Week 2 HW: DNA READ WRITE AND EDIT
Part 1: Benchling & In-silico Gel Art
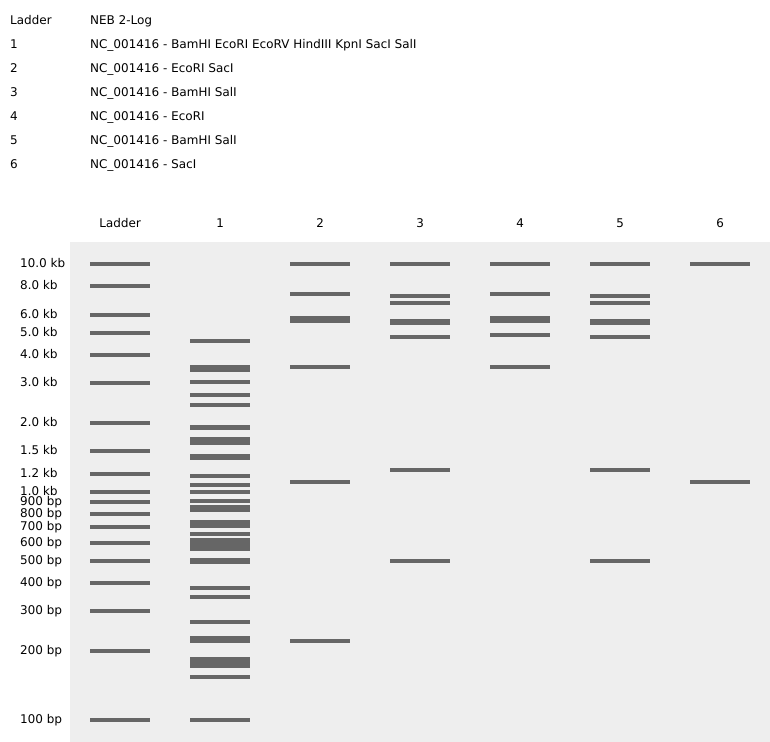
In-Silico Gel Art: Latent Figure Protocol
Project Overview
For this week’s assignment, I used Benchling to simulate restriction enzyme digests on the Lambda Phage genome (NC_001416). My goal was to move beyond simple data analysis and create “Gel Art” in the style of Paul Vanouse’s Latent Figure Protocol.
The Visual Design
I designed a zigzag pattern that emerges from a complex reference lane. By selecting specific enzymes, I was able to control the migration height of the DNA bands to create a deliberate visual W shape.
Enzyme Key and Lane Setup
| Lane | Enzyme Combination | Visual Goal |
|---|---|---|
| Ladder | NEB 2-Log | Size reference for the DNA bands. |
| Lane 1 | All 7 Enzymes | The Master Key: EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, SalI. |
| Lane 2 | SacI | Low Point: Sharp band near the bottom. |
| Lane 3 | BamHI + SalI | Mid-Point: Moving the pattern upward. |
| Lane 4 | EcoRI | High Point: The peak of the zigzag. |
| Lane 5 | BamHI + SalI | Mid-Point: Symmetric return to the middle. |
| Lane 6 | SacI | Low Point: Completing the zigzag at the bottom. |
Final Result
Reflection
Working with EcoRV was a challenge because it cuts the genome 21 times, resulting in a significant amount of noise. By isolating simpler cutters, such as SacI and EcoRI, in the later lanes, I was able to make the intended artwork much clearer.
View my Benchling Virtual Digest Project
Part 3: DNA Design Challenge
** 3.1. Choose Your Protein**
- Protein Chosen: Insulin (Homo sapiens)
- Why: I chose Insulin because it is a vital hormone for glucose regulation and holds historical significance as the first human protein to be manufactured using recombinant DNA technology.
- Protein Sequence (FASTA format):
sp|P01308|INS_HUMAN Insulin OS=Homo sapiens OX=9606 GN=INS PE=1 SV=1 MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAED LQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
** 3.2. Reverse Translate**
Process: Using the Sequence Manipulation Suite, I reverse-translated the Insulin amino acid sequence into a DNA sequence. I used the most likely codons based on the genetic code to ensure a usable, non-degenerate sequence.
Insulin DNA Sequence (Naive/Initial):
atggcgctgtggatgcgcctgctgccgctgctggcgctgctggcgctgtggggcccggatccggcggcggcgtttgtgaaccagcatctgtgcggcagccatctggtggaagcgctgtatctggtgtgcggcgaacgcggctttttttataccccgaaaacccgccgcgaagcggaagatctgcaggtgggccaggtggaactgggcggcggcccgggcgcgggcagcctgcagccgctggcgctggaaggcagcctgcagaaacgcggcattgtggaacagtgctgcaccagcatttgcagcctgtatcagctggaaaactattgcaactaa
3.3. Codon optimization.
Chosen Organism: Escherichia coli (E. coli)
Why do we need to optimize? Different organisms have different “preferences” for which codons they use to build proteins. If we put the human insulin DNA sequence directly into E. coli, the bacteria might lack the necessary tRNA “building blocks” to read it efficiently. By using the IDT Codon Optimization Tool, I have swapped the human codons for the ones that E. coli prefers, ensuring the fastest and most reliable production of the protein.
Optimized Insulin DNA Sequence (for E. coli): ATG GCA CTG TGG ATG CGC CTG CTG CCG TTG TTA GCT CTG CTG GCG TTA TGG GGG CCG GAT CCG GCG GCG GCC TTC GTG AAT CAG CAT TTA TGT GGC TCA CAC CTG GTC GAA GCC TTG TAC TTA GTC TGT GGT GAA CGT GGT TTT TTT TAC ACA CCG AAA ACC CGC CGT GAA GCG GAG GAC CTT CAG GTG GGC CAG GTT GAA CTG GGC GGC GGT CCG GGC GCG GGA TCT CTT CAG CCT CTG GCT TTA GAA GGA AGC CTG CAG AAA CGC GGC ATT GTG GAG CAG TGC TGT ACC TCT ATT TGC TCC CTG TAT CAG TTG GAA AAC TAT TGT AAT TAA
3.4. You have a sequence! Now what?
To turn my digital sequence into a physical protein, I would use the following technologies:
- Chemical DNA Synthesis: I would send my optimized sequence to a vendor like IDT to synthesize the physical DNA strands.
- Recombinant Expression: I would insert this DNA into a plasmid and transform it into E. coli cells. The bacteria act as a biological factory, using transcription and translation to manufacture the insulin.
- Cell-Free Synthesis: Alternatively, I could use a X-TL system, which uses cellular machinery in a test tube to produce the protein without needing a living host.
3.5. Biological Systems
How can a single gene code for multiple proteins? Nature is far more efficient than a simple 1:1 “one gene, one protein” rule. Through Alternative Splicing, a cell can choose which sections of an RNA transcript to keep and which to discard. This allows the same gene to produce several different versions of a protein, known as isoforms, which can have different functions in the body.
Case Study: Human Insulin (P01308)
- Isoforms: This gene produces 2 isoforms via alternative splicing.
- Maturation: Insulin also undergoes Post-translational processing, where it is trimmed from a long Preproinsulin chain into the final active hormone.
The Biomolecular Flow: Below is the full breakdown of how my digital DNA sequence becomes a functional protein.
| Level | Sequence | Key Change |
|---|---|---|
| DNA | ATG GCA CTG TGG... | Optimized for E. coli host |
| RNA | AUG GCA CUG UGG... | Transcribed copy; T is now U |
| Protein | M A L W ... | Translated amino acid sequence |
Part 4: Prepare a Twist DNA Synthesis Order
Project: Insulin_v1.0_System_Architecture
Developer: [Elsa Muleya]
Status: Compiled & Verified
Target Environment: E. coli OS
1. The Source Code (DNA)
The circular plasmid represents the permanent Read-Only Memory (ROM) of the biological system.
- ENTRY_POINT (promoter): Executes the START command. It signals the system’s hardware to begin data processing at position 1.
- DATA_PACKET (RBS): The Ribosome Binding Site acts as the Buffer. It prepares the hardware to load the upcoming instructions.
- MAIN_APP (Insulin CDS): The primary logic gate. This is the raw sequence that defines the structure of the final output (Insulin).
- METADATA_TAG (7x His Tag): An attached Header. This 7-histidine string acts as a unique ID for downstream sorting and purification.
- EOF_MARKER (Terminator): The exit(0) command. It forces the system to stop reading and release the hardware resources.
2. The Compiler (Transcription)
This is the process of converting the High-Level Code (DNA) into Machine Code (mRNA).
- The system’s compiler (RNA Polymerase) docks at the ENTRY_POINT.
- It generates a temporary copy of the data. This is equivalent to loading an application from the Hard Drive (DNA) into RAM (mRNA) for active execution.
3. The Execution (Translation)
The system hardware (Ribosome) executes the instructions stored in the RAM (mRNA).
- BIT_READING: The hardware reads the code in 3-bit segments called Codons.
- OUTPUT_GENERATION: For every 3 bits read, the system adds one unit (amino acid) to the physical product.
- FRAME_CHECK: I have verified the 7x His Tag is in-frame, ensuring the Metadata Header is correctly attached to the Main App without data corruption.
4. System Security & Multi-Threading (The Vector)
The design uses the pTwist Amp High Copy backbone for optimized performance.
- FIREWALL (AmpR): Provides Ampicillin resistance. This acts as a security filter; any cell that does not contain the “authorized” plasmid is deleted by the antibiotic.
- MULTI-THREADING (colE1_high_copy): Forces the cell to run hundreds of instances of the program simultaneously. This maximizes the Data Throughput, resulting in high-volume insulin production.
Build Logs:
- Coordinates: 1-2761 bp
- Topology: Circular
- Resistance: Ampicillin
- Integrity: Verified

**Part 5: 5.1 DNA READ | 5.2 DNA WRITE | 5.3 DNA EDIT
5.1 DNA READ: PALEOVIROMICS & PERMAFROST SURVEILLANCE
(i) WHAT DNA AND WHY? I intend to sequence ancient viral DNA/RNA (eDNA) extracted from Siberian permafrost cores (Reference: Alempic et al., 2023). As climate change accelerates, dormant pathogens like Pithovirus or Pandoravirus are resurfacing. Sequencing these allows for the creation of a Pre-emptive Pandemic Library to identify ancestral motifs and develop vaccine scaffolds before zoonotic spillover occurs.
(ii) TECHNOLOGY & METHODOLOGY: Technology: Oxford Nanopore Technologies (ONT) Ultra-Long Read Sequencing.
- GENERATION: 3rd Generation (Single-molecule, real-time sequencing).
- INPUT: Environmental DNA (eDNA) from permafrost meltwater.
- PREPARATION STEPS:
- EXTRACTION: Bead-based magnetic isolation of fragmented ancient DNA.
- REPAIR: End-repair and A-tailing to fix degraded DNA termini.
- TARGETED ENRICHMENT: Hybrid capture using RNA-probe baits to isolate viral sequences from bacterial/fungal background.
- ADAPTER LIGATION: Attaching motor proteins to pull DNA through pores.
- DECODING (BASE CALLING): DNA passes through a protein nanopore, disrupting an ionic current. Each base creates a specific squiggle (electrical signature). Recurrent Neural Networks (RNNs) like the ‘Dorado’ basecaller translate these signals into ATCG sequences.
- OUTPUT: FastQ files containing Long Reads (10kb - 2Mb), enabling high-fidelity de novo assembly of unknown viral genomes.
5.2 DNA WRITE: DE NOVO ANTIFREEZE GLYCOPROTEINS (AFGPs)
(i) WHAT DNA AND WHY? I want to synthesize DNA encoding a De Novo Synthetic Antifreeze Glycoprotein (AFGP), inspired by Arctic Notothenioids (Reference: Zhuang, 2014).
- SEQUENCE: [Ala-Ala-Thr]n repeats, optimized for human tissue compatibility.
- WHY: To enable “Supercooling” in organ transplantation. This DNA would produce proteins that prevent ice crystal formation, extending the viability of donor organs from hours to several days.
(ii) TECHNOLOGY & METHODOLOGY: Technology: Silicon-based Phosphoramidite Synthesis (e.g., Twist Bioscience).
- ESSENTIAL STEPS:
- DE-BLOCKING: Acidic removal of the DMT protective group from the silicon-bound nucleotide.
- COUPLING: Addition of the next phosphoramidite monomer (A,T,C, or G).
- CAPPING: Acetification of failed strands to prevent truncation errors.
- OXIDATION: Stabilizing the phosphite triester bond.
- LIMITATIONS:
- SPEED: Chemical synthesis is a multi-day process involving logistics.
- SCALABILITY: Individual oligos are limited to ~300bp; longer constructs require Gibson Assembly, which is difficult for repetitive sequences like [Ala-Ala-Thr]n.
5.3 DNA EDIT: MUTATION-AGNOSTIC PROGERIA CORRECTION
(i) WHAT DNA AND WHY? I want to edit the LMNA gene in human fibroblasts to treat Hutchinson-Gilford Progeria Syndrome (HGPS).
- THE EDIT: Deletion of the CAAX box motif at the C-terminus.
- WHY: Instead of fixing a patient-specific mutation, removing the CAAX box prevents the toxic protein (progerin) from anchoring to the nuclear membrane. This is a Mutation-Agnostic therapeutic approach applicable to all HGPS patients.
(ii) TECHNOLOGY & METHODOLOGY: Technology: Prime Editing (PE).
- HOW IT EDITS: Uses an engineered Cas9 nickase fused to a Reverse Transcriptase (RT). It uses a Search-and-Replace mechanism without causing double-strand breaks.
- ESSENTIAL STEPS:
- SEARCH: The pegRNA (prime editing guide RNA) targets the LMNA site.
- NICK: Cas9 nicks only the target DNA strand.
- REPLACE: The RT enzyme synthesizes new DNA directly from the pegRNA template into the nicked site.
- INPUTS & PREPARATION:
- INPUT: Plasmids/mRNA encoding the PE protein, pegRNA, and a nick-gRNA.
- DESIGN: Computational modeling of the Primer Binding Site (PBS) thermodynamics to ensure stable hybridization.
- LIMITATIONS:
- EFFICIENCY: Prime editing often has lower “on-target” efficiency in primary cells compared to standard CRISPR.
- DELIVERY: The PE complex is too large for many standard viral delivery vectors (AAVs).
REFERENCES & RESOURCES
- Alempic, J. M., et al. (2023). “An update on eukaryotic viruses revived from ancient permafrost.” Viruses.
- Zhuang, X. (2014). “Creating sense from non-sense DNA: de novo genesis and evolutionary history of antifreeze glycoprotein gene.” UIUC.
- Anzalone, A. V., et al. (2019). “Search-and-replace genome editing without double-strand breaks or donor DNA.” Nature.
- Twist Bioscience Technical Documentation (2024). “Silicon-based DNA Synthesis.”