ipTM.
This peptide: WLNYAVAVAWWE has a ipTM of 0.48 which is slightly higher than the previous one.
Here are it’s peptiverse results:
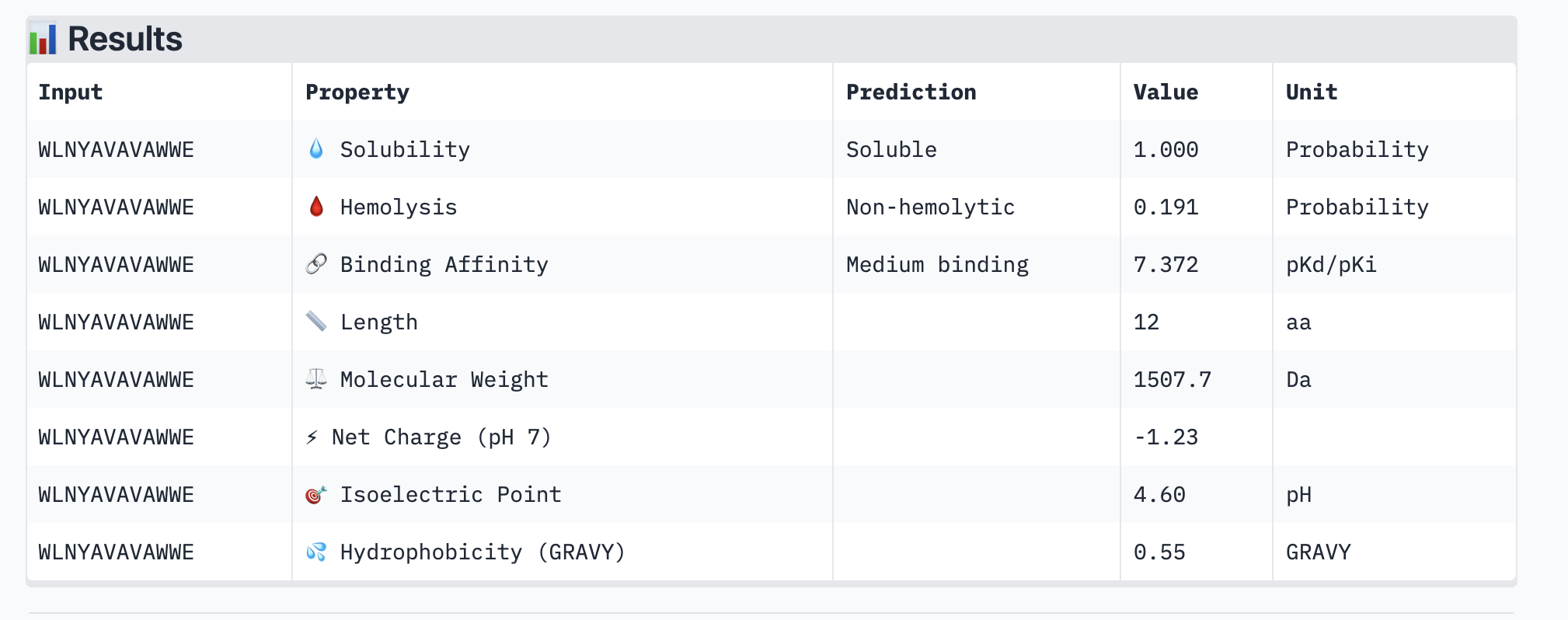
This is a better peptide because it is medium-binding so a bit stronger than the typical peptide I’ve been generating. However it is still water soluble and non-hemolyctic which is important and necessary for therapies. I will move forward with this peptide then.
Part 4: Generate Optimized Peptides with moPPIt
PepMLM generates peptides conditioned on the whole protein sequence, while moPPit lets you target specific residues, giving you more control over where the peptide binds.
Open the moPPit Colab linked from the HuggingFace moPPIt model card Make a copy and switch to a GPU runtime. In the notebook: Paste your A4V mutant SOD1 sequence. Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch). Set peptide length to 12 amino acids. Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides. After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
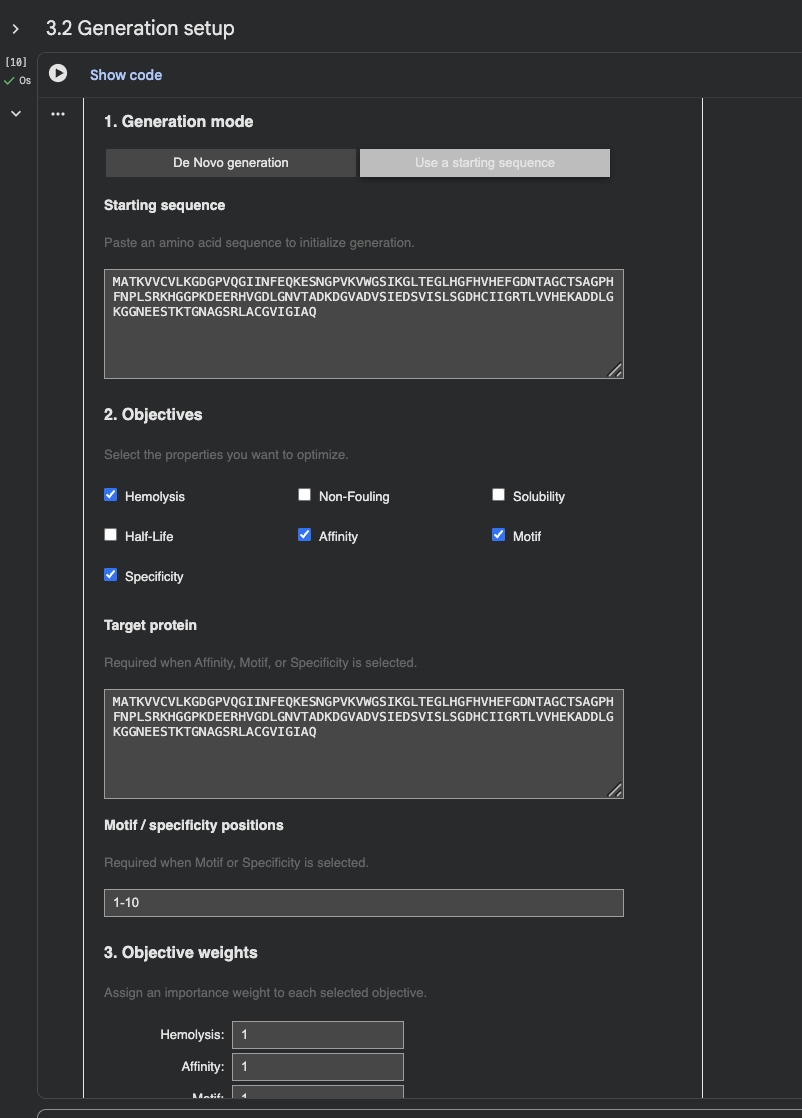
Some samples:
moPPit peptides are more targeted than PepMLM peptides because they are designed to bind a specific region near the A4V mutation, rather than the whole protein. Before clinical studies, you would haveto evaluate them using AlphaFold3 for binding confirmation. Then PeptiVerse for solubility and hemolysis, and eventually in-vitro binding and test for toxicity.
Final Project: L-Protein Mutants
Objective: to improve the stability and autofolding of the lysis protein
Stage 1: Engineer novel L-protein mutants using protein design tools
Here, I ran the notebook to be able to “score” for what would happen to the protein if you mutated into another amino acid.
From the Colab:
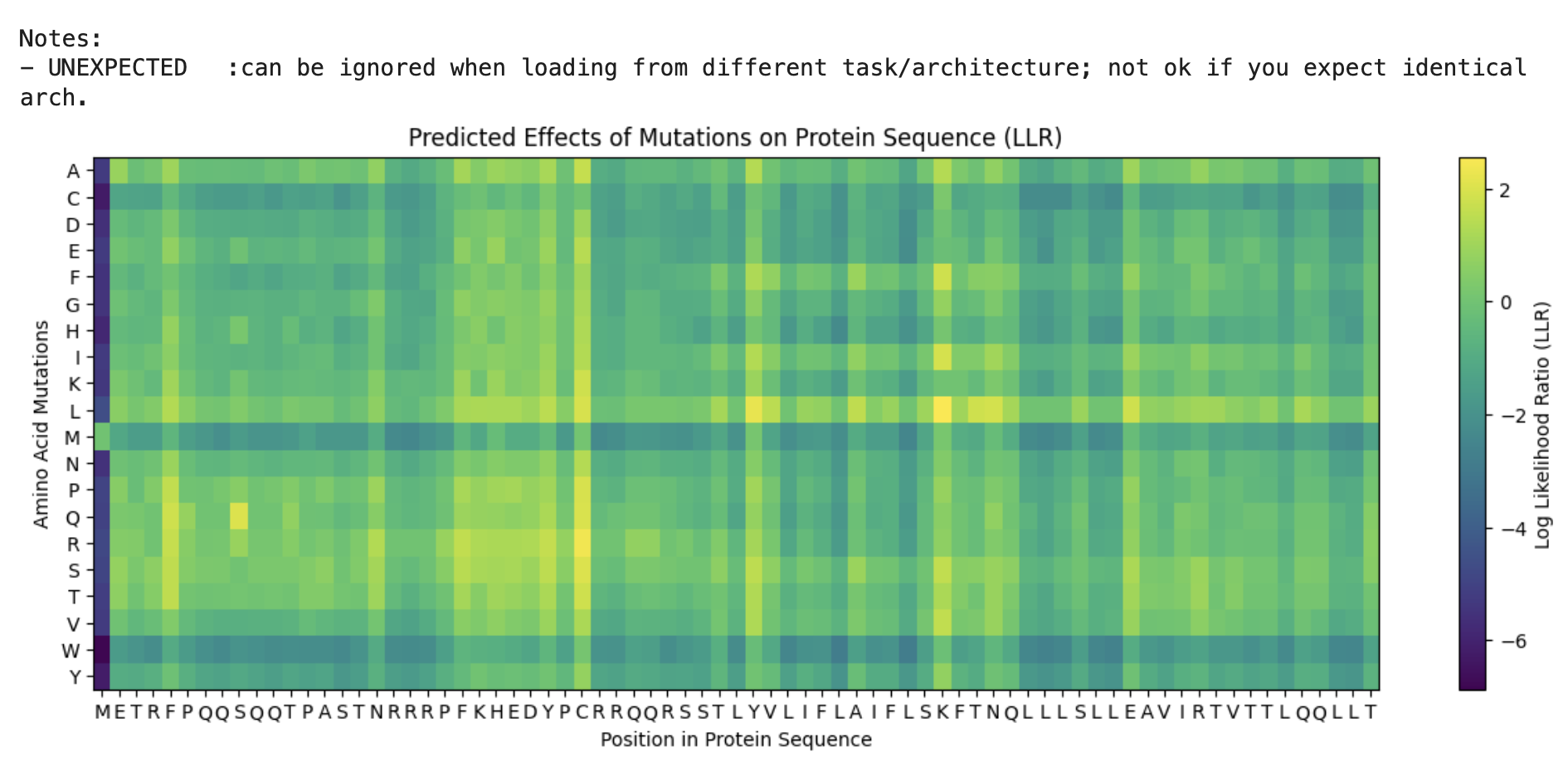
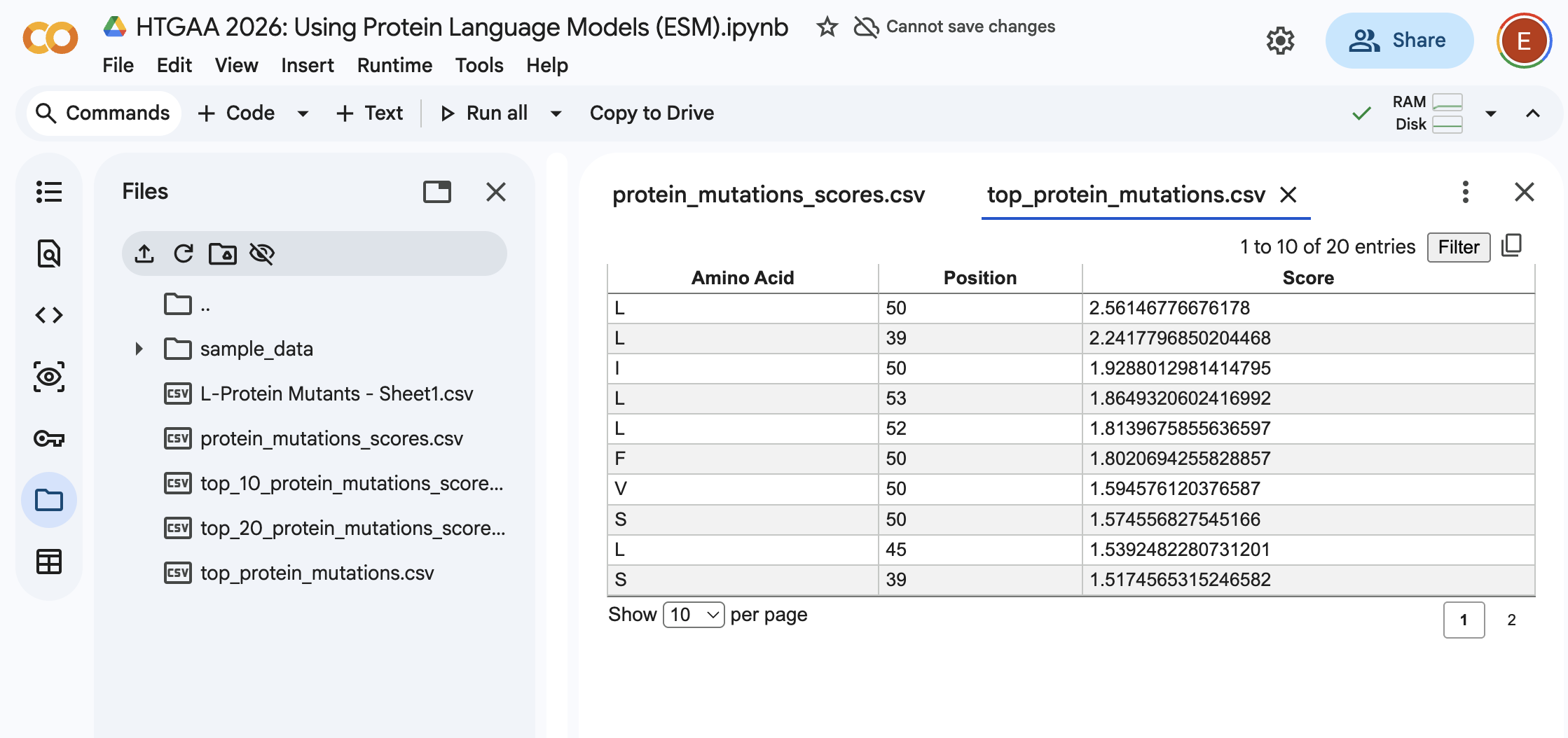
I’ll be honest I’m not seeing a lot of correlation between the 2 datasets. Essentially I can see that for example, for position 50 there is a high score for the mutation. However in the provided dataset I’m seeing a 1 for protein level but not for lysis.
Stage 2: Synthesize the L-protein mutant gene via Twist
Stage 3: Clone the L-protein mutant gene into a plasmid using Gibson Assembly
Stage 4: Test the L-protein mutant’s structural integrity using the Nuclera system
Stage 5: Test the L-protein in E. coli with plaque assays