Week 4 HW: Protein Design I
Homework: Protein Design I
Part A. Conceptual Questions
1.# Assignment: Proteins and Amino Acids
1. Amino Acids in 500g of Meat
To calculate the total molecules, we first look at the protein density. Meat is roughly 20% protein by mass.
- Protein Mass: 500g 0.20 = 100g
- Average Molecular Weight (MW): 100 Daltons (g/mol)
- Moles of AA: 100g / 100g/mol= 1 mole
Using Avogadro’s number, 1 mole contains approximately 6.022 * 10^23 molecules. That is sextillion amino acids in a single large steak.
2. Metabolic Identity: Why don’t we turn into cows?
When we ingest beef or fish, our digestive system performs proteolysis. Enzymes such as pepsin and trypsin break down foreign proteins into their constituent amino acids. Our ribosomes then take those bricks and reassemble them into human-specific proteins according to the instructions in our DNA. We don’t become the cow because we recycle the parts, not the blueprints.
3. The Standard 20
While there are hundreds of amino acids found in nature, only 20 are universally encoded.
- The Frozen Accident Theory: Francis Crick proposed that once life settled on a set of 20 that covered the necessary chemical functionalities (acidic, basic, polar, non-polar), the translation machinery became too complex to change. Adding a new one would have required re-coding the entire genome, which would be evolutionarily lethal.
4. Non-Natural Amino Acids (nAAs)
We can expand the genetic code. By engineering aminoacyl-tRNA synthetases, we can incorporate synthetic amino acids.
- Design Proposal: p-Azidophenylalanine (pAzF).
- Function: It contains an azide group (N3) that allows for Click Chemistry. This lets us chemically staple drugs or fluorescent dyes to a protein at a precise location that nature never intended.
5. Pre-Biotic Origins
Before life and enzymes, amino acids were produced through abiotic synthesis.
- The Miller-Urey Experiment: Demonstrated that simple gases (methane, ammonia, hydrogen) plus an energy source (lightning/sparks) could spontaneously generate glycine and alanine.
- Astrobiology: Analysis of the Murchison meteorite proved that amino acids can form in space via Strecker synthesis, suggesting the ingredients for life are ubiquitous in the solar system.
6. Handedness of D-amino Acid Helices
In biology, we use L-amino acids, which form right-handed α-helices. If you synthesize a peptide using D-amino acids (the mirror image), the resulting helix will be left-handed. The steric hindrance of the D-side chains makes a right-handed twist energetically impossible.
7. Discovering New Helices
Beyond the common α-helix, we find the Φ-helix and the π. We “discover” these by plotting the dihedral angles Ψ on a Ramachandran Plot. By using β-peptides (which have an extra carbon in the backbone), we can create entirely new foldamers with geometries that nature hasn’t explored.
8. Why the Right-Handed Preference?
It comes down to the L-configuration of the alpha-carbon. In a right-handed helix, the side chains (R-groups) point away from the centre, minimising steric clashes. In a left-handed helix made of L-amino acids, the side chains would bump into the backbone and each other, making the structure unstable.
9. β-sheet Aggregation
β-sheets are inherently sticky because they have hydrogen bond donors and acceptors along their edges that are exposed.
- Driving Force: The Hydrophobic Effect. When β-strands come together, they bury their oily (hydrophobic) side chains away from water. The formation of inter-strand hydrogen bonds then locks them into place. Stacking β-sheets together gives them a crystalline-like lattice.
10. Amyloids: From Disease to Materials
Amyloid plaques (associated with Alzheimer’s) are essentially β-sheets that have aggregated out of control.
- Utility: These structures are incredibly stable—stronger than steel in some cases. Scientists are now using amyloid-inspired β-sheets to create functional nanomaterials, such as conductive nanowires or ultra-stable drug-delivery scaffolds.
Part B: Protein Analysis and Visualization
1. Protein Selection: The Bacterial Buster
For this assignment, I chose Hen Egg-White Lysozyme (HEWL). I selected this protein because it is a classic example of structure-equals-function. It acts as a biological weapon by physically slicing through bacterial cell walls. It was also the first enzyme ever to have its 3D structure solved by X-ray crystallography, making it a landmark in biotechnology. It was discovered by Alexander Fleming (before he found penicillin) because he noticed his own nasal mucus could kill bacteria.
2. Sequence Analysis
- Amino Acid Sequence:
KVFGRCELAAAMKRHGLDNYRGYSLGNWVCAAKFESNFNTQATNRNTDGSTDYGILQINSRWWCNDGRTPGSRNLCNIPCSALLSSDITASVNCAKKIVSDGNGMNAWVAWRNRCKGTDVQAWIRGCRL - Length: 129 amino acids.
- The most Frequent Amino Acids are Asparagine (N), followed by Alanine (A) and Glycine (G), but the Cysteine (C) residues are the most structurally significant, as they form 4 disulfide “staples” that keep the protein stable.**
| Amino Acid | Count | Percentage |
|---|---|---|
| Asparagine (N) | 14 | 10.85% |
| Glycine (G) | 12 | 9.30% |
| Alanine (A) | 12 | 9.30% |
- Why Asparagine (N)? In the structure of Lysozyme, Asparagine is crucial for its function as a Bacterial Buster. Because Asparagine is excellent at forming hydrogen bonds, these 14 residues act like molecular velcro on the surface of the protein. They help the enzyme stick to the bacterial cell wall (peptidoglycan) so it can stay in place long enough to perform its catalytic cut.
Homologs and Evolutionary Relatives
Using the UniProt BLAST tool, I searched for sequences similar to my Lysozyme query.
- Homolog Count: The search returned over 250 homologs.
- Diverse Species: Homologs were found across a wide range of vertebrates, including:
- Birds: Quail (Colinus virginianus), Pheasants (Phasianus colchicus), and Turkeys (Meleagris gallopavo).
- Reptiles: Turtles (Chelydra serpentina) and Alligators (Alligator sinensis).
- Mammals: Humans (Homo sapiens), Gorillas, and even Milk isozymes in Cattle (Bos taurus).
Protein Family Classification
My protein belongs to the Glycosyl Hydrolase Family 22 (GH22). Members of this family are specialized enzymes that identify and break the $\beta(1\rightarrow4)$ glycosidic bonds in the peptidoglycan of bacterial cell walls. Essentially, being part of this family means the protein’s primary job is to act as a highly specific pair of molecular scissors.
3. Protein Structure & Bioinformatics Analysis: 1LZ1
a. RCSB PDB Structure Overview
I identified the structural data for my protein using the RCSB Protein Data Bank.
- PDB ID: 1LZ1
- Structure Title: Refinement of Human Lysozyme at 1.5 Angstroms Resolution.
- Release Date: The structure was officially released on 1985-01-02.
- Resolution: 1.50 Å.
- Quality Assessment: This is an excellent quality structure. Since 1.50 Å is significantly lower (better) than the standard 2.70 Å benchmark, it provides high-atomic detail, allowing us to see precise hydrogen-bond interactions.
b. Composition and Non-Protein Molecules
Apart from the protein chain, the solved crystal structure contains:
- Nitrate Ions ($NO_3^-$): Found in the crystallization buffer.
- Water ($H_2O$): Essential for understanding the protein’s stability in a liquid environment. meaning its biological function is to use water to break chemical bonds in sugars.
Structural Classification Analysis
I analyzed the structural hierarchy of my protein using the SCOP2 (Structural Classification of Proteins) database.
- PDB ID: 1LZ1
- SCOP Representative: 2NWD X
- Class: Alpha and beta proteins (a+b)
- Fold: Lysozyme-like
- Superfamily: Lysozyme-like
- Family: C-type lysozyme
Summary: My protein belongs to the C-type lysozyme family. This structural classification is significant because it groups my protein with other evolutionary relatives (like the chicken lysozyme found in my BLAST search) that share a specific Alpha+Beta fold. This specific shape is what creates the cleft or active site that allows the protein to function as a Hydrolase, breaking down bacterial cell walls. On the RCSB PDB page, the protein is formally classified as a HYDROLASE (O-GLYCOSYL), which confirms its mechanical family—enzymes that use a water molecule to break the sugar bonds in bacterial cell walls.
4. PyMOL Protein Analysis: Hen Egg-White Lysozyme (1LZ1)
a. Protein Visualizations
I visualized the Lysozyme protein using three different representation methods to understand its structure at various scales.
- Cartoon: Shows the overall 3D folding architecture and secondary structure flow.
- Ribbon: Simplifies the view by tracing only the polypeptide backbone.
- Ball and Stick (Sticks): Reveals the precise location of every atom and the chemical bonds connecting them.
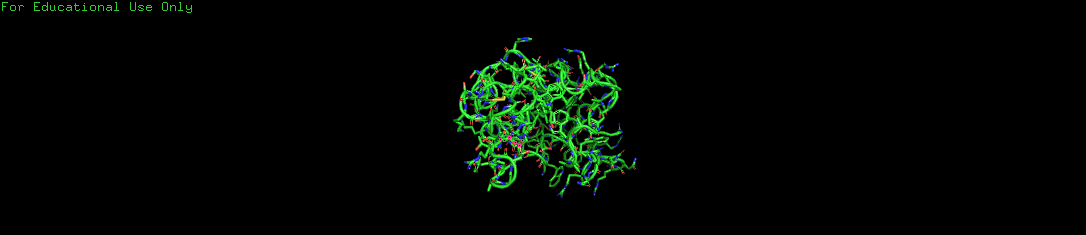
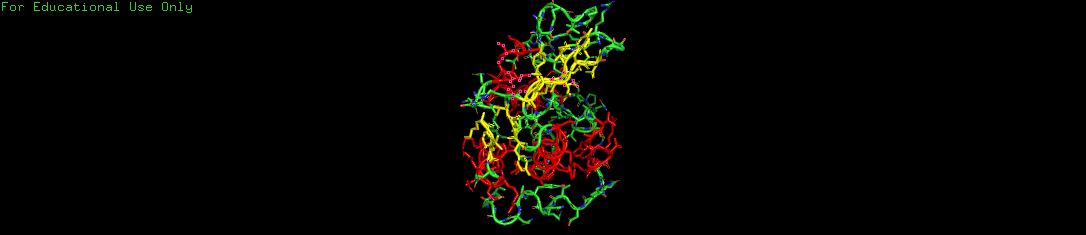
b. Secondary Structure: Helices vs. Sheets
By coloring the protein by its secondary structure, I analyzed the building blocks of its shape.
- Observation: The protein is dominated by alpha-helices (colored red).
- Analysis: Several prominent spiral helices form the core of the protein. In contrast, there is only one small anti-parallel beta-sheet (colored yellow) acting as a structural wing on the side.
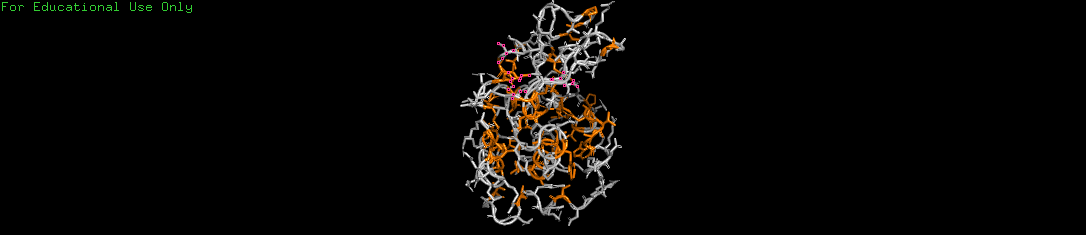
c. Residue Distribution: Hydrophobic vs. Hydrophilic
I colored the residues to see how the protein interacts with its watery environment in an egg white.
- Hydrophobic (Orange): These water-fearing residues are almost entirely tucked away inside the protein’s core.
- Hydrophilic (Gray): These water-loving residues dominate the outer surface.
- Conclusion: This follows the oil drop model of protein folding, where the hydrophobic core is shielded from water to maintain stability.
My PyMOL Protein Views
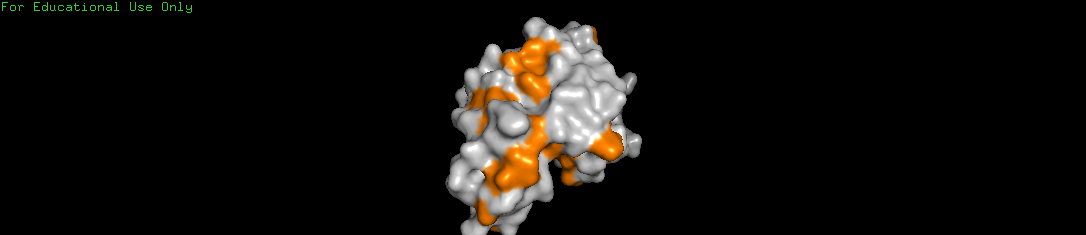
d. Surface Analysis and Binding Pockets
Visualizing the molecular surface allows us to see how the protein grabs its targets.
- Observation: The protein is not a solid sphere; it has a very distinct binding pocket.
- Analysis: A deep canyon or cleft is clearly visible cutting across the center of the molecule.
- Function: This hole is the active site where the lysozyme captures and breaks down the sugar chains (polysaccharides) of bacterial cell walls
 ---
---C1. Protein Language Modeling
In this section, I explored the capabilities of modern protein AI models using Bacteriorhodopsin (PDB: 1C3W) as a model system. Bacteriorhodopsin is a sophisticated light-driven proton pump found in Halobacterium salinarum, characterized by its iconic seven-transmembrane alpha-helical structure.
a. Deep Mutational Scanning with ESM2
Using the ESM2 language model (specifically the esm2_t6_8M_UR50D variant), I generated an unsupervised deep mutational scan of the 1C3W sequence. The model predicts the “fitness” of every possible single-point mutation by calculating the log-likelihood of each amino acid at every position in the sequence.
The Heatmap Analysis
The heatmap visualizes the model scores, where the x-axis represents the residue position and the y-axis represents the 20 standard amino acids:
- High Scores (Yellow/Light Green): Indicate mutations the AI predicts are favorable or neutral.
- Low Scores (Dark Purple/Blue): Indicate mutations predicted to be destabilizing or functionally detrimental.
Identifying a Standout Mutation
A particularly interesting pattern emerged at Position 168:
- The Observation: While many transmembrane residues are highly constrained (visible as dark vertical columns), position 168 shows a high tolerance for Proline (P), with a model score of 5.394987.
- The Interpretation: In the context of a 7-helix bundle, Proline usually acts as a helix breaker. However, the AI’s high score suggests that at this specific coordinate, the structural “kink” or rigidity introduced by Proline is actually beneficial for the protein’s native fold or its conformational light-cycle.
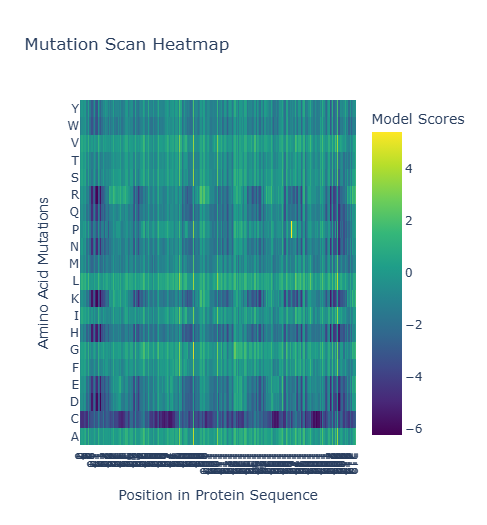
(Bonus) Experimental Comparison
Experimental data for Bacteriorhodopsin highlights critical residues like D85 and D96 as essential for proton transport. My ESM2 scan accurately reflects this: these positions appear as dark vertical stripes, meaning the language model assigned low likelihoods to almost all mutations at these sites. This demonstrates that the AI has learned functional biological constraints purely from evolutionary sequence data.
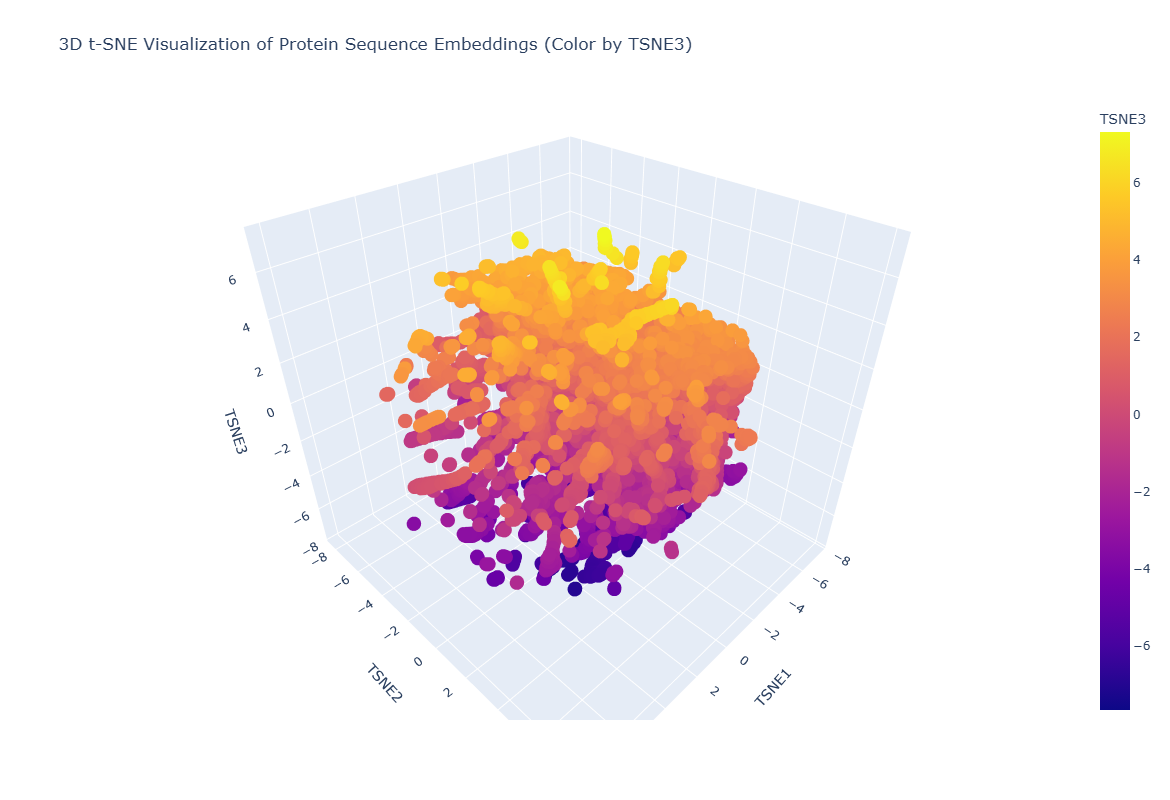
Latent Space Analysis: Mapping the Protein Universe
After processing 15,177 sequences from the ASTRAL dataset through the ESM2 transformer, I projected the resulting high-dimensional embeddings into a 3D latent space using t-SNE. This visualization allows us to see how the AI categorizes proteins without any human-labeled data.
Neighborhood Analysis: Structural Peer Groups
Looking at the 3D scatter plot, it is clear that the neighborhoods are not random. The clusters represent distinct structural architectures:
- The Neighborhoods: The map forms a dense central mass of globular, soluble proteins with distinct arms extending outward. These arms represent specialized folds, such as all-beta sheets or long alpha-helical bundles.
- Biological Logic: Proteins in the same neighborhood share similar biophysical properties. By hovering over the data points, I found that proteins clustered near my target are often involved in energy transduction or membrane transport.
1C3W Position & Neighborhood
I placed my protein, Bacteriorhodopsin (1C3W), into this map to see who its neighbors are.
- The Neighbors: My protein landed in a cluster populated by other transmembrane proteins, such as Vacuolar ATP synthase subunits (visible in my analysis as the yellow cluster).
- Position Significance: 1C3W sits in a specialized island on the periphery of the main protein cloud. This position is highly significant because it reflects the protein’s hydrophobic nature.
- Conclusion: The AI successfully grouped Bacteriorhodopsin with other membrane-embedded proton pumps and synthases. Even though the sequence identity might be low, the model recognizes the shared “structural grammar” required to span a lipid bilayer. This proves that the ESM2 latent space effectively approximates biological function and fold-topology purely from sequence data.
C2. Protein Folding with ESMFold
In this stage, I used ESMFold to predict the 3D atomic structure of Bacteriorhodopsin (1C3W) directly from its amino acid sequence. This test determines if the AI can accurately recreate the physical geometry of a complex membrane protein.
Fold Results & Structural Accuracy
The ESMFold prediction was highly successful. The model generated a clear, seven-transmembrane alpha-helical bundle that aligns almost perfectly with the original experimental structure from the PDB.
- The Verdict: The predicted coordinates match the original structure with high confidence. The AI correctly identified the hydrophobic nature of the sequence and packed the helices into the characteristic barrel shape required for its function as a proton pump.

Sequence Resilience & Mutation Testing
I performed two separate “stress tests” on the sequence to see how much change the structure could tolerate before it collapsed.
1. Small Mutations (The Point Test)
I first introduced minor point mutations into the loop regions of the protein.
- Observation: The protein was remarkably resilient. The overall 7-helix bundle remained intact, with only tiny shifts in the flexible loops. This shows the fold is robust against minor “noise” in non-structural areas.

2. Large Segments (The Collapse Test)
I then replaced a large, 20-residue segment of a core transmembrane helix with flexible Glycines to break the structural pillar.
- Observation: The structure was not resilient to this change. The helical bundle was significantly distorted, and the parallel arrangement of the “barrel” caved in.
- Conclusion: Bacteriorhodopsin is resilient to surface-level mutations but highly dependent on the integrity of its transmembrane helices. The “grammar” of this protein requires these rigid pillars to stay upright; once a pillar is removed, the entire architecture fails.

–
C3. Protein Generation (Inverse Folding)
In the final stage of my project, I moved beyond studying natural proteins to De Novo Design. I used ProteinMPNN to perform Inverse Folding the process of providing the AI with a fixed 3D backbone and asking it to “dream up” a brand-new amino acid sequence that would stabilize that specific shape.
The Inverse-Folding Process
- The Blueprint: I provided the high-confidence 3D coordinates (PDB file) of my Bacteriorhodopsin fold as the structural input.
- Sequence Analysis: The model generated several candidate sequences. My top-ranked design had a Sequence Recovery of 47.3%.
- Observations: This means that the AI completely redesigned 52.7% of the protein’s sequence. While the “letters” changed significantly, the predicted sequence probabilities remained high for residues that maintain the hydrophobic core of the helices.
`> Generating sequences…
tmp, score=1.6136, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020 TGRPEWIWLALGTALMGLGTLYFLVKGMGVSDPDAKKFYAITTLVPAIAFTMYLSMLLGYGLTMVPFGGEQNPIYWARYADWLFTTPLLLLDLALLVDADQGTILALVGADGIMIGTGLVGALTKVYSYRFVWWAISTAAMLYILYVLFFGFSMRPEVASTFKVLRNVTVVLWSAYPVVWLIGSEGAGIVPLNIETLLFMVLDVSAKVGFGLILLRSRAIFG T=0.1, sample=0, score=0.9199, seq_recovery=0.4730 APDLSKPWWAIGTIVFLLGTIFFAVRGLLVTDPRARVFYILCTLIPLIMFICYLAILLGFGWVLVPVDGKLRKVPVARYVGWLLTGPLILLCLARLVDAPAGLVALLVALWLVSVLTALLGALSTVPPLRFVFLAISTAALLLILAILLFGFTLDPRVRPTYLVLVALFVVLWLLYPVVLLLGPEGLNVIPLHVFEALVTVLDILLTVGFGLILVSSPAIFS T=0.1, sample=0, score=0.9118, seq_recovery=0.4459 APRLWAPWVALGTAVMAAGAVYFAARGARVTDPRARWFYVLATLIPLIMAVCFLAILLGLGVVLVPKDGKLRPIPVMLFVGWLLTGPLILLCLARLVDASPALIALLVALWVIAVLSALIGALSTIPPLRFVFLAISTLALLIILYILLFGFTLDPRVRPTYLVLVTLFIILWSLYPIILLLGPFGLNLIPLSVFMALITVLDILLTVGFGLILLASPAIRA
New Sequence:APDLSKPWWAIGTIVFLLGTIFFAVRGLLVTDPRARVFYILCTLIPLIMFICYLAILLGFGWVLVPVDGKLRKVPVARYVGWLLTGPLILLCLARLVDAPAGLVALLVALWLVSVLTALLGALSTVPPLRFVFLAISTAALLLILAILLFGFTLDPRVRPTYLVLVALFVVLWLLYPVVLLLGPEGLNVIPLHVFEALVTVLDILLTVGFGLILVSSPAIFS`
Validation: AI Sequence vs. Natural Shape
To prove the design worked, I took the AI-generated sequence (APDLSKPWWAIGTIVFLLGTIFFAVRGLLVTDPRARVFYILCTLIPLIMFICYLAILLGFGWVLVPVDGKLRKVPVARYVGWLLTGPLILLCLARLVDAPAGLVALLVALWLVSVLTALLGALSTVPPLRFVFLAISTAALLLILAILLFGFTLDPRVRPTYLVLVALFVVLWLLYPVVLLLGPEGLNVIPLHVFEALVTVLDILLTVGFGLILVSSPAIFS) and fed it back into ESMFold to see if it would still form the seven-transmembrane bundle.
- The Result: The validation was a total success. Despite being less than half-identical to the natural sequence found in nature, the synthetic sequence folded into the identical 7-helix architecture.
- Comparison: The predicted structure for the synthetic sequence matches the original 1C3W backbone almost perfectly, demonstrating that the AI successfully captured the “structural grammar” of the protein.

Final Project Conclusion
This journey from sequence analysis to de novo design highlights a fundamental principle of modern bioengineering: Structure is more conserved than sequence. Through this lab, I have demonstrated that:
- Language Models (ESM2) can organize the protein universe by structural similarity without being explicitly taught physics.
- Folding models (ESMFold) can accurately predict complex transmembrane architectures.
- Inverse-folding models (ProteinMPNN) allow us to design entirely new, non-natural sequences that fulfill specific geometric goals.
This capability is the cornerstone of the next generation of drug discovery and synthetic biology, allowing us to build custom molecular machines from the ground up.
link to protein language modeling work: https://colab.research.google.com/drive/1TyJ7DqysYyLd2P1MPcW8_aDekQIB6x07?usp=sharing
HTGAA 2026: Bacteriophage Engineering Project
Topic: Engineering the MS2 L-Protein for Enhanced Lytic Kinetics
1. Project Goals
Our team is focusing on two primary engineering objectives for the MS2 bacteriophage L-protein:
- Increased Toxicity (Hard): Optimize lytic kinetics to trigger faster host cell lysis by bypassing the DnaJ-dependent “damping” mechanism.
- Increased Stability (Easy): Redesign the N-terminal and transmembrane domains to prevent proteolytic degradation, ensuring robust protein accumulation.
2. Proposed Computational Pipeline
Step 1: Generative Sequence Design (Evo 2)
- Approach: We will utilize the Evo 2 genome language model to generate a library of novel MS2 L variants. We will specifically prompt the model to design “L-odj-like” variants (L-overcomes-DnaJ) by modifying the N-terminal Domain 1.
- Reasoning: Evo 2 can navigate novel evolutionary spaces beyond the 67 unique mutations identified in natural screens, accessing sequence diversity that purely experimental methods might miss.
Step 2: Sequence Stability Optimization (ProteinMPNN)
- Approach: Use ProteinMPNN to perform inverse folding on the core Transmembrane Domain (TMD) of the generated candidates.
- Reasoning: ProteinMPNN redesigns sequences to fit the specific 3D backbone required for membrane insertion while optimizing for thermodynamic stability, preventing accumulation defects.
Step 3: Functional Motif Tuning (ESM-2 / ESM-3)
- Approach: Use ESM-2/3 protein language models to extract embeddings and perform in silico mutagenesis on the essential Leu48-Ser49 (LS) motif.
- Reasoning: ESM models identify which substitutions in the surrounding Domain 2 and Domain 4 preserve the critical hydrophobic and polar character necessary for function.
Step 4: Oligomerization Verification (AlphaFold-Multimer)
- Approach: Use AlphaFold-Multimer to predict the ability of designed variants to assemble into high-order oligomeric complexes (decamers or higher).
- Reasoning: MS2 L must form large membrane-disrupting clusters. This step validates if mutations at the TMD interface promote or hinder essential assembly.
3. Pipeline Schematic: From Sequence to Pore
To engineer the MS2 L-protein, we utilize a tiered computational pipeline. This workflow moves from broad “sequence discovery” to high-resolution “structural validation,” ensuring each candidate is both stable and functional before experimental testing.
Phase 1: Sequence Discovery via Evo 2
The Architect We initiate the pipeline using Evo 2, a genomic-scale language model. By providing the MS2 genome as context, we prompt the model to generate novel L-protein sequences. Unlike traditional mutagenesis, Evo 2 identifies long-range dependencies within the genome, allowing us to design “L-odj” (overcomes DnaJ) variants that can bypass host inhibitory mechanisms while maintaining the integrity of the viral life cycle.
Phase 2: Stability Refinement via ProteinMPNN
The Reinforcer Generative models can sometimes produce “orphan” sequences that are theoretically toxic but physically unstable. We use ProteinMPNN to perform inverse folding on the Transmembrane Domain (TMD). By fixing the 3D backbone required for membrane insertion and “redesigning” the amino acid side chains, we maximize the thermodynamic stability of the protein. This ensures the L-protein accumulates in the E. coli membrane rather than being degraded by host proteases.
Phase 3: Functional Filtering via ESM-2/3
The Evaluator To ensure our redesigned sequences haven’t lost their “killing power,” we use ESM-2/3 (Evolutionary Scale Models). We extract embeddings to perform zero-shot fitness predictions, specifically focusing on the essential Leu48-Ser49 (LS) motif. This step acts as a filter: any sequence that deviates from the hydrophobic and polar requirements of the LS-motif—the core engine of MS2-induced lysis—is discarded.
Phase 4: Quaternary Validation via AlphaFold-Multimer
The Gatekeeper The final and most rigorous check involves AlphaFold-Multimer. MS2 L-protein does not work in isolation; it must oligomerize into high-order clusters (likely decamers) to create a pore large enough for cytoplasmic leakage. We model the top 10 candidates in a 10-mer configuration to verify that our mutations haven’t disrupted the protein-protein interfaces required for assembly. Only candidates that show a stable, pore-forming geometry are selected for synthesis.
4. Potential Pitfalls
The Suicide Problem
If our engineered L protein is too toxic and bypasses DnaJ entirely, it might lyse the E. coli before the phage has finished replicating its genome. This would result in “lysis from without” but zero phage progeny, making the engineering a failure for phage therapy applications.
- Membrane Complexity: Most of these tools (like AlphaFold and ProteinMPNN) were trained on soluble proteins. Modeling a protein that lives entirely inside a lipid bilayer is computationally noisy, and the predicted oligomers might not behave the same way in a real, pressurized bacterial membrane.
References
- Nelson, D. L., & Cox, M. M. (2021). Lehninger Principles of Biochemistry. 8th Ed.
- Miller, S. L. (1953). “A Production of Amino Acids Under Possible Primitive Earth Conditions.” Science.
- Dobson, C. M. (2003). “Protein folding and misfolding.” Nature.
- Crick, F. H. (1968). “The origin of the genetic code.” Journal of Molecular Biology.